
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantitative structure determination of adsorbed formate and surface hydroxyls on Fe3O4(001)†
P. T. P.
Ryan
 ab,
D. J.
Payne
b,
T.-L.
Lee
a and
D. A.
Duncan
*a
ab,
D. J.
Payne
b,
T.-L.
Lee
a and
D. A.
Duncan
*a
aDiamond Light Source, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK. E-mail: david.duncan@diamond.ac.uk
bDepartment of Materials, Imperial College London, SW7 2AZ, UK
First published on 13th December 2021
Abstract
Using the chemically specific techniques of normal incidence X-ray standing waves and photoelectron diffraction, we have investigated the dissociative adsorption of formic acid on the Fe3O4(001) surface, specifically probing the local structures of both the adsorbed formate and resulting surface hydroxyl. Using model independent direct methods, we reinforce the observations of a previous surface X-ray diffraction study that the formate molecule adsorbs with both oxygens atop octahedrally coordinated surface Fe cations and that ∼60% of the formate is adsorbed in the so called tet site. We additionally determine, for the first time, that the surface hydroxyl species are found at the so called int site. This confirms previous DFT predictions and reinforces the pivotal role the surface hydroxyl plays in lifting the subsurface cation vacancy termination of the Fe3O4(001) surface.
Introduction
Magnetite, Fe3O4, an abundant and cheap metal-oxide, has found increased scientific interest in recent decades in a host of different scientific fields. These range from the use of the mineral in both lithium ion batteries1 and as an electrocatalyst in fuel cells2 as well as a support material for single atom catalysts (SACs).3 Many of these applications arise due to the rich catalogue of surface structures for Fe3O4 which, in turn, can be attributed to its interesting redox chemistry. For example, the subsurface cation vacancy (SCV) reconstruction of the Fe3O4(001) surface is responsible for kinetically stabilising a range of different metal adatom phases, thus providing model catalytic systems with a well-defined single reactive site.3–6Notably, for device applications, citrate or olate stabilised nanoparticles of Fe3O4 have been used for tissue specific drug therapy where the negative charge of the modified nanoparticle surface can facilitate the adsorption of positively charged drugs. These drugs are then found to be released in the acidic environments of tumours, thereby creating tumour specific chemotherapy treatments.7,8 In the same vein, citrate stabilised nanoparticles of Fe3O4 are utilised as magnetic resonance imaging (MRI) contrast mediums, and, in many cases, are preferred over more conventional contrast mediums due to their excellent magnetic properties, tuneable biocompatibility and, importantly, their low toxicity.9,10 Often, control of these properties is achieved via the organic functionalisation of the Fe3O4 surface, by the adsorption of appropriate surface ligands.
Though a wide range of molecular and supramolecular species are used as coatings to such magnetite nanoparticles, they commonly contain functional groups that are thought to form ionic interactions with the surface of the nanoparticle. In particular, the carboxylate functional group is widespread in surface functionalization in general,11–17 and magnetite surface functionalisation specifically,18–20 due to how strongly it adsorbs on the surface and its relative robustness.
Thus, understanding the interaction of carboxylate containing compounds with Fe3O4 surfaces could facilitate the bottom up approach to designing and developing new organic functionalised Fe3O4 materials. More generally such knowledge could also provide a fundamental insight into the structures of the Fe3O4 surfaces under different conditions providing vital information for studies utilising Fe3O4 in applications such as SAC, which rely heavily on such information.
The Fe3O4(001) facet, the second most energetically stable facet after the (111), reconstructs under oxidation giving the subsurface cation vacancy (SCV) termination with (√2 × √2) R45° periodicity.21 Gamba et al. investigated the interaction of formic acid with the Fe3O4(001) SCV reconstructed surface, using X-ray photoelectron spectroscopy (XPS), low energy electron diffraction (LEED), scanning tunnelling microscopy (STM), infrared reflection adsorption spectroscopy (IRRAS) and density functional theory (DFT) calculations.22 XPS and IRRAS indicated that formic acid underwent dissociative adsorption at room temperature; the O–H bond of the formic acid breaks producing a surface adsorbed formate and a proton that, itself, is believed to form a surface hydroxyl. The dissociative adsorption of formic acid, according to LEED, lifted the SCV reconstruction giving a LEED pattern indicative of a bulk-like termination. A recently published surface X-ray diffraction (SXRD) study by Arndt et al.23 has shown that the formate is bound to two surface Feoct cations through its two carboxylate oxygens, OHCOO, which is consistent with how formate24–29 and more broadly the carboxylate functional group,30–37 bonds with most surfaces. Moreover, they were able to determine that the formate molecule preferentially binds to one of the two bidentate adsorption sites, the ‘tet’ adsorption site, with the formate carbon atoms coincident with the 1st subsurface Fetet in the [110] crystallographic direction, shown in Fig. 1. However, as SXRD is both chemically “blind” and insensitive to H atoms, this study was unable to probe the location of the surface hydroxyl. Furthermore, SXRD analyses is model dependent, requiring prospective structures to be theoretically modelled and compared against experimental results, thus if the correct structure is not modelled, it will not be found. Understanding the adsorption site of this hydroxyl species has significant importance in understanding how the SCV reconstruction is lifted upon hydroxylation, which is of interest not only to fundamentally understand the chemistry of Fe3O4, but also for investigations concerning the use of Fe3O4 as a support for SACs, where transient hydroxylation of the support surface will occur as part of the catalytic cycle.
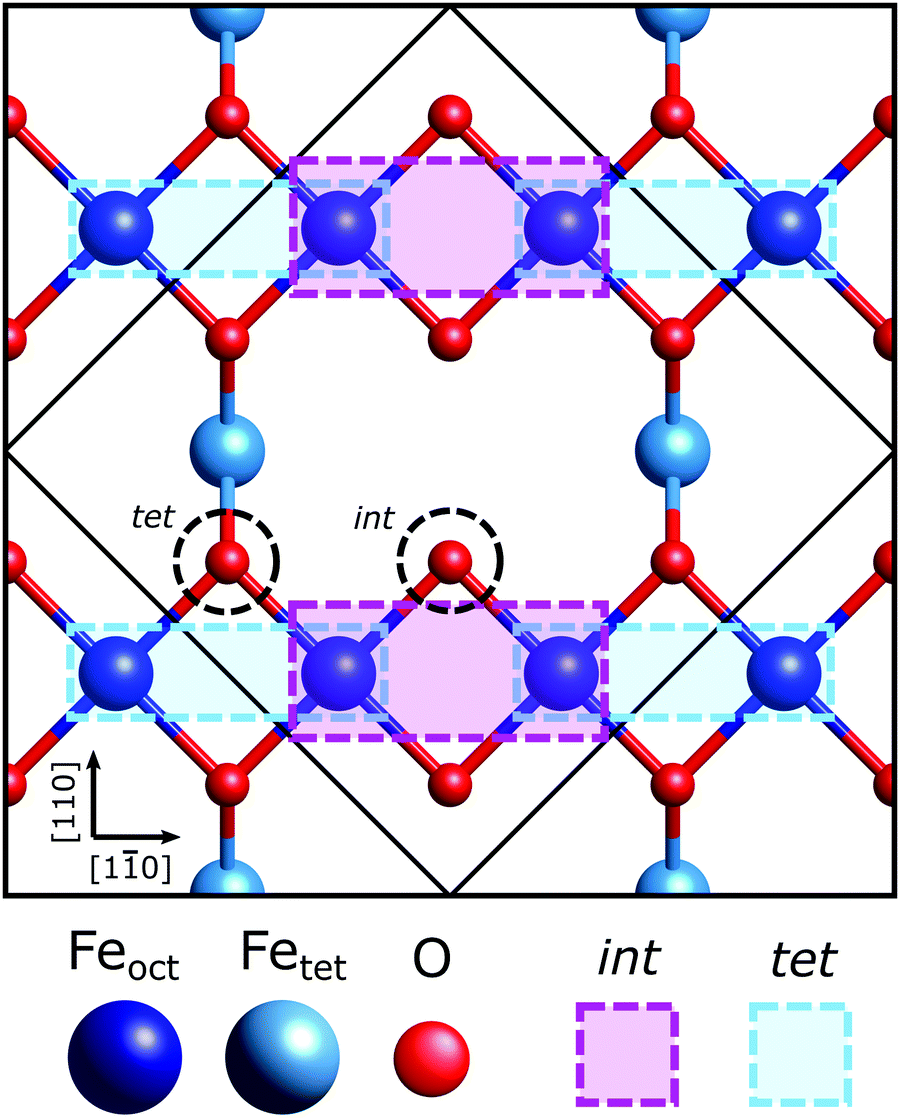 | ||
| Fig. 1 Schematic of the outermost octahedral and tetrahedral layers of the bulk terminated Fe3O4(001) surface. The light blue boxes mark the surface Feoct cations that the OHCOO bond to while in the tet site and the larger, purple boxes are for those of the int site. The black dotted circles mark the oxygens of the possible surface hydroxyl (OOH) sites (tet, int). | ||
As such, in this work we exploit the chemical specificity of energy-scanned photoelectron diffraction (PhD) and normal incidence X-ray standing wave (NIXSW) techniques to determine the adsorption geometry of the adsorbed formate and the location of OOH. We determine the adsorption site of formate on Fe3O4(001) to sub-ångstrom resolution, confirming prior studies, and identify the preferred adsorption site of the surface hydroxyl. We determine that the surface hydroxyl does indeed occupy the interstitial site (int) of the surface. Ultimately, this experimentally reinforces the important role the surface hydroxyl plays in the mechanism for the lifting of the SCV reconstruction.
Experimental details
All measurements were conducted at 300 K in the permanent ultra-high vacuum (UHV – ∼3 × 10−10 mbar) end-station on the I09 beamline38 at the Diamond Light Source. Beamline I09 utilises two separate undulators which are monochromated by a double Si(111) crystal monochromator and a plane grating monochromator. These two separate lines provide simultaneous access to both ‘hard’ and ‘soft’ X-ray energies. Specifically, we have used incident photon energies of 650 eV and 435 eV for all the O 1s and C 1s soft X-ray photoelectron spectroscopy (SXPS) measurements, respectively. A photon energy range of 600–890 eV was used for the O 1s PhD measurements. For the NIXSW, ranges of ∼2950–2960 eV, ∼2950–2960 eV and ∼4170–4180 eV were used for the {113}, (004) and (044) reflections, respectively. The absolute binding energy scale of all XP spectra were calibrated by subsequent measurements of the Fermi edge, which defined the binding energy scale origin.All photoelectron spectra were acquired using a VG Scienta EW4000 HAXPES hemispherical electron analyser (angular acceptance range ± 28°) mounted perpendicular to the incident radiation and in plane with the polarisation of the incident photon (linear-horizontal). All peaks in the XP spectra were fitted using a numerical convolution of a Gaussian and a Lorentzian peak shape. Within each core-level the peaks were allowed to take different fitted Gaussian widths but the same fitted Lorentzian width. For all peaks in the XPS data, the binding energy was a fitting parameter expect for the bulk O 1s core-level peak whose binding energy was fixed at the literature value of 530.0 eV.39
A Fe3O4(001) single crystal (±0.1°, purchased from and polished by the Surface Preparation Laboratory and PI-KEM) was cleaned in situ via ten cycles of sequential sputtering (Ar+, voltage: 1 keV, emission current: 3 mA, 10 min) and annealing (∼600 °C, 10 min). The annealing was alternated between annealing in UHV and in an oxygen partial pressure of 1 × 10−6 mbar with the final anneal in an oxygen partial pressure. The prepared samples showed a sharp (√2 × √2) R45° LEED pattern indicating the presence of SCV surface reconstruction.
Formic acid sourced from Sigma Aldrich was purified by several freeze–pump–thaw cycles. The clean Fe3O4(001) surface was exposed to 5 × 10−8 mbar of formic acid for 200 seconds (∼10 L, where 1 L is 1 × 10−6 mbar s).
The details of the NIXSW and PhD techniques are given in the electronic ESI† (Sections 1 and 2).
Results
SXPS
Fig. 2a shows the peak fitted C 1s core-level SXPS data after exposure to formic acid. Three species are observed and assigned as a carbon shake up feature (CS) at 289.4 eV, carbon from adsorbed formate (CHCOO) at 289.1 eV and adventitious carbon (Cad) at 285.0 eV. Intact formic acid is not expected on the surface as, in the previous work by Gamba et al.,22 the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OH stretching modes were absent above 190 K.
O and C–OH stretching modes were absent above 190 K.
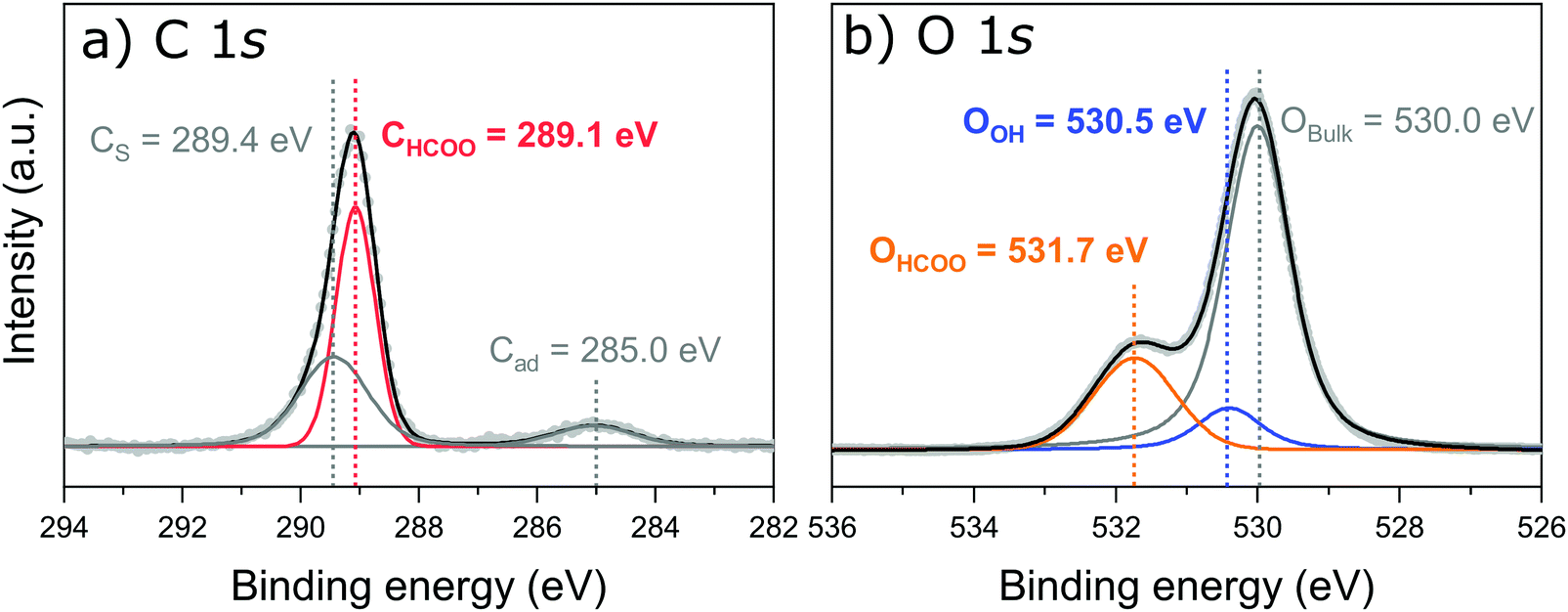 | ||
| Fig. 2 Peak fitted soft XPS spectra after exposing the Fe304(001) surface to formic acid. (a) The C 1s core-level, measured with a photon energy of 450 eV, fitted with three peaks; a carbon shakeup feature, CS, carbon from adsorbed formate, CHCOO, and adventitious carbon, Cad. (b) The O 1s core-level, measured with a photon energy of 680 eV, with three identified species; oxygen from the bulk metal–oxide, OBulk, formate oxygen, OHCOO, and a surface hydroxyl oxygen, OOH. | ||
The O 1s SXP spectrum, shown in Fig. 2b, exhibits three features: bulk oxide, (Obulk) at 530.0 eV, a surface hydroxyl oxygen (OOH) at 531.7 eV, and adsorbed formate, (OHCOO) at 531.7 eV. The assignments are in close agreement with those of Gamba et al.22 as well as for carboxylates on other metal–oxide surfaces.40,41 These SXPS results were used to fit the corresponding spectra for both the PhD and NIXSW measurements. A direct comparison of the O 1s region both before and after dosing formic acid is given in Fig. S1 in Section 3 (ESI†).
NIXSW – adsorption heights
Fig. 3 shows the fitted photoelectron intensity profiles of the (004) reflection for the CHCOO, OHCOO and OOH species after saturating the SCV surface with formic acid. Also inset to the figures are the values of the corresponding coherent fraction, f004, and position, P004, for each of the fits.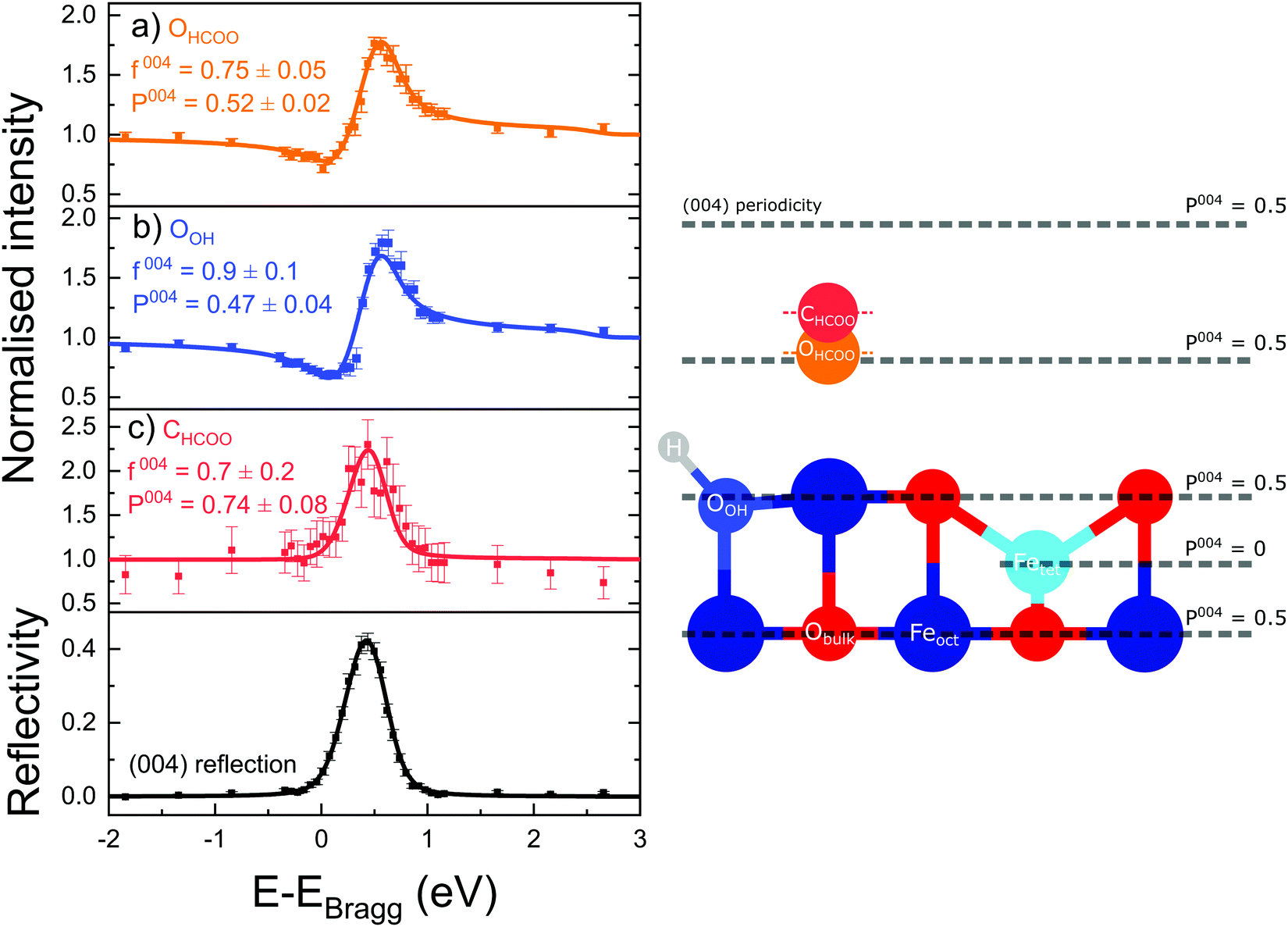 | ||
| Fig. 3 Depicted is the (004) reflection data and the corresponding fitted photoelectron profiles for the (a) OHCOO, (b) OOH and (c) CHCOO species. The coherent positions, P004, and coherent fractions, f004, for each fit are also given. The OHCOO, OOH and CHCOO positions are shown schematically on the right with respect to a bulk truncated Fe3O4(001) surface (no surface relaxations are shown). The OHCOO and OOH coherent positions are very similar though the OOH atom is assumed to be coincident with a lattice plane in the surface. These results are consistent with the formate bidentate bonding to the surface. | ||
Both the OHCOO and OOH photoelectron profiles (Fig. 3a and b) are similar indicating that both these species occupy near identical positions relative to the bulk (004) planes. This understanding is reinforced by the similarity in the fitted coherent positions for the OOH and OHCOO species 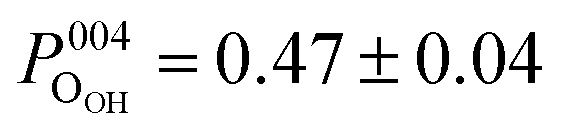 and
and 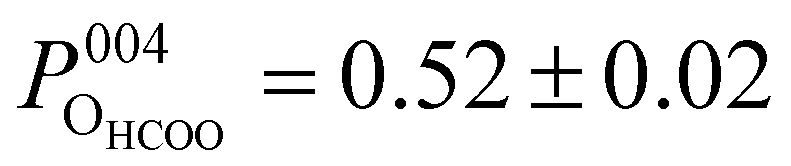 , respectively. Since, when fitting the NIXSW data, the origin of the substrate's unit cell was defined to be centred on a tetrahedrally coordinated Fe cation layer, these values of P004 place either species close to coincidence with a bulk or bulk-projected octahedrally coordinated Fe cation layer, i.e. P004 ≈ 0.5. The corresponding heights relative to a bulk-like octahedrally terminated surface are 2.14 ± 0.04 Å and −0.04 ± 0.04 Å for the OHCOO and OOH atoms respectively. Note that the height difference between the OHCOO and OOH atoms is similar to the (004) bulk spacing as the OOH atom is assumed as being in the surface and the OHCOO atom is assumed as existing above the surface at a projected (004) lattice plane.
, respectively. Since, when fitting the NIXSW data, the origin of the substrate's unit cell was defined to be centred on a tetrahedrally coordinated Fe cation layer, these values of P004 place either species close to coincidence with a bulk or bulk-projected octahedrally coordinated Fe cation layer, i.e. P004 ≈ 0.5. The corresponding heights relative to a bulk-like octahedrally terminated surface are 2.14 ± 0.04 Å and −0.04 ± 0.04 Å for the OHCOO and OOH atoms respectively. Note that the height difference between the OHCOO and OOH atoms is similar to the (004) bulk spacing as the OOH atom is assumed as being in the surface and the OHCOO atom is assumed as existing above the surface at a projected (004) lattice plane.
The photoelectron intensity profile for the CHCOO species (Fig. 3c) is clearly different from that of the oxygen species’, identifying that it sits at a significantly different position relative to the bulk Bragg diffraction planes. The fitted coherent position, 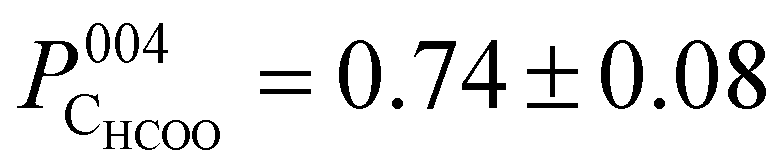 , places the C atom 0.6 ± 0.2 Å above the OHCOO atoms as is expected for a formate molecule adsorbed in a bidentate configuration.
, places the C atom 0.6 ± 0.2 Å above the OHCOO atoms as is expected for a formate molecule adsorbed in a bidentate configuration.
The associated f004 for the OOH, OHCOO and CHCOO species, respectively, are 0.9 ± 0.1, 0.78 ± 0.05 and 0.7 ± 0.2. The coherent fraction for the atoms of the formate (CHCOO and OHCOO) are significantly lower than that of the bulk O and the OOH species. Were these coherent fractions the result of thermal vibrations, they would suggest a root mean square (rms) vibrational amplitude of 0.28 Å for the CHCOO atoms and 0.25 Å for the OHCOO atoms. This is confirmed by the PhD results presented below which were fitted with a rms vibrational amplitude of approximately 0.3 Å. This could indicate a particularly large vibrational amplitude normal to the surface for the adsorbed formate.
NIXSW – real space imaging
The photoelectron yield profiles for the other measured reflections ((044), (113), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 13), (131) and (
13), (131) and (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 11)) for the carbon and oxygen species can be found in Fig. S2 and S3 in Section 4 (ESI†) and their corresponding coherent fractions and positions are given in Table S1 in Section 4 (ESI†). The results from these reflections, along with the (004) data, were used to produce three-dimensional real space density maps for each species via a Fourier expansion.42Fig. 4 shows the real space density map for the CHCOO species at its corresponding (004) height above the unit cell. Note that, as the number of Fourier components in the expansion is non-infinite, the presence of artefacts in these real space images is to be expected. Thus, we must be careful in deciding which maxima in the real space images are real, and which are not. However, in the density map of the C species, there are clear maxima at the sites halfway between the lateral positions of the Feoct atoms along the [110] direction and a clear global maximum at the tet site. Therefore, we immediately confirm the results of the SXRD study of Arndt et al.23 in a model independent manner.
11)) for the carbon and oxygen species can be found in Fig. S2 and S3 in Section 4 (ESI†) and their corresponding coherent fractions and positions are given in Table S1 in Section 4 (ESI†). The results from these reflections, along with the (004) data, were used to produce three-dimensional real space density maps for each species via a Fourier expansion.42Fig. 4 shows the real space density map for the CHCOO species at its corresponding (004) height above the unit cell. Note that, as the number of Fourier components in the expansion is non-infinite, the presence of artefacts in these real space images is to be expected. Thus, we must be careful in deciding which maxima in the real space images are real, and which are not. However, in the density map of the C species, there are clear maxima at the sites halfway between the lateral positions of the Feoct atoms along the [110] direction and a clear global maximum at the tet site. Therefore, we immediately confirm the results of the SXRD study of Arndt et al.23 in a model independent manner.
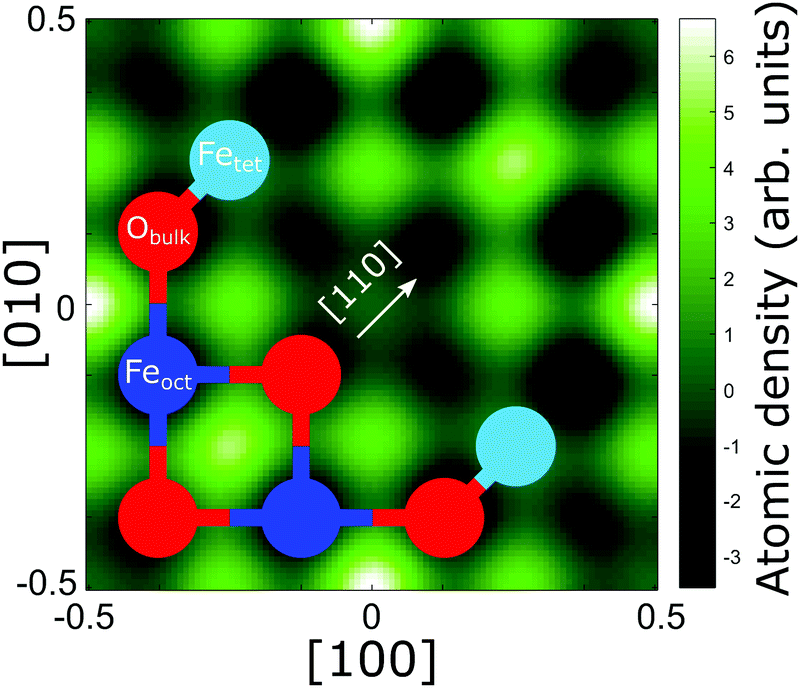 | ||
Fig. 4 The NIXSW real space image for the CHCOO species taken at its corresponding (004) adsorption height 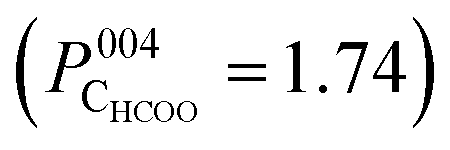 above the Fe3O4(001) surface. For comparison, overlaid is half of the octahedrally (P004 = 0.5) and tetrahedrally (P004 = 0.0) coordinated layers of the bulk Fe3O4 unit cell. The colour scale represents the probability of atomic density at that site, with white representing maximal probability, black minimal probability. above the Fe3O4(001) surface. For comparison, overlaid is half of the octahedrally (P004 = 0.5) and tetrahedrally (P004 = 0.0) coordinated layers of the bulk Fe3O4 unit cell. The colour scale represents the probability of atomic density at that site, with white representing maximal probability, black minimal probability. | ||
In the SXRD results of Arndt et al.23 a partial co-occupation of the int site (15 ± 10%) was observed. Our NIXSW results presented here favour adsorption of the formate molecule in the tet site (where the global maximum density occurs in Fig. 4), but do also suggest that partial occupation of the int site (where the second highest density occurs in Fig. 4) cannot be excluded. As such, we undertook a fitting of the total CHCOO unit cell density with the calculated total unit cell densities for a formate molecule in the tet and int sites with the aim of determining the percentage occupancy of both sites. A full description of this procedure is given in Section 5 (ESI†) and the corresponding example images for the calculated tet, int and best fit densities at the CHCOO (004) height above the unit cell are also shown in the Section 5 (Fig. S4, ESI†). The best fit comprised a model with 62 (±5)% of CHCOO in the tet site and 38 (±5)% CHCOO in the int site.
The imaging for the OHCOO species (see Fig. S5a in Section 6, ESI†) have peak intensities in sites directly atop Feoct and O atoms in the (004) layer below the OHCOO. This imaging corresponds to the OHCOO being found atop the surface Feoct atoms with the extra maxima above the surface O atoms being due to the symmetry of the bulk crystal. This adsorption site for the OHCOO agrees well with the sites identified by Arndt et al.23 The imaging of the OOH species (Fig. S5b in Section 6, ESI†) places it in a bulk oxygen site on the surface. However, it is not possible, solely from the NIXSW, to differentiate between the int or tet sites for the OOH adsorption site.
PhD – qualitative analysis
Though it is necessary to perform multiple scattering calculations in order to obtain quantitative results for PhD data, a direct inspection of the data can provide a qualitative insight into the adsorption structure. The O 1s PhD spectra for the OBulk, OOH and OHCOO species in both the [100] and [110] azimuthal directions are shown in Fig. 5. The presence of strong modulations at low emission angles, with respect to normal emission (NE), for both the OOH and OHCOO species indicates that these species are located atop or near-atop strongly scattering surface atoms, i.e. Fe atoms.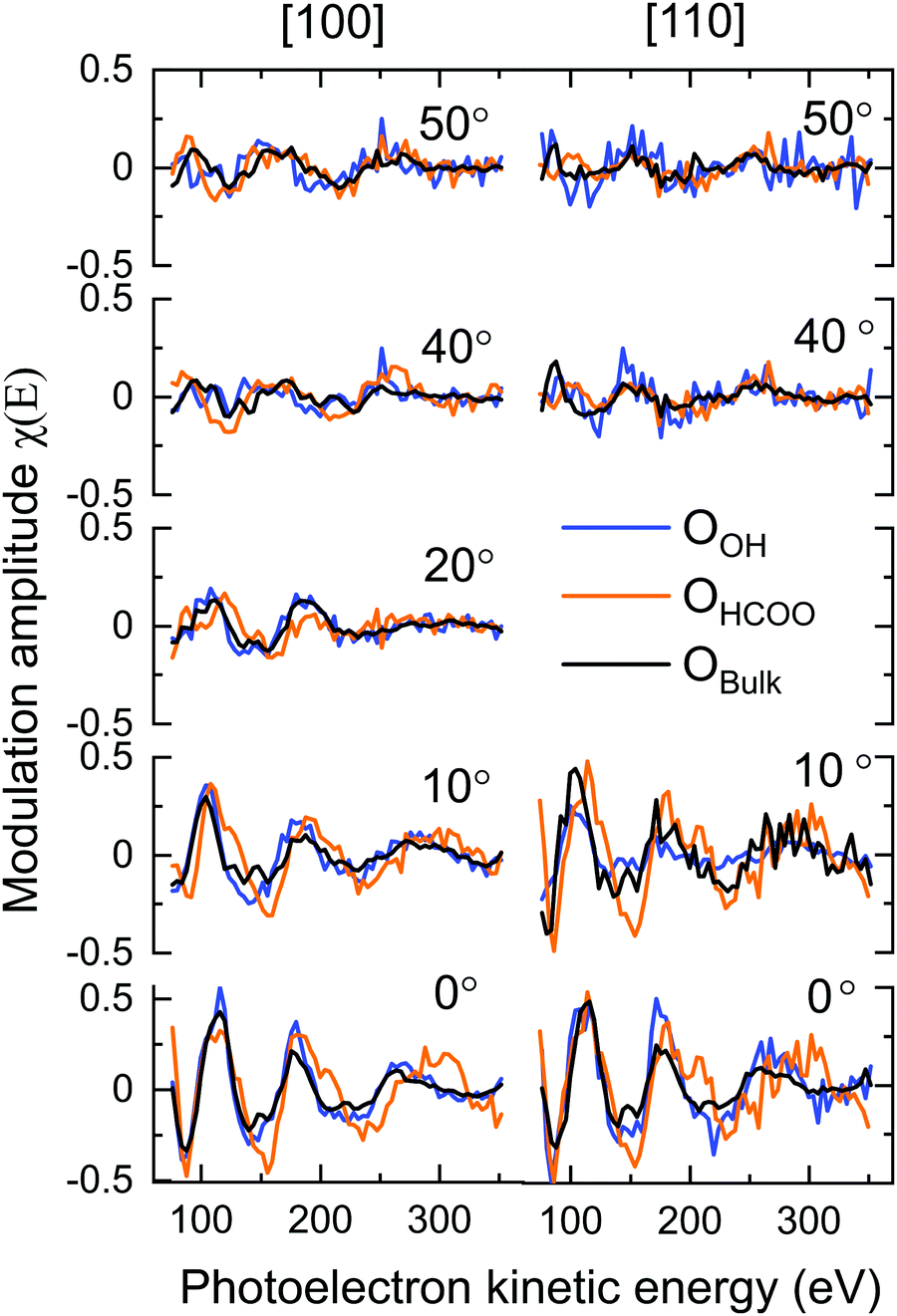 | ||
| Fig. 5 The experimental modulations for the 18 beams used in the PhD analysis for the OHCOO (orange), OOH (blue) and OBulk (black) species. Note, only the OHCOO and OOH species were used as emitters in the multiple scattering calculations and the OBulk beams are provided here for comparison only. Polar emission angles are given with respect to normal emission. The OHCOO and OOH modulations are very similar to the those of the OBulk indicating that the OHCOO and OOH species sit in bulk or bulk projected positions on the surface. | ||
More generally, the modulations present in the OOH and OHCOO spectra are similar, though with key differences, to those found for the OBulk demonstrating that both the OOH and OHCOO sit at bulk or bulk-like oxygen sites. Whereas the OBulk and OOH modulations remain well correlated with one another with increasing photoelectron kinetic energy, the period of those modulations for the OHCOO species become longer particularly for the emission angles close to NE. This difference suggests that the d(Fe–OHCOO) bond length is shorter than that of the d(Fe-OBulk) or d(Fe–OOH).
The greatest difference, between the OOH and OBulk modulations, occur for the spectra measured at an emission angle of 10° off NE along the [110] azimuth direction. Notably, the OBulk modulations are much stronger, suggesting the existence of a strong scattering atom in this direction for the bulk oxygen atoms which is not situated adjacent to the OOH. The [110] azimuthal direction points down the axis of the surface O–Fetet bonds and so the weak modulations found for OOH in this direction could indicate that the OOH sits in the int surface site, not adjacent to the Fetet atoms, which is confirmed by the quantitative fitting below.
PhD – quantitative analysis
Table 1 gives the R-factors of all four single-site optimised structures on a bulk terminated surface. These R-factors were determined via optimising fits to the experimental modulations using a particle swarm optimisation (PSO) algorithm.43 See the ESI† for further details. Other surface terminations than bulk terminated were also considered (see the Section 7, ESI†), however the bulk terminated surface, as determined by Arndt et al.23 was found to have the best agreement with the experimental data (see the Section 8, ESI†). The single structure that best fits the PhD modulations has the formate in the tet position and with the OOH in the int position situated above a subsurface Feoct cation. The fits of the optimised structure to the experimental modulations for both the OOH and OHCOO species are given in Fig. 6 and the structure is shown in Fig. 7. After parameter optimisation (see the Section 9, ESI†), this model has an R-factor = 0.22 (variance of 0.02). After considering substrate layer relaxations, the height of the carboxylate oxygens above the surface in this optimised structure is consistent with the height determined by NIXSW. Moreover, the bond length between the OHCOO and Feoct (dOHCOO–Feoct) atoms, and the intermolecular OHCOO–OHCOO distance (dOHCOO–OHCOO) for this best fit optimised structure are consistent with the results of Arndt et al.23 (see Table 2), and also agree well with other structural studies of carboxylates on both metal25–29 and metal–oxide24 surfaces. Further structural parameters are given in Table S3 in Section 8 (ESI†). The bond length between OOH and the Feoct directly below it (dOOH–Feoct) is also included in Table 2 and is discussed below.| HCOO position | OOH position | R factor |
|---|---|---|
| tet | int | 0.22(2) |
| tet | 0.32(3) | |
| int | int | 0.30(3) |
| tet | 0.36(3) |
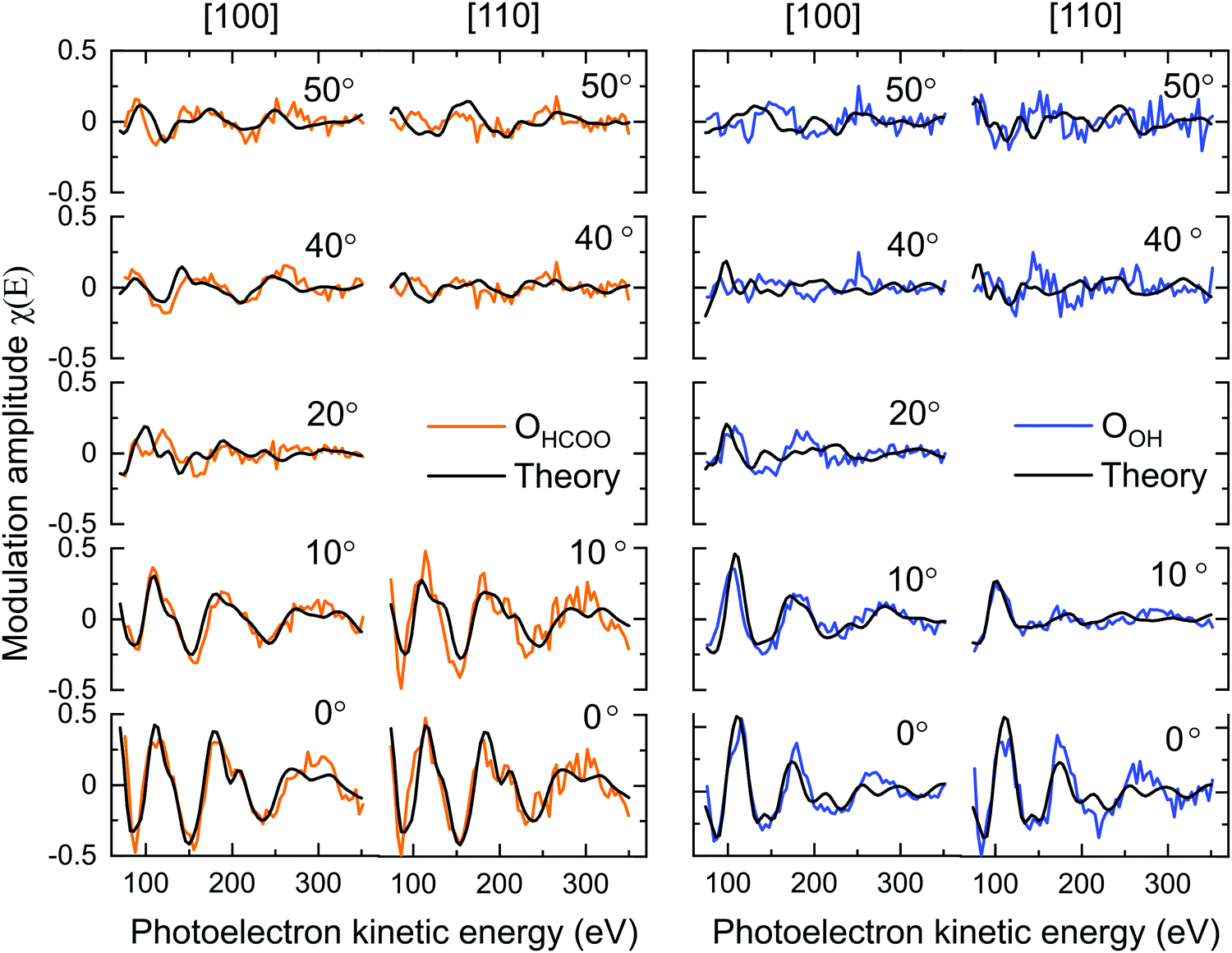 | ||
| Fig. 6 Fits to the PhD experimental modulations along the two azimuthal directions ([100] and [110]). The polar angle is provided for each beam. | ||
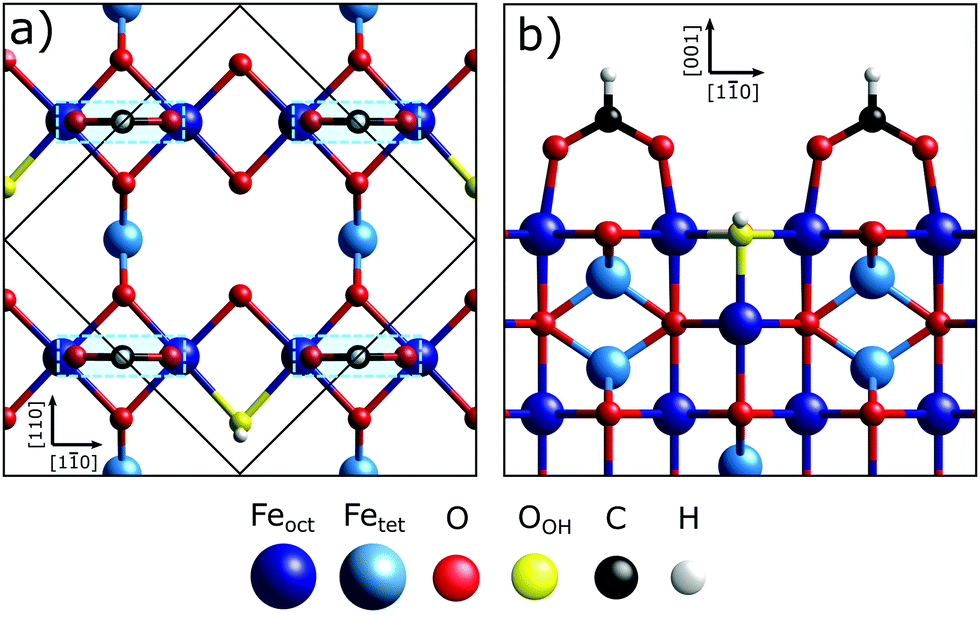 | ||
| Fig. 7 (a) Plan view and (b) side view (along the [110] direction) for the best fit bulk structure. The formate is in the tet position, as marked by the overlaid light blue boxes, while the OOH atom is in the int position. | ||
| Model | d OHCOO–Feoct (Å) | d OOH–Feoct (Å) | d OHCOO–OHCOO (Å) |
|---|---|---|---|
| PhD | 1.98 (±0.05) | 2.2 (+0.2/−0.1) | 2.4 (±0.1) |
| SXRD23 | 1.96 (±0.07) | — | 2.51 (±0.05) |
The prior work by Arndt et al.23 and the NIXSW results point to a co-occupation of the tet and int sites by the formate. For the PhD, the percentage occupation of the formate in the tet and int sites was determined by undertaking domain averaged PSO optimisations of the PhD measurements. In such a calculation, an additional structural parameter determining the fraction of occupation for one of the adsorption sites was optimised. These PhD calculations found that 70 ± 50% of the formate occupies the tet site which agrees well with both the NIXSW results above, and Arndt et al.23 Moreover, an improvement of the R-factor to 0.19 (0.02) is seen when including domain averaging over both of the sites.
Discussion
In this study the adsorption site of the formate has been directly probed in a model independent manner by NIXSW real space imaging, finding that a majority of the formate, 62 ± 5%, is adsorbed in the tet site on the surface coincident with the 1st layer Fetet atoms. These results confirm the model dependent results of Arndt et al.23 (75 ± 10%) and our PhD optimisations (70 ± 50% for the bulk terminated structure). It could be that the preference for the tet site is patterned by the SCV reconstruction with the mechanism of its lifting consistently causing the formate to be located in the tet site on the surface. This would not be hard to rationalise as the reactivity of the Fe3O4(001) surface to various adsorbates is dictated by the atomic and electronic structure of the SCV reconstruction. Notably, the SCV reconstruction contains two highly reactive oxygen anions which DFT predicts have charges of −1,21 and it is likely that the dissociative adsorption of the formic acid preferentially occurs at these sites. These reactive oxygen anions act as H-acceptors during the dissociation of the formic acid's O–H bond, accepting the H+ while the resulting HCOO− binds to surface Feoct cations.That H+ dissociation must occur at the int site, may indicate the origin of the formate's adsorption site preference at the tet site. Intact formic acid molecules that transiently bind to the tet site would have their carboxylic H atom directed towards an int site; transient adsorption of formic acid in the int site would, instead, direct this H atoms towards a tet site. Previous STM measurements (Gamba et al.22) have indicated that formate is mobile on the surface. Thus, while all formate molecules may adsorb onto the surface at a tet site, the mobility of the molecule would result in a stochastic mixture of adsorption in the two sites.
The formate molecule is found to adsorb on the surface with two different lateral arrangements, as indicated by STM.22 These were assigned to a (1 × 1) and (2 × 1) periodicities on the surface, with the (2 × 1) arrangement consisting of two apparent formate molecules offset along the [![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 10] direction on neighbouring Feoct rows on the surface. This would be consistent with one formate molecule in an int site and one in a tet site. It is important to note that no such overlayer mesh has been observed in LEED studies, likely because the islands sizes of the (2 × 1) periodicity are too small. Were the two periodicities of approximately equal coverage on the surface, as STM indicates, then the relative coverage of tet to int would be 75
10] direction on neighbouring Feoct rows on the surface. This would be consistent with one formate molecule in an int site and one in a tet site. It is important to note that no such overlayer mesh has been observed in LEED studies, likely because the islands sizes of the (2 × 1) periodicity are too small. Were the two periodicities of approximately equal coverage on the surface, as STM indicates, then the relative coverage of tet to int would be 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25. The observed ∼60:40 ratio implies a 1:2 ratio of (1 × 1):(2 × 1), such a high amount of (2 × 1) on the surface would not be consistent with the STM studies nor the lack of additional spots in the LEED. Therefore, it is possible that some of the formate in the (1 × 1) periodicity are occupying int sites.
:25. The observed ∼60:40 ratio implies a 1:2 ratio of (1 × 1):(2 × 1), such a high amount of (2 × 1) on the surface would not be consistent with the STM studies nor the lack of additional spots in the LEED. Therefore, it is possible that some of the formate in the (1 × 1) periodicity are occupying int sites.
The only other quantitative surface structural study into the adsorption of a hydroxyl species on a metal oxide surface is OH on TiO2(110).44 In this study the d(M–OOH) was found to be 1.97 ± 0.05 Å, corresponding well with the bulk Ti–O bond length. Here, however, d(Fe–OOH) was found to be systematically longer than the bulk d(Fe–O) distance of 2.06 Å. This corroborates with bulk bond length differences between d(Fe–O) and d(Fe–OOH) in iron oxide-hydroxide minerals goethite (α-FeO(OH)),45 akageneite (β-FeO(OH))46 and lepidocrocite (λ-FeO(OH)),47 all of which contain both Fe–O and Fe–OOH bonds. Additionally, Fe cation complexes containing either a hydroxy ligand, OH, or an oxo ligand, O, are also found to follow this trend with the d(Fe–OOH) bond length consistently being found to be longer.48–50
Within this work we have determined the OOH to occupy an int site. This is consistent with DFT calculations performed by Arndt et al.23 and was also the site predicted by Bourgund et al.51 for native surface hydroxyls on the Fe3O4(001) surface due to the adsorption of background gas phase hydrogen. Moreover, DFT calculations into the formation of water monolayers on the Fe3O4(001) surface indicate that these monolayers are initiated by the formation of surface hydroxyls which occupy the int site.52 Our results are the first structural determination of hydroxyl occupation in the int site, strongly supporting the DFT calculations of these prior works. These results are also consistent with the behaviour of deposited metal adatoms on the SCV reconstruction, which are found to adsorb at the two highly reactive surface oxygen anions of charge −1.4
Conclusions
In this study we have used the chemical specificity of the PhD and NIXSW techniques to determine the adsorption geometry of both the formate and surface hydroxyl formed during room temperature dissociation of formic acid on Fe3O4(001). Using model independent direct methods, we have confirmed previous assignments22,23 of the formate adsorption site as bidentate bonding to the surface through its two OHCOO atoms to two surface Feoct cations. Additionally, we find that a majority of the formate, 62 ± 5%, is in the tet adsorption site (see Fig. 1) with a minority of the formate, 38 ± 5%, in the int adsorption site. This is consistent with the PhD measurements which find that 70 ± 50% of the formate is in the tet site on a bulk-like substrate.We have also determined experimentally, for the first time, the adsorption site of the surface hydroxyl as the int site corroborating several DFT calculations and STM measurements into the site of the hydroxyl species.23,51,52 Through determining experimentally the surface hydroxyl adsorption site we have reinforced the pivotal role that hydroxyl plays in the lifting of the SCV reconstruction. The preference of the surface hydroxyl to be situated in a bulk-like int site likely drives the Feint cation into the first subsurface cation vacancy, initiating the destabilisation of the SCV reconstruction to cation diffusion from the bulk. This study adds to the growing understanding of how hydroxyl forming species interact with the Fe3O4(001) surface, and particularly how they influence the chemistry of the SCV reconstruction. This work is therefore of interest in many fields including those concerning the functionalisation of oxide nanoparticles for a range of applications but also within the field of catalysis where Fe3O4 is utilised as a promising substrate material for single atom catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Diamond Light Source for the award of beam time (SI17635-1). The computing resource was provided by the STFC Scientific Computing Department's SCARF cluster. PTPR was supported by the Advance Characterisation of Materials (ACM) CDT.References
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, Carbon-encapsulated Fe3O4 nanoparticles as a high-rate lithium ion battery anode material, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- Z. S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, 3D nitrogen-doped graphene aerogel-supported Fe3O4 nanoparticles as efficient electrocatalysts for the oxygen reduction reaction, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- Z. Novotný, G. Argentero, Z. Wang, M. Schmid, U. Diebold and G. S. Parkinson, Ordered array of single adatoms with remarkable thermal stability: Au/Fe3O4(001), Phys. Rev. Lett., 2012, 108, 1–5 CrossRef PubMed.
- M. Meier, Z. Jakub, J. Balajka, J. Hulva, R. Bliem, P. K. Thakur, T. L. Lee, C. Franchini, M. Schmid, U. Diebold, F. Allegretti, D. A. Duncan and G. S. Parkinson, Probing the geometry of copper and silver adatoms on magnetite: Quantitative experiment: Versus theory, Nanoscale, 2018, 10, 2226–2230 RSC.
- G. S. Parkinson, Unravelling single atom catalysis: The surface science approach, Cuihua Xuebao/Chinese J. Catal., 2017, 38, 1454–1459 Search PubMed.
- P. T. P. Ryan, Z. Jakub, J. Balajka, J. Hulva, M. Meier, J. T. Küchle, P. J. Blowey, P. K. Thakur, C. Franchini, D. J. Payne, D. P. Woodruff, L. A. Rochford, F. Allegretti, T. L. Lee, G. S. Parkinson and D. A. Duncan, Direct measurement of Ni incorporation into Fe3O4(001), Phys. Chem. Chem. Phys., 2018, 20, 16469–16476 RSC.
- S. Nigam, K. C. Barick and D. Bahadur, Development of citrate-stabilized Fe3O4 nanoparticles: Conjugation and release of doxorubicin for therapeutic applications, J. Magn. Magn. Mater., 2011, 323, 237–243 CrossRef CAS.
- A. Friedman, S. Claypool and R. Liu, The Smart Targeting of Nanoparticles, Curr. Pharm. Des., 2013, 19, 6315–6329 CrossRef CAS PubMed.
- Z. R. Stephen, F. M. Kievit and M. Zhang, Magnetite nanoparticles for medical MR imaging, Mater. Today, 2011, 14, 330–338 CrossRef CAS PubMed.
- C. Y. Haw, F. Mohamed, C. H. Chia, S. Radiman, S. Zakaria, N. M. Huang and H. N. Lim, Hydrothermal synthesis of magnetite nanoparticles as MRI contrast agents, Ceram. Int., 2010, 36, 1417–1422 CrossRef CAS.
- R. Han, F. Blobner, J. Bauer, D. A. Duncan, J. V. Barth, P. Feulner and F. Allegretti, Toward interfacing organic semiconductors with ferromagnetic transition metal substrates: Enhanced stability: Via carboxylate anchoring, Chem. Commun., 2016, 52, 9805–9808 RSC.
- N. E. Schlotter, M. D. Porter, T. B. Bright and D. L. Allara, Formation and structure of a spontaneously adsorbed monolayer of arachidic on silver, Chem. Phys. Lett., 1986, 132, 93–98 CrossRef CAS.
- L. H. Dubois, B. R. Zegarski and R. G. Nuzzo, Spontaneous Organization of Carboxylic Acid Monolayer Films in Ultrahigh Vacuum. Kinetic Constraints to Assembly via Gas-Phase Adsorption, Langmuir, 1986, 2, 412–417 CrossRef CAS.
- Q. Guo, I. Cocks and E. M. Williams, The adsorption of benzoic acid on a TiO2(110) surface studied using STM, ESDIAD and LEED, Surf. Sci., 1997, 393, 1–11 CrossRef CAS.
- M. Wühn, J. Weckesser and C. Wöll, Bonding and orientational ordering of long-chain carboxylic acids on Cu(111): Investigations using X-ray absorption spectroscopy, Langmuir, 2001, 17, 7605–7612 CrossRef.
- D. L. Allara and R. G. Nuzzo, Spontaneously Organized Molecular Assemblies. 1. Formation, Dynamics, and Physical Properties of n-Alkanoic Acids Adsorbed from Solution on an Oxidized Aluminum Surface, Langmuir, 1985, 1, 45–52 CrossRef CAS.
- T. Bauer, T. Schmaltz, T. Lenz, M. Halik, B. Meyer and T. Clark, Phosphonate- and carboxylate-based self-assembled monolayers for organic devices: A theoretical study of surface binding on aluminum oxide with experimental support, ACS Appl. Mater. Interfaces, 2013, 5, 6073–6080 CrossRef CAS PubMed.
- M. Z. Ansar, S. Atiq, S. Riaz and S. Naseem, Magnetite Nano-crystallites for Anti-cancer Drug Delivery, Mater. Today Proc., 2015, 2, 5410–5414 CrossRef.
- E. Calì, J. Qi, O. Preedy, S. Chen, D. Boldrin, W. R. Branford, L. Vandeperre and M. P. Ryan, Functionalised magnetic nanoparticles for uranium adsorption with ultra-high capacity and selectivity, J. Mater. Chem. A, 2018, 6, 3063–3073 RSC.
- W. Li, J. T. Mayo, D. N. Benoit, L. D. Troyer, Z. A. Lewicka, B. J. Lafferty, J. G. Catalano, S. S. Lee, V. L. Colvin and J. D. Fortner, Engineered superparamagnetic iron oxide nanoparticles for ultra-enhanced uranium separation and sensing, J. Mater. Chem. A, 2016, 4, 15022–15029 RSC.
- R. Bliem, E. McDermott, P. Ferstl, M. Setvin, O. Gamba, J. Pavelec, M. A. Schneider, M. Schmid, U. Diebold, P. Blaha, L. Hammer and G. S. Parkinson, Subsurface cation vacancy stabilization of the magnetite (001) surface, Science, 2014, 346, 1215–1218 CrossRef CAS PubMed.
- O. Gamba, H. Noei, J. Pavelec, R. Bliem, M. Schmid, U. Diebold, A. Stierle and G. S. Parkinson, Adsorption of Formic Acid on the Fe3O4(001) Surface, J. Phys. Chem. C, 2015, 119, 20459–20465 CrossRef CAS.
- B. Arndt, K. Sellschopp, M. Creutzburg, E. Grånäs, K. Krausert, V. Vonk, S. Müller, H. Noei, G. B. V. Feldbauer and A. Stierle, Carboxylic acid induced near-surface restructuring of a magnetite surface, Commun. Chem., 2019, 2, 92 CrossRef.
- D. I. Sayago, M. Polcik, R. Lindsay, R. L. Toomes, J. T. Hoeft, M. Kittel and D. P. Woodruff, Structure determination of formic acid reaction products on TiO2(110), J. Phys. Chem. B, 2004, 108, 14316–14323 CrossRef CAS.
- A. Sotiropoulos, P. K. Milligan, B. C. C. Cowie and M. Kadodwala, Structural study of formate on Cu(111), Surf. Sci., 2000, 444, 52–60 CrossRef CAS.
- D. Kreikemeyer-Lorenzo, W. Unterberger, D. A. Duncan, M. K. Bradley, T. J. Lerotholi, J. Robinson and D. P. Woodruff, Face-dependent bond lengths in molecular chemisorption: The formate species on Cu(111) and Cu(110), Phys. Rev. Lett., 2011, 107, 046102 CrossRef CAS PubMed.
- M. D. Crapper, C. E. Riley and D. P. Woodruff, The structure of formate on Cu(100) and Cu(110) surfaces, Surf. Sci., 1987, 184, 121–136 CrossRef CAS.
- D. P. Woodruff, C. F. McConville, A. L. D. Kilcoyne, T. Lindner, J. Somers, M. Surman, G. Paolucci and A. M. Bradshaw, The structure of the formate species on copper surfaces: new photoelectron diffraction results and sexafs data reassessed, Surf. Sci., 1988, 201, 228–244 CrossRef CAS.
- J. Stöhr, J. L. Gland, W. Eberhardt, D. Outka, R. J. Madix, F. Sette, R. J. Koestner and U. Doebler, Bonding and bond lengths of chemisorbed molecules from near-edge x-ray-absorption fine-structure studies, Phys. Rev. Lett., 1983, 51, 2414–2417 CrossRef.
- K. U. Weiss, R. Dippel, K. M. Schindler, P. Gardner, V. Fritzsche, A. M. Bradshaw, A. L. D. Kilcoyne and D. P. Woodruff, Chemical shift photoelectron diffraction from molecular adsorbates, Phys. Rev. Lett., 1992, 69, 3196–3199, DOI:10.1103/PhysRevLett.69.3196.
- M. Pascal, C. L. A. Lamont, M. Kittel, J. T. Hoeft, R. Terborg, M. Polcik, J. H. Kang, R. Toomes and D. P. Woodruff, Quantitative structural determination of the high coverage phase of the benzoate species on Cu(110), Surf. Sci., 2001, 492, 285–293 CrossRef CAS.
- J. H. Kang, R. L. Toomes, M. Polcik, M. Kittel, J. T. Hoeft, V. Efstathiou, D. P. Woodruff and A. M. Bradshaw, Structural investigation of glycine on Cu(100) and comparison to glycine on Cu(110), J. Chem. Phys., 2003, 118, 6059–6071 CrossRef CAS.
- N. A. Booth, D. P. Woodruff, O. Schaff, T. Gießel, R. Lindsay, P. Baumgärtel and A. M. Bradshaw, Determination of the local structure of glycine adsorbed on Cu(110), Surf. Sci., 1998, 397, 258–269 CrossRef CAS.
- D. I. Sayago, M. Polcik, G. Nisbet, C. L. A. Lamont and D. P. Woodruff, Local structure determination of a chiral adsorbate: Alanine on Cu(110), Surf. Sci., 2005, 590, 76–87 CrossRef CAS.
- D. A. Duncan, W. Unterberger, D. C. Jackson, M. K. Knight, E. A. Kröger, K. A. Hogan, C. L. A. Lamont, T. J. Lerotholi and D. P. Woodruff, Quantitative local structure determination of R,R-tartaric acid on Cu(110): Monotartrate and bitartrate phases, Surf. Sci., 2012, 606, 1435–1442 CrossRef CAS.
- T. J. Lerotholi, E. A. Kröger, M. J. Knight, W. Unterberger, K. Hogan, D. C. Jackson, C. L. A. Lamont and D. P. Woodruff, Adsorption structure of glycine on TiO2(110): A photoelectron diffraction determination, Surf. Sci., 2009, 603, 2305–2311 CrossRef CAS.
- W. Busayaporn, D. A. Duncan, F. Allegretti, A. Wander, M. Bech, P. J. Møller, B. P. Doyle, N. M. Harrison, G. Thornton and R. Lindsay, Structure of a model dye/titania interface: Geometry of benzoate on rutile-TiO2 (110)(1 × 1), J. Phys. Chem. C, 2016, 120, 14690–14698 CrossRef CAS.
- T. L. Lee and D. A. Duncan, A Two-Color Beamline for Electron Spectroscopies at Diamond Light Source, Synchrotron Radiat. News, 2018, 31, 16–22 CrossRef.
- N. S. McIntyre and D. G. Zetaruk, X-ray photoelectron spectroscopic studies of iron oxides, Anal. Chem., 2002, 49, 1521–1529 CrossRef.
- D. Wilson and M. A. Langell, XPS analysis of oleylamine/oleic acid capped Fe3O4 nanoparticles as a function of temperature, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS.
- J. Schnadt, J. N. O’Shea, L. Patthey, J. Schiessling, J. Krempaský, M. Shi, N. Mårtensson and P. A. Brühwiler, Structural study of adsorption of isonicotinic acid and related molecules on rutile TiO2(110) II: XPS, Surf. Sci., 2003, 544, 74–86 CrossRef CAS.
- L. Cheng, P. Fenter, M. J. Bedzyk and N. C. Sturchio, Fourier-Expansion Solution of Atom Distributions in a Crystal Using X-Ray Standing Waves, Phys. Rev. Lett., 2003, 90, 4 Search PubMed.
- D. A. Duncan, J. I. J. Choi and D. P. Woodruff, Global search algorithms in surface structure determination using photoelectron diffraction, Surf. Sci., 2012, 606, 278–284 CrossRef CAS.
- W. Unterberger, T. J. Lerotholi, E. A. Kröger, M. J. Knight, D. A. Duncan, D. Kreikemeyer-Lorenzo, K. A. Hogan, D. C. Jackson, R. Włodarczyk, M. Sierka, J. Sauer and D. P. Woodruff, Local hydroxyl adsorption geometry on TiO2(110), Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115461 CrossRef.
- A. F. Gualtieri and P. Venturelli, In situ study of the goethite-hematite phase transformation by real time synchrotron powder diffraction, Am. Mineral., 1999, 84, 895–904, DOI:10.2138/am-1999-5-625.
- J. E. Post, P. J. Heaney, R. B. Von Dreele and J. C. Hanson, Neutron and temperature-resolved synchrotron X-ray powder diffraction study of akaganéite, Am. Mineral., 2003, 88, 782–788 CrossRef CAS.
- A. N. Christensen, M. S. Lehmann, P. Convert, L. Beyer, O. Bastiansen, G. Braathen, L. Fernholt, G. Gundersen, C. J. Nielsen, B. N. Cyvin and S. J. Cyvin, Deuteration of Crystalline Hydroxides. Hydrogen Bonds of gamma-AlOO(H,D) and gamma-FeOO(H,D), Acta Chem. Scand., 1982, 36a, 303–308 CrossRef.
- L. Nagy, H. Ohtaki, T. Yamaguchi and M. Nomura, EXAFS study of iron(III) complexes of sugar-type ligands, Inorganica Chim. Acta, 1989, 159, 201–207 CrossRef CAS.
- C. E. MacBeth, A. P. Golombek, J. Young, C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik, O2 activation by nonheme iron complexes: A monomeric Fe(III)-Oxo complex derived from O2, Science, 2000, 289, 938–941 CrossRef CAS PubMed.
- C. E. MacBeth, R. Gupta, K. R. Mitchell-Koch, V. G. Young, G. H. Lushington, W. H. Thompson, M. P. Hendrich and A. S. Borovik, Utilization of Hydrogen Bonds to Stabilize M–O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands, J. Am. Chem. Soc., 2004, 126, 2556–2567 Search PubMed.
- A. Bourgund, B. A. J. Lechner, M. Meier, C. Franchini, G. S. Parkinson, U. Heiz and F. Esch, Influence of Local Defects on the Dynamics of O–H Bond Breaking and Formation on a Magnetite Surface, J. Phys. Chem. C, 2019, 123, 19742–19747 CrossRef CAS.
- M. Meier, J. Hulva, Z. Jakub, J. Pavelec, M. Setvin, R. Bliem, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Water agglomerates on Fe3O4(001), Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5642–E5650 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp04241f |
| This journal is © the Owner Societies 2022 |