
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon–carbon bond activation by B(OMe)3/B2pin2-mediated fragmentation borylation†
Li
Wang‡
ab,
Qi
Zhong‡
ab,
Youliang
Zou
b,
Youzhi
Yin
b,
Aizhen
Wu
b,
Quan
Chen
b,
Ke
Zhang
b,
Jiachen
Jiang
b,
Mengzhen
Zhao
b and
Hua
Zhang
 *ab
*ab
aKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education, Hubei Key Laboratory of Catalysis and Materials Science, South-Central University for Nationalities, Wuhan 430074, China. E-mail: huazhang@scuec.edu.cn
bCollege of Chemistry, Nanchang University, Nanchang 330031, China
First published on 2nd November 2021
Abstract
Selective carbon–carbon bond activation is important in chemical industry and fundamental organic synthesis, but remains challenging. In this study, non-polar unstrained Csp2–Csp3 and Csp2–Csp2 bond activation was achieved by B(OMe)3/B2pin2-mediated fragmentation borylation. Various indole derivatives underwent C2-regioselective C–C bond activation to afford two C–B bonds under transition-metal-free conditions. Preliminary mechanistic investigations suggested that C–B bond formation and C–C bond cleavage probably occurred in a concerted process. This new reaction mode will stimulate the development of reactions based on inert C–C bond activation.
Introduction
Carbon–carbon bond activation, which enables the direct production of valuable functionalized products from widely available and inexpensive raw materials obtained from natural resources, is the foundation of many important industrial processes and has wide-ranging applications in complex molecule synthesis.1–14 Owing to the ubiquity of carbon–carbon bonds in organic molecules, the study of transformations based on selective carbon–carbon bond activation will greatly help realize free molecular editing, thus stimulating growing interest in the synthetic community. In recent decades, the rapid development of synthetic methods based on strained C–C bond cleavage via strain release,15–23 polar C–C bond cleavage24–36via transition-metal-catalysed oxidative addition and β-carbon elimination, and oxidation induced C–C cleavage of alkylarenes,37–39 among others, has been witnessed (Scheme 1a). Owing to a high bond dissociation energy and competitive C–H bond activation, the selective activation of non-polar unstrained C–C bonds is challenging, with reports remaining rare. Reported successes mainly rely on the directed transition-metal-catalysed reductive hydrogenolysis of substrates bearing two directing groups (Scheme 1b). In this context, Milstein and co-workers contributed the pioneering work, wherein the hydrogenolysis of non-polar unstrained Csp2–Csp3 bonds in pincer-type substrates with bidentate phosphine as the directing group was achieved via Rh catalysis (Scheme 1b).40–43 Recently, Dong and co-workers made a significant advancement, achieving the activation of Csp2–Csp2 bonds in a wide range of biaryl compounds bearing two ortho-directing groups using Rh and Ru catalysis (Scheme 1b).44–46 Although these studies proved that the selective cleavage of non-polar unstrained C–C bonds is feasible, the development of new systems to advance this field is needed. Herein, we describe the development of a B(OMe)3/bis(pinacolato)diborane(B2pin2)-mediated non-polar unstrained C–C bond activation reaction (Scheme 1c). In this reaction, both Csp2–Csp3 and Csp2–Csp2 bond activation occurred regioselectively at the C2-position in various substituted indoles, affording two C–B bond products under transition-metal-free conditions. This study not only provides a new approach to highly challenging selective carbon–carbon bond cleavage, but also provides new opportunities for further applications.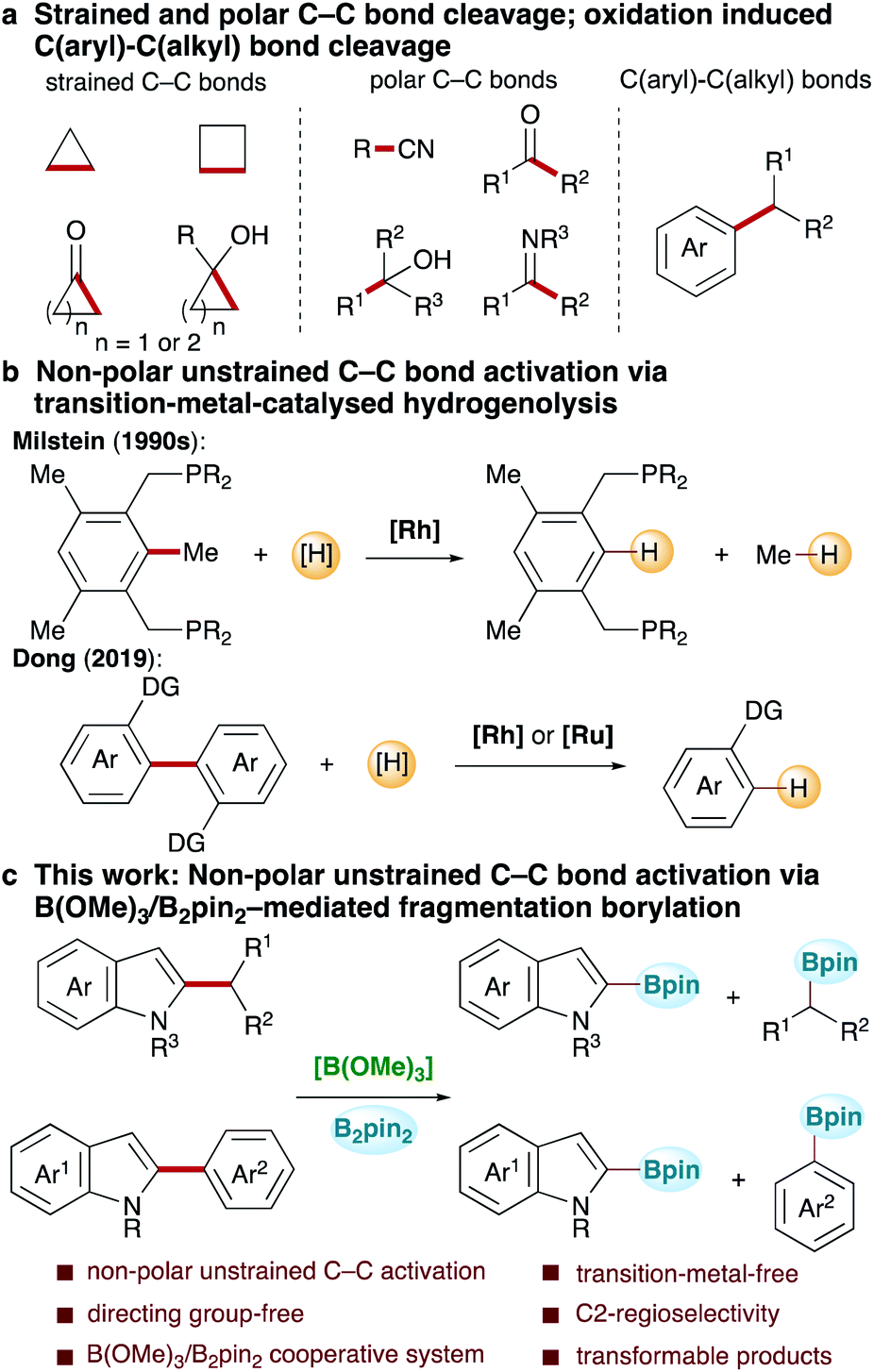 | ||
| Scheme 1 (a) Strained and polar C–C bond cleavage; oxidation induced C(aryl)–C(alkyl) bond cleavage. (b) Non-polar unstrained C–C bond activation via transition-metal-catalysed hydrogenolysis. (c) This work: non-polar unstrained C–C bond activation via B(OMe)3/B2pin2-mediated fragmentation borylation. | ||
Results and discussion
As an extension of our previous study towards the Lewis acid-promoted C–H borylation of indoles,47 we envisaged that installing substituents on the pyrrole core of indoles would provide opportunities for C–H borylation of the benzenoid moiety of indoles.48,49 This concept was initially investigated using the reaction of 1,2-dimethyl indole (1aa) and B2pin2. Surprisingly, no desired benzenoid C–H borylation products were obtained in the reaction, but C–C bond activation product 2a was observed. Owing to the importance of C–C bond activation, we continued to optimize this Csp2–Csp3 bond activation reaction. Extensive screening showed that the best result was obtained when the reaction was conducted in the presence of B(OMe)3 (30 mol%) and Et3N (30 mol%) in n-octane at 160 °C for 48 h, affording 1-methyl-2-indolyl boronic pinacol ester (2a) in 75% isolated yield (Table 1, entry 1).50 The results of varying the standard conditions are shown in Table 1. Compound 2a was not detected in the absence of B(OMe)3, indicating that B(OMe)3 catalysed this reaction (Table 1, entry 2). Using other boron Lewis acids, such as B(OiPr)3, BPh3, PhB(OH)2, and BF3·Et2O, as catalysts gave lower yields (entries 3–7). Furthermore, BBr3 and B(C6F5)3 were ineffective (entries 8 and 9). No. 2a was obtained using other metal Lewis acids as catalysts (entry 10). Lower yields of 2a were obtained in the absence of Et3N or in the presence of nBu3N (entries 11 and 12). Non-polar n-hexane was also a suitable solvent for this reaction (entry 13). Decreasing the reaction time resulted in only trace product, unless accompanied by an increased reaction temperature, which resulted in a moderate yield of 2a (entries 14 and 15). Lowering the reaction temperature to 140 °C led to trace product formation (entry 16).|
|
||
|---|---|---|
| Entry | Deviation from standard reaction conditionsa | Yieldb (%) |
| a Standard reaction conditions: 1aa (0.20 mmol), B2pin2 (0.50 mmol), B(OMe)3 (0.06 mmol), Et3N (0.06 mmol), n-octane (1.0 mL), 160 °C, and 48 h; HCl (1.0 mL, 6.0 M) and 0.5 h. b Yields determined by GC using naphthalene as the internal standard. c Isolated yield in parentheses. d THF (0.1 mL) was added. e No Et3N. f 10 mol%. g Metal Lewis acids: Zn(OTf)2, ZnCl2, Sc(OTf)3, AlCl3, InCl3, FeCl3, CuCl2, and NiCl2. | ||
| 1 | None | 83(75)c |
| 2 | No B(OMe)3 | 0 |
| 3 | B(OiPr)3, instead of B(OMe)3 | 70 |
| 4 | BPh3, instead of B(OMe)3 | 59 |
| 5 | PhB(OH)2, instead of B(OMe)3 | 66 |
| 6d | BF3·Et2O, instead of B(OMe)3 | Trace |
| 7d,e | BF3·Et2O, instead of B(OMe)3 | 62 |
| 8 | BBr3, instead of B(OMe)3 | 0 |
| 9f | B(C6F5)3, instead of B(OMe)3 | Trace |
| 10g | Metal Lewis acids, instead of B(OMe)3 | 0 |
| 11 | No Et3N | 59 |
| 12 | n Bu3N, instead of Et3N | 78 |
| 13 | n-Hexane, instead of n-octane | 68 |
| 14 | 16 h, instead of 48 h | Trace |
| 15 | 180 °C, 16 h instead of 160 °C, 48 h | 67 |
| 16 | 140 °C, instead of 160 °C | Trace |
Having established optimal reaction conditions, the substrate scope of this C–C bond fragmentation borylation reaction was investigated. As shown in Scheme 2, the site-selectivity of this reaction was first studied. Exclusive C2-regioselectivity was observed, with the reactions of C4-, C5-, C6-, and C7-substituted 1,2-dimethyl indoles uniformly producing C2-borylated products (2b–2e) in good yields. Furthermore, C3-substituted 1,2-dimethyl indole afforded C2-borylated product 2f, albeit in a lower yield under more harsh conditions. Next, the functional group tolerance of the indolyl fragment was examined. 1,2-Dimethyl indoles bearing tert-butyl, methoxy, piperidyl, and trifluoromethyl groups reacted well with B2pin2, giving the corresponding borylation products (2g–2j) in good yields. Halogen substituents, such as F, Cl, Br, and I (2k–2n), were all tolerated in this C–C bond fragmentation borylation reaction, making further functionalization feasible. Transformable trimethylsilyl (2o) and (pinacolato)boryl (2p) groups, and a phenyl group (2q), were also well tolerated. Generally, both electron-donating and electron-withdrawing substituents on the indole ring did not affect the reaction efficiency. Different N-substituted indoles were also tested. Both N-butyl and N-benzyl indoles reacted well, giving the corresponding products (2r and 2s) in good yields. However, electron-withdrawing protecting groups, such as acetyl and tert-butoxycarbonyl, resulted in no desired product formation. 2-Methyl-1-phenyl indole, 1-benzyl-2,5-dimethyl pyrrole, and 2-methyl benzothiophene gave trace or no desired products. The borylation of 5-methoxy-1-methyl indoles bearing alkyl C2-substituents other than a methyl group was performed to examine the scope of the alkyl fragment (Scheme 2). 5-Methoxy-1-methyl indoles bearing primary alkyl C2-substituents, such as hexyl and benzyl groups, were suitable substrates, affording the corresponding indolyl boronic pinacol ester 2h and alkyl boronic pinacol esters 3b and 3c in moderate yields under modified conditions. 5-Methoxy-1-methyl indole bearing a secondary alkyl C2-substituent (cyclohexyl) gave lower yields (2h and 3d), while no desired products (2h and 3e) were obtained when 5-methoxy-1-methyl indole bearing a tertiary alkyl C2-substituent (tert-butyl) was used as the substrate.
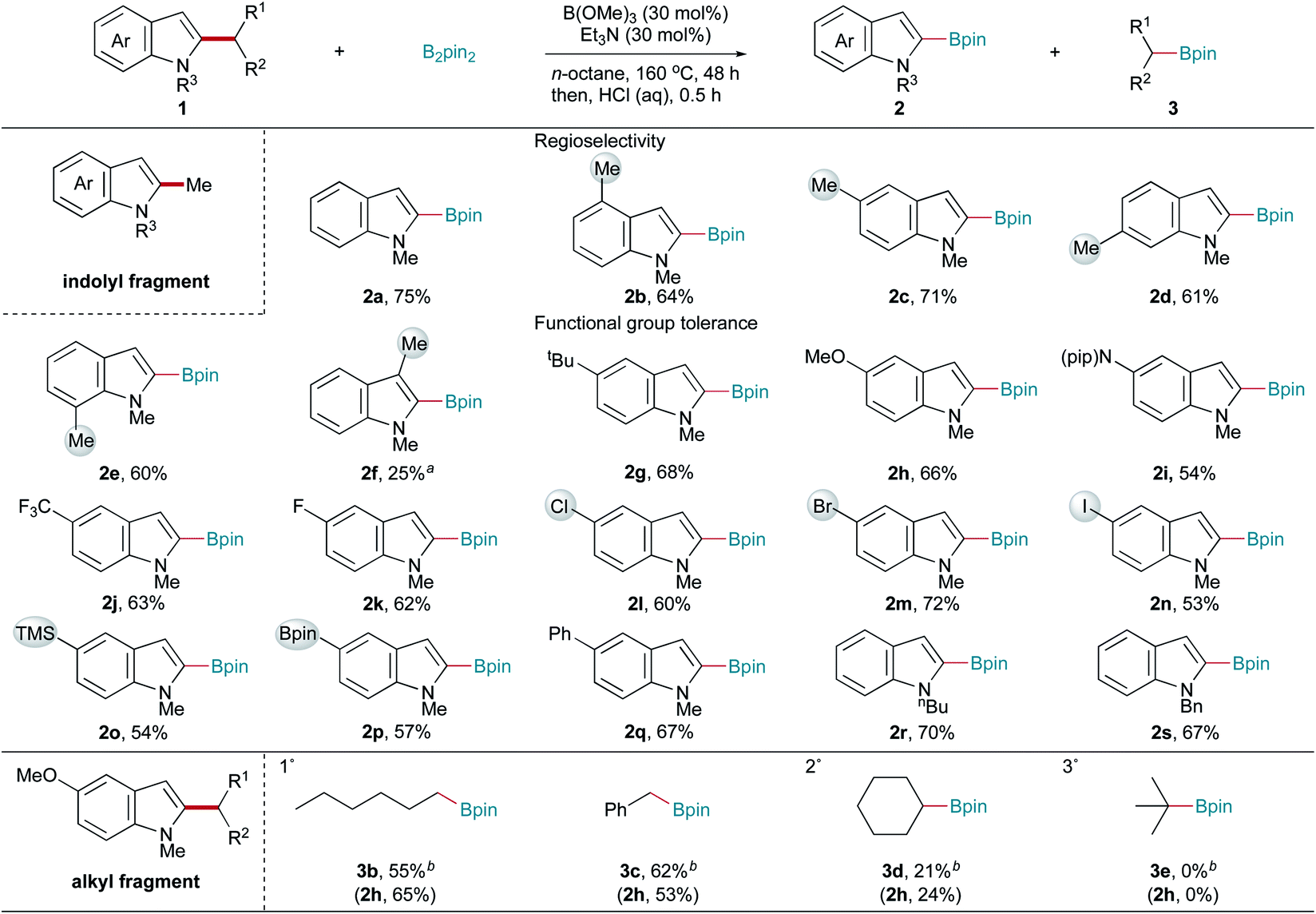 | ||
| Scheme 2 Scope of Csp2–Csp3 bond fragmentation borylation. Reaction conditions: 1 (0.20 mmol), B2pin2 (0.50 mmol), B(OMe)3 (0.06 mmol), Et3N (0.06 mmol), n-octane (1.0 mL), 160 °C, and 48 h; HCl (1.0 mL, 6.0 M) and 0.5 h. Isolated yields. a B(OMe)3 (0.20 mmol), B2pin2 (0.80 mmol), and 180 °C. b B(OMe)3 (0.20 mmol), B2pin2 (0.80 mmol), Et3N (0.06 mmol), n-octane (1.0 mL), 160 °C, and 48 h. | ||
Encouraged by the above results, we investigated whether this protocol could be extended to Csp2–Csp2 bonds. To our delight, this transition-metal-free system also promoted Csp2–Csp2 bond cleavage. The reaction conditions were slightly optimized to ensure a satisfactory yield (for details, see Table S1†). The substrate scope was also carefully examined, with the results shown in Scheme 3. Various C2-aryl-substituted 5-methoxy-1-methyl indoles were subjected to this borylation reaction to examine the aryl fragment scope. Aryl moieties bearing electronically diverse substituents all underwent C–C bond fragmentation borylation smoothly, affording the corresponding indolyl (2h) and aryl (3f–3m) boronic pinacol esters in moderate yields. Notably, the substrate with a (1,1′-biphenyl)-4-yl substituent (1hn) underwent C2-selective C–C bond activation, with the biphenyl moiety remaining intact (3m). Similarly, a C(indolyl)–C(2-naphthalenyl) bond (1oh) was also cleaved to give the corresponding boronic pinacol esters. Next, the indolyl fragment scope was investigated. 1-Methyl-2-(3,4,5-trimethoxyphenyl) indole (1ap) reacted well, affording the corresponding products 2a and 3p in moderate yields. The reactions of C4-, C5-, C6-, and C7-substituted indoles all produced the corresponding indolyl (2b–2e) and 3,4,5-trimethoxyphenyl (3p) boronic acid pinacol esters in moderate yields. The desired products were obtained in low yields from C3-substituted indole 1fp, probably due to steric hindrance. Electron-donating (1hp) and electron-withdrawing (1jp and 1kp) substituents on the indole ring were compatible with this C–C bond activation reaction. Transformable chloride (1lp) and trimethylsilyl (1op) groups were also tolerated. 1-Methyl-5-phenyl-2-(3,4,5-trimethoxyphenyl) indole (1qp) also reacted well with B2pin2, affording the corresponding products 2q and 3p in moderate yields. Trace desired products were obtained using 1,2-diphenyl indole, 1-benzyl-2,5-diphenyl pyrrole, and 2-phenyl benzothiophene as the substrate.
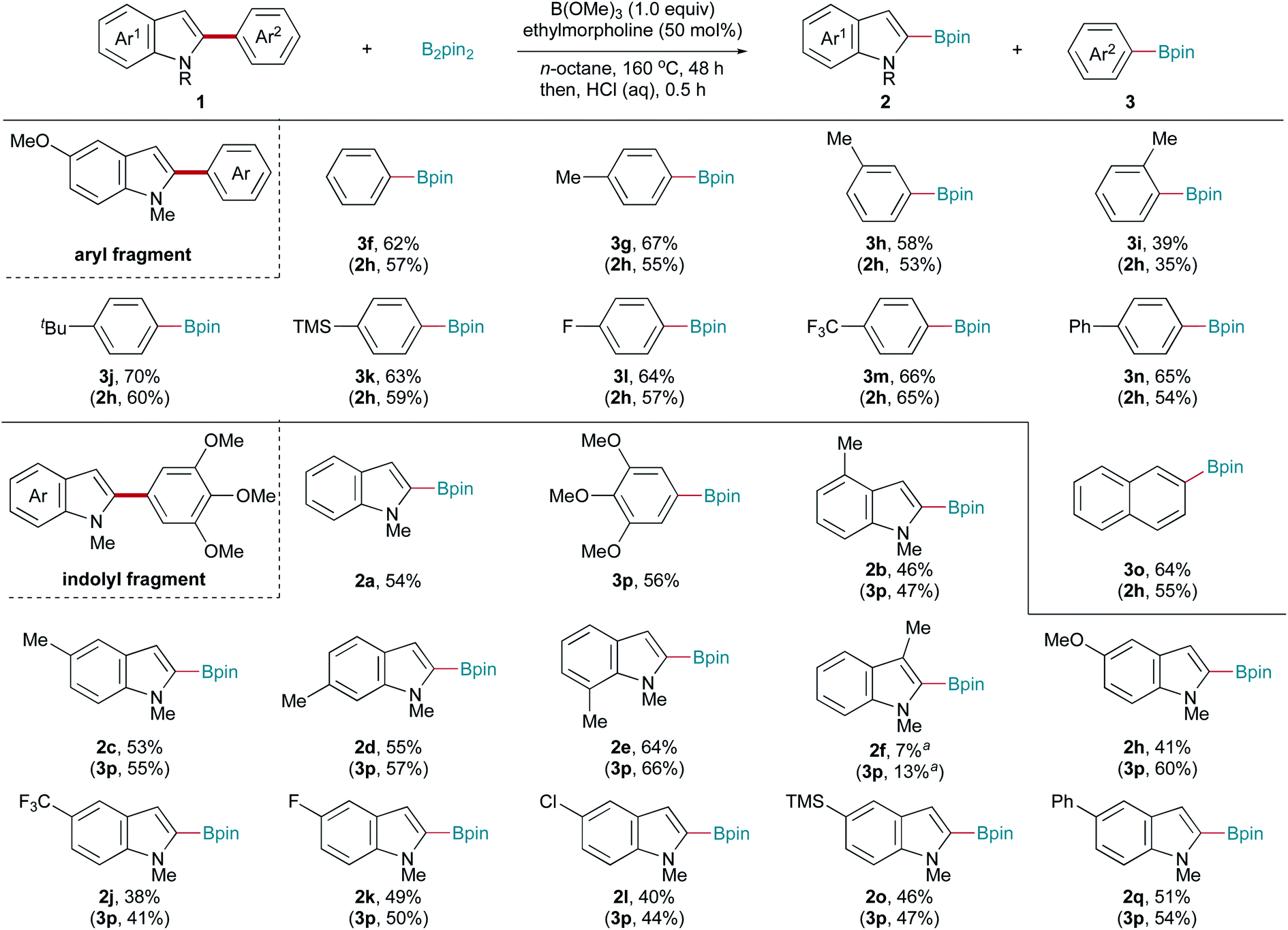 | ||
| Scheme 3 Scope of Csp2–Csp2 bond fragmentation borylation. Reaction conditions: B2pin2 (0.80 mmol), B(OMe)3 (0.20 mmol), ethylmorpholine (0.10 mmol), n-octane (1.0 mL), 160 °C, and 48 h; HCl (1.0 mL, 6.0 M) and 0.5 h. Isolated yields. a 1H NMR spectroscopic yield. | ||
The scalability and practicability of this C–C bond fragmentation borylation reaction were also studied. Good yields were obtained from gram-scale experiments under both standard and non-anhydrous conditions (Scheme 4a). Furthermore, C–C bond fragmentation borylation was performed with bis(neopentyl glycolato)diboron (B2neop2) and bis(hexylene glycolato)diboron (B2hg2) as the boron source (Scheme 4b).
To obtain mechanistic insight into this B(OMe)3/B2pin2-mediated C–C bond activation reaction, a series of control experiments were designed and performed. When radical clock substrate 1aq was used, only indolyl (2a) and cyclopropylmethyl (3q) boronic pinacol esters were obtained (Scheme 4c). The reaction of 1hf with B2pin2 in the presence of a stoichiometric amount of radical scavenger 9,10-dihydroanthracene (6) afforded 2h and 3f in 64% and 67% yields, respectively (Scheme 4c). These results ruled out the possibility of this transformation involving a radical pathway. When 1ar or 1as was subjected to this C–C bond activation reaction, only trace amounts of iso-propyl (3r′) or tert-butyl (3e) boronic pinacol esters were detected (Scheme 4d). These results further ruled out the possibility of the mechanism involving a carbocation intermediate.
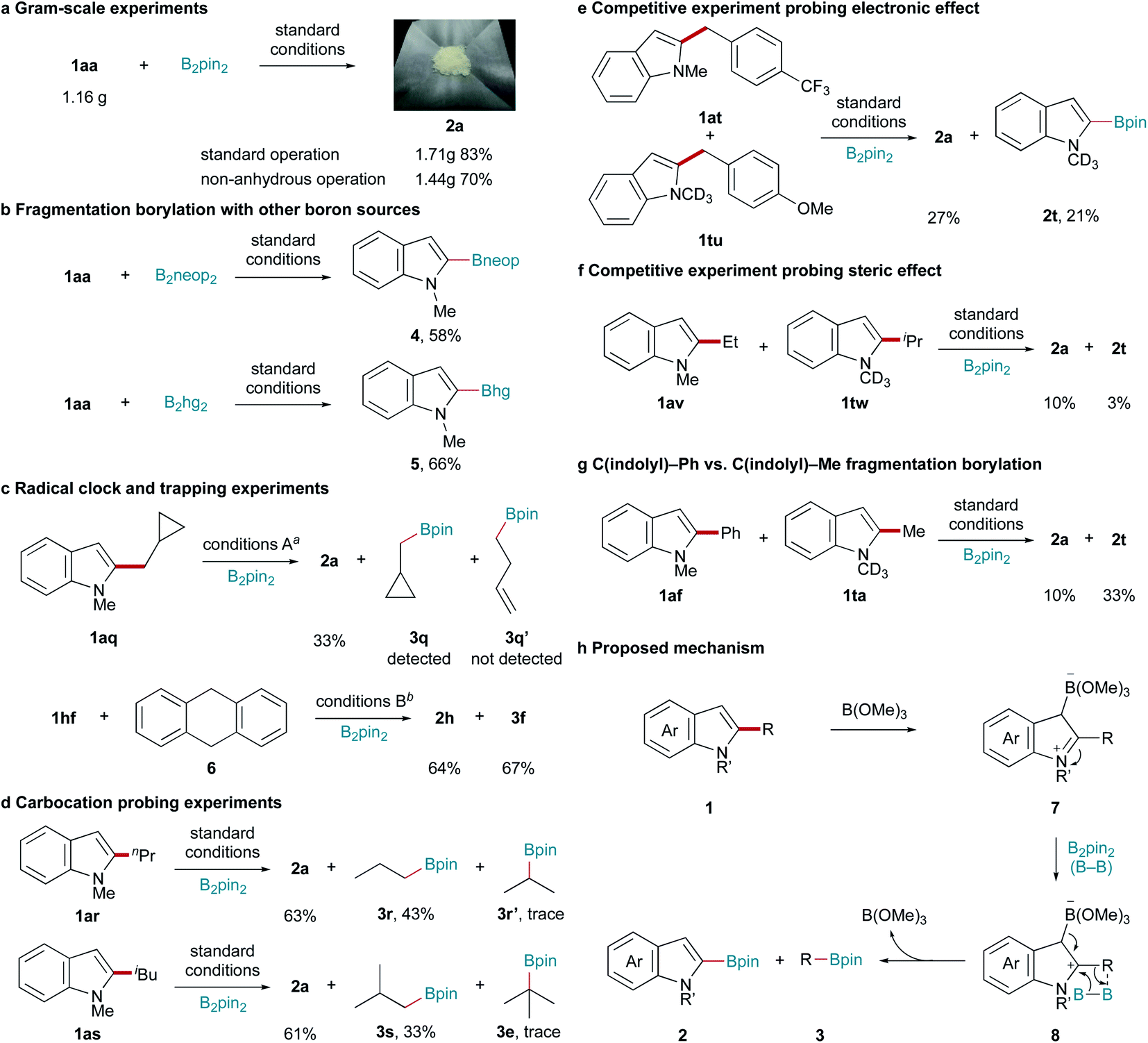 | ||
| Scheme 4 Control experiments and proposed mechanism. (a) Gram-scale experiments. (b) Fragmentation borylation with other boron sources. (c) Radical clock and trapping experiments. (d) Carbocation probing experiments. (e) Competition experiment probing the electronic effect. (f) Competition experiment probing the stereo effect. (g) C(indolyl)–Ph vs. C(indolyl)–Me fragmentation borylation. (h) Proposed mechanism. a Conditions A: B2pin2 (0.80 mmol), B(OMe)3 (0.20 mmol), n-octane (1.0 mL), 180 °C, 24 h; HCl (1.0 mL, 6.0 M), and 0.5 h. b Conditions B: B2pin2 (0.80 mmol), B(OMe)3 (0.20 mmol), ethylmorpholine (0.10 mmol), n-octane (1.0 mL), 160 °C, and 48 h; HCl (1.0 mL, 6.0 M) and 0.5 h. | ||
The competitive experiment of 1at and 1tu indicated that the electronic effect of the C2 substituent had no obvious influence on the C–C bond fragmentation borylation (Scheme 4e). However, the competitive experiment of 1av and 1tw indicated that the steric effect of the C2 substituent clearly influenced the C–C bond fragmentation borylation (Scheme 4f). The competitive experiment of 1af and 1ta indicated that C(indolyl)–Me bond activation was more favoured than C(indolyl)–Ph bond activation under the same conditions (Scheme 4g). Based on the above results and previous reports,51 we proposed the following mechanism for this B(OMe)3/B2pin2-mediated C–C bond activation reaction. B(OMe)3 initially attacks the C3-position of 1, affording intermediate 7, which further reacts with B2pin2 to give products 2 and 3via intermediate 8. The cleavage of the C–C and B–B bonds and C–B bond formation probably occurred in a concerted process (Scheme 4h).
Conclusions
In conclusion, B(OMe)3/B2pin2-mediated C–C bond fragmentation borylation has been developed as a new transition-metal-free protocol for non-polar unstrained C–C bond activation. In this system, C2-regioselective C–C bond cleavage of indole derivatives was achieved, producing the corresponding boronic pinacol esters in moderate to good yields. Preliminary mechanistic studies suggested that C–C bond cleavage and C–B bond formation probably occurred in a concerted process. These results will help stimulate further interest in the exploration of C–C bond activation using transition-metal-free techniques. Efforts to gain further mechanistic insights and expand the reaction mode to other C–C bonds are ongoing in our laboratories.Data availability
All the experimental data have been included in the ESI.†Author contributions
L. W., Q. Z., and H. Z. conceived and designed the experiments. L. W., Q. Z., Y. Z., Y. Y., A. W., Q. C., K. Z., J. J., and M. Z. performed the experiments. L. W. and Q. Z. analysed the data. L. W., Q. Z., and H. Z. prepared the manuscript with feedback from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by the Natural Science Foundation of China (21602096). The authors thank Prof. Guoyin Yin (Wuhan University), Prof. Chao Liu (Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences), Prof. Yu Lan (Chongqing University), Prof. Gang Li (Utah State University) and Prof. Ruopeng Bai (Chongqing University) for constructive discussion.References
- B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef PubMed.
- C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC.
- A. S. Goldman, Nature, 2010, 463, 435–436 CrossRef CAS PubMed.
- M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100–1105 RSC.
- K. Ruhland, Eur. J. Org. Chem., 2012, 2683–2706 CrossRef CAS.
- G. Dong, C–C Bond Activation, Springer, Berlin, 2014 Search PubMed.
- L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed.
- I. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414–429 CrossRef CAS PubMed.
- M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed.
- D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed.
- Z. Nairoukh, M. Cormier and I. Marek, Nat. Rev. Chem., 2017, 1, 0035 CrossRef.
- M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS PubMed.
- B. Zhao, T. Rogge, L. Ackermann and Z. Shi, Chem. Soc. Rev., 2021, 50, 8903–8953 RSC.
- B. Zhao, B. Prabagar and Z. Shi, Chem, 2021, 7, 2585–2634 CAS.
- T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835–2840 RSC.
- T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS PubMed.
- C. Aïssa, Synthesis, 2011, 3389–3407 CrossRef.
- D. J. Mack and J. T. Njardarson, ACS Catal., 2013, 3, 272–286 CrossRef CAS.
- P.-h. Chen and G. Dong, Chem.–Eur. J., 2016, 22, 18290–18315 CrossRef CAS PubMed.
- P.-h. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed.
- G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
- H. M. Ko and G. Dong, Nat. Chem., 2014, 6, 739–744 CrossRef CAS PubMed.
- Y. Wang, J. Bai, Y. Yang, W. Zhao, Y. Liang, D. Wang, Y. Zhao and Z. Shi, Chem. Sci., 2021, 12, 3599–3607 RSC.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567–581 RSC.
- H. Liu, M. Feng and X. Jiang, Chem.–Asian J., 2014, 9, 3360–3389 CrossRef CAS PubMed.
- F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC.
- Y. Xia and G. Dong, Nat. Rev. Chem., 2020, 4, 600–614 CrossRef CAS PubMed.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed.
- C. He, S. Guo, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273–8275 CrossRef CAS PubMed.
- Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546–550 CrossRef CAS PubMed.
- J. B. Roque, Y. Kuroda, L. T. Goettemann and R. Sarpong, Science, 2018, 361, 171–174 CrossRef CAS PubMed.
- J. B. Roque, Y. Kuroda, L. T. Gottemann and R. Sarpong, Nature, 2018, 564, 244–248 CrossRef CAS PubMed.
- V. T. Tran, J. A. Gurak Jr, K. S. Yang and K. M. Engle, Nat. Chem., 2018, 10, 1126–1133 CrossRef CAS PubMed.
- Y. Xu, X. Qi, P. Zheng, C. C. Berti, P. Liu and G. Dong, Nature, 2019, 567, 373–378 CrossRef CAS PubMed.
- A. J. Smaligo, M. Swain, J. C. Quintana, M. F. Tan, D. A. Kim and O. Kwon, Science, 2019, 364, 681–685 CrossRef CAS PubMed.
- Y. Adeli, K. Huang, Y. Liang, Y. Jiang, J. Liu, S. Song, C.-C. Zeng and N. Jiao, ACS Catal., 2019, 9, 2063–2067 CrossRef CAS.
- J. Liu, X. Qiu, X. Huang, X. Luo, C. Zhang, J. Wei, J. Pan, Y. Liang, Y. Zhu, Q. Qin, S. Song and N. Jiao, Nat. Chem., 2019, 11, 71–77 CrossRef CAS PubMed.
- S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed.
- M. Gozin, A. Weisman, Y. Ben-David and D. Milstein, Nature, 1993, 364, 699–701 CrossRef CAS.
- M. Gozin, M. Alzenberg, S.-Y. Liou, A. Weisman, Y. Ben-David and D. Milstein, Nature, 1994, 370, 42–44 CrossRef CAS.
- S.-Y. Liou, M. Gozin and D. Milstein, J. Am. Chem. Soc., 1995, 117, 9774–9775 CrossRef CAS.
- S.-Y. Liou, M. E. van der Boom and D. Milstein, Chem. Commun., 1998, 687–688 RSC.
- J. Zhu, J. Wang and G. Dong, Nat. Chem., 2019, 11, 45–51 CrossRef CAS PubMed.
- J. Zhu, P.-h. Chen, G. Lu, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 18630–18640 CrossRef CAS PubMed.
- J. Zhu, R. Zhang and G. Dong, Nat. Chem., 2021, 13, 836–842 CrossRef CAS PubMed.
- Q. Zhong, S. Qin, Y. Yin, J. Hu and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 14891–14895 CrossRef CAS PubMed.
- J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723–1736 CrossRef CAS PubMed.
- B. Prabagar, Y. Yang and Z. Shi, Chem. Soc. Rev., 2021, 50, 11249–11269 RSC.
- Considerable yield of unstable C2,C3-diborylated indole 2a′ was obtained under fragmentation borylation conditions. C2,C3-diborylated indole 2a′ could occur C3-protodeboronation by the treatment of aqueous hydrochloride solution to produce 2a quantitatively. Methyl boronic pinacol ester 3a was not detected by GCMS due to its instability under fragmentation borylation conditions. For details of 2a′, see the ESI†.
- F. Focante, I. Camurati, D. Nanni, R. Leardini and L. Resconi, Organometallics, 2004, 23, 5135–5141 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04487g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |