
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRitter-type iodo(III)amidation of unactivated alkynes for the stereoselective synthesis of multisubstituted enamides†
Jinkui
Chai‡
ab,
Wei
Ding‡
bc,
Chen
Wang
 d,
Shingo
Ito
b,
Junliang
Wu
*a and
Naohiko
Yoshikai
*be
d,
Shingo
Ito
b,
Junliang
Wu
*a and
Naohiko
Yoshikai
*be
aCollege of Chemistry, Institute of Green Catalysis, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: wujl@zzu.edu.cn
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
cDivision of Molecular Catalysis and Synthesis, Henan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450001, P. R. China
dZhejiang Key Laboratory of Alternative Technologies for Fine Chemical Process, Shaoxing University, Shaoxing 312000, P. R. China
eGraduate School of Pharmaceutical Sciences, Tohoku University, 6-3 Aoba, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: naohiko.yoshikai.c5@tohoku.ac.jp
First published on 4th November 2021
Abstract
The Ritter reaction, Brønsted- or Lewis acid-mediated amidation of alkene or alcohol with nitrile via a carbocation, represents a classical method for the synthesis of tertiary amides. Although analogous reactions through a vinyl cation or a species alike may offer a route to enamide, an important synthetic building block as well as a common functionality in bioactive compounds, such transformations remain largely elusive. Herein, we report a Ritter-type trans-difunctionalization of alkynes with a trivalent iodine electrophile and nitrile, which affords β-iodanyl enamides in moderate to good yields. Mediated by benziodoxole triflate (BXT), the reaction proves applicable to a variety of internal alkynes as well as to various alkyl- and arylnitriles. The benziodoxole group in the product serves as a versatile handle for further transformations, thus allowing for the preparation of various tri- and tetrasubstituted enamides that are not readily accessible by other means.
Introduction
Enamides are versatile intermediates for the synthesis of nitrogen-containing molecules.1 As stable and electron-rich olefins, they participate in diverse transformations such as asymmetric hydrogenation,2 cycloaddition,1a,b addition to carbonyls and imines,3 and halofunctionalization.4 As for transition metal-catalyzed C–C coupling, enamides have been extensively used in the Heck reaction, and more recently have attracted attention as substrates for C–H activation5 and enantioselective hydroalkynylation.6 The enamide moiety is also prevalent in bioactive natural products as well as in drug candidates.1 Conventional methods for the enamide synthesis include condensation of amides and aldehydes, Curtius rearrangement of unsaturated acyl azides, and Wittig or Horner–Wadsworth–Emmons olefination, which often suffer from narrow scope, low stereocontrol, and/or harsh reaction conditions.7 More recently, methods based on the functionalization of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds have been developed, such as hydroamidation to alkynes8,9 and carbometalation to ynamides,10 enabling stereocontrol over the enamide C
C bonds have been developed, such as hydroamidation to alkynes8,9 and carbometalation to ynamides,10 enabling stereocontrol over the enamide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.11 However, the former method is currently limited to terminal alkynes and inherently does not allow access to tetrasubstituted enamides, whereas the latter requires specific types of amide moieties to promote the desired carbometalation. The Cu-catalyzed amidation of alkenyl halides represents another well-established stereoselective route to enamides,12 but the synthesis of the prerequisite, stereodefined alkenyl halides hinders its application to highly substituted enamides. As such, the chemical space of tetrasubstituted enamides, especially those containing biologically more relevant secondary amide moieties, remains largely inaccessible.13
C bond.11 However, the former method is currently limited to terminal alkynes and inherently does not allow access to tetrasubstituted enamides, whereas the latter requires specific types of amide moieties to promote the desired carbometalation. The Cu-catalyzed amidation of alkenyl halides represents another well-established stereoselective route to enamides,12 but the synthesis of the prerequisite, stereodefined alkenyl halides hinders its application to highly substituted enamides. As such, the chemical space of tetrasubstituted enamides, especially those containing biologically more relevant secondary amide moieties, remains largely inaccessible.13
The Ritter reaction, Brønsted- or Lewis acid-mediated reaction of olefins or alcohols with nitrile via carbocation intermediates, represents a classical but important synthetic approach to tertiary alkyl amides (Scheme 1a).14 In analogy to the Ritter amidation, one would conceive a possible approach to enamides involving the addition of nitrile to an acid- or electrophile-activated alkyne as a vinyl cation-like species, followed by capture of the nitrilium intermediate with water (Scheme 1b). Despite the apparent simplicity, to our knowledge, such transformation on simple unactivated alkynes has remained elusive, while Liu and Yang recently reported a gold-catalyzed Ritter-type amidation of chloro- and bromoalkynes to give trisubstituted, β-haloenamides.15 Herein, we report that benziodoxole triflate (BXT; 1),16 a cyclic trivalent iodine electrophile, promotes the Ritter-type difunctionalization of unactivated alkynes to afford tetrasubstituted β-iodanyl enamides in a regio- and stereoselective fashion (Scheme 1c). The reaction tolerates a variety of internal alkynes as well as alkyl- and arylnitriles. The iodanyl group in the product serves as a handle for the synthesis of tri- and tetrasubstituted enamides that are otherwise difficult to access by existing methods.
 | ||
| Scheme 1 Ritter-type amidation. | ||
Results and discussion
Recently, we reported a trans-iodo(III)etherification reaction of alkynes with BXT and alcohol or with BF3-activated fluorobenziodoxole (FBX) and dialkyl ether, which allowed for the stereoselective synthesis of multisubstituted vinyl ethers.17 During this study, we failed in engaging primary and secondary amides as nucleophiles in place of alcohol, observing no desired enamide product. Given this result, we turned our attention to the use of nitrile and water to achieve the putative iodo(III)amidation. In fact, trivalent iodine-mediated functionalization of alkynes with nitrile and water has a precedent. Saito and co-workers reported a synthesis of oxazoles from alkyne, nitrile, and water mediated by iodosylbenzene (PhIO) and a Brønsted acid such as TfOH (Scheme 2a).18 However, their mechanistic study indicated that this reaction was initiated by the addition of a triflate anion, rather than nitrile, to the I(III)-activated alkyne, and in fact no iodanyl-substituted enamide intermediate was observed. During our study on the iodo(III)etherification, we also observed oxazole formation from 1-phenyl-1-butyne and FBX/BF3 in wet, HPLC-grade acetonitrile, which was assumed to involve the addition of the nitrile to the BX-activated alkyne (Scheme 2b). Despite these precedent and observation, given the stability of the BX group among other iodanyl groups,19 we surmised that the desired Ritter-type iodo(III)amidation would be feasible under careful control of water content and other conditions.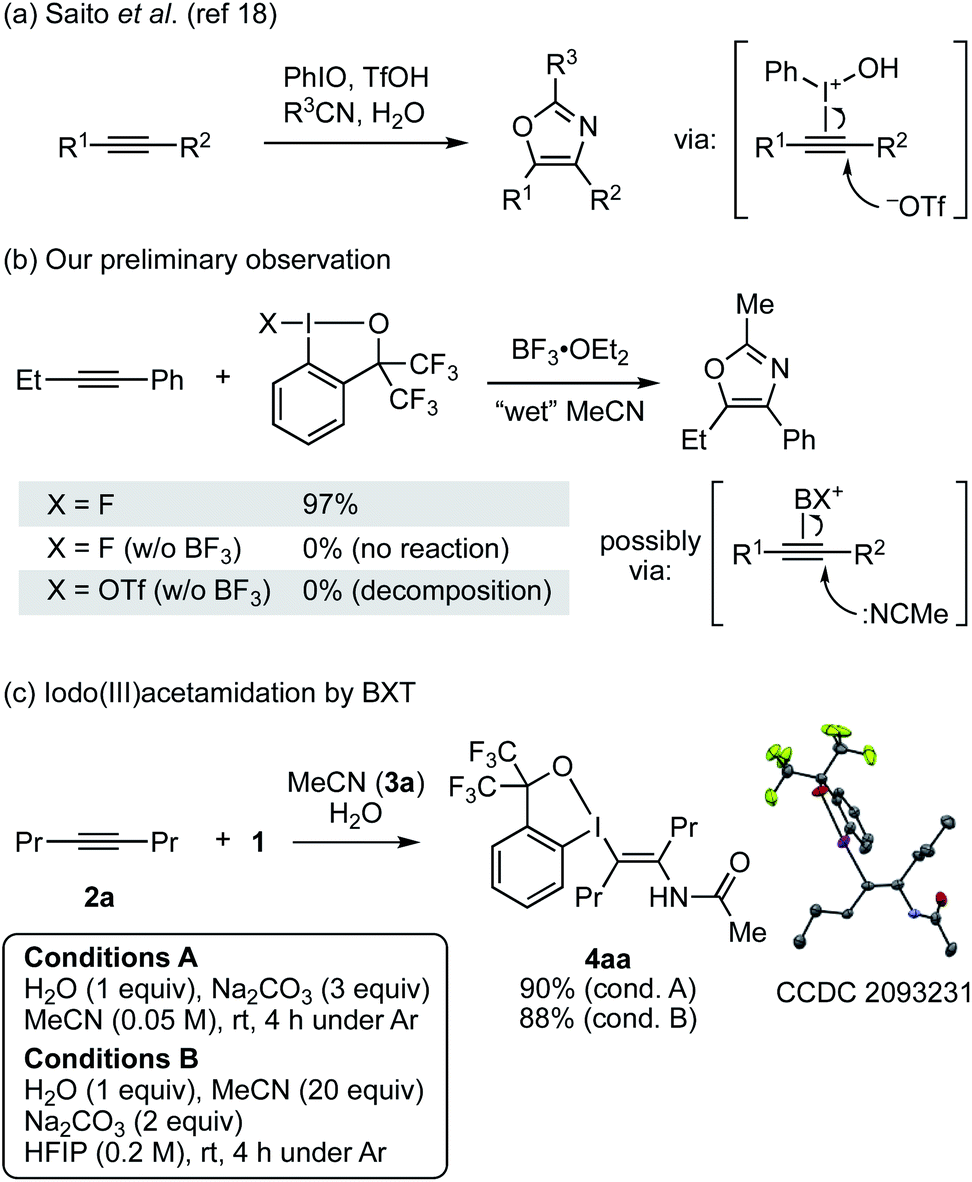 | ||
| Scheme 2 Iodine(III)-mediated reaction of alkyne with nitrile and water. | ||
Upon extensive screening of reaction conditions (see Tables S1 and S2†), we succeeded in the desired iodo(III)amidation of 4-octyne (2a) with 1, acetonitrile, and water under two sets of optimized conditions (Scheme 2c). Under conditions A, 2a was reacted with 1 (2 equiv.) and H2O (1 equiv.) in the presence of Na2CO3 (3 equiv.) in dry MeCN (3a) as the solvent at room temperature, affording the desired trans-enamide 4aa in 90% yield with exclusive stereoselectivity. Alternatively, conditions B employed 20 equiv. of MeCN in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), which also efficiently promoted the reaction to afford 4aa in 88% yield. Lower equivalents (2–10 equiv.) of MeCN led to diminished yields. The yield dropped significantly in trifluoroethanol (TFE), and no desired product was obtained in other solvents such as 1,2-dichloroethane and toluene. Thus, HFIP appeared to benefit the reaction through its cation-stabilizing nature and low nucleophilicity.20 The molecular structure of 4aa was unambiguously established by X-ray crystallographic analysis. Note that both the reaction conditions were established under an argon atmosphere in a glovebox. The reaction could also be set up in open air, but the yield was lower, varying by 50–70%, and less reproducible presumably due to the difficulty in controlling the water content from the moisture. In fact, the reaction set up in open air, even without added water, could afford a substantial yield (ca. 30%) of 4aa.
Scheme 3 summarizes the scope of alkynes for the iodo(III)acetamidation using MeCN and H2O. A variety of dialkylalkynes smoothly participated in the reaction under either conditions A or B to afford the corresponding β-iodanylenamides 4aa–4ia in moderate to good yields. The preparation of 4aa could be performed on a 2 mmol scale, albeit in somewhat lower yield. For the reactions of unsymmetrically substituted dialkylalkynes, amidation took place preferentially at the sterically more hindered acetylenic carbon, while the degree of steric discrimination was moderate, with a regioisomer ratio of ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 3:1, across alkynes containing a methyl group at one end and a primary or secondary alkyl group at the other end (see 4ea–4ha). Exclusive regioselectivity was observed in the reaction of 2,2-dimethyloct-3-yne, where the amidation occurred at the carbon proximal to the tert-butyl group (see 4ia and its X-ray structure). Interestingly, this regioselectivity trend marks a sharp contrast with that observed in the iodo(III)etherification of dialkylalkynes with MeOH, where the addition of a methoxy group occurred selectively at the sterically less hindered position.17a This difference between the iodo(III)amidation and the iodo(III)etherification would be attributed to the sterically less sensitive nature of the nitrile nucleophile compared to the alcohol nucleophile. As such, the reaction would have taken place to avoid the steric repulsion between the BX cation and the alkyne substituent rather than between the nitrile and the alkyne substituent (vide infra). Functionalized dialkylalkynes such as 1,4-bis(benzyloxy)but-2-yne failed to give the desired product; it was recovered under conditions A and completely decomposed under conditions B. Besides dialkylalkynes, a series of aryl(alkyl)alkynes underwent iodo(III)acetamidation. Thus, amidation expectedly occurred at the position proximal to the cation-stabilizing aryl group, thus affording the corresponding enamide products 4ja–4pa in moderate yields. As a general trend, conditions A proved suitable for alkynes bearing an electron-donating or neutral substituent; they reacted much more sluggishly under conditions B. The opposite was the case for alkynes bearing electron-withdrawing substituents such as ester, ketone, and CF3 groups. An enyne could also be transformed into dienamide 4qa in 65% yield under conditions A. Note that terminal alkynes failed to give the iodo(III)amidation product under the present conditions.
:1 to 3:1, across alkynes containing a methyl group at one end and a primary or secondary alkyl group at the other end (see 4ea–4ha). Exclusive regioselectivity was observed in the reaction of 2,2-dimethyloct-3-yne, where the amidation occurred at the carbon proximal to the tert-butyl group (see 4ia and its X-ray structure). Interestingly, this regioselectivity trend marks a sharp contrast with that observed in the iodo(III)etherification of dialkylalkynes with MeOH, where the addition of a methoxy group occurred selectively at the sterically less hindered position.17a This difference between the iodo(III)amidation and the iodo(III)etherification would be attributed to the sterically less sensitive nature of the nitrile nucleophile compared to the alcohol nucleophile. As such, the reaction would have taken place to avoid the steric repulsion between the BX cation and the alkyne substituent rather than between the nitrile and the alkyne substituent (vide infra). Functionalized dialkylalkynes such as 1,4-bis(benzyloxy)but-2-yne failed to give the desired product; it was recovered under conditions A and completely decomposed under conditions B. Besides dialkylalkynes, a series of aryl(alkyl)alkynes underwent iodo(III)acetamidation. Thus, amidation expectedly occurred at the position proximal to the cation-stabilizing aryl group, thus affording the corresponding enamide products 4ja–4pa in moderate yields. As a general trend, conditions A proved suitable for alkynes bearing an electron-donating or neutral substituent; they reacted much more sluggishly under conditions B. The opposite was the case for alkynes bearing electron-withdrawing substituents such as ester, ketone, and CF3 groups. An enyne could also be transformed into dienamide 4qa in 65% yield under conditions A. Note that terminal alkynes failed to give the iodo(III)amidation product under the present conditions.
 | ||
| Scheme 3 Iodo(III)acetamidation of various alkynes (0.2 mmol scale). The symbol BX in the product refers to the benziodoxole moiety. For reactions that produced a mixture of regioisomers, the major isomer is shown along with rr (regioisomer ratio) determined by 1H NMR. | ||
We next explored the scope of nitriles in the iodo(III)amidation of 4-octyne (Scheme 4). Here, conditions B proved effective across a wide variety of organonitriles. Thus, aliphatic nitriles including deuterated acetonitrile ([D3]-4aa), primary alkyl nitriles (4ab–4ae), benzyl and allyl nitriles (4af–4ah), secondary alkyl nitriles (4ai–4ak), and tertiary alkyl nitriles (4al and 4am) underwent the desired iodo(III)amidation to afford the corresponding β-iodanyl enamides in moderate to high yields. The yields were relatively insensitive to the steric size of the nitrile substituent, except that 1-adamanyl nitrile afforded the product 4am in a modest 26% yield. In these reactions, unlike the iodo(III)acetamidation with MeCN, the nitrile substrates were neither dried nor purified prior to the reaction but used as received. Attempted reactions using unpurified propionitrile or butyronitrile as the solvent under conditions A afforded only low yields of the desired enamide, presumably due to the water content in such reagent-grade nitriles. Aromatic nitriles including parent and substituted benzonitriles and thiophene-2-carbonitrile also participated in the reaction with 2a to afford the desired products 4an–4ar in moderate yields.
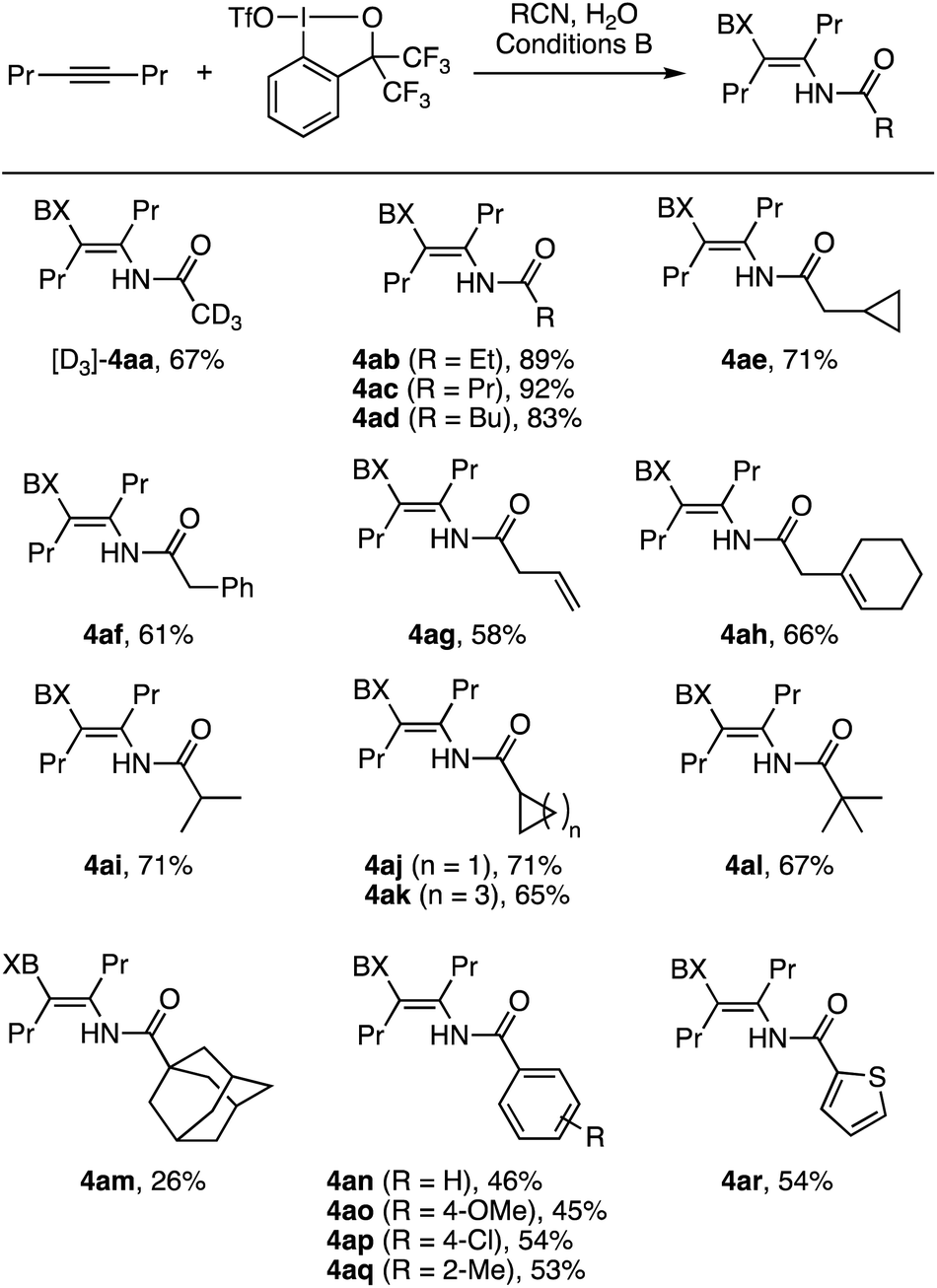 | ||
| Scheme 4 Iodo(III)amidation of 4-octyne with various nitriles (0.2 mmol scale). The symbol BX in the product refers to the benziodoxole moiety. | ||
As discussed earlier, the present reaction is considered to proceed via the nucleophilic attack of nitrile to benziodoxole cation (BX+)-activated alkyne, followed by the addition of water to the thus-formed nitrilium cation. The role of water as the nucleophile was supported by control reactions using 18O-labeled water, which resulted in a substantial degree of 18O incorporation into the enamide product (see the ESI†). Another, less obvious observation was that the reaction in the absence of water afforded N-vinylacetimidate derivative 4aa′ in 30% yield (Scheme 5). This product likely formed via the addition of poorly nucleophilic 1,1,1,3,3,3-hexafluoro-2-(2-iodophenyl)propan-2-ol, formed via decomposition of BXT, to the nitrilium intermediate.
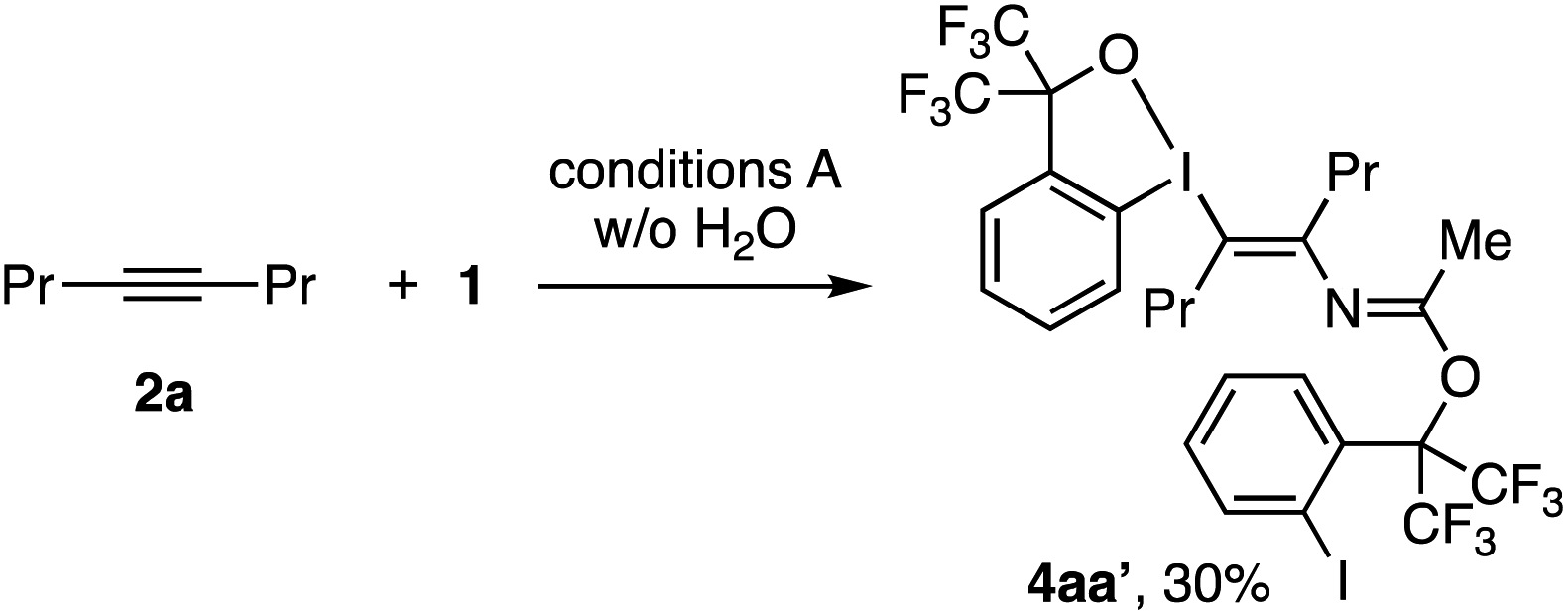 | ||
| Scheme 5 Formation of the acetimidate derivative in the absence of H2O. | ||
To gain insight into the nature of the transition state for the iodo(III)amidation as well as the possible origin of the regioselectivity for unsymmetrically substituted dialkylalkyne, we performed DFT calculations.21 Thus, regioisomeric transition states (TSs) for the addition of acetonitrile to 4,4-dimethylpent-2-yne activated by BX+ were explored (Fig. 1). Consistent with the experimental observation (see 4ia), TS1b, where acetonitrile attacks the more hindered acetylenic carbon, was calculated to be more stable than its regioisomeric TS (TS1a) by 1.7 kcal mol−1. The overall activation free energy from the separated alkyne, BXT, and MeCN toward TS1b and TfO− was 22.9 kcal mol−1. The lengths of the forming C–I(III) (2.34 Å/2.30 Å in TS1a/TS1b) and C–N (2.09 Å/2.16 Å in TS1a/TS1b) bonds and the angles around the acetylenic carbons indicate that TS1b features more advanced C–I(III) bond formation and less advanced C–N bond formation as compared with TS1a. This was also consistent with the calculated Mayer bond orders for the C–I(III) (0.531/0.560 in TS1a/TS1b) and C–N (0.228/0.203 in TS1a/TS1b) bonds. The magnitude of these bond orders, along with the geometry of the bent alkyne in the TS as revealed by the bond angles, illustrates that the C–I(III) bond formation is intrinsically much earlier than the C–N bond formation. Given this nature of the iodo(III)functionalization TS,17 it appears reasonable that the sterically undemanding nitrile nucleophile has preference toward the addition to the more hindered acetylenic carbon so that the steric repulsion between the bulky BX and tBu groups can be minimized. The entire energy profile for the reaction between 4,4-dimethylpent-2-yne, BXT, and MeCN is provided in the ESI,† which supports kinetic control of the regioselectivity of the reaction.
 | ||
| Fig. 1 Regioisomeric transition states for the addition of MeCN to 4,4-dimethylpent-2-yne activated by the BX cation. The bond distances are in Å. | ||
The product of the present reaction, β-iodanyl enamide, represents an addition to the repertoire of functionalized vinylbenziodoxoles (vinyl-BXs)9,17,19d,22,23 and provides opportunities for further synthetic transformations via cleavage of the BX group (Scheme 6a). Sonogashira and Stille couplings of 4aa afforded the conjugate enamides 5 and 6, respectively, in good yields. While Sonogashira coupling took place with retention of the olefin stereochemistry, Stille coupling afforded a mixture of E/Z isomers in ca. 5:1 ratio. The CuCN/proline-mediated cyanation of 4aa proceeded smoothly at 105 °C to afford the products (E)-7 and (Z)-7 as separable isomers in ca. 3:1 ratio. At a lower temperature of 50 °C, the reaction furnished the β-iodoenamide 8 with an E/Z ratio of 5:1. Hydrodehalogenation of 4aa with the Pd/formic acid system afforded the trisubstituted enamide 9 (E/Z = 5:1) in good yield, whereas reductive homocoupling of 4aa mediated by the Pd catalyst and Zn afforded the novel diene–diamide 10 (E,E/E,Z = 4:1) in excellent yield. It should be pointed out that, unlike the partial loss of the stereochemical integrity in the Pd-catalyzed coupling reactions of 4aa, analogous reactions of β-iodanyl vinyl ether took place with retention of the stereochemistry.17a While the reason for this difference remains unclear, we speculate that the vinylpalladium species formed upon oxidative addition of 4aa has a partial carbene-like character due to the electron-donating effect of the amide group, thus allowing rotation of the olefinic C–C bond. The chelating ability of the amide group might also facilitate the isomerization of the vinylpalladium species.
 | ||
| Scheme 6 Downstream transformations of β-iodanyl enamides. | ||
Many of the multisubstituted enamides synthesized above are not readily accessible by other means, and hence would open numerous opportunities for downstream synthetic transformations. For demonstration, we explored the application to the pyridine/pyrimidine synthesis developed by Movassaghi (Scheme 6b).24 Thus, trisubstituted enamides obtained through the iodo(III)amidation/hydrodehalogenation sequence were subjected to Tf2O, 2-chloropyridine, and electron-rich alkyne or nitrile, affording multisubstituted pyridines 11–14 and pyrimidine 15 in good yields.
Conclusions
In conclusion, we have developed a Ritter-type trans-difunctionalization of alkynes mediated by the trivalent iodine electrophile BXT for the synthesis of multisubstituted enamides. The reaction was achieved with a careful choice of the reaction conditions including the water content and the reaction medium, and proved applicable to a variety of internal alkynes as well as nitriles, thus affording trans-iodanyl enamides in moderate to good yields. The versatility of the BX group allows for the preparation of various tri- and tetrasubstituted enamides through cross-coupling. The multisubstituted enamides made accessible by the present method would inspire further development of enamide-based transformations for the synthesis of nitrogen-containing molecules.Data availability
All experimental, computational and crystallographic data is available in the ESI.†Author contributions
J. W. and N. Y. conceptualized the project. J. C. and W. D. performed and analyzed the experiments with assistance of S. I., J. W. and N. Y. C. W. performed DFT calculations. N. Y. prepared the manuscript with contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agency for Science, Technology and Research (A*STAR) AME IRG Grant (A2083c0056). J. C. thanks the China Scholarship Council for the support of the overseas visiting student program. We thank Dr Yongxin Li (Nanyang Technological University) for his assistance with the X-ray crystallographic analysis.Notes and references
- (a) T. Courant, G. Dagousset and G. Masson, Synthesis, 2015, 47, 1799–1826 CrossRef CAS; (b) G. Bernadat and G. Masson, Synlett, 2014, 25, 2842–2867 CrossRef CAS; (c) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455–3460 RSC.
- K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599–4657 CrossRef CAS PubMed.
- R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292–301 CrossRef CAS PubMed.
- A. Alix, C. Lalli, P. Retailleau and G. Masson, J. Am. Chem. Soc., 2012, 134, 10389–10392 CrossRef CAS PubMed.
- T. Zhu, S. Xie, P. Rojsitthisak and J. Wu, Org. Biomol. Chem., 2020, 18, 1504–1521 RSC.
- (a) X.-Y. Bai, Z.-X. Wang and B.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 9007–9011 CrossRef CAS PubMed; (b) X.-Y. Bai, W.-W. Zhang, Q. Li and B.-J. Li, J. Am. Chem. Soc., 2018, 140, 506–514 CrossRef CAS PubMed; (c) S.-L. Zhang, W.-W. Zhang and B.-J. Li, J. Am. Chem. Soc., 2021, 143, 9639–9647 CrossRef CAS PubMed.
- G. Evano, A.-C. Gaumont, C. Alayrac, I. E. Wrona, J. R. Giguere, O. Delacroix, A. Bayle, K. Jouvin, C. Theunissen, J. Gatignol and A. C. Silvanus, Tetrahedron, 2014, 70, 1529–1616 CrossRef CAS.
- L. B. Huang, M. Arndt, K. Goossen, H. Heydt and L. J. Goossen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- D. Shimbo, T. Maruyama, N. Tada and A. Itoh, Org. Biomol. Chem., 2021, 19, 2442–2447 RSC.
- (a) B. Gourdet and H. W. Lam, J. Am. Chem. Soc., 2009, 131, 3802–3803 CrossRef CAS PubMed; (b) Y. Minko, M. Pasco, H. Chechik and I. Marek, Beilstein J. Org. Chem., 2013, 9, 526–532 CrossRef CAS PubMed.
- For enamide synthesis through other types of functionalization of CC bonds, see:
(a) J. A. Mulder, K. C. M. Kurtz, R. P. Hsung, H. Coverdale, M. O. Frederick, L. Shen and C. A. Zificsak, Org. Lett., 2003, 5, 1547–1550 CrossRef CAS PubMed;
(b) M. Li, H. Yuan, B. Zhao, F. Liang and J. Zhang, Chem. Commun., 2014, 50, 2360–2363 RSC;
(c) M. Ide, Y. Yauchi and T. Iwasawa, Eur. J. Org. Chem., 2014, 3262–3267 CrossRef CAS.
- T. Kuranaga, Y. Sesoko and M. Inoue, Nat. Prod. Rep., 2014, 31, 514–532 RSC.
- B. M. Trost, J. J. Cregg and N. Quach, J. Am. Chem. Soc., 2017, 139, 5133–5139 CrossRef CAS PubMed.
- (a) A. Guérinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 19–28 CrossRef; (b) D. Jiang, T. He, L. Ma and Z. Wang, RSC Adv., 2014, 4, 64936–64946 RSC; (c) G. M. Ziarani, F. S. Hasankiadeh and F. Mohajer, ChemistrySelect, 2020, 5, 14349–14379 CrossRef; (d) M.-E. Chen, X.-W. Chen, Y.-H. Hu, R. Ye, J.-W. Lv, B. Li and F.-M. Zhang, Org. Chem. Front., 2021, 8, 4623–4664 RSC.
- C. Liu and F. Yang, Eur. J. Org. Chem., 2019, 6867–6870 CrossRef CAS.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547–6551 CrossRef CAS PubMed.
- (a) W. Ding, J. Chai, C. Wang, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2020, 142, 8619–8624 CrossRef CAS PubMed; (b) J. Chai, W. Ding, J. Wu and N. Yoshikai, Chem.–Asian J., 2020, 15, 2166–2169 CrossRef CAS PubMed.
- A. Saito, A. Taniguchi, Y. Kambara and Y. Hanzawa, Org. Lett., 2013, 15, 2672–2675 CrossRef CAS PubMed.
- (a) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef CAS PubMed; (b) J. Waser, Synlett, 2016, 27, 2761–2773 CrossRef CAS; (c) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212–3225 CrossRef CAS PubMed; (d) N. Declas, G. Pisella and J. Waser, Helv. Chim. Acta, 2020, 103, e2000191 CrossRef CAS.
- V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Lebœuf, Chem. Commun., 2020, 56, 11548–11564 RSC.
- See the ESI† for the detail of the computational methods..
- (a) J. Wu, X. Deng, H. Hirao and N. Yoshikai, J. Am. Chem. Soc., 2016, 138, 9105–9108 CrossRef CAS PubMed; (b) B. Wu, J. Wu and N. Yoshikai, Chem.–Asian J., 2017, 12, 3123–3127 CrossRef CAS PubMed; (c) J. Wu, K. Xu, H. Hirao and N. Yoshikai, Chem.–Eur. J., 2017, 23, 1521–1525 CrossRef CAS PubMed; (d) W. Ding, C. Wang, J. R. Tan, C. C. Ho, F. Leon, F. Garcia and N. Yoshikai, Chem. Sci., 2020, 11, 7356–7361 RSC; (e) R. A. Laskar, W. Ding and N. Yoshikai, Org. Lett., 2021, 23, 1113–1117 CrossRef CAS PubMed.
- (a) E. Stridfeldt, A. Seemann, M. J. Bouma, C. Dey, A. Ertan and B. Olofsson, Chem.–Eur. J., 2016, 22, 16066–16070 CrossRef CAS PubMed; (b) P. Caramenti, N. Declas, R. Tessier, M. D. Wodrich and J. Waser, Chem. Sci., 2019, 10, 3223–3230 RSC; (c) D. Shimbo, A. Shibata, M. Yudasaka, T. Maruyama, N. Tada, B. Uno and A. Itoh, Org. Lett., 2019, 21, 9769–9773 CrossRef CAS PubMed; (d) L. Castoldi, E. M. Di Tommaso, M. Reitti, B. Grafen and B. Olofsson, Angew. Chem., Int. Ed., 2020, 59, 15512–15516 CrossRef CAS PubMed; (e) N. Declas and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 18256–18260 CrossRef CAS PubMed.
- (a) M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096–10097 CrossRef CAS PubMed; (b) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254–14255 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1981452, 2093231 and 2100970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05240c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |