
Combination of ultrashort PCR and Pyrococcus furiosus Argonaute for DNA detection†
Ruyi
He
 ab,
Longyu
Wang
b,
Fei
Wang
b,
Jun
Yang
b,
Xiao
Yu
c,
Yuan
Wang
d,
Zhiguo
Liu
*a,
Chunhua
Li
*b and
Lixin
Ma
*b
ab,
Longyu
Wang
b,
Fei
Wang
b,
Jun
Yang
b,
Xiao
Yu
c,
Yuan
Wang
d,
Zhiguo
Liu
*a,
Chunhua
Li
*b and
Lixin
Ma
*b
aSchool of Life Science and Technology, Wuhan Polytechnic University, China. E-mail: zhiguo_l@126.com
bState Key Laboratory of Biocatalysis and Enzyme Engineering, Hubei Collaborative Innovation Center for Green Transformation of Bio-resources, Hubei Key Laboratory of Industrial Biotechnology, School of Life Sciences, Hubei University, Wuhan, Hubei, People's Republic of China. E-mail: xgspring72@hubu.edu.cn; malixing@hubu.edu.cn
cHubei Provincial Center for Disease Control and Prevention, Wuhan, Hubei, People's Republic of China
dMedical College of Hubei University of Arts and Sciences, Xiangyang, Hubei, People's Republic of China
First published on 30th November 2021
Abstract
A simple and user-friendly nucleic acid sensing platform with 10 aM sensitivity, named USPCRP (combines ultrashort PCR with Pyrococcus furiosus Argonaute cleavage for nuleic acids detection) is reported. The product of this ultrashort PCR could be directly used as a DNA guide to mediate PfAgo cleavage of molecular beacons.
Used in crucial applications in multiple fields – including medicine, human health, food quality control and the environment – nucleic acid detection technology has been given extensive research attention.1,2 Over the past decades, dozens of nucleic acid sensing systems, such as quantitative PCR,3 isothermal amplification,4 nucleic acid hybridization,5 and next generation sequencing (NGS),6 have been well established and widely applied. Among others, the newly emerged CRISPR/Cas-based nucleic acid biosensing systems, including SHERLOCK (Cas13a),7 DETECTR (Cas12a),8 HOLMES (Cas12a),9 and Cas14-DETECTR,10 are regarded as highly innovative approaches that could satisfy various detection goals, such as determination of pathogen-specific nucleic acids, discrimination of single nucleotide polymorphisms (SNPs), and quantification of target nucleic acids, and exhibited an impressive sensitivity, specificity and efficiency. Despite outstanding performance, the existing CRISPR/Cas biosensing systems still have some inherent limitations. For example, Cas9- or Cas12-based biosensing methods require specific PAM (protospacer adjacent motif) for target recognition,11,12 which may limit their wider applications or add some complexity. Besides, those that employ RAP (recombinase polymerase amplification)13 or LAMP (loop-mediated amplification)14 to preamplify the target nucleic acids – such as SHERLOCK and HOLMES – suffer from the demand for multiple enzymes or primer sets, and further complicate the system. Therefore, simpler and user-friendly strategies are highly desired.
Analogous to CRISPR/Cas, Argonaute proteins (Agos) are also a kind of nucleic acid-guided endonuclease and are ubiquitously found in eucaryotes and procaryotes.15 Among the well-studied prokaryotic Argonautes (pAgos), archaeon Pyrococcus furiosus Argonaute (PfAgo) is a typical DNA-guided endonuclease which utilizes small interfering DNAs (siDNAs) with a length of 15–31 nt to target cognate ssDNA without the requirement of a PAM. Featuring this no PAM requirement and shorter DNA guides, PfAgo offers a tempting opportunity to develop novel nucleic acid sensing platforms.
We previously established a PfAgo-based nucleic acid detection method, PAND,17 which enables genotyping and multiplexing detection of DNA. In PAND, three input-guides were used to direct PfAgo to cleave the double-stranded DNA (dsDNA) target, and the newly generated ssDNA product mediated PfAgo to cleave complementary molecular beacons. The demand for three elaborately designed DNA guides increased the costs and design complexity and PAND needs a total time of up to 2 hours.17 These limited its wider applications especially when skilled personnel were not available. Herein, on the basis of the finding by Swarts, et al.16 that the cleavage activity of PfAgo could only be triggered by siDNAs longer than 15 nt, we proposed a simpler, and user-friendly nucleic acid sensing platform, which has no PAM limitation and no input-guides. This platform directly uses PCR products as effective guides for cleaving molecular beacons. One of the primers designed was 5′-phosphorylated with a length of <15 nt, while the other was a common one. After PCR, the 5′-phosphorylated DNA strand of the dsDNA products served as a functional DNA guide to direct the PfAgo-catalyzed cleavage of the reporter.
Firstly, we confirmed our platform16 using a molecular beacon as a target. We found that only a slight fluorescence signal was detected with the guide DNAs of 14 or 13 nt, but no signal with those of <13 nt, and longer guide DNAs did not observe stronger fluorescence (Fig. S1†). Based on these results, we synthesized a 5′-phosphorylated primer of 11–13 nt, named primer-g, for generating functional DNA guides from the target DNA. Considering the limited yield of guide DNA given by a single piece of primer, we then synthesized another conventional primer named primer-h, whose annealing temperature is close to that of primer-g, to allow for exponential amplification generating a high yield of guides. The desired PCR products, confirmed by sequencing, were obtained using these primers, which were then successfully used for the PfAgo-mediated specific cleavage of molecular beacons (Fig. S2†). The PCR product was designed to be 22–28 bp long, thus this PCR was named ultrashort PCR (usPCR). In this system, the 5′-phosphorylated DNA strand of the usPCR product could be used as gDNA directly to mediate PfAgo cleavage of molecular beacons, and no additional gDNA was required. This method was called USPCRP (combines the ultrashort PCR with Pyrococcus furiosus Argonaute cleavage for nuleic acids detection) (Scheme 1), which was easy to design, prepare and implement, omitting the requirement for well-trained personnel. The short primers and products mean lower annealing/extension temperature and denaturation temperature, while PCR products used directly as gDNA could decrease cost.
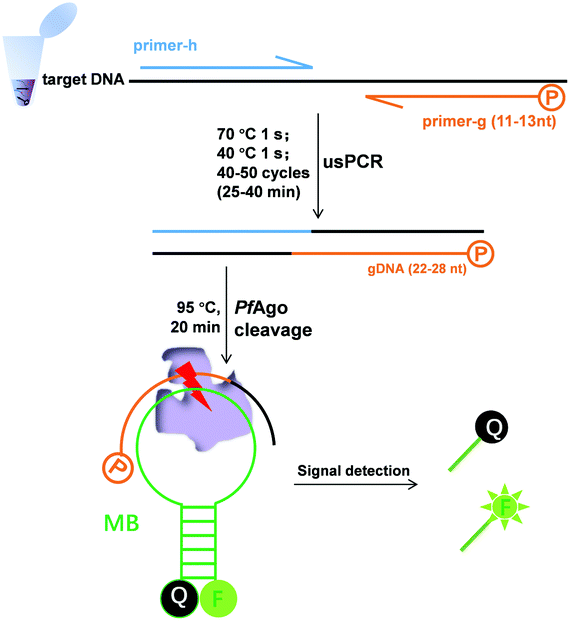 | ||
| Scheme 1 Principle of nucleic acid detection via the combination of ultrashort PCR and Pyrococcus furiosus Argonaute. The 5′-phosphorylated primer (primer-g), another primer (primer-h), and molecular beacons (MB) are highlighted in different colors. | ||
Given that the desired PCR fragment is fairly short, we next explored if a two-step PCR with lower denaturation temperature would fulfil a time-efficient amplification of guide DNAs. We investigated various combinations of denaturation temperatures ranging from 60 °C to 90 °C with annealing/extension temperatures ranging from 30 °C to 50 °C with ssDNA of SARS-CoV-2 ORF1ab (ssDNA-O2) as the target (Fig. S4†). Both desired bands and considerable fluorescence signals were observed under all the tested temperature conditions except at the denaturation temperature of 60 °C, and neither desired bands nor obvious fluorescence signals were detected at the annealing/extension temperature of 50 °C (Fig. 1A, and Fig. S3A†). These results indicated that amplification of guide DNAs could be well achieved via a two-step PCR with lower denaturation temperature. The temperature combination of 70 °C/40 °C was thereby selected as the optimal temperature condition for the two-step PCR.
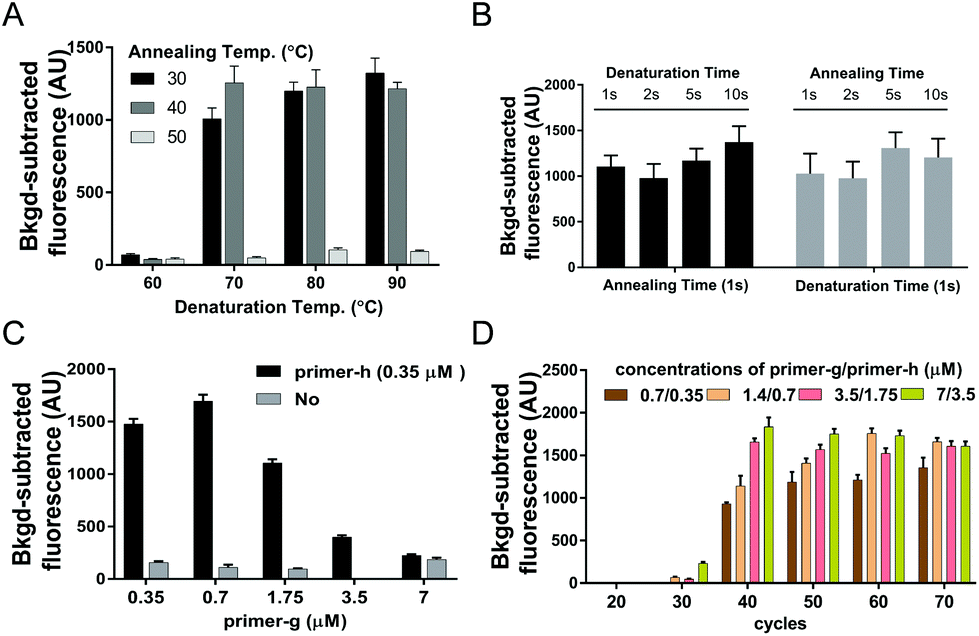 | ||
| Fig. 1 Optimization of USPCRP. Synthesized ssDNA of SARS-CoV-2 ORF1ab gene was used as the template. (A) Results of USPCRP under various usPCR temperature combinations as indicated. (B) Results of USPCRP with various usPCR cycle durations as indicated under the optimal temperature combination of 70 °C/40 °C. (C) Influence of various primer ratios on USPCRP under determined optimal usPCR conditions. (D) Comparison of the USPCRP under various primer concentrations and cycle numbers. Data are presented as mean ± standard deviation of three independent replicates. | ||
To further improve the time-efficiency of amplification, we then optimized the time spent on denaturation and annealing/extension. We found that a short time of 1 s for denaturation and annealing/extension, is enough to give a considerable yield of the guide DNA within 30 min (Fig. 1B and Fig. S3B†). Compared to a conventional PCR procedure, this optimized two-step PCR could save ∼50% time.
With the aim of yielding more guide DNAs, we evaluated various elevated concentrations of primer-g pairing with a constant concentration of primer-h.18 The highest yield of guide DNA was achieved under a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of primer-g to primer-h, giving the highest fluorescence signal value as well (Fig. 1C and Fig. S3C†). The higher primer ratio, such as 10:1 or 20:1, was found to give a much lower yield of guide DNA and a weaker fluorescence signal, which may be due to a badly compromised polymerization speed caused by severely unbalanced concentrations of the primer pair. These results indicated that asymmetric usPCR with a proper primer ratio could facilitate guide yields and a resulting higher cleavage activity of PfAgo. Moreover, neither visible desired PCR products nor a high fluorescence signal value could be detected with no primer-h added (Fig. 1C and Fig. S3C†), suggesting that an exponential amplification of guide DNA is a prerequisite for achieving a higher sensitivity detection.
:1 ratio of primer-g to primer-h, giving the highest fluorescence signal value as well (Fig. 1C and Fig. S3C†). The higher primer ratio, such as 10:1 or 20:1, was found to give a much lower yield of guide DNA and a weaker fluorescence signal, which may be due to a badly compromised polymerization speed caused by severely unbalanced concentrations of the primer pair. These results indicated that asymmetric usPCR with a proper primer ratio could facilitate guide yields and a resulting higher cleavage activity of PfAgo. Moreover, neither visible desired PCR products nor a high fluorescence signal value could be detected with no primer-h added (Fig. 1C and Fig. S3C†), suggesting that an exponential amplification of guide DNA is a prerequisite for achieving a higher sensitivity detection.
An appropriate primer concentration is essential for a high yield of PCR products.20 We investigated the influence of primer concentration and the cycle numbers on the yield of guide DNAs. We tested various primer concentrations and cycle numbers. The results indicated that considerable fluorescence intensity was observed with a cycle number of >30, and a positive correlation between the fluorescence intensity and the primer concentrations was detected at 40 or 50 cycle numbers (Fig. 1D). In view of the sufficiently high fluorescence intensity obtained under primer pair concentrations of 0.7/0.35 μM together with a cycle number of 40, we chose these as the optimal conditions for usPCR.
To investigate if USPCRP could detect double-stranded DNA (dsDNA), we selected mock ASFV VP72 gene and HPV16E6 as targets, respectively, the corresponding sequences are shown in Fig. S4.† Both the specific desired PCR products and the significant fluorescence signals demonstrated the successful detection of dsDNA of DNA viruses with USPCRP (Fig. S5†).
To assess the sensitivity of USPCRP, we tested various nucleic acid templates. Serial dilutions of plasmid pUC19-HPV16E6 (from 1 nM to 1 aM) and ssDNA of SARS-CoV-2 ORF1ab (ssDNA-OS2) (from 1 nM to 1 aM) were used as targets. As for HPV16E6 gene detection, the results showed a detection limit of 10 aM and when pUC19-HPV16E6 concentration was in the range of 10 pM to 10 aM, a strong linear relationship between the fluorescence intensity and target concentration (Fig. 2A and B). Similarly, for ORF1ab gene detection, the limit of detection was 100 aM and a strong linear relationship between the fluorescence intensity and the ssDNA-OS2 concentration in the range of 10 pM to 100 aM was obtained (Fig. 2C and D).
 | ||
| Fig. 2 Analysis of USPCRP sensitivity. The usPCR was carried out at 70 °C for 1 s, 40 °C for 1 s, for 40 thermal cycles, followed with PfAgo digestion for 20 min. (A) The BKgd-subtracted fluorescence (AU) of USPCRP for various concentrations of pUC19-HPV16E6. (B) Calibration plot of the BKgd-subtracted fluorescence (AU) versus logarithmic concentration of HPV16E6 (from 10 aM to 10 pM). (C) The BKgd-subtracted fluorescence (AU) of USPCRP for various concentrations of ORF1ab. (D) Calibration plot of the BKgd-subtracted fluorescence (AU) versus logarithmic concentration of ORF1ab (from 100 aM to 10 pM). Data are presented as mean ± SD of three technical replicates (*P < 0.05; **P < 0.01; ***P < 0.001). | ||
The specificity of USPCRP was also evaluated. A conserved gene among Coronaviruses, ORF1ab, which is considered as an indicator of virus evolution, was selected for the test. A conserved segment of each ORF1ab from SARS-CoV-2, MERS-CoV and SARS-CoV was synthesized in the form of ssDNA, which served as the target DNA (Fig. 3A). Only in the presence of a primer pair specific to the target was any desired PCR product detected (Fig. 3B). When the primers together with the molecular beacon specific to the SARS-CoV-2 ORF1ab were used, only the target from SARS-CoV-2 could be detected and no obvious fluorescence signals were observed for either SARS-CoV ORF1ab or MERS-CoV ORF1ab (Fig. 3C).
 | ||
| Fig. 3 Analysis of USPCRP specificity. Synthesized ssDNAs of ORF1ab (ssDNA-O) from different Coronaviruses were used as the targets. (A) Diagram of the sequences of a conserved domain within ORF1ab of SARS-CoV-2, MERS-CoV and SARS-CoV. The orange shows the bases that are the same among the three sequences; blue means the bases were the same in two out of the three sequences; black means the bases were different in the three sequences. (B) TBE-PAGE analysis of PCR products; primer S2, M, S are specific for ORF1ab of SARS-Cov2, SARS-CoV, MERS-CoV, respectively. M is the ssDNA marker. (C) Results of target DNA detection. The primers and molecular beacon specific to the SARS-CoV-2 ORF1ab were applied for detection of all targets as indicated. Data are presented as mean ± standard deviation of three independent replicates. **p < 0.01. NC = negative control. | ||
Our previous study found that a mismatched gDNA with one mutation at 8–15 nt from its 5′ end could not direct PfAgo to cleave target DNA.17 To investigate if USPCRP was competent in detecting a single-base mutation, two tightly linked SNPs sites of SARS-CoV-2 at nt 8782 (C/T) and nt 28144 (T/C)19 were selected and the corresponding ssDNAs were used as the template. In line with our previous finding, USPCRP was found to be capable of distinguishing these two single-base mutations with high specificity (Fig. S6†). These results demonstrated the capability of USPCRP to provide consistent discrimination among sequences of high similarity, indicating its satisfying specificity.
To evaluate the application potential of USPCRP, clinical samples were analyzed. A rapid detection of HPV16 in the clinical samples was achieved by USPCRP (Fig. 4A), and the results are consistent with those of conventional PCR (Fig. S7A†). Similarly, detection of SARS-CoV-2 from nasopharyngeal and oropharyngeal swabs was also easily achieved using USPCRP (Fig. 4B), with the results highly consistent with that of RT-qPCR provided by Wuhan CDC (Fig. S7B†). These results demonstrate the feasibility and effectiveness of USPCRP in clinical diagnosis applications.
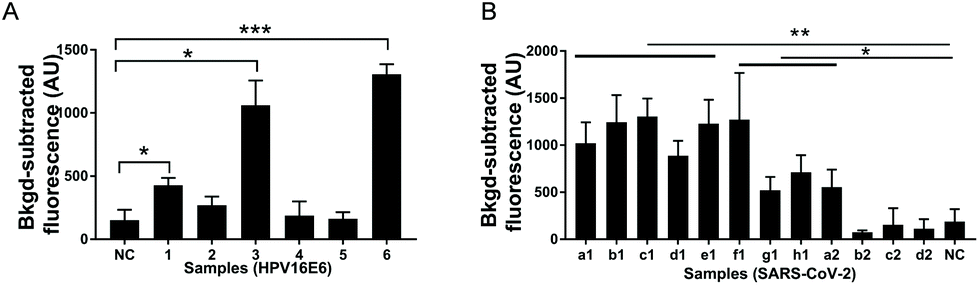 | ||
| Fig. 4 Virus detection in clinical samples using USPCRP. (A) Detection of HPV16 from clinical samples (1, 2, 3, 4, 5, 6) using USPCRP, the E6 gene was used as the target; (B) detection of SARS-CoV-2 from clinical samples (a1, b1, c1, d1, e1, f1, g1, h1, a2, b2, c2, d2) using USPCRP, the ORF1ab gene was used as the target. Data are presented as mean standard deviation of three independent replicates. *p < 0.05, **p < 0.01, ***p < 0.001. NC = negative control. | ||
Conclusions
In this study, we developed a novel nucleic acid detection system, USPCRP, which is simple, rapid and has high sensitivity. Due to the unique properties of PfAgo, an ultrashort asymmetric PCR (usPCR) was able to be established for generating functional guide DNAs, which dramatically reduced the detection time and added significant simplicity to this platform. Moreover, the whole system was further simplified with no primer screening, no PAM constraint and no more than 2 enzymes included (Taq DNA polymerase and PfAgo). By combining usPCR with PfAgo-mediated specific cleavage, USPCRP achieved detection of nucleic acids with a high sensitivity of 10 aM and high specificity single-base resolution, as well as the successful detection of viruses from clinical samples. What should be noticed is that DNA polymerase without 3′–5′ exonuclease activity (such as Taq DNA polymerase) should be chosen due to the short length of usPCR products to avoid degradation, and for RNA detection, reverse transcription is necessary. Besides, there are still some limitations, such as compared to PAND, USPCRP is difficult to achieve multiplexing detection, short primers might increase the risk of nonspecific amplification. Fortunately PfAgo could help to guarantee the specificity of USPCRP. Meanwhile, we could increase the specificity of amplification by selecting optimized primer-h. We believe with increasing understanding of Argonautes, these unique proteins would represent a new avenue for the development of next-generation nucleic acid sensing platforms.Author contributions
Ruyi He: methodology, data curation, validation, visualization, writing. Longyu Wang: methodology. Fei Wang: methodology. Jun Yang: resources. Xiao Yu: obtain SARS-CoV-2 nucleic acid extracts. Yuan Wang: obtained the HPV16 nucleic acid extract. Zhiguo Liu: modified the language, checked and proofread. Chunhua Li: resources, investigation, writing original draft. Lixin Ma: conceptualization, validation, resources, funding acquisition, supervision.Conflicts of interest
Hubei University has patents pending for these method on which the authors are the listed inventors.Acknowledgements
This work was funded by China National Key Research and Development (R&D) Program (2021YFC2100100).Notes and references
- L. Wu and X. Qu, Chem. Soc. Rev., 2015, 44(10), 2963–2997, 10.1039/c4cs00370e.
- T. Kuchta, R. Knutsson, A. Fiore, E. Kudirkiene, A. Hohl, D. Horvatek Tomic, V. Gotcheva, B. Popping, S. Scaramagli, A. To Kim, M. Wagner and D. De Medici, Lett. Appl. Microbiol., 2014, 59(3), 263–271, DOI:10.1111/lam.12283.
- A. Krumbholz, M. Schafer, T. Lorentz and A. Sauerbrei, Med. Microbiol. Immunol., 2019, 208(2), 197–204, DOI:10.1007/s00430-019-00580-2.
- L. Yan, J. Zhou, Y. Zheng, A. S. Gamson, B. T. Roembke, S. Nakayama and H. O. Sintim, Mol. BioSyst., 2014, 10(5), 970–1003, 10.1039/c3mb70304e.
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270(5235), 467–470, DOI:10.1126/science.270.5235.467.
- E. L. van Dijk, H. Auger, Y. Jaszczyszyn and C. Thermes, Trends Genet., 2014, 30(9), 418–426, DOI:10.1016/j.tig.2014.07.001.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356(6336), 438–442, DOI:10.1126/science.aam9321.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360(6387), 436–439, DOI:10.1126/science.aar6245.
- S. Y. Li, Q. X. Cheng, J. M. Wang, X. Y. Li, Z. L. Zhang, S. Gao, R. B. Cao, G. P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20, DOI:10.1038/s41421-018-0028-z.
- L. B. Harrington, D. Burstein, J. S. Chen, D. Paez-Espino, E. Ma, I. P. Witte, J. C. Cofsky, N. C. Kyrpides, J. F. Banfield and J. A. Doudna, Science, 2018, 362(6416), 839–842, DOI:10.1126/science.aav4294.
- F. Jiang and J. A. Doudna, CRISPR-Cas9 Structures and Mechanisms, Annu. Rev. Biophys., 2017, 46, 505–529, DOI:10.1146/annurev-biophys-062215-010822.
- T. Jacobsen, F. Ttofali, C. Y. Liao, S. Manchalu, B. N. Gray and C. L. Beisel, Nucleic Acids Res., 2020, 48(10), 5624–5638, DOI:10.1093/nar/gkaa272.
- R. K. Daher, G. Stewart, M. Boissinot and M. G. Bergeron, Clin. Chem., 2016, 62(7), 947–958, DOI:10.1373/clinchem.2015.245829.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28(12), E63, DOI:10.1093/nar/28.12.e63.
- J. Wu, J. Yang, W. C. Cho and Y. Zheng, J. Adv. Res., 2020, 24, 317–324, DOI:10.1016/j.jare.2020.04.017.
- D. C. Swarts, J. W. Hegge, I. Hinojo, M. Shiimori, M. A. Ellis, J. Dumrongkulraksa, R. M. Terns, M. P. Terns and J. van der Oost, Nucleic Acids Res., 2015, 43(10), 5120–5129, DOI:10.1093/nar/gkv415.
- R. He, L. Wang, F. Wang, W. Li, Y. Liu, A. Li, Y. Wang, W. Mao, C. Zhai and L. Ma, Chem. Commun., 2019, 55(88), 13219–13222, 10.1039/c9cc07339f.
- J. S. Farrar and C. T. Wittwer, Clin. Chem., 2015, 61(1), 145–153, DOI:10.1373/clinchem.2014.228304.
- X. L. Tang, C. C. Wu, X. Li, Y. H. Song, X. M. Yao, X. K. Wu, Y. G. Duan, H. Zhang, Y. R. Wang, Z. H. Qian, J. Cui and J. Lu, Natl. Sci. Rev., 2020, 7(6), 1012–1023, DOI:10.1093/nsr/nwaa036.
- J. S. Farrar and C. T. Wittwer, Clin. Chem., 2015, 61(1), 145–153, DOI:10.1373/clinchem.2014.228304.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an01521d |
| This journal is © The Royal Society of Chemistry 2022 |