
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic amine mediated cleavage of Caromatic–Cα bonds in lignin and its platform molecules†
Yu
Xin
ab,
Xiaojun
Shen
 ac,
Minghua
Dong
ab,
Xiaomeng
Cheng
ab,
Shulin
Liu
ab,
Junjuan
Yang
a,
Zhenpeng
Wang
a,
Huizhen
Liu
*ab and
Buxing
Han
ab
ac,
Minghua
Dong
ab,
Xiaomeng
Cheng
ab,
Shulin
Liu
ab,
Junjuan
Yang
a,
Zhenpeng
Wang
a,
Huizhen
Liu
*ab and
Buxing
Han
ab
aBeijing National Laboratory for Molecular Science, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Centre for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuhz@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Catalysis (SKLC), Dalian National Laboratory for Clean Energy (DNL), Dalian, China
First published on 3rd November 2021
Abstract
The activation and cleavage of C–C bonds remains a critical scientific issue in many organic reactions and is an unmet challenge due to their intrinsic inertness and ubiquity. Meanwhile, it is crucial for the valorization of lignin into high-value chemicals. Here, we proposed a novel strategy to enhance the Caromatic–Cα bond cleavage by pre-functionalization with amine sources, in which an active amine intermediate is first formed through Markovnikov hydroamination to reduce the dissociation energy of the Caromatic–Cα bond which is then cleaved to form target chemicals. More importantly, this strategy provides a method to achieve the maximum utilization of the aromatic nucleus and side chains in lignin or its platform molecules. Phenols and N,N-dimethylethylamine compounds with high yields were produced from herbaceous lignin or the p-coumaric acid monomer in the presence of industrially available dimethylamine (DMA).
Introduction
With the depletion of fossil fuels as a source of fuels, chemicals, and energy, the fraction of energy and chemicals provided by renewable resources such as biomass can be expected to increase in the foreseeable future.1–3 As the second most abundant form of biomass, the valorization of lignin has attracted more and more attention.4–7Lignin consists of three main parts: methoxy groups, aromatic nucleus, and side chains.8–10 The depolymerization of lignin into small molecular compounds by breaking the C–C bond and C–O bond is an important strategy to realize lignin transformation. Substantial progress has been made in C–O bond cleavage of lignin model compounds and native lignin.11–16 However, the catalytic activation of C–C bonds remains an unmet challenge due to their intrinsic inertness and ubiquity.17,18 So far, catalytic oxidation of lignin has been one of the most widely used methods to obtain oxygen-containing valuable compounds by breaking the interunit C–C linkages of lignin.19,20 One strategy to achieve oxidative cleavage of the Cα–Cβ bond involves the initial oxidation of Cα–OH to Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O which reduces the bond dissociation energy followed by Cα–Cβ cleavage to generate acid or aldehyde products.21–23 Another strategy for oxidative cleavage of the Cα–Cβ bond is the initial selective oxidation of Cγ–OH to an aldehyde, followed by a retro-aldol reaction.24–26 Additionally, Caromatic–Cα bond cleavage can also be realized via oxidative decarboxylation or Baeyer–Villiger (BV) oxidation.27,28 Although some fine chemicals, such as aromatic aldehydes and acids, have been produced from lignin or its platform compounds by oxidation reaction via cleavage of the C–C bond, the side chains were usually oxidized to CO2, which thus reduces the atom economy of lignin. In addition, some relevant studies on C–C bond activation in lignin by using electrocatalytic methods have been published.29–31 Recently, it was reported that zeolites showed remarkable performance in the selective elimination of the alkyl group, generating phenol and propylene as desired products, which achieved the utilization of the aromatic nucleus and side chains in lignin.32–35 So, there are two important issues to realize the valorization of lignin: (1) developing effective strategies to achieve the breakage of C–C bonds; (2) transforming the aromatic nucleus and side chains in lignin to valuable compounds to improve the atom economy.
O which reduces the bond dissociation energy followed by Cα–Cβ cleavage to generate acid or aldehyde products.21–23 Another strategy for oxidative cleavage of the Cα–Cβ bond is the initial selective oxidation of Cγ–OH to an aldehyde, followed by a retro-aldol reaction.24–26 Additionally, Caromatic–Cα bond cleavage can also be realized via oxidative decarboxylation or Baeyer–Villiger (BV) oxidation.27,28 Although some fine chemicals, such as aromatic aldehydes and acids, have been produced from lignin or its platform compounds by oxidation reaction via cleavage of the C–C bond, the side chains were usually oxidized to CO2, which thus reduces the atom economy of lignin. In addition, some relevant studies on C–C bond activation in lignin by using electrocatalytic methods have been published.29–31 Recently, it was reported that zeolites showed remarkable performance in the selective elimination of the alkyl group, generating phenol and propylene as desired products, which achieved the utilization of the aromatic nucleus and side chains in lignin.32–35 So, there are two important issues to realize the valorization of lignin: (1) developing effective strategies to achieve the breakage of C–C bonds; (2) transforming the aromatic nucleus and side chains in lignin to valuable compounds to improve the atom economy.
Herbaceous lignin is an important non-edible biomass which contains two specific important monomers, namely p-coumaric acid and ferulic acid (Fig. S1†).7,8 The remarkable high content of p-coumaric acid was discovered and p-coumaric acid units could account for 63.61 per 100 aromatic rings in some herbage.36 Moreover, p-coumaric acid with 99.5% purity could be tailored from corn GVL-lignin in a recent report.37 The selective transformation of methoxy groups in lignin has been reported to produce acetic acid.38 It is attractive and reasonable to regard side chains in lignin as the ethyl donor to generate valuable compounds. Phenols are important commodity chemicals with a broad range of applications, such as in producing resin, bisphenol A and pharmaceuticals.39,40 Currently, phenol is petrochemically produced in industry by a cumene process. Utilization of the renewable lignin fraction as a raw material to produce phenols can liberate us from our reliance on fossil resources.8 Several research studies have been conducted to obtain phenol or catechol from lignin and its monomers,17,41 while the conversion of side chains in lignin is not considered during these transformations. For example, it has been reported that catechol could be prepared from bio-renewable ferulic acid and its derivatives via Brønsted acid-catalyzed defunctionalization while side chains are lost.41N,N-Dimethylethylamine is an important intermediate mainly used in the production of pharmaceuticals, pesticides and other organic chemicals. The production of N,N-dimethylethylamine from lignin has not been reported.
Herein, we proposed a novel strategy to enhance the Caromatic–Cα bond cleavage by pre-functionalization with amine sources. Phenols and N,N-dimethylethylamine compounds were produced from herbaceous lignin and the p-coumaric acid monomer in the presence of industrially available DMA (Scheme 1). First, the p-coumaric acid monomer was decarboxylated to 4-vinylphenol, which then reacted with DMA to form an active amine intermediate via Markovnikov hydroamination. The Caromatic–Csp2 bond was transformed into a Caromatic–Csp3 (–N(CH3)) bond and the bond dissociation energy reduced from 124 kcal mol−1 to 97 kcal mol−1 during the process. The active amine intermediate was converted to phenol and N,N-dimethylethylamine compounds via Caromatic–Csp3 (–N(CH3)) bond cleavage. The yields of phenol and N,N-dimethylethylamine could be up to 89.3% and 85.2%, respectively. This strategy provides a method to achieve the maximum utilization of the aromatic nucleus and side chains in lignin or its platform molecules.
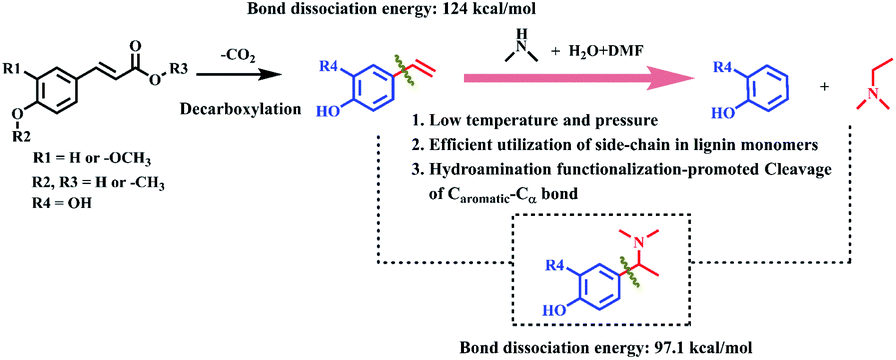 | ||
| Scheme 1 The transformation of lignin monomers into phenols and N,N-dimethylethylamine. | ||
Results and discussion
p-Coumaric acid, featuring carbon–carbon double bond groups, is present in abundance in herbaceous lignin.42–44 We commenced our investigation using p-coumaric acid as a model substrate. Phenol and N,N-dimethylethylamine were produced in the presence of DMA and their yields were strongly dependent on the dosage of DMA. With the increased addition of DMA, the yield of 4-ethylphenol decreased while the production of phenol increased, indicating that a high DMA dosage favors the formation of phenol. The conversion and yields of phenol and N,N-dimethylethylamine were very low when the mole ratios of DMA and p-coumaric acid were 4 and 8, which sharply increased to 77.8% and 70.9%, respectively, when the mole ratio of DMA and p-coumaric acid increased to 20. The yields of phenol and N,N-dimethylethylamine could reach 89.3% and 85.2%, respectively, when the mole ratio of DMA and p-coumaric acid was 40 (Table 1, entries 1–4; Fig. S2–10†). However, p-coumaric acid was mainly converted to 4-ethylphenol in the absence of DMA at 473 K for 1 h with only 14.5% conversion (Table 1, entry 8). It was found that the optimized reaction temperature was 473 K. The decrease of reaction temperature from 448 K to 423 K resulted in a sharp decline in conversion and product yields. Nevertheless, the yields of phenol and N,N-dimethylethylamine at 473 K and 498 K were nearly the same. For all the reactions, the yield of N,N-dimethylethylamine is slightly lower than that of phenol, probably due to its high volatility.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | T [K] | DMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :substrate [mol/mol] :substrate [mol/mol] |
Conv. [%] | Yield [%] | ||
| 2 | 3 | 4 | ||||
| a Reaction conditions: 0.6 mmol substrate, 40 wt% DMA in water (0–2.5 g), 1.5 mL DMF, 1 MPa Ar, 1 h. | ||||||
| 1 | 473 | 4 | 26.4 | 7.3 | 17.2 | 3.2 |
| 2 | 473 | 8 | 34.5 | 21.2 | 11.3 | 14.7 |
| 3 | 473 | 20 | 95.2 | 77.8 | 14.9 | 70.9 |
| 4 | 473 | 40 | >99 | 89.3 | 6.2 | 85.2 |
| 5 | 423 | 40 | 17.1 | 11.6 | 2.8 | 4.7 |
| 6 | 448 | 40 | 78.2 | 71.7 | 4.5 | 69.2 |
| 7 | 498 | 40 | >99 | 83.1 | 5.7 | 80.4 |
| 8 | 473 | 0 | 14.5 | 0.7 | 12.1 | Trace |
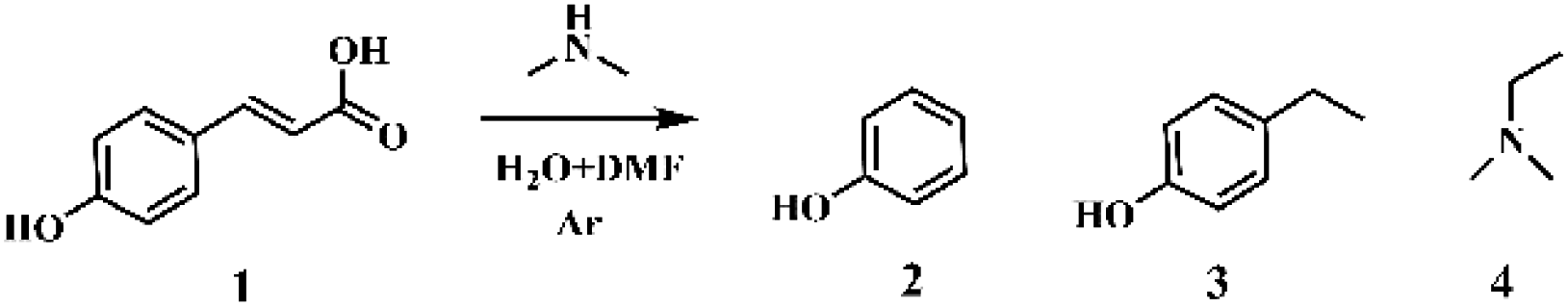
The amine source is not limited to DMA. Other amines, such as primary alkylamines, secondary alkylamines, cyclic amines and aniline, could also afford the transformation of p-coumaric acid to phenol and N,N-dimethylethylamine (Table S1†). The final products are all phenol and N,N-dimethylethylamine in the presence of different amines since all amine sources could react with DMF by transamidation reaction, producing a large amount of DMA with stronger basicity and nucleophilicity. Furthermore, the transamidation reaction occurred prior to the target reaction. Notably, amines with stronger nucleophilic and basicity favor the production of phenol and N,N-dimethylethylamine, due to the rapid transamidation reaction resulting in rapid DMA production. In addition, cyclic amines were less reactive than aliphatic amines. Aniline afforded the lowest yields of phenol and N,N-dimethylethylamine due to its weakest nucleophilicity and basicity. In addition, the conversion of p-coumaric acid is increased 52.1% in the presence of ammonia, and the alkylamine product is still N,N-dimethylethylamine (Table S2†). The reason is that the presence of ammonia enhanced the decomposition of DMF to produce dimethylamine, which further reacted with p-coumaric acid to generate target products.
To understand the reaction mechanism, control experiments were studied to identify the key intermediates. The transformation of p-coumaric acid in the presence of water, DMA and DMF was performed at 448 K and trace amounts of 4-vinylphenol (1c) and 4-(1-(dimethylamino)ethyl)phenol (5) as intermediates were detected during the reaction process (Fig. S11†). Furthermore, the reactivity of other possible reaction intermediates was checked (Scheme 2). 4-Propylphenol (1a) was not reactive since the Caromatic–Csp3 bond with no amine functionalization could not be directly broken in the reaction system. The decarboxylation reaction could not occur for the substrate 3-(4-hydroxyphenyl)propanoic acid (1b), indicating that the presence of a conjugated double bond is crucial to the decarboxylation. 4-Vinylphenol (1c) could be transformed into phenol and N,N-dimethylethylamine with yields of 93.5% and 89.6%, respectively. To confirm the Markovnikov or anti-Markovnikov hydroamination product as the reactive amine intermediate, 1d and 1e were used as substrates to perform the reaction. 4-[2-(dimethylamino)ethyl]phenol (1e) was also nonreactive, while 4-(1-aminoethyl)phenol (1d) could be efficiently transformed into phenol with 83.1% yield. Based on the experimental results and related knowledge from the literature, a possible pathway of this reaction was proposed, which is shown schematically in Fig. 1.
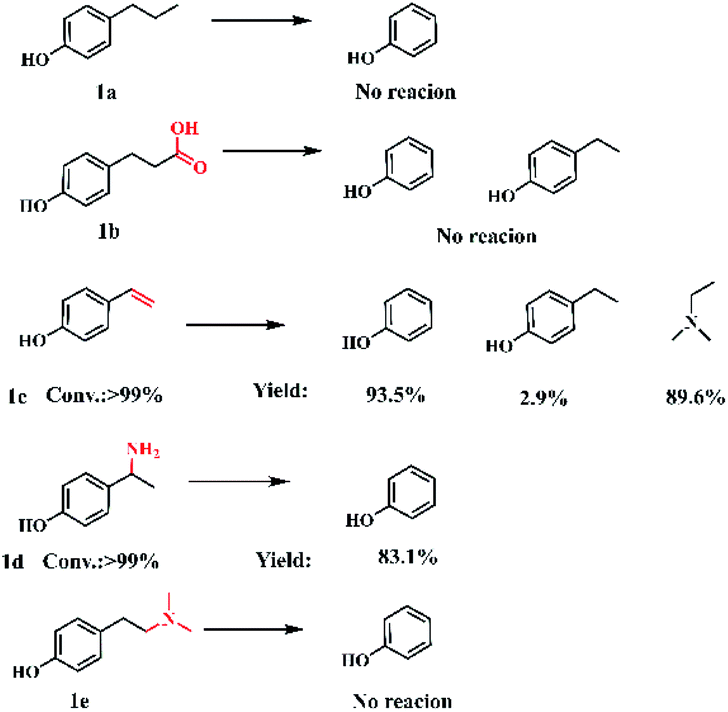 | ||
| Scheme 2 Control experiments. Reaction conditions: 0.6 mmol substrate, 40 wt% DMA in water (2.5 g), 1.5 mL DMF, 473 K, 1 MPa Ar, 1 h. | ||
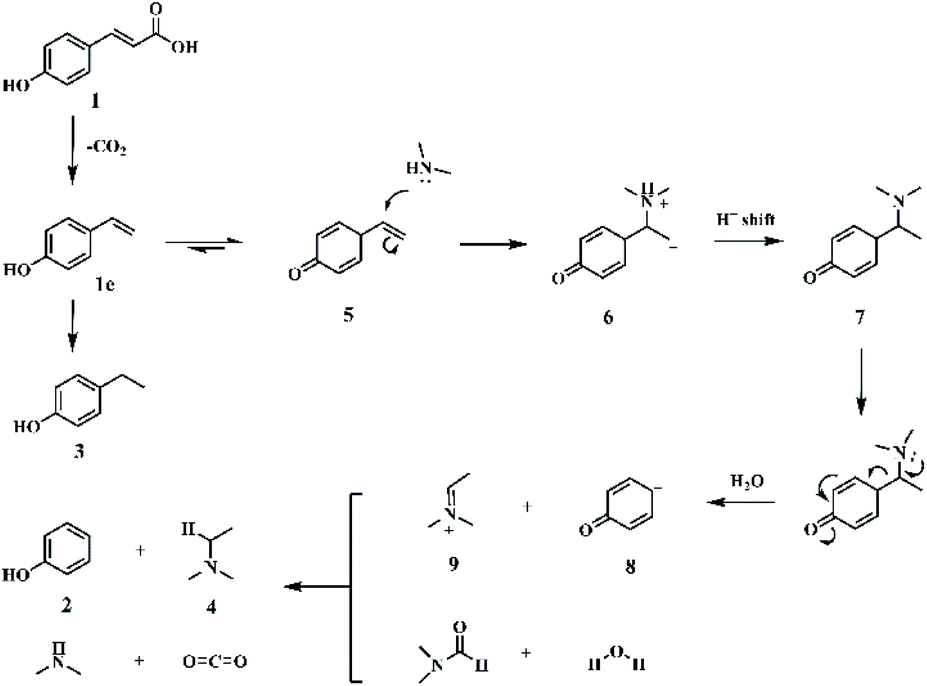 | ||
| Fig. 1 Proposed reaction mechanism. | ||
Initially, the substrate, p-coumaric acid (1), was transformed into 4-vinylphenol (1c) by decarboxylation.45 Then, two parallel routes exist in the following process. One is the hydrogenation of carbon–carbon double bonds in 4-vinylphenol to produce 4-ethylphenol as a by-product (3). The other route involves the Markovnikov hydroamination of 4-vinylphenol to generate 4-[1-(dimethylamino)ethyl]phenol (5), followed by the scission of a Caromatic–Cα bond to form final target compounds with H2O and/or DMF as the hydrogen donors. During the reaction process, anti-Markovnikov hydroamination of 4-vinylphenol didn't occur since no 4-[2-(dimethylamino)ethyl]phenol (1e) was detected. The bond dissociation energy (BDE) of Caromatic–Cα in 4-vinylphenol was found to be 124 kcal mol−1, which reduced to 97.1 kcal mol−1 in 4-[1-(dimethylamino)ethyl]phenol (5) (Fig. S12†), suggesting that the Markovnikov C–N formation promoted the cleavage of the Caromatic–Cα bond.
To further explore the role of DMA except as the reactant during the reaction, the reactivity of 4-(1-aminoethyl)phenol (1d) in the presence or absence of DMA was checked. The results showed that the yield of phenol was only 5.3% and sharply increased to 85.3% in the presence of DMA (Fig. S13†), indicating that DMA promoted the cleavage of carbon–carbon bonds in the intermediate 1d.
It should be mentioned that CO2 and H2 were detected in the gaseous products after reaction (Fig. S14†). To confirm the hydrogen donor for the scission of the Caromatic–Cα bond, controlled experiments for the transformation of 4-(1-aminoethyl)phenol (1d) were performed in the presence of deuterated water (D2O) (Fig. S15–17†), and C6H4DOD (m/z = 96) was detected. However, C6H5OD (m/z = 95) was formed because of hydrogen–deuterium exchange when phenol was treated under the reaction conditions in the presence of D2O. These results reveal that water is the hydrogen donor for the cleavage of the Caromatic–Cα bond. Moreover, 16OC = 18O (m/z = 46) was observed in the gaseous product after the reaction with 4-vinylphenol (1c) as a substrate when H218O was added in the reaction system (Fig. S18†). During the reaction process, DMF reacted with H2O to produce DMA, CO2 and H2. However, the yield of the product was very low when DMA is absent from the starting material because of the vital role of the sufficient amount of DMA and the basic environment in the reaction (Table 1, entry 8).
DMF could facilitate the decarboxylation reaction,45 which is the first step in the conversion of p-coumaric acid into phenol and N,N-dimethylethylamine. The effect of DMF dosage in water on the product distribution was investigated (Table S3†). In the absence of DMF, only 54.3% yield of phenol was obtained and no N,N-dimethylethylamine was produced. Furthermore, no CO2 was detected in the gaseous products, indicating that decarboxylation reaction didn't occur in the absence of DMF (Fig. S19†). The introduction of DMF in water obviously enhanced the yields of phenol and N,N-dimethylethylamine. However, the effect of DMF dosage on the product distribution from 0.5 to 1.5 mL is negligible.
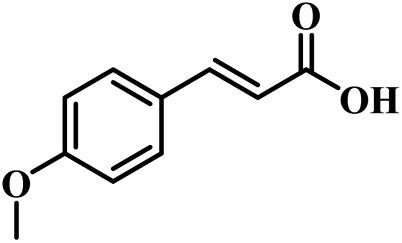
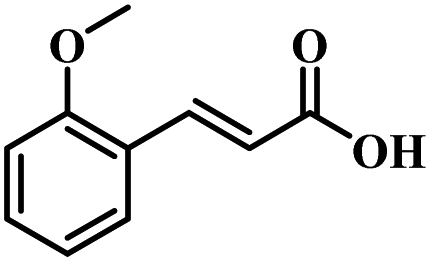
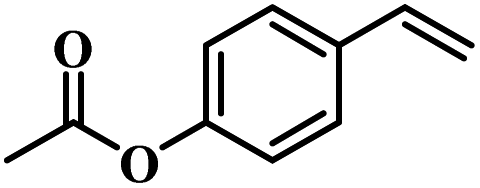
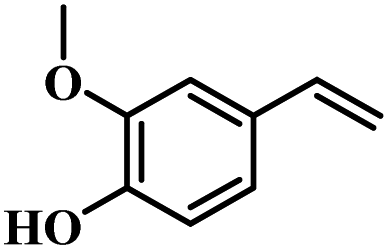
The reactivity of other lignin derivatives with carbon–carbon double bonds and phenolic hydroxyl/methoxy groups was also checked under the optimal reaction conditions (Table 2). Interestingly, 4-methoxystyrene (1f) could also be transformed into phenol and N,N-dimethylethylamine with a satisfactory yield, indicating that the methoxy groups could be hydrolyzed to phenolic hydroxyl groups in this reaction system. Anethole (1g) can be converted to phenol and N,N-dimethylpropylamine with 86.9% and 83.1% yields, respectively. 3-(4-Methoxyphenyl)acrylic acid (1h) afforded 89.2% and 85.2% yields of phenol and N,N-dimethylpropylamine in 4 h. The effect of reaction time on the transformation of 3-(4-methoxyphenyl)acrylic acid was conducted to further confirm the reaction route (Fig. S20†). It was shown that 4-methoxystyrene (1f) and 4-vinylphenol (1c) are key reaction intermediates, which further confirmed that decarboxylation is the first step in the cascade reaction. In addition, the position of the methoxy group in the aromatic rings had a significant effect on the reactivity of the substrate. >99% conversion of 3-(2-methoxyphenyl)acrylic acid (1i) could be obtained and the yield of phenol and N,N-dimethylethylamine could reach 95.4% and 90.6%, respectively. Unfortunately, no phenol or N,N-dimethylethylamine was detected for the transformation of 3-(3-methoxyphenyl)acrylic acid (1j). A possible reason may be that the methoxy group in the m-position didn't activate the Caromatic atom in the Caromatic–Csp2 bond. Moreover, 4-vinylphenyl acetate (1k) could also be converted into targeted compounds with high yields, meaning that acetoxy functional groups could be hydrolyzed to phenolic hydroxyl groups. Considering that p-coumaric acid mainly exists in native lignin in its ester forms, we checked the reactivity of methyl 3-(4-hydroxyphenyl)acrylate (1l), which could be transformed into phenol and N,N-dimethylethylamine with yields of 86.1% and 87.8%, respectively. 2-Methoxy-4-vinylphenol (1m) afforded catechol with yields of only 66.4%, while the yield of alkylamine could reach 82%. The instability of catechol in DMA aqueous solution resulted in lower yield of catechol. Additionally, ferulic acid (1n), an important structural unit in herbaceous lignin, could be converted to catechol with 58.8% yield and N,N-dimethylethylamine with 79.2% yield.
Inspired by the results obtained from the experiments with lignin derivatives, we applied the methodology to the conversion of bamboo lignin containing a substantial number of p-coumaric acid and ferulic acid units. The structure of bamboo lignin was determined from 2D HSQC NMR spectra (Fig. S20†). Interestingly, 4 wt% phenols and 3.6 wt% N,N-dimethylethylamine were obtained. Furthermore, the disappearance of the p-coumaric acid, ferulic acid and methoxy group peaks in the 2D HSQC NMR spectra after reaction confirmed the successful transformation of bamboo lignin. For real lignin, the yields of target products are not too high, which was ascribed to the natural recalcitrance and complexities of lignin. In the catalytic system, two specific important monomers, namely p-coumaric acid and ferulic acid, were first tailored through the quick hydrolysis of the ester bonds. Then, the two monomers were transformed into target chemicals. However, the rest of the lignin fraction may be re-polymerized.
|
|
||
|---|---|---|
| a Reaction conditions: 0.6 mmol substrate, 40 wt% DMA in water (2.5 g), 1.5 mL DMF, 473 K, 1 MPa Ar, 4 h. b Reaction conditions: 0.6 mmol substrate, 40 wt% DMA in water (1.5 g), 1 g water, 1.5 mL DMF, 473 K, 1 MPa Ar, 4 h. | ||

|

|
|
| 1f: 91.7% 2 | 1g: 86.9% 2 | 1h: 89.2% 2 |
| 93.4% 4 | 83.1% 7 | 85.2% 4 |
| Conv.: >99% | Conv.: >99% | Conv.: >99% |
|
|
|
| 1i: 95.4% 2 | 1j: 0% 2 | 1k: 89.4% 2 |
| 90.6% 4 | 0% 4 | 87.1% 4 |
| Conv.: >99% | Conv.: >99% | Conv.: >99% |

|
|

|
| 1l: 87.8% 2 | 1m: 66.4% 6b | 1n: 58.8% 6b |
| 86.1% 4 | 82.0% 4 | 79.2% 4 |
| Conv.: >99% | Conv.: >99% | Conv.: >99% |
Conclusions
In summary, we have proposed a novel strategy to cleave Caromatic–Cα bonds, which involves pre-functionalization of the Cα with amine and then cleavage of Caromatic–Cα bonds. This new strategy has been successfully applied in the selective transformation of lignin platform chemicals, and both aromatic rings and side-chains are transformed into valuable chemicals with high yields. Further research demonstrated that the reaction pathway involves the initial decarboxylation of p-coumaric acid into 4-vinylphenol, followed by Markovnikov hydroamination of 4-vinylphenol with DMA to give the active amine intermediate, and the final scission of the Caromatic–Cα bond in the amine intermediate. We believe that the new method to cleave Caromatic–Cα bonds has wide application in exploring efficient organic reactions, and the strategy of simultaneously transforming aromatic rings and side-chain moieties in lignin platform molecules into corresponding valuable chemicals with high yields has promising potential for application in valorization of lignin.Experimental section
Preparation of bamboo lignin
The bamboo lignin was prepared as follows.46 Bamboo was milled for 1 h at 450 rpm in a planetary ball mill (Fritsch GMBH, Idar-Oberstein, Germany). The ball-milled wood (5.0 g) was then suspended in acetate buffer (0.05 mol L−1, 100 mL, pH 4.8), with the loading of 100 FPU cellulase (Celluclast 1.5 L) and 100 FPU β-glucosidase (Novozyme 188). The reaction mixture was incubated at 50 °C in a rotary shaker (150 rpm) for 48 h. After enzymatic hydrolysis, the residue was collected by centrifugation and washed with acidified water (pH 2) followed by freeze-drying. The residues were then treated with 0.05 M HCl in acidic dioxane–water (85:15, v/v) at 86 °C under a nitrogen atmosphere for 2 h. The resulting mixture was filtered and the filtrate containing lignin was collected. The solid residue was washed with fresh dioxane–water (85:15, v/v) until the filtrate became clear. The filtrate and washings were combined and then neutralized with solid sodium bicarbonate. The neutralized solution was concentrated and finally precipitated in a large quantity of acidified water (10 volumes, pH 2). The precipitated lignin was collected by centrifugation and freeze-dried under high vacuum. To remove the carbohydrate remaining in the lignin, the lignin was dissolved in 90% acetic acid (20 mL), and then the lignin solution was dropped into 10 volumes of acidic water (pH 2) producing a lignin precipitate. The precipitated lignin was washed with acidified water several times and then freeze-dried.
Catalytic performance
Typically, the reaction of p-coumaric acid with dimethylamine (DMA) was conducted in a 10 mL Teflon-lined stainless-steel autoclave. p-Coumaric acid (0.1 g, 0.6 mmol), DMF (1.5 mL) and 40 wt% DMA aqueous solution (2.5 g) were transferred into the autoclave. The reactor was charged to 1 MPa Ar pressure. The reaction was then performed at 473 K under magnetic stirring for 1 h. After reaction, the autoclave was quenched in an ice-water bath to room temperature. The liquid solution was analyzed qualitatively by GC-MS (Agilent, 5977A) and Nuclear Magnetic Resonance (NMR) and quantitatively with a GC equipped with a flame ionization detector (FID, Agilent, 4890D) using 1,4-dioxane as the internal standard. Conversion of p-coumaric acid and yield of products were calculated by using the equations| p-Coumaric acid conversion = (moles of reacted p-coumaric acid)/(moles of starting p-coumaric acid) × 100% |
| Phenol yield = (moles of phenol)/(moles of starting p-coumaric acid) × 100% |
| N,N-Dimethylethylamine yield = (moles of N,N-dimethylethylamine)/(moles of starting p-coumaric acid) × 100% |
Data availability
The experimental data has been placed in ESI.†Author contributions
Y. X. and H. Z. L. conceived and designed the present work. Y. X. and H. Z. L. wrote the manuscript and B. X. H. reviewed the manuscript. Y. X. conducted all of the experimental work. Z. P. W., X. J. S. and M. H. D. assisted with GC-MS, NMR measurement and calculation of bond energy. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (21871277, 22179132, 22003069).References
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed
.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed
- M. Stcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed
- C. Xu, R. A. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed
- H. Li, A. Bunrit, N. Li and F. Wang, Chem. Soc. Rev., 2020, 49, 3748–3763 RSC
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed
- X. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499–11503 CrossRef CAS PubMed
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8, 16104 CrossRef PubMed
- C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935–5940 CrossRef CAS PubMed
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yua and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 6310 CrossRef
- M. Wang, J. Ma, H. Liu, N. Luo, Z. Zhao and F. Wang, ACS Catal., 2018, 8, 2129–2165 CrossRef CAS
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed
- M. Wang and F. Wang, Adv.
Mater., 2019, 31, 1901866 CrossRef
- H. Luo, L. Wang, S. Shang, G. Li, Y. Lv, S. Gao and W. Dai, Angew. Chem., Int. Ed., 2020, 59, 19268–19274 CrossRef CAS
- L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303–11307 CrossRef CAS PubMed
- C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528–6532 CrossRef CAS PubMed
- H. Liu, C. Dong, Z. Zhang, P. Wu and X. Jiang, Angew. Chem., Int. Ed., 2012, 51, 12570–12574 CrossRef CAS PubMed
- S. Dabral, P. C. J. Kamer and C. Bolm, ChemSusChem, 2017, 10, 2707–2713 CrossRef CAS PubMed
- T. Stein, T. Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef PubMed
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135(17), 6415–6418 CrossRef CAS
- M. Wang, M. Liu, H. Li, Z. Zhao, X. Zhang and F. Wang, ACS Catal., 2018, 8, 6837–6843 CrossRef CAS
- Y. Wang, Q. Wang, J. He and Y. Zhang, Green Chem., 2017, 19, 3135–3141 RSC
- L. Ma, H. Zhou, X. Kong, Z. Li and H. Duan, ACS Sustainable Chem. Eng., 2021, 9, 1932–1940 CrossRef CAS
- T. Cui, L. Ma, S. Wang, C. Ye, X. Liang, Z. Zhang, G. Meng, L. Zheng, H. Hu, J. Zhang, H. Duan, D. Wang and Y. Li, J. Am. Chem. Soc., 2021, 143, 9429–9439 CrossRef CAS
- H. Zhou, Z. Li, S. Xu, L. Lu, M. Xu, K. Ji, R. Ge, Y. Yan, L. Ma, X. Kong, L. Zheng and H. Duan, Angew. Chem., Int. Ed., 2021, 60, 8976–8982 CrossRef CAS PubMed
- Y. Liao, S. F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolai, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385 CrossRef CAS PubMed
- Y. Liao, M. d'Halluin, E. Makshina, D. Verboekend and B. F. Sels, Appl. Catal., B, 2018, 234, 117–129 CrossRef CAS
- X. Huang, J. M. Ludenhoff, M. Dirks, X. Ouyang, M. D. Boot and E. J. M. Hensen, ACS Catal., 2018, 8, 11184–11190 CrossRef CAS PubMed
- J. Yan, Q. Meng, X. Shen, B. Chen, Y. Sun, J. Xiang, H. Liu and B. Han, Sci. Adv., 2020, 6, eabd1951 CrossRef CAS PubMed
- T. Chen, Z. Li, X. Zhang, D. Min, Y. Wu, J. Wen and T. Yuan, Polymers, 2018, 10, 1157 CrossRef PubMed
- V. I. Timokhin, M. Regner, A. H. Motagamwala, C. Sener, S. D. Karlen, J. A. Dumesic and J. Ralph, ACS Sustain. Chem. Eng., 2020, 8, 17427–17438 CrossRef CAS
- Q. Q. Mei, H. Z. Liu and B. X. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS
- S. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105–107 CrossRef CAS
- Y. Morimoto, S. Bunno, N. Fujieda, H. Sugimoto and S. Itoh, J. Am. Chem. Soc., 2015, 137, 5867–5870 CrossRef CAS
- J. Bomon, E. Van Den Broeck, M. Bal, Y. Liao, S. Sergeyev, V. Van Speybroeck, B. F. Sels and B. U. W. Maes, Angew. Chem., Int. Ed., 2020, 59, 3063–3068 CrossRef CAS PubMed
- Y. Xi, S. Ma and J. F. Hartwig, Nature, 2020, 588, 254–260 CrossRef CAS PubMed
- A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef CAS
- S. Streiff and F. Jerome, Chem. Soc. Rev., 2021, 50, 1512–1521 RSC
- D. Kong, P. J. Moon, E. K. J. Lui, O. Bsharat and R. J. Lundgren, Science, 2020, 369, 557 CrossRef CAS PubMed
- X. Shen, Q. Meng, Q. Mei, H. Liu, J. Yan, J. Song, D. Tan, B. Chen, Z. Zhang, G. Yang and B. Han, Chem. Sci., 2020, 11, 1347–1352 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2, and Fig. S1–S15. See DOI: 10.1039/d1sc05231d |
| This journal is © The Royal Society of Chemistry 2021 |