
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceType I macrophage activator photosensitizer against hypoxic tumors†
Guang
Yang‡
,
Song-Bo
Lu‡
 ,
Chong
Li
,
Feng
Chen
,
Jen-Shyang
Ni
,
Menglei
Zha
,
Yaxi
Li
,
Ji
Gao
,
Tianyi
Kang
,
Chao
Liu
and
Kai
Li
*
,
Chong
Li
,
Feng
Chen
,
Jen-Shyang
Ni
,
Menglei
Zha
,
Yaxi
Li
,
Ji
Gao
,
Tianyi
Kang
,
Chao
Liu
and
Kai
Li
*
Shenzhen Key Laboratory of Smart Healthcare Engineering, Department of Biomedical Engineering, Southern University of Science and Technology (SUSTech), Shenzhen 518055, China. E-mail: lik@sustech.edu.cn
First published on 20th October 2021
Abstract
Photodynamic immunotherapy has emerged as a promising strategy to treat cancer. However, the hypoxic nature of most solid tumors and notoriously immunosuppressive tumor microenvironment could greatly compromise the efficacy of photodynamic immunotherapy. To address this challenge, we rationally synthesized a type I photosensitizer of TPA-DCR nanoparticles (NPs) with aggregation-enhanced reactive oxygen species generation via an oxygen-independent pathway. We demonstrated that the free radicals produced by TPA-DCR NPs could reprogram M0 and M2 macrophages into an anti-tumor state, which is not restricted by the hypoxic conditions. The activated M1 macrophages could further induce the immunogenic cell death of cancer cells by secreting pro-inflammatory cytokines and phagocytosis. In addition, in vivo anti-tumor experiments revealed that the TPA-DCR NPs could further trigger tumor immune response by re-educating tumor-associated macrophages toward M1 phenotype and promoting T cell infiltration. Overall, this work demonstrates the design of type I organic photosensitizers and mechanistic investigation of their superior anti-tumor efficacy. The results will benefit the exploration of advanced strategies to regulate the tumor microenvironment for effective photodynamic immunotherapy against hypoxic tumors.
Introduction
Photodynamic immunotherapy is an emerging anti-cancer therapeutic strategy with low systemic toxicity and the absence of initial resistance, which harnesses the innate immune system to attack tumor cells.1–4 In general, the reactive oxygen species (ROS) that are generated during photodynamic therapy (PDT) can induce the immunogenic cell death (ICD) of tumor cells and activate the immune cells, triggering the immune response in the tumor microenvironment.5–8 However, the efficacy of photodynamic immunotherapy has always been limited by the harsh immunosuppressive nature of the tumor microenvironment. For instance, tumor-associated macrophage (TAM) is one of the critical drivers of an immunosuppressive tumor microenvironment, as it acts as a powerhouse for tumor angiogenesis and metastasis by secreting pro-tumor cytokines.9–14 In addition, the hypoxic conditions in solid tumors are another important restriction in realizing desired efficacy of photodynamic immunotherapy, because PDT processes are always oxygen-dependent and require oxygenated conditions.15–18 Therefore, there remains an urgent demand for rationally designed photosensitizers, which can efficiently generate ROS under hypoxic conditions and induce the polarization of TAMs towards anti-tumor phenotypes (M1), to improve the outcome of advanced photodynamic immunotherapy.Currently, it is still extremely challenging to overcome both immunosuppression and hypoxia in the tumor microenvironment using photodynamic immunotherapy, due to the lack of mechanistic understanding and shortage of strategic guidelines. The mechanism of PDT can be generally classified into two categories based on different photochemical reaction processes: type I and type II.19–23 In the type I pathway, hydrogen abstraction and electron transfer between photosensitizers (PSs) in the first excited triplet state (T1) and biological substrates may occur to produce free radicals, which can interact with oxygen and water to produce superoxide anions (˙O2−) and hydroxyl radicals (˙OH), respectively.24–27 During the type II photochemical reaction, PSs in the T1 state convert the surrounding 3O2 into cytotoxic singlet oxygen (1O2) via direct energy transfer.28,29 Therefore, type II PDT requires well-oxygenated environments, while type I PDT can be operated under hypoxic conditions for a broader range of application scenarios. Despite the successful commercialization of several organic PSs (e.g., rose bengal and chlorin e6), most of the reported organic PSs generally favor type II PDT, because they do not possess the feature of inorganic counterparts that can facilitate the generation of electron–hole pairs to induce charge separation as in the type I pathway.30,31 Therefore, the exploration of highly efficient organic type I PSs and elucidation of their working mechanism in hypoxic tumor environments will be of great benefit to the future development of photodynamic immunotherapy.
Herein, we have rationally designed and synthesized an aggregation-induced emission (AIE) photosensitizer, TPA-DCR, with a donor–acceptor (D–A) structure. The photosensitizer has shown aggregation-enhanced ROS generation upon light irradiation, mainly via the oxygen-independent type I mechanism. Theoretical calculation indicated that the small energy barrier between singlet and triplet states (ΔEST) may favor the formation of highly active T3 and T4 excitons in excited TPA-DCR molecules, thereby benefiting the electron transfer process to produce type I ROS. More importantly, we have demonstrated that the TPA-DCR nanoparticles (NPs) could effectively polarize the M0 and M2 macrophages toward the M1 phenotype under hypoxic conditions, leading to efficient killing of the 4T1 cancer cells through phagocytosis and secretion of pro-inflammatory cytokines. Interestingly, unlike traditional ICD caused by PDT, we discovered that the activated M1 macrophages could upregulate the expression levels of damage-associated molecular patterns (DAMPs) in 4T1 cells undergoing ICD. As a result, extensive in vivo experiments showed that our macrophage-mediated photodynamic immunotherapeutic strategy could induce the ICD of hypoxic tumor tissues and promote the infiltration of T cells, thereby significantly inhibiting tumor growth in a mice model.
Results and discussion
Synthesis and characterization of the photosensitizer
First, we designed and synthesized a donor–acceptor structured AIEgen, 2-(5-(4-(bis(phenyl)amino)benzylidene)-4-oxo-3-phenylthiazolidin-2-ylidene)malononitrile (TPA-DCR), in a one-step strategy. The structure of TPA-DCR was confirmed by molecular characterization in the ESI (Scheme S1 and Fig. S1–S3†). The photophysical results in tetrahydrofuran (THF) showed that TPA-DCR exhibited the main absorption and emission peaks at 460 nm and 608 nm, respectively (Fig. 1a). To better understand the photophysical properties of TPA-DCR, we carried out further theoretical calculation by using Gaussian 16 (ref. 32) with time-dependent density functional theory (TD-DFT) at the level of PBE1PBE/6-31G(d). The distribution of the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of TPA-DCR in the S1 state (Fig. 1b) indicated an energy gap of 2.24 eV. In addition, the excited geometries of TPA-DCR pointed to its twisted structure, having a dihedral angle of 1.87° between the hexatomic ring and five-membered ring (Fig. S4†). As an important indicator to evaluate the feasibility of the ISC process, the ΔEST values of TPA-DCR were calculated and are shown in Fig. 1c. The results indicated small ΔEST values (<0.3 eV) of TPA-DCR of S1T3 and S1T4 ISC channels. Based on Kohn–Sham frontier orbital analysis, the ISC process is more vulnerable to be triggered when the energy gap is below 0.3 eV.33 As a result, the twisted structure of TPA-DCR can result in small ΔEST values in the excited state, ensuring good ROS-generating ability. Especially, the ΔEST values of S1T3 and S1T4 channels are 0.191 eV and 0.161 eV, respectively. According to the literature, type I ROS can be generated after the occurrence of intersystem crossing (ISC) processes between S1 and high-level triplet states.20 Thus, the formation of highly active T3 and T4 excitons is promoted in excited TPA-DCR molecules, which might benefit electron transfer processes for type I ROS generation. In addition, the T1–S0 energy gap of TPA-DCR is lower than the energy required to produce 1O2 through excitation energy transfer (0.651 versus 1.12 eV).34 Therefore, the type I mechanism dominates during the ROS generation of TPA-DCR.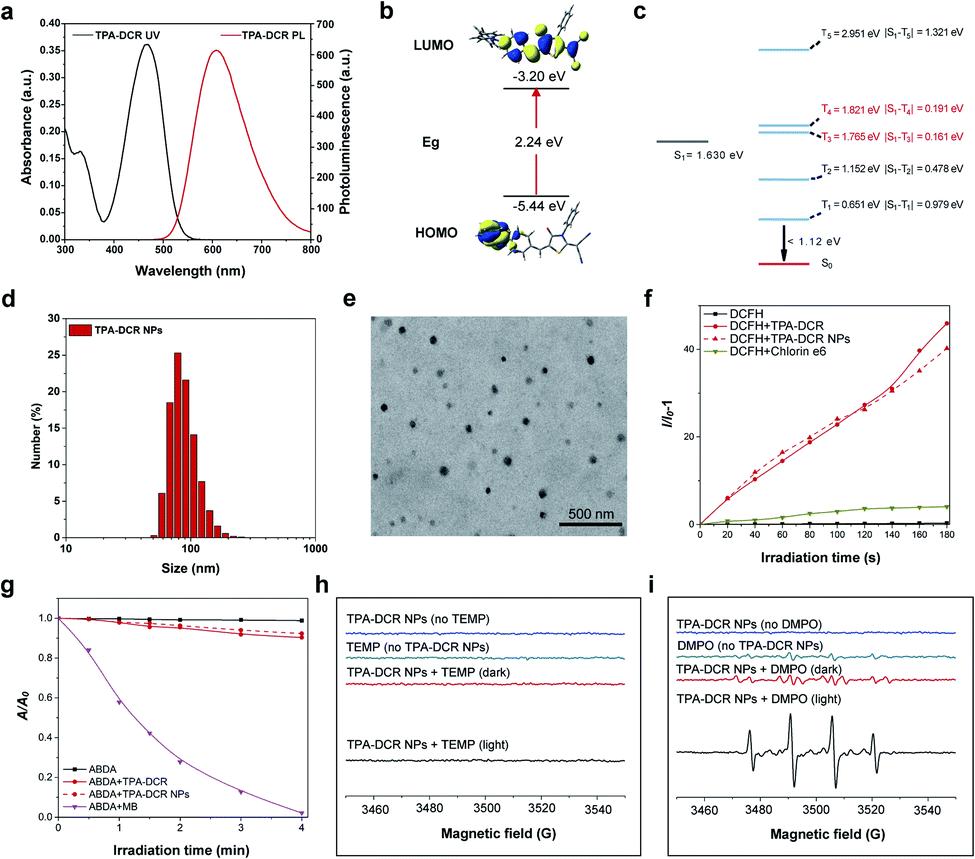 | ||
| Fig. 1 (a) Ultraviolet-visible (UV-Vis) and photoluminescence (PL) spectra of TPA-DCR in THF. (b) The HOMO–LUMO distribution of TPA-DCR calculated by TD-DFT at the level of PBE1PBE/6-31G(d). (c) Relative energy levels of TPA-DCR calculated by TD-DFT. S1, S0 and Tn represent the first-singlet, ground and multi-triplet states, respectively. (d) DLS and (e) TEM results of TPA-DCR NPs, and the scale bar is 500 nm. (f) Time-dependent changes of fluorescence intensities (I/I0 − 1) at 525 nm for the DCFH indicator with TPA-DCR aggregates (90% PBS fraction in a PBS/THF mixture) and NPs, using the commercial photosensitizer chlorin e6 as a benchmark. (g) Time-dependent changes of absorption intensities (A/A0) at 379 nm for ABDA mixed with methylene blue (MB), TPA-DCR aggregates (90% water fraction in a H2O/THF mixture) and TPA-DCR NPs. EPR spectra of TPA-DCR NPs (1 mM) using (h) TEMP (100 mM) and (i) DMPO (100 mM) as the indicators. The white light irradiation of light-treated groups was conducted for 2 min at 40 mW cm−2. | ||
The AIE characteristic of TPA-DCR was verified by measuring the fluorescence spectra in THF-water mixtures. The results indicated that TPA-DCR was faintly emissive with water fractions below 70%, while the emission intensity started to increase upon increasing the water fraction to 90% (Fig. S5†). However, TPA-DCR could barely produce ROS in THF solution with 0 vol% of PBS, but promoted ROS generation gradually with increased PBS fractions along with the formation of aggregates (Fig. S6†). To improve the biocompatibility of the photosensitizer for biomedical applications, TPA-DCR was further encapsulated in DSPE-PEG2000 to create water-dispersible TPA-DCR NPs by nanoprecipitation following our established protocols.35 The size and shape of these NPs were then measured using dynamic light scattering (DLS) and transmission electron microscopy (TEM). These NPs had an average hydrodynamic diameter of 80–90 nm with a spherical shape (Fig. 1d and e). The UV-Vis absorption and PL spectra of TPA-DCR NPs were similar to those of the THF solution (Fig. S7†). The time-resolved fluorescence spectra showed that the fluorescence lifetime of TPA-DCR aggregates (H2O/THF = 9/1) and TPA-DCR NPs (in water) were 2.33 ns and 2.02 ns, respectively (Fig. S8†). The fluorescence quantum yield (QY) of TPA-DCR NPs was determined to be 29.3% using rhodamine B in ethanol (97%) as the standard (Fig. S9†),36 suggesting their suitability in in vivo imaging evaluation.
After confirming the optical properties of TPA-DCR NPs, their total ROS, type II ROS and type I ROS production rates were then measured using the indicators of 2′,7′-dichlorodihydrofluo-rescein (DCFH), 2,2′-(anthracene-9,10-diylbis(methylene))dimalonic acid (ABDA) and 3′-p-(hydroxyphenyl)fluorescein (HPF), respectively. The total ROS-generating rate from TPA-DCR NPs was ∼10-fold that of the commercial photosensitizer of chlorin e6 (Fig. 1f). In contrast, the type II ROS produced under the same experimental conditions was only 10% of that generated from commercial methylene blue (MB, singlet oxygen quantum yield of 52%) (Fig. 1g). Meanwhile, TPA-DCR can generate a large number of free radicals upon light irradiation, using HPF as the indicator (Fig. S10†).37,38 We further characterized the in vitro ROS generation capability of the photosensitizer by electron paramagnetic resonance (EPR) spectroscopy, using 2,2,6,6-tetramethyl-4-piperidone (TEMP) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as spin-trap agents for 1O2 and ˙OH, respectively. As shown in Fig. 1h, the acquired EPR spectrum showed no signal in the presence of TEMP and TPA-DCR NPs under white light irradiation (40 mW cm−2) for 2 min, indicating negligible generation of 1O2 from the type II mechanism. On the other hand, the spectrum of DMPO + TPA-DCR NPs irradiated under white light for 2 min showed obvious EPR signals from the DMPO/˙OH adduct (Fig. 1i), displaying four-line resonances with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 intensity (small fluctuations on the baseline indicate the signal of oxidized DMPO). Under both conditions, no obvious ROS signals could be observed in other control groups. These data suggested that the TPA-DCR NPs generated free radicals mainly via the type I PDT mechanism upon light irradiation, with negligible ability to produce singlet oxygen.
:2:2:1 intensity (small fluctuations on the baseline indicate the signal of oxidized DMPO). Under both conditions, no obvious ROS signals could be observed in other control groups. These data suggested that the TPA-DCR NPs generated free radicals mainly via the type I PDT mechanism upon light irradiation, with negligible ability to produce singlet oxygen.
In vitro anti-tumor effect of activated macrophages
Prior to cell and animal experiments, the cytotoxicity of TPA-DCR NPs towards macrophages was first evaluated at varied concentrations on RAW264.7 cells using the CCK-8 assay. The results clearly indicated that TPA-DCR NPs had no significant light and dark cytotoxicity (Fig. S11†). In previous work, we designed an AIE photosensitizer that could efficiently activate macrophages by generating type I ROS.39 Herein, we analyzed the macrophage activation by TPA-DCR NPs using ELISA (Fig. S12 and S13†), immunofluorescence staining (Fig. S14†) and western blotting (Fig. S15†). In addition, we have examined the expression level of the pro-inflammatory cytokines of TNF-α from M0 and M2 macrophages treated with PBS, TPA-DCR NPs, PBS + light, and TPA-DCR NPs + light, showing significantly higher TNF-α expression (Fig. S16†). These data collectively confirmed that TPA-DCR NPs could polarize M0 and M2 macrophages to M1 phenotypes via activating the NF-κB signaling pathway.Considering that most tumor microenvironments are hypoxic,40,41 we further investigated the validity and efficacy of our strategy to employ a type I photosensitizer for triggering macrophage activation under similar conditions. To establish the hypoxic environment during the polarization of M0 and M2 macrophages, the TPA-DCR NP-supplemented culture medium was charged with argon to expel oxygen before light irradiation. Therefore, the sample was under anaerobic conditions during the light irradiation for 3 min. After the light irradiation, the aerobic medium was replaced by a normal culture medium and the cells were cultured for 24 h under normoxic condition before ELISA analysis. The results showed that the efficacy of TPA-DCR NPs in activating M0 and M2 macrophages toward M1 remains consistent under hypoxic and aerobic conditions (Fig. 2a, b and S17†), suggesting that the TPA-DCR NPs are not oxygen-dependent owing to their type I PDT signature.
In order to further verify the effectiveness of activated M1 macrophages in killing tumor cells, transwell chambers (0.3 μm), which only allow cytokines to pass (Fig. 2c), were used to analyze the anti-tumor effect of pro-inflammatory cytokines produced by M1 macrophages. First, RAW264.7 cells were added in the lower layer after being treated with TPA-DCR NPs and light (10 mW−2 white light, 3 min) for activation. The 4T1 cells were subsequentially added to the upper layer of M0 (RAW264.7), M1 macrophages (RAW264.7 pre-treated with TPA-DCR NPs and light for polarization), and only TPA-DCR NPs. The cell viability of 4T1 cells was subsequentially measured by CCK-8 after 24 h. The results indicate that ∼40% of the 4T1 cells in the upper layer were killed by soluble factors (including pro-inflammatory cytokines) secreted by the activated macrophages. But there was no death in 4T1 cells treated with only M0 macrophages or those treated with TPA-DCR NPs + light irradiation in the absence of macrophages (Fig. 2d). Then, we chose transwell chambers with a larger pore size (0.6 μm, allowing cells to pass) to verify whether the TPA-DCR NP-activated macrophages could phagocytize 4T1 cells. To facilitate the study of cellular interactions, the 4T1 cells were prelabeled with a green-emissive fluorophore BTPETD, while M0 or M1 macrophages were prelabeled with a red-emissive fluorophore TPETPAFN.42 The 4T1 cells were added to the lower layer while M0 and M1 macrophages (RAW264.7 pre-treated with TPA-DCR NPs) or a blank sample was added to the upper layer. After 24 h, the cells in the lower layer from all three groups were imaged under a confocal microscope. Fluorescence imaging results indicated that M1 macrophages could phagocytize 4T1 cells with a clear overlay of red and green fluorescence signals. On the other hand, only green fluorescence from 4T1 cells could be observed in the other two groups (4T1 and 4T1 + M0), suggesting that the absence of phagocytosis by macrophages (Fig. 2e). According to the above experiments, we conclude that macrophages could kill tumor cells through phagocytizing and secreting soluble factors (including pro-inflammatory cytokines) that can pass the transwell barriers after activation by ROS from light-irradiated TPA-DCR NPs.
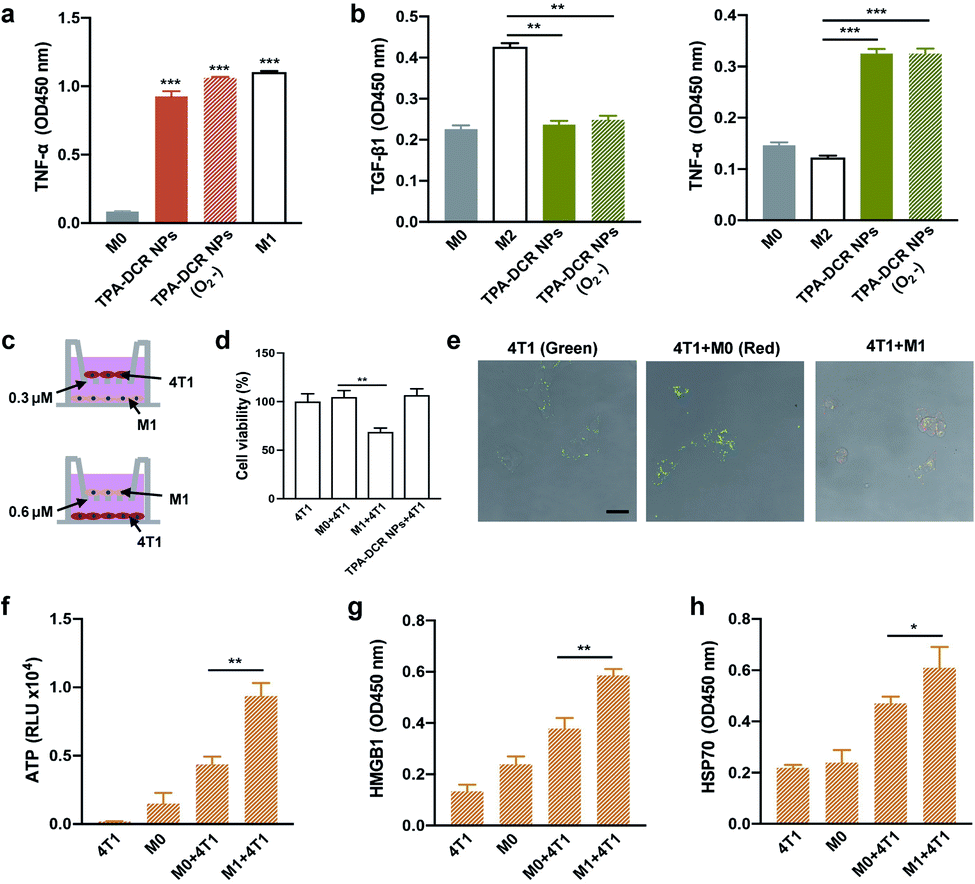 | ||
| Fig. 2 (a) The level of proinflammatory cytokine TNF-α secreted by M0 macrophages upon activation by TPA-DCR NPs and light under aerobic or hypoxic conditions (***p < 0.001 vs. the M0 group, n = 3). (b) The level of anti-inflammatory cytokine TGF-β1 and proinflammatory cytokine TNF-α secreted by M2 macrophages upon activation by TPA-DCR NPs and light under aerobic or hypoxic conditions, **p < 0.01, ***p < 0.001 vs. the M2 group, and n = 3. (c) Schematic illustration of the transwell experimental procedures. (d) Cell viability of 4T1 cells treated with M0 macrophages, TPA-DCR NP-activated M1 macrophages or TPA-DCR NPs and light (**p < 0.01 vs. the M0 + 4T1 group, n = 3). (e) The phagocytosis effect of M1 macrophages on 4T1 cells as detected by confocal microscopy, and the scale bar is 25 μm. ELISA measurement of the expression levels of (f) ATP, (g) HMGB1 and (h) HSP70 in 4T1 cells treated with M0 macrophages or TPA-DCR NP-activated M1 macrophages (*p < 0.05, **p < 0.01 vs. the M0 + 4T1 group, and n = 3). The concentration of TPA-DCR NPs is 50 μM, and white light irradiation is set to be 10 mW cm−2 for 3 min. | ||
ICD can transform tumor cells from being non-immunogenic to immunogenic by releasing damage-associated molecular patterns (DAMPs) that promote antigen identification and uptake, further enhancing the immune response.43–46 Taking into account the profound ability of activated M1 macrophages towards killing 4T1 cells, we wondered whether the M1 macrophages that were activated in our strategy could cause the ICD of tumor cells. To answer this question, the DAMPs (ICD markers, e.g., ATP, HMGB1, and HSP70) of 4T1 cells were investigated after being treated with activated M1 macrophages. The 4T1 cells were added to M0 (pre-treated with PBS) and M1 (pre-treated with TPA-DCR NPs + light) macrophages. After 24 h, the ATP in the supernatant and the proteins (HMGB1 and HSP70) in the cell lysate were analyzed by ELISA. Remarkably, the expression levels of secreted ATP from the M1 + 4T1 group were 2-fold higher than those from the M0 + 4T1 group (Fig. 2f). In contrast, the expression levels of HMGB1 (Fig. 2g) and HSP70 (Fig. 2h) in the M1 + 4T1 group were significantly higher than those in the M0 + 4T1 group. Together, these data clearly indicated that M1 macrophages activated with TPA-DCR NPs could induce the ICD of 4T1 tumor cells, which would potentially benefit anti-cancer efficacy in vivo for better tumor elimination.
In vivo anti-tumor effect of photodynamic immunotherapy
Encouraged by the findings of our in vitro studies, we evaluated the in vivo antitumor effect of TPA-DCR NPs in tumor-bearing mice. Firstly, a 4T1 subcutaneous tumor model was established, and the tumor-bearing mice were randomly divided into six groups (n = 5) (Fig. 3a). The hypoxic tumor environment was confirmed by immunofluorescence staining, which showed high expression levels of hypoxia-associated protein HIF-1α in the tumor tissue (Fig. 3b). The mice were intravenously injected with PBS (group i and group ii) or TPA-DCR NPs (groups iii, iv, v, and vi), and the groups ii, iv, vi were treated with light irradiation. To evaluate the role of macrophages in photodynamic immunotherapy, the mice from group v and vi were pre-treated with clodronate liposomes to deplete macrophages 24 h before injecting TPA-DCR NPs. According to in vivo fluorescence imaging results (Fig. S18 and S19†), mice were treated with or without white light irradiation at 12 h post injection to maximize the accumulation of NPs in tumor tissues. Tumor volumes were recorded every two days after treatments, demonstrating that the treatment with TPA-DCR NPs + light in group iv could almost completely eliminate the tumors. However, after depleting macrophages, the anti-tumor effect of TPA-DCR NPs in group vi was completely reversed and greatly compromised (Fig. 3c–e). This could be attributed to the fact that the sole ROS generated from PDT could not eliminate all tumor cells in the absence of macrophages, without stimulating the immune response in the tumor microenvironment. Hematoxylin and eosin (H&E) staining results also revealed that the tumor tissues suffered the most damage after treatment with TPA-DCR NPs + light (group iv) (Fig. 3f).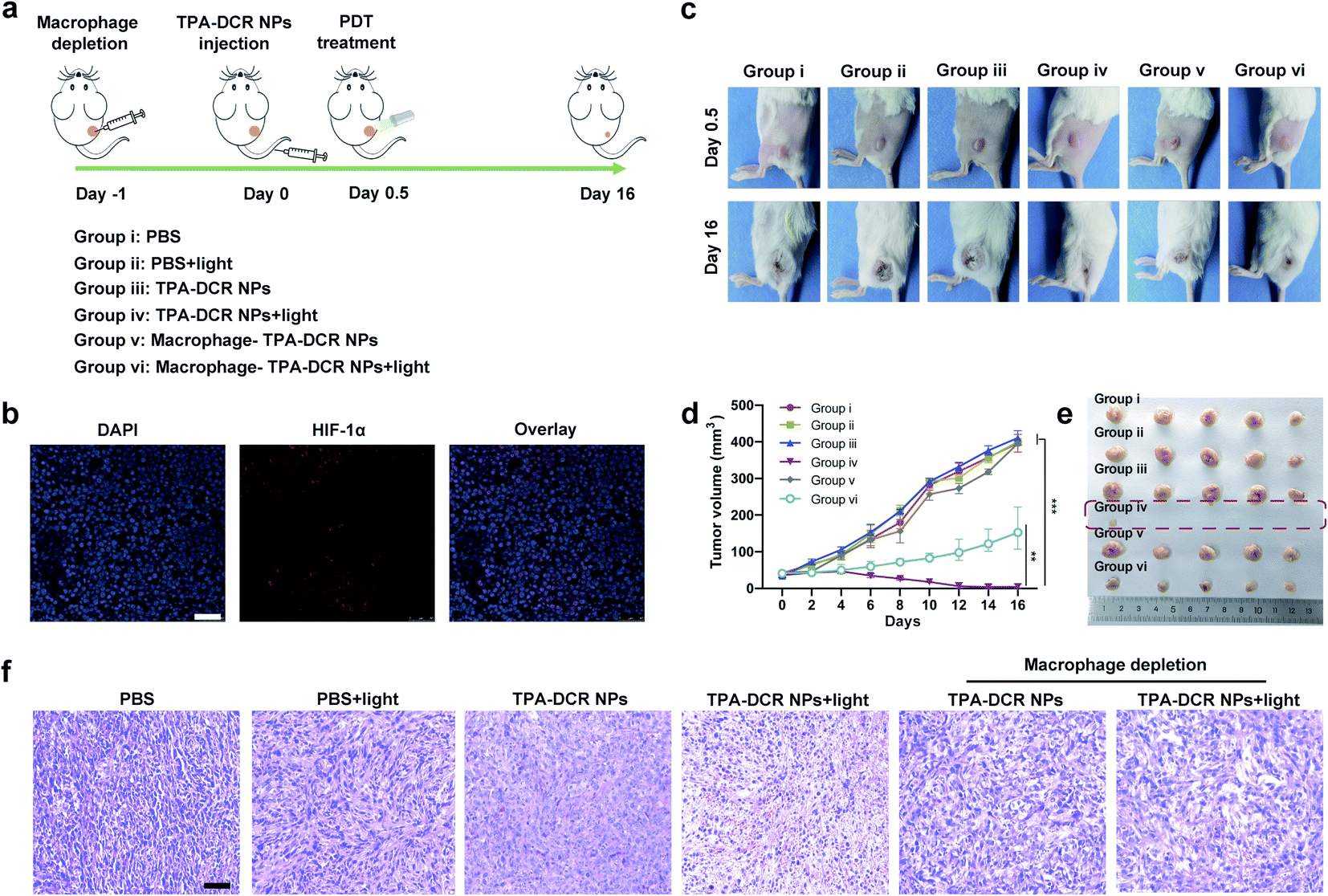 | ||
| Fig. 3 (a) Schematic illustration of the in vivo experimental procedures. The 4T1 tumor-bearing mice were randomly divided into six groups. 1× PBS (200 μL) or TPA-DCR NPs (200 μL, 1 mg mL−1) were intravenously injected, and the groups ii, iv, vi were treated with light irradition (300 mW cm−2, 5 min). In groups v and vi, clodronate liposomes were used to deplete macrophages in tumors. (b) Immunofluorescence imaging of 4T1 tumor sections. Cell nuclei and hypoxia-related protein HIF-1α were stained with DAPI (blue) and HIF-1α antibodies (red), respectively. (c) Representative photos of mice from different groups. (d) The tumor growth curves in all groups. (e) Photos of the 4T1 tumors collected from all groups on the 16th day post treatment. (f) Representative H&E staining of tumor tissues from all groups. Scale bar is 100 μm. **p < 0.01, ***p < 0.001 vs. group iv, and n = 5. | ||
To validate whether the type I photosensitizer of TPA-DCR NPs could reverse the immunosuppression of the hypoxic tumor microenvironment, the populations of M1 and M2 macrophages in tumor tissues were measured by flow cytometry and immunofluorescence staining. Macrophages in tumor tissues were sorted out using magnetic beads at 24 h after PDT treatment. Specific biomarkers were used to individually label M1 (F4/80-APC, CD11c-FITC) and M2 (F4/80-APC, CD206-Alexa 488) macrophages. Results indicated that the number of M1 cells in TPA-DCR NPs + light treatment increased by ∼3-fold, and the number of M2 cells decreased by ∼42%, compared with the PBS group (group i) (Fig. 4a, b, S20 and S21†). In tumor tissues, the expression levels of ICD biomarkers: ATP, HMGB1 and HSP70, showed a significant boost in group iv. Interestingly, the expression levels of ICD biomarkers in mice with macrophage depletion (group vi) were lower than those of group iv (Fig. 4c–e). These findings suggested the importance of TAMs in photodynamic immunotherapy, in which the TPA-DCR NPs could activate TAMs to M1 upon light irradiation to further induce the ICD of tumor tissues. Considering the importance of T cells in anti-tumor immune response,47–49 we also analyzed CD4+ T cells and CD8+ T cells in tumor tissues and the spleen by immunofluorescence staining and flow cytometry. The results suggested that the numbers of CD4+ T cells and CD8+ T cells from the spleen in the PDT group (group iv) were ∼4-fold and ∼3-fold, respectively, compared with those in the control (group i) (Fig. 4f). Meanwhile, the tumor infiltration of CD4+ T cells and CD8+ T cells also significantly increased by ∼3 times after PDT treatment in group iv (Fig. S22 and S23†). According to the literature,50–52 such a treatment could significantly inhibit the growth of abscopal tumors and recurrent tumors, when the CD8+ T cells and CD4+ T cells in the tumor treatment group were ∼2–3 times higher than those in the control group. Based on these results, we speculated that type I photosensitizer-triggered macrophage-mediated photodynamic immunotherapy could directly promote the tumor infiltration of T cells by inducing tumor ICD, which could contribute to the profound anti-tumor efficacy and complete tumor ablation without recurrence in the testing period.
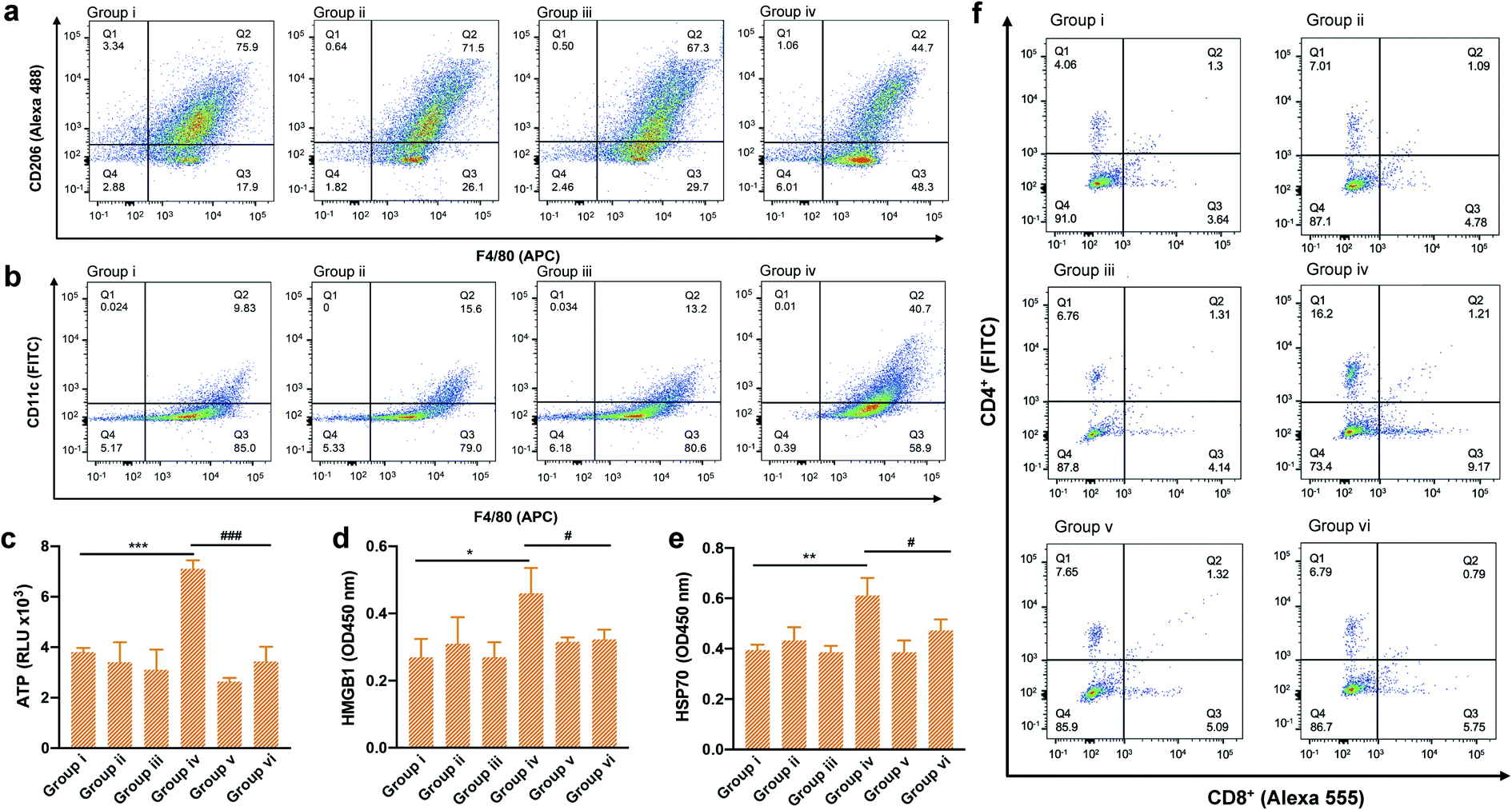 | ||
| Fig. 4 (a and b) Flow cytometric analysis of the M2 (F/480, CD206) and M1 (F4/80, CD11c) cells in tumor tissues from groups i to iv. ELISA results showed the expression of (c) ATP, (d) HMGB1 and (e) HSP70 in tumor tissues from different groups, *p < 0.05, **p < 0.01, ***p < 0.001 vs. the groups i, #p < 0.05, ###p < 0.001 vs. group vi, and n = 3. (f) Flow cytometric analysis of the populations of CD4+ and CD8+ T cells in tumor tissues collected from different groups. | ||
In vivo biocompatibility of TPA-DCR NPs
To evaluate the safety of the photosensitizer, we recorded the body weight of the mice during the therapeutic period, and observed no significant difference in all groups (Fig. S24†). The in vivo toxicity of TPA-DCR NPs was further evaluated in healthy mice, which were intravenously injected with PBS or TPA-DCR NPs (10 mg kg−1). After 7 days, the morphology of major organs was investigated with H&E staining. No abnormal histology was observed in the heart, liver, spleen, lung or kidney sections from mice that were treated with either PBS or TPA-DCR NPs (Fig. S25†). Furthermore, the results of serum biochemistry (Fig. S26†) and blood routine test of samples from both groups (Fig. S27†) indicated no significant difference in all the key indicators. These results demonstrated the in vivo safety of this type I photosensitizer.Conclusions
In summary, we have rationally synthesized a D–A structured type I photosensitizer of TPA-DCR with aggregation-enhanced ROS generation, and unveiled its working mechanism in photosensitizer-triggered, macrophage-mediated photodynamic immunotherapy (Scheme 1). The type I ROS produced by TPA-DCR NPs could activate the NF-κB signaling pathway, further promoting the polarization of M0 and M2 macrophages to the M1 state even under hypoxic conditions. Interestingly, unlike the traditional ICD caused by PDT, we discovered that the photosensitizer-polarized M1 macrophages could effectively increase the expression levels of ATP, HMGB1 and HSP70, inducing the ICD of tumor cells, thus enhancing the treatment outcome of photodynamic immunotherapy. Overall, in vivo experiments demonstrated that TPA-DCR NPs could reverse the immunosuppressive microenvironment in hypoxic tumors by re-educating TAMs to the M1 phenotype, inducing the ICD of tumor cells, and promoting the tumor infiltration of CD4+ and CD8+ T cells. Eventually, almost complete tumor elimination has been achieved by single PDT treatment without repeated injection of the photosensitizer or in combination with an immunoadjuvant. Taken together, we demonstrated a strategy of using type I PDT for macrophage activation to overcome the intrinsic hypoxic conditions in tumors and reverse the tumor immunosuppressive environment, shining light on the future development of photodynamic immunotherapy to maximize the potential of the patient's own immune system. Considering that the absorption and emission of TPA-DCR NPs are in the visible region, we will explore a series of type I photosensitizers that can be effectively excited with light sources in the near-infrared region for improved tissue penetration ability, to facilitate their anti-tumor applications in more sophisticated biological studies (e.g., treating deep-seated tumors).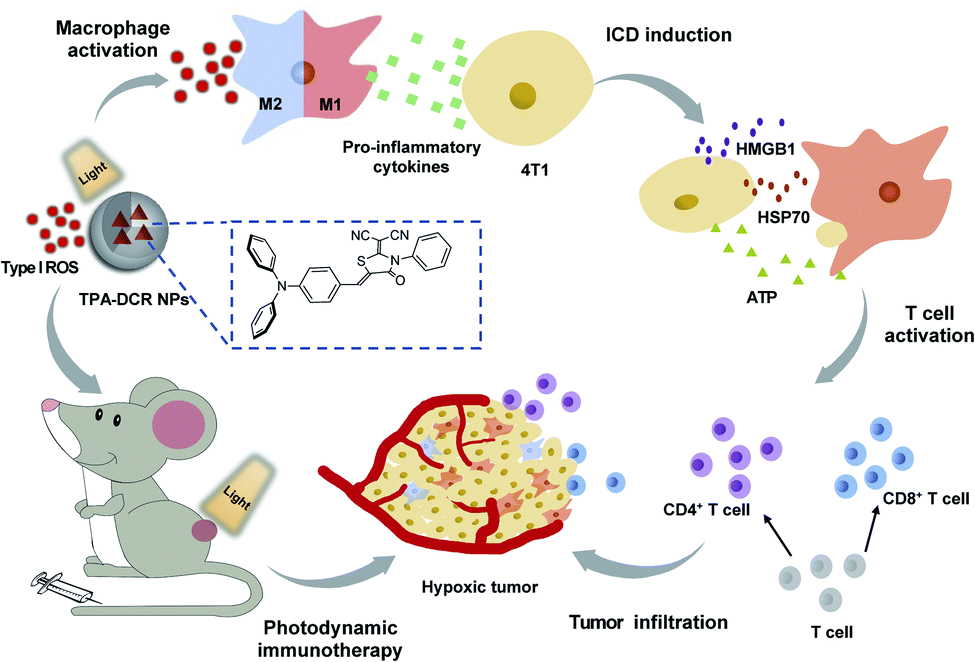 | ||
| Scheme 1 Illustration of using the type I photosensitizer of TPA-DCR NPs for macrophage-mediated photodynamic immunotherapy to treat hypoxic tumors. | ||
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
K. L. devised the method and conceived the project. G. Y. designed and performed the experiments, and analyzed the data. S.-B. L. and J.-S. Ni contributed to the chemical experiments. C. L. contributed to the calculation. F. C., M. Z., Y. L., J. G. and T. K. performed the experiments. All authors contributed to writing the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal experiments were performed in strict accordance with the guidelines for the Care and Use of Laboratory Animals of SUSTech and approved by the Animal Ethics Committee of the Center for Experimental Animal Research of SUSTech (SUSTC-2018-115), China. The authors are grateful to the Ministry of Science and Technology of China (2020YFA0908900), the National Natural Science Foundation of China (31870991), the Department of Science and Technology of Guangdong Province (2019ZT08Y191), the Shenzhen Key Laboratory of Smart Healthcare Engineering (ZDSYS20200811144003009), and the Shenzhen Science and Technology Program (JSGG20200225151916021, KQTD20190929172743294, and JCYJ20190809154011696) for financial support. The time-resolved fluorescence characterization was supported by the Shenzhen Key Laboratory of Full Spectral Solar Electricity Generation (FSSEG) from the Shenzhen Key Laboratory Project (No. ZDSYS201602261933302). The authors thank Dr Yang Y., Ren Z., Li H. and Lin L. at the SUSTech Core Research Facilities for the EPR and HRMS testing, respectively. The authors acknowledge the Center for Computational Science and Engineering at SUSTech for theoretical calculation support and SUSTech Core Research Facilities for technical support. All in vivo procedures have been approved by the Animal Ethics Committee of the Center for Experimental Animal Research of SUSTech, China.References
- B. Ding, S. Shao, C. Yu, B. Teng, M. Wang, Z. Cheng, K.-L. Wong, P. a. Ma and J. Lin, Adv. Mater., 2018, 30, 1900927 Search PubMed.
- H.-T. Feng, Y. Li, X. Duan, X. Wang, C. Qi, J. W. Y. Lam, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 15966–15974 CrossRef CAS PubMed.
- A. Gao, B. Chen, J. Gao, F. Zhou, M. Saeed, B. Hou, Y. Li and H. Yu, Nano Lett., 2020, 20, 353–362 CrossRef CAS PubMed.
- H. Zhu, Y. Fang, Q. Miao, X. Qi, D. Ding, P. Chen and K. Pu, ACS Nano, 2017, 11, 8998–9009 CrossRef CAS PubMed.
- S. He, J. Li, Y. Lyu, J. Huang and K. Pu, J. Am. Chem. Soc., 2020, 142, 7075–7082 CrossRef CAS PubMed.
- P. Cheng, J. Zhang, J. Huang, Q. Miao, C. Xu and K. Pu, Chem. Sci., 2018, 9, 6340–6347 RSC.
- J. Li, Y. Fang, Y. Zhang, H. Wang, Z. Yang and D. Ding, Adv. Mater., 2021, 33, 2008518 CrossRef CAS PubMed.
- D.-W. Zheng, F. Gao, Q. Cheng, P. Bao, X. Dong, J.-X. Fan, W. Song, X. Zeng, S.-X. Cheng and X.-Z. Zhang, Nat. Commun., 2020, 11, 1985 CrossRef CAS PubMed.
- A. A. Barkal, K. Weiskopf, K. S. Kao, S. R. Gordon, B. Rosental, Y. Y. Yiu, B. M. George, M. Markovic, N. G. Ring, J. M. Tsai, K. M. McKenna, P. Y. Ho, R. Z. Cheng, J. Y. Chen, L. J. Barkal, A. M. Ring, I. L. Weissman and R. L. Maute, Nat. Immunol., 2018, 19, 76–84 CrossRef CAS PubMed.
- Z. Gu, T. Liu, J. Tang, Y. Yang, H. Song, Z. K. Tuong, J. Fu and C. Yu, J. Am. Chem. Soc., 2019, 141, 6122–6126 CrossRef CAS PubMed.
- J.-J. Hu, Q. Lei and X.-Z. Zhang, Prog. Mater. Sci., 2020, 114, 100685 CrossRef CAS.
- Q. Hu, H. Li, E. Archibong, Q. Chen, H. Ruan, S. Ahn, E. Dukhovlinova, Y. Kang, D. Wen, G. Dotti and Z. Gu, Nat. Biomed. Eng., 2021, 5, 1038–1047 CrossRef CAS PubMed.
- J. Li, X. Jiang, H. Li, M. Gelinsky and Z. Gu, Adv. Mater., 2021, 33, 2004172 CrossRef CAS PubMed.
- H. Ruan, Q. Hu, D. Wen, Q. Chen, G. Chen, Y. Lu, J. Wang, H. Cheng, W. Lu and Z. Gu, Adv. Mater., 2019, 31, 1970120 CrossRef.
- L. Li, C. Shao, T. Liu, Z. Chao, H. Chen, F. Xiao, H. He, Z. Wei, Y. Zhu, H. Wang, X. Zhang, Y. Wen, B. Yang, F. He and L. Tian, Adv. Mater., 2020, 32, 2003471 CrossRef CAS PubMed.
- J. Sun, K. Du, J. Diao, X. Cai, F. Feng and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 12122–12128 CrossRef CAS PubMed.
- H. Zhu, J. Li, X. Qi, P. Chen and K. Pu, Nano Lett., 2018, 18, 586–594 CrossRef CAS PubMed.
- M.-Z. Zou, W.-L. Liu, H.-S. Chen, X.-F. Bai, F. Gao, J.-J. Ye, H. Cheng and X.-Z. Zhang, Natl. Sci. Rev., 2021, 8, nwaa160 CrossRef CAS PubMed.
- Z. Liu, H. Zou, Z. Zhao, P. Zhang, G.-G. Shan, R. T. K. Kwok, J. W. Y. Lam, L. Zheng and B. Z. Tang, ACS Nano, 2019, 13, 11283–11293 CrossRef CAS PubMed.
- Q. Y. Li, J. Y. Gong, Y. Li, R. Y. Zhang, H. R. Wang, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, M.-H. Li, J. G. Wang and B. Z. Tang, Chem. Sci., 2021, 12, 709–717 RSC.
- M. Wu, X. Liu, H. Chen, Y. Duan, J. Liu, Y. Pan and B. Liu, Angew. Chem., Int. Ed., 2021, 60, 9093–9098 CrossRef CAS PubMed.
- M. Kang, Z. Zhang, N. Song, M. Li, P. Sun, X. Chen, D. Wang and B. Z. Tang, Aggregate, 2020, 1, 80–106 CrossRef.
- C. Ji, L. Lai, P. Li, Z. Wu, W. Cheng and M. Yin, Aggregate, 2020, 2, e114 Search PubMed.
- J.-S. Ni, T. Min, Y. Li, M. Zha, P. Zhang, C. L. Ho and K. Li, Angew. Chem., Int. Ed., 2020, 59, 10179–10185 CrossRef CAS PubMed.
- M. Oszajca, M. Brindell, L. Orzel, J. M. Dabrowski, K. Spiewak, P. Labuz, M. Pacia, A. Stochel-Gaudyn, W. Macyk, R. van Eldik and G. Stochel, Coord. Chem. Rev., 2016, 327–328, 143–165 CrossRef CAS.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- D. Wang, H. Su, R. T. K. Kwok, X. Hu, H. Zou, Q. Luo, M. M. S. Lee, W. Xu, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2018, 9, 3685–3693 RSC.
- Z. Lv, H. Wei, Q. Li, X. Su, S. Liu, K. Y. Zhang, W. Lv, Q. Zhao, X. Li and W. Huang, Chem. Sci., 2018, 9, 502–512 RSC.
- Y. Zheng, H. Lu, Z. Jiang, Y. Guan, J. Zou, X. Wang, R. Cheng and H. Gao, J. Mater. Chem. B, 2017, 5, 6277–6281 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov and J. L. Sonnenberg, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- W. Wu, D. Mao, S. Xu, Kenry, F. Hu, X. Li, D. Kong and B. Liu, Chem, 2018, 4, 1937–1951 CAS.
- K.-X. Teng, W.-K. Chen, L.-Y. Niu, W.-H. Fang, G. Cui and Q.-Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 19912–19920 CrossRef CAS PubMed.
- J.-S. Ni, X. Zhang, G. Yang, T. Kang, X. Lin, M. Zha, Y. Li, L. Wang and K. Li, Angew. Chem., Int. Ed., 2020, 59, 11298–11302 CrossRef CAS PubMed.
- G. Weber and F. W. J. Teale, Trans. Faraday Soc., 1957, 53, 646–655 RSC.
- M. Nemoto, H. Kokubun and M. Koizumi, Bull. Chem. Soc. Jpn., 1969, 42, 1223–1230 CrossRef CAS.
- X. He, Y. Yang, Y. Guo, S. Lu, Y. Du, J.-J. Li, X. Zhang, N. L. C. Leung, Z. Zhao, G. Niu, S. Yang, Z. Weng, R. T. K. Kwok, J. W. Y. Lam, G. Xie and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 3959–3969 CrossRef CAS PubMed.
- G. Yang, J.-S. Ni, Y. Li, M. Zha, Y. Tu and K. Li, Angew. Chem., Int. Ed., 2021, 60, 5386–5393 CrossRef CAS PubMed.
- Y. Cheng, X. Kong, Y. Chang, Y. Feng, R. Zheng, X. Wu, K. Xu, X. Gao and H. Zhang, Adv. Mater., 2020, 32, 1908109 CrossRef CAS PubMed.
- G. Yang, S. Z. F. Phua, W. Q. Lim, R. Zhang, L. Feng, G. Liu, H. Wu, A. K. Bindra, D. Jana, Z. Liu and Y. Zhao, Adv. Mater., 2019, 31, 1901513 CrossRef PubMed.
- K. Li, Z. Zhu, P. Cai, R. Liu, N. Tomczak, D. Ding, J. Liu, W. Qin, Z. Zhao, Y. Hu, X. Chen, B. Z. Tang and B. Liu, Chem. Mater., 2013, 25, 4181–4187 CrossRef CAS.
- C. Chen, X. Ni, S. Jia, Y. Liang, X. Wu, D. Kong and D. Ding, Adv. Mater., 2019, 31, 1904914 CrossRef CAS PubMed.
- B. Hou, L. Zhou, H. Wang, M. Saeed, D. Wang, Z. Xu, Y. Li and H. Yu, Adv. Mater., 2020, 32, 1907210 CrossRef CAS PubMed.
- J. Li, H. Gao, R. Liu, C. Chen, S. Zeng, Q. Liu and D. Ding, Sci. China: Chem., 2020, 63, 1428–1434 CAS.
- L. Sun, F. Shen, L. Tian, H. Tao, Z. Xiong, J. Xu and Z. Liu, Adv. Mater., 2021, 33, 2007910 CrossRef CAS PubMed.
- J. Xu, L. Xu, C. Wang, R. Yang, Q. Zhuang, X. Han, Z. Dong, W. Zhu, R. Peng and Z. Liu, ACS Nano, 2017, 11, 4463–4474 CrossRef CAS PubMed.
- C. Wang, Y. Ye, Q. Hu, A. Bellotti and Z. Gu, Adv. Mater., 2017, 29, 1606036 CrossRef PubMed.
- C. Zhang, F. Gao, W. Wu, W.-X. Qiu, L. Zhang, R. Li, Z.-N. Zhuang, W. Yu, H. Cheng and X.-Z. Zhang, ACS Nano, 2019, 13, 11249–11262 CrossRef CAS PubMed.
- B. Hou, L. Zhou, H. Wang, M. Saeed, D. Wang, Z. Xu, Y. Li and H. Yu, Adv. Mater., 2020, 32, 1907210 CrossRef CAS PubMed.
- J. Xu, B. Zheng, S. Zhang, X. Liao, Q. Tong, G. Wei, S. Yu, G. Chen, A. Wu, S. Gao, Y. Qian, Z. Xiao and W. Lu, Adv. Funct. Mater., 2021, 31, 2008022 CrossRef CAS.
- Y. Wang, Z. Wang, B. Chen, Q. Yin, M. Pan, H. Xia, B. Zhang, Y. Yan, Z. Jiang, Q. Zhang and Y. Wang, Nano Lett., 2021, 21, 4371–4380 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04124j |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |