
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenation of a twisted non-polar π-system as a tool to modulate phosphorescence at room temperature†
Giliandro
Farias
a,
Cristian A. M.
Salla
b,
Murat
Aydemir
cd,
Ludmilla
Sturm
e,
Pierre
Dechambenoit
e,
Fabien
Durola
 e,
Bernardo
de Souza
*a,
Harald
Bock
*e,
Andrew P.
Monkman
*c and
Ivan H.
Bechtold
*b
e,
Bernardo
de Souza
*a,
Harald
Bock
*e,
Andrew P.
Monkman
*c and
Ivan H.
Bechtold
*b
aDepartment of Chemistry, Universidade Federal de Santa Catarina, 88040-900 Florianópolis, SC, Brazil. E-mail: bernadsz@gmail.com
bDepartment of Physics, Universidade Federal de Santa Catarina, 88040-900 Florianópolis, SC, Brazil. E-mail: ivan.bechtold@ufsc.br
cDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: ap.monkman@durham.ac.uk
dErzurum Technical University, Department of Fundamental Sciences, Erzurum, Turkey
eCentre de Recherche Paul Pascal, CNRS, Université de Bordeaux, 115, av. Schweitzer, 33600 Pessac, France. E-mail: harald.bock@crpp.cnrs.fr
First published on 1st November 2021
Abstract
Halogenation of a twisted three-fold symmetric hydrocarbon with F, Cl or Br leads to strong modulation of triplet–triplet annihilation and dual phosphorescence, one thermally activated and the other very persistent and visible by eye, with different relative contributions depending on the halide. The room temperature phosphorescence is highly unusual given the absence of lone-pair-contributing heteroatoms. The interplay between the spin–orbit coupling matrix elements and the spatial configuration of the triplet state induces efficient intersystem crossing and thus room temperature phosphorescence even without relying on heteroatomic electron lone pairs. A ninefold increase of the ISC rate after introduction of three bromine atoms is accompanied by a much higher 34-fold increase of phosphorescence rate.
Introduction
Persistent room temperature phosphorescence (RTP) has drawn considerable attention in the past few years, and its potential application has extended to bioimaging,1–3 encryption,4 light sources,5–7 and nonlinear optics.8 However, phosphors have been mainly limited to inorganic or organometallic materials,2,9,10 while pure organic phosphors with RTP are extremely rare but of great practical interest.11,12 This scarcity of organic phosphors is mainly due to the usually very weak spin–orbit coupling (SOC) and the instability of triplet excitons, combined with an only sketchy understanding of the inherent mechanisms involved.11,13–16SOC is the dominant mechanism that induces the spin-flip transition and generates the triplet states through intersystem crossing (ISC).17,18 In general, the SOC is much greater in heavy element-containing systems due to its dependence on the nuclear charge, which is referred to as the heavy atom effect (HAE).19 Thus, in many systems containing atoms such as Ir and Pt, ISC and phosphorescence (Ph) are dominant.10,20,21 However, enhancing SOC in purely organic chromophores through a non-metal HAE using Cl, Br, I,22,23 and most recently S, Se, and Te24–26 has attracted attention for developing sustainable strategies to induce ISC.
Several groups have employed different methodologies to develop efficient organic RTP systems. However, only a few examples of organic materials with persistent RTP in the air with a long lifetime (τ) (>10 ms) have been described.4,13,16,27–34 The incorporation of heteroatoms with electron lone pairs, such as N and O, has been the most common strategy to induce RTP due to the effective SOC related to transitions between (π,π*) and (n,π*) states, following El-Sayed's rule.35 Halogenation of such systems has been shown to be a versatile tool to control lifetime and quantum yield (Φ).23,30,31 Recently, alternative strategies to induce RTP and enhance its efficiency were investigated, including molecular aggregation,16,27,36 energy-gap narrowing37,38 and twisting of the π-system.39
In order to elucidate these aspects, we investigated our recently discovered twisted hydrocarbon homotruxene (HTX)39 through the introduction of three new triply halogenated homologs HTX-F, HTX-Cl, and HTX-Br (Fig. 1a), which are highly unusual as they show pronounced RTP in the absence of (heteroatom) electron lone pairs. The RTP of HTX, HTX-F, and HTX-Cl is long-lived and readily visible by the eye after switching off the excitation source, while HTX-Br shows strong Ph under UV-illumination (Fig. 1b). Time-dependent measurements and density functional theory (DFT) calculations reveal that these twisted π-systems can effectively induce ISC and thus RTP. Using the halogenated homologs, we evaluated the HAE in terms of radiative decay rates and spin–orbit coupling matrix elements (SOCME). The HAE has distinct, independent effects on ISC and Ph depending on the configuration of the low-lying excited states, which depends on the halide atom. Our results show the relations between emission mechanisms, the spatial configurations of the triplet state wavefunctions, and SOCME, which control both the independent access and the deactivation of the triplet state in twisted π-systems, allowing the design of new and efficient purely organic RTP emitters.
 | ||
| Fig. 1 Molecular structures and RTP under air from the powder. (a) Homotruxenes with increasingly heavy halogens. (b) Emission from HTX, HTX-F, HTX-Cl and HTX-Br (left to right) captured during the excitation under UV light (∼365 nm) and after switching off the excitation. The time delay was estimated to be approximately 10, 20, 30 and, 70 ms for these selected images. See movie of the emission in the ESI.† | ||
Results and discussion
Synthesis and photophysical properties
The three trihalogenated derivatives were synthesized by TiCl4-induced trimerization of the corresponding tetralone, as previously described for the synthesis of unsubstituted HTX.39 Single crystals of HTX-Cl and HTX-Br for X-ray crystallography were obtained as colorless plates by slow diffusion of ethanol into chloroform solutions. The packing structure of both exhibits π–π interactions, which directly impact their photophysical properties in the solid state (Fig. 2a for HTX-Cl packing and ESI†). | ||
| Fig. 2 Packing of HTX-Cl in the crystal and Absorption of the HTX and halogenated homologs in different solvents. (a) Packing of HTX-Cl in the crystal. Stick representation of the packing in the (ab) plane (left) and along the c axis (right); C grey, Cl green, hydrogen atoms omitted for clarity. (b) Absorption of the HTX and halogenated homologs in dilute chloroform (left) and 2-MeTHF (right) solutions, 10−5 mol L−1 and 10−3 mol L−1, respectively. | ||
In 2-methyltetrahydrofuran (2-MeTHF) solution at room temperature (RT), HTX and its halogenated derivatives show a weak absorption band (ε = 100–200 cm−1 M−1) at ca. 340–370 nm (Fig. S2†). A further, even weaker band (ε = 10–30 cm−1 M−1) is observed between 370 nm and 400 nm. The band at 340–370 nm can be assigned to the S0 → S2 transition by comparison of the theoretical and experimental oscillator strength (OS) (Table S2†), showing an acceptable error, which is mainly due to overestimation of the integrated area of the combined S1 and S2 absorptions in the experimental spectra. The identity of the absorption at 370–400 nm with the S0 → S1 transition was likewise verified by comparing the theoretical and experimental OS and by dilute solution measurements (Fig. S3a†). The absorptions centered at 370–400 nm are extremely weak for very dilute solutions, but excitation at these wavelengths gives strong emission (Fig. S3b–d†). Absorption spectra of the four homologs in dilute chloroform solution exhibit a maximum at 275 nm with a shoulder-like band at 290 nm for HTX, while with the halide derivatives, the band close to 290 is less intense (ε < 4 × 104 L mol−1 cm−1) but more resolved and extends plateau-like with some vibrational resolution to approximately 305 nm. All these intense high energy bands are attributed to π,π* transitions originating mainly from locally excited (LE) states, as confirmed by time-dependent density functional theory (TD-DFT) analysis. Fig. 2b shows the absorption of the four homologs in dilute chloroform (left) and 2-MeTHF (right) solutions.
The steady-state emission spectra of all compounds in dilute 2-MeTHF solution consist of one structured emission peak, with two maxima at about 398 and 412 nm and a shoulder at about 434 nm (Fig. 3) having an average spacing of 190–200 meV (ca. 1620 cm−1). Excitation spectra were measured from these two emission bands, 390 nm and 410 nm (Fig. S3†). The emission at 410 nm is much stronger when excited in the 370–400 nm absorption band, whereas excitation at 340–370 nm yields primarily 390 nm emission. This we assign to two lowest-lying excitation bands giving rise to two overlapping but different emission bands with different vibronic spacings. We further measured the excitation spectra at low concentration (Fig. S3d†). Monitoring at 390 nm reveals excitation peaks at ca. 320–330 nm and 345 nm, which could be a vibronic progression with vibronic spacing of 110 meV, whilst monitoring at 410 nm, we observe a distinct excitation band with the electronic 0–0 emission peak at ca. 331 nm and three well-resolved vibronic peaks with spacing of 169 meV. These observations fully support our assignment of absorption and emission from both the S0 → S1 and S0 → S2 transitions and agree with our theoretical predictions (see below). The emission of all compounds is slightly dependent on the solvating environment (Fig. S4†). In toluene solution, a slight red shift of approximately 1 nm is observed compared to 2-MeTHF. For the halogenated compounds, an extra emission feature on the blue edge at ca. 375 nm can be observed, which is weaker in toluene than in 2-MeTHF. As a further distinction between HTX and its halogenated derivatives, the mode related to the maxima at 412 nm in HTX is slightly more intense than the mode related to the maxima at 398 nm, suggesting a larger distortion in the excited state than for the halogenated systems where the 398 nm mode is strongest, which was confirmed analyzing their optimized geometries (compare S2 RMSD in Table S10†). At 80 K, a new emission peak is observed for all compounds with two maxima in the range of 555–616 nm, which may be assigned to Ph (Fig. 3, dashed line).
 | ||
| Fig. 3 Emission spectra in dilute 2-MeTHF solution. The solid lines and filled areas are the steady-state spectra in degassed and in aerated solution at RT. Emission intensity increases by 87% for HTX, 30% for HTX-F, 27% for HTX-Cl and 18% for HTX-Br in degassed solution compared to aerated solution. The dashed lines are the emission spectra collected at 90 K. | ||
The whole emission band is oxygen-dependent (Fig. 3, filled area and Fig. S5†), and the part suppressed in air is assigned to delayed fluorescence (DF) from triplet–triplet annihilation (TTA), whereas the part observed in air is assigned to prompt fluorescence (PF). The measured PF quantum yields (ΦPF) (Table S3†) are in the range of 32.8% (HTX-F) to 1.1% (HTX-Br) in 2-MeTHF, and 17.9% (HTX) to 1.2% (HTX-Br) in toluene. The DF quantum yields (ΦDF) determined by the comparison of degassed to aerated emission intensity show that the contribution of the (oxygen-sensitive) delayed emission decreases from HTX (18.5%) to HTX-Br (0.2%) in 2-MeTHF, while in toluene, it is highest for HTX-F (2.1%) and lowest for HTX-Br (0.4%). HTX-Br presents the lowest ΦPF and ΦDF values, and it is the only compound where DF is higher in toluene than in 2-MeTHF. The decrease of ΦDF in 2-MeTHF with increasing substituent weight is due the increasingly fast HAE-assisted deactivation of the triplet state by Ph or non-radiative decay. As DF from TTA depends on the triplet concentration, a strong decrease of the DF intensity is observed as the triplet population is quenched by these other decay channels.
Time-correlated single-photon counting (TCSPC) emission decays obtained from 2-MeTHF solutions and fitted with tri-exponentials (Fig. S6†) revealed one dominant fast decay lifetime close to 1 ns (typically, >90%) and one ranging from 2.5 ns (HTX-Cl) to 13 ns (HTX) ns (typically, 5–10%). These two intense components with lower lifetimes perfectly match with the observed emission band from two overlapping states. The third component with a lifetime close to 23 ns has a very low contribution (typically 0.2%) and potentially could indicate a very small aggregate content.
Nanosecond time-resolved delayed emission data were obtained using a gated detection technique,40 in degassed 2-MeTHF solution. The decay curves of all compounds are characterized by a rapid component (PF) followed by a slower one (Fig. 4a). For HTX at RT, from time delay (TD) = 1.1 ns to 100 ns (Fig. 4b, black line), an emission peak at 415 nm (τ = 12.7 ns) with a weak vibronic structure is observed. At TD = 1.6–27 μs, emission from the same peak is observed, ascribed to TTA-DF. At an even longer delay, beyond TD = 27 μs, a continuous red-shift is observed, and the band becomes Gaussian-shaped, which is associated with the contribution from a relatively fast Ph at 440–450 nm (τ = 89.6–293.7 μs) (Fig. 4b, light grey line). The decay curves as a function of the temperature show only a slight difference in the PF (Fig. 4c). At 90 K the emission at TD = 1.1 ns is slightly blue-shifted, centered at 412 nm, due to the decrease of vibrational relaxation at low temperature. The long-lived emission component in the μs to ms range is almost completely gone, in line with the DF arising from collisional TTA, which is frozen out at 90 K and the faster Ph being thermally activated. However, a clear emission peak ascribed to a slower Ph emission (τ = 498 ms) is dominant from TD = 376 μs to 8.4 ms (Fig. 4b, dashed line). The slower Ph emission is well-structured and shows two main peaks at 565 and 604 nm and a shoulder close to 650 nm. The decay curve in toluene at RT (Fig. 4d) is very similar to the curve in 2-MeTHF in the ns window, whereas the long-lived emission is more intense in toluene than in 2-MeTHF. According to our theoretical predictions, this is attributed to the CT character of the fast Ph species, which is less quenched in the non-polar solvent through a non-radiative pathway to the longer lived species, thus the intensity of the long-lived component in toluene solution is higher. In aerated solution, the DF and Ph components disappear, confirming the triplet state contribution (see ESI and Fig. S7† for fitting parameters and peak positions as a function of TD). To confirm the TTA mechanism, the intensity dependence of the DF emission was analyzed as a function of the laser excitation dose, where a slope close to 2 (1.70 ± 0.06) was found, indicating a bimolecular process (Fig. 4e and f) as required for TTA.
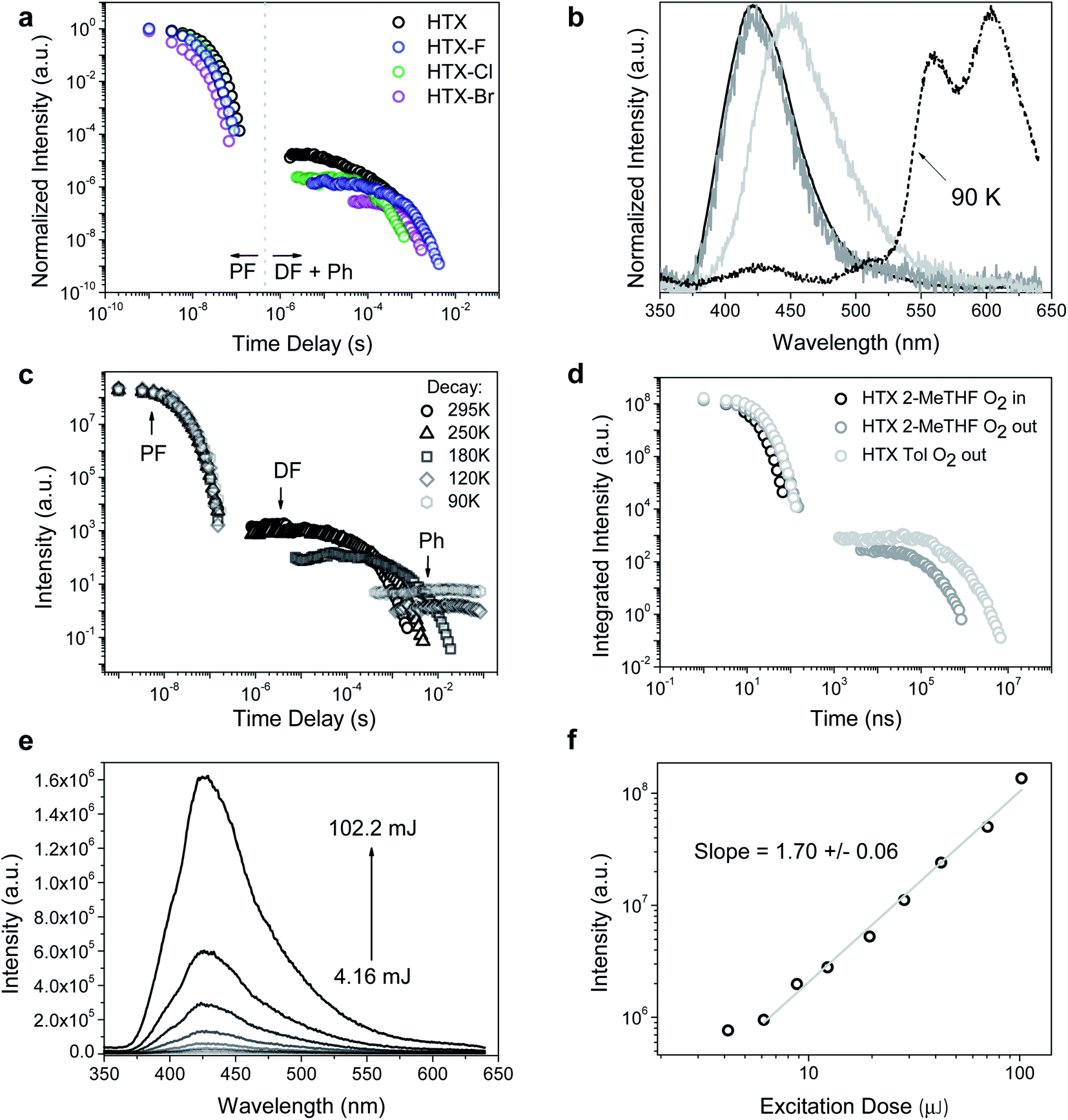 | ||
| Fig. 4 Time-resolved measurements for HTX and halogenated homologs. (a) Time-resolved decay in 2-MeTHF at RT. (b) Normalized emission spectra in degassed solution at 295 K for HTX (black line: TD = 1.1–100 ns; grey line: TD = 1.6–27 μs; light grey line: TD = 422 μs to 1.2 ms) and normalized emission spectra of the long-lived component collected at 90 K (TD = 376 μs to 8.4 ms, dashed line). (c) Time-resolved decay of HTX in 2-MeTHF as function of temperature (see ESI† for fitting parameters). (d) Time-resolved decay of HTX as function of the environment. (e) Emission spectra and (f) intensity as a function of the laser excitation dose for HTX in the absence of oxygen (excitation at 337 nm, TD = 13 μs, time of integration (Ti) = 100 μs). The excitation wavelength is 355 nm. | ||
By measuring the decay curves at decreasing concentrations (Fig. S8a†) the PF decay can be resolved into a triexponential decay at RT, very similar to the TCSPC results. This is attributed to decreasing non-radiative decay pathways through molecular collision, thus increasing the S1 and S2 states lifetime. The normalized spectra taken over the entire measurement confirmed the existence of different emitting species. At TD = 1.1 ns in dilute conditions, a well-resolved emission peak centered at 432 nm is observed related to S1 emission (Fig. S8b†). Around TD = 5.85 ns for all concentrations, the dominance of the S2 emission is observed at 412 nm (Fig. S8c†). No evident relaxation of this state is seen over ns time frame for all concentrations. For the long-lived decay, the TTA DF from the same excited state can be observed for all concentrations (Fig. S8d†). The spectra taken at low temperature show a similar concentration dependence. The spectra taken at low temperature confirm low energy PF from S1. By cooling down, very well-structured spectra are obtained in dilute solutions in the fast TD regime (Fig. S8e†). The Ph measured in dilute conditions shows a slight blue-shift of the emission and an increase of the 0–0 mode with decreasing concentration (Fig. S8f†). The Ph shift to low energy in more concentrated solutions can be attributed to the triplet state stabilization through intermolecular interactions.
We further investigated the initial prompt emission spectra, given that it decays biexponentially. We observe a rapidly evolving emission band at the earliest times that we can measure (Fig. S9†). We observe a relatively red-shifted red band that peaks around 440–450 nm in the first few measurement time frames around time zero. This emission peak vanishes within 1 ns, giving the fast decay component observed. We are left with a stronger band peaking at 410 nm, having a lifetime of ca. 12 ns. The time-resolved emission from this band shows no significant vibrational structure, compatible with hot emission from unrelaxed states.41 These observations fit with the emission-dependent excitation profiles we observe and confirm the mixed nature of the prompt emission.
The emission behavior of HTX-F and HTX-Cl is similar to HTX, with the emission decay curves showing a dual prompt and a delayed emission at RT and long-lived Ph becoming dominant on cooling (Fig. S10 and S11†). For HTX-F the emission curves shift from 440 nm (TD = 1.1 ns, τ = 9.4 ns) to 411 nm (TD = 101 ns, τ = 48.9 ns) and for HTX-Cl the emission curve shifts from 444 nm (TD = 1.1 ns, τ = 2.46 ns) and stabilizes at 412 nm (TD = 30 ns, τ = 31.5 ns). For HTX-F the faster Ph is observed from TD = 75 μs to 2.12 ms and for HTX-Cl from TD = 133–670 μs. These differences observed between HTX and the lighter halogenated homologs are due to the distinct decay rates of low-lying states upon halogenation. The DF analysis as a function of the laser excitation dose also showed quadratic gradients of 2.06 ± 0.05 (HTX-F) and 2.22 ± 0.07 (HTX-Cl) coherent with TTA (Fig. S14†). On cooling down to 90 K the long-lived decay is mainly due to slow Ph emission with τ = 766 ms for HTX-F and τ = 315 ms for HTX-Cl.
Compared to the three other compounds, HTX-Br shows in the PF a blue-shift of the red edge of the emission peak and stabilizes at 416 nm (TD = 12 ns) (Fig. 5b). The slower components of the emission show distinct features: the slow Ph can be seen at RT along with the TTA, the Ph being the dominant component and increasing on cooling down to 90 K (Fig. 5b and S12†). On cooling down, the lifetime of this Ph increases from 805 μs (RT) to 14.8 ms (90 K). Fig. 5c shows emission spectra as a function of the laser excitation dose, where the DF (435 nm) and Ph (590 nm) peaks are clearly visible. In Fig. 5d the quadratic gradient of 2.01 ± 0.15 at low excitation dose turns to 1.44 ± 0.03 at higher excitation doses for the 435 nm peak, where not yet triplet-excited molecules become scarce, again confirming that the DF arises from a TTA mechanism.42 One the other hand, the 1.01 ± 0.04 slope of the 590 nm peak confirms (monomolecular) Ph emission at long wavelengths.
 | ||
| Fig. 5 Time-resolved measurements for HTX-Br. (a) Time-resolved decay of HTX-Br in 2-MeTHF as function of temperature. (b) Normalized spectra taken after different TD at RT; TD = 1.1–9 ns (black line); TD = 12–66 ns (grey line); TD = 12–699 μs (light grey line); TD = 751 μs to 3.7 ms (purple line). The excitation wavelength was 355 nm. All measurements were performed in the absence of oxygen. Emission spectra (c) and intensity (d) as a function of the laser excitation dose, excitation at 337 nm, TD = 20 μs, Ti = 1 ms. | ||
In the solid state, the PF is observed in the time-resolved emission spectra at earlier times with biexponential decay for HTX, HTX-Cl and HTX-Br due to a dimmer/aggregation emission contribution (Fig. S16†). For all compounds, the slow Ph pathway dominates. Only a small contribution of the fast Ph pathway is seen. The slow Ph at seen at low energy in the three lighter homologs is long-lived, with lifetimes of 0.21, 0.20, and 0.11 s for HTX, HTX-F, and HTX-Cl, respectively. The ΦPh was not measurable in solution, but in powder, at RT it is 6.2%, 3.3%, 2.7% and 6.3% for HTX, HTX-F, HTX-Cl and HTX-Br (Table S5†), respectively, which allows the Ph in the solid to be visible by eye after switching off the excitation (except for HTX-Br, whose Ph is too fast for visible afterglow). This increased efficiency of the Ph emission in the solid state is attributed to the molecular aggregation due to dense stacking as observed in the X-ray structures.16 The ΦPh of HTX-Br is the same as of HTX, but with a much shorter lifetime of 8.14 ms, not being observable by eye after switching-off the excitation. But as the relative intensity of the orange phosphorescence versus the blue fluorescence of HTX-Br is much higher than with HTX, the Ph of HTX-Br is apparent to the human eye in the dark by the deep orange color of the emission from the powder under UV excitation, whereas the emission of HTX is bluish in the same conditions.
With the usual assumption that the quantum yield for internal conversion (ΦIC) is negligible, we computed the decay rates of (prompt) fluorescence (kPF), phosphorescence (kPh) and intersystem crossing (kISC). In solution the highest kPF is obtained for HTX-F, while the substitution of halides increases the kISC (Table 1). In solid state the PF rate decreases from HTX to HTX-Br while kISC increases. kPh is particularly affected by the heavier bromide atom. This large enhancement of kPh while kISC is not much affected by the Br substituent can be explained by different heavy-atom effects operating in the ISC and the Ph processes (see below).
| HTX | HTX-F | HTX-Cl | HTX-Br | ||
|---|---|---|---|---|---|
| a Obtained from TCSPC measurements at RT and defined as τ = ∑τi2Ai/∑τiAi, for a tri-exponential profile. b Obtained from time-resolved measurements at 90 K. c k PF = ΦPF/τPF. d k ISC = ΦISC/τPF, considering ΦISC = 1 − ΦPF and assuming, as usual, that the Φ for internal conversion is nil, ΦIC = 0. e k Ph ∼ τPh−1. f Obtained from time-resolved measurements at RT. g k Ph = ΦPh/τPh. | |||||
| Solution | τ PF (ns)a | 8.19 | 2.60 | 1.36 | 1.06 |
| Φ PF | 0.211 | 0.328 | 0.078 | 0.011 | |
| τ Ph (ms)b | 498 | 766 | 315 | 14.8 | |
| k PF × 107 (s−1)c | 2.58 | 12.61 | 5.74 | 1.04 | |
| k ISC × 108 (s−1)d | 0.96 | 2.58 | 6.79 | 9.33 | |
| k Ph (s−1)e | 2.01 | 1.30 | 3.17 | 67.57 | |
| Powder | τ PF (ns)f | 11.40 | 8.90 | 5.97 | 2.26 |
| Φ PF | 0.410 | 0.197 | 0.103 | 0.012 | |
| τ Ph (ms)b | 210 | 200 | 111 | 8.34 | |
| Φ Ph | 0.062 | 0.033 | 0.027 | 0.063 | |
| k PF × 107 (s−1)c | 3.60 | 2.21 | 1.73 | 0.53 | |
| k ISC × 107 (s−1)d | 5.18 | 9.02 | 15.03 | 43.72 | |
| k Ph (s−1)g | 0.29 | 0.17 | 0.24 | 7.55 | |
Theoretical investigations
DFT calculations were performed to obtain the ground state geometries (Fig. S17†), showing a good agreement with the X-ray structures. The calculated frontier orbitals for all compounds are shown in Fig. S18,† where the HOMO and HOMO−1 are almost degenerate π orbitals located mainly at the central ring, while the LUMO and LUMO+1 are the corresponding π* orbitals, with contributions both on the inner and part of the outer rings. The HOMO−2 and the LUMO+2, in contrast, are located quite selectively at the outer rings. For the halide substituted compounds, a significant contribution of the heteroatoms to the HOMO−2, HOMO−1 and HOMO is observed.Based on these geometries, the theoretical absorption spectra were modeled using TD-DFT (Fig. S19†). TD-DFT density difference plots indicate that the S1 and S2 excited states arise from π,π* transitions at the central ring. S1 has characteristics of a charge-transfer (CT) resonance state,43 while S2 is more likely a LE state. This agrees with the experimental data with the S1 → S0 transition having very low extinction and low OS. The low-lying triplet states are essentially combinations of the same excitations from these frontier orbitals, however with additional contributions from the outer rings to T1–T3, with implications on the SOCME.
To evaluate both the Ph and the ISC pathways, SOCME were calculated for the S1 and T1 geometries (Fig. 6a, b, S20 and S21†).44 As the ISC should occur mainly from S1, the ISC decay rate is proportional to the magnitude of the SOCME between S1 and all the triplets below (T1–T6). From HTX to HTX-Br, the sum of the SOCME between S1 and T1–T6 increases approximately by a factor of four. The Ph can be correlated to the SOC between all the low-lying triplets and the ground state. Compared to HTX, substitution by the light F atom has, as expected, relatively little impact on the SOCME. However, the substitution by the heavier Cl and Br atoms leads to an increase. Whilst the SOCME for HTX-Cl are about two times larger than for HTX, for HTX-Br it is fourteen times larger. This fits extremely well with our experimental observations. S1 has a very short lifetime, which is likely due to rapid non-radiative quenching by ISC, especially for HTX-Br where emission from the S1 state is almost completely quenched. However, S2 is not affected by the non-radiative decay in the same way. The time-resolved spectra also indicate that there is little IC from S2 to S1, indicating zero crossing between the respective potential energy surfaces, even with vibronic coupling. This is further supported by the fact that S1 and S2 have different natures, with S2 being a LE state (strongly coupled to S0), whereas S1 is a resonant CT configuration and therefore weakly coupled to S0.
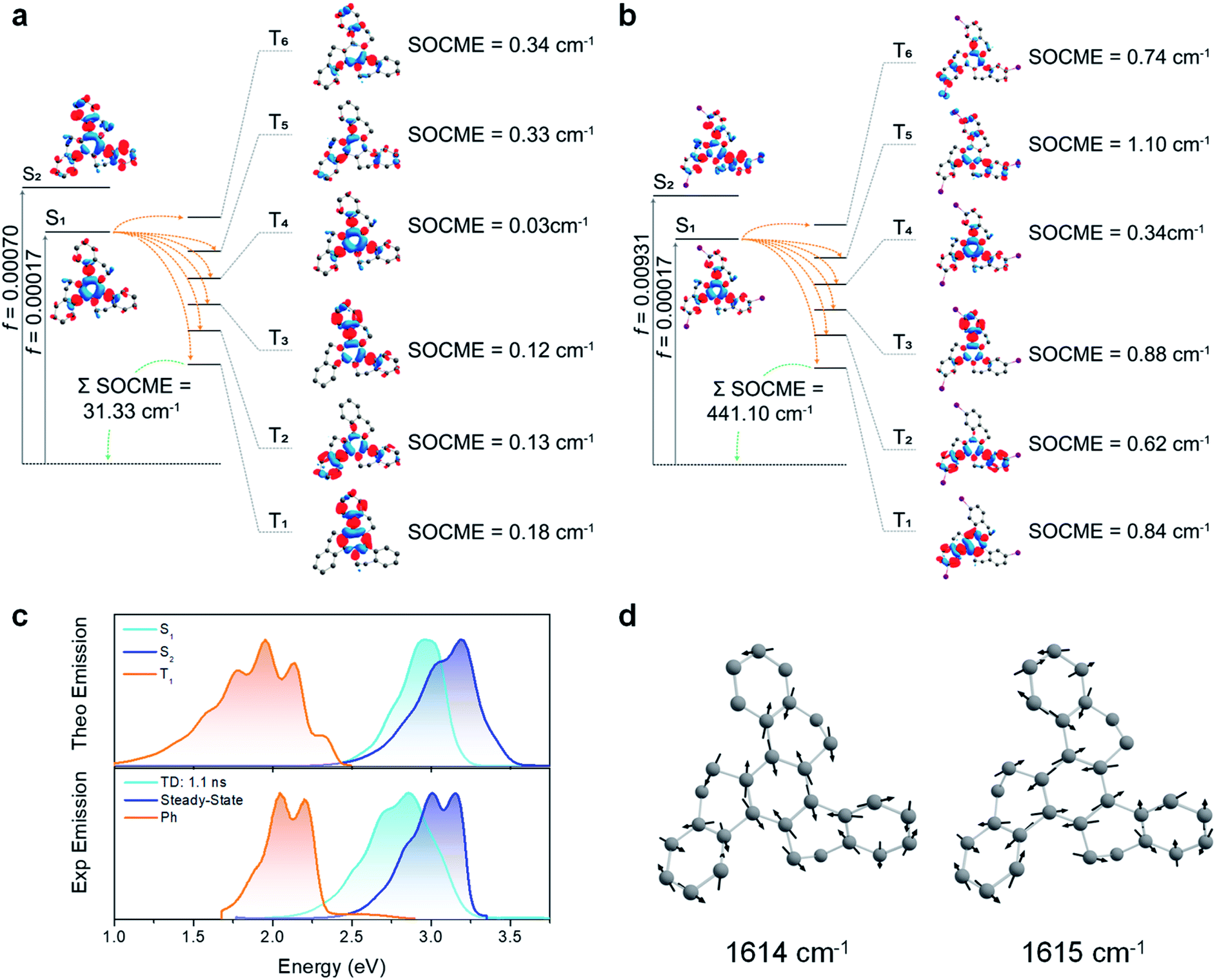 | ||
| Fig. 6 SOC-TD-DFT and predicted emission data. (a) Jablonski diagram for HTX and calculated SOCME for S1 → Tn transitions (orange arrows) at the S1 optimized geometry and for the T1 → S0 transition (green arrow) at the optimized T1 geometry. The SOCME were calculated using √∑〈Tj (MS=0,±1)|HSO|Si〉2 and the ∑SOCME is the sum of all matrix elements between the first ten singlet and triplet states, ∑〈T1–10|HSO|S0–10〉. The OS for S1 and S2 are indicated on the vertical excitation arrows. (b) Analogous Jablonski diagram for HTX-Br showing a larger SOCME between the low-lying states. (c) Experimental and predicted normalized fluorescence and phosphorescence using PBE0/Def2-TZVP(-f) and the path-integral approach. The singlet 0–0 energy difference was red-shifted by 0.30 eV to match the experimental data for both S1 and S2 emission. (d) Normal modes close to 1600 cm−1 (200 meV) for HTX. An animation of these normal modes can be seen in the ESI.† | ||
Combining the path-integral approach with the SOC results, we obtained individual emission rates for S1, S2, and T1 (Table S10†). These decay rates are predominantly governed by the Herzberg–Teller (HT) vibronic coupling effect. The predicted emission spectra obtained from the simulated emission rates are in good agreement with the experimental emission spectra (Fig. 6c and S22–S24†), confirming the coexistence of long-wavelength Ph with short-wavelength dual PF from S1 and S2 states. Furthermore, the vibronic structure in the emission spectra related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration modes close to 1600 cm−1 confirms that these modes are strongly coupled to the electronic transitions. These vibrations break the symmetry of the inner and other rings and increase the dihedral angle between them (Fig. 6d).
C stretching vibration modes close to 1600 cm−1 confirms that these modes are strongly coupled to the electronic transitions. These vibrations break the symmetry of the inner and other rings and increase the dihedral angle between them (Fig. 6d).
Relationship between experimental and theoretical photophysical properties
The experimental and theoretical results together confirm a mixed PF from S1 and S2 states. Fluorine substitution yields an improved PF decay rate (Table 1) and thus enhances the PF. This occurs through the significant contribution of HOMO → LUMO (82%) for S2 in HTX-F (Table S7†), giving a strong LE character and a high OS. On the other hand, a relatively weak OS for the lowest two excited singlet states (Tables S6 and S8–S10†) is observed for the other three compounds. Due to the weak OS and consequently a lower deactivation rate for the fluorescence channel for the other three compounds, ISC is competitive to PF, especially in quenching the S1 → S0 resonant CT transition.In organic molecules, efficient SOC usually occurs only in transitions between (π,π*) and (n,π*) states, following El-Sayed's rule.35 By contrast, when the donor and acceptor orbitals have the same (π,π*) configuration, ISC is disfavored in planar systems. However, as observed in the compounds investigated here, another possibility to efficiently induce a spin-flip arises if the initial and final (π,π*) states are spatially twisted with respect to each other in a non-planar molecular configuration (Fig. 7a–c). Then, due to the angle between the states, there is a change of orbital angular momentum on excitation (de-excitation) associated with a non-zero SOC operator allowing access to the triplet state.
 | ||
| Fig. 7 Possible ISC pathways and emission mechanism. (a) Representation of transitions from 1(π,π*) to 3(n,π*) states, in agreement with El-Sayed's rule, from px to py orbitals that efficiently overlap under SOC, inducing an efficient ISC mechanism. 1(n,π*) to 3(π,π*) transitions are likewise allowed by El-Sayed's rule. (b) By contrast, ISC from 1(π,π*) to 3(π,π*) (or from 1(n,π*) to 3(n,π*)) is disfavored in a planar aromatic system because orbital overlap is poor and SOC is inefficient. (c) In a twisted aromatic molecule, a transition between two (π,π*) states comes with nonvanishing SOC matrix elements, allowing an efficient ISC. (d) Proposed mechanism for RTP in HTX and its halogenated derivatives. After excitation, dual PF deactivates the S1 and S2 excited states to the ground state (S0) (Fig. S8–S12†). Considering that kPF (S2) ≫ kISC, the excited molecule may also transform from S1 to T1 through ISC, enabling a faster Ph decay and the slower Ph with μs to ms lifetimes at RT. Due to the presence of low-lying excited triplet states well below S1, the HAE enhances only slightly kISC, whereas kPh is strongly affected. | ||
The HAE on SOC in such twisted π-systems is potentially an efficient way to modulate RTP in organic molecules. The measured kISC (Table 1) is not much affected by the introduction of F into HTX but increases by about 6 times for HTX-Cl and nine times for HTX-Br. Whereas the substitution with fluorine and chlorine does not significantly affect the Ph, HTX-Br shows a remarkable 34-fold increase compared to HTX. This larger enhancement of Ph decay rate (kPh) compared to kISC can be rationalized from the observation that the higher triplet states that are energetically close in energy to S1, which should contribute most to kISC, have large components on the inner ring, similarly to S1. Due to the similar configuration, the SOCME is small, and due to the orbital localization far from the halogen atoms, the HAE is small as well. Nevertheless, even small values of SOCME between the low-lying states close to 1–5 cm−1 efficiently induce a spin-flip transition. The halide attached to the outer rings contributes significantly to the lowest triplet orbitals and thus causes a strong enhancement of the SOCME, making it possible for a strong HAE on kPh, most prominently from bromine. Therefore, the twisted geometry allows an efficient ISC and RTP, while the HAE mostly speeds up Ph (Fig. 7d). A long-lived triplet state allows collisional TTA to compete with Ph to give TTA DF. For HTX-F and HTX-Cl, the smaller increase of SOC is not enough to efficiently favor Ph emission over TTA and we observe similar DF as in the parent HTX. On the other hand, for HTX-Br, the large SOC increase results in a strongly enhanced Ph dominating over the TTA. This difference between HTX-Br and the lighter homologs is clearly observed in power dependence measurements, where at high power, the power dependence of the DF intensity flattens to 1.44 implying a mixture of TTA and competing monomolecular decay. Furthermore, the calculated Ph rate shows that even for heavy HTX-Br, the Ph path is dominated by the vibronic coupling, related to the “twisting” mode around 1600 cm−1. At low temperature, TTA vanishes because intermolecular hopping of triplet excitons is greatly reduced, whereas even though the Ph rate decreases as the 1600 cm−1 modes are frozen out, Ph still occurs as no TTA depletes the triplet population.
For all four compounds, a weak emission in the 100 μs to ms window at 440–450 nm is observed (Fig. S7 and S10–S12†). This emission is temperature sensitive and the lifetime increases on cooling. However, the larger energy gap between the S1 and T1 states for these molecules at ca. 0.90 eV rules out the assignment to thermally activated DF. Even if the T1 and T2 states vibronically couple to drive second order SOC, T2 is still far below the S1 as well, resulting in a highly unlikely TADF mechanism for these molecules. This is why we assign the fast Ph to emission from the triplet CT state T4, due to its large OS and similar orbital geometry compared to S1.31,45–48 This monomolecular decay process spectrally overlaps with the TTA and for HTX-Br starts to compete with TTA, influencing the power-dependent decay described above.
Conclusion
In summary, efficient ISC between two (π,π*) states with a SOCME of up to 5 cm−1 is enabled by a twisted molecular geometry, leading to RTP in purely organic molecules without lone-pair-providing heteroatoms such as N or O. For the twisted compounds described here we find that a dual fluorescence emission competes with the ISC. Substitution with the lightest halogen fluorine mainly impacts the PF, while with chlorine and bromine, the HAE enhances the ISC and Ph mechanisms in distinct ways. This enhancement depends on the strength of the HAE on the states involved in the spin-flip transition and the spatial position of the relevant triplet orbitals on the molecule: As the higher excited triplet states that dominate in ISC are less centered on the heavy atoms than the lower excited triplet states that dominate Ph, the HAE impacts Ph much more than ISC. The ISC rate is increased only by about nine times on the introduction of three heavy Br atoms into HTX, while the Ph rate is increased by about 34-fold. Thus, a molecular design based on twist, HAE, and threefold symmetry providing a nonvanishing SOC in the absence of typical electron lone-pair (i.e. n,π-orbital) heteroatoms is found to be a fruitful approach to triplet-emissive materials.Experimental section
Synthesis
HTX and its trihalogenated derivatives were synthesized in 3 to 5% yield by RT trimerization of the corresponding 1-tetralone in the presence of TiCl4 and triethylamine in dichloromethane (DCM). This procedure gives about two times lower yields than the solvent-free trimerization with TiCl4 alone at higher temperatures previously used for the synthesis of HTX,39 but it proved to be more adapted to the handling of moderate quantities of 7-halo-1-tetralones as starting materials, which are far more expensive than unsubstituted 1-tetralone. The RT trimerization used here is a modification of the procedure of Pyrko49 for the trimerization of 6-methoxy-1-tetralone. Whereas Pyrko's procedure, where the tetralone is added first under cooling to a solution of TiCl4 in DCM and triethylamine is added last, fails with unsubstituted tetralone, HTX is formed when the tetralone and triethylamine are rapidly added simultaneously without cooling, and the three trihalogenated homologs are obtained similarly (see ESI†). In spite of the low yields, the products could be purified easily due to their distinct apolarity compared to starting material and side products, and due to their much stronger tendency to crystallize from solution.HTX was synthesized as previously described.39 Alternatively, it could also be obtained in 5.0% (193 mg, 0.50 mmol) yield by the following procedure used for the synthesis of the three halogenated homologs.
HTX-F, HTX-Cl and HTX-Br were synthesized as follows:
Under exclusion of moisture, TiCl4 (8 mL, 73 mmol) is added to dry DCM (40 mL) under stirring. Then a solution of 7-halo-1-tetralone (30 mmol) and triethylamine (3.0 g, 30 mmol) in DCM (30 mL) is added quickly from a dropping funnel, such that the reaction mixture boils during addition. The mixture is stirred for 16 h at RT and then poured into water. After extraction with DCM, the organic phase is evaporated, the residue is chromatographed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DCM and petroleum ether (PE) on silica, whereby the apolar trimeric product is eluted quickly, whereas large quantities of the dimeric ketone side product elute more slowly. The product crystallizes upon concentration of the DCM-PE solution, is filtered off, and boiled out with acetone. All three homologs show the same characteristic broadening of the four aliphatic 1H NMR signals due to moderately fast conformational fluctuations of the twisted aliphatic bridge, as previously observed for HTX.39
:1 mixture of DCM and petroleum ether (PE) on silica, whereby the apolar trimeric product is eluted quickly, whereas large quantities of the dimeric ketone side product elute more slowly. The product crystallizes upon concentration of the DCM-PE solution, is filtered off, and boiled out with acetone. All three homologs show the same characteristic broadening of the four aliphatic 1H NMR signals due to moderately fast conformational fluctuations of the twisted aliphatic bridge, as previously observed for HTX.39
Single crystal X-ray diffraction
Single crystals of HTX-Br and HTX-Cl were obtained as colourless plates by slow diffusion of ethanol into chloroform solutions. They were coated with CargilleTM NHV immersion oil and mounted on a fiber loop, followed by data collection at 120 K. The crystallographic data were collected with a Bruker APEX II Quasar diffractometer, equipped with a graphite monochromator centred on the path of MoKα radiation. The program SAINT was used to integrate the data, which was thereafter corrected using SADABS.50 The structure was solved using SHELXT51 and refined by a full-matrix least-squares method on F2 using SHELXL-2018.52 All non-hydrogen atoms were refined with anisotropic displacement parameters, whereas hydrogen atoms were assigned to ideal positions and refined isotropically using suitable riding models. The CIF files have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2081280 and 2081281 for HTX-Br and HTX-Cl, respectively.†Photophysical measurements
Absorption spectra were collected using a UV-3600 double beam spectrometer (Shimadzu), and fluorescence spectra were collected using Fluoromax fluorescence spectrometer (Jobin Yvon). The solutions were degassed in a long-necked quartz cuvette using three freeze–thaw cycles and then mounted in a liquid nitrogen cryostat (Janis Research) for the measurements at low temperatures. Time-resolved photoluminescence spectra (including Ph) decays were measured using either a time-correlated single photon counting set-up (TCSPC, Horiba Deltaflex) with a range of nanoLED (357 nm) and laser diode (405 nm) excitation sources, or a nanosecond gated spectrograph-coupled iCCD (Stanford, 4Picos) and a high energy pulsed Nd:YAG laser emitting at 355 nm (SL312, EKSPLA); the pulse duration was approximately 150 ps and the energy of per pulse was chosen around 100 μJ. Emission was focused onto a spectrograph and detected on a sensitive gated iCCD camera (Stanford Computer Optics) having sub-nanosecond resolution. The quantum yields were determined using a Hamamatsu Photonics Absolute Quantum Yield Measurement System model c9920-02G. For powder measurements, a cylindrical quartz cuvette was used.Theoretical modeling
The geometry of the emitter molecules was optimized in vacuum, using the Orca 4.2.1 (ref. 53) software package. For the modeling of the electronic structures, DFT with the PBE0 functional was chosen, and the ZORA-Def2-TZVP(-f) basis was used for all atoms, including ZORA scalar-relativistic corrections. Dispersion effects were included using Grimme's D3 correction with Becke–Johnson (BJ) damping.54 The evaluation of the four-center integrals was accelerated with the RIJCOSX algorithm, using the resolution of identity approximation for the Coulomb part (RIJ), and the chain of spheres approach for the Fock exchange (COSX).55,56 RIJ requires the specification of an auxiliary basis set for the Coulomb part (Def2/J) and a numerical integration grid for the exchange part (GRID4 and GRIDX4), as discussed elsewhere.53 The vibrational frequencies computed at the optimized geometry for all molecules included no imaginary ones, confirming that they were true minima. The DFT ground state geometries were compared to the X-ray structure and the maximum average error found was of the order of 0.44% for bond lengths and of 0.65% for bond angles, legitimizing the use of the PBE0 functional and def2-TZVP(-f) basis set in subsequent investigations. TD-DFT under Tamn–Dancoff approximation (TDA) was employed to obtain the first 30 singlet and spin-adapted triplet excited states. The same calculation protocol was used to optimize the geometry and calculate the Hessian of the first excited state. The first triplet was optimized from the ground state of a UKS calculation. To include solvent effects in the excited state energies, a conductor-like polarizable continuum model (CPCM) was used, using 2-MeTHF where the refractive index and dielectric constant were used as published by Aycock57 and toluene as solvent. SOC on top of the TD-DFT results was included by using quasi-degenerate perturbation theory.57 The SOC integrals used here are the ones calculated using a mean-field named RI-SOMF(1X), described elsewhere.58 In order to compute the fluorescence and phosphorescence spectra, the path integral approach implemented in the ORCA_ESD module44,59 was used with the same protocol as above for the SCF and TD-DFT. Defaults were used unless mentioned. The normal modes chosen for the model were those with frequencies above 300 cm−1 and a Gaussian line shape was set, with a line width of 350 cm−1 for phosphorescence and for fluorescence spectra. Images of the complex geometries were obtained using the Chemcraft program.Author contributions
GF, CAMS, APM, HB and IHB elaborated the manuscript. LS, FD and HB synthesized the compounds. FD grew the single crystals. PD determined the structure of the single crystals by X-ray analysis. GF, CAMS, and MA measured the photophysical characteristics. GF and BS performed the DFT calculations. BS, HB, APM, and IHB supervised the RTP research. All authors discussed the progress of the research and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to CNPq, FINEP, CAPES, FAPESC, INCT-INEO, CAPES-COFECUB (#937/20 and #Ph-C 962/20) and H2020-MSCA-RISE-2017 (OCTA, #778158) for financial support of this work.References
- X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- Y. He, N. Cheng, X. Xu, J. Fu and J. Wang, Org. Electron., 2019, 64, 247–251 CrossRef CAS.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 139, 6376–6381 CrossRef CAS PubMed.
- S. Hirata, J. Mater. Chem. C, 2018, 6, 11785–11794 RSC.
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- Y. Wang, J. Yang, Y. Tian, M. Fang, Q. Liao, L. Wang, W. Hu, B. Z. Tang and Z. Li, Chem. Sci., 2020, 11, 833–838 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- N. J. Turro, V. Ramamuthy and J. C. Scaiano, Modern Molecular Photochemistry of Organics Molecules, University Science Books, Sausalito, 2010 Search PubMed.
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coord. Chem. Rev., 2020, 417, 213371 CrossRef CAS.
- K. Li, Y. Chen, J. Wang and C. Yang, Coord. Chem. Rev., 2021, 433, 213755 CrossRef CAS.
- Y. P. Rey, D. G. Abradelo, N. Santschi, C. A. Strassert and R. Gilmour, Eur. J. Org. Chem., 2017, 2017, 2170–2178 CrossRef CAS.
- Y. Wen, H. Liu, S. Zhang, Y. Gao, Y. Yan and B. Yang, J. Mater. Chem. C, 2019, 7, 12502–12508 RSC.
- L. Xu, G. Li, T. Xu, W. Zhang, S. Zhang, S. Yin, Z. An and G. He, Chem. Commun., 2018, 54, 9226–9229 RSC.
- G. He, W. TorresDelgado, D. J. Schatz, C. Merten, A. Mohammadpour, L. Mayr, M. J. Ferguson, R. McDonald, A. Brown, K. Shankar and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 4587–4591 CrossRef CAS PubMed.
- Z.-Y. Liu, J.-W. Hu, C.-H. Huang, T.-H. Huang, D.-G. Chen, S.-Y. Ho, K.-Y. Chen, E. Y. Li and P.-T. Chou, J. Am. Chem. Soc., 2019, 141, 9885–9894 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- C. Sun, X. Ran, X. Wang, Z. Cheng, Q. Wu, S. Cai, L. Gu, N. Gan, H. Shi, Z. An, H. Shi and W. Huang, J. Phys. Chem. Lett., 2018, 9, 335–339 CrossRef CAS PubMed.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- B. Ding, L. Ma, Z. Huang, X. Ma and H. Tian, Sci. Adv., 2021, 7, eabf9668 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- L. Zhang, M. Li, Q.-Y. Gao and C.-F. Chen, Chem. Commun., 2020, 56, 4296–4299 RSC.
- F. Li, S. Guo, Y. Qin, Y. Shi, M. Han, Z. An, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2019, 7, 1900511 CrossRef CAS.
- Y. Hu, Z. Wang, X. Jiang, X. Cai, S.-J. Su, F. Huang and Y. Cao, Chem. Commun., 2018, 54, 7850–7853 RSC.
- C. A. M. Salla, G. Farias, M. Rouzières, P. Dechambenoit, F. Durola, H. Bock, B. deSouza and I. H. Bechtold, Angew. Chem., Int. Ed., 2019, 58, 6982–6986 CrossRef CAS PubMed.
- C. Rothe, S. King and A. Monkman, Nat. Mater., 2006, 5, 463–466 CrossRef CAS PubMed.
- B. I. Greene, R. M. Hochstrasser and R. B. Weisman, J. Chem. Phys., 1979, 70, 1247–1259 CrossRef CAS.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- W. Kim, A. Nowak-Król, Y. Hong, F. Schlosser, F. Würthner and D. Kim, J. Phys. Chem. Lett., 2019, 10, 1919–1927 CrossRef CAS PubMed.
- B. de Souza, G. Farias, F. Neese and R. Izsák, J. Chem. Theory Comput., 2019, 15, 1896–1904 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- V. Jankus, M. Aydemir, F. B. Dias and A. P. Monkman, Adv. Sci., 2016, 3, 1500221 CrossRef PubMed.
- P. Pander, R. Motyka, P. Zassowski, M. K. Etherington, D. Varsano, T. J. da Silva, M. J. Caldas, P. Data and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 23934–23942 CrossRef CAS.
- A. Pyrko, Zh. Org. Khim., 1992, 28, 215 CAS.
- G. M. Sheldrick, SADABS Version 2.03, Bruker Analytical X-Ray Systems, Madison, WI, USA, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- R. Izsák, F. Neese and W. Klopper, J. Chem. Phys., 2013, 139, 094111 CrossRef PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- B. de Souza, F. Neese and R. Izsák, J. Chem. Phys., 2018, 148, 034104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2081280 and 2081281. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04936d |
| This journal is © The Royal Society of Chemistry 2021 |