
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular assemblies of organo-functionalised hybrid polyoxometalates: from functional building blocks to hierarchical nanomaterials
Jamie M.
Cameron
 a,
Geoffroy
Guillemot
b,
Theodor
Galambos
b,
Sharad S.
Amin
a,
Elizabeth
Hampson
a,
Kevin
Mall Haidaraly
b,
Graham N.
Newton
*a and
Guillaume
Izzet
*b
a,
Geoffroy
Guillemot
b,
Theodor
Galambos
b,
Sharad S.
Amin
a,
Elizabeth
Hampson
a,
Kevin
Mall Haidaraly
b,
Graham N.
Newton
*a and
Guillaume
Izzet
*b
aNottingham Applied Materials and Interfaces (NAMI) Group, The GSK Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, UK. E-mail: graham.newton@nottingham.ac.uk
bSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, IPCM, 4 Place Jussieu, F-75005 Paris, France. E-mail: guillaume.izzet@sorbonne-universite.fr
First published on 10th December 2021
Abstract
This review provides a comprehensive overview of recent advances in the supramolecular organisation and hierarchical self-assembly of organo-functionalised hybrid polyoxometalates (hereafter referred to as hybrid POMs), and their emerging role as multi-functional building blocks in the construction of new nanomaterials. Polyoxometalates have long been studied as a fascinating outgrowth of traditional metal-oxide chemistry, where the unusual position they occupy between individual metal oxoanions and solid-state bulk oxides imbues them with a range of attractive properties (e.g. solubility, high structural modularity and tuneable properties/reactivity). Specifically, the capacity for POMs to be covalently coupled to an effectively limitless range of organic moieties has opened exciting new avenues in their rational design, while the combination of distinct organic and inorganic components facilitates the formation of complex molecular architectures and the emergence of new, unique functionalities. Here, we present a detailed discussion of the design opportunities afforded by hybrid POMs, where fine control over their size, topology and their covalent and non-covalent interactions with a range of other species and/or substrates makes them ideal building blocks in the assembly of a broad range of supramolecular hybrid nanomaterials. We review both direct self-assembly approaches (encompassing both solution and solid-state approaches) and the non-covalent interactions of hybrid POMs with a range of suitable substrates (including cavitands, carbon nanotubes and biological systems), while giving key consideration to the underlying driving forces in each case. Ultimately, this review aims to demonstrate the enormous potential that the rational assembly of hybrid POM clusters shows for the development of next-generation nanomaterials with applications in areas as diverse as catalysis, energy-storage and molecular biology, while providing our perspective on where the next major developments in the field may emerge.
 Jamie M. Cameron | Jamie M. Cameron is a Research Associate in the Nottingham Applied Materials and Interfaces Group at the University of Nottingham (UK). He completed his PhD with Prof. Lee Cronin at the University of Glasgow (UK) on the self-assembly of metal-oxide clusters, before becoming a Postdoctoral Team Leader in the same group. In 2015, he was awarded a JSPS Postdoctoral Fellowship to work with Prof. Hiroki Oshio at the University of Tsukuba (Japan) on organic–inorganic hybrid materials. His current research explores the use of molecular design strategies to prepare new hierarchical nanomaterials for applications in next-generation energy technologies. |
 Geoffroy Guillemot | Geoffroy Guillemot is a graduate of the Université Pierre et Marie Curie (UPMC). He obtained his PhD in Chemistry from the Ecole Polytechnique Fédérale de Lausanne in 2002 under the supervision of Prof. Carlo Floriani. After postdoctoral stays with Prof. Dr A. Pfaltz (University of Basel), Dr J.-M. Basset (Université Claude Bernard à Lyon), Prof. O. Reinaud (Université René Descartes) and Dr P. LeFloch (Ecole Polytechnique), he returned to the UPMC in 2008 where he was appointed Assistant Professor and later Associate Professor of Inorganic Chemistry. His research focuses on the design of isolated site models using polyoxometalates for applications in metal-catalyzed atom or group transfer reactions. |
 Theodor Galambos | Theodor Galambos studies chemistry at the Georg-August-University Göttingen where he also obtained his BSc in 2018 under the supervision of Prof. P. Vana. During his master's studies he joined the group of Prof. M. M. Hansmann in late 2019 for a short-term research internship before leaving for a six-month stay at Sorbonne Université, Paris, including a three-month internship under the supervision of Dr Guillaume Izzet. After his return to Germany, he completed an internship at Covestro AG, Leverkusen and is currently working on his master's thesis at Volkswagen AG, Wolfsburg. |
 Sharad Amin | Sharad S. Amin is a Research Associate at the University of Nottingham (UK) in the Nottingham Applied Materials and Interfaces group. He completed his PhD in 2021 on the supramolecular assembly of redox-active polyoxometalate soft-materials under the supervision of Dr Graham Newton. Following his PhD, Sharad was awarded an EPSRC doctoral prize to conduct research on the development of these redox-active hybrid organic–inorganic soft materials to towards solid-state nanostructured metal-oxide architectures for energy devices. |
 Elizabeth Hampson | Elizabeth Hampson is a Research Associate in the Nottingham Applied Materials and Interfaces Group at the University of Nottingham (UK). She received an MSci in Medicinal and Biological Chemistry in 2017 at the university before she carried out her PhD under the supervision of Dr Graham Newton, on the synthesis of asymmetric organic–inorganic hybrid polyoxometalates. Following completion of her PhD earlier in 2021, she is now researching electrolytes and additives for next-generation batteries. |
 Kevin Mall Haidaraly | Kevin Mall Haidaraly was born and grew up in Madagascar. He travelled to France to pursue a degree in chemistry where he spent the majority of his scholarship at the Université de Nantes but moved to Canada during to complete a final year project with Prof. Garry Hanan at the University of Montreal. He is currently studying towards a PhD under the guidance of Dr Guillaume Izzet and Dr Fabrice Mathevet, where he works on the development of a new family of mesomorphic polyoxometale-based hybrids for photonics applications. |
 Graham N. Newton | Graham N. Newton obtained his PhD from the University of Glasgow (UK) in 2009. After carrying out a JSPS postdoctoral research fellowship he was appointed Assistant Professor at the University of Tsukuba, Japan, in 2011. In 2015 he moved to the University of Nottingham, UK, where he is now Associate Professor of Inorganic and Materials Chemistry and a leader of the Nottingham Applied Materials and Interfaces group. His research interests include the synthesis and characterisation of redox-active materials, from molecular species through to complex nanomaterials. He has a particular interest in the development of organic–inorganic hybrid molecular metal oxides for applications in energy storage and photocatalysis. |
 Guillaume Izzet | Guillaume Izzet is a graduate of the Ecole Nationale Supérieure de Chimie de Paris. He obtained his PhD at the University Paris-Sud under the guidance of Prof. O. Reinaud in 2004. He was then appointed as a postdoctoral research fellow in the group of Prof. A. Harriman in Newcastle University. In 2006 he moved to Prague to join the research group of Prof. J. Michl at the Academy of Sciences of the Czech Republic. He finally became CNRS associate researcher in 2008 at the Institut Parisien de Chimie Moléculaire in the e-POM group. His research interests are focused on inorganic supramolecular chemistry, polyoxometalate functionalization, artificial photosynthesis, molecular electronics & photonics. |
1. Introduction
The elaboration of well-defined nanostructured architectures through the self-assembly of individual molecular building blocks is a challenging and contemporary research issue, and increasingly vital to the ongoing development of new nanotechnologies.1 Nanostructured materials obtained via the hierarchical assembly of molecular units show promising applications in various domains, including catalysis,2 medicine,3 molecular electronics,4 information processing,5,6 gas storage7,8 and energy conversion.9,10 Classical bottom-up strategies use preformed entities as elementary building blocks, which are combined through self–assembly processes to form the target material.11 Obtaining these complex, artificial assemblies relies on the use of molecular components that are capable of displaying directional, orthogonal and/or switchable interactions (e.g. metal coordination, electrostatic pairing, hydrophobic effects). Indeed, the cooperation of multiple non-covalent interactions is typically the key to forming ordered extended assemblies with emergent properties.12–14 Although often difficult to predict, the emergence of novel properties frequently accompanies the formation of complex superstructures from the combination of even the most simple of components.15Polyoxometalates (POMs) are nanosized molecular metal-oxides with diverse, albeit very well-defined, structures. They are often seen as key intermediates between small, soluble metal oxo complexes and insoluble solid-state bulk metal oxides.16,17 POMs have attracted considerable interest in recent years thanks to their attractiveness as transferable building blocks for the elaboration of multi-functional materials,18,19 which is in large part thanks to their unique structural and chemical properties. POMs are large and charge-delocalised polyanionic species, which develop strong electrostatic interactions with cationic species. They are also prone to associate with hydrophobic and neutral polar phases and frequently behave as chaotropic agents.20,21 The highly charged and chaotropic nature of POMs often results in their self-assembly into larger supramolecular architectures and the emergence of new structures and even functionalities. Beside the spontaneous formation of single-walled hollow vesicles known as “blackberry” structures,22 the formation of POM-based supramolecular assemblies often relies on their association to organic components through electrostatic interactions. Some remarkable examples such as artificial quantasomes (i.e. photoactive 2D nanostructured amphiphilic assemblies),23 nanoporous 2D frameworks,24,25 hexagonally ordered nanocomposite films,26 well-ordered supramolecular hydrogels,21 polypseudorotaxanes,27 or liquid crystalline materials,28 were developed using this approach. In most of these cases, organisation at both the molecular level, and the long-range ordering of these systems, mostly relies on the combination of the close electrostatic interactions between the POM and the cationic species with an additional driving force (e.g. the chaotropic effect, void filling behaviour or nanophase segregation). As such, these approaches are often highly unpredictable due to the lack of any directionality imparted by the isotropic structure of many POMs. Typically, the self-assembly processes are predominantly governed by the ratio of the volumes occupied by the organic and inorganic components. Synthetic control of the organisation of these materials therefore relies almost entirely on the structure-directing effect of the associated organic components (either cationic or neutral), which surround the POM and behave as a steering ‘glue’ to direct the assembly of the system.
Covalently functionalised hybrid polyoxometalates (hybrid POMs) provide an attractive alternative route for the integration of POMs into complex molecular architectures.29–33 Although the design and synthesis of hybrid POMs is typically much more synthetically demanding than the preparation of systems based on discreet organic and inorganic components, these species allow a far greater degree of control and directionality between the tethered organic and inorganic sub-units. POM functionalisation not only allows combination of the POM's properties with those of the appended subunit,34 and in some cases can even modify the POMs own electronic structure,35 but also greatly facilitates integration of the POM into more complex supramolecular assemblies. Indeed, the communities’ expertise in the synthesis, functionalisation and characterisation of hybrid POMs has now reached a sufficient level of maturity to envision their integration into increasingly elaborate multi-functional supramolecular architectures.
Here, we will describe the current state of the art in the design and synthesis of hybrid POMs, with a particular focus on how this facilitates their assembly into larger supramolecular nanostructures. In particular, we will structure our discussion around the two key routes which have emerged as a means to incorporate hybrid POMs into larger nanostructures: (i) the direct self-assembly of hybrid POMs as unique building blocks, either in solution (via metal-directed self-assembly, or by exploiting the tuneable amphiphilic properties of some hybrid POMs), or in the solid-state (as liquid crystals, or other nanostructured soft materials); (ii) the combination of hybrid POMs with a secondary substrate (macrocyclic hosts, carbon nanotubes, or biological macromolecules/assemblies), where specific interactions between the organic sub-unit of the hybrid POM and the substrate predominate. This review will aim to comprehensively demonstrate the enormous potential that the rational design and functionalisation of hybrid POMs shows as a means to access the next-generation of multi-functional, hierarchical supramolecular nanostructures. Note that the preparation of crystalline POM-based coordination networks is an emerging field, which has been discussed elsewhere recently,36,37 but mostly remains out of the scope of this review (the examples given here will only concern systems in which the hybrid POM is pre-formed prior to preparation of the crystalline network). Similarly, we will not provide detailed descriptions of the synthetic approaches to each hybrid POM platform, nor any associated post-functionalisation techniques, since these have been discussed in detail in several excellent recent reviews.31,38,39
2. Hybrid polyoxometalate platforms
This section briefly describes the hybrid POM platforms which have been explored thus far as molecular components or precursors for the self-assembly of larger supramolecular architectures (Fig. 1), in order to establish a common terminology which will be used throughout the remainder of this review. For the purposes of this review, each ‘platform’ described below simply reflects the most commonly occurring structural archetypes in the literature at present, and (though these are, of course, highly significant in the supramolecular assembly of each hybrid POM platform) we will consider neither the specifics of the covalently grafted ligands or the associated cations when briefly introducing each type of hybrid-POM platform below. It should also be noted that the array of hybrid POM platforms described in the following is by no means a complete account of all possible hybrid POM scaffolds, and again, these have been largely reviewed elsewhere.31,38,39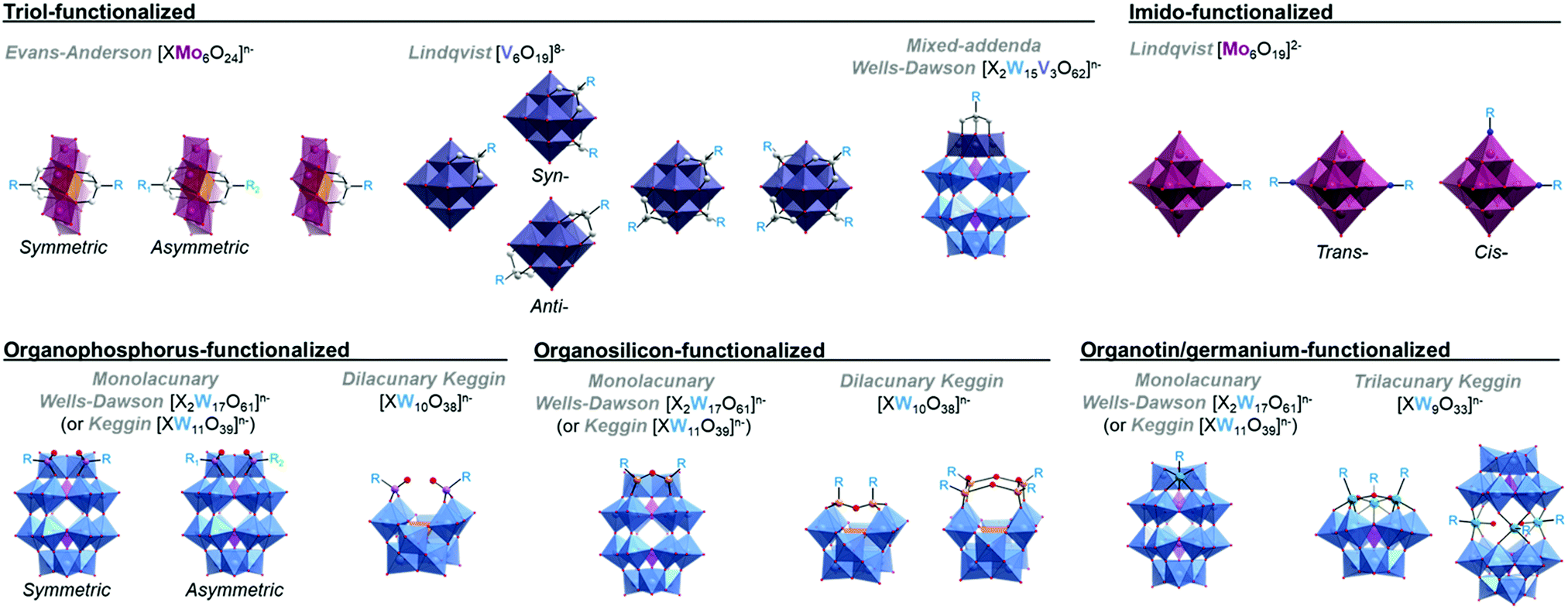 | ||
| Fig. 1 Library of the most commonly studied hybrid POM platforms and functionalisation strategies. Colour code: {MoO6} = purple polyhedra, {MnO6} = orange polyhedra, {VO6} = grey/blue polyhedra, {WO6} = blue polyhedra, {PO4} = purple tetrahedra, {SiO4} = orange tetrahedra, O = red, N = blue, P = pink, Si = orange, Sn = green. | ||
2.1 Triol-functionalised hybrid POMs
2.2 Imido-functionalised hexamolybdates
It is possible to incorporate nitrogen-based ligands through the formal substitution of terminal oxo-groups on the hexamolybdate Lindqvist-structure, [Mo6O19]2−. Either one or two terminal oxo sites may be exchanged, resulting in both monotopic hybrid Lindqvist platforms as well as ditopic species which preferentially adopt a cis conformation with a 90° angle between the two linker units (though a trans configuration with a 180° orientation of the organic moieties has previously been reported).2.3 Organophosphorus-functionalised POMs
Functionalisation with organophosphorus groups is also a viable route for preparation of hybrid POM platforms, typically exploiting the monovacant [P2W17O61]6− Wells–Dawson cluster. Unlike some other hybrid POM platforms, caution must be taken to avoid hydrolysis of the anchoring P–O–W bonds,43–45 with Wells–Dawson clusters typically proving more stable than their Keggin-type analogues. Though functionalisation of multivacant clusters has been demonstrated, functionalisation of monovacant species is by far the more common approach. This yields ditopic hybrid POM platforms with two phosphonyl moieties incorporated on either edge of the single lacunary site. Interestingly, the new bis-phosphonate site can even coordinate additional metal cations.46 As in the case of Anderson–Evans based platforms, it has also recently been shown to be possible to prepare and purify asymmetrically functionalised species. An interesting feature of organophosphorus based POM platforms is the very high angle of ca. 160° between the organic moieties, providing a close to linear functionalisation topology.2.4 Organosilicon-functionalised POMs
2.5 Organotin-functionalised POMs
Incorporation of organotin ligands into several POM frameworks has been observed for a variety of lacunary POM species.52–55 A particular benefit of the organotin functionalisation of POMs is the similar ionic radius of SnIV compared to WVI (and MoVI), which facilitates the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion of organotin fragments at lacunary sites, corresponding to the formal replacement of a WVI atom by the corresponding SnIV ion. Though, as in the organophosphorus and organosilicon examples above, the functionalisation of monovacant Keggin and Wells–Dawson clusters yielding monotopic platforms is by far the most common approach, the aforementioned capacity of SnIV to formally replace vacant WVI positions on the POM allows for the relatively simple formation of trifunctionalised species as well.
:1 inclusion of organotin fragments at lacunary sites, corresponding to the formal replacement of a WVI atom by the corresponding SnIV ion. Though, as in the organophosphorus and organosilicon examples above, the functionalisation of monovacant Keggin and Wells–Dawson clusters yielding monotopic platforms is by far the most common approach, the aforementioned capacity of SnIV to formally replace vacant WVI positions on the POM allows for the relatively simple formation of trifunctionalised species as well.
3. Direct self-assembly strategies
3.1 Metal coordination
Coupled to the aforementioned tuneable topologies of hybrid POM platforms, metal-driven self-assembly is an extremely powerful tool for the rational synthesis of discrete 0D, through to extended 3D assemblies, as it exploits strong and highly directional metal–ligand bonds.56–58 3D crystalline networks (including MOFs) have a wide range of properties and applications.59,60 In comparison to crystalline networks, discrete supramolecular coordination complexes display well-defined structures that are prone to form elaborate hierarchical systems based on weak interactions between the molecular units. Indeed, the construction of self-assembled systems with multiple levels of organisation is often achieved following a stepwise synthetic strategy relying on the design of a preassembled structural motif that can further self-assemble into more complex nanostructures through additional noncovalent interactions.61,62 These hierarchical self-assemblies of supramolecular complexes are at the basis of nanostructured soft materials with multifunctional properties for material science.11 Besides the formation of crystalline POM-based coordination networks, the elaboration of supramolecular coordination complexes containing POMs is thus particularly attractive to develop hierarchically organised species that could be at the basis of future soft multi-functional material with emergent properties.A considerable number of POM-supported transition metal complexes have been obtained by reaction of POMs with either mixtures of transition metal salts and organic ligands or with pre-formed metal–organic compounds. Such reactions most often lead to unpredictable results with the notable exception of some POM-based metal–organic frameworks (POMOFs),63,64 or ionic crystals.65 In contrast to those mentioned above, there have been relatively few reports of the synthetic strategy of metal coordination to chelating ligands on a pre-formed covalent POM hybrid. This approach can enable effective control in the preparation of supramolecular architectures, ranging from discrete nano-oligomers to coordination polymer frameworks and macroions, by fine-tuning in the POM hybrid design, selection of the metal linker and the reaction conditions. The plan of the following section will be oriented according to the topology of the hybrid ligand and that of the metal linker, which are crucial parameters to determine the nature of the resulting supramolecular assembly.
In a study comparing the coordination of four metal cations – MnII, CoII, NiII and ZnII with a bis-capped hexavanadate, Hill and co-workers reported almost isostructural 3D networks assembled for all, comprising polymer chains linked by H-bonding pyridyl and coordinated solvent moieties.67 The group later prepared a carboxylate terminated oxocluster analogue, which, when coordinated to TbIII ions in the presence of an organic linker, 4,4′-bis(pyridine-N-oxide), assembled into a porous 3D network that displayed catalytic activity in aerobic oxidation.68
Coordination networks demonstrating magnetic properties were built from the complexation of 2,6-bis(pyrazol-1-yl)pyridine (bpp) functionalised Mn-Anderson oxoclusters, [MnMo6O18{(OCH2)3CNHCO-(bpp)}2]3−, with FeII,69 by altering the stoichiometry of the metallic cation. A coordination polymer material formed with equimolar FeII, which demonstrated significant spin-crossover photoconversion (ca. 8%), although its structure could not be sufficiently characterised. A crystalline cationic 2D network was isolated, however, when FeII was added in excess. In the latter compound, two additional FeII centres were shown to be coordinated to each POM via the oxo-ligands, and with complexed solvent in place of a second bpp. Yet, no significant spin conversion was observed in this compound as a result of the weaker coordination sphere around the FeII centres in comparison to the bis(bpp) complex. The authors also suggest that some sensitivity to intermolecular arrangements, regarding the nature of counterions and labile solvent molecules in the materials, limited spin transition.
More recently, a more complex covalent terpyridine hybrid system demonstrated orthogonal chelation and photo-cross-linking into a multi-responsive supramolecular polymer.70 The coordination of 1 to 1.2 eq. FeII to the hybrid POM and subsequent formation of gels or fibres was shown to be reversible using a competitive ligand, EDTA. An additional reversible pathway to polymerisation by the photo-dimerisation of a coumarin-terminated quaternary ammonium surfactant in place of the Mn-Anderson oxocluster cations could be used separately or together with coordination to tune supramolecular polymerisation (Fig. 2).
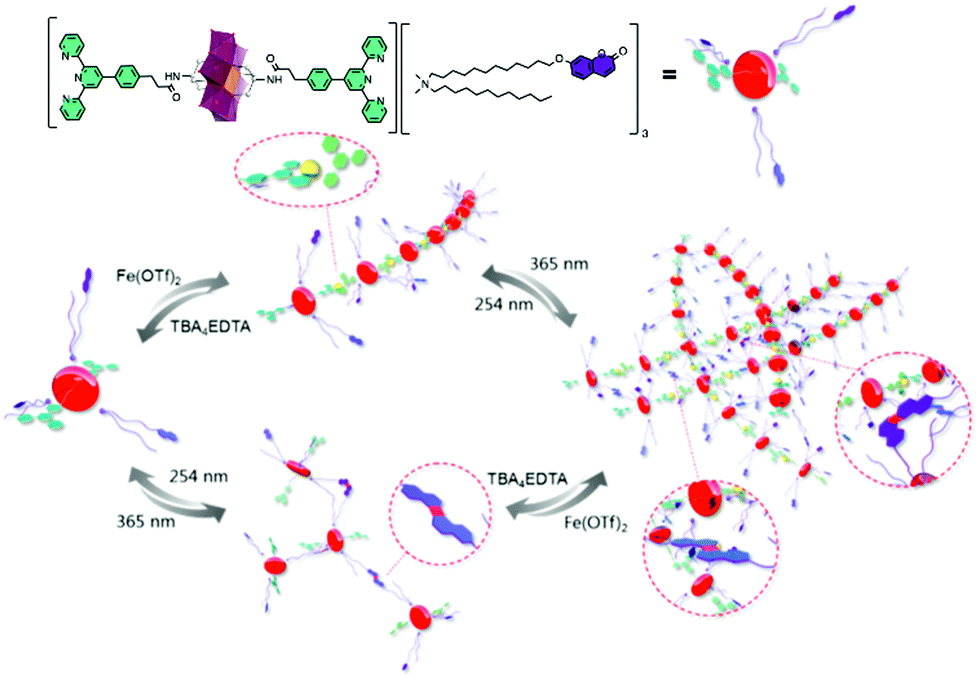 | ||
| Fig. 2 Schematic illustration of the elaboration of supramolecular polymer via orthogonal self-assembly strategy combining metal coordination and photo-cross-linking. This figure has been adapted from ref. 70 with permission from the American Chemical Society, copyright 2019. | ||
Covalently bonded PtII and HgII acetylide units connected to bisphosphonate functionalised Dawson derivatives forming oligomeric hybrids were reported by Liu, Li, Wong and co-workers.71,72 The oligomeric compounds, processed as thin films by the Langmuir-Blodgett technique, displayed photoelectric properties. Interestingly, oligomer chains of a comparable length comprising five repeating organometallic units were observed according to MALDI-TOF experiments for both the PtII and HgII materials.
Hybrid POMs with ca. 180° ligand coordination vectors have also been associated to metal acceptors with higher connectivity. The formation of porous POM-based organic frameworks by reaction of the ditopic bis-pyridine Mn-Anderson POM hybrid with CuI was reported by Yang and co-workers.73 This reaction yielded two kinds of crystalline oxocluster organic framework differing by the nature of coordinating cuprous iodide cluster (CuxIy) formed in situ. Cu4I4 and Cu2I2 halide clusters, which were obtained by varying the synthetic conditions, were shown to link to four remote pyridine ligands in tetrahedral and square planar geometries respectively, allowing the construction of an interpenetrated anionic diamondoid structure and a 2D anionic layer (Fig. 3).
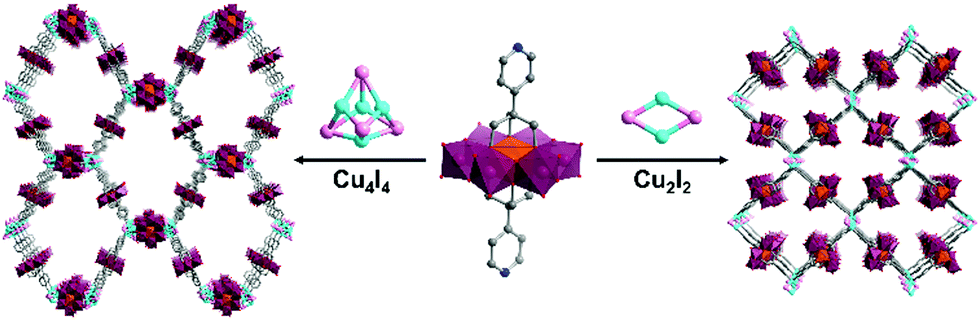 | ||
| Fig. 3 Structures of the POM-based crystalline organic frameworks associating bis pyridyl Anderson hybrids connected to a CuxIx halide cluster. This figure has been adapted from ref. 73 with permission from John Wiley and Sons, copyright 2016. | ||
Using similar bis-pyridine terminated ditopic Anderson hybrids, Forgan and co-workers explored several design considerations for the elaboration of coordination framework materials. In addition to the reaction conditions, the combination of metal cations, co-ligands, and the nature of the remote pyridyl ligand capping the Mn-Anderson oxocluster, were varied to serendipitously isolate six kinds of structural frameworks.74
The pyridyl N-position has also been explored as a directing factor in a recent study reported by Guo, Wang and co-workers. The authors showed that while two 2D coordination polymers were obtained by reaction of [Cu(PPh3)2(CH3CN)2]ClO4 with [MnMo6O18((OCH2)3CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–3-Py)2]3− using different molar ratios between the POM hybrid and Cu complex, only one structure, a 1D double chain, was obtainable using the (4)-pyridyl terminated analogue hybrid.75 In all structures, CuI centres were similarly coordinated to one pyridine function and a terminal oxygen atom of an adjacent oxocluster. Metal-directed assembly has also been demonstrated using octamolybdates that were previously functionalised with amino acids (proline and lysine). Carboxyl or amino binding sites on proline or lysine moieties were coordinated with CuII, ZnII and CoII cations to form a range of extended framework structures, some with antitumor selectivities.76
CH–3-Py)2]3− using different molar ratios between the POM hybrid and Cu complex, only one structure, a 1D double chain, was obtainable using the (4)-pyridyl terminated analogue hybrid.75 In all structures, CuI centres were similarly coordinated to one pyridine function and a terminal oxygen atom of an adjacent oxocluster. Metal-directed assembly has also been demonstrated using octamolybdates that were previously functionalised with amino acids (proline and lysine). Carboxyl or amino binding sites on proline or lysine moieties were coordinated with CuII, ZnII and CoII cations to form a range of extended framework structures, some with antitumor selectivities.76
The first example of the metal-driven self-assembly of a ditopic hybrid displaying 90° coordination vector is reported by Peng and co-workers.79 They observed the precipitation of a FeII bis-terpyridine hybrid after the coordination of FeII to a hexamolybdate hybrid. The low solubility of the supramolecular species was due to the neutralisation of the POM charge by the metal linker, which precluded its further characterisation. The authors also proposed the plausible formation of cyclic oligomers but could not go deeper in the investigation of the supramolecular species.
The first characterisation of a metallomacrocycle by metal coordination of a hybrid POM was reported by Hasenknopf and co-workers.80 In this study they used a bis-capped hybrid hexavanadate that was post-functionalised with two remote pyridine ligands at the metal position, forming a 60° coordination vector. Upon reaction with 1 equiv. trans-[PdCl2(CH3CN)2], they managed to isolate a molecular triangle comprised of three POMs and three metal linkers and characterised it by combination of DOSY NMR and mass spectrometry (Fig. 4).
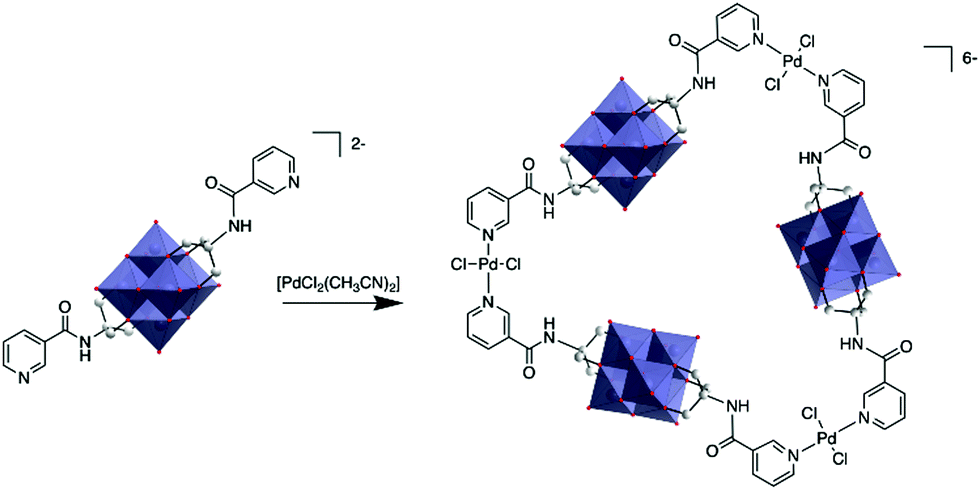 | ||
| Fig. 4 Synthesis of a POM-based trimer from a bis(pyridine-trisalkoxo)-hexavanadate anion and trans-[PdCl2(CH3CN)2]. This figure has been adapted from ref. 80 with permission from the Royal Society of Chemistry, copyright 2012. | ||
Izzet and co-workers then reported several similar metal-directed cyclic oligomers from organosilyl derivatives of POMs (for which the angle between the two arms is ca. 90°) using the same ligand (pyridine) and metal linker (trans-[PdCl2(CH3CN)2]).81,82 They characterised the resulting supramolecular species by combination of DOSY NMR, mass spectrometry and SAXS. SAXS is a powerful technique to characterise structures of large synthetic molecules, molecular assemblies, nanoparticles78 and aggregates with sizes ranging from 1 to 100 nm.77,83 This technique is particularly well-suited to characterise nanosized metal–oxo clusters assemblies.84–86 In order to understand the thermodynamics of the formation of the discrete metallomacrocycle, they compared the self-assembly behaviour of two analogue ditopic building units differing by the nature (and hence the charge) of the POM (i.e. a Keggin and a Dawson). While Dawson-type hybrids selectively led to the formation of molecular triangles, the same reaction with Keggin-type analogues provided a mixture of molecular triangles and squares. These studies outlined the decisive effect of the charge of the POM disfavouring the formation of large assemblies probably owing to an important entropic contribution. This was confirmed by an isothermal titration calorimetry (ITC) experiment that confirmed the stronger association in the case of the Keggin hybrids. They also showed that the formation of the cyclic assembly was a noncooperative process, since the entropic term was not compensated by the chelate effect. They also observed that while the POM-based molecular building block and related coordination oligomers displayed different shapes, a power law between the diffusion coefficient D and the molecular mass M was applicable to this series of hybrids,81 indicating that the diffusion coefficient of these compounds was mainly determined by their occupied volume rather than by their shape. Izzet and co-workers also used the Dawson organosilyl platform functionalised with remote terpyridine ligands to study the metal driven self-assembly in the presence of cationic metal linkers. In the presence of 1 equiv. of divalent cations (Fe2+ or Co2+),87,88 they observed the formation of discrete metallomacrocycles in a highly dissociating solvent (i.e. DMSO). While the formation of molecular triangles was initially postulated,87 the authors concluded that a mixture of molecular triangles and squares was more likely.88 By contrast to the self-assembly in the presence of neutral “PdCl2” metal linker, which only afforded discrete species, the presence of charged subunits in the metallomacrocycle (POM and metal linker) can drive their aggregation, which results from a competition between the solvation energy of the discrete species and intermolecular electrostatic interactions. Consequently, in a less dissociating solvent (MeCN) or a protic solvent (H2O), the primary supramolecular structures that combine negatively charged POMs and cationic metal linkers, further self-assembled through intermolecular electrostatic interactions in a reversible process. The solvent composition and the charge of the metal linker were found to be key parameters that steer the supramolecular organisation. Different types of hierarchical self-assemblies, zero-dimensional (0D) dense nanoparticles, and 1D worm-like nanoobjects, could be selectively formed owing to different aggregation modes of the metallomacrocycles (Fig. 5). Similar worm-like nanoaggregates were also formed in the presence of H2O and displayed promising molecular carrier properties with a polyaromatic guest.
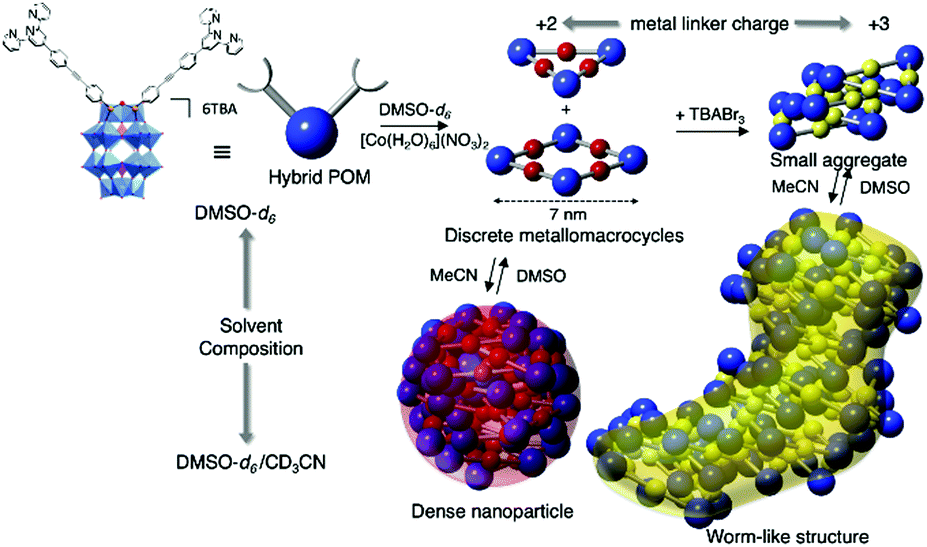 | ||
| Fig. 5 Schematic representation of the formation of nanosized aggregates by hierarchical metal-driven self-assembly of a bis-terpyridine terminated hybrid POM according to the solvent composition and the metal linker charge. This figure has been adapted from ref. 88 under Creative Commons Licence 4.0 (CC-BY-NC-ND). | ||
As ditopic organosilyl derivatives of POMs afforded mixtures of triangle- and square-shaped secondary building units upon reaction with a linear metal linker, some mismatches and defects in the stacking of the metallomacrocycles occurred, which can in part explain the formation of the worm-like structures. Following their initial work, Izzet and co-workers investigated the self-assembly of a bis-pyridine terminated Keggin hybrid with a cationic 90° Pd(II) acceptor, since a unique type of square-shaped metallomacrocycle (containing two hybrid POMs and two Pd nodes) could be formed using this strategy.89 Again, in DMSO only discrete species were observed which formed hierarchical species upon solvent change. Strikingly different hierarchical assemblies were then observed, with the dimeric cycle organising into either branched worms-like structures or multi-layer vesicles dependent on the nature of the solvent.
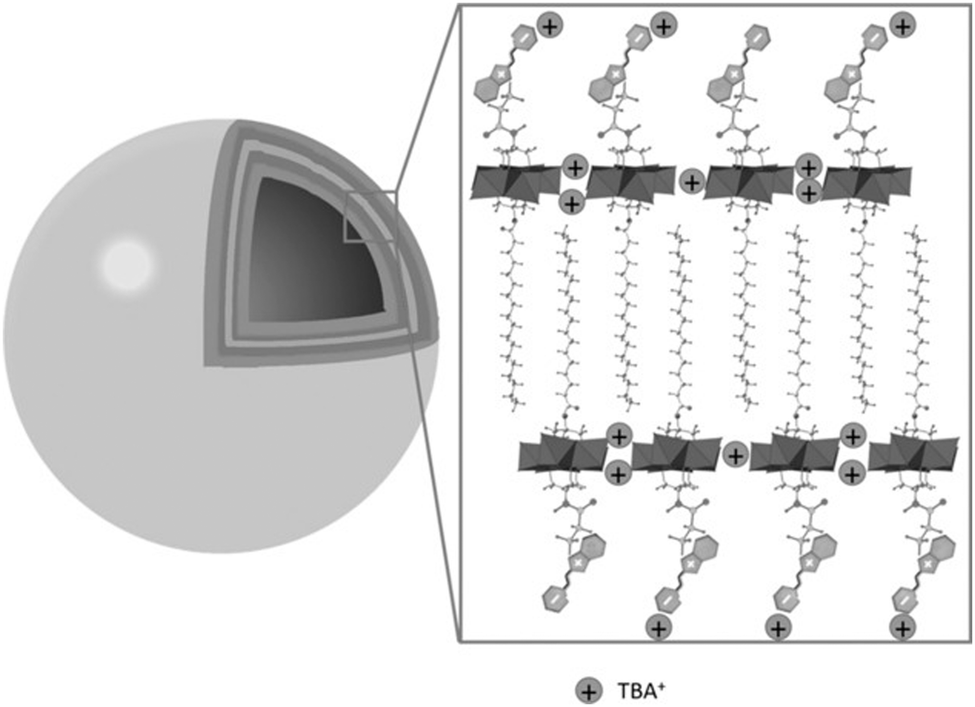
Singly-organofunctionalised hybrid POMs have also be used as building blocks, forming dumbbell-like species in the presence of a linear metal linker. Following their initial work with ditopic hybrids, Izzet and co-workers reported the aggregation of dumbbell-shaped polyoxometalate hybrids, using organotin derivatives of monovacant Keggin- or Dawson-type POMs terminated with a remote terpyridine function.90 They observed the formation of dumbbell-like species in DMSO and showed using ITC that the metal coordination to the POM-based building unit was reinforced by coulombic interactions between the POMs and the metal linker and counter balanced by an entropic cost associated to the POM (the entropic contribution being higher for the Dawson than for the Keggin). They also evaluated the effect of the nature of the POM (Keggin vs Dawson) and the charge of the metal linker (2+ and 3 +) on the aggregation of the dumbbell species. In MeCN, they characterised by SAXS the formation of nanostructured aggregates of different sizes according to the nature of the POM and the charge of the cobalt linker and concluded that aggregation was favoured (bigger size and shorter POM–POM distances) when the charge of the metal linker approached that of the POM. They also observed the formation of organogels retaining some nanostructuration of the aggregates in the presence of EtOH owing to a low solvation energy in this solvent. They showed that the redox state and the nano-organisation of the gels could be photoswitched, providing potentiality to these compounds as redox-responsive smart materials. Finally, molecular dynamics simulations combined to DFT calculations corroborated that the dumbbell-like species can aggregate according to the solvent composition. Interestingly, the computational results outlined the non-innocent role of the TBA counter ions in the aggregation process (Fig. 6).
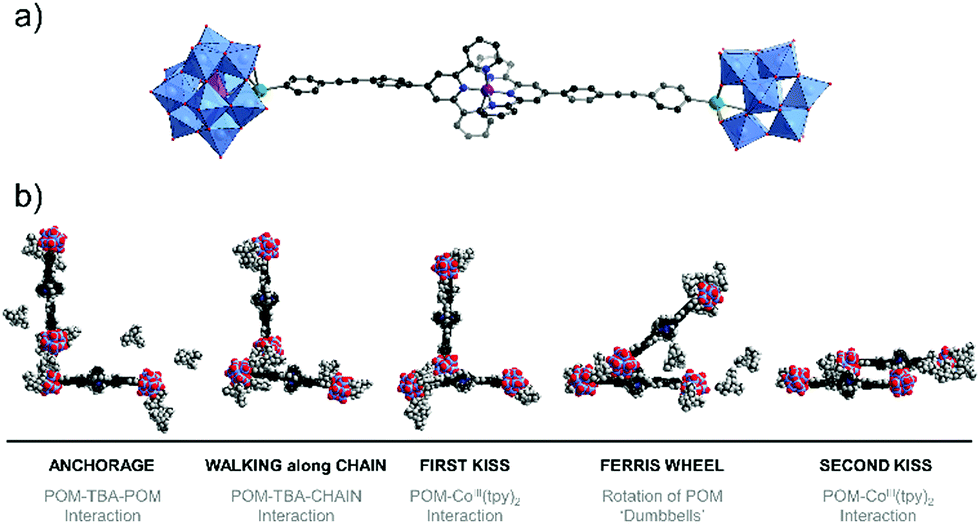 | ||
| Fig. 6 Molecular dynamic study of Keggin-type organotin dumbbell-like species in H2O outlining the role of the TBA counter-cations in the aggregation process (Sn = green spheres, Co = purple sphere). This figure has been adapted from ref. 90 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Several single-side functionalised oxoclusters have demonstrated directed assembly into complex supramolecular assemblies using higher connectivity metal acceptors. Yang and co-workers used a monopyridyl-terminated Anderson-type oxocluster [CrMo6O18(–OH)3{(OCH2)3C–4-Py}]4− building unit to develop different crystalline frameworks by metal coordination.91 Upon reaction with CuI they obtained a cube-like nanocomposite structure, which differed from their coordination frameworks made with a ditopic Anderson hybrid.73 An uncommon octanuclear [Cu8I6]2+ halide cluster was formed around which eight hybrid POMs were coordinated by the terminal pyridine ligand. Furthermore, the nanocomposite compound itself self-assembled via H-bonding into a crystalline 3D porous supramolecular framework, in which large tetragonal channels were constructed from effectively edge-sharing octahedron-like cages comprised of 16 macroclusters (Fig. 7).
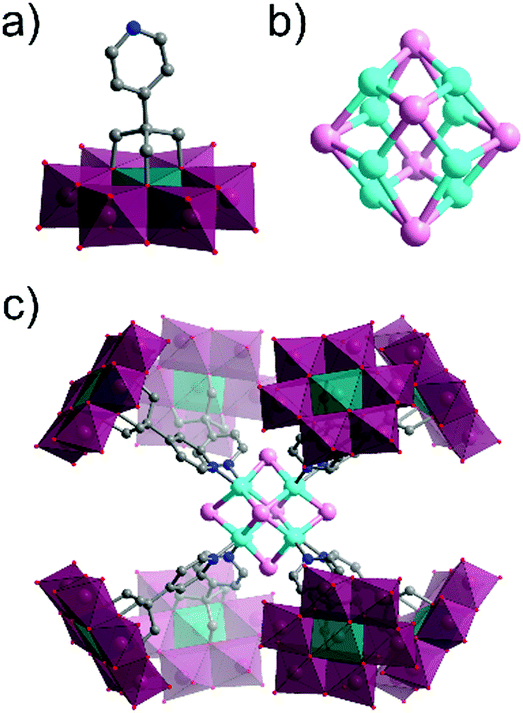 | ||
| Fig. 7 Structure of the monotopic Anderson–Evans-type metal halide cluster, highlighting: (a) the {CrMo6} hybrid POM building block ({CrO6} = teal green octahedra); (b) the Cu8I6 metal halide core and; (c) the assembled octameric nanostructure. This figure has been adapted from ref. 91 with permission from the American Chemical Society, copyright 2016. | ||
A mono-carboxylate functionalised hexamolybdate, [(Mo6O18)NC6H4–4-COOH]2−, assembled into a tetrameric species with a paddle-wheel formation upon coordination with CuII, in which two metal ions resided in the centre. Liu, Wang, Wei and co-workers demonstrated their hierarchical assembly as a secondary building unit into large “blackberry” structures (Fig. 8).92 The formation and size of the latter could, to some extent, be tuned by adjusting the polarity of the solvent composition. Blackberry size was in fact found to depend linearly on solvent dielectric constant (1/ε), within a certain polarity range. Outside this polarity range, too low or too high surface charge of the tetrameric species prevented the aggregation due to unfavourable electrostatic interactions.
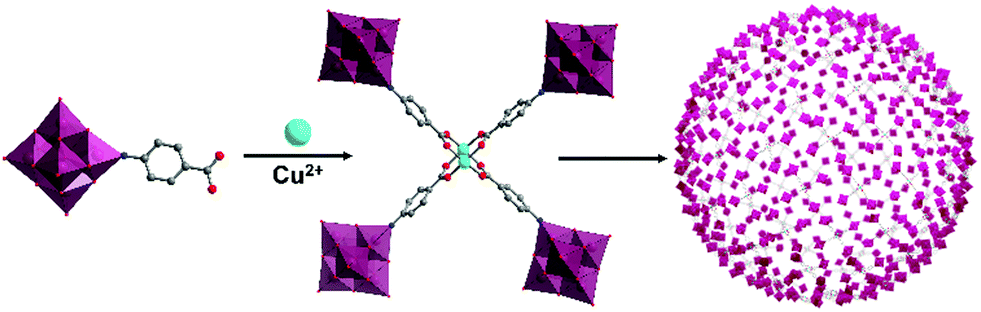 | ||
| Fig. 8 Model of the electrostatically assembled “blackberry” structure formed by the self-assembly of tetrameric hexamolybdate hybrids. This figure has been adapted from ref. 92 with permission from the American Chemical Society, copyright 2013. | ||
3.2 Hybrid POM amphiphiles
In solution, self-assembly of large and charge-delocalised polyanionic POMs can be triggered by their association with hydrophobic tails, i.e. long alkyl chains. Early strategies relied on the exchange of charge balancing counterions by cationic ammonium surfactants. This electrostatic approach historically led to the emergence of a family of core–shell like assemblies, called surfactant-encapsulated-POMs (SEPs). Driven by solvophobic interactions, these SEPs can self-assemble at the air/water interface to form Langmuir–Blodgett films as optical or catalytic materials,93,94 while in solution they may form nanostructures such as vesicles and onion like spheres by controlling the solvent polarity.95 The field of SEPs has been widely explored by Kurth and Wu and recently reviewed.96 A main drawback associated to this electrostatic approach is the formation of compounds that display poor solubility in aqueous solutions. Keggin-type polyoxometalates when combined to amphiphilic cations have been reported to preferentially form POM-based nanoparticles able to stabilise Pickering type emulsions in water–oil mixtures.97 Bauduin and co-workers also reported that POM-surfactant may spontaneously form in water through self-assembly of Keggin type anions and non-ionic surfactant leading to micelles containing POMs at the interface.98 Such unexpected self-assembly process has been associated to the tendency of POMs as super chaotropic anions to adsorb on hydrophilic neutral surfaces. As fascinating these results are, the lack of solubility in water restricts the scope of their potential applications.The alternative approach consists in chemically grafting hydrophobic tails through covalent linkage to POM scaffolds, through either direct or post-functionalisation paths. This leads to a class of POM-based surfactants that is commonly called covalent amphiphilic POM–organic hybrids. While being more sophisticated and requiring multistep synthesis as explained in former sections, this procedure allows the development of tailor-made amphiphilic POMs by controlling the number and the relative orientation of the hydrophobic chains, as sketched in Fig. 9. This variety of functionalisation produces a very unique family of POM-based surfactants containing polar heads that are adjustable in size, charge and shape and allows controlled self-assembly formation as a function of the packing parameter, a characteristic number related to the shape of one single surfactant molecule (Fig. 10).99,100 Another key point brought by the covalent approach is to enable the substitution of alkaline cations (or even protons) for the conventional bulky ammonium, thus improving the hydrophilicity of the POM polar head and increasing water solubility (i.e. increasing the critical aggregation concentration, CAC). While covalent POM-based surfactants have already been the subject of several reviews,22,99,101 the next sections aim to emphasize the peculiarities of the POMs as a large inorganic polyanionic head in the self-assembly processes and to highlight recent results in which self-assembly of POM surfactants provided new opportunities to produce nanofunctional materials with potential to transfer POM properties to the supramolecular domain. As with any smart materials, the challenge is to organise these inorganic objects into supramolecular structures with long-range order; allowing preparation of micellar structures or fibers with potential as nanocatalysts, or switchable materials.
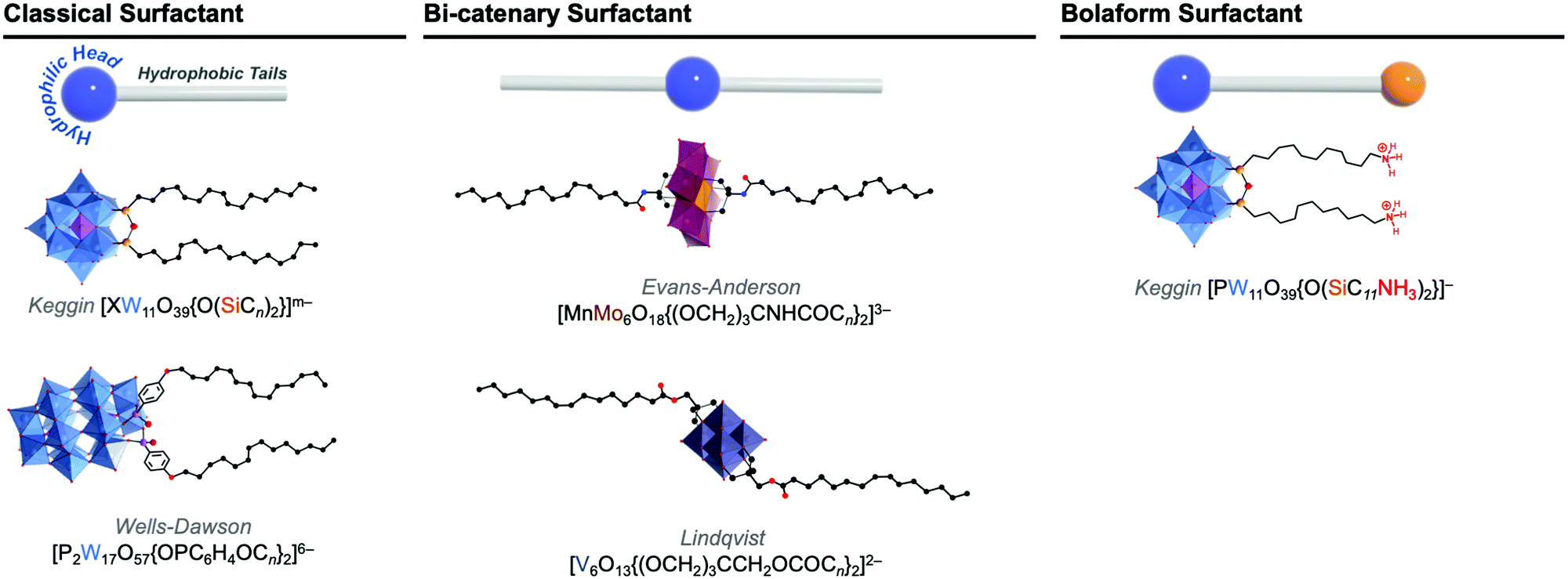 | ||
| Fig. 9 Molecular structures of some representative amphiphilic POMs and their comparison to: classical ‘head–tail’-type surfactants, bi-catenary-type surfactants, and bolaform type surfactants. | ||
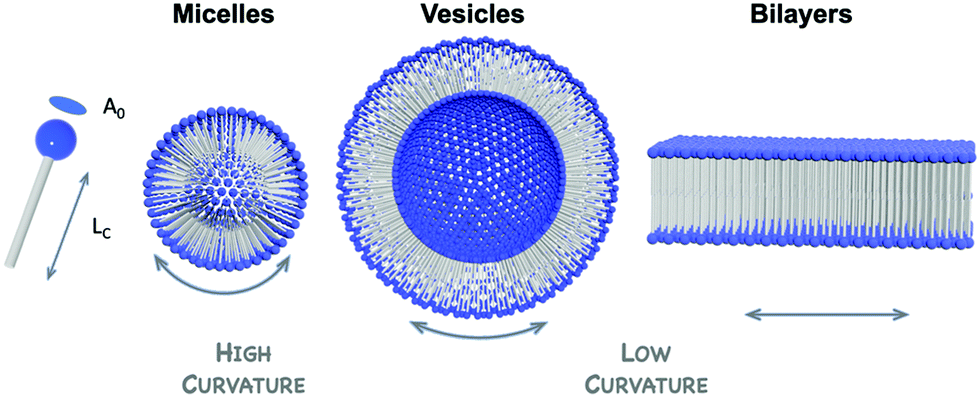 | ||
| Fig. 10 Schematic highlighting the common structures of amphiphilic hybrid-POM supramolecular assemblies, which depend on both the shape of the POM-surfactant and the packing parameter (defined as P = V/(A0LC), where V represents the volume occupied by the hydrophobic tails, A0 the area of the polar head and LC the length of the hydrophobic tails). | ||
Alkaline counter ions are also playing a crucial role, not only in screening the charge of the POMs, but also in displaying specific interactions.109 The attractive electrostatic forces between POMs and these counterions are actually involved in the formation and stability of self-assembled structures, be they organised monolayers on planar surfaces or nanoparticles, or “blackberry” structures.110–112 Substitution of protons or alkali counterions for quaternary ammoniums in covalent amphiphilic hybrid POMs has therefore rapidly emerged as a privileged method for amplifying the dipolar character of the POM surfactants, favouring the self-assembly kinetics and promoting micellisation and emulsifying properties.
A relevant example has been reported by Wang and co-workers. The authors depicted the preparation of a triol-functionalised mixed-addenda Wells–Dawson [P2W15V3O59{(OCH2)3C–R}]9− bearing an ATRP initiator group, from which polymerisation of styrene was initiated.113,114 The reaction led to a well-defined POM–polystyrene hybrid (Fig. 11). Upon proton exchange for TBA, the DMF solution became opaque revealing the amphiphilic character of the H+–POM–polystyrene compound, which displayed supramolecular assembly into vesicular aggregates. TEM and SEM analysis allowed to determine that the aggregate structure is similar to a reversed vesicle, the POM polar head being located in the middle of the membrane, which size varies from 150 to 350 nm depending on the experimental conditions.
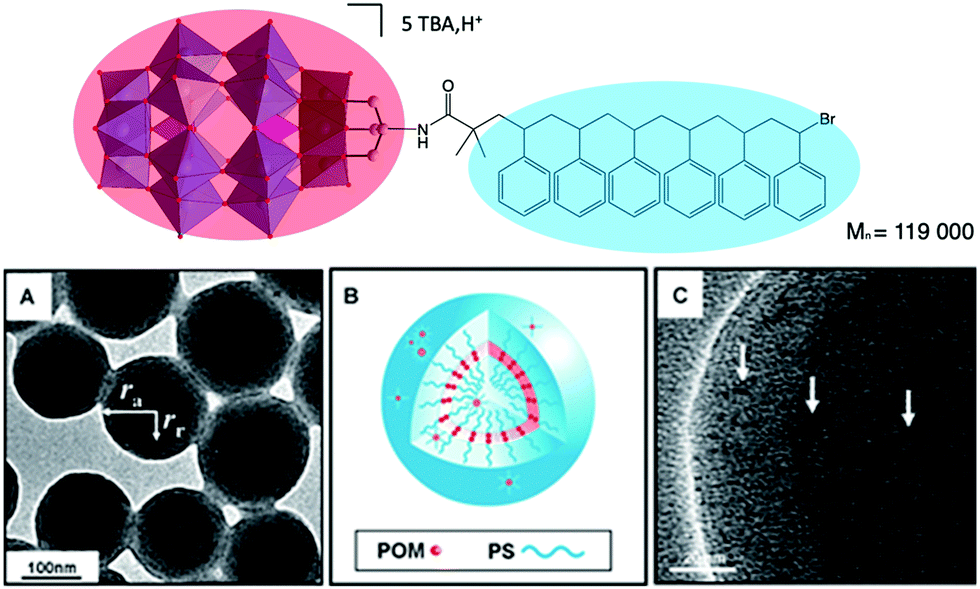 | ||
| Fig. 11 (Top) Molecular structure of a POM–polystyrene hybrid; (bottom) representations of its vesicular aggregates, showing: (a,c) high-resolution TEM images and, (b) a schematic representation of the vesicle. This figure has been adapted from ref. 114 with permission from John Wiley and Sons, copyright 2012. | ||
The formation and size of vesicles is triggered by the nature of the counterions. This has been highlighted by two independent reports by the groups of Wei and Hill. In 2011, Wei and co-workers reported the preparation of [V6O13{(OCH2)3CCH2OOC(CH2)16CH3}2]2− by covalently grafting two C18 chains through esterification reaction between stearic acid and a triol-functionalised hexavanadate cluster (Fig. 9).103 As a TBA salt this amphiphilic POM self-assembles into vesicles only when water is added to acetone solutions (RH = 180–220 nm) whereas the proton and sodium derivatives, prepared by ion-exchange chromatography, readily self-assemble in pure aqueous solutions into vesicles of much smaller sizes (RH = 57 nm and 55 nm respectively).
The assemblies display remarkable fluorescence properties and the origin of the emission is thought to involve an emissive state derived from a ligand to metal charge transfer (LMCT). The authors speculated that the exchange of cations triggers a structural change in the self-assembly, with an increased interaction between the POM heads resulting in an increase of the fluorescence intensity. Although POMs are known to be luminescence quenchers due to their ability to readily store electrons, the study of these assemblies using fluorescence microscopy revealed that the intensity did not decay over time demonstrating a high stability compared to organic fluorophores that undergo photo-bleaching when exposed to UV light. The effect of the cation on the packing structure and fluorescence profile of hybrid POM vesicles was also nicely exemplified by Hill and co-workers when studying similar Lindqvist type hexavanadate clusters functionalised by pyrene fluorescent groups (Fig. 12).115 The presence of two pyrene moieties led to smaller assemblies (RH =50 nm) in H2O/DMSO (80:20 v/v) mixture as a result of additional bending energy required, which led to increased curvature in the packing. The fluorescent profile of the pyrene groups was employed as a useful tool to investigate the molecular packing within the superstructure. Changes in the pyrene fluorescence were notably observed when TBA cations were exchanged for protons, which induced the formation of smaller aggregates (RH = 30 nm), as a result of a decrease of alkyl chains in the hydrophobic layer of the vesicles. Hill et al. observed the formation of an excimer fluorescence peak (480 nm) that is ascribed to the close packing of two pyrene molecules. This unique result was further tested by varying the pH of the solution where a decrease in RH was observed with increasing pH, the process being reversible. The works of the groups of Wei and Hill demonstrate interesting luminescence properties associated with the self-assembly of amphiphiles hybrid POMs in solution. The combination of the photo-activity and controlled self-assembly displays great potential to fabricate state-of-the-art photo-active colloidal materials.
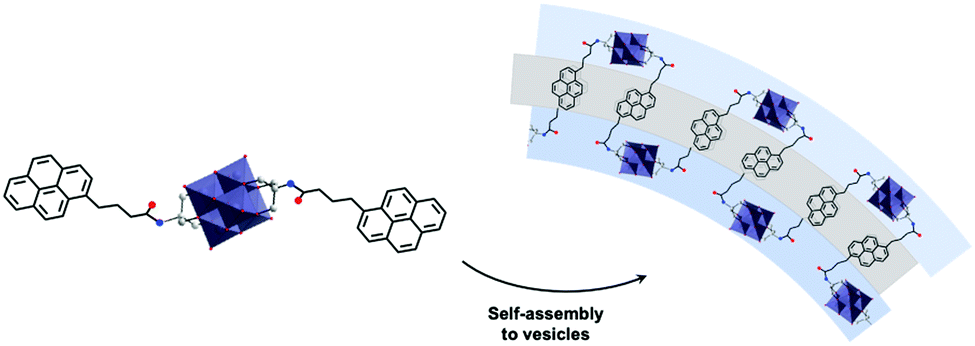 | ||
| Fig. 12 The molecular structure of a hexavanadate hybrid cluster bearing two pendant arms functionalised by pyrene groups showing the proposed arrangement of the vesicular structures it forms in solution. This figure has been adapted from ref. 115 with permission from the American Chemical Society, copyright 2011. | ||
In 2012, Polarz and co-workers reported an original bolaform surfactant bearing a Keggin-type POM and an antipodal positively charge ammonium group connected by a hydrophobic spacer.116 The asymmetric size of the polar heads creates highly curved interfaces leading to the formation of monolayer membrane vesicles in water with unusually small diameter (15 nm). The authors also reported that the membrane of the vesicle is highly impermeable favoring the encapsulation of hydrophilic or ionic compounds in the confined aqueous nanophase.
The bi-catenary POM hybrids based on Anderson–Evans and Lindqvist oxoclusters and the bolaform-type hybrid POMs displayed structures that are not well suited for the formation of micelles. Special interests have therefore also been devoted to POM surfactants whose shapes are more related to classical surfactant sodium dodecyl sulfate (SDS). These compounds exhibit an anionic polar head, alkali-metals counter ions and long alkyl chains directed on one single side. An elegant study has been nicely detailed in the case of double-tailed surfactant derived from monovacant Keggin anions [XW11O39]q−. A pioneering work by Chambers et al. in 2003 detailed the ability of the bis(silyl) derivative [SiW11O39{O(SiC12H25)2}]4−, as tetrabutylammonium salts, to reversibly form thin monolayers at the air/water interface by using Langmuir–Blodgett technique.117 This work highlighted the fact that such covalent amphiphilic POMs are robust and gave an impetus to further studies in the field. Polarz and co-workers prepared alkaline (Na+, K+) and acid derivatives of the analogous (TBA)3[PW11O39{O(SiR)2}] by postpreparative ion-exchange chromatography in order to study their aggregation in water. Using X-ray reflectivity studies, Polarz and co-workers confirmed that the interfacial self-assembly of these bi(silyl) derivatives in the LB films was driven by the packing of the polar heads in a hexagonal lattice, whereas the hydrophobic tails did not relevantly contribute to the overall process, an unusual behavior for surfactants.118 More importantly, these authors reported the formation of lyotropic liquid crystalline phases, whose structures depend on the alkyl chain lengths and counterions associated with POM surfactants: while an hexagonal phase of packed cylinders is obtained with sodium as counterions, the acid salt leads to a lamellar structure (see Part 3.3.1. for more details). By studying aqueous solution at lower surfactant concentration, Polarz et al. also observed the formation of micelles, whose sizes scale with the length of the alkyl chain (based on DLS and SAXS measurements). The authors finally demonstrated that this class of POM surfactant could act as emulsifying agents, i.e. decreasing surface and interfacial tensions.119,120 These results again highlighted the crucial role played by the nature of the counterions in the kinetics of aggregate formation in solution.
The structure of the aggregates formed in solution also strongly depends on the geometry of the surfactant in its complete environment, which can be described by using the packing parameter P.101 To this respect amphiphilic POMs are very uniquely polarisable agents with a charge/volume ratio at the polar head that can be modified without changing the overall shape of the surfactant. Polarz and co-workers extended their series of POM surfactants [XW11O39{O(SiC16H33)2}]n− to X = Si(IV) and B(III) and as a consequence n = 4 and 5 respectively. Unusual self-assembled structures such as a new type of micelle with dumbbell shape was reported in the case of higher charged POMs. Such structuration was tentatively attributed by the authors to the presence of stronger repulsive electrostatic while the contribution of attractive, hydrophobic (van der Waals) interactions remains constant (same size and shape of the hydrophobic tails).121
In 2014, Proust et al. also reported the preparation of double-tailed POM surfactants TMA3K[γ-SiW10O36(CnH2n+1PO)2] (TMA = tetramethylammonium) based on the divacant [γ-SiW10O36]8− anion (Fig. 13).122 Proton and alkaline cations can substitute TMA in these systems using a cation-exchange resin leading to water soluble derivative A4[γ-SiW10O36(CnH2n+1PO)2] (A = K, Na, H). SWAXS measurements on water solutions of A4[γ-SiW10O36(C12H25PO)2] allowed estimation of the critical aggregation concentration and, in combination with DOSY NMR, indicated the formation of micelles with an aggregation number nagg ≈ 33 and a hydrodynamic radius RH = 1.8 nm.123 A peculiarity stems from the presence of a proton as counter ion in the potassic derivatives K3H[γ-SiW10O36(OPCnH2n+1)2] making these amphiphilic POMs weak acids (pKa of 3.0). As a result, the apparent negative charge was found to be pH dependent, strongly affecting the self-assembly process. A 10 mM solution of K3H[γ-SiW10O36(OPC12H25)2] (>CAC) resulted in a pH of 2.6 and formation of micelles was observed by NMR. Increase of the pH forced a disassembling process to monomers due to the increase of the apparent negative charge and consequently an increase of the CAC (Fig. 13). Note that special attention should always be addressed to the stability of the covalent amphiphilic POMs in aqueous solution. Whereas highly basic pH conditions are known to promote the disassembly of the polyoxometalate framework, highly acidic conditions may also catalyze the chemical degradation of the W–O–P linkage, a mixed anhydride of tungstic acid and phosphoric acid, hence sensitive to hydrolysis. Overall, the formation of micelles when using these bis(silylated) or bis(phosphonylated) POM derivatives, which are related to SDS structures, reveals the low packing parameter (less than 1/3) exhibited by these POMs surfactants that is consistent with a large polar head and a rather low volume of hydrophobic tails, thus exhibiting a cone-like shape.100 The emulsifying ability of this series of POM surfactants opened the scope of applications to catalytic reactions in complex media.
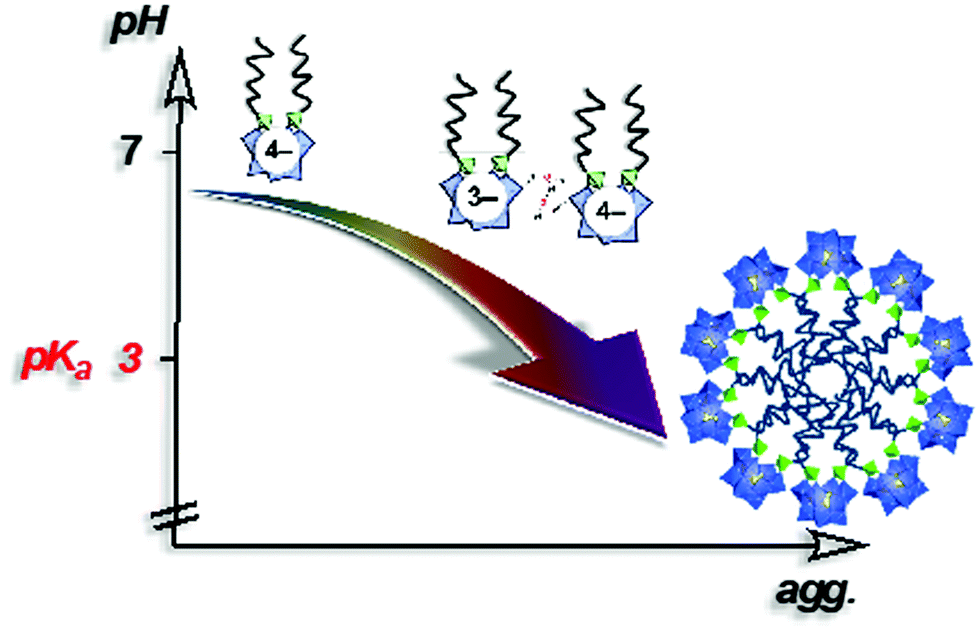 | ||
| Fig. 13 Schematic showing aggregation as a function of the pH in a 10 mM aqueous solution of K3H[γ-SiW10O36(POC12H25)2] (>CMC).122 | ||
 | ||
| Fig. 14 The catalytic surfactant H3[PW11O39{O(SiC16H33)2}] acts as an emulsifying agent and a catalyst for (a) the acid-catalyzed polymerisation of styrene, and (b) the polymerisation of siloxane to polysiloxane and formation of nanocomposites as promising candidates for proton-conducting materials. This figure has been adapted from ref. 119 with permission from the American Chemical Society, Copyright 2010, and ref. 120 with permission from the Centre National de la Recherche Scientifique (CNRS) and the Royal Society of Chemistry, copyright 2016. | ||
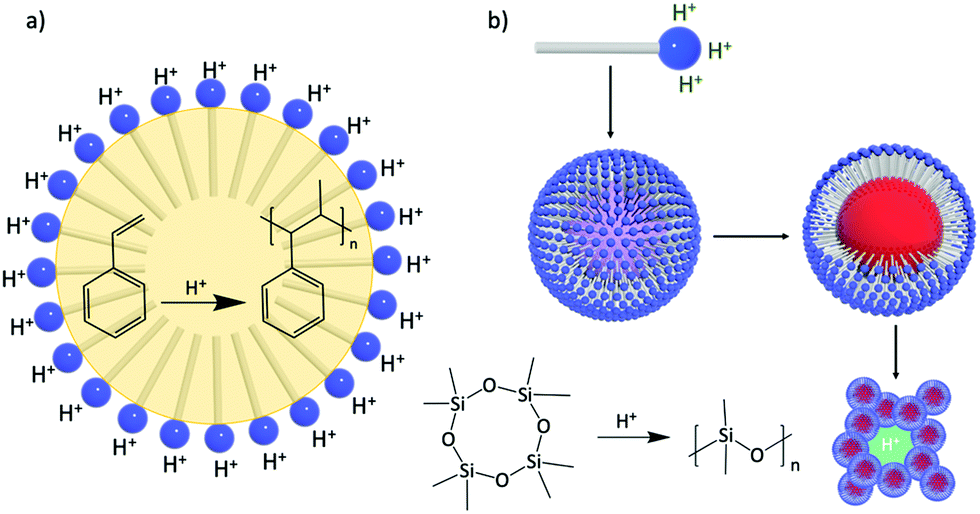
Design of amphiphilic hybrid POMs reported in Fig. 9 was also driven by their use as a catalytic surfactant able to (i) stabilise (micro)emulsion or micelles, and (ii) enhance catalytic activity at the interfaces or in compartmentalised environments. The emulsifying properties of Polarz's POM surfactants H3[PW11O39{O(SiC16H33)2}] have been used in acid-based catalysis for the polymerisation of styrene. Indeed, stable styrene/water emulsions can be prepared and stirring under gentle heating (50 °C) produced white solids containing POM surfactants and polystyrene. DLS and TEM measurements revealed the formation of spherical objects with diameters in the size range 300–350 nm with POM surfactants located on the surface of the polymer particle (Fig. 14a).119
The ability of these POM surfactants to fulfill multiple tasks was further demonstrated by Polarz and co-workers.132 Similarly, H3[PW11O39{O(SiC16H33)2}] was used as a catalytic surfactant for, first, the formation of stable emulsions of octamethylcyclotetrasiloxane in water and, secondly, the acid-catalyzed generation of polydimethylsiloxane (PDMS) upon the addition of PhSi(OEt)3 as cross linker. DLS and TEM measurements of the resulting dispersion confirmed the formation of spherical large nanoparticles (around 155 nm) with narrow size distribution. The dispersion also exhibited high stability for weeks and no leaching of tungstate species or degradation of the surfactant was detected, which was tentatively attributed to the fact that the alkyl chains of the surfactant are tightly buried into the polysiloxane matrix. Of particular interest, Polarz and co-workers also reported that solvent removal allowed to irreversibly assemble a nanoporous network from these particles (Fig. 14b). Since the POM polar heads of the surfactant are still located at the surface of the network, the overall materials are characterised by proton-conductivity inside its channels, which make them promising candidates for applications as membranes in fuel cells.
Double-tailed covalent POM surfactants have also been investigated in biphasic oxidation reactions following the same strategy to both stabilise polydisperse media and catalyze an oxidation reaction using aqueous hydrogen peroxide. From the series of compounds reported by Proust and co-workers, K3H[γ-SiW10O36(C12H25PO)2] was shown to provide a Winsor I system (WI) in water–ether mixtures, i.e. oil-in-water microemulsion (μem), in equilibrium with an excess of the organic phase.122 Quantification by NMR spectroscopy clearly confirmed that the amphiphilic POM-based hybrid was mainly localised in the μem phase and 2D heteronuclear chemical shift correlation between 31P and 183W nuclei observed by HMQC experiments proved the structural integrity of the hybrid POM. Distribution of the POM surfactant in the phases can also be ascertained by using small and wide-angle X-ray scattering (SWAXS). The SWAXS results indicate that both diethylether and water coexist as two distinct phases in the solution, as expected in μems, and that the ether droplets of the WI μem have a diameter of ca. 5.0 nm (Fig. 15). Stabilisation of a WI type μem confirms that K3H[γ-SiW10O36(C12H25PO)2] belongs to the class of hydrophilic surfactant, i.e. with large polar head and a rather small hydrophobic chain. The use of microemulsions for catalysis is particularly interesting since it provides a thermodynamically stable nanostructured phase and an extended oil–water interface (much higher than classical biphasic system)51 that favors the exchange of reagents and products between the nano-domains. As an example, the WI-type system in Fig. 15 was able to conduct the oxidation of organic sulfide to a sulfone/sulfoxide mixture at room temperature using hydrogen peroxide without demixing phenomenon.
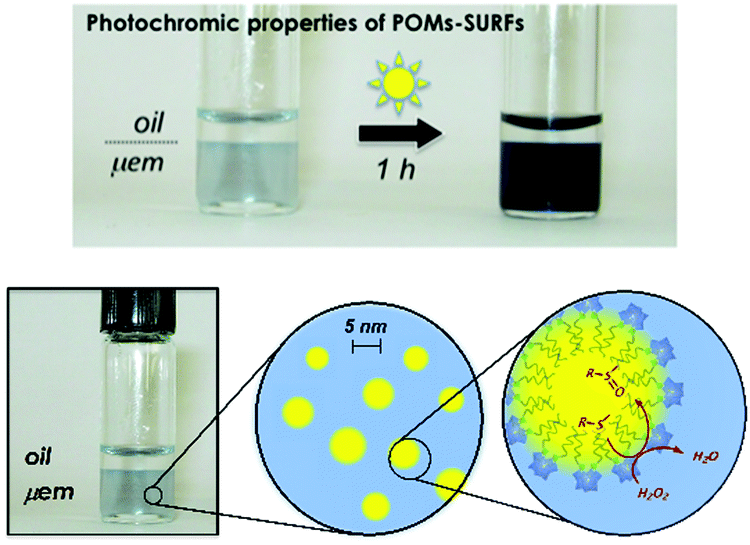 | ||
| Fig. 15 (a) The partitioning of POM surfactants between the two phases of a WI microemulsion is easily achieved by irradiation with UV or even sunlight; (b) microemulsion is made of 5 nm diameter oil-containing micelles. Adapted from ref. 122 with permission from the Royal Society of Chemistry, copyright 2014. | ||
The water soluble sodium and acid derivatives of the bi catenary hexavanadate [V6O13{(OCH2)3CCH2OC(O)C17H35}2]2− were also used by Liu, Wei and co-workers to stabilise hexane/water emulsions and catalyze the oxidation of thiophene with hydrogen peroxide, as a model reaction for oxidation desulfurisation of diesel.136 Enhanced catalytic activity was described as a consequence of high concentration of the catalytic surfactant at the water–oil interface. Liu and Wei also reported that the single-tailed hexavanadate–organic hybrid surfactant was able to self-assemble into micelle structures in water solutions, and to further coagulate into a 1D anisotropic structure with high surface area. These hierarchical self-assemblies were reported to be a highly efficient heterogeneous catalyst for the oxidation of organic sulfides.137 Segregation of reactants in specific nanodomains have been more recently reported by the same authors, who demonstrated that bilayer vesicles formed from double-tailed hexavanadate–organic hybrid surfactant with long alkyl chains were efficient catalysts for the oxidation of TMB (3,3′,5,5′-tetramethylbenzidine) with H2O2, a model reaction mimicking a peroxidase activity. The authors claimed that the increase in the oxidation rate of TMB compared to reaction carried out with unassembled systems was promoted by the bilayer self-assembly that create hydrophobic domains that segregate the TMB and favors its accessibility to the hydrophilic catalytic POM head.138
The versatility of this concept was further exploited by the same group to produce photo-switchable materials based on the self-assembly of an organotin-functionalised POM.140 In this inorganic–organic hybrid system, a propargyl azobenzene moiety was connected using a copper-catalysed Huisgen reaction to a lacunary Keggin-type POM functionalised by an organo-tin chain containing an amido group and terminated by an azido function. The POM-Azo hybrid was prepared as a TBA salt and substitution with protons by an ionic exchange reaction over a silica led to a water-soluble hybrid POM exhibiting amphiphilic character. Spontaneously self-assembly into hollow vesicles with a bilayer structure was demonstrated by dynamic light scattering and transmission electron microscopy (D = 60 nm). Photo-switching of the azobenzene function to the cis-configuration upon UV-light irradiation induced a modulation of the POM-Azo assemblies, the regular spherical assembly being transformed into an irregular morphology. Cis–trans isomerisation under light irradiation (450 nm) brought the assembly back to regular spherical vesicles, thus proving the dynamic reversible transformation. An expansion of this work, in which the authors extended their photoisomerisation strategy to include the formation of host–guest complexes with cyclodextrin hosts is described in more detail in Section 4.1 below.
In 2016, Mialane, Liu et al. reported the synthesis of asymmetrical Evans–Anderson hybrid POM, (TBA)3[MnMo6O18{(OCH2)3CNHC21H19N2O4}{(OCH2)3CNHCOC15H31}], by covalently grafting a photoswitchable spiropyran (SP) fragment and a long hydrophobic alkyl chain.141 Under UV irradiation (λ = 365 nm) colourless and hydrophobic SP underwent a C–O bond cleavage resulting in the formation of a zwitterionic merocyanine (MC) isomer, which is hydrophilic and strongly absorbs in the visible regions. This conversion triggered the self-assembly processes in mixed solvents (Fig. 16). In water/acetonitrile mixture (2 to 2.5 v/v%) hollow vesicles were formed, which sizes depended on the water content (RH from 13 to 110 nm, respectively). The aggregation processes are reported to reach equilibrium after ca. 30 h. In less polar solvent mixtures, 84 v/v% toluene/acetonitrile, an equilibrium was reached after three days, leading to spherical assemblies thought to be reverse vesicles due to solvent polarity, exhibiting a RH = 112 nm. These assembly processes are reported to be reversible and sizes of the assemblies were found to be inversely proportional to the dielectric constant of the mixed solvent. The system reported by the groups of Mialane and Liu represented an original example of a light and solvent-controlled self-assembly of a photoresponsive hybrid POM.
 | ||
| Fig. 16 Self-assembly of the photoswitchable asymmetric POM hybrid, SP–POM–C16 (SP = spiropyran, C16 = decahexane) in polar solvents to form hollow vesicles. This figure has been adapted from ref. 141 with permission from John Wiley and Sons, copyright 2016. | ||
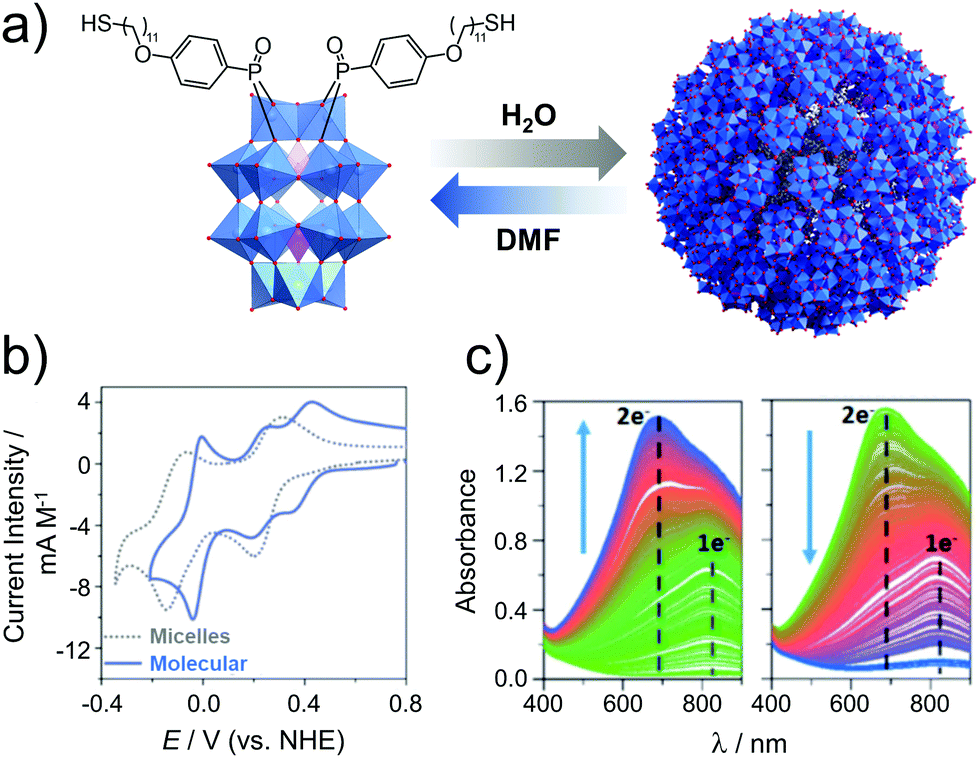 | ||
| Fig. 17 (a) Structure of the amphiphilic hybrid POM, H6[P2W17O61(PO2C17H27S)2], showing its reversible assembly into micelles (Davg = 9 nm); (b) cyclic voltammograms of the hybrid POM micelles in 0.1 M H2SO4 (aq.) and the molecular cluster species (after addition of DMF), and; (c) spectroelectrochemical analysis of the micellar system showing reduction (left, E = 0.05 V) and reoxidation (right, E = 0.61 V). This figure has been adapted from ref. 142 with permission from the Royal Society of Chemistry, copyright 2017. | ||
The concept of redox active assemblies was further developed by Newton et al. to understand the relationship between the structure of the surfactant on the structure and redox chemistry of its supramolecular assembly. Here, the authors studied the effects of chain-length on the structure and the electronic properties of the supramolecular assembly.143 A library of POM-surfactants were synthesised based on Wells–Dawson grafted to alkyl chains of varying lengths, K6[P2W17O61(POC6H4O(CnH2n+1))2] (n = 10, 12, 14, 16, 18 and 20). Based on increasing chain lengths, the assemblies seemed to decrease in size, owing to a larger hydrophobic driving force to pack tighter together. Furthermore, the electrochemical properties of these hybrids were investigated in 0.1 M H2SO4. Observation aligned accurately with the typical two redox processes assigned to the assembly state established in the previous study for longer chain lengths C16–20, however shorter chain lengths C10–14 displayed 3 redox processes, with the first redox process shifting closer to the second as the chain length increased. Based on the assembly morphology, C10 displayed polydisperse aggregates, which is attributed to the lack of hydrophobic interactions which may also increase its solubility in the molecular state, this reason is speculated to be why a molecular electrochemical profile is observed. Overall, it was established that longer chain-lengths lead to smaller assemblies, with a clear contrast in the CV vs. the molecule. This fundamental study clarified the correlation between the level of hydrophobicity and the electrochemical profile of the superstructure.
More recently, Newton et al. reported a simple synthetic procedure to tailor a multifunctional hybrid system using an asymmetrically functionalised Dawson type POM hybrid.144 The authors described the preparation of the mono-lacunary Wells–Dawson bearing two distinct aryl-phosphonic acids, a terpyridine-based ligand (tpy) and a long alkyl chain group (C18). As a potassium salt, the POM hybrid displayed amphiphilic properties and the presence of the C18 alkyl chain enabled self-assembly into micellar structures (DH = 6 nm). Similar electrochemical trends to that reported above were observed in this new system, as a typical fingerprint of these assemblies. By using 0.5 eq. of an FeII linker a dimeric POM–FeII–POM compound could be isolated, which, lacking the distinct “head–tail” polarity of the uncoordinated precursor, formed variably shaped non-hollow aggregates in a H2O–MeCN (9:1 v/v) mixture in comparison to the micelles assembled by the precursor. In contrast, in a successive study on the PtII coordinated oxocluster,145 micellar assemblies were enabled by the retained amphiphilic polarity that can be considered metal-decorated, as, unlike in the aforementioned bis-organosilyl Dawson oxoclusters, a high angle (approx. 160°)44 exists between the two distinct arylphosphonate ligands. Notably, individual micelles appeared to coalesce into small worm-like structures likened to those of Izzet and co-workers,88,89 suggestive of some degree of further supramolecular aggregation.
The concept of redox-active assemblies has been applied to energy storage, specifically redox flow batteries (RBFs) as POM assemblies possess the ability to carry large amounts of charge and are less susceptible to membrane leaching due to their size.146 Polarz and co-workers reported a fully conjugated surfactant by grafting a redox active ligand, anthraquinone (AQ), to a lacunary Keggin type POM K7[PW11O39] via a conjugated chain and siloxane linkers, to afford AQπPOM.147,148 The motive behind this novel design was to fabricate a redox active amphiphile, where the hydrophobic moiety could also be employed as a charge carrier, therefore increasing the charge loading capacity of these assemblies. DFT calculations suggested that the HOMO is located on the centre of the conjugated ligand and extends to the AQ, while the LUMO is located on the POM.148 At very low concentrations (0.01 g L−1) in aqueous solutions, AQπPOM assembled into micelles (DH = 8.7 nm). The CV data of the AQπPOM micelle displays reduction of the POM with the electron transferring to the core AQ and then reversibly oxidised back. Furthermore, the size of the micelles was analysed by DLS post bulk electrolysis of the micelles where a 3 nm increase was observed. This proved that the structural integrity of the battery like micelles was maintained and therefore, electrochemically stable. This supramolecular electron storage characteristic is a great opportunity to create innovative nano-scale micellar batteries or develop the field of RBFs.
A similar strategy has also been employed by Rieger and co-workers in 2015 using a Wells–Dawson POM hybrid substituted with a hydrophobic dodecylamide chain [α1-P2W17O61{Sn(CH2)2CONHC12H25}]7− and ammonium (NH4+) as counterions.150 Aqueous emulsion polymerisation of styrene was also reported to lead to the formation of stable latexes in which the amphiphilic POMs act as stabiliser adsorbed at the surface of the PS particles. This strategy actually reproduced the seminal work by Judeinstein151 that was successively developed by Maatta152 and Thouvenot153,154 for the preparation of organically modified POMs bearing polymerisable functionalities (vinyl, allyl, methacryl, styryl), some of which behaving as surfactants when the counterions are alkaline cations. These POM hybrids may act either as a co-monomer or as reticulating agent or cross-linkers in the polymerisation of organic monomers promoting the formation of either hybrid polymers with POM pendant units or hydrogels.155 It is worth mentioning that the inclusion of POMs in polymeric networks has been the mean of many studies within the aim to develop catalytic, magnetic and photo- or electrochromic materials from inert matrices.156–158 The works by Yin, Liu and Rieger nevertheless represent a new methodology for the preparation of POM–polymer conjugates with core–shell structures. Major objectives were to synthetically control and stabilise nanoparticles and to generate covalent interactions between the NPs and the POM in order to avoid leaching of the latter in solution under extreme conditions and finally to overcome the poor processability of pristine POMs.158 A more challenging objective was to associate integrated functions brought by the polyoxometalate entity itself. Rieger and co-workers described their POM–surface decorated polystyrene particles as photochromic materials due to the photoreduction of the POM core shell under UV irradiation. The POM mediated surface photoactivity was also observed by the photoreduction of silver ions which led to spatially controlled nucleation of small silver nanoparticles exclusively at the surface of POM–Polystyrene conjugate.150
The use of POMs as protecting ligands for electrostatically stabilised metal(0) nanoparticles and nanocomposites has been nicely reviewed by Weinstock et al.159 Few examples of covalently bound POMs to gold (AuNPs)160–162 and platinum163 nanoparticles have also been reported using on-purpose designed hybrid POMs. Towards this end, hybrid POMs featuring thiol-terminated alkyl chains have been prepared as capping agents in order to create a strong interaction with the nanoparticles through a thiolate-type binding mode. Such strategy ensured the formation of ordered assemblies of POMs at the gold surface and as expected to prevent agglomeration of gold nanoparticles. Newton et al. prepared the orgonophosphoryl-modified Wells–Dawson POM, K6[P2W17O61{PO2C17H27S}2] (Fig. 18), and reported that the nanocomposites that were obtained following this strategy exhibited much higher stability to temperature and pH values compared to their electrostatically stabilised analogues.162 Nanomaterials that combine optical properties of gold nanoparticles with the electro- and photochemical features of the POM framework are of particular interest to obtain multifunctional nanomaterial with potential applications in catalytic and sensing technologies. Polarz et al. reported the preparation of the Keggin derivatives [SiW11O39{Si(CH2)n–SH}2O]4− which were also attached to the surface of gold nanoparticles using thiolate-type binding mode.161 With protons as the counter ions, the POM surfactant AuNp composites exhibited Brønsted acid catalytic activity in esterification reactions that could be significantly enhanced by radiation with light. The authors attributed these results to the synergetic interaction between the attached hybrid POM and the surface-plasmon resonance of AuNp as the effect was proven to depend on the distance between the POM head and the AuNp surface (by varying the alkyl chain length, n value). This example featured how synergetic interactions can arise through association of POM and nanoparticles and how rational preparation methodologies are central issues to prepare nanomaterials with controllable and tailored properties.
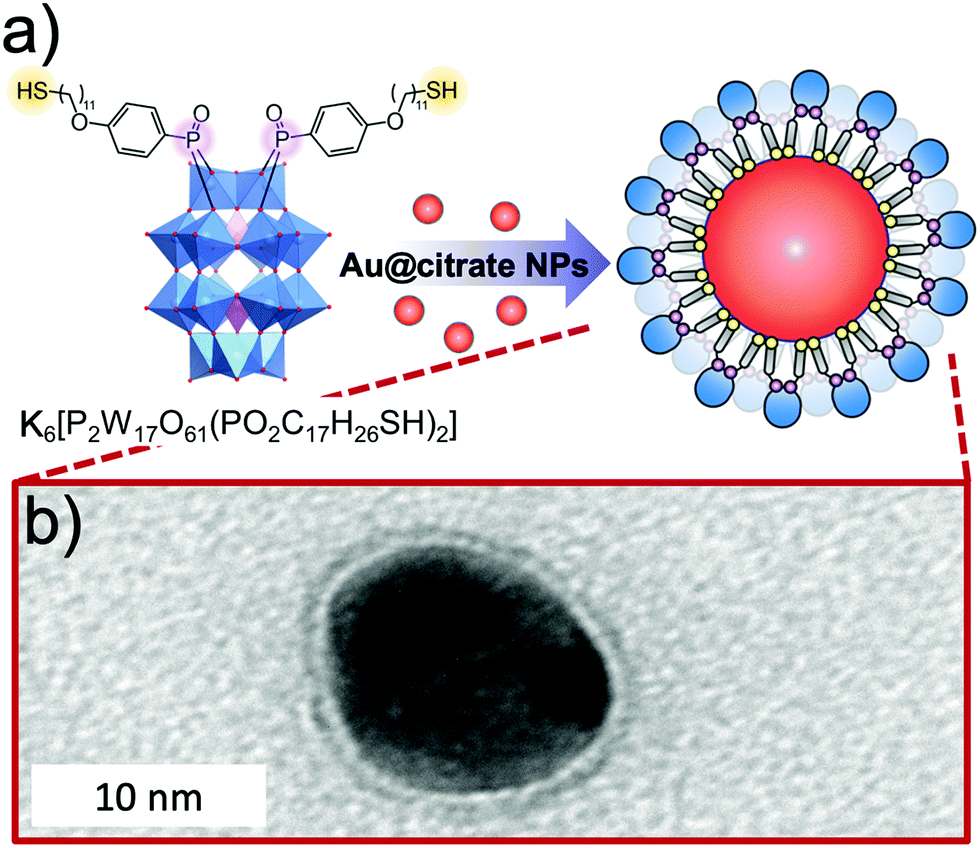 | ||
| Fig. 18 (a) Structure of the thiolated hybrid POM, K6[P2W17O61{PO2C17H26SH}2], showing its spontaneous exchange with citrate to produce covalently functionalised Au nanoparticles; (b) HR-TEM micrograph of a single Au@POM nanoparticle. This figure has been adapted from ref. 162 under the Creative Commons CC BY License. | ||
3.3 Nanostructured POM-based hybrid materials
As pristine POMs are often difficult to handle owing to their crystalline character, they often form inhomogeneous films with aggregated particles.164 Their processing can be achieved by counterion exchange which allows their immobilisation on surfaces,165–168 their embedding in polymers26 or the formation of membranes22 through self-assembly processes. Yet, as mentioned in the introduction the resulting soft materials rarely display internal nanostructuration owing to the isotropic character of pristine POMs, despite some notable exceptions.21,23–28 The controlled assembly of hybrid POMs through combination of electrostatic interactions between POM units and another specific interaction between the organic moieties is thus an appealing strategy to process POMs into nanostructured soft materials. Among them, mesomorphic materials (including liquid crystals) are appealing since these organised and oriented nanostructures display a liquid state allowing an efficient control of the thin film's morphology for further device fabrication steps.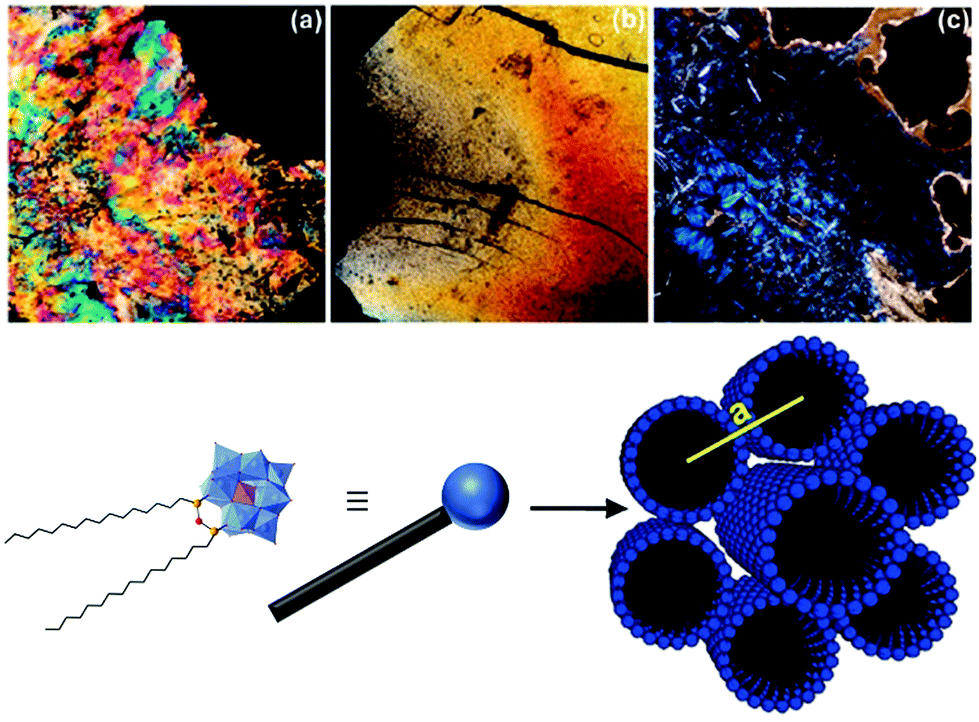 | ||
| Fig. 19 Top: Polarisation microscopy images of (a) Na3[PW11O39(SiC12H25)2O], (b) Na3[PW11O39(SiC16H33)2O] and (c) (TBA)3[PW11O39(SiC16H33)2O]. Bottom: Schematic representation of the [PW11O39(SiC16H33)2O]3− structure. This figure has been adapted from ref. 119 with permission from the American Chemical Society, Copyright 2010. | ||
With regards to the [PW11O39(SiC16)2](Na)3 liquid crystal the authors found it to self-assemble into hexagonally packed cylinders in the space group P6/mm. While the C18 analogue exhibited a similar structure the authors observed a hexagonal close packing (P6/mmc) of spherical micelles for the hybrid- POM derivative of shorter chain length. Furthermore, upon changing the respective counter ions from sodium cations to protons and organic ammonium cations the authors observed the formation of lamellar structures. More recently Polarz and co-workers also reported the synthesis of a hybrid POM with a π-conjugated tail based on the same organosilane POM-based platform employed in their previous study using para-phenylene ethynylene remote units.147 The authors determined a hexagonally ordered cylindrical structure for the liquid crystal phase in the space group P6/mm for the resulting lyotropic liquid crystals formed by the self-assembly of their compound. Moreover, the formed liquid crystals displayed semiconductor properties as shown by photosensor experiments. Finally, the authors also concluded that the decreased conformational freedom of the organic chains in the liquid crystal state enhanced the delocalisation of electrons compared to the micellar state of the hybrid POM compound in solution.
The synthesis of hybrid POM-based thermotropic liquid crystalline materials was recently pioneered by Song and co-workers.170 In their study the authors coupled gallic acid derivatives with long alkyl chains to an amino-functionalised Keggin-POM derivative resulting in a fan-shaped structure. The liquid crystalline materials were determined to assemble into layered smectic A phases. In the model structure, the authors proposed that the organic chains are located between POM layers. More recently, the first example of a photoactive donor–acceptor POM-based material was reported by Izzet, Heinrich, Mathevet and co-workers. In their work the authors coupled two moieties of a novel bis(thiophene)thienothiophene (BTTT) derivative to the bis-iodo aryl terminated Keggin POM-based hybrid platforms.171 The authors reported a multi-layered lamellar structure of the mesophase consisting in the alternation of POM/TBA counter ion double layers and double sub-layers of tilted BTTT mesogens, isolated from each other by a continuum of molten aliphatic chains (Fig. 20). Interestingly, the organisations of the POM and BTTT sub-units were found to be similar to that of BTTT and hybrid POM parent reference compounds. For instance, the branched terminal aliphatic chains impose high tilting of BTTT units in the hybrid as well as in the organic precursor and POM precursor and hybrid exhibit close quasi-hexagonal rectangular arrangements of POM and TBA. Finally, the authors were able to confirm the donor–acceptor properties of this material via photophysical studies making it a promising starting point for future investigations. The predictability of the individual sub-unit organisations in this system should facilitate the design of future generations of hybrids. Notably, using the same hybrid, the modification of the counter-ion should offer an effective way to tune the LC/conductive properties of POM-based mesogens.
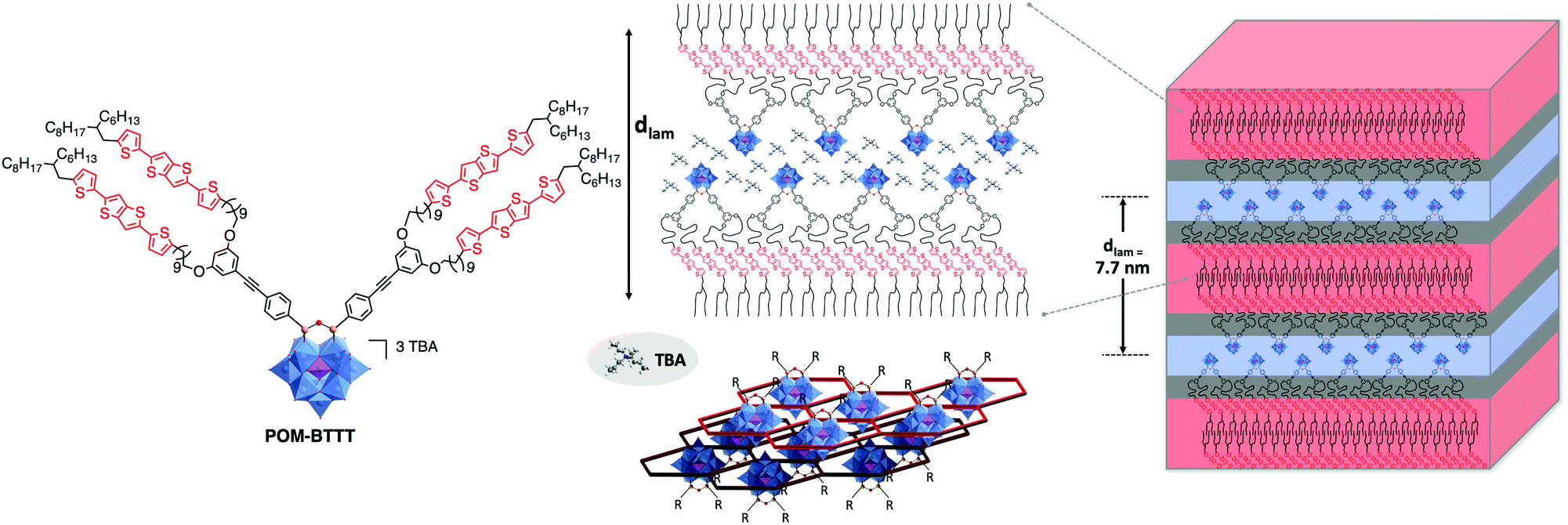 | ||
| Fig. 20 Left: Molecular structure of the branched POM–BTTT hybrid oxocluster; middle: a schematic view of multi-layered self-assembly of the lamellar hybrid material, highlighting the specific arrangement of the POM/TBA double-layers (bottom, centre: showing POM clusters in blue and the surrounding TBA honeycomb structure in red – note the laterally offset bottom layer is shown in darker colours); Right: 3D schematic representation of lamellar nanomaterial, respecting the molecular segment sizes and shapes. This figure has been adapted from ref. 171 with permission from John Wiley and Sons, copyright 2021. | ||
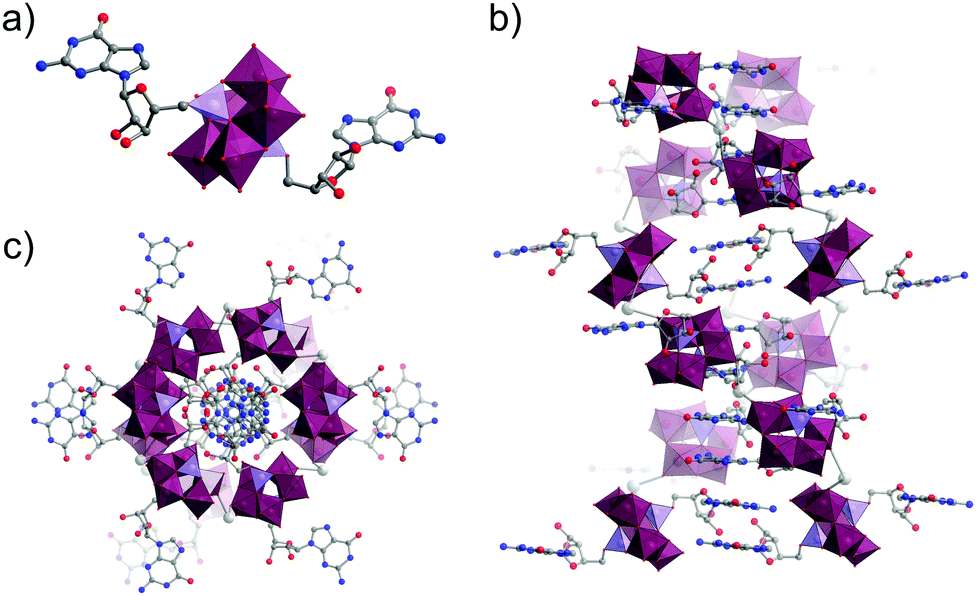 | ||
| Fig. 21 (a) Molecular structure of the guanosine-functionalised Strandberg-type hybrid oxocluster; (b) Stacking of clusters into a Z-DNA like double helix through a combination of hydrogen bonding and Na± mediated interactions (not shown here for clarity) as viewed along the crystallographic b-axis and; (c) viewed along the crystallographic c-axis. This figure has been adapted from ref. 172 under Creative Commons Licence CC-BY. | ||
Very recently Zheng, Streb, Chen and co-workers.173 reported the synthesis of novel hybrid POMs via functionalisation with organic boronic acids that can act as tritopic linkers. In their study the authors employed Wells–Dawson POMs of the general formula [M3P2W15O62]9− incorporating NbV and TaV ions respectively. The resulting hybrid POMs were shown to self-assemble in the solid state into nanocapsules with a diameter of 2.8 nm to 4 nm depending on the organic moiety of the boronic acid. These nanocapsules were characterised by single-crystal X-ray diffraction. In case of 3-pyridineboronic acid the authors observed tetrahedral aggregates in which four POM core units are linked by four organoboron moieties. When employing 5-pyrimidinylboronic acid the authors observed the formation of dodecameric nanocapsules based on trimeric subunits connected by K+ ions displaying an inner cavity of ca. 1.3 nm (Fig. 22). Certainly, this new functionalisation strategy could pave the way for a new class of hybrid POM architectures and applications.
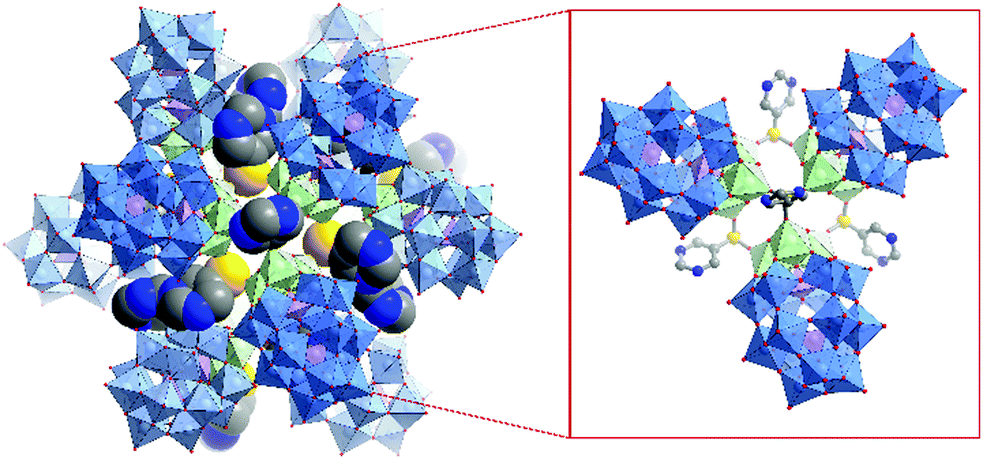 | ||
| Fig. 22 Structure of the supramolecular nanocapsule formed by the self-assembly of four trimeric {P2W15Ta3} units linked by boronic acid groups; inset: detailed view of the boronic acid (5-pyrimidinylboronic acid) mediated linkage of the ({P2W15Ta3})3 trimers. This figure has been adapted from ref. 173 under Creative Commons Licence CC-BY. | ||
Polymer–polyoxometalate hybrids are another class of compounds that may be considered of interest for the self-assembly of nanostructures. Most notably Wang and co-workers demonstrated the possibility to tune the self-assembly behaviour of nanostructures by varying the molecular weight of the hybrid's polymer component.174 For their studies they linked linear poly(ε-caprolactone) (PCL) of two different molecular weights to a Wells–Dawson type POM. The polymer–POM hybrid of low molecular weight self-assembled into nanotubes consisting of a hybrid membrane in which an amorphous PCL-layer is sandwiched between two layers of the POM. In contrast, the polymer–POM hybrid of high molecular weight self-assembled into hexagonal nanosheets. Besides single-layer nanosheets the authors also observed multilayer nanosheets composed of a stack of single-layer nanosheets. With regards to the structure of the observed hexagonal nanosheets, the authors propose a crystalline PCL layer, which is sandwiched by two single POM layers. With respect to the molecular weight dependence of the observed nano-objects, the authors argued that the rigidity of the crystalline PCL layer formed by the polymer POM hybrids with the higher molecular weight prevents the formation of nanotubes, which would be the preferred structure as it further minimises the free energy of the structure in question. In addition to the characterisation of these individual nano-objects the authors were also able to synthesise tube-graft-sheet nano-objects via stepwise self-assembly.
Another class of hybrid POMs that has shown a remarkable variety of nanostructures in recent years are polyoxometalate–silsesquioxane (POM–POSS) hybrids.175 These kind of heterocluster Janus molecules were pioneered by Wang and co-workers. In one of their early studies176 they used a Wells–Dawson-type POM ([P2W15V3O62]9−) tethered to an aminopropylisobutyl derivative of polyhedral oligomeric silsesquioxane via an organic linker and observed the self-assembly of hexagonal crystals with a filled honeycomb superstructure. In this case the POM moieties constitute the hexagonal honeycomb framework, while the POSS form close packed hexagonal cylinders within the honeycomb framework.
In a different study Liu, Yue, Cheng and co-workers177 investigated the influence of molecular topology, surface functionality of the POSS blocks and solvent polarity on the nature of the self-assembled nanostructures. In their work they used organosilyl and organotin derivatives of Keggin-type POMs in combination with isobutyl (BPOSS) or cyclohexyl (CPOSS) functionalised POSSs (Fig. 23). By employing a solvent mixture of chloroform/methanol the POM–BPOSS hybrid self-assembled into multiple-layered lamellae in which POM- and BPOSS-layers are stacked in an alternating fashion. However, by using a water/acetonitrile mixture first the formation of colloidal spheres with an internal lamellar structure was observed followed after 10 additional days by the formation of extended nanobelts with internal lamellar crystals. The authors propose a mechanism of nucleation of the first observed colloidal spheres and subsequent growth into the thermodynamically more favourable nanobelt structure. However, the authors concluded that in both cases the molecular packing in the crystal lattice is dominated by the POM subunits. Regarding the CPOSS analogue of the POM hybrid in question the authors observed the formation of nanosheets that are composed of two internal crystalline POM layers sandwiched between two amorphous CPOSS layers when using a chloroform/methanol mixture as the solvent for self-assembly. When employing the water/acetonitrile mixture for self-assembly the authors observed, again, the formation of spherical aggregates with a very similar internal lamellar structure to their BPOSS counterpart, however they did not observe any further structural evolution into nanobelts. The authors attributed this different behaviour in both cases to the non-crystalline nature of the CPOSS moieties. With regard to the POM–2BPOSS hybrid the authors investigated in their studies, they found that these hybrid molecules also formed nanosheets, however in this case the internal layer consists of BPOSS single crystals sandwiched between two POM layers. This structure was observed in solvents of different polarity. This was attributed to the increased ratio of BPOSS-moieties to the POM; the self-assembly is hence no longer governed by the POM, instead the crystallisation of the BPOSS cages is the main driving force for the formation of the nanosheets.
 | ||
| Fig. 23 Self-assembly of a series of molecular Janus particles constructed by chemically linking Keggin-type POMs with functionalised POSS cages. This figure has been adapted from ref. 177 with permission from the American Chemical Society, Copyright 2016. | ||
Wang and co-workers expanded the concept of tethering multiple POSS blocks to a single POM unit.175 The authors reported the self-assembly of mesoscale graphene-like honeycomb mono- and multilayers of a four-fold POSS-substituted Wells–Dawson type POM (4BPOSS–POM).178 Like for graphene the mesoscale honeycomb monolayers of the 4BPOSS–POM hybrid were found to form multilayers via different stacking modes. The self-assembly mechanism was investigated via coarse-grained molecular simulations in which the authors identified three key steps of the self-assembly: First, the formation of trimers driven by the counterion-mediated electrostatic interactions between the POM blocks, second, the assembly of six such trimers into cells and third, the aggregation of cells into the observed honeycomb structure. In a subsequent study Wang and co-workers also investigated the self-assembly of similar POM–POSS hybrid molecules into cubosomes.179 The authors observed the self-assembly of cubosomes with internal double diamond structure from an acetone/n-decane mixture (Fig. 24). They employed dynamic conditions in which the acetone was allowed to evaporate slowly over time, decreasing the solubility of the hybrid molecules and facilitating aggregation. Based on their observations of intermediates the authors proposed a mechanism for the self-assembly of cubosomes with an internal double diamond structure. According to their findings the formation of cubosomes takes place via a four-step process. The self-assembly of vesicles with a dual bilayer in which a POM double layer is sandwiched between two POSS monolayers occurs first. Subsequently the so formed vesicles accumulate into larger aggregates. Topological reorganisation takes place within larger aggregates, starting from the centre until the double diamond structure occupies the entire interior of the aggregates forming spherical cubosomes. Further growth occurs on the facets of the cubosome via crystal nucleation and growth.
 | ||
| Fig. 24 Formation processes and mechanism of cubosomes. (A) TEM images demonstrating the size-dependent shapes and inner nanostructures of cubosomes growing under the dynamic conditions. (A-1–A-7) A vesicle, a hemi-fused vesicle, particles with a foam-like structure, an aggregate with a sponge-like nanostructure, an ordered nanostructure in the centre of the aggregate (highlighted by the yellow circle), a spherical cubosome, and faceted cubosomes. (B) Evolution from molecules to faceted cubosomes through different mechanisms. This figure has been adapted from ref. 179 with permission from the American Chemical Society, Copyright 2019. | ||
4. Combination of hybrid POMs with substrates through non-covalent interactions
In addition to their ability to act as discrete molecular building blocks in the preparation of larger molecular and supramolecular nanosystems, hybrid POMs have also recently been explored as functional components in combination with a range of other species capable of acting as either hosts or guests in relation to the POM. As in other well-established host–guest systems, this relies strongly on non-covalent interactions though, in this instance, these are usually more heavily directional and/or specific than in many of the other systems described above. Exploiting these host–guest interactions offers broad scope for the careful design of new supramolecular nanostructures and nanomaterials, which can display unique, dynamic responses (e.g. self-healing, switchable properties, chiral recognition or transfer). Hybrid POMs are particularly well-suited here, as their near-limitless scope for molecular design can be used to judiciously control both the self-assembly and physicochemical properties of the resulting complex. This is not necessarily trivial or, indeed, predictable however it does present enormous potential for novel supramolecular design strategies. The following section thus describes recent examples of the formation of more complex supramolecular systems, comprising a hybrid POM in combination with one or more additional components whereby the attractive properties of each component combine to present new functionalities.4.1 Hybrid POM complexes with cavitands
The combination of both all-inorganic and hybrid POMs with macrocyclic hosts is a relatively recent area of interest, with the vast majority of studies reported only in the last decade. The most common cavitands – cyclodextrins, calixarenes, curcubiturils and pillar-arenes – have been widely studied for their remarkable inclusion properties,180–183 which typically rely on their hydrophobic inner cavities. Owing to their chaotropic character, classical inorganic POMs (such as the Lindqvist, Keggin or Wells–Dawson anions) can readily form inclusion (or partial-inclusion) complexes with suitable cyclodextrins (CDs). This is largely thanks to the complementarity of their molecular structures and is driven predominantly by favourable steric-fitting between the host and the guest oxocluster, and some hydrogen-bonding interactions.21,86,184 Such inclusion complexes are stable in both solution and the solid-state and have recently been studied for their phase-transfer catalytic properties,185 and as a novel tool for probing the self-assembly behaviours of small POMs by mass spectrometry.186,187 That said, the formation of stable host–guest complexes between conventional POMs and cavitands (particularly curcubiturils and pillar-arenes, which do not share the excellent complementarity found in POM⊂CD systems) remains the exception rather than the rule, and in many cases, POM@cavitand complexes form crystalline networks in which the POM remains external to the macrocyclic cavity and often requires additional functionalisation of the macrocycle (e.g. appending cationic groups to electrostatically associate with the POM).188–190 The development of hybrid POM@cavitand inclusion complexes therefore presents several potential advantages, allowing far greater synthetic control and access to a broad range of prospective new properties through the engineering of dynamic or stimuli-responsive host–guest interactions. This was exemplified by the pioneering 2012 study of Izzet, Ménand, Sollogoub, Proust and co-workers, who demonstrated the first example of hybrid POM@cavitand complexes between an organotin-functionalised Wells–Dawson and both α- and β-CDs.191 Here, the iodoaryl moiety which has been anchored to the POM via the iodoaryl unit is found to be either partially (α-CD) or fully (β-CD) included within the host, forming a stable 1:1 complex which could be characterised in both the solution- and solid-state using a combination of 1H NMR and single crystal X-ray diffraction studies. The binding constants were similar to that of iodobenzene with α-CD and β-CD, indicating the absence of specific interaction between the POM and the CD (Fig. 25).192 Interestingly, in the presence of γ-CD the complexation of the hybrid turned out to be more complicated,193 probably owing the possible binding of the γ-CD to the iodoaryl moiety and to the sidewall of the POM, as further observed with pristine Wells–Dawson POMs.21 The authors also demonstrated a remarkable “self-healing” effect in the POM:CD complexes. When exposed to base, the organotin moiety is hydrolytically cleaved from the monolacunary POM (as is common to many hybrid POM species). This process is partially reversible on neutralisation of the solution, however after 4 cycles of hydrolysis and re-condensation of the organic group, only 20% of the original hybrid complex is found to remain, with most being lost as insoluble stannous salts and plenary (and thus unreactive) Wells–Dawson oxocluster, [P2W18O62]6−. Conversely, after four cycles of hydrolysis/neutralisation, POM@α–CD and POM@β–CD inclusion complexes are found to be more than 95% and 99% intact respectively, thanks to the protective influence of the cavitand, which prevents the organotin species from oligomerisation or disproportionation when exposed to base.
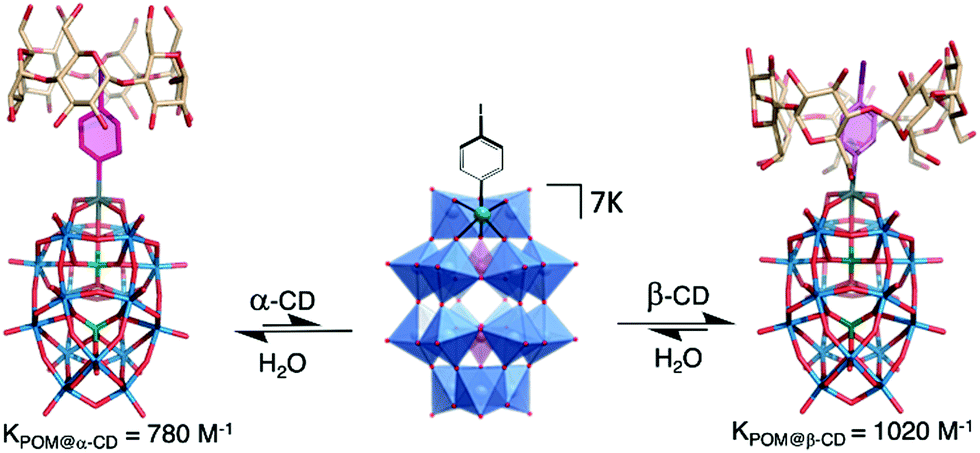 | ||
| Fig. 25 Formation of the POM@α-CD (left) and POM@β-CD (right) inclusion complexes from an iodo-aryl functionalised Wells–Dawson hybrid (note: aryl group is highlighted pink in wireframe models for clarity). This figure has been adapted from ref. 191 with permission from John Wiley and Sons, copyright 2012. | ||
Further evidence for the potential to engineer unique supramolecular materials using hybrid POM design strategies was provided shortly thereafter by Wu and co-workers, who developed a switchable material based on the combination of azobenzene functionalised Anderson oxoclusters and a pyridinium-functionalised β-CD host (Fig. 26).194 Exploiting the well-known photo-switching properties of the azobenzene–CD insertion complex,195 alongside the electrostatic interaction of the cation-functionalised CD with the polyoxoanions, the authors successfully demonstrated the self-assembly of a new chiral, photo-switchable supramolecular network. In this case, the hybrid POM in the trans-conformation forms a 1:2 inclusion complex with the CD host, allowing chiral-transfer from the intrinsically chiral β-CD molecules to the azobenzene units, which can then propagate throughout the whole material. This leads the trans-POM@CD complex to self-assemble into remarkable right-handed twisted lamellar structures as observed by SEM. When irradiated with UV-light, cis-isomerisation of the azobenzene units results in disassembly of the host–guest complex, resulting in a loss of chirality (as confirmed by circular dichroism spectroscopy (CDS)) and solubilisation of the merely electrostatically bound POM@CD complex which loses its unique morphology. The authors later built on this study by developing a new complex using a similar approach.196 In this case, they designed a new single-sided, asymmetric Anderson hybrid in order to construct a three-component supramolecular material which included a cationic methylene blue (MB) dye alongside the azobenzene functionalised hybrid POM@α-CD inclusion complex. This allowed the authors to directly study chiral transfer from the CD host to the electrostatically bound dye molecules, using the hybrid POM as a bridging unit. By observing formation of an induced Cotton signal from the MB chromophores, CDS was used to confirm that formation of the POM inclusion complex is vital to transfer chirality from the hydrophobic cavity of the α-CD unit to the MB dye, which does not form an inclusion complex in the absence of the azobenzene-functionalised Anderson oxocluster.
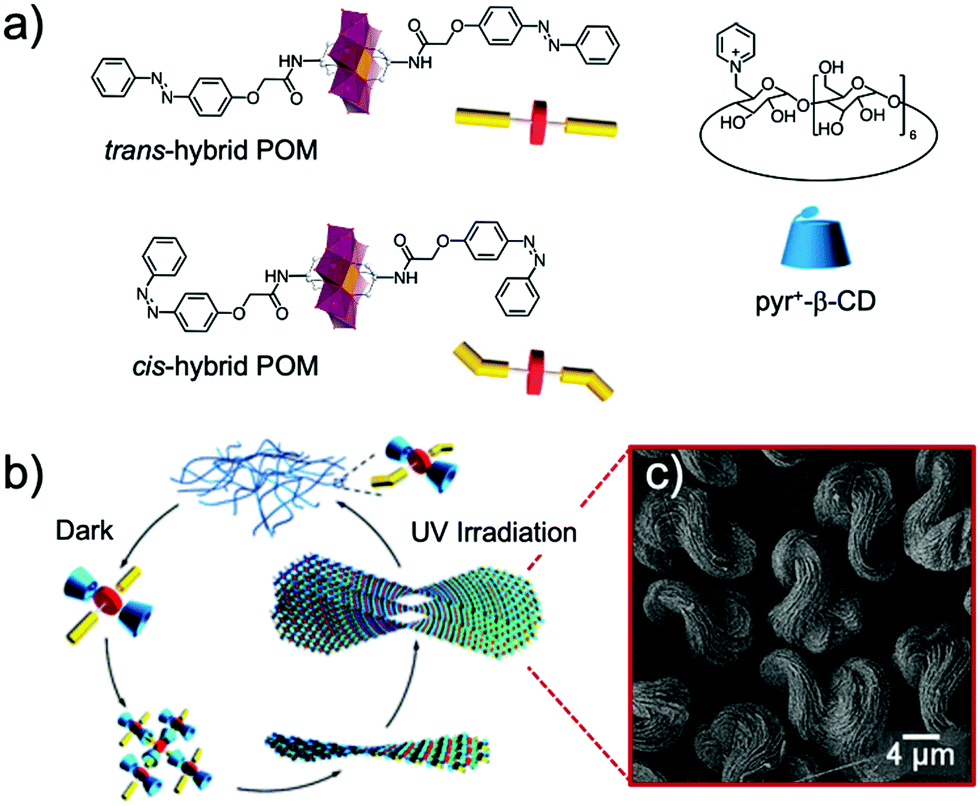 | ||
| Fig. 26 (a) Molecular structures and simplified representations of the trans- and cis-isomers of an azobenzene functionalised Anderson-type hybrid-POM anion and a pyridinium-functionalised β-CD cation; (b) schematic showing the self-assembly of the trans-isomer of the hybrid-POM:β-CD complex into nanosized, chiral aggregates upon inclusion of the trans-azobenzene groups into the β-CD cavity and its subsequent disassembly into irregular nanofibres under UV-irradiation, driven by the photo-isomerisation of the azobenzene unit, and: (c) an SEM image of the right-handed chiral nanostructures formed by the extended hybrid-POM:CD inclusion complex. This figure has been adapted from ref. 194 with permission from the Royal Society of Chemistry, copyright 2013. | ||
The same group also recently reported a series of dynamic, photo-switchable materials based on the self-assembly of an organotin-functionalised Keggin POM containing a remote azobenzene moity.140 In aqueous solution, this hybrid spontaneously self-assembles into vesicles thanks to the amphiphilic nature of the hybrid oxocluster (see Section 3.2.3). When mixed with equimolar quantities of either α-CD or β-CD, an induced Cotton signal appears in the CDS corresponding to the azobenzene groups, indicative of chiral transfer from the internal cavity of the cavitand to the POM bound azobenzene moieties and dynamic light scattering measurements indicate rapid dissociation of the vesicles (hydrodynamic diameter of ca. 60 nm) to the monodispersed state which has an average hydrodynamic diameter of ca. 3.6 nm, closely matching the expected size of the POM@CD inclusion complex. Photo-switching of the azobenzene moieties with 365 nm light leads to the dissociation of the POM@CD inclusion complex, which reforms the self-assembled spherical particles, this phenomenon being entirely reversible. Finally, competitive binding studies with adamantane-1-carboxylic acid also allow control of the self-assembly behaviour of the hybrid POM, selectively binding with CD over the azobenzene–POM hybrid and thus favouring assembly of vesicles over the monodisperse POM@CD complex. In combination with the authors previous work, this study shows the impressive potential of this tailored building block approach in the design of novel supramolecular materials with new and remarkable features arising as a result of the precise control of the host–guest interaction between the POM and the cavitand host molecule.
The dynamic properties of host–guest systems also have significant implications for the design of new smart materials, and the direct integration of hybrid POM@CD inclusion complexes into polymers has also recently been explored. Song and co-workers demonstrated the preparation of new host–guest hydrogels, combining adamantane-functionalised Anderson-oxoclusters with an acrylamide functionalised β-CD monomer species.197 Radical copolymerisation with additional acrylamide monomer forms a new supramolecular polymer, yielding a series of hydrogels with varying amounts of incorporated POM. Rheological analysis found that increasing concentrations of POM, which acts here as a supramolecular cross-linking species, leads to materials with higher elastic modulus due to an increased network density. Wu and co-workers have also recently utilised the combination of an anthracene-functionalised Anderson POM with γ-CD to facilitate the templated polymerisation of the hybrid POM units into linear chain polymers using the photo-dimerisation of the anthracene groups to form new C–C bonds between the oxoclusters (Fig. 27).198 Here, the authors report two distinct new materials: a supramolecular polyrotaxane-type complex in which the anthracene units stack within the γ-CD cavity to form uniform and parallel linear strands of alternating POM and CD; and a covalent polymer in which the photo-dimerised anthracene reacts within the cavity of the γ-CD cavity to form highly linear polymer chains. Crucially, however, in the absence of the templating effect of the γ-CD unit, the hybrid POM complex forms globular assemblies, indicating the importance of the supramolecular templating effect provided by the host–guest interaction between the POM and the CD cavitand.
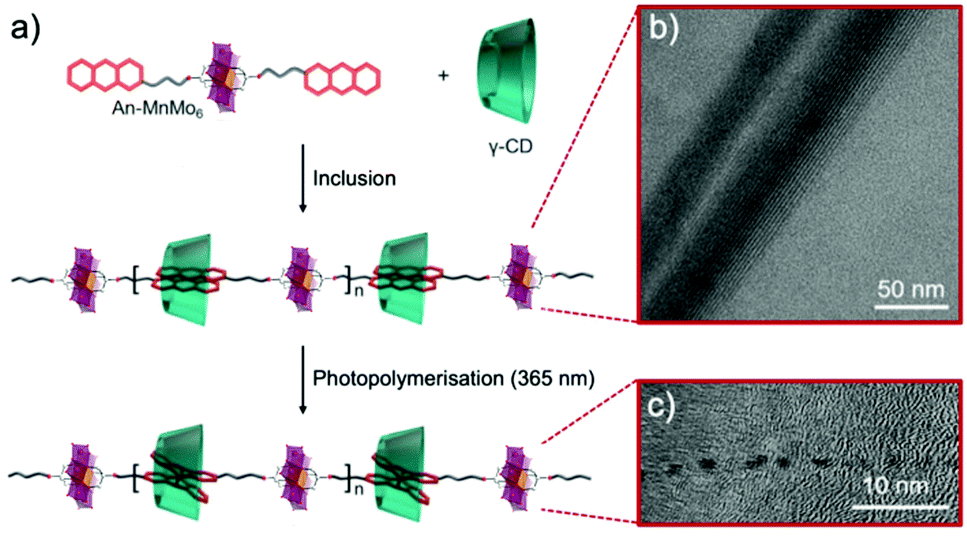 | ||
| Fig. 27 (a) Schematic representations of the anthracene functionalised Anderson-type hybrid POM and γ-cyclodextrin, showing their self-assembly into a supramolecular polyrotaxane which can then be photopolymerised into a covalent linear polymer chain; (b) TEM imaging of the supramolecular polyrotaxane, showing a bundle of highly linear adjacent polymer strands, and; (c) TEM imaging of an isolated single-chain of the covalent polymer (dark spots show individual POM anions arrayed along the polymer backbone). This figure has been adapted from ref. 198 with permission from the Royal Society of Chemistry, copyright 2019. | ||
The effect of the host–guest interaction between hybrid POMs and suitable cavitands can also play a more subtle role in helping to moderate functionality. A clear example of this was reported recently by Kögerler and co-workers, who prepared a 1:2 POM:α-CD inclusion complex using a phenyl-arsonate functionalised [MIIO8Pd12(C6H5AsO3)8]6− (where M = Co, Ni, Zn) polyoxopalladate.199 In this complex, the formation of the inclusion complex between the terminal phenyl-groups on the polyoxopalladate and the α-CD units was found to lead to a small but significant distortion to the cubic ligand-field of the centrally bound metal ion owing to the “chemical pressure” applied to the POM in the solid state through the formation of an inclusion complex. In the case of the Co analogue particularly, this structural distortion was significant enough to generate a slight single-ion magnetic anisotropy, which the authors studied via EPR, SQUID magnetometry and X-ray absorption techniques.
4.2 Hybrid POM complexes with carbon nanotubes
The rich, stable and highly tuneable multi-redox activity of POMs is one of the key properties driving interest in their use in the next-generation of energy and electronics technologies. As the primary limiting factors in their wider use relate mainly to their low surface area and lack of electrical conductivity in the solid state, a great deal of research effort has been invested in recent years into strategies to overcome these specific challenges.18,200 Of particular interest has been the combination of POMs with various high surface area carbons, and especially low dimensionality nanostructures such as carbon nanotubes (CNTs), which have the potential for very high surface areas and electrical conductivity. Similar to the host–guest complexes discussed above, this field is still very much in its infancy though a number of different approaches have already emerged for the preparation of new POM@CNT composite materials. Promising results were reported very recently for example by Walsh, Khlobystov, Newton and co-workers, who demonstrated the direct encapsulation of [P2W18O62]6− within single-walled carbon nanotubes (SWCNTs), which formed new electroactive composites with high POM loadings and notable chemical and electrochemical stability.201 Far more commonly, POM@CNT composites have typically been formed by deposition of various POMs onto the exterior of the nanotube, either by aggregation/crystallisation of the oxocluster directly onto the CNT,202,203 or by surface functionalisation of the carbon surface – either using direct chemical approaches or through combination with a modifier such as a cationic polymer – in order to facilitate stronger covalent or electrostatic interactions between the POM and the CNT.168,204 Examples of the covalent coupling of hybrid POMs to CNTs via a peptide bond formation and their study as promising anode materials for Li-ion batteries were also reported.205,206 Several challenges remain in each case however, the most notable of which being that direct modification of the sp2 carbon framework can often have a negative effect on the optical and/or conductive properties of the CNT. Moreover, electrostatic association of the POM and CNT is highly susceptible to unwanted ion exchange reactions and can lead to issues centred around the long-term stability of the composite material.The potential for carefully designed hybrid POMs to selectively and non-covalently interact with CNTs is therefore a promising route to develop stable, tuneable nanocomposites, which retain the attractive properties of both the POM and CNT components. This was first demonstrated by Song and co-workers in 2013, who used a non-covalent sidewall functionalisation approach in order to couple a pyrene-appended Keggin POM to SWCNTs by exploiting the strong π–π stacking interaction between the ligand and nanotube surface.207 The new hybrid nanomaterial was characterised by high-resolution TEM (HR-TEM) imaging, which are decorated with discrete particles of ca. 1–1.5 nm diameter, corresponding well to the hybrid POM. Both Raman and fluorescence spectroscopy are also demonstrated to be useful tools for the characterisation of these supramolecular composites, particularly as association of the POM with the nanotube leads to a strong and characteristic quenching of the pyrene emission. The authors also studied this material as a novel Li-ion electrode material and found that it displays a high discharge capacity (580 mA h g−1 at a current density of 0.5 mA cm−2), which is retained over at least 100 charge–discharge cycles with close to 100% coulombic efficiency. Notably, this is significantly better than the performance of the hybrid POM when used as an electrode material alone (i.e. in the absence of CNTs). This study was followed in 2015 by Song, Streb and co-workers, who prepared a similar composite material using a pyrene-functionalised Mn-Anderson POM hybrid combined with multi-walled carbon nanotubes.208 In this case, the composite was employed as an anode material in a prototypical Li-ion cell, which displayed improved performance with a retained capacity of 665 mA h g−1 (at 0.5 mA cm−2) after 100 cycles. More detailed electrochemical analysis via AC electrochemical impedance studies revealed that the supramolecular POM/CNT composite also showed higher performance for both Li+ ion transport and electrical conductivity than either the CNTs alone or a physical mixture of both POM and CNT components, helping to explain its superior electrochemical properties as a potential Li-ion anode material.
Proust, Guldi, Marti-Gastaldo and Coronado and co-workers also explored the use of π–π interactions to immobilise an organosilane derivative of Wells–Dawson POM onto SWCNTs in order to study the photophysical properties of POM/CNT composites (Fig. 28).209 Whilst no obvious ground-state electronic communication between the POMs and CNTs was discernible, fluorescence spectra of the POM/CNT composites reveals both red-shifting and a strong quenching effect on the emission maxima associated with the nanotubes. This was rationalised as being due to an unusual photoinduced charge transfer interaction between the SWCNTs and the POM, as observed by transient absorption spectroscopy, resulting from the transient oxidation of the nanotube and concomitant reduction of the POM (unusual in this case because both SWCNTs and POMs are typically employed as electron acceptors). Given the stable and well-established multi-redox activity of many hybrid POM species, the authors note the potential of such composites in the design of new advanced photochemical dyads.
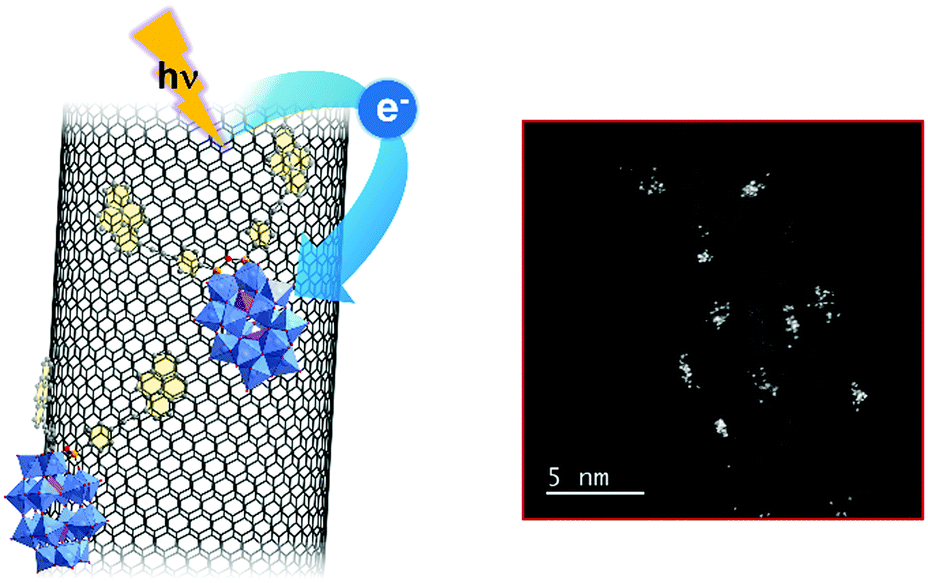 | ||
| Fig. 28 (a) Non-covalent functionalisation of single-walled carbon nanotubes with a pyrene-functionalised Wells–Dawson oxocluster, highlighting the transient photo-driven charge transfer from the SWCNT to the appended POM complex, and: (b) HAADF-STEM imaging of the resultant nanocomposite (bright spots correspond to individual tungsten atoms in each POM). This figure has been adapted from ref. 209 with permission from the Royal Society of Chemistry, copyright 2014. | ||
Aside from the electrochemical and photochemical energy applications described above, the potential for the design of hybrid POMs as a means to selectively disperse, solubilise and enrich CNTs has been studied recently by Prato, Bonchio and co-workers. The authors first explored the interaction of a bis-pyrene organosilane derivative of [γ-SiW10O36]8− with both C60 and C70 fullerenes, and with SWCNTs, in order to yield a range of new supramolecular nanostructures.210 In the case of the fullerenes, both 0-D globular nano-aggregates and 2-D Langmuir films were prepared, with the hybrid POMs found to have a ten-times higher binding affinity for the larger fullerene due to the larger π-surface of the C70 guest. The hybrid POM was also combined with commercially available SWCNTs and analysis of the Raman spectra, and in particular the ‘radial breathing mode’ (RBM) of the nanotubes (which can be associated with SWCNTs of different diameters/chiralities), was used to observe a preferential interaction of the bis-pyrene POM hybrid with larger diameter nanotubes. Crucially, when the supramolecular composite was subjected to thorough washing with acid and base, the hybrid POM could be removed to regenerate the bare nanotube, indicating that this strategy may have some promise for CNT separation and processing.
More recently, the authors also explored the use of a pair of bis-tryptophan functionalised [γ-SiW10O36]8− enantiomers in comparison to the previously reported pyrene-functionalised analogue.211 In this study, each of the three hybrid POM molecular ‘tweezers’ was combined with a commercially available mixture of SWCNTs and the interaction between each POM ‘host’ and the nanotube guests was characterised using a combination of UV-vis, fluorescence and Raman spectroscopies, alongside a computational analysis of the possible binding modes. As before, the normalised RBM intensities in the Raman spectra are highly diagnostic of the nature of the interaction of the POM with the CNT and find that the smaller, more polar tryptophan binding pocket favours enrichment of nanotubes with diameters below 1.1 nm, whilst the larger, more hydrophobic bis-pyrene binding pocket enriches nanotubes with diameters between 1.1–1.2 nm. As such, the results of this study are proposed to offer useful information with regards to engineering possible SWCNT/peptide interactions for use in drug delivery and future CNS-based therapies. Interestingly, a 2014 study by Song and co-workers can be related to this concept as it also explores the use of a bis-pyrene functionalised hybrid POM to facilitate the interaction of SWCNTs with human serum albumin protein.212 In this work, the authors use high-resolution electron microscopy alongside XPS to demonstrate that the prior non-covalent functionalisation of the CNT with the hybrid POM greatly improves the nanotubes absorption capacity for human serum albumin as compared to either of the individual components. As such, this strategy may have interesting implications for the development of future biosensors or lab-on-a-chip type applications.
4.3 Hybrid POM complexes with biological structures
The interactions of POMs with biomolecules and their corresponding biological activity has been a target of study for many years.213–216 Their high charge and strong hydrogen bonding character allows POMs to interact in a variety of interesting ways with biological substrates such as proteins, lipids and nucleic acids, whilst the extraordinary tuneability of POM structure and composition provides near limitless opportunities for researchers to tailor these properties.Perhaps the most striking recent example of the application of POM–biomolecule interactions can be found in the award of the 2009 Nobel Prise in Chemistry to Ada Yonath, who was awarded a share of the prise for her seminal work in protein crystallography and uncovering the structure of the ribosome. In part, this work exploited the interaction of a simple (NH4)6[P2W18O62] Wells–Dawson oxocluster with specific sites in the ribosomal subunits of a model organism, Thermus Thermophilus, to dramatically extend the resolution of the diffraction map from ca. 7–9 Å to 3 Å.217 In recent years, the interaction of POMs with biological systems has expanded to encompass a range of different interests, including their exploration as protein crystallisation agents,214,218 artificial enzyme mimics,215 and metallodrugs.216
The potential for hybrid POMs in this area is also obvious. The ability to graft almost any organic functionality onto an inorganic POM with real synthetic control could, in some cases, represent a ‘best-of-both-worlds’ approach, where the functionality of the POM (be it as an imaging tool, a metallodrug, an enzyme mimic, etc.) could be selectively introduced to the intended substrate in a site-specific fashion by carefully controlling its specific binding interactions via the appended organic side-chains. In addition, the enhanced stability of covalently bound hybrid POMs could be a real advantage in elucidating mechanistic details of the mode of action of POMs in biological systems such as drug assays, where the relative ease with which the electrostatically-bound hybrids (where the organic functionality is appended to the POM as a charge balancing cation) can dissociate under biologically relevant conditions may make identifying the true active species all the more challenging.219,220 As such, it is worth noting that while this section will attempt to serve as a brief overview of this growing field, it is difficult to fully encompass all research activities within the particular scope of this review and also that the exact nature of the non-covalent or supramolecular interactions which predominate in POM–biomolecule interactions is often, understandably, not as clear-cut as in many of the examples reported above.
As discussed above, one of the clearest success stories in this field in recent years is the application of POMs as crystallisation agents in protein crystallography. The interaction of POMs with proteins often serves two purposes: solving the “phase problem”, and helping to stabilise the folded protein structure, aiding both crystallisation and improving the resolution of the crystal structure. In recent years, much of this work has been pioneered by Rompel and co-workers, who have studied the use of the tellurium-centred Anderson oxocluster [TeMo6O24]6− to resolve the previously unknown structures of a mushroom tyrosinase and aurone synthase.221,222 Though most research in this area has focused exclusively on pristine POMs, the same group also recently explored the potential of a series of hybrid Anderson oxoclusters to be used as additives in the crystallisation of human and bovine serum albumin proteins (Fig. 29). In these studies, the authors prepared a series of Anderson oxoclusters with a range of both mono- and bis-substituted hydrophilic ligands (e.g. –NH3+ or –OH terminated tris-bases),223 and bis-substituted hydrophobic ligands (phenyl- or cinnamic acid-terminated groups),224 and studied their stability under biologically relevant aqueous conditions in the presence of the relevant protein. In both cases, the oxoclusters showed excellent stability in a pH range of 4 to 9 and, critically, showed no hydrolytic reactivity towards the serum albumin proteins. The more hydrophilic species all showed clear interaction with the surface of BSA, leading to a charge inversion from positive to negative zeta potentials, indicating their broad suitability as additives. In the more hydrophobic hybrid POMs, fluorescence quenching measurements were employed to study the specific binding of the hybrid with HSA and from this the authors were able to propose a 1:1 interaction between the protein and the POM, identifying a specific binding interaction on a particular subdomain site in the protein structure. Though it is important to recognise that the use of hybrid POMs as macromolecular crystallisation agents is still at a very early stage, it is worth noting the authors’ own perspective (expressed in a recent review)225 that hybrid POMs may be particularly useful as additives in the notoriously challenging crystallisation of membrane proteins. This is because these proteins are typically highly hydrophobic, precluding the use of pristine POM additives, which bind to proteins mainly through polar interactions. As exemplified in Section 3.2, there are already numerous examples of surfactant-like hybrid POM amphiphiles, which may be interesting to explore for this application in the future.
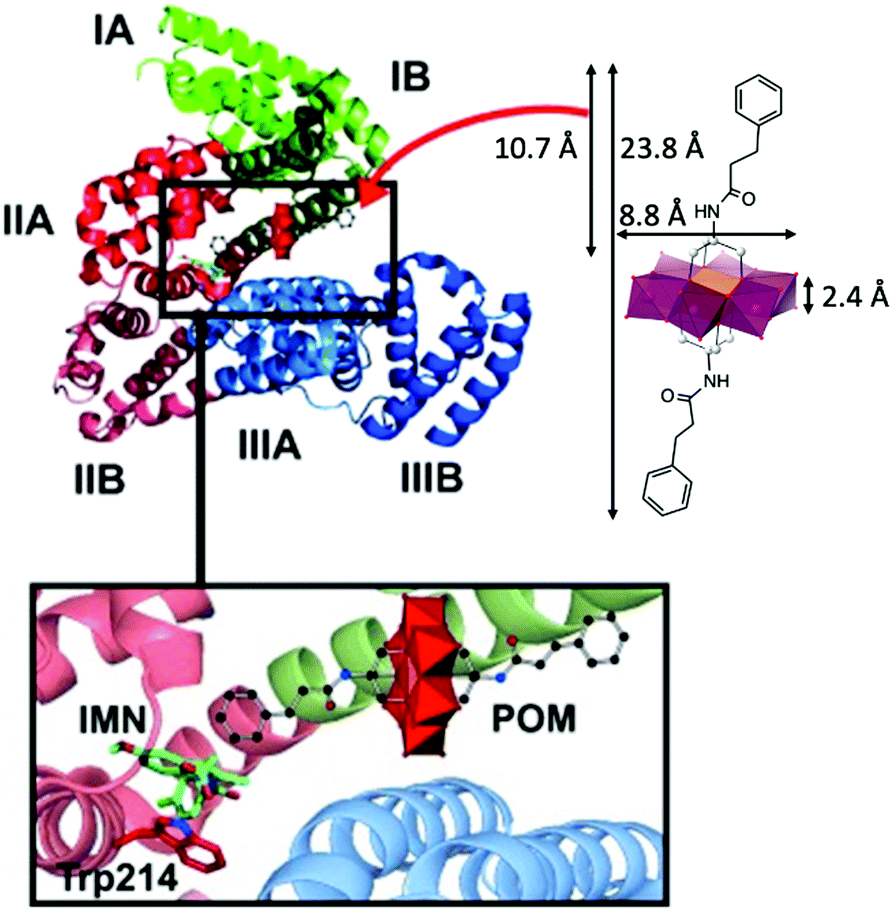 | ||
| Fig. 29 Representation of the hypothetical binding site of a benzene-functionalised Anderson-type hybrid POM in Human Serum Albumin protein (shown as a ribbon structure with each subdomain depicted in a different colour). Shown inset is a closer view of the position of the hybrid POM within the HSA binding site alongside indomethacin (IMN) and tryptophan (Trp214) groups which are used to confirm binding of the POM through fluorescence studies. This figure has been adapted from ref. 224 with permission from John Wiley and Sons, copyright 2015. | ||
Alongside these crystallisation studies, several other examples of interactivity between hybrid POMs and peptide or protein units have also been discussed recently in the literature. Recently, Bonchio and Carraro demonstrated the interaction of a bis-biotinylated [γ-SiW10O36]8− POM with the tetrameric avidin protein, exploiting the well-known host–guest chemistry of the biotin–avidin complex.226 This study is interesting in that it clearly shows a dual-mode interaction between the hybrid-POM and the protein, where both weak and non-specific electrostatic association, and strong and highly site-specific host–guest binding can be determined by a combination of fluorimetry, CDS, UV-vis and surface-plasmon resonance studies.
In addition to these studies, the intra-molecular association of peptide chains when bound to POMs has also been explored. For instance, Cronin et al. demonstrated the use of a bis-functionalised Anderson oxocluster as an “inorganic amino acid” in the solid-supported preparation of a range of oligopeptide chains.227 When used to prepare a symmetrical peptide sequence in which the POM is grafted to two identical 15-mer chains, an interesting structural feature was observed by CDS, whereby the 15-mer itself showed no obvious chirality however the new hybrid POM shows clear features characteristic of α-helix formation. Although the authors were unable to probe this feature further due to the limited solubility of the hybrid oligopeptide conjugate, it does suggest an interesting interplay between the POM and the appended biomolecules which leads to the formation of some secondary structure. A more recent study by Dumont, Lelli and Lacôte has explored the intra-molecular interactions between POMs and peptides in more detail.228 In this work, the authors prepared a family of hybrid Wells Dawson oxoclusters to which a series of short polyglycine chains are appended and then used a combination of spectroscopic characterisation and molecular dynamics simulations to study the conformation of the peptide and its specific interaction with the surface of the POM. This found that the polyglycine chains fold towards the POM surface, forming “zipper-like” hydrogen bond networks where the specific structure is determined by the topology of the POM (the authors used different POM isomers), and that hydrogen-bonding interactions between the POM and the peptide chain serve to rigidify the normally conformationally flexible peptide, even when longer chain structures are used.
The interaction of hybrid POMs with oligonucleotides/DNA has also been explored recently by Hasenknopf, O’Sullivan and co-workers as a strategy for the electrochemical detection of polymerase chain reaction (PCR) products. The authors prepared organotin-functionalised Wells–Dawson and Keggin oxoclusters and conjugated them with a 5′-NH2 terminated 21-mer oligonucleotide primer using an amide-coupling approach.229 Though the small quantities of product are challenging for typical POM characterisation, the new conjugate was successfully analysed using a combination of electrospray ionisation mass spectrometry (ESI-MS) and vibrational spectroscopies. The resulting conjugates were then used in the PCR amplification of a sequence of Francisella tularensis and the resulting POM-bearing single-stranded DNA (ssDNA) was electrochemically characterised by hybridisation with a complimentary sequence appended to a gold electrode surface (Fig. 30). All hybrid POM/DNA conjugates were found to have detection limits in the sub-10 nM range, with the hybrid Wells–Dawson found to have the higher sensitivity thanks to its bulkier structure facilitating easier electron transfer. The authors then built on this study using a modified approach, coupling a 7-deaza-7-propargylamino purine base directly to the organotin-functionalised Keggin and Wells–Dawson POMs in order to facilitate direct incorporation of the redox tagged purine into the PCR product for improved electrochemical detection.230 Most recently, the authors expanded the latter approach to explore the multiplexed detection of single nucleotide polymorphisms on a microfluidic electrode array.231
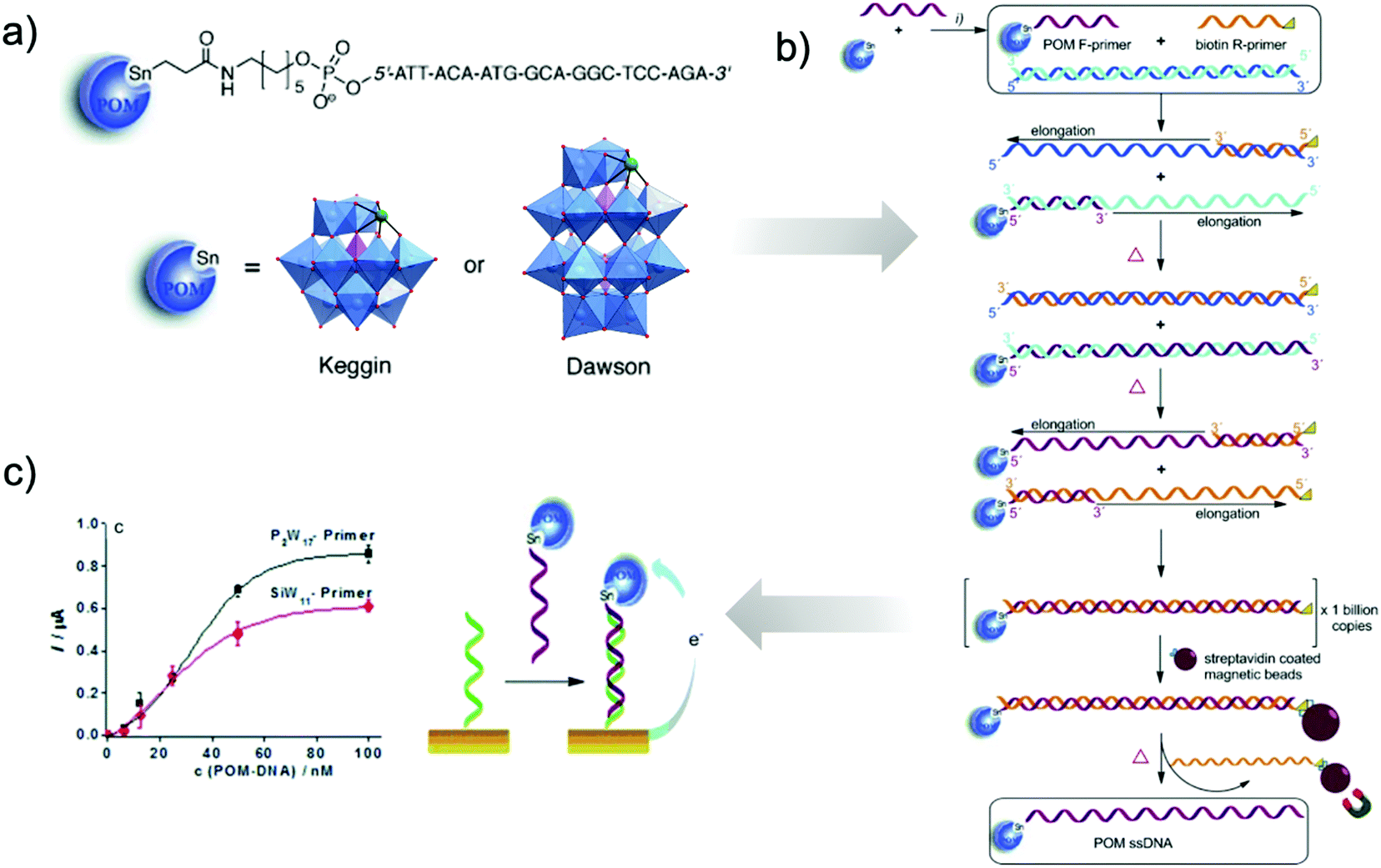 | ||
| Fig. 30 Schematic of: (a) the bio-functionalisation of either Keggin- or Wells–Dawson-type POMs with an oligonucleotide for use as a forward-primer (F-primer) in PCR amplification (b) and the subsequent electrochemical detection of POM-labelled PCR products (c). This figure has been adapted from ref. 229 with permission from John Wiley and Sons, copyright 2015. | ||
Hybrid POMs have also been explored in combination with a range of live cell types (e.g. viruses, bacteria and/or mammalian) and are of obvious specific interest in the design and targeting of these species as novel metallodrugs in particular (vide infra). As might be expected, these interactions are often complex, difficult to characterise and can even be somewhat counterintuitive. As an example, Cronin et al. reported the preparation of an asymmetric Mn-Anderson hybrid POM self-assembled monolayer (SAM) functionalised with pyrene groups which was used for cell-adhesion studies with human fibroblast cells.232 In this work, the hybrid POM SAM was found to be an effective adherent, however equivalent SAMs terminated in either pyrene groups or the amino-tris-base functionalised POM alone showed no cell adhesion properties, indicating the special utility of the hybrid POM nanostructure. POMs have also been shown to both disrupt,233,234 and penetrate,235,236 a variety of microbial and mammalian cell membranes, which allows their targeted use in a range of imaging, theranostic and metallodrug applications.216 For instance, Patzke et al. have shown the covalent attachment of fluorescein to a Wells–Dawson oxocluster in order to study the cellular uptake and distribution of the hybrid POM in HeLa cells via confocal fluorescence microscopy.237 The hybrid oxocluster was found to be stable at physiological pH, non-cytotoxic and had good cell uptake over 24 h, with the oxoclusters showing various dispersion modes throughout the cytoplasm.
Fabretti, Bonchio, Carraro and co-workers recently reported a similar study, preparing a family of fluorophore-functionalised [γ-SiW10O36]8− and [A-γ-PW9O34]9− hybrid oxoclusters to study their internalisation into model HEK cells.238 Interestingly, the comparatively hydrophobic fluorophores (i.e. dansyl-, pyrene- and fluorescein-groups) make these hybrid POMs amphiphilic, and they self-assemble into spherical aggregates of ca. 80–200 nm in size when dispersed in the aqueous phase. This is highly significant because, as will be discussed in more detail below, the formation of vesicular/micellular structures appears to be particularly important in mediating interactions between the POM and the lipid bilayer of the cell membrane.234,239 As such, the authors find that their hybrid POMs show good cell uptake and negligible cytotoxicity but in this case, that the localisation of the hybrid POM within the cells is dependent on both the nature of the oxocluster and fluorophore. For instance, dansyl- and pyrene-functionalised {SiW10} oxoclusters are mainly co-localised with the mitochondria, whereas the fluorescein–{SiW10} and {PW9} hybrids showed preferential targeting of the cell nuclei. This clearly indicates the multifaceted interplay between both the inorganic and organic components of the imaging agent with its cellular uptake behaviour, and highlights both the complexity of the challenge in elucidating clear design rules for bioactive hybrid POM species but also their immense potential for developing tuneable and highly specific biological interactions.
The final application for which POMs have been explored in a biological context is as novel metallodrugs and anti-microbial agents. The broader context of this field lies well outside the scope of this review, and readers are directed to several excellent recent summaries for a more detailed overview,215,216,240 however it is fair to say that the vast majority of these approaches have relied on the use of the direct action of purely inorganic POMs (or their synergistic combination with more conventional antibiotics) to elicit a pharmacological effect. Broadly speaking, these approaches have found some initial successes although persistent issues with low activity (at least in comparison with other conventional medicines), poor stability under physiological conditions, and the long-term and significant toxicity of POMs have been identified as the most pressing problems. The use of hybrid POMs thus represents a unique opportunity to design more tailored systems with improved uptake/activity, stability and – most importantly – reduced cytotoxicity. This has been shown in recent studies where hybrid POMs are found to have superior properties in both antiviral applications, such as inhibition studies against HIV protease,241 and in antibacterial studies against a broad range of Gram-positive and Gram-negative organisms.242
Pristine POMs have been known to have inhibitory activity against tumour cells since the 1960s,243 however the use of hybrid POMs in this field has been an increasingly active area of research over the last decade in particular. For instance, a number of studies using a range of organoimido- and organobenzoyldiazenido-functionalised hexamolybdate Lindqvist-type POMs have shown significant inhibitory activity towards a range of human leucocythemia and breast cancer cell lines.244–246 Dolbecq, Oldfield and co-workers also reported comparable trends in a series of bisphosphonate–polyoxomolybdate hybrids,247,248 the most effective of which was complexed to the bisphosphonate-based drug zolendronate and showed promising activity against human sarcoma cells in a mouse xenograft, decreasing tumour volume by ca. 85% against the control whilst showing no adverse toxicity towards the mice. This trend follows that previously reported for an organotin-functionalised Keggin ion, [GeW9O34{Sn(OH)(n-Bu)}3]4−, which has substantial inhibitory in vivo activity against hepatocarcinoma mice models with markedly lower toxicity than the clinical chemotherapeutic, cyclophosphamide.249
The impressive pharmaceutical activity attainable by hybrid POMs was also observed recently in a tris-functionalised hexavanadate oxocluster grafted to amino acid esters, which was assayed against a range of human cancer cell lines and showed higher inhibitory activity than the clinically available chemotherapy drug 5-fluorouracil.250 Hybrid Anderson oxoclusters have also shown promise as therapeutic agents, as recently demonstrated by Wang et al., who found that a cholic-acid functionalised Mn-Anderson oxocluster showed promising inhibition against breast cancer cells in vitro, whilst being significantly less toxic to a non-cancerous control cell line.251 It is important to note that in each of the cases highlighted above, a synergistic effect between both the POM and the organic group appears to be crucial, as the hybrid species are universally more active than either the parent oxocluster and/or the appended organic group (even if the exact mechanism still remains a mystery). That said, a recent report by Carraro, Ruzza and co-workers also highlights the difficulties of teasing out structure–activity relationships in biological systems and that much further work is required. In this study, a Mn-Anderson oxocluster was functionalised with a short-chain peptide sequence (demobensin-1, which is specific to receptors on cancer cells) in order to study targeted therapy with the POM-based metallodrug.252 In this case, however, the hybrid oxocluster is found to have similar activity towards HeLa cells as the parent tris-base Mn-Anderson, indicating a non-receptor mediated uptake mechanism. Interestingly, the authors note that this may be due to conformational effects caused by incorporation of the POM into the peptide sequence, as discussed in examples given above.227,228
The use of hybrid polyoxomolybdate metallodrugs also opens pathways to more complex or strategic molecular design strategies in order to enhance targeting and efficacy or add additional functionality. A notable example of this was reported by Chen, Hao and Wei, who demonstrated a novel in situ degradation strategy using a hexamolybdate oxocluster linked to a cleavable N-acylureido (acy) group by a 2-amino-3-methylbenzoxyl (AMB) linker.253 The POM–AMB–acy hybrid was found to be metastable when tested in cell incubation medium, decomposing within 40 min to form MoO42− as the primarily active species. Most remarkably, the hybrid POM was shown to have a strong inhibitory effect against malignant glioma cells (ca. 20× more effective than the clinically used antiglioma medication temozolomide) and was also found to be able to cross the blood–brain barrier in vivo. Another example by Yadollahi and co-workers exploit a hybrid [GeW9V3O37((CH2O)3N–Boc–Cys)]4− oxocluster (Boc = tert-butyloxycarbonyl, Cys = cysteine) which is then post-synthetically coupled with thiolated mesoporous silica nanoparticles (via a disulphide bond) loaded with the anticancer drug doxorubicin, and the imaging agent fluorescein isothiocyanate (via deprotection of the Boc group).254 The disulphide bond is cleavable within the unique environment of cancerous cells, causing decoupling of the hybrid POM from the nanoparticles, releasing both the fluorescent-tagged POM and the doxorubicin into the cellular environment. The nanocomposites were found to have superior activity against glioblastoma cells in vitro when compared to either the POM or chemotherapy drug alone.
In line with the broader themes of this review, the complex supramolecular assembly and non-covalent interactions between hybrid POM species and biological entities cannot be underestimated. This is most clearly demonstrated in an elegant study by Liu et al., who developed an unusual long-chain alkoxysilane-modified Preyssler anion (Fig. 31), [NaP5W30O110{Si(CH2)3NHCON(C16H33)2}]14−, which spontaneously self-assembled into a liposome-like structure due to the amphiphilic character of the hybrid POM.239 Whilst both the hybrid {P5W30} oxocluster and the parent {P5W30} Preyssler anion showed significantly higher antiproliferative properties against human colorectal cancer cells vs. 5-fluorouracil, the supramolecular assembly of the hybrid into vesicular structures was found to improve its inhibitory activity, particularly over shorter incubation times (e.g. <48 h). The authors used a biotinylated model membrane structure to elucidate the interaction between the both the hybrid POM liposomes and the parent inorganic POM with the cell membrane. This found that whilst both compounds rapidly associate with the lipid bilayer of the cell wall, the inorganic POM can easily dissociate back into the bulk whilst the hybrid oxocluster tends to strongly intercalate with the bilayer, forming a stable POM/lipid complex which facilitates its uptake into the cytoplasm. This model is therefore consistent with the greater activity of the hybrid complex, particularly over shorter incubation times.
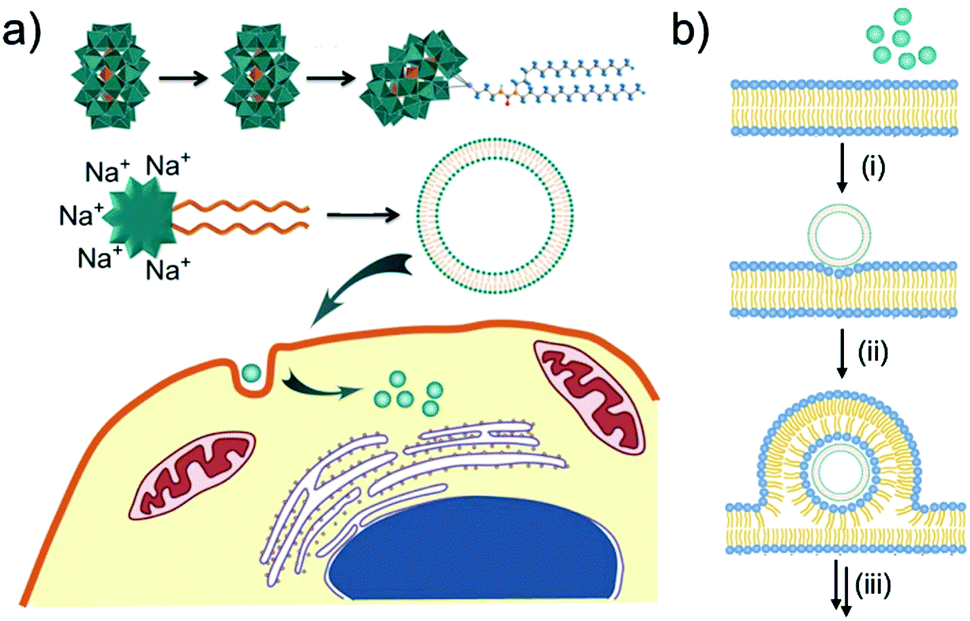 | ||
| Fig. 31 (a) Schematic of the synthesis of a lipid-modified Preyssler-type hybrid POM and its self-assembly into vesicles which facilitate its membrane crossover and intracellular localisation within human colorectal tumour cells (in vitro), and; (b) proposed model of the interaction between hybrid POM vesicles and the phospholipid bilayer of the cell-membrane, showing: (i) binding of the POM-vesicle to the lipid membrane, (ii) trapping/intercalation of the vesicle within the membrane bilayer (preventing dissociation), and (iii) membrane crossover and uptake into cell interior. Colour code: {WO6} = teal polyhedra; {PO4} = orange polyhedra; Si = blue/grey; C = grey; N = orange; O = red; H = blue. This figure has been reproduced from ref. 239 with permission from John Wiley and Sons, copyright 2015. | ||
In similarly elegant work, the authors built on this study to demonstrate how a tweezer-like organoplatinum-functionalised Keggin could be deployed as a potent pro-drug against human colorectal cancers when encapsulated into a block copolymer shell.255 The resulting nanoparticles are able to intercalate with and penetrate the cell membrane as before, and the hybrid POM nanocomposite shows both superior anti-tumour activity and improved biocompatibility vs. cisplatin in mice models.
5 Perspectives
The organofunctionalisation of POMs offers a direct and highly controllable means modify both the electronic structure and physical character of the molecular species. Simultaneously, it entails the chemist to explore and subsequently tune the nature of the non-covalent interactions between neighbouring molecules and between these molecules and their environment. This highly specific supramolecular chemistry and its potential to extend the self-assembly of POMs into materials that bridge the molecular, nano, and macro length scales, will lead to new, as yet, unexplored potential applications in a range of technologies, spanning energy storage, data processing, site-specific catalysis, and biomolecular transformations.To this end, the elaboration of hybrid POM nanoassemblies, whether as colloids or as nanomaterials, necessitates the implementation of additional characterisation techniques beyond those classically used to probe the characterisation of POMs (e.g. multinuclear NMR or single crystal X-ray diffraction). To highlight just a few of these approaches, NMR diffusion experiments can give access to the self-diffusion coefficient (this technique being yet only adapted to discrete systems as colloids have T2 relaxation times which are too short to probe adequately), dynamic light scattering can provide insights into the size and solution behaviours of POM-based nanoaggregates and X-ray scattering measurements allow solution phase structure determination of molecular assemblies or aggregates of sizes ranging from 1 to 100 nm, while also providing information on the molecular-level nanostructure. Microscopy techniques, especially TEM and cryo-TEM (despite the technical challenges posed by damage to samples by the high energy electron beam), have been widely and successfully employed owing to the very high electron density (and thus achievable imaging contrast) of most polyoxometalates. Cryo-TEM is particularly useful since it allows the direct observation of complex supramolecular nanostructures without being altered by the interaction with the substrate and/or the ultra-high vacuum required with classical electron microscopy techniques. That said, cryo-TEM is mostly limited to high freezing point solvents such as water or DMSO and it is still not widely employed in the POM community.
Though the application of advanced analytical techniques for the characterisation of POM-based assemblies and nanomaterials will surely continue to improve, it is also worth highlighting that theoretical studies also remain a considerable challenge owing to the time-consuming calculations required for such large systems containing a large number of heavy atoms.256 As an example, density functional theory simulations are usually limited to systems containing just 4 discrete POM clusters. The combination of DFT with molecular dynamics simulation methods is, however, particularly well-suited for investigating aggregation processes at the molecular level. Promising first attempts, which outlined the importance of both solvation and the association of the POM with counter cations in the aggregation process have been recently reported,90,257 though it must be noted that this approach is still limited to modest numbers of POM clusters. To address this, coarse-grained molecular dynamics simulations have proven effective as a method to considerably increase the size of simulated supramolecular assemblies. This approach has, for example, been investigated to study the formation of large Keplerate-type POM assemblies,258,259 though further development is required in order to properly parametrise and account for the peculiar chaotropic character of POMs in particular.176,178 As ever, the combination of experiment and theory is critical in order to provide the fundamental understanding of hybrid POM assembly in order to guide the further design of complex, multifunctional molecular architectures.
This review article outlines the enormous diversity in both the physical nature (e.g. molecular and supramolecular aggregates, colloidal micelles and vesicles, gels, liquid crystals and crystalline networks) and structure of hybrid POM-based self-assembled nanostructures. However, while materials using pristine (i.e. unmodified) POMs and transition metal substituted POMs are increasingly well-developed for application across a range of different fields (e.g. electrochemical storage,200 dye-sensitised solar cells,260 catalysis,261 proton exchange membranes,262 electrochromic devices,164 to highlight but a few), the implementation of self-assembled hybrid POM nanostructures into tangible device configurations remains essentially unexplored, despite the indisputable advantages of these nanomaterials. This may, in part at least, be due to the typically more demanding synthesis of these materials in comparison to the often ‘one-pot’ nature of traditional POMs, but the potentially lower chemical and thermal robustness of these species must also be considered. Closer cooperation between experts in POM chemistry and materials scientists in particular, will be critical in closing this gap.
The advantages presented by the unique properties accessible through the continued design and development of hybrid POMs and their related nanostructures are, however, clear. Numerous examples of emerging areas of study have been presented above – ranging from the formation of dynamic nanocapsules, of interest for catalysis or energy storage, to the development of photoswitchable nanomaterials showing cooperative properties. Indeed, the continued development of nanostructured soft materials based on hybrid POMs, which combine long-range ordering with distinct, switchable functionalities would constitute a major advance and lead to a range of interesting potential applications as new nanoscale sensors or actuators. Furthermore, although POMs have very well-established (and highly tuneable) multi-redox activities – marking them as being of considerable interest as unique energy storage, electronic or catalytic species – POMs often exhibit low or negligible conductivity in the solid state.263–265 Consequently, unless processed as thin films or embedded in a conductive matrix (e.g. conductive polymers or bulk or nanostructured carbons, etc.), full exploitation of the attractive redox chemistry of POM-based materials is highly challenging to achieve. The supramolecular-assembly of POM units via their hybridisation with a range of structure-directing and/or electronically active organic groups is therefore an especially interesting avenue for further study, and it will be particularly important to develop clear structure–property relationships and new design rules to govern the fabrication of new electronically active materials. Finally, there is unquestionably scope to develop the concept of tuneable POM-biomolecule interactions, perhaps in tandem with studies on the photo/electrochemical properties of the assembly. This challenging issue remains mostly unexplored, while the development of hybrid POMs with organic groups targeting site specific binding or molecular recognition for precise diagnosis and therapy would also constitute a major breakthrough in this field.
6 Conclusion
In summary, this review provides a comprehensive discussion of the supramolecular self-assembly of hybrid nanomaterials based on pre-defined organofunctionalised hybrid-polyoxometalate synthons/building blocks. Hybrid-POMs offer almost unparalleled opportunities to seamlessly integrate both organic and inorganic functionalities into new, hierarchically structured assemblies, where the inherent redox, photochemical and catalytic reactivity of the metal oxide sub-unit can be combined with the highly tuneable structure-directing properties (either via covalent or non-covalent interactions) of the appended organic groups. As several excellent reviews have recently described the synthesis and functionalisation of hybrid POMs themselves, this review focuses exclusively on how the clearly defined structure, chemistry and directionality of these unique molecular building blocks allows for new semi-rational design approaches to be applied in the construction of larger molecular and supramolecular assemblies. Numerous synthetic strategies have emerged as the field has rapidly grown over the past decade or so, which we have broadly broken down into two distinct categories: (i) direct self-assembly, whereby metal-directed, amphiphilic, electrostatic or hydrogen-bonding interactions are used to generate new nanomaterials in both the solution and solid state, or; (ii) self-assembly mediated by a substrate, whereby non-covalent interactions between the hybrid POM synthon(s) and a range of possible host/guest species (ranging from other small molecules such as cavitands, through carbon nanomaterials and even large biomolecules) is used to generate new species or facilitate integration of the hybrid POM into larger biological systems such as microbes or mammalian cells. It is also important to note that while this field is still very much in its infancy, with rapid progress being made over only the last decade or so, several key examples of emerging or potential application areas have also been discussed, including in catalysis, energy storage, and biochemistry.Author contributions
All authors contributed to data curation of the literature and writing the original draft. JMC, GG, GNN and GI conceptualised the review, performed reviewing and editing of the manuscript and contributed to visualisation of the figures. SA contributed additional visualisation of some 3D graphical assets.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors thank the EPSRC International Network on Polyoxometalate Science for Advanced Functional Energy Materials (EP/S031170/1). GNN and JMC thank the Leverhulme Trust (RPG-2016-442) and the University of Nottingham Propulsion Futures Beacon of Excellence for support. E. H. thanks the Low Dimensional Materials and Interfaces Doctoral Training Programme at the University of Nottingham. S. A. thanks the EPSRC for funding through the Centre for Doctoral Training in Sustainable Chemistry (EP/L015633/1). KMH thanks the French National Research Agency (MESOMORPHICS project Grant ANR-20-CE06-0021) for PhD funding.Notes and references
- G. M. Whitesides, J. K. Kriebel and B. T. Mayers, Self-Assembly and Nanostructured Materials, Springer, Berlin, 2005 Search PubMed.
- N. Rahimi and R. Karimzadeh, Appl. Catal., A, 2011, 398, 1–17 CrossRef CAS.
- E. Ruiz-Hitzky, P. Aranda, M. Darder and M. Ogawa, Chem. Soc. Rev., 2011, 40, 801–828 RSC.
- E. Moulin, J. J. Cid and N. Giuseppone, Adv. Mater., 2013, 25, 477–487 CrossRef CAS PubMed.
- A. S. Tayi, A. K. Shveyd, A. C. H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS.
- H. B. Yu, Y. Luo, K. Beverly, J. F. Stoddart, H. R. Tseng and J. R. Heath, Angew. Chem., Int. Ed., 2003, 42, 5706–5711 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- G. Ferey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. De Weireld, A. Vimont, M. Daturi and J. S. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- Y. L. Wu, K. E. Brown and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 13322–13325 CrossRef CAS.
- Y. Sun, C. Y. Chen and P. J. Stang, Acc. Chem. Res., 2019, 52, 802–817 CrossRef CAS.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS.
- C. Rest, R. Kandanelli and G. Fernandez, Chem. Soc. Rev., 2015, 44, 2573 RSC.
- C. J. Bruns, D. Fujita, M. Hoshino, S. Sato, J. F. Stoddart and M. Fujita, J. Am. Chem. Soc., 2014, 136, 12027–12034 CrossRef CAS.
- P. Luisi, Found. Chem., 2002, 4, 183–200 CrossRef CAS.
- H. N. Miras, J. Yan, D. L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS.
- Y. F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC.
- H. L. Li and L. X. Wu, Polym. Int., 2020, 69, 665–667 CrossRef CAS.
- K. I. Assaf and W. M. Nau, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- M. A. Moussawi, N. Leclerc-Laronze, S. Floquet, P. A. Abramov, M. N. Sokolov, S. Cordier, A. Ponchel, E. Monflier, H. Bricout, D. Landy, M. Haouas, J. Marrot and E. Cadot, J. Am. Chem. Soc., 2017, 139, 12793–12803 CrossRef CAS.
- P. C. Yin, D. Li and T. B. Liu, Chem. Soc. Rev., 2012, 41, 7368–7383 RSC.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Commun., 2019, 11, 146–153 CAS.
- G. H. Zhang, B. Y. Li, Y. Zhou, X. F. Chen, B. Li, Z. Y. Lu and L. X. Wu, Nat. Commun., 2020, 11, 425 CrossRef CAS.
- L. Yue, S. Wang, D. Zhou, H. Zhang, B. Li and L. X. Wu, Nat. Commun., 2016, 7, 10742 CrossRef CAS PubMed.
- T. Lunkenbein, M. Kamperman, Z. H. Li, C. Bojer, M. Drechsler, S. Forster, U. Wiesner, A. H. E. Muller and J. Breu, J. Am. Chem. Soc., 2012, 134, 12685–12692 CrossRef CAS PubMed.
- M. A. Alam, Y. S. Kim, S. Ogawa, A. Tsuda, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2008, 47, 2070–2073 CrossRef CAS.
- W. Li and L. X. Wu, Polym. Int., 2014, 63, 1750–1764 CrossRef CAS.
- J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47, 5120–5136 RSC.
- G. Izzet, F. Volatron and A. Proust, Chem. Rec., 2017, 17, 250–266 CrossRef CAS.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- M. P. Santoni, G. S. Hanan and B. Hasenknopf, Coord. Chem. Rev., 2014, 281, 64–85 CrossRef CAS.
- C. P. Pradeep, D.-L. Long, G. N. Newton, Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47(23), 4388–4391 CrossRef CAS.
- In this article, we define the POM functionalization as the covalent grating of one or several organic moieties onto the polyoxometalate framework.
- A. J. Kibler and G. N. Newton, Polyhedron, 2018, 154, 1–20 CrossRef CAS.
- J. X. Liu, X. B. Zhang, Y. L. Li, S. L. Huang and G. Y. Yang, Coord. Chem. Rev., 2020, 414, 213260 CrossRef CAS.
- L. Vila-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef CAS.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- J. Spandl, C. Daniel, I. Brudgam and H. Hartl, Angew. Chem., Int. Ed., 2003, 42, 1163–1166 CrossRef CAS.
- S. Chakraborty, E. Schreiber, K. R. Sanchez-Lievanos, M. Tariq, W. W. Brennessel, K. E. Knowles and E. M. Matson, Chem. Sci., 2021, 12, 12744–12753 RSC.
- B. E. Petel, R. L. Meyer, M. L. Maiola, W. W. Brennessel, A. M. Muller and E. M. Matson, J. Am. Chem. Soc., 2020, 142, 1049–1056 CrossRef CAS.
- G. S. Kim, K. S. Hagen and C. L. Hill, Inorg. Chem., 1992, 31, 5316–5324 CrossRef CAS.
- S. Fujimoto, J. M. Cameron, R. J. Wei, K. Kastner, D. Robinson, V. Sans, G. N. Newton and H. Oshio, Inorg. Chem., 2017, 56, 12169–12177 CrossRef CAS.
- J. M. Cameron, S. Fujimoto, K. Kastner, R.-J. Wei, D. Robinson, V. Sans, G. N. Newton and H. H. Oshio, Chem. – Eur. J., 2016, 23(1), 47–50 CrossRef PubMed.
- J. M. Cameron, S. Fujimoto, R.-J. Wei, G. N. Newton and H. Oshio, Dalton Trans., 2018, 47(31), 10590–10594 RSC.
- B. Matt, S. Renaudineau, L. M. Chamoreau, C. Afonso, G. Izzet and A. Proust, J. Org. Chem., 2011, 76, 3107–3112 CrossRef CAS.
- K. Nomiya, Y. Togashi, Y. Kasahara, S. Aoki, H. Seki, M. Noguchi and S. Yoshida, Inorg. Chem., 2011, 50, 9606–9619 CrossRef CAS PubMed.
- C. N. Kato, Y. Kasahara, K. Hayashi, A. Yamaguchi, T. Hasegawa and K. Nomiya, Eur. J. Inorg. Chem., 2006, 4834–4842 CrossRef CAS.
- S. Aoki, T. Kurashina, Y. Kasahara, T. Nishijima and K. Nomiya, Dalton Trans., 2011, 40, 1243–1253 RSC.
- C. R. Mayer, I. Fournier and R. Thouvenot, Chem. – Eur. J., 2000, 6, 105–110 CrossRef CAS.
- F. B. Xin and M. T. Pope, Inorg. Chem., 1996, 35, 5693–5695 CrossRef CAS.
- F. B. Xin, M. T. Pope, G. J. Long and U. Russo, Inorg. Chem., 1996, 35, 1207–1213 CrossRef CAS.
- F. B. Xin and M. T. Pope, Organometallics, 1994, 13, 4881–4886 CrossRef CAS.
- R. Cao, K. P. O'Halloran, D. A. Hillesheim, S. Lense, K. I. Hardcastle and C. L. Hill, CrystEngComm, 2011, 13, 738–740 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- T. Yamada, K. Otsubo, R. Makiura and H. Kitagawa, Chem. Soc. Rev., 2013, 42, 6655–6669 RSC.
- A. Schmidt, A. Casini and F. E. Kuhn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- R. Ricco, C. Pfeiffer, K. Sumida, C. J. Sumby, P. Falcaro, S. Furukawa, N. R. Champness and C. J. Doonan, CrystEngComm, 2016, 18, 6532–6542 RSC.
- See special issue of The 7th International Conference on Metal-Organic Frameworks & Open Framework Compounds (MOF2020), Chem. Soc. Rev., 2020.
- X. Z. Yan, S. J. Li, J. B. Pollock, T. R. Cook, J. Z. Chen, Y. Y. Zhang, X. F. Ji, Y. H. Yu, F. H. Huang and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15585–15590 CrossRef CAS.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- B. Nohra, H. El Moll, L. M. R. Albelo, P. Mialane, J. Marrot, C. Mellot-Draznieks, M. O'Keeffe, R. N. Biboum, J. Lemaire, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2011, 133, 13363–13374 CrossRef CAS PubMed.
- L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, A. Sampieri, D. W. Lewis, A. Gomez, B. Nohra, P. Mialane, J. Marrot, F. Secheresse, C. Mellot-Draznieks, R. N. Biboum, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2009, 131, 16078–16087 CrossRef.
- S. Uchida and N. Mizuno, Coord. Chem. Rev., 2007, 251, 2537–2546 CrossRef CAS.
- S. Favette, B. Hasenknopf, J. Vaissermann, P. Gouzerh and C. Roux, Chem. Commun., 2003, 2664–2665 RSC.
- J. W. Han, K. I. Hardcastle and C. L. Hill, Eur. J. Inorg. Chem., 2006, 2598–2603 CrossRef CAS.
- J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129, 15094–15095 CrossRef CAS.
- A. Abhervé, M. Palacios-Corella, J. M. Clemente-Juan, R. Marx, P. Neugebauer, J. van Slageren, M. Clemente-León and E. Coronado, J. Mater. Chem. C, 2015, 3, 7936–7945 RSC.
- J. Yan, H. Huang, Z. Miao, Q. Zhang and Y. Yan, Macromolecules, 2019, 52, 9545–9554 CrossRef CAS.
- L. Liu, L. Hu, Q. Liu, Z.-L. Du, F.-B. Li, G.-H. Li, X.-J. Zhu, W.-Y. Wong, L. Wang and H. Li, Dalton Trans., 2015, 44, 306–315 RSC.
- L. Liu, L. Hu, Q. Liu, S.-Z. Liu, F.-B. Li, L. Li, L. Peng, D.-H. Yao, W.-Y. Wong and Z.-L. Du, J. Organomet. Chem., 2015, 792, 102–108 CrossRef CAS.
- X.-X. Li, Y.-X. Wang, R.-H. Wang, C.-Y. Cui, C.-B. Tian and G.-Y. Yang, Angew. Chem., Int. Ed., 2016, 55, 6462–6466 CrossRef CAS.
- F.-J. Yazigi, C. Wilson, D.-L. Long and R. S. Forgan, Cryst. Growth Des., 2017, 17, 4739–4748 CrossRef CAS.
- M.-C. Zhu, Y.-Y. Huang, J.-P. Ma, S.-M. Hu, Y. Wang, J. Guo, Y.-X. Zhao and L.-S. Wang, Cryst. Growth Des., 2019, 19, 925–931 CrossRef CAS.
- Z. Chen, H. An, H. Zhang and Y. Hu, CrystEngComm, 2013, 15, 4711–4720 RSC.
- J. Als-Nielsen and D. McMorrow, Elements of modern x-ray physics, Wiley, New York, 2001 Search PubMed.
- J. Maes, N. Castro, K. De Nolf, W. Walravens, B. Abecassis and Z. Hens, Chem. Mater., 2018, 30, 3952–3962 CrossRef CAS.
- J. Kang, B. Xu, Z. Peng, X. Zhu, Y. Wei and D. R. Powell, Angew. Chem., Int. Ed., 2005, 44, 6902–6905 CrossRef CAS.
- M.-P. Santoni, A. K. Pal, G. S. Hanan, M.-C. Tang, K. Venne, A. Furtos, P. Ménard-Tremblay, C. Malveau and B. Hasenknopf, Chem. Commun., 2012, 48, 200–202 RSC.
- M. Piot, S. Hupin, H. Lavanan, C. Afonso, L. Bouteiller, A. Proust and G. Izzet, Inorg. Chem., 2017, 56, 8490–8496 CrossRef CAS PubMed.
- G. Izzet, A. Macdonell, C. Rinfray, M. Piot, S. Renaudineau, E. Derat, B. Abécassis, C. Afonso and A. Proust, Chem. – Eur. J., 2015, 21, 19010–19015 CrossRef CAS.
- A. Guinier and G. Fournet, Small Angle Scaterring of X-Rays., Wiley, New York, 1955 Search PubMed.
- M. Li, W. Y. Wang and P. C. Yin, Chem. – Eur. J., 2018, 24, 6639–6644 CrossRef CAS.
- M. Nyman, Coord. Chem. Rev., 2017, 352, 461–472 CrossRef CAS.
- Y. L. Wu, R. F. Shi, Y. L. Wu, J. M. Holcroft, Z. C. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 4111–4118 CrossRef CAS PubMed.
- G. Izzet, B. Abécassis, D. Brouri, M. Piot, B. Matt, S. A. Serapian, C. Bo and A. Proust, J. Am. Chem. Soc., 2016, 138, 5093–5099 CrossRef CAS.
- M. Piot, B. Abécassis, D. Brouri, C. Troufflard, A. Proust and G. Izzet, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8895–8900 CrossRef CAS.
- R. Salles, B. Abecassis, E. Derat, D. Brouri, A. Bernard, Q. C. Zhang, A. Proust, C. Desmarets and G. Izzet, Inorg. Chem., 2020, 59, 2458–2463 CrossRef CAS.
- M. S. Centellas, M. Piot, R. Salles, A. Proust, L. Tortech, D. Brouri, S. Hupin, B. Abecassis, D. Landy, C. Bo and G. Izzet, Chem. Sci., 2020, 11, 11072–11080 RSC.
- X.-X. Li, X. Ma, W.-X. Zheng, Y.-J. Qi, S.-T. Zheng and G.-Y. Yang, Inorg. Chem., 2016, 55, 8257–8259 CrossRef CAS PubMed.
- Y. Zhu, P. C. Yin, F. P. Xiao, D. Li, E. Bitterlich, Z. C. Xiao, J. Zhang, J. Hao, T. B. Liu, Y. Wang and Y. G. Wei, J. Am. Chem. Soc., 2013, 135, 17155–17160 CrossRef CAS.
- T. Ito, H. Yashiro and T. Yamase, Langmuir, 2006, 22, 2806–2810 CrossRef CAS PubMed.
- L. de Viguerie, A. Mouret, H. P. Brau, V. Nardello-Rataj, A. Proust and P. Bauduin, CrystEngComm, 2012, 14, 8446–8453 RSC.
- H. L. Li, H. Sun, W. Qi, M. Xu and L. X. Wu, Angew. Chem., Int. Ed., 2007, 46, 1300–1303 CrossRef CAS.
- B. Li, W. Li, H. L. Li and L. X. Wu, Acc. Chem. Res., 2017, 50, 1391–1399 CrossRef CAS PubMed.
- L. Leclercq, A. Mouret, A. Proust, V. Schmitt, P. Bauduin, J. M. Aubry and V. Nardello-Rataj, Chem. – Eur. J., 2012, 18, 14352–14358 CrossRef CAS.
- B. Naskar, O. Diat, V. Nardello-Rataj and P. Bauduin, J. Phys. Chem. C, 2015, 119, 20985–20992 CrossRef CAS.
- S. Polarz, S. Landsmann and A. Klaiber, Angew. Chem., Int. Ed., 2014, 53, 946–954 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, 3rd edn, 2011 Search PubMed.
- D. Li, P. C. Yin and T. B. Liu, Dalton Trans., 2012, 41, 2853–2861 RSC.
- J. Zhang, Y. F. Song, L. Cronin and T. B. Liu, J. Am. Chem. Soc., 2008, 130, 14408–14409 CrossRef CAS.
- P. C. Yin, P. F. Wu, Z. C. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. B. Liu and Y. G. Wei, Angew. Chem., Int. Ed., 2011, 50, 2521–2525 CrossRef CAS PubMed.
- M. H. Rosnes, C. Musumeci, C. P. Pradeep, J. S. Mathieson, D. L. Long, Y. F. Song, B. Pignataro, R. Cogdell and L. Cronin, J. Am. Chem. Soc., 2010, 132, 15490–15492 CrossRef CAS PubMed.
- C. P. Pradeep, M. F. Misdrahi, F. Y. Li, J. Zhang, L. Xu, D. L. Long, T. B. Liu and L. Cronin, Angew. Chem., Int. Ed., 2009, 48, 8309–8313 CrossRef CAS.
- J. Zhang, Y. F. Song, L. Cronin and T. B. Liu, Chem. – Eur. J., 2010, 16, 11320–11324 CrossRef CAS.
- J. Zhang, T. B. Liu, S. S. Mal and U. Kortz, Eur. J. Inorg. Chem., 2010, 3195–3200 CrossRef CAS.
- M. F. Misdrahi, M. H. Wang, C. P. Pradeep, F. Y. Li, C. Lydon, L. Xu, L. Cronin and T. B. Liu, Langmuir, 2011, 27, 9193–9202 CrossRef CAS.
- A. Chaumont and G. Wipff, Eur. J. Inorg. Chem., 2013, 1835–1853 CrossRef CAS.
- J. M. Pigga, M. L. Kistler, C. Y. Shew, M. R. Antonio and T. B. Liu, Angew. Chem., Int. Ed., 2009, 48, 6538–6542 CrossRef CAS PubMed.
- T. B. Liu, Langmuir, 2010, 26, 9202–9213 CrossRef CAS PubMed.
- Y. F. Wang and I. A. Weinstock, Dalton Trans., 2010, 39, 6143–6152 RSC.
- Y. K. Han, Y. Xiao, Z. J. Zhang, B. Liu, P. Zheng, S. J. He and W. Wang, Macromolecules, 2009, 42, 6543–6548 CrossRef CAS.
- Y. Xiao, Y. K. Han, N. Xia, M. B. Hu, P. Zheng and W. Wang, Chem. – Eur. J., 2012, 18, 11325–11333 CrossRef CAS PubMed.
- D. Li, J. Song, P. C. Yin, S. Simotwo, A. J. Bassler, Y. Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. B. Liu, J. Am. Chem. Soc., 2011, 133, 14010–14016 CrossRef CAS PubMed.
- S. Landsmann, M. Luka and S. Polarz, Nat. Commun., 2012, 3, 1299 CrossRef PubMed.
- R. C. Chambers, E. J. O. Atkinson, D. McAdams, E. J. Hayden and D. J. A. Brown, Chem. Commun., 2003, 2456–2457 RSC.
- J. J. Giner-Casares, G. Brezesinski, H. Mohwald, S. Landsmann and S. Polarz, J. Phys. Chem. Lett., 2012, 3, 322–326 CrossRef CAS.
- S. Landsmann, C. Lizandara-Pueyo and S. Polarz, J. Am. Chem. Soc., 2010, 132, 5315–5321 CrossRef CAS.
- A. Klaiber, S. Landsmann, T. Loffler and S. Polarz, New J. Chem., 2016, 40, 919–922 RSC.
- A. Klaiber, C. Lanz, S. Landsmann, J. Gehring, M. Drechsler and S. Polarz, Langmuir, 2016, 32, 10920–10927 CrossRef CAS.
- V. Jallet, G. Guillemot, J. Lai, P. Bauduin, V. Nardello-Rataj and A. Proust, Chem. Commun., 2014, 50, 6610–6612 RSC.
- V. Jallet, Conception de Polyoxométallates Amphiphiles Pour La Catalyse d’oxydation En Microémulsion, Université Pierre et Marie Curie, 2014, vol. 6 Search PubMed.
- I. Kozhevnikov, Catalysis by Polyoxometalates, Chichester, England, 2002 Search PubMed.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310–8311 CrossRef CAS.
- C. Venturello, R. Daloisio, J. C. J. Bart and M. Ricci, J. Mol. Catal., 1985, 32, 107–110 CrossRef CAS.
- R. Neumann and A. M. Khenkin, J. Org. Chem., 1994, 59, 7577–7579 CrossRef CAS.
- R. Ishimoto, K. Kamata and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 4662–4665 CrossRef CAS PubMed.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587–3593 CrossRef CAS.
- R. Yahya, M. Craven, E. F. Kozhevnikova, A. Steiner, P. Samunual, I. V. Kozhevnikov and D. E. Bergbreiter, Catal. Sci. Technol., 2015, 5, 818–821 RSC.
- J. M. Bregeault, M. Vennat, L. Salles, J. Y. Piquemal, Y. Mahha, E. Briot, P. C. Bakala, A. Atlamsani and R. Thouvenot, J. Mol. Catal. A: Chem., 2006, 250, 177–189 CrossRef CAS.
- G. Maayan, R. Popovitz-Biro and R. Neumann, J. Am. Chem. Soc., 2006, 128, 4968–4969 CrossRef CAS PubMed.
- A. Mouret, L. Leclercq, A. Muhlbauer and V. Nardello-Rataj, Green Chem., 2014, 16, 269–278 RSC.
- P. C. Yin, J. Wang, Z. C. Xiao, P. F. Wu, Y. G. Wei and T. B. Liu, Chem. – Eur. J., 2012, 18, 9174–9178 CrossRef CAS.
- P. C. Yin, A. Bayaguud, P. Cheng, F. Haso, L. Hu, J. Wang, D. Vezenov, R. E. Winans, J. Hao, T. Li, Y. G. Wei and T. B. Liu, Chem. – Eur. J., 2014, 20, 9589–9595 CrossRef CAS.
- K. Chen, A. Bayaguud, H. Li, Y. Chu, H. C. Zhang, H. L. Jia, B. F. Zhang, Z. C. Xiao, P. F. Wu, T. B. Liu and Y. G. Wei, Nano Res., 2018, 11, 1313–1321 CrossRef CAS.
- Y. Yan, H. B. Wang, B. Li, G. F. Hou, Z. D. Yin, L. X. Wu and V. W. W. Yam, Angew. Chem., Int. Ed., 2010, 49, 9233–9236 CrossRef CAS PubMed.
- H. B. Li, F. R. Jiang, G. H. Zhang, B. Li and L. X. Wu, Dalton Trans., 2019, 48, 5168–5175 RSC.
- Y. Chu, A. Saad, P. C. Yin, J. Y. Z. Wu, O. Oms, A. Dolbecq, P. Mialane and T. B. Liu, Chem. – Eur. J., 2016, 22, 11756–11762 CrossRef CAS.
- K. Kastner, A. J. Kibler, E. Karjalainen, J. A. Fernandes, V. Sans and G. N. Newton, J. Mater. Chem. A, 2017, 5, 11577–11581 RSC.
- S. Amin, J. M. Cameron, J. A. Watts, D. A. Walsh, V. Sans and G. N. Newton, Mol. Syst. Des. Eng., 2019, 4, 995–999 RSC.
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Angew. Chem., Int. Ed., 2019, 58, 18281–18285 CrossRef CAS PubMed.
- E. Hampson, J. M. Cameron, J. A. Watts and G. N. Newton, Chem. Commun., 2020, 56, 8237–8240 RSC.
- C. L. Peake, A. J. Kibler, G. N. Newton and D. A. Walsh, ACS Appl. Energy Mater., 2021, 4(9), 8765–8773 CrossRef CAS.
- A. Klaiber and S. Polarz, ACS Nano, 2016, 10, 10041–10048 CrossRef CAS PubMed.
- A. Klaiber, T. Kollek, S. Cardinal, N. Hug, M. Drechsler and S. Polarz, Adv. Mater. Interfaces, 2018, 5, 1701430 CrossRef.
- X. Y. Chen, H. Li, P. C. Yin and T. B. Liu, Chem. Commun., 2015, 51, 6104–6107 RSC.
- J. Lesage de La Haye, J. M. Guigner, E. Marceau, L. Ruhlmann, B. Hasenknopf, E. Lacote and J. Rieger, Chem. – Eur. J., 2015, 21, 2948–2953 CrossRef CAS.
- P. Judeinstein, Chem. Mater., 1992, 4, 4–7 CrossRef CAS.
- A. R. Moore, H. Kwen, A. M. Beatty and E. A. Maatta, Chem. Commun., 2000, 1793–1794 RSC.
- C. R. Mayer, R. Thouvenot and T. Lalot, Chem. Mater., 2000, 12, 257–260 CrossRef CAS.
- C. R. Mayer, R. Thouvenot and T. Lalot, Macromolecules, 2000, 33, 4433–4437 CrossRef CAS.
- C. R. Mayer, V. Cabuil, T. Lalot and R. Thouvenot, Angew. Chem., Int. Ed., 1999, 38, 3672–3675 CrossRef CAS.
- B. Keita and L. Nadjo, Mater. Chem. Phys., 1989, 22, 77–103 CrossRef.
- K. Nomiya, H. Murasaki and M. Miwa, Polyhedron, 1986, 5, 1031–1033 CrossRef CAS.
- W. Qi and L. X. Wu, Polym. Int., 2009, 58, 1217–1225 CrossRef CAS.
- Y. F. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC.
- C. R. Mayer, S. Neveu and V. Cabuil, Angew. Chem., Int. Ed., 2002, 41, 501–503 CrossRef CAS.
- S. Sutter, B. Trepka, S. Siroky, K. Hagedorn, S. Theiss, P. Baum and S. Polarz, ACS Appl. Mater. Interfaces, 2019, 11, 15936–15944 CrossRef CAS.
- C. Martin, K. Kastner, J. M. Cameron, E. Hampson, J. A. Fernandes, E. K. Gibson, D. A. Walsh, V. Sans and G. N. Newton, Angew. Chem., Int. Ed., 2020, 59, 14331–14335 CrossRef CAS PubMed.
- A. Gillet, S. Cher, M. Tasse, T. Blon, S. Alves, G. Izzet, B. Chaudret, A. Proust, P. Demont, F. Volatron and S. Tricard, Nanoscale Horiz., 2021, 6, 271–276 RSC.
- S. M. Wang, J. Hwang and E. Kim, J. Mater. Chem. C, 2019, 7, 7828–7850 RSC.
- B. Fleury, M. Billon, F. Duclairoir, L. Dubois, A. Fanton and G. Bidan, Thin Solid Films, 2011, 519, 3732–3738 CrossRef CAS.
- D. Fattakhova-Rohlfing, J. Rathousky, Y. Rohlfing, O. Bartels and M. Wark, Langmuir, 2005, 21, 11320–11329 CrossRef.
- D. Velessiotis, A. M. Douvas, P. Dimitrakis, P. Argitis and N. Glezos, Microelectron. Eng., 2012, 97, 150–153 CrossRef CAS.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Commun., 2010, 2, 826–831 CAS.
- Y. Martinetto, B. Pegot, C. Roch-Marchal, B. Cottyn-Boitte and S. Floquet, Eur. J. Inorg. Chem., 2020, 228–247 CrossRef CAS.
- C. G. Lin, W. Chen, S. Omwoma and Y. F. Song, J. Mater. Chem. C, 2015, 3, 15–18 RSC.
- X. L. Zhu, C. Hessin, A. Salame, L. Sosa-Vargas, D. Kreher, C. Adachi, A. Proust, P. Mialane, J. Marrot, A. Bouchet, M. Sliwa, S. Mery, B. Heinrich, F. Mathevet and G. Izzet, Angew. Chem., Int. Ed., 2021, 60, 8419–8424 CrossRef CAS.
- V. Kulikov, N. A. B. Johnson, A. J. Surman, M. Hutin, S. M. Kelly, M. Hezwani, D. L. Long, G. Meyer and L. Cronin, Angew. Chem., Int. Ed., 2017, 56, 1141–1145 CrossRef CAS.
- S. J. Li, Y. F. Zhou, N. N. Ma, J. Zhang, Z. P. Zheng, C. Streb and X. N. Chen, Angew. Chem., Int. Ed., 2020, 59, 8537–8540 CrossRef CAS.
- J. Tang, X. Y. Li, H. Wu, L. J. Ren, Y. Q. Zhang, H. X. Yao, M. B. Hu and W. Wang, Langmuir, 2016, 32, 460–467 CrossRef CAS.
- L. J. Ren, H. K. Liu, H. Wu, M. B. Hu and W. Wang, Adv. Mater., 2020, 32, 1805863 CrossRef CAS.
- C. Ma, H. Wu, Z. H. Huang, R. H. Guo, M. B. Hu, C. Kubel, L. T. Yan and W. Wang, Angew. Chem., Int. Ed., 2015, 54, 15699–15704 CrossRef CAS.
- H. Liu, J. C. Luo, W. P. Shan, D. Guo, J. Wang, C. H. Hsu, M. J. Huang, W. Zhang, B. Lotz, W. B. Zhang, T. B. Liu, K. Yue and S. Z. D. Cheng, ACS Nano, 2016, 10, 6585–6596 CrossRef CAS PubMed.
- X. S. Hou, G. L. Zhu, L. J. Ren, Z. H. Huang, R. B. Zhang, G. Ungar, L. T. Yan and W. Wang, J. Am. Chem. Soc., 2018, 140, 1805–1811 CrossRef CAS PubMed.
- H. K. Liu, L. J. Ren, H. Wu, Y. L. Ma, S. Richter, M. Godehardt, C. Kubel and W. Wang, J. Am. Chem. Soc., 2019, 141, 831–839 CrossRef CAS PubMed.
- Y. Yu and J. Rebek, Acc. Chem. Res., 2018, 51, 3031–3040 CrossRef CAS PubMed.
- J. Murray, K. Kim, T. Ogoshi, W. Yao and B. C. Gibb, Chem. Soc. Rev., 2017, 46, 2479–2496 RSC.
- N. L. Strutt, H. C. Zhang, S. T. Schneebeli and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2631–2642 CrossRef CAS PubMed.
- J. A. Foster and J. W. Steed, Angew. Chem., Int. Ed., 2010, 49, 6718–6724 CrossRef CAS PubMed.
- M. A. Moussawi, M. Haouas, S. Floquet, W. E. Shepard, P. A. Abramov, M. N. Sokolov, V. P. Fedin, S. Cordier, A. Ponchel, E. Monflier, J. Marrot and E. Cadot, J. Am. Chem. Soc., 2017, 139, 14376–14379 CrossRef CAS PubMed.
- L. B. Ni, H. Li, H. J. Xu, C. Shen, R. Z. Liu, J. Xie, F. M. Zhang, C. Chen, H. X. Zhao, T. F. Zuo and G. W. Diao, ACS Appl. Mater. Interfaces, 2019, 11, 38708–38718 CrossRef CAS.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198–11201 CrossRef CAS.
- P. Su, A. J. Smith, J. Warneke and J. Laskin, J. Am. Soc. Mass Spectrom., 2019, 30, 1934–1945 CrossRef CAS.
- X. Fang, P. Kogerler, L. Isaacs, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2009, 131, 432–433 CrossRef CAS.
- J. Tian, Z. Y. Xu, D. W. Zhang, H. Wang, S. H. Xie, D. W. Xu, Y. H. Ren, H. Wang, Y. Liu and Z. T. Li, Nat. Commun., 2016, 7, 11580 CrossRef CAS PubMed.
- F. Zhou, L. L. Zeng, W. Guo, Y. Z. Yan, F. Wu and M. Pan, Int. J. Energy Res., 2019, 43, 7095–7106 CrossRef CAS.
- G. Izzet, M. Ménand, B. Matt, S. Renaudineau, L. M. Chamoreau, M. Sollogoub and A. Proust, Angew. Chem., Int. Ed., 2012, 51, 487–490 CrossRef CAS.
- Q. X. Guo, S. H. Luo and Y. C. Liu, J. Inclusion Phenom., 1998, 30, 173–182 CrossRef CAS.
- Unpublished results.
- L. Yue, H. Ai, Y. Yang, W. J. Lu and L. X. Wu, Chem. Commun., 2013, 49, 9770–9772 RSC.
- A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed.
- B. Zhang, L. Yue, Y. Wang, Y. Yang and L. X. Wu, Chem. Commun., 2014, 50, 10823–10826 RSC.
- C. G. Lin, G. D. Fura, Y. Long, W. M. Xuan and Y. F. Song, Inorg. Chem. Front., 2017, 4, 789–794 RSC.
- W. M. Guan, G. X. Wang, J. B. Ding, B. Li and L. X. Wu, Chem. Commun., 2019, 55, 10788–10791 RSC.
- M. Stuckart, N. V. Izarova, J. van Leusen, A. Smekhova, C. Schmitz-Antoniak, H. Bamberger, J. van Slageren, B. Santiago-Schubel and P. Kogerler, Chem. – Eur. J., 2018, 24, 17767–17778 CrossRef CAS.
- Y. C. Ji, L. J. Huang, J. Hu, C. Streb and Y. F. Song, Energy Environ. Sci., 2015, 8, 776–789 RSC.
- J. W. Jordan, G. A. Lowe, R. L. McSweeney, C. T. Stoppiello, R. W. Lodge, S. T. Skowron, J. Biskupek, G. A. Rance, U. Kaiser, D. A. Walsh, G. N. Newton and A. N. Khlobystov, Adv. Mater., 2019, 31, 1904182 CrossRef CAS.
- N. Kawasaki, H. Wang, R. Nakanishi, S. Hamanaka, R. Kitaura, H. Shinohara, T. Yokoyama, H. Yoshikawa and K. Awaga, Angew. Chem., Int. Ed., 2011, 50, 3471–3474 CrossRef CAS PubMed.
- J. Hu, Y. C. Ji, W. Chen, C. Streb and Y. F. Song, Energy Environ. Sci., 2016, 9, 1095–1101 RSC.
- M. Genovese and K. Lian, ACS Appl. Mater. Interfaces, 2016, 8, 19100–19109 CrossRef CAS.
- W. Chen, L. J. Huang, J. Hu, T. F. Li, F. F. Jia and Y. F. Song, Phys. Chem. Chem. Phys., 2014, 16, 19668–19673 RSC.
- Y. C. Ji, J. Hu, L. J. Huang, W. Chen, C. Streb and Y. F. Song, Chem. – Eur. J., 2015, 21, 6469–6474 CrossRef CAS.
- D. Ma, L. Y. Liang, W. Chen, H. M. Liu and Y. F. Song, Adv. Funct. Mater., 2013, 23, 6100–6105 CrossRef CAS.
- L. J. Huang, J. Hu, Y. C. Ji, C. Streb and Y. F. Song, Chem. – Eur. J., 2015, 21, 18799–18804 CrossRef CAS PubMed.
- C. Bosch-Navarro, B. Matt, G. Izzet, C. Romero-Nieto, K. Dirian, A. Raya, S. I. Molina, A. Proust, D. M. Guldi, C. Marti-Gastaldo and E. Coronado, Chem. Sci., 2014, 5, 4346–4354 RSC.
- G. Modugno, Z. Syrgiannis, A. Bonasera, M. Carraro, G. Giancane, L. Valli, M. Bonchio and M. Prato, Chem. Commun., 2014, 50, 4881–4883 RSC.
- Z. Syrgiannis, G. Trautwein, M. Calvaresi, G. Modugno, F. Zerbetto, M. Carraro, M. Prato and M. Bonchio, Eur. J. Inorg. Chem., 2019, 374–379 CrossRef CAS.
- Y. C. Ji, T. F. Li and Y. F. Song, Ind. Eng. Chem. Res., 2014, 53, 11566–11570 CrossRef CAS.
- P. F. Gao, Y. Q. Wu and L. X. Wu, Soft Matter, 2016, 12, 8464–8479 RSC.
- A. Bijelic and A. Rompel, Chemtexts, 2018, 4, 10 CrossRef.
- L. S. Van Rompuy and T. N. Parac-Vogt, Curr. Opin. Biotechnol, 2019, 58, 92–99 CrossRef CAS.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS PubMed.
- F. Schluenzen, A. Tocilj, R. Zarivach, J. Harms, M. Gluehmann, D. Janell, A. Bashan, H. Bartels, I. Agmon, F. Franceschi and A. Yonath, Cell, 2000, 102, 615–623 CrossRef CAS.
- A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38 CrossRef CAS.
- H. Liu, Y. L. Zou, L. Zhang, J. X. Liu, C. Y. Song, D. F. Chai, G. G. Gao and Y. F. Qiu, J. Coord. Chem., 2014, 67, 2257–2270 CrossRef CAS.
- J. Q. Sha, X. Li, Y. H. Zhou, P. F. Yan, G. M. Li and C. Wang, Solid State Sci., 2011, 13, 1972–1977 CrossRef CAS.
- C. Molitor, A. Bijelic and A. Rompel, Chem. Commun., 2016, 52, 12286–12289 RSC.
- S. G. Mauracher, C. Molitor, R. Al-Oweini, U. Kortz and A. Rompel, acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 2301–2315 CrossRef CAS.
- A. Blazevic, E. Al-Sayed, A. Roller, G. Giester and A. Rompel, Chem. – Eur. J., 2015, 21, 4762–4771 CrossRef CAS.
- E. Al-Sayed, A. Blazevic, A. Roller and A. Rompel, Chem. – Eur. J., 2015, 21, 17800–17807 CrossRef CAS.
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS.
- V. A. Zamolo, G. Modugno, E. Lubian, A. Cazzolaro, F. Mancin, L. Giotta, D. Mastrogiacomo, L. Valli, A. Saccani, S. Krol, M. Bonchio and M. Carraro, Front. Chem., 2018, 6, 278 CrossRef PubMed.
- C. Yvon, A. J. Surman, M. Hutin, J. Alex, B. O. Smith, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 3336–3341 CrossRef CAS.
- D. Vilona, D. Lachkar, E. Dumont, M. Lelli and E. Lacôte, Chem. – Eur. J., 2017, 23, 13323–13327 CrossRef CAS.
- A. M. Debela, M. Ortiz, V. Beni, S. Thorimbert, D. Lesage, R. B. Cole, C. K. O'Sullivan and B. Hasenknopf, Chem. – Eur. J., 2015, 21, 17721–17727 CrossRef CAS.
- M. Ortiz, A. M. Debela, M. Svobodova, S. Thorimbert, D. Lesage, R. B. Cole, B. Hasenknopf and C. K. O'Sullivan, Chem. – Eur. J., 2017, 23, 10597–10603 CrossRef CAS.
- N. Chahin, L. A. Uribe, A. M. Debela, S. Thorimbert, B. Hasenknopf, M. Ortiz, I. Katakis and C. K. O'Sullivan, Biosens. Bioelectron., 2018, 117, 201–206 CrossRef CAS.
- Y. F. Song, N. McMillan, D. L. Long, S. Kane, J. Malm, M. O. Riehle, C. P. Pradeep, N. Gadegaard and L. Cronin, J. Am. Chem. Soc., 2009, 131, 1340–1341 CrossRef CAS PubMed.
- B. X. Jing, M. Hutin, E. Connor, L. Cronin and Y. X. Zhu, Chem. Sci., 2013, 4, 3818–3826 RSC.
- A. Sakamoto, K. Unoura and H. Nabika, J. Phys. Chem. C, 2018, 122, 1404–1411 CrossRef CAS.
- Z. M. Zhang, X. P. Duan, S. Yao, Z. S. Wang, Z. K. Lin, Y. G. Li, L. S. Long, E. B. Wang and W. B. Lin, Chem. Sci., 2016, 7, 4220–4229 RSC.
- L. Ni, P. Greenspan, R. Gutman, C. Kelloes, M. A. Farmer and F. D. Boudinot, Antiviral Res., 1996, 32, 141–148 CrossRef CAS.
- G. Geisberger, E. B. Gyenge, D. Hinger, P. Bosiger, C. Maake and G. R. Patzke, Dalton Trans., 2013, 42, 9914–9920 RSC.
- G. Modugno, E. Fabbretti, A. Dalle Vedove, T. Da Ros, C. Maccato, H. S. Hosseini, M. Bonchio and M. Carraro, Eur. J. Inorg. Chem., 2018, 4955–4961 CrossRef CAS.
- L. Fu, H. Q. Gao, M. Yan, S. Z. Li, X. Y. Li, Z. F. Dai and S. Q. Liu, Small, 2015, 11, 2938–2945 CrossRef CAS.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- A. Flutsch, T. Schroeder, M. G. Grutter and G. R. Patzke, Bioorg. Med. Chem. Lett., 2011, 21, 1162–1166 CrossRef PubMed.
- P. Yang, Z. G. Lin, G. Alfaro-Espinoza, M. S. Ullrich, C. I. Rat, C. Silvestru and U. Kortz, Inorg. Chem., 2016, 55, 251–258 CrossRef CAS.
- H. N. Mukherjee, J. Indian Med. Assoc., 1965, 44, 477–479 CAS.
- L. S. Wang, P. C. Yin, J. Zhang, F. P. Xiao, Z. K. Fang, W. W. Fu, Y. G. Wei and S. J. Xue, Eur. J. Inorg. Chem., 2017, 5475–5484 CrossRef CAS.
- H. Yu, S. L. Le, X. H. Zeng, J. Y. Zhang and J. L. Xie, Inorg. Chem. Commun., 2014, 39, 135–139 CrossRef CAS.
- S. She, S. T. Bian, J. Hao, J. W. Zhang, J. Zhang and Y. G. Wei, Chem. – Eur. J., 2014, 20, 16987–16994 CrossRef CAS.
- A. Boulmier, X. X. Feng, O. Oms, P. Mialane, E. Riviere, C. J. Shin, J. Q. Yao, T. Kubo, T. Furuta, E. Oldfield and A. Dolbecq, Inorg. Chem., 2017, 56, 7558–7565 CrossRef CAS.
- J. D. Compain, P. Mialane, J. Marrot, F. Secheresse, W. Zhu, E. Oldfield and A. Dolbecq, Chem. – Eur. J., 2010, 16, 13741–13748 CrossRef CAS.
- Z. X. Dong, R. K. Tan, J. Cao, Y. Yang, C. F. Kong, J. Du, S. Zhu, Y. Zhang, J. Lu, B. Q. Huang and S. X. Liu, Eur. J. Med. Chem., 2011, 46, 2477–2484 CrossRef CAS PubMed.
- X. K. Hu, H. Wang, B. Huang, N. Li, K. H. Hu, B. L. Wu, Z. C. Xiao, Y. H. Wei and P. F. Wu, J. Inorg. Biochem., 2019, 193, 130–132 CrossRef CAS.
- H. K. Yang, Y. X. Cheng, M. M. Su, Y. Xiao, M. B. Hu, W. Wang and Q. Wang, Bioorg. Med. Chem. Lett., 2013, 23, 1462–1466 CrossRef CAS.
- D. Ventura, A. Calderan, C. Honisch, S. Krol, S. Serrati, M. Bonchio, M. Carraro and P. Ruzza, Peptide Sci., 2018, 110, e24047 CrossRef.
- S. She, S. T. Bian, R. C. Huo, K. Chen, Z. H. Huang, J. W. Zhang, J. Hao and Y. G. Wei, Sci. Rep., 2016, 6, 33529 CrossRef CAS PubMed.
- D. Karimian, B. Yadollahi and V. Mirkhani, Microporous Mesoporous Mater., 2017, 247, 23–30 CrossRef CAS.
- T. Sun, W. Cui, M. Yan, G. Qin, W. Guo, H. Gu, S. Liu and Q. Wu, Adv. Mater., 2016, 28(34), 7397–7404 CrossRef CAS.
- G. Toupalas, J. Karlsson, F. A. Black, A. Masip-Sanchez, X. Lopez, Y. Ben M'Barek, S. Blanchard, A. Proust, S. Alves, P. Chabera, I. P. Clark, T. Pullerits, J. M. Poblet, E. A. Gibson and G. Izzet, Angew. Chem., Int. Ed., 2021, 60, 6518–6525 CrossRef CAS PubMed.
- E. Nikoloudakis, K. Karikis, M. Laurans, C. Kokotidou, A. Sole-Daura, J. J. Carbo, A. Charisiadis, G. Charalambidis, G. Izzet, A. Mitraki, A. M. Douvas, J. M. Poblet, A. Proust and A. G. Coutsolelos, Dalton Trans., 2018, 47, 6304–6313 RSC.
- R. Carr, I. A. Weinstock, A. Sivaprasadarao, A. Muller and A. Aksimentiev, Nano Lett., 2008, 8, 3916–3921 CrossRef CAS PubMed.
- Z. N. Liu, T. B. Liu and M. Tsige, Sci. Rep., 2016, 6, 26595 CrossRef CAS PubMed.
- L. Chen, W. L. Chen, X. L. Wang, Y. G. Li, Z. M. Su and E. B. Wang, Chem. Soc. Rev., 2019, 48, 260–284 RSC.
- C. T. Buru and O. K. Farha, ACS Appl. Mater. Interfaces, 2020, 12, 5345–5360 CrossRef CAS.
- L. Zhai and H. L. Li, Molecules, 2019, 24, 3425 CrossRef CAS.
- M. J. Turo, L. F. Chen, C. E. Moore and A. M. Schimpf, J. Am. Chem. Soc., 2019, 141, 4553–4557 CrossRef CAS.
- R. Tsunashima, Y. Iwamoto, Y. Baba, C. Kato, K. Ichihashi, S. Nishihara, K. Inoue, K. Ishiguro, Y. F. Song and T. Akutagawa, Angew. Chem., Int. Ed., 2014, 53, 11228–11231 CrossRef CAS.
- R. Tsunashima, I. Nakamura, R. Oue, S. Koga, H. Oki, S. Noro, T. Nakamura and T. Akutagawa, Dalton Trans., 2017, 46, 12619–12624 RSC.
| This journal is © The Royal Society of Chemistry 2022 |