
Oriented attachment and activated dipoles leading to anisotropic H-bond-free self-assembly of n-acene derivatives into organic nanoribbons emitting linearly polarised blue light†
Philip
Schäfer
a,
Leire
Gartzia-Rivero
 ab,
Min-Tzu
Kao
a,
Christian
Schäfer
a,
Stéphane
Massip
c,
Christiaan
de Vet
a,
Guillaume
Raffy
a and
André
Del Guerzo
*a
ab,
Min-Tzu
Kao
a,
Christian
Schäfer
a,
Stéphane
Massip
c,
Christiaan
de Vet
a,
Guillaume
Raffy
a and
André
Del Guerzo
*a
aUniv Bordeaux, CNRS, Bordeaux INP, Institut des Sciences Moléculaires, UMR5255, 33400 Talence, France. E-mail: andre.del-guerzo@u-bordeaux.fr
bDepartment of Physical Chemistry, University of the Basque Country (UPV/EHU), Apartado 644, 48080 Bilbao, Spain
cIECB, Univ Bordeaux, CNRS UMS3033, INSERM US001, F-33607 Pessac, France
First published on 18th November 2020
Abstract
The design of organic self-assembled nanomaterials requires a more complete understanding of all mechanisms leading to anisotropic and low-defect growth in solvent-processing, in particular in the vast case of polyaromatic compounds. 2,3-Dihexadecyloxy-9,10-diphenylanthracene (DPA16) self-assembles from solution into fluorescent nanoribbons without the addition of surfactants or polymers. These very high aspect-ratio anisotropic objects display a deep-blue emission with a remarkable linear polarisation P reaching a value of 0.6. The emission colour of ribbons can be further tuned by doping with a tetracene analogue. Crystallography, spectroscopy, atomic force and fluorescence polarisation microscopy techniques reveal that the growth mechanism not only involves a common ‘Ostwald ripening’, but also two commonly unforeseen features. First, an ‘oriented attachment’ mechanism contributes to the growth of the ribbons preventing packing mismatches. This mechanism has so far solely been demonstrated for inorganic nanoparticles. Second, a thermally activated dipole in less stable asymmetric conformations of DPA16 is proposed to contribute majorly to directional intermolecular interactions determining the main growth axis of the ribbons. These dipole–dipole interactions would contribute to explain the very high anisotropy of growth, in the absence of directional H-bonds predominating in many reported studies. This work provides new insights into the design of self-assembled nanoobjects with very high aspect ratios.
Introduction
Organic materials have evolved in recent years into significant alternatives to inorganic materials for devices, especially in the fields of optics and electronics, commercialized organic light-emitting diode (OLED) screens constituting the current epitome. In order to comply with actual constraints set for their developments, such as solvent-processability, flexibility of substrates and related thinness of active layers, control of optical and/or electronic properties through their anisotropy and crystallinity, nanoscience can propose innovative solutions.1–4 For example, currently the light output in LCD, OLED and QLED screens is polarised, also due to the antiglare layers.5,6 Optical linear polarisers are used to polarise the light-source, but simultaneously induce major transmission losses.7 It would constitute an essential advance to use sources generating linearly polarised light, whether it is a backlit or an emissive pixel, to avoid transmission losses and improve overall efficiency. Anticipated 3D-screens exploiting circularly polarised luminescence (CPL)8 also implies control of polarisation, either directly generated or obtained by traditional transformation of linearly polarised light with an optical layer acting as a quarter wave-plate. Whereas the latter technology is available, CPL-generating materials are being developed to provide in the future thin-layer, flexible alternatives.A bottom-up approach to achieve promising nanomaterials with aforementioned specificities is self-assembly. Among this vast family of structures, self-assembled anisotropic nano-objects with very high aspect ratios show demonstrated potential for applications. Among these, functional nanofibres9 are known to display intense and tuneable fluorescence,10,11 interesting charge mobility,12,13 birefringence,14 linear or circular dichroism,15,16 linearly17 or circularly6,18 polarised luminescence. This adds up to the strong and flexible mechanical properties exploited for example in gels for industrial applications.19 Nanofibres can also act as structural templates, for example for therapeutics,20 or as an orienting matrix for luminescent nanorods leading to polarised emission.21 Recently, it has been also shown that multi-scale hierarchical self-assembly can be achieved on surfaces by photo-controlled patterning: indeed, nanofibres can be aligned and oriented at will within a micrometric pattern, with each pattern displaying collective linearly polarised fluorescence.22
The fundamental mechanism of self-assembly into nanofibres has remarkably been described by Schenning and Meijer,23–27 with a particular focus on the nucleation and cooperative growth mechanism. So far, anisotropic growth has convincingly been described when directional H-bonds are involved, since the proposed molecular packing structures tend to correlate with the preferential growth direction and the main direction of H-bonds. It is of common understanding that nanofibres are kinetically trapped states.28 It also has been shown previously that through a ripening stage nanofibres can turn into nanoribbons.29 In that study, the differences in the optical properties were correlated with the evolution of the molecular packing, and nanoribbons were displaying larger cross-sections, whereas nanofibres showed more curvatures, entanglements and branching: these latter features led to the formation of a fibrillar network and gelation of the solvent. These anisotropic nanostructures are usually considered as meta-stable states, since crystals would be expected to be the thermodynamically most stable form. Crystals are however not always attainable, or do not constitute the most stable form.30,31 However, for aforementioned applications, molecules with H-bonding capacities are often not preferred. In the absence of H-bonds, what governs the main growth direction yielding a tremendous anisotropy? An additional unsuspected question appears when nucleation/growth of organic materials is compared to that of inorganic nanomaterials. Indeed, in the inorganic case, an oriented attachment mechanism can lead to the formation of nano-objects.32 This mechanism has however been neglected in the case of organic nanomaterials and its impact on materials’ properties has not been described yet, most probably due to the lack of evidence. This experimental work is set to address the fundamental questions on the generation of highly anisotropic nano-objects from polyaromatic derivatives and their related linearly polarised fluorescence.
A founding family of molecules for electronic and photonic applications includes n-acenes,33 used over the years for sensors,34 OPVs,35 OLEDs36–38 and OFETs.39–43 Many acene derivatives have been synthesized to induce anisotropic self-assembly, and strikingly in the case of alkoxy-substituted anthracenes, only the 2,3-disubstitution leads to self-assembly despite numerous variants being tested.17,44 Exploiting this broad know-how, the novel derivative 2,3-dihexadecyloxy-9,10-diphenylanthracene (DPA16) has been designed to form anisotropic nanomaterials with an emission that is polarised, deep-blue, and colour-tunable,17,45 and offer a playground to reveal the structure–property relationship. Deep-blue emissive materials have in general been particularly difficult to obtain and can be a bottleneck in the development of some technologies, in part due to common structural defect-induced red-shifting.46,47 The DPA core was chosen for its renowned high quantum yield of emission (a unity standard for many years) and photostability in anoxia, whereas alkoxy-chains were added to tune the solubility, induce the self-assembly and influence the molecular packing in the solid state. In this work, fluorescence anisotropy is not only the main entailed optical property, but it also constitutes a decisive handle to determine the orientation of molecules and therefore indications on molecular packing and crystalline unit cell orientation. Non-destructive fluorescence microscopy techniques (polarisation, lifetimes, and hyper-spectral) were essential to reveal the key elements of anisotropic growth of nano-ribbons of DPA16, and constitute an original approach contrasting with previous studies.48–51
Results and discussion
Molecular design and synthesis
DPA16 (2,3-dioxyhexadecyl-9,10-diphenylanthracene) has been designed based on the robust capacity of 2,3-dialkoxy-n-acene derivatives to self-assemble into nanofibres.17 DPA16 displays a very high fluorescence quantum yield of 75% in polar solvent solution (Table S1, ESI†), a deep-blue absorption and emission (λmax = 416 nm, ‘Commission Internationale de l’Eclairage’ CIE coordinates x = 0.154, y = 0.050, Fig. S1, ESI†) and high photo-stability. Indeed, as compared to anthracene, the deactivating singlet S1 to triplet T2 intersystem conversion (ISC) and the oxidation in the 9,10 position into anthraquinone are known to be negligible in diphenyl-anthracenes.52 Hexadecyl chains have been shown to induce nanoribbon formation, in contrast to shorter chain derivatives. The synthesis of DPA16, shown in Scheme 1, is based on the alkylation of 2,3-dihydroxyanthraquinone,53 subsequent addition of phenyl-lithium and final reduction.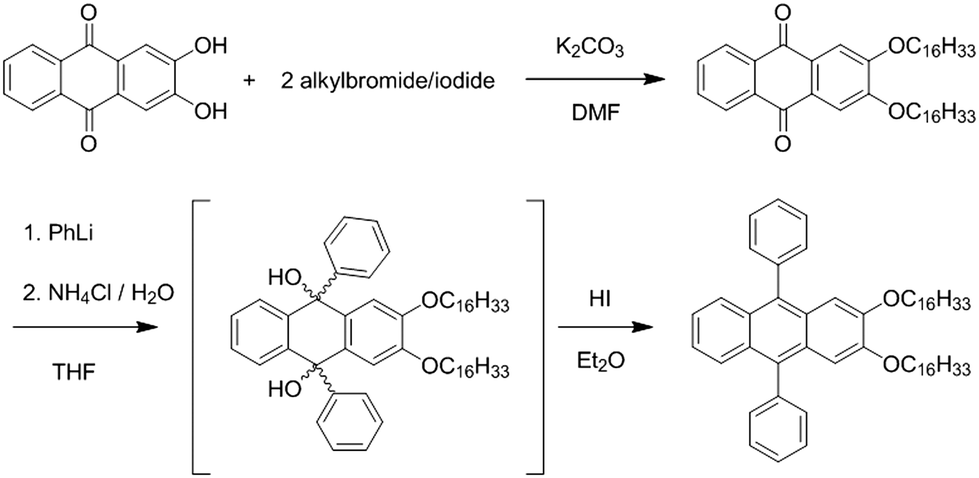 | ||
| Scheme 1 Reaction sequence for 2,3-dihexadecyloxy-9,10-diphenyl-anthracene (DPA16). | ||
Ribbon self-assembly and optical properties
The self-assembly of DPA16 into nano-ribbons is triggered by rapidly injecting a dichloromethane (DCM) solution of DPA16 into the less favourable solvent methanol (MeOH), resulting in a decrease of solubility and an increase of DPA16 concentration above saturation. The properties of the ribbons and the kinetics of self-assembly are influenced by the concentration of DPA16, the composition of the solvent mixture and the temperature. Ribbons obtained at 23 °C are called “type A”, and some properties depend on the kinetics of their formation. In the slower process explored, the so-called “ripe” type-A ribbons (AR) are formed in >24 hours. Indeed, the highest proportion of good solvent and lowest concentration of DPA16 used (1 × 10−4 M in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM 20:1) result in the formation of 0.5–2 μm wide, 50–500 nm thick ribbons showing a deep blue emission with a narrow band structured spectrum (Fig. 1a and c, CIE coordinates x = 0.158 and y = 0.066, see Fig. S2, ESI†). The latter is similar to that of the monomer in solution, with an average fluorescence lifetime of 13.1 ± 2.6 ns that is higher than that in solution (6.9 ns in THF, Fig. 1d, f and Fig. S3, Table S1, ESI†). Confocal fluorescence microscopy (CFM) shows that the ribbons display homogeneous spectra and fluorescence decays.
:DCM 20:1) result in the formation of 0.5–2 μm wide, 50–500 nm thick ribbons showing a deep blue emission with a narrow band structured spectrum (Fig. 1a and c, CIE coordinates x = 0.158 and y = 0.066, see Fig. S2, ESI†). The latter is similar to that of the monomer in solution, with an average fluorescence lifetime of 13.1 ± 2.6 ns that is higher than that in solution (6.9 ns in THF, Fig. 1d, f and Fig. S3, Table S1, ESI†). Confocal fluorescence microscopy (CFM) shows that the ribbons display homogeneous spectra and fluorescence decays.
 | ||
| Fig. 1 (a and b) Confocal hyperspectral image (RGB colours calculated from spectra at each pixel), (a) of blue “ripe” type-A AR DPA16 ribbons formed at 22 °C, scale bar 10 μm, (b) “fast” type-A AF DPA16 ribbons formed at 22 °C, scale bar 5 μm. (c) Emission spectra from an AR ribbon in (a, dark blue) and two AF ribbons from (b), one short-lived (blue) and one long-lived (lightest blue). (d and e) FLIM-images (false colour code: average lifetimes) of (d) same AR ribbons as (a), (e) same AF ribbons as (b). (f) Fluorescence decays of AR ribbon (dark blue) and a cyan-emitting AF ribbon. (g and h) Fluorescence polarisation image of AR and AF ribbons, respectively (false colour-code: P). (i) Polarisation plot (P vs. orientation angle) of a selection of straight ribbon segments from: + crosses (g), × crosses (h). | ||
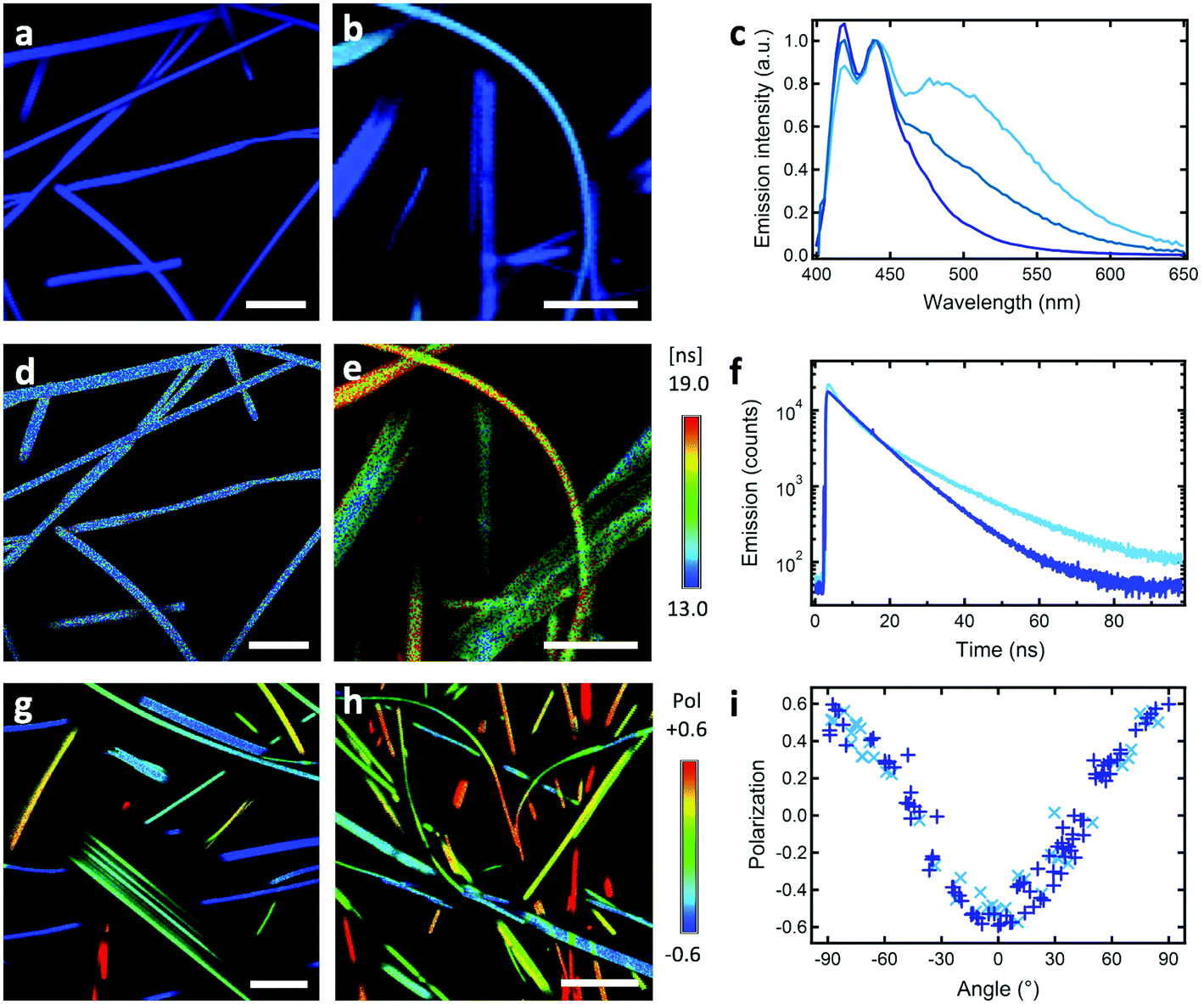
A remarkable property is that ribbons emit mostly linearly polarised light (Pmax = 0.60, eqn (1) and (2)) oriented perpendicular to their main growth axis (θphase = 90°), as revealed by polarisation-CFM (Fig. 1g and h). The polarisation P varies according to a (co)sinusoidal function (see fitting curve in Fig. 3). P is defined as follows:
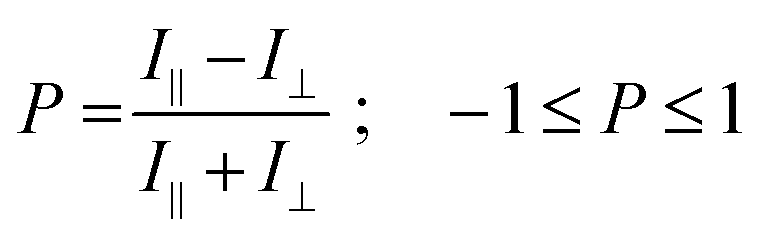 | (1) |
| P = Pmax × cos2(θ + θphase) | (2) |
The so-called “fast” type-A ribbons (AF), more practical since growing in only a few hours, are obtained by increasing the oversaturation with a lower proportion of DCM (in most cases MeOH:DCM 50:1, see the experimental section). CFM and atomic force microscopy (AFM) reveal that the blue fluorescent ribbons named AF are at least tens to hundreds of μm long, but only 100–1000 nm wide and 40–500 nm thick, hence narrower and thinner than AR. Their bulk emission quantum yield is high, 40%, and the radiative constant is 6 × 107 s−1. In the ribbons, excitation (revealing the absorption features) and emission spectra are bathochromically shifted relative to dilute solutions of DPA16 in MeOH and DCM (Fig. 1 and Fig. S1, ESI†), as typically observed upon aggregation of fluorophores.54–56 The excitation spectrum of DPA16-ribbons reveals also a moderate broadening of the transition bands, indicating some weak dipolar coupling of transition dipoles. This lowers the precision on the determination of the Stokes shift in the aggregated state, which remains in agreement with those measured in solution (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ribbon ∼ 1000–1450 cm−1vs. DCM = 1214 cm−1, MeOH = 1244 cm−1) and with a commonly reduced value attributed to conformational restriction in the solid state.57 This leads to emission – reabsorption in the bulk, as demonstrated by the difference of spectra in bulk and individual ribbons (Fig. S1, ESI†). CFM further reveals that emission spectra and fluorescence lifetimes can vary from ribbon to ribbon (Fig. 1). Some AF ribbons emit deep blue similar to AR ribbons, whereas other AF ribbons emit with a colour tending towards cyan due to a larger contribution of a broad red-shifted excimer-like emission band at λmax = 490 ± 20 nm (Fig. 1 and Fig. S1–S3, ESI†). The intensity-weighted average CIE coordinates for the representative AF ribbons in Fig. 1 are x = 0.19 and y = 0.21 (blue colour, Fig. S2, ESI†). In addition, the overall fluorescence lifetime distribution is broadened around a larger average value of 16.1 ± 4.0 ns (Fig. 1 and Fig. S4, ESI†). FLIM reveals that some ribbons emit with a long fluorescence lifetime τ of 19 ± 4 ns whereas others with a τ of 14 ± 3 ns, akin to the ripe AR ribbons. This could also be at the origin of a slightly higher quantum yield for AF compared to AR ribbons (40% vs. ∼30%). CFM shows that cyan emission and longer lifetimes are correlated (Fig. S4 and S5, ESI†). An important feature is that AF ribbons show almost the same polarisation of emission as ripe AR ribbons, with Pmax = 0.50 ± 0.05 (Fig. 1h and i).
ribbon ∼ 1000–1450 cm−1vs. DCM = 1214 cm−1, MeOH = 1244 cm−1) and with a commonly reduced value attributed to conformational restriction in the solid state.57 This leads to emission – reabsorption in the bulk, as demonstrated by the difference of spectra in bulk and individual ribbons (Fig. S1, ESI†). CFM further reveals that emission spectra and fluorescence lifetimes can vary from ribbon to ribbon (Fig. 1). Some AF ribbons emit deep blue similar to AR ribbons, whereas other AF ribbons emit with a colour tending towards cyan due to a larger contribution of a broad red-shifted excimer-like emission band at λmax = 490 ± 20 nm (Fig. 1 and Fig. S1–S3, ESI†). The intensity-weighted average CIE coordinates for the representative AF ribbons in Fig. 1 are x = 0.19 and y = 0.21 (blue colour, Fig. S2, ESI†). In addition, the overall fluorescence lifetime distribution is broadened around a larger average value of 16.1 ± 4.0 ns (Fig. 1 and Fig. S4, ESI†). FLIM reveals that some ribbons emit with a long fluorescence lifetime τ of 19 ± 4 ns whereas others with a τ of 14 ± 3 ns, akin to the ripe AR ribbons. This could also be at the origin of a slightly higher quantum yield for AF compared to AR ribbons (40% vs. ∼30%). CFM shows that cyan emission and longer lifetimes are correlated (Fig. S4 and S5, ESI†). An important feature is that AF ribbons show almost the same polarisation of emission as ripe AR ribbons, with Pmax = 0.50 ± 0.05 (Fig. 1h and i).
The spectral features of cyan emission are reminiscent of those reported for DPA or anthracenes in some aggregated states.56,58 The broad emission band is attributed to pairs of molecules in structural defects, acting as exciton traps, emitting from longer-lived excimer-like states with dipolar coupling strengths depending on their relative orientation.54,55,59,60 This suggests that ribbons with cyan emission and longer lifetimes contain some packing defects. An estimation of the amount of defects is provided by the controlled addition of an exciton trap,61 the energy acceptor 6,11-diphenyl-2,3-dihexadecyltetracene, described in previous studies.17,45 With only 0.1 mol% doping, substantial quenching and sensitization of the tetracene occurs (Fig. 2). This suggests that the undoped AF ribbons have <0.1 mol% of defects and the AR ribbons, evidencing no measurable cyan emission, thus have remarkably negligible amounts of structural defect. This is an indication of a crystalline-like order, such as in previously studied correlations in nanofibres.17,62 Besides, the homogeneity of the optical properties within AF ribbons, but with variations among different sub-sets of ribbons suggests that the self-assembling mechanism somewhat diverges along its progression (see below).
 | ||
| Fig. 2 (a) Photography under UV light irradiation of pristine DPA16 AF ribbons (blue) and 2 mol% tetracene doped AF ribbons (green); (b) emission spectrum of 0.1 mol% doped ribbons; (c) structure of the tetracene dopant; (d) polarisation plot of 0.5 mol% doped ribbons showing both blue emission and green emission, with respective P values represented by blue and green crosses. | ||
This experiment also preludes to an extension of the family of AF ribbons to DPA16-based nanoribbons displaying a wide range of colours, for example tuneable from blue to green (Fig. 2).63 This tuning is achieved by the dispersion within the DPA16 ribbons of up to ∼2 mol% of tetracene (preparation is described in the ESI†), and defect-emission becomes unobservable and has very low impact on the colour. The principles of colour-tuning with this tetracene were reported previously in 2,3-didecyloxyanthracene (DDOA) nanofibres.17,45 The doped green ribbons also emit linearly polarised light very similar to the blue ribbons (Fig. 2d), showing that the DPA16 host nanostructure orients the guest and maintains a high anisotropy of both structure and emission.
Molecular packing model of DPA16-ribbons
The molecular packing of DPA16 determines the optical properties of all type-A nanoribbons. In order to determine this structure in the absence of crystallization of DPA16 and hence X-ray crystallographic data, a correlation was first established between the highly sensitive fluorescence properties of a family of shorter chain 2,3-dialkoxy-9,10-diphenylanthracene analogues and their respective crystal structures.63 As compared to unsubstituted DPA, and as reported in other cases,64–69 with long alkyl chains a segregation appears between chains and aromatic cores forming alternating layers (Scheme 2 and Fig. S6, ESI†). In 2,3-dioctyloxy-9,10-diphenylanthracene (DPA8), the anthracene-cores are separated by 17–18 Å along the c-axis, out of the range of possible π–π interactions.70 This is accompanied by a quasi-coplanar stacking of the DPA anthracene cores with a distance of ∼3.7 Å. However, they are systematically oriented almost orthogonally to each other due to the steric hindrance induced by the lateral phenyl rings. It can be proposed that the packing of DPA8 in crystals and that of DPA16 in nanoribbons tend to be essentially the same (Scheme 2 and Fig. S7, ESI†), with deviations described below. This hypothesis is corroborated by the comparison of the polarisation of the emissions and AFM measurements.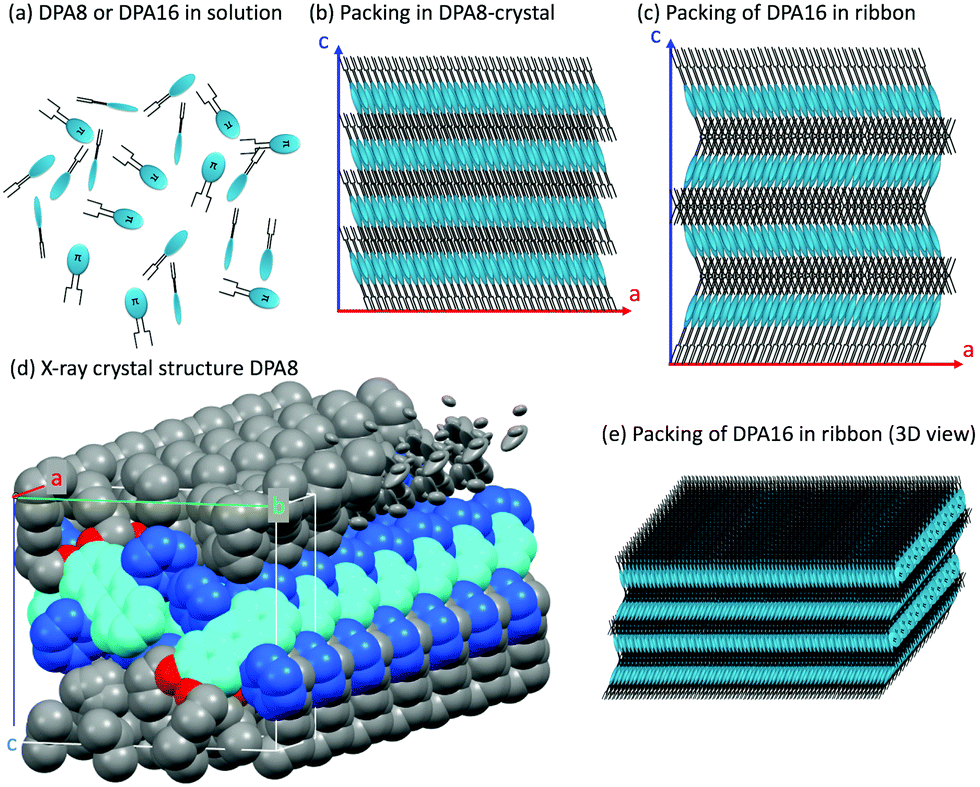 | ||
| Scheme 2 Schematic representation of: (a) DPA8 and DPA16 molecules used in (b, c and e), (b) packing of DPA8 in crystals (ac plane); (c) proposed packing of DPA16 in ribbons (ac plane): layers are packed with alternating orientations and are 0.7 nm thicker along the c-axis than in DPA8; (d) X-ray crystal structure of DPA8 showing seven unit cells stacked along the a-axis. The chain layer contains interdigitated chains from neighbour layers and is partially represented in elliptical representation to highlight the uncertainty on the position of some chain ends (indicative of disorder); (e) 3D-view of (c) and nanoparticle of DPA16. | ||
AFM measurements show that typically the AR ribbons lay flat on the surface, with the shorter axis upwards. The layer-like morphology appears clearly (Fig. 3 and Fig. S8, S9, ESI†) in both DPA16-ribbons and DPA8-crystals with reproducible layer heights of 2.8 ± 0.3 nm and 2.1 ± 0.3 nm, respectively. The difference in layer thickness of 7 ± 3 Å can be attributed to the additional alkyl-chain length of DPA16, suggesting that the chains in both cases are oriented essentially along the shorter axis (Scheme 2 and Fig. S7, ESI†). The orientation of the anthracene cores can be proposed based on fluorescence polarisation microscopy. Indeed, the emission polarisation P is determined by the orientation of the fluorophore and its packing. The polarisation plots (P vs. angle of orientation of ribbon) of DPA16 ribbons and DPA8 crystals lying flat on a surface are identical and can be both described by a model simulating the P values using the same orientation of the anthracene cores as in the crystal structure of DPA8 (Fig. 3c and Fig. S10, ESI†). The crystalline unit cell is thereby determined to be oriented with the c-axis along the shortest dimension and the a-axis along the infinite growth direction (Scheme 2; symmetry P21/a; a = 0.94 nm, b = 2.05 nm, and c ≈ 2.5 nm). However, the analysis of some rarer ribbons that lay on their edge (a/c-plane) requires a small modification of this model. In this case, the polarisation plots (Fig. 3d and Fig. S11, ESI†) can only be simulated with a model in which the DPA16 molecules within a layer are organized as in DPA8 crystals, but successive layers along the c-axis are statistically oriented 50% along the (a)-axis and 50% along the opposite (−a)-axis (Scheme 2c). This contrasts with the DPA8 packing (all layers oriented along the a-axis), which is certainly energetically the most favourable in a large crystal. However, the structure proposed for DPA16 suggests that a less specific inter-layer interaction can occur at a negligible energetic “cost”, due to a lesser specificity of the longer alkyl chain interactions. A certain disorder of the chain packing can already be observed in the DPA8 crystal structure, appearing as an uncertainty of the positions of some terminal CH2 of the alkyl chains (Scheme 2d). This non-specific layer orientation and additional disorder of the chains may be critical features for the growth of anisotropic nanoribbons, rather than crystals. The deviation from perfectly crystalline packing does however not induce reduction of the polarisation of the emission observed along the c-axis direction and makes the polarisation along both minor axes essentially identical.
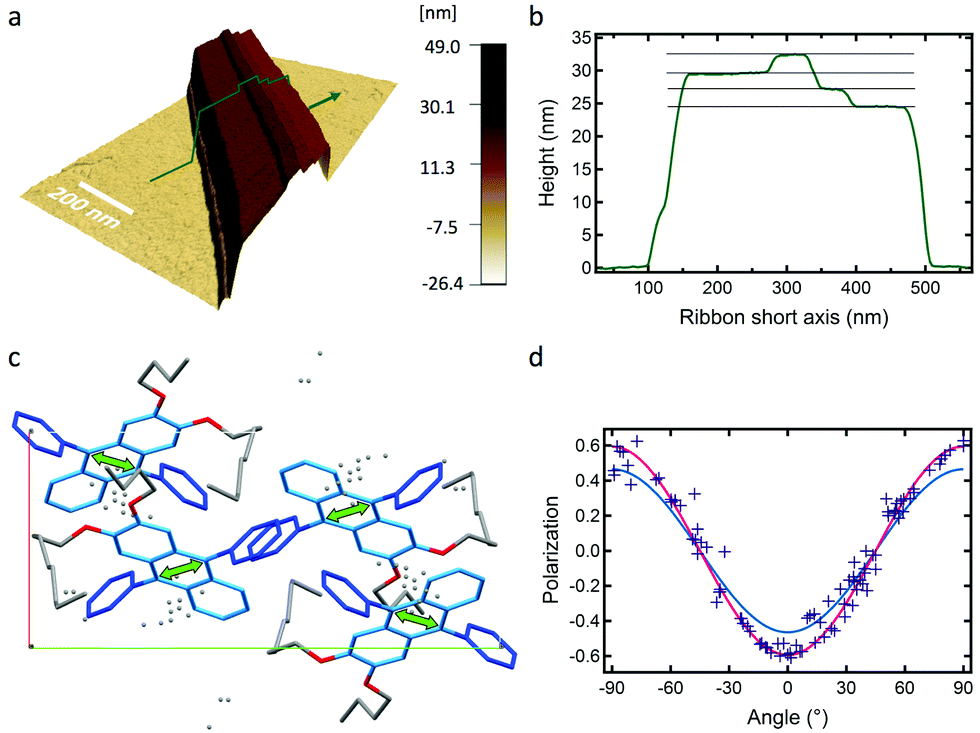 | ||
| Fig. 3 (a) AFM topography image in 3D of an AR ribbon (green arrow, profile in (b)). (b) Profile along the short axis of the ribbon in (a) and top-layer heights of 2.56, 2.53 and 2.68 nm (±0.08 nm), (c) top-view of molecular packing unit cell proposed for a ribbon lying flat oriented with an angle θ of 90°, and the transition dipole moment orientation (green arrow), (d) P vs. θ polarisation plot for AR ribbons and (full lines) simulations of ribbons lying flat (red line, Pmax = 0.60, same as (c)) and on-edge (blue line, Pmax = 0.47), both θphase = 90°. | ||
In order to verify whether emission polarisation is influenced by possible intermolecular transition dipole coupling, Kasha's exciton theory71 was applied, although small spectral red-shifts and Stokes shifts suggest a very weak coupling.54–56 As illustrated in more details in the ESI† (Fig. S12), only very weak coupling at <20 cm−1 is obtained due to inter-dipole angles that are close to 90° or too large distances, and a moderate oscillator strength. Calculations also confirm that an exciton hopping can occur through homo-FRET (Förster Resonance Energy Transfer) with efficiencies >90% in the first shells. This results in only a slight broadening of the absorption bands, a negligible impact of photoselection (see eqn (2)) and an overall emission polarisation not affected by coupling.
Oriented attachment growth and Ostwald ripening
The understanding of nanoribbons’ properties requires a clear knowledge of the growth mechanisms, which is not limited to a simple one-step aggregation process. Optical transmission video microscopy of the “fast” type-A ribbon formation in a small sample holder without stirring (Fig. 4a, b and Fig. S13, ESI†) reveals the initial generation of a high density of diffusing aggregates, 400–2000 nm wide, as well as larger static aggregates. Progressively, the density of observable slowly diffusing particles decreases while ribbons grow. The gap between ribbons and observable particles widens over time, possibly due to the down-sizing of the particles in proximity of the up-taking ribbons, as would be expected for Ostwald ripening, or accelerated/hindered diffusion. A time-lapse observation of growth in more typical volumes of 5 mL under stirring was conducted by fluorescence microscopy, exploiting lifetime- and hyper-spectral imaging to reveal structural heterogeneities (Fig. 4c, d and Fig. S14, S15, ESI†). In the first 2 minutes, very few big (1–5 μm) strongly heterogeneous aggregates and small more homogeneous aggregates are observed. The first sparse anisotropic 8 μm-long heterogeneous object appears after 3 min. After 4 min the first heterogeneous ribbons can easily be found in addition to aggregates. Meanwhile, the biggest aforementioned aggregates are still present but do not grow quickly towards ribbons even within 7 min (Fig. 4e, can be compared to ribbons observed in Fig. 4b). Their contribution of cyan emission indicates larger amounts of defects which might be responsible for their halting growth and subsequent slow dissociation. After 30 min, only ribbons without the presence of other aggregates are observed (Fig. 4f). The ripening can continue slowly over days inducing a slight reduction of the cyan emission.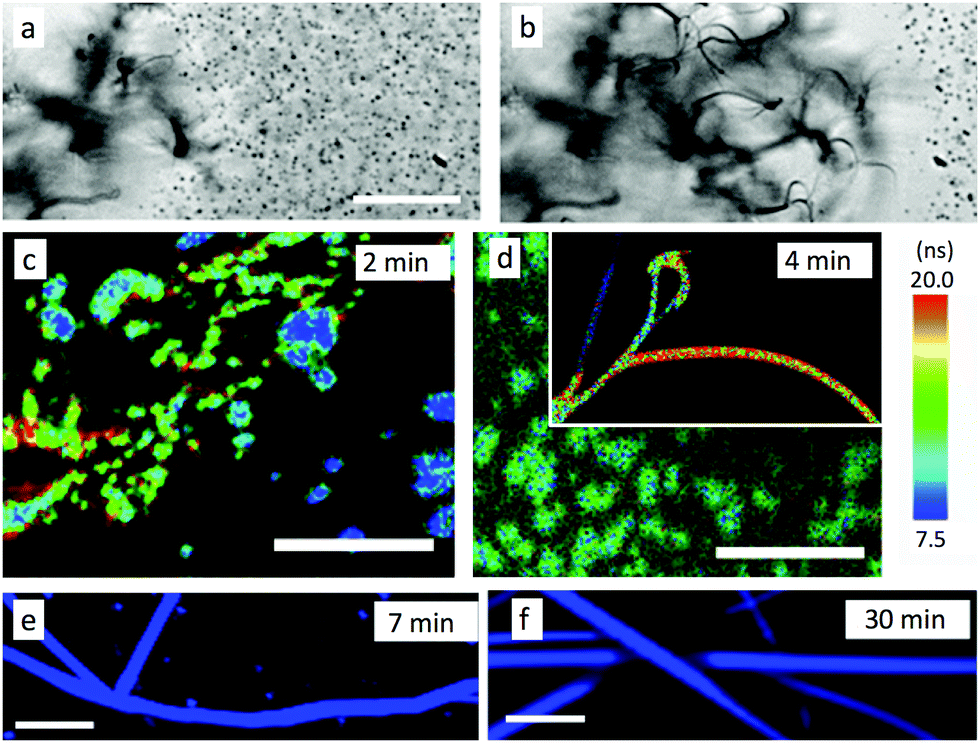 | ||
| Fig. 4 Evolution of AF ribbon ripening, with preparation conditions that slightly differ in (a and b) as compared to (c and f), see text: (a and b) optical transmission microscopy of diffusing aggregates and almost immobile ribbons, scale bar 10 μm, (a) at t0, <30 s after injection, (b) at t0 + 30 s; (c and d) FLIM-images of aggregates (average lifetime, scale bar 5 μm), (c) at 2 minutes, (d) at 4 min (inset: different area, same scale); (e and f) hyperspectral RGB-colour converted image of ribbons, scale bar 10 μm, (e) at 7 min, (f) at 30 min. | ||
In order to investigate more precisely the self-assembly mechanism, the evolution of the growth was slowed down using lower temperatures and additionally stopped by drying the samples after 2 h and before observation (DCM:MeOH 1:50). This induces a stepwise inhibition of the growth, revealing clearly three different stages (in contrast to the mixture of objects in the experiments above). Formed at 5 °C, nanoparticles displaying sizes from 20 to 80 nm wide are observed by AFM (Fig. 5a), but no AF ribbons are observed. At 10 °C, meso-aggregates of similar nanoparticles are observed (Fig. 5b). These display some rather straight edges and are quite flat with uniform layer heights of 3.6 ± 0.3 nm, with a first basis of 10.8 ± 0.8 nm of height, which corresponds to 3 layers of 3.6 nm. This might be the threshold number for a stable layer-aggregate. These aggregates are larger and sufficiently emissive for polarisation microscopy, which reveals that meso-aggregates emit highly polarised light with Pmax = 0.50 ± 0.05 (Fig. 5d and f). This approaches the highest values obtained even for the AR ribbons and the previously proposed molecular packing. This is only possible if all of the individual nanoparticles composing the meso-aggregate are oriented the same way, rather than randomly in which case the polarisation would be averaged to zero. This demonstrates an across-the-aggregate identical and homogeneous molecular packing.
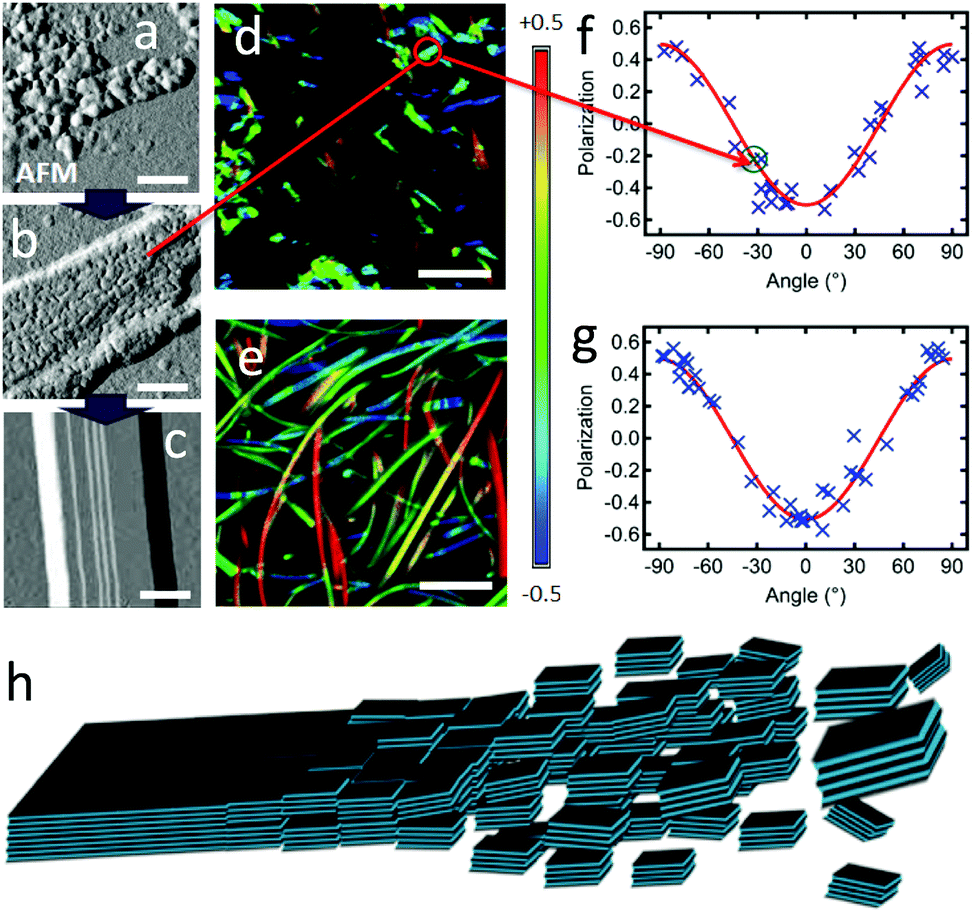 | ||
| Fig. 5 (a–c) AFM amplitude images, with scale bar of 200 nm, of (a) nano-particles formed at 5 °C (same concentrations as AF); (b) assembly of nanoparticles (meso-particle) formed at 10 °C; (c) AF ribbon formed at >15 °C. (d and e) Polarisation images, with scale bar of 5 μm, of (d) meso-particles, same area as (b); (e) ribbons. (f and g) Polarisation plots from images (d) and (e), respectively (highlighted in (f), the P value for the meso-particle measured in (b)); (red curves): fitting according to eqn (2) yielding Pmax = 0.50 ± 0.05 and θphase = 90° for both data sets. (h) Scheme of the self-assembly process through oriented attachment growth. Aromatic cores are light blue, alkoxy chains are black lines (see also Scheme 2). | ||
The ordered assembly of nanoparticles into meso-aggregates clearly supports the intervention of an unanticipated growth mechanism, oriented attachment growth. This mechanism was described first by R. L. Penn and J. F. Banfield72 for attachment of nanocrystals upon recognition of the same crystalline faces and subsequent fusion in the assembly of inorganic materials.32,72–75 This growth mechanism has however not been proposed yet for pure organic systems, although mediation by organic ligands of inorganic particles76,77 and organic systems with meta-stable aggregates had been observed.78,79 In the final case, observed in samples grown at >15 °C the aggregates form clear uniform ribbons with reduced layer heights ranging from 2.5 nm to 3.1 nm. This can be rationalized by fusion of the particles with partial reorganization and denser packing of the alkoxychains, possibly finalizing the connection by the expulsion of solvent molecules. The oriented attachment mechanism is also supported by the clear differences of the three facets of the monoclinic unit cell proposed for DPA16. Indeed, the a/b-face exposes only alkyl chains, the a/c-plane partially alkyl chains and phenyl groups and the b/c-plane exposes the alkoxychains and anthracene-cores. As a consequence, and as proposed in Fig. 5h, specifically oriented interactions are favoured between colliding nanoparticles and consequent fusion (see also the Discussion section).
It is most likely that a competition occurs between oriented attachment growth and Ostwald type ripening. Such a hypothetical combination has so far only been mentioned by Li and co-workers for inorganic nanocrystals.32 Although the oriented attachment growth results in coherent interfaces and single crystals, defects at the plane of attachment occur in inorganic materials and can therefore be expected in the ribbons.72 Thus, the contribution of red-shifted longer-lived emission in the AF ribbons suggests some presence of defects due to oriented attachment growth mediated self-assembly. The proportions of both mechanisms could change during the progression of the assembly process, leading to ribbons with individually slightly different spectral properties. Initially, under high super-saturation, ‘cyan’ AF ribbons with more defects would be formed. Progressively, the uptake of nanoparticles induces a decrease in the concentration of particles and possibly disfavours oriented attachment growth or allows a more defect-free oriented attachment. Simultaneously, Ostwald ripening could progressively prevail, favouring defect-free AF ribbons growth with deeper blue emission. In the case of AR ribbons, due to higher DCM content the super-saturation is lower even at the initial stages, and therefore the relative contribution of Ostwald ripening could be more important at all stages. This would lead to slower and more defect-free growth.
Emission polarisation orientation and dipole moment switching
Slowing down the self-assembly at 10 °C with low DPA16 concentration (1 × 10−4 M) and decreased MeOH:DCM ratio (20:1) results in the formation in >24 h of ripe “type B” ribbons (BR), being shorter than type A (non-infinite) and often laterally aggregated (Fig. 6a). The unambiguous, although partial, polarisation curve obtained from isolated ribbon-fragments yields a polarisation amplitude of Pmax = 0.5 ± 0.1, but with θphase = 0°, thus 90° phase-shifted relative to the case of type A ribbons (Fig. 6b, see also Fig. S16, ESI†). This suggests a similar packing of the emissive aromatic core in both types of ribbons, but with a long-axis transposed by 90° (b-axis in type B, and a-axis in type A). According to the model herein, the a/b planes constitute the largest surface, and the b/c-planes are exposed on the sides of the ribbon and grow to a limited extent at 10 °C. However, re-thermalization to 23 °C during fast drying of the ribbon sample in the microscopy setup most likely promotes the interactions of these b/c-planes and resulting lateral attachment of ribbons, but also more disorganized entanglement of type B ribbons along this direction (equivalent to the main growth direction for type A ribbons). This behaviour implies that attachment along the a-axis is favoured by a thermal activation, provided at 23 °C but not at 10 °C. Slight differences in the packing of the side chains could occur at 10 °C and 23 °C.
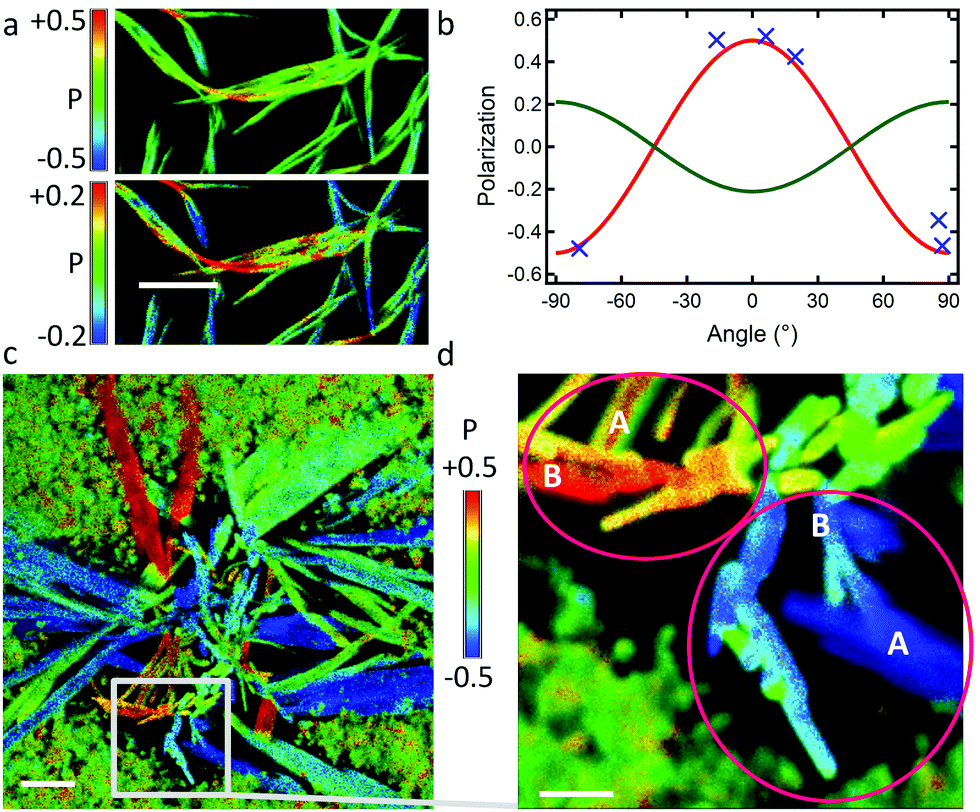 | ||
| Fig. 6 (a) Polarisation image of BR ribbons; scale bar is 5 μm; (top) and (bottom) are identical images with two different scales of P; (b) polarisation plot (blue crosses) from isolated ribbon sections in image (a); (red curve) simulated plot for ribbons lying flat on the surface with the a/b plane and θphase = 0°; (green curve) simulated plot for ribbons on their edge with the b/c plane on the surface and θphase = 90°; in entangled ribbons, the twisting of ribbons leads to a contribution of both oppositely polarised emissions and a resulting attenuation of the overall detected polarisation (green colour code in (a-top)) (see also Fig. S16 in ESI†). (c and d) Nano-aggregates, prepared at 5 °C, then exposed to DCM vapor. (c) Polarisation image of a sprout of ribbons; scale bar is 5 μm. (d) Excerpt of (c) showing sites (pink ellipses) where the polarisation of type-A ribbons (marked as “A”) and of adjacent perpendicularly oriented type-B ribbons (“B”) are similar. Scale bar is 2 μm. | ||
The similarity in molecular packing can be highlighted under experimental conditions favouring epitaxial-growth. This could not be provoked in solution, however, slow growth of ribbons on a microscope glass coverslip by applying a DCM solvent-vapor annealing on dry DPA16 nano-aggregates shows an interesting behaviour. Polarisation microscopy (Fig. 6c) reveals that B-ribbons form in the centre of the growth region, whereas wide A-ribbons form towards the periphery, apparently at a later stage. Most interestingly, A-ribbons start growing from a B-ribbon and elongate nearly perpendicular to it (Fig. 6d and Fig. S17, ESI†), suggesting an epitaxial growth and thus an only small difference of unit cells in type A- and B-ribbons.
A finer observation of the fluorescence spectra reveals that A-type ribbons emit like DPA8 crystals, whereas B-type ribbons and nanoaggregates emit similar to DPA16 molecules in THF solution (Fig. 7 and Fig. S18, ESI†). Some unexpected refinement of the molecular packing model is required to accommodate these differences, considering that the orientations of the aromatic cores are suggested by polarisation microscopy. A more red-shifted blue emission in the ripe type-A ribbons can be explained by a larger van der Waals term based on Kasha's exciton dipolar coupling theory, meaning that the excited states are stabilized by a strong permanent dipole of adjacent molecules. Its origin cannot evidently be attributed to the modest dipole of 0.68 debye of DPA16 in its more stable conformation. Therefore DFT-calculations have been conducted and revealed that asymmetric conformations display a much larger dipole moment. In particular, an asymmetric conformation with a higher energy minimum displays a dipole of 3.30 debye with a significant out-of-(aromatic) plane component (Fig. 8 and Fig. S19, S20, ESI†). The variations in dihedral angles and in electron density distribution is coupled with a slight torsion of the side-phenyl rings. The energy landscape shows that barriers between symmetric and asymmetric states can be easily overcome at 23 °C, allowing regular switching between conformations in solution (Fig. 8a). Although higher in energy, an asymmetric conformation can be observed for DPA8 in crystals, and in this geometry the dipole is calculated to be 1.25 debye. As a result, as can be seen in Fig. 8c and d, favourable head-to-tail dipole–dipole interactions occur amongst adjacent DPA8 successively packed along the a-axis. By analogy, the same interactions are proposed to favour and accelerate the assembly of DPA16 along the a-axis during growth.
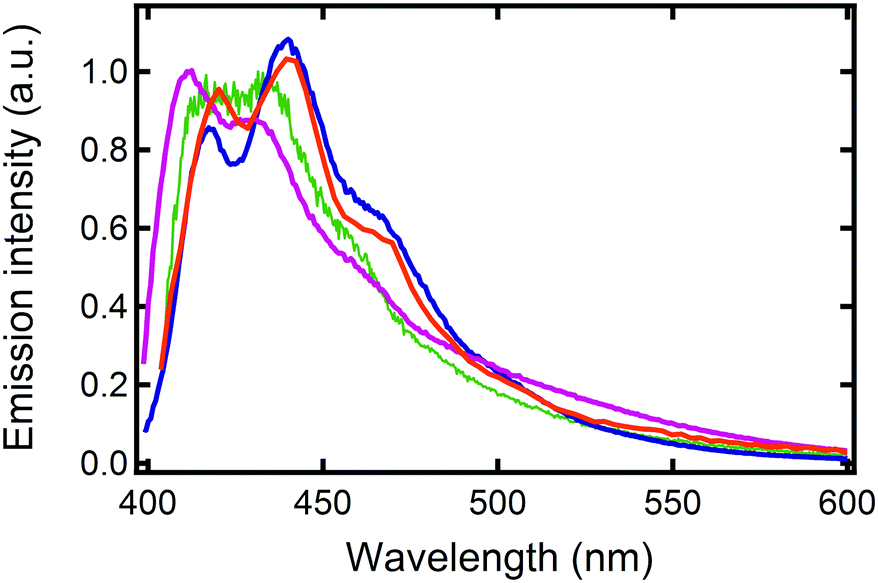 | ||
| Fig. 7 Emission spectra at 23 °C of DPA16 ribbons: BR prepared at 10 °C (violet), AR at 22 °C (blue), of DPA16 nanoparticles (AF conditions, green) and a DPA8 crystal (orange). | ||
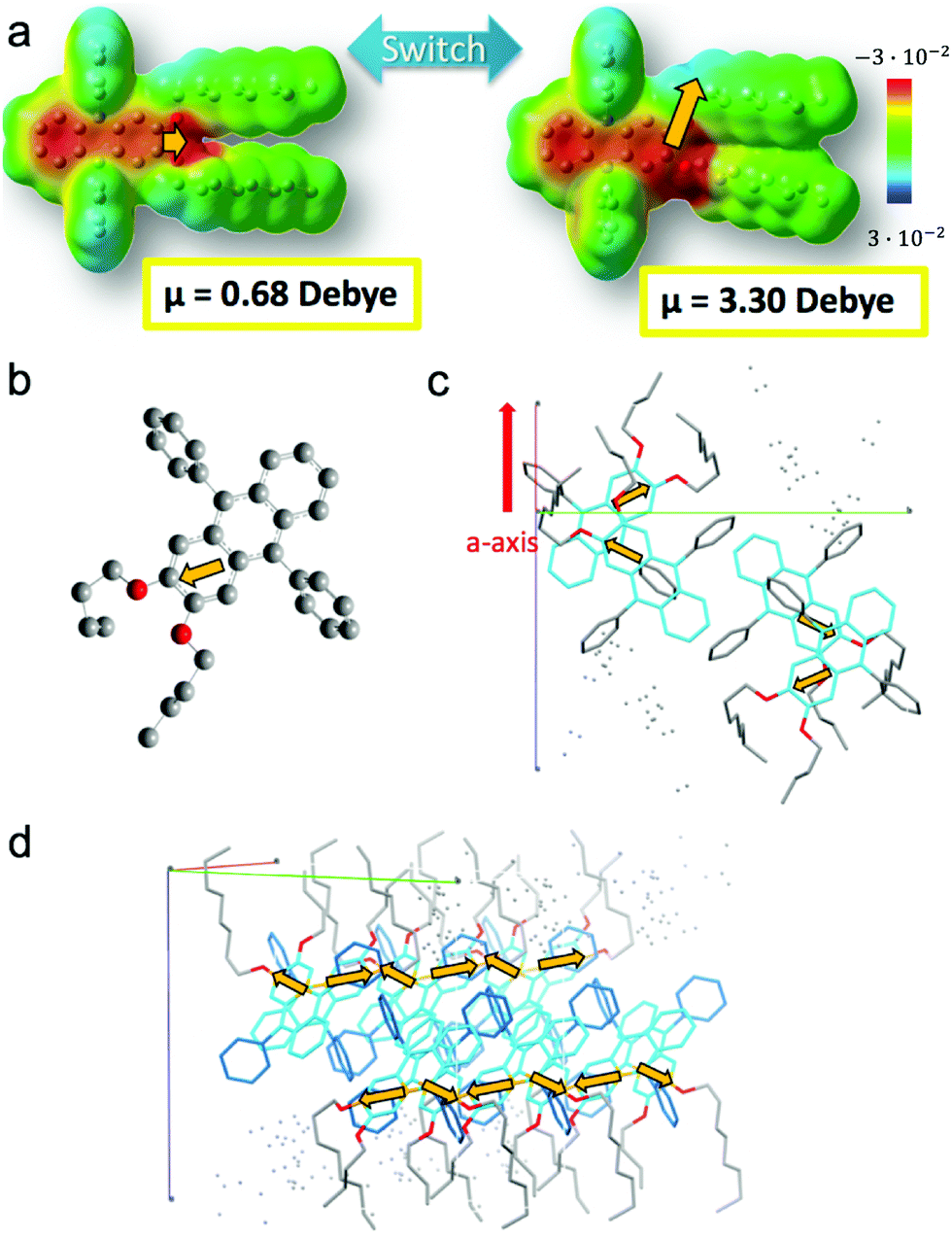 | ||
| Fig. 8 (a) Charge density distribution in DPA8 in the most stable symmetric and a meta-stable asymmetric configuration. The switch induces an increase of dipolar moment μ (orange arrow) from 0.68 debye to 3.30 debye, with an out-of-(aromatic plane) orientation. (b) Dipole moment calculated for a DPA4 in a conformer identical to crystallized DPA8. (c) Inter-dipole interactions of neighbouring molecules in a unit cell. abc axis are represented. (d) Packing of several unit cells along the a-axis showing the alignment of dipoles (arrows in orange not represented in perspective). | ||
In contrast, this higher energy conformation must be scarcer at lower temperatures and B-type ribbons are proposed to form under conditions, with higher DCM content, in which the concentration of asymmetric conformers is sub-critical for aggregation. This may favour the slower assembly of more stable conformers with a smaller dipole. Therefore, the emission is less red-shifted in type-B ribbons as compared to type-A ribbons, in which excitons are stabilized by a more polar environment (Fig. 7). In the case of nanoaggregates, a combination of both types of molecular conformations could be at the origin of a broadening of the emission spectrum. It is conceivable that in small nanoaggregates, which are unripe structures, some molecules can still switch from the symmetric to the asymmetric conformation depending on the temperature. Attaining a certain threshold of high dipoles in the nanoaggregates could trigger a collective switching of conformations or promote a cooperative binding of molecules in the higher energy conformation (e.g. during Ostwald ripening). This conformational change could also occur upon nano-particles’ oriented attachment. Besides, the incorporation of solvent molecules which influence dipole moment interactions cannot be completely excluded, as observed in several organic crystals,80–82 and could play a dynamic role.
Discussion and conclusions
Scheme 3 highlights the correlations between ribbon growth conditions, their properties and the mechanism proposed.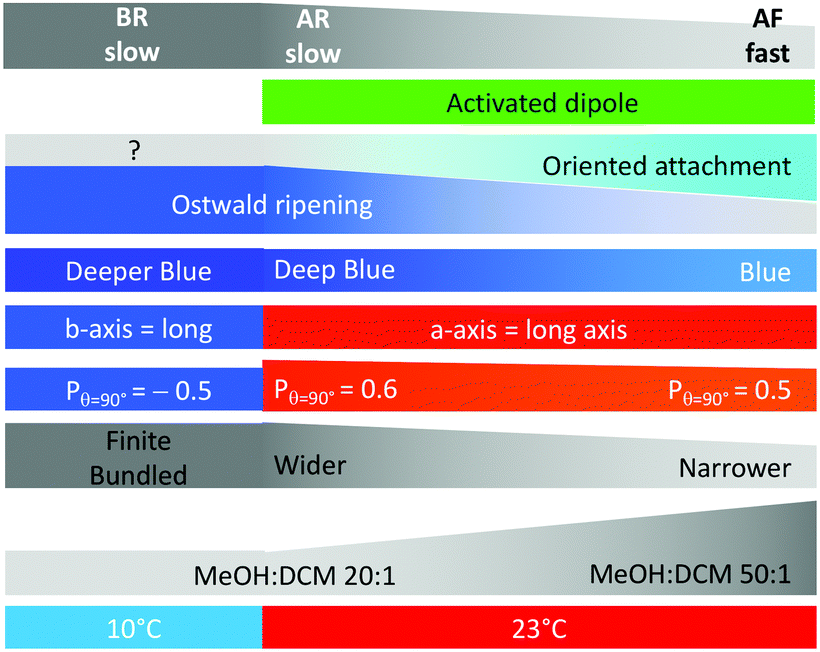 | ||
| Scheme 3 Summary of results and interpretations, including (from bottom to top): growth temperature, solvent mixture, ribbon size, fluorescence polarisation, main growth axis, emission colour, proportional contribution of growth mechanism, presence of activated dipoles, and growth speed. | ||
The high linearly polarised fluorescence and anisotropic growth of nano-ribbons are directly correlated with the molecular packing of the molecule of interest, DPA16. A fundamental understanding of the impact of the molecular design of hydrogen bonding-free self-assembly providing both aforementioned properties can be deduced from this study. The anthracene and alkyl chain moieties are well-separated in the molecular structure, facilitating a phase-segregation in the solid state between the aromatic and aliphatic moieties. Albeit side phenyl rings introduce steric hindrance, the anthracenes still pack preferentially in stacked configurations. The packing of alkyl chains is difficult due to their great length, and thus is partially disordered and reduces the self-assembly kinetics. This leads to the formation of layers that stack along the shorter ribbon axis (c-axis), but are limited to nano- or mesoscale dimensions.
The axial 2,3-di-alkoxy substitution on the aromatic core plays an essential role in the molecular packing and in the dynamics of the self-assembly, although through an initially unsuspected way. Indeed, the distortion of the Aryl–O–CH2 bonds contribute to establish a stronger dipole estimated to be at least 1.25 debye (up to 3.30 debye) in less stable asymmetric conformers that are reached by thermal activation. The resulting dipole–dipole intermolecular interactions strengthen the binding of the b/c facets in the packed state with (i) diffusing molecules (Ostwald ripening mechanism) or (ii) nano-particles (oriented attachment mechanism). In these two cases, cooperativity could affect the DPA16 conformational change upon binding and increase the ‘degree of polymerization’.23 This creates a strong anisotropy along the three spatial directions:
(i) The a-axis is characterized in type-A ribbons by strong interactions and faster infinite growth due to strong directional dipole–dipole interactions between b/c facets, in addition to aromatic core interactions; whereas the absence of dipole–dipole interactions may limit its growth in type-B ribbons at lower temperature.
(ii) The c-axis grows the slowest and to the least extent, due to weaker aliphatic interactions. The a/b facet is also the least specific, as demonstrated by the fact that sheets can grow with both (a) and (−a) orientations.
(iii) The b-axis grows to an intermediate extent, and although it supplants the a-axis growth in B-type ribbons, it does not lead to infinite growth in contrast to a-axis growth in A-type ribbons. This could be a result of insufficiently strong interactions between a/c facets, such as missing dipole–dipole interactions combined to a reduced cooperativity in the binding.
Thus, a correlation between a molecular feature, not involving H-bonds, and a privileged growth direction could be demonstrated. The intervention of a conformational switching inducing directional bonding through dipoles could be proposed to occur more generally in the directional self-assembly of H-bond free aromatic compounds.
Additional factors are related to the solubility of DPA16 and the driving force thereby induced. The higher degree of supersaturation reached due to lower solubility leads to faster self-assembly kinetics and thinner ribbons. This can induce a larger contribution of the oriented attachment growth mechanism and the appearance of some defect-related emission, although preserving in majority the deep-blue emission component. Therefore, blue-emissive ribbons with quantum yields of 40% are formed even under more practical “fast” growth conditions. A deeper blue emission can, if necessary, be achieved by a slower ripening into larger ribbons. In both cases, the polarisation of emission is remarkably high (Pmax = 0.5–0.6). This can be compared for example to the state-of-the-art aligned nanorods displaying a degree of polarisation P of 0.62 that were developed to improve transmission efficiency in emissive layers for QLED screens.83 In a comparable application of DPA16 ribbons, with 0.5 < P < 0.6, it can be extrapolated that the light emitted by ribbons fully aligned on a surface would be transmitted through a clean-up linear polariser with a high efficiency of 75–80%, in the difficult blue colour range.
Concerning the direct relationship between anisotropic growth and linearly polarised fluorescence, it can be concluded from this study that a relationship exists, although more indirect as intuitively expected. Indeed, at the level of a single nanoribbon, since the molecular packing governs the polarisation of emission, theoretically even objects growing identically in the a and b directions would yield a linearly polarised emission with the same amplitude as if growth occurred essentially only along a. However, the highly different interactions of the unit cells in the three directions a, b and c guarantees to lead to anisotropic growth and therefore avoids ‘fatal’ misorientations in the self-assembly in the a/b plane. For example, it could easily be imagined that attachment of nanoparticles during growth could occur with ‘failed’ orientation, leading to disordered packing and attachment of oppositely oriented ‘patches’. This would lead to an averaging of the polarisation tending towards overall unpolarised emission of an object. Another important impact of anisotropic growth resides in the capacity to exploit the properties of individual objects. Indeed, first, due to the high aspect ratio, the ribbons tend to lay flat on the surface, thus all in the same plane. Second, it has been shown that highly anisotropic nanoobjects, such as nanofibres, can be collectively oriented on a surface. The mechanism proposed in previous studies show that the anisotropic feature is important to collectively align the nanoobjects, resulting in micro- to macroscopic collective anisotropic optical properties, such as linearly polarised emission or birefringence.14 This can also be exploited to achieve hierarchical self-assembly and patterning.22
In conclusion, this study provides a better understanding of the unexpected mechanisms involved in anisotropic growth of H-bond free aromatic compounds, involving oriented attachment as well as a directional conformational switching induced dipole–dipole interaction. The resulting nanoribbons display attractive deep-blue and highly linearly polarised emission, solvent-processed surface coverage and possible broad colour-tuning.
Experimental
Additional details for the synthesis and characterization of DPA8 and DPA16 are available in the ESI.† The DPA8 crystal structure was registered as CCDC (2013401).AR ribbons were obtained by dissolving DPA16 in DCM at a concentration of 2 mM, and then injecting 250 μL of this solution into 5 mL of N2-purged MeOH in a glass vial at r.t. and briefly stirred (final [DPA16] = 9.5 × 10−5 M). The vial was left for 24 hours with very gentle stirring, in the dark under N2. A glass pipette was used to gently pick up ribbons with very little solvent and dripped onto a rotating (200–1000 rpm) microscopy coverslip (glass, 170 μm thick, 2 × 2 cm). The solvent evaporates quickly and after 30 additional seconds the slide was placed in a closed sample holder purged with N2 in the microscope. A similar procedure was used for AF ribbons. In most experiments, 100 μL of a 5 mM DCM solution were injected into 5 mL of MeOH ([DPA16] = 9.8 × 10−5 M) kept at 23, 10 or 5 °C within a thermostated water-bath. Ribbons were typically prepared the day before characterization. In a set of spectroscopic measurements, AF ribbons were obtained by injecting 100 μL of a 10 mM DCM solution into 3 mL of MeOH ([DPA16] = 3.2 × 10−4 M). This yielded ribbons in <5 hours.
A modified PicoQuant MT200 confocal microscopy setup was used in a variety of configurations. Excitation was performed with a Picoquant 5 MHz pulsed diode laser at 375 nm. For hyperspectral imaging, as described elsewhere,21 a confocal configuration was used to acquire emission spectra (Andor spectrometer with Newton electron multiplying charge coupled device (EM-CCD) camera) and spectra were converted first to CIE colour coordinates and then displayed in RGB colour code. Spectral intensity correction was performed with an ARGO-U SLB8 slide (Argolight). FLIM was achieved with a standard Picoquant MT200 confocal setup and Symphotime software. Polarisation microscopy was performed, as described elsewhere,22 using the confocal setup and homemade Labview VIs. The G-factor calibration was achieved using a dilute solution of the studied compound as the reference. For wide field video-microscopy, the standard Olympus IX71 transmission setup was used, with detection by a Hamamatsu ImagEM EM-CCD (160 nm pixel−1). Atomic force microscopy was performed in intermittent-mode scan using an Asylum Research MFP-3D (now Bruker, tips: 70–75 kHz, radius ∼7 nm, 1.7–3 N m−1). Samples were prepared on glass like for CFM.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support by the European Commission 7th Framework Programme Marie Curie Actions FP7 via the ITN-2012 No. 316656 called SMARTNET, the French National Agency for Research (ANR), the Centre National de la Recherche Scientifique (CNRS), the French Ministry of Education and Research, and the Région Aquitaine. L. G.-R. acknowledges the financial support (postdoctoral grant and project IT912-16) received by the Department of Education, Science and Universities of the Basque Country Government. The authors thank SGIker of UPV/EHU for technical support with the computational calculations, carried out in the “arina” informatic cluster. PS and CdV were funded by SMARTNET, CS by the ANR, MTK by the Région Aquitaine. This work has benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform (BPCS) at IECB, CNRS UMS3033, INSERM US001, Univ Bordeaux. http://www.iecb.ubordeaux.fr/index.php/fr/plateformestechnologiques.Notes and references
- Y. Wang, L. Sun, C. Wang, F. Yang, X. Ren, X. Zhang, H. Dong and W. Hu, Chem. Soc. Rev., 2019, 48, 1492–1530 RSC.
- C. Zhang, Y. Yan, Y. S. Zhao and J. Yao, Acc. Chem. Res., 2014, 47, 3448–3458 CrossRef CAS.
- X. K. Chen, D. Kim and J. L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS.
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS.
- P. Boher, T. Leroux, V. Collomb-Patton and T. Bignon, J. Soc. Inf. Disp., 2015, 23, 429–437 CrossRef CAS.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2019, 1900110, 1–33 Search PubMed.
- A. K. Srivastava, W. Zhang, J. Schneider, A. L. Rogach, V. G. Chigrinov and H. S. Kwok, Adv. Mater., 2017, 29, 1–6 CrossRef.
- M. Li, S. H. Li, D. Zhang, M. Cai, L. Duan, M. K. Fung and C. F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260–6265 CrossRef CAS.
- X. Zhang, D. Gçrl, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 1270–1274 CrossRef CAS.
- S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766–1776 CrossRef CAS.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS.
- I. O. Shklyarevskiy, P. Jonkheijm, P. C. M. Christianen, A. P. H. J. Schenning, A. Del Guerzo, J. P. Desvergne, E. W. Meijer and J. C. Maan, Langmuir, 2005, 21, 2108–2112 CrossRef CAS.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS.
- S. Banerjee, R. K. Das, P. Terech, A. de Geyer, C. Aymonier, A. Loppinet-Serani, G. Raffy, U. Maitra, A. Del Guerzo and J.-P. Desvergne, J. Mater. Chem. C, 2013, 1, 3305 RSC.
- C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M. T. Kao, A. G. L. Olive and A. Del Guerzo, J. Am. Chem. Soc., 2011, 133, 316–325 CrossRef CAS.
- H. Jintoku, M.-T. Kao, A. Del Guerzo, Y. Yoshigashima, T. Masunaga, M. Takafuji and H. Ihara, J. Mater. Chem. C, 2015, 3, 5970–5975 RSC.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539–7551 RSC.
- A. Chakrabarty, G. Raffy, M. Maity, L. Gartzia-Rivero, S. Marre, C. Aymonier, U. Maitra and A. Del Guerzo, Small, 2018, 14, 1802311 CrossRef.
- C. de Vet, L. Gartzia-Rivero, P. Schäfer, G. Raffy and A. Del Guerzo, Small, 2020, 1906723, 1–10 Search PubMed.
- P. Jonkheijm, P. Van Der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS.
- P. A. Korevaar, T. F. A. De Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef CAS.
- J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS.
- P. A. Korevaar, C. Schaefer, T. F. A. De Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS.
- C. H. Chen, L. C. Palmer and S. I. Stupp, Nano Lett., 2018, 18, 6832–6841 CrossRef CAS.
- M. T. Kao, C. Schäfer, G. Raffy and A. Del Guerzo, Photochem. Photobiol. Sci., 2012, 11, 1730–1736 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. De La Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS.
- I. Ramos Sasselli, P. J. Halling, R. V. Ulijn and T. Tuttle, ACS Nano, 2016, 10, 2661–2668 CrossRef CAS.
- D. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. F. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS.
- D. Koylu, S. Sarrafpour, J. Zhang, S. Ramjattan, M. J. Panzer and S. W. Thomas III, Chem. Commun., 2012, 48, 9489–9491 RSC.
- S. Karak, J. A. Lim, S. Ferdous, V. V. Duzhko and A. L. Briseno, Adv. Funct. Mater., 2014, 24, 1039–1046 CrossRef CAS.
- D. Y. Kondakov, J. Appl. Phys., 2007, 102, 114504 CrossRef.
- S. A. Odom, S. R. Parkin and J. E. Anthony, Org. Lett., 2003, 5, 4245–4248 CrossRef CAS.
- B. Yang, J. Xiao, J. I. Wong, J. Guo, Y. Wu, L. Ong, L. L. Lao, F. Boey, H. Zhang, H. Y. Yang and Q. Zhang, J. Phys. Chem. C, 2011, 115, 7924–7927 CrossRef CAS.
- B. Park, I. In, P. Gopalan, P. G. Evans, S. King and P. F. Lyman, Appl. Phys. Lett., 2008, 92, 133302 CrossRef.
- C. S. Kim, S. Lee, E. D. Gomez, J. E. Anthony and Y.-L. Loo, Appl. Phys. Lett., 2008, 93, 103302 CrossRef.
- A. L. Briseno, R. J. Tseng, M.-M. Ling, E. H. L. Falcao, Y. Yang, F. Wudl and Z. Bao, Adv. Mater., 2006, 18, 2320–2324 CrossRef CAS.
- L. Zhang, A. Fonari, Y. Liu, A.-L. M. Hoyt, H. Lee, D. Granger, S. Parkin, T. P. Russell, J. E. Anthony, J.-L. Brédas, V. Coropceanu and A. L. Briseno, J. Am. Chem. Soc., 2014, 136, 9248–9251 CrossRef CAS.
- J. B. Chang and V. Subramanian, Appl. Phys. Lett., 2006, 88, 233513 CrossRef.
- J. Reichwagen, H. Hopf, A. Del Guerzo, C. Belin, H. Bouas-Laurent and J.-P. Desvergne, Org. Lett., 2005, 7, 971–974 CrossRef CAS.
- C. Giansante, C. Schäfer, G. Raffy and A. Del Guerzo, J. Phys. Chem. C, 2012, 116, 21706–21716 CrossRef CAS.
- H. J. Bolink, E. Barea, R. D. Costa, E. Coronado, S. Sudhakar, C. Zhen and A. Sellinger, Org. Electron., 2008, 9, 155–163 CrossRef CAS.
- A. Kadashchuk, S. Schols, Y. Skryshevski, I. Beynik, C. Teichert, G. Hernandez-Sosa, H. Sitter, A. Andreev, P. Frank and A. Winkler, in Interface Controlled Organic Thin Films, ed. K. Al-Shamery and G. Horowitz, H. Sitter and H.-G. Rubahn, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, pp. 121–125 Search PubMed.
- H.-G. H.-G. Liao, L. Cui, S. Whitelam and H. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS.
- D. Dasgupta, S. Srinivasan, C. Rochas, A. Ajayaghosh and J.-M. Guenet, Soft Matter, 2011, 7, 9311–9315 RSC.
- Y. Liu, R.-Y. Wang, J.-L. Li, B. Yuan, M. Han, P. Wang and X.-Y. Liu, CrystEngComm, 2014, 16, 5402–5408 RSC.
- T. E. Kaiser, I. G. Scheblykin, D. Thomsson and F. Würthner, J. Phys. Chem. B, 2009, 113, 15836–15842 CrossRef CAS.
- C.-H. Ting, Chem. Phys. Lett., 1967, 1, 335–336 CrossRef CAS.
- J.-P. Desvergne, T. Brotin, D. Meerschaut, G. Clavier, F. Placin, J.-L. Pozzo and H. Bouas-Laurent, New J. Chem., 2004, 28, 234–243 RSC.
- Z. Zhang, Y. Zhang, D. Yao, H. Bi, I. Javed, Y. Fan, H. Zhang and Y. Wang, Cryst. Growth Des., 2009, 9, 5069–5076 CrossRef CAS.
- P. K. K. Lekha and E. Prasad, Chem. – Eur. J., 2010, 16, 3699–3706 CrossRef CAS.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS.
- A. Bloess, Y. Durand, M. Matsushita, R. Verberk, E. J. J. Groenen and J. Schmidt, J. Phys. Chem. A, 2001, 105, 3016–3021 CrossRef CAS.
- X. X. Zhang, G. Yuan, Q. Li, B. Wang, X. X. Zhang, R. Zhang, J. C. Chang, C. S. Lee, S. T. Lee, G. Yuan, Q. Li, B. Wang, R. Zhang, J. C. Chang, C. S. Lee and S. T. Lee, Chem. Mater., 2008, 20, 6945–6950 CrossRef CAS.
- G. Zhang, G. Yang, S. Wang, Q. Chen and J. S. Ma, Chem. – Eur. J., 2007, 13, 3630–3635 CrossRef CAS.
- S. Hashimoto, S. Ikuta, T. Asahi and H. Masuhara, Langmuir, 1998, 14, 4284–4291 CrossRef CAS.
- A. Del Guerzo, A. G. L. Olive, J. Reichwagen, H. Hopf and J. P. Desvergne, J. Am. Chem. Soc., 2005, 127, 17984–17985 CrossRef CAS.
- P. Terech, C. Aymonier, A. Loppinet-Serani, S. Bhat, S. Banerjee, R. Das, U. Maitra, A. Del Guerzo and J. P. Desvergne, J. Phys. Chem. B, 2010, 114, 11409–11419 CrossRef CAS.
- M.-T. Kao, PhD thesis, Université de Bordeaux, 2012.
- Y. Che, X. Yang, G. Liu, C. Yu, H. Ji, J. Zuo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2010, 132, 5743–5750 CrossRef CAS.
- P. S. Hariharan, D. Moon and S. P. Anthony, CrystEngComm, 2017, 19, 6489–6497 RSC.
- M. Kopeć, W. Niemiec, A. Laschewsky, M. Nowakowska and S. Zapotoczny, J. Phys. Chem. C, 2014, 118, 2215–2221 CrossRef.
- J. Tsutsumi, S. Matsuoka, S. Inoue, H. Minemawari, T. Yamada and T. Hasegawa, J. Mater. Chem. C, 2015, 3, 1976–1981 RSC.
- Y. Xu, S. Leng, C. Xue, R. Sun, J. Pan, J. Ford and S. Jin, Angew. Chem., Int. Ed., 2007, 46, 3896–3899 CrossRef CAS.
- J. Zhang, F. Li, B. Yuan, Q. Song, Z. Wang and X. Zhang, Langmuir, 2013, 29, 6348–6353 CrossRef CAS.
- M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2004, 108, 10200–10207 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549–1557 CrossRef CAS.
- Q. Zhang, S.-J. Liu and S.-H. Yu, J. Mater. Chem., 2009, 19, 191 RSC.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350–2365 CrossRef.
- X. Zhang and M. Takeuchi, Angew. Chem., Int. Ed., 2009, 48, 9646–9651 CrossRef CAS.
- T. Lei, Z.-H. Guo, C. Zheng, Y. Cao, D. Liang and J. Pei, Chem. Sci., 2012, 3, 1162–1168 RSC.
- X. Zhang, X. Zhang, B. Wang, C. Zhang, J. C. Chang, C. S. Lee and S.-T. Lee, J. Phys. Chem. C, 2008, 112, 16264–16268 CrossRef CAS.
- J. Yang, H. Park and L. J. Kaufman, Angew. Chem., 2018, 1–6 Search PubMed.
- J. L. Atwood and J. W. Steed, Organic nanostructures, Wiley-VCH Verlag GmbH, Weinheim, 2008 Search PubMed.
- S. Zheng, M. Xu and X. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 20285–20291 CrossRef CAS.
- A. Pick, M. Klues, A. Rinn, K. Harms, S. Chatterjee and G. Witte, Cryst. Growth Des., 2015, 15, 5495–5504 CrossRef CAS.
- T. Du, J. Schneider, A. K. Srivastava, A. S. Susha, V. G. Chigrinov, H. S. Kwok and A. L. Rogach, ACS Nano, 2015, 9, 11049–11055 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopy and microscopy data, calculations, and crystal structure. CCDC 2013401. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc04789a |
| This journal is © The Royal Society of Chemistry 2021 |