
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperator-independent high-throughput polymerization screening based on automated inline NMR and online SEC†
Joren
Van Herck
,
Iyomali
Abeysekera
,
Axel-Laurenz
Buckinx
,
Kewei
Cai
,
Jordan
Hooker
 ,
Kirti
Thakur
,
Emma
Van de Reydt
,
Pieter-Jan
Voorter
,
Dries
Wyers
and
Tanja
Junkers
*
,
Kirti
Thakur
,
Emma
Van de Reydt
,
Pieter-Jan
Voorter
,
Dries
Wyers
and
Tanja
Junkers
*
Polymer Reaction Design Group, School of Chemistry, Monash University, 19 Rainforest Walk, Clayton, Victoria 3800, Australia. E-mail: tanja.junkers@monash.edu
First published on 5th July 2022
Abstract
Traditional protocols for high-throughput screening and experimentation are inherently time-consuming and cost-ineffective. Herein, we present a continuous flow-based automated synthesis platform that allows for rapid screenings of polymerizations. The platform uses online monitoring to acquire real time analytic data. Software is developed to guide data acquisition, and most importantly, to carry out reactions and their analysis autonomously. Further algorithms automatically detect experimental inaccuracies, and clean data. Data is aggregated and provided directly in a machine-readable manner, opening pathways towards creation of ‘big data’ sets for kinetic information that is independent of individual user biases and systematic errors. We demonstrate this platform on reversible-addition fragmentation chain transfer polymerization (RAFT). 8 different operators, ranging from PhD students with no prior experience in flow chemistry or RAFT polymerization, up to the professor of the research group created in this way a coherent dataset spanning 8 different monomers containing 3600 NMR spectra and about 400 molecular weight distribution analyses. Coherence of the dataset is demonstrated by reducing key kinetic information that describe the whole covered reaction space in a single parameter.
Introduction
The chemical space is vast and complex and it is often hard, if not impossible, to have a full notion of a studied system without performing detailed empirical studies. Discovery and optimization steps are tedious tasks and are often ‘bottlenecks’ for applied research and subsequently commercialisation of final products. Too often detailed studies beyond acquiring a handful of datapoints is deemed incremental research, despite a wealth of knowledge waiting to be harnessed. Performing multiple experiments in parallel can drastically increase the efficiency of chemical synthesis and robotics has emerged to fill this gap. Robotic high-throughput experimentation is a topic of research for many years already, both in small molecule chemistry and in polymer research.1,2High-throughput screening in polymer chemistry serves two purposes. First, structure–property relationships can be studied in a highly efficient way by producing and testing of polymer libraries.3–6 Second, the kinetics of polymerisations under various conditions can be determined.7–10 While the first improves the final material design, the latter gives the opportunity to explore and understand the underlying reaction mechanisms and find the optimal conditions for efficient reaction protocols (on laboratory scale, but also in intensified industrial processes). Nevertheless, a common feature of both is an efficient workflow and the collection of reliable and reproducible data. Throughout the years, multiple innovations have helped chemists reach these goals.11–14
Flow chemistry has seen a rapid development over the past decade.15,16 Besides well-known benefits such as increased heat dissipation, improved mixing and safer synthesis protocols, continuous flow can be of added value for kinetic screening of reactions. Unlike the reaction time in batch reactions, the residence and reaction time of chemicals inside a flow reactor is related to the feed flowrate, and can hence be shifted towards larger or shorter reaction times dynamically.17 Consequently, different reaction conditions can be screened with high time precision, all in the same reactor space. By connecting multiple feeds to the reactors and simply changing the flowrates accordingly, stoichiometric ratios can be adjusted and screened in an efficient way. The most important aspect of flow chemistry is, however, the high reproducibility of reactions. The good thermal conductivity of flow reactors allows one to minimize batch-to-batch variations, and hence provides more reliable data in general when compared to batch-wise probing of reaction conditions.
Other steps towards the modernisation of high-throughput screenings concern the analysis of reaction products. When performing screenings in batch, samples have to be physically taken from the reactor and specifically prepared for the technique used before measurement. This workflow easily becomes the bottleneck in high-throughput experimentation. Automated sample handling methods do exist but are usually very specific to one characterisation technique and are not readily available in a standard chemistry lab.10 Likewise, in situ methods speed up the process but such methods are limited to a restricted reaction space.18,19 Moreover, for each series of conditions to be analysed, parallel reactors are still required, limiting the number of experiments possible. Flow reactors, however, are able to integrate real-time data acquisition by placing analytic devices in the stream of synthesis, i.e. inline or in a parallel flow, i.e. online.20 By using characterisation techniques directly in the flow of synthesis, reaction products can be continuously monitored without the need of manual sampling. Additionally, they reduce the risk of human sampling errors or chemical alteration upon change in the environment, for instance from light degradation. Since reaction times can be dynamically chosen, reactions are screened in a single experiment in a very broad window of conditions, including temperature, concentration ratios, dilution and time. Common monitoring techniques in polymer research include spectroscopic techniques i.e. NMR7,8,21 or IR,22 mass spectrometry23 or size exclusion chromatography (SEC).24–26
Last, but not least, a crucial advance towards efficient data gathering is presented by automation and digitalization of chemical processes.27Via automation, reactor setups can be pre-programmed to screen a variety of conditions, thereby minimizing interference of the operator (and thus removing a further cause for variance in gathered data). Furthermore, acquired data can be processed, analysed and interpreted by software in order to develop a fully autonomous system. In theory, such advanced setups could run for 24 hours a day, 7 days a week.
It is exactly this feature that makes the combination of flow chemistry with online monitoring and automated software so attractive for high-throughput screenings (Fig. 1). Firstly, the use of software to start reactions, alter conditions and analyse data outputs facilitates (untrained) researchers to perform standardized protocols. Secondly, since changes in conditions are programmed and executed with digital precision, batch-to-batch variations or human inaccuracies are vastly reduced. Similarly, automated data cleaning and interpretation reduces human bias. Both methods ensure more reliable and consistent data generation. Perhaps the most important point of the digital transformation of chemical process is the amount of data that can be generated with automated flow reactors in combination with real-time analysis. Thanks to continuous collection and the theoretically endless run time of such platforms, the achievable data density is considerably higher compared to offline methods. This in turn contributes to an increased quality of kinetic reaction models by itself.
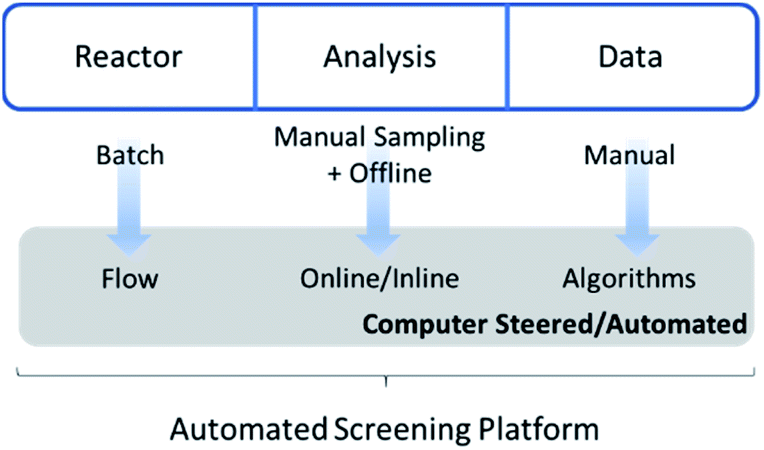 | ||
| Fig. 1 Chemical workflow efficiency increase via automated high-throughput flow chemistry screening methods. | ||
In this work, we present a fully automated polymer synthesis platform for high-throughput kinetic screenings of reversible-addition fragmentation chain transfer (RAFT) radical polymerizations. The developed setup is summarized in Fig. 2. We have demonstrated certain aspects of automation for this type of polymerization earlier, but until now all reactions still either required human interaction at every crucial step, or lacked the ability to acquire systematic data. The key development here to make the next evolutionary step is software development. Only if an interface is developed that allows for simple guidance of reactors, the full potential described above can be called in. The ease of use, consistency, reliability and efficiency of software we developed for this purpose is demonstrated by a screening of 8 different monomers, performed by 8 different operators (each having a different educational and academic background). With this data we can not only show fully consistent and operator-independent data acquisition, but via automated data cleaning and modelling of the acquired data, we also provide subtle insights in polymer kinetics, and provide new, machine-learning based approaches towards predicting future reactions.
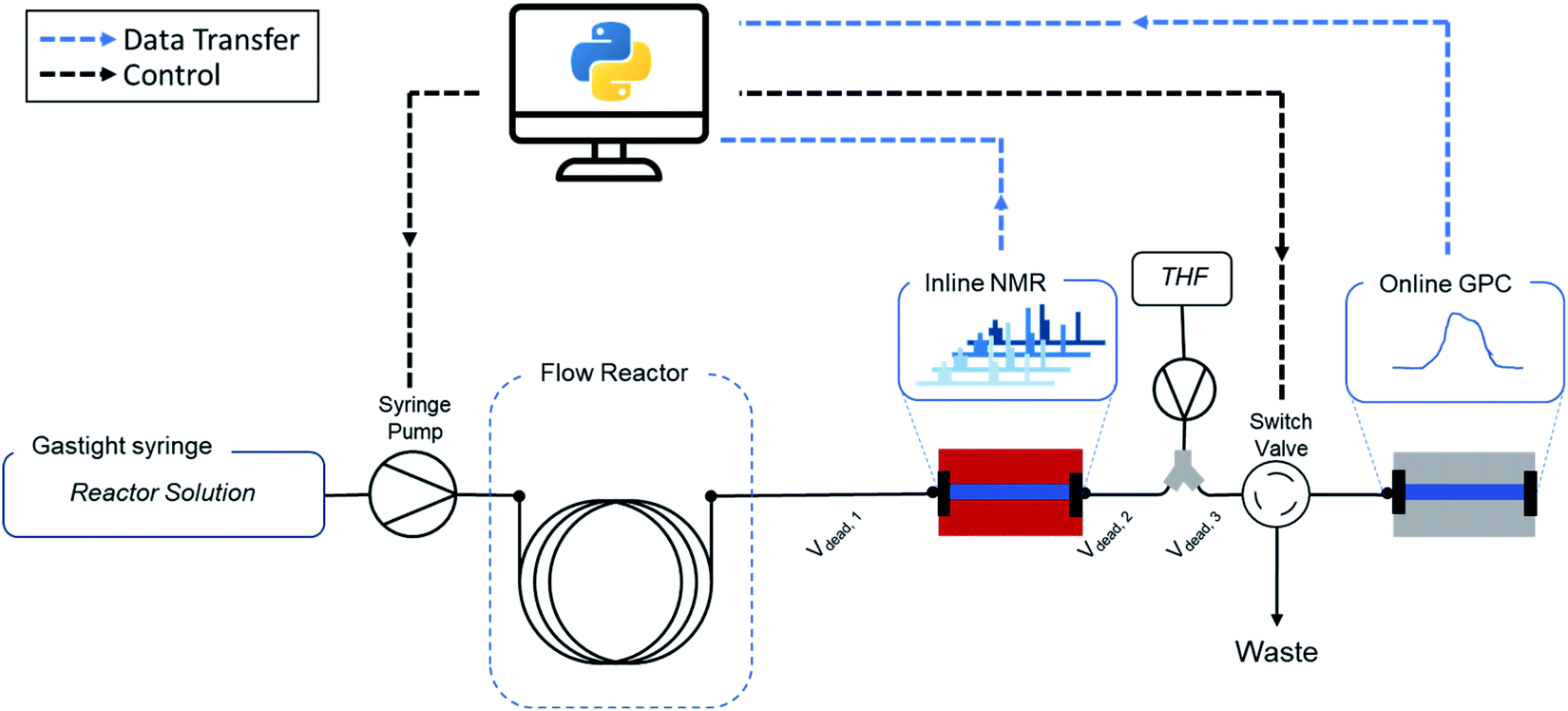 | ||
| Fig. 2 Automated polymer screening platform used in this work. The combination of flow chemistry, real-time analysis and automation yield high volume data in an efficient and consistent way. Custom software makes the setup accessible to operators without background in flow chemistry or online monitoring. | ||
Results and discussion
Automated polymer screening platform
Today, reversible addition–fragmentation chain transfer (RAFT) polymerisation has become one of the most relevant techniques for the synthesis of complex macromolecular materials.28 Although the general reaction mechanism is known, uncertainties persist about the importance and rate of certain reactions, making kinetic modelling of these polymerizations still challenging.29,30 Further kinetic studies of individual systems are – and will be – still needed for the time to come. Having said this, control over RAFT polymerisation is affected by many parameters, such as choice of the RAFT agent, radical initiator, type of monomer, temperature, stoichiometric ratios of reactants and absolute concentrations, etc. Manual systematic screenings are therefore tedious and time-consuming. High-throughput screening methods accelerate these protocols while maintaining, or even improving, the data quality.31 Since we have a long standing history in kinetic investigations into RAFT,7,32,33 we used this as a model reaction to validate and demonstrate our automated screening platform.As useful as flow already is for high throughput screening, one significant extra benefit is the ability to screen reaction conditions in transient fashion.17 Starting from a stable reactor flowrate A, corresponding to a specific reaction time, the flowrate can be abruptly changed to an alternate flow rate B. In consequence, different plugs in the reactor will then experience different residence times. By close monitoring of all plugs in the reactor, the complete range between residence times A and B can thus be analysed during the stabilisation period. The stabilization time itself only takes as long as the residence time of flow rate B. These so-called timesweeps are powerful experiments for ultra-fast screening of reactions.7,8,23,33 Compared to manual batch and steady-state flow sampling, timesweeps can decrease experiment time and waste production by over 200%.17 Moreover, using online and inline analysis, the amount of datapoints can be drastically increased, since acquisition is only limited by the time resolution of the characterisation technique employed rather than material resources and research operator time.
The two most important indicators to follow RAFT polymerizations are monomer conversion and the average molecular weight of the residual polymer. Both can be measured online in continuous flow, conversion via NMR spectroscopy, and average molecular weight via size exclusion chromatography (SEC).8,17,24,34 Thanks to the miniaturization of NMR devices, coupling of flow reactors to benchtop models are made simple.35 SEC is a little bit more tricky to couple online, as it relies on spaced injections into HPLC systems, and is hence non-continuous. Traditional offline GPC systems are associated with a relatively long analysis time of 20–40 minutes. Sampling time can, however, be reduced to about 12 minutes per sample. Time resolution can be further brought down by overlapping elugrams, making SEC suitable for routine online screening.24–26 It should be noted that the overlapping of elugrams needs to be adjusted to the molecular weight range of interest, and is pre-set before the experiments. In previous works, we successfully used the above-mentioned monitoring tools separately.7,24 In our upgraded version, we combined both characterisation techniques – inline NMR and online GPC – to increases the data density and to yield a more thorough kinetic profile of the polymerization under investigation.
As mentioned above, a key development is though the software platform of our setup. Via Labview and Python, all parts of the setup are interconnected and communicating (see Fig. 2 and ESI† for details). The software controls the flowrates, collects the acquired online monitoring data and bundles all the parameters into a single experiment output. The software allows to follow each individual data acquisition precisely in time and brings all data ‘in tune’, accounting for individual dead volumes and detector delay times. In short, it allows the operator to ignore all engineering aspects of the setup, and to focus on outcome of the experiments rather than on the intricacies of data acquisition. A graphical user interface (GUI) guides the user through every step of the reaction, initializes the experiment and executes all required steps autonomously. This minimizes the need for knowledge of every underlaying technical feature of the setup and makes the method easy to use and accessible for researchers of all academic backgrounds, including those who have no background in flow chemistry at all.
Data acquisition: consistency is key
One of the objectives for the design of the platform was a straightforward workflow. This was achieved by development of control software that is as self-explanatory as possible. The only chemical task that is required to be carried out manually is preparing the reaction solution and filling a syringe as feed for the reactant pump. From there on, the operator only needs to provide key information to the software (such as weighed in amount of monomer, initiator, etc.) to start the experiments. Automating workflows as such, and thereby minimizing human interference, increases the consistency and reliability of screenings. To demonstrate the robustness of the procedures, we screened RAFT polymerizations by different operators. All members of our research group at the time of project execution, from undergraduate student to professor, were given the task to screen a monomer of their choice for three different reaction conditions. It is important to note that this includes students who have not performed any flow reaction, or even RAFT polymerization before in their work. In a conventional setting, this effort would inevitably result in scattered, inconsistent data due to variation in systematic errors, batch-to-batch variation, or possibly procedural errors being made. Most importantly though, such screening across operators would take usually very significant amount of time, as experimentation inevitably would include familiarization and training of operators.Next to deviations in experiments, also data analysis can interfere with data consistency. Between different individuals, variations in parameter settings or interpretation of the data can give a biased conclusion of the experiment and analysis of a dataset is by far not as standardized as it may seem on first glance. An example for human bias is setting spectral integration limits, or various assumptions being made when analysing data. The platform therefore features standardized and pre-programmed settings in combination with automated data collection and analysis. NMR spectra are measured in a 17 seconds interval and monomer conversions are calculated in real-time. Likewise, a switch valve injects a polymers sample onto the SEC column every 3 minutes. Molecular weight distributions are extracted and directly interpreted by the platform's software. Since the volumes of setup and timepoints of flowrate changes are known, exact residence times can be calculated for each acquired datapoint. The molecular weight range measurable is limited by the solution viscosity, which depends on the exact monomer under investigation and the weight–average molecular weight of the residual polymer. While high MW polymer is feasible to synthesize in flow reactors, we limited our work to few tens of thousands of Dalton to avoid inconsistencies.
Both data series from NMR and SEC are then combined in one standardized comma-separated values (CSV) file. The importance of standardized data formats cannot be underestimated. Structured data is easy to work with, both for humans, but also for computer software. With the digital transformation of chemistry, databases, artificial intelligence and machine learning algorithms are becoming more and more part of everyday chemical research. Since these technologies rely on big data, easy machine-readable readouts facilitate their integration into modern chemistry.
Based on the residual csv file, summary plots are automatically generated and updated in real-time. This creates an overview of the reaction progress and gives the operator a chance to stop the process in case any abnormality is detected.
An example for the automatically generated output of a reaction is given for the RAFT polymerization of a 1 molar solution of cyclohexyl acrylate (1 M cyclohexyl acrylate, 0.005 M AIBN, DPtarget of 50, 80 °C, butyl acetate as solvent) (see Fig. 3). Raw NMR data is summarized as scan-conversion plots. In the scan–conversion plot, a clear distinction can be made between the timesweep phase (Fig. 3A – red) and the stabilisation time related to the dead volumes (Fig. 3A – blue). After deriving the residence time (tres) of each timesweep datapoint, a perfect connection of the timesweeps can be observed, illustrating the robustness of the concept as a fast screening method (Fig. 3B).17 A similar trend is seen in the tres–DP (degree of polymerization) plot, where the last injection of the first timesweep has the same molecular weight as the first polymer trace of the second timesweep (Fig. 3C). Finally, the high linear correlation between monomer conversion and degree of polymerization (R2 = 0.984) in the conversion–DP plot indicates a successful RAFT polymerisation.28 Extrapolating the fit reveals a DP of 51 at 100% conversion, which is in good agreement with the targeted DP of 50 (Fig. 3D) for the respective experiment. This result is much in line with theoretical expectations, but yet again, achieving high data quality to this accuracy is – without automation – far from trivial, and usually requires well trained and experienced researchers to produce comparable results (not to mention that human sampling would result in much lower number of data points, and hence higher statistical uncertainty).
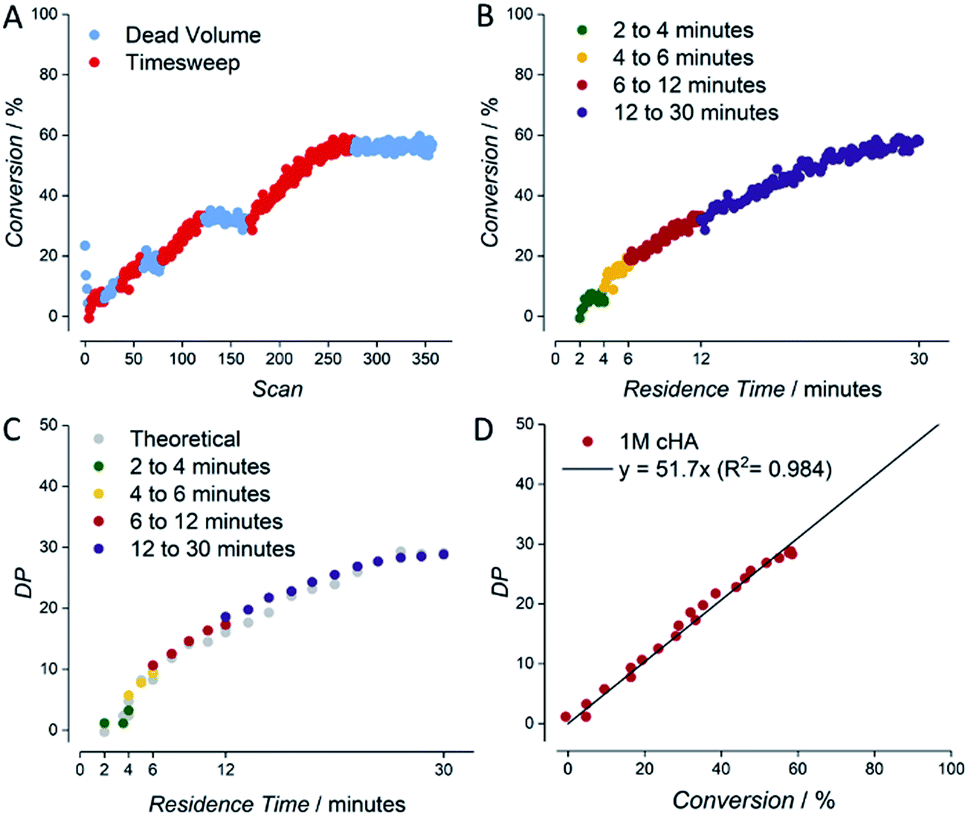 | ||
| Fig. 3 Example for data derived by the setup shown in Fig. 2 for the kinetic screening of a 1 molar cyclohexyl acrylate RAFT polymerization. (A) NMR scan–conversion plot. (B) tres–conversion plot. (C) tres–DP plot. (D) Conversion–DP plot. | ||
Data cleaning towards ‘big data’ use
The automated platform drastically increases the volume of data. During one screening reaction from 3 to 30 minutes residence time, more than 250 NMR spectra and 16 GPC samples are collected over a time span of 1.5 hours. Manually batch screening would in most cases not exceed 6–10 data points for each dimension.Multiplying this number for 3 different conditions per operator, this results in about 3600 NMR spectra and close to 400 SEC chromatograms, taken within less than 8 workdays in the laboratory. It is quite obvious that this methodology indeed is a high-throughput screening method, exceeding typical robotic screening methods available in the market. In turn, having access to such an amount of data, opens the pathway towards so-called “big data” analysis. Big data analysis allows for deep learning methods in machine learning, and creates pathways so far not taken in chemical synthesis. Having said that (use and processing of big data starts with even more data than discussed in here), the next important step in big data treatment is that screening methods are required that test the reliability of acquired data. Even with the shown methodology, not always perfect data is obtained. Glitches may still occur as in any sophisticated process control, and despite all its advantages, data should not be taken for granted with respect to its accuracy. Standardized data cleaning processes must be applied as manual review of every single data point becomes increasingly more tedious and carries the risk of introducing human bias in error analysis.
As mentioned, outliers in data cannot be completely avoided. These originate from faulty experiment initialization or a short malfunction of software or hardware. The first can be corrected by reviewing the relevant experiment, while the latter problem mostly leads to the removal of the “faulty” data. To this end, we introduced automatic checking of the data stored in csv format. The data cleaning algorithm used to inspect each experimental output file was programmed to detect four kinds of deviations, for both the NMR data as well as the SEC data. NMR data was flagged for ‘timesweep jumps’ and negative conversions. Timesweep jumps are defined as a discontinuity of two consecutive timesweeps (Fig. 4C). If these were detected, we decided to remove the first timesweep from the dataset, since the second timesweep is always measured at lower flowrates and hypothesised to yield more reliable data (high flowrates can lead to NMR accuracies if mass transport is faster than spin relaxation times). Secondly, negative conversion – obviously impossible – is sometimes reported when working at lower monomer concentrations due to working close to the detection limit of the benchtop NMR (Fig. 4B). In both cases, deleting of these datapoints increases the overall quality of the experiment.
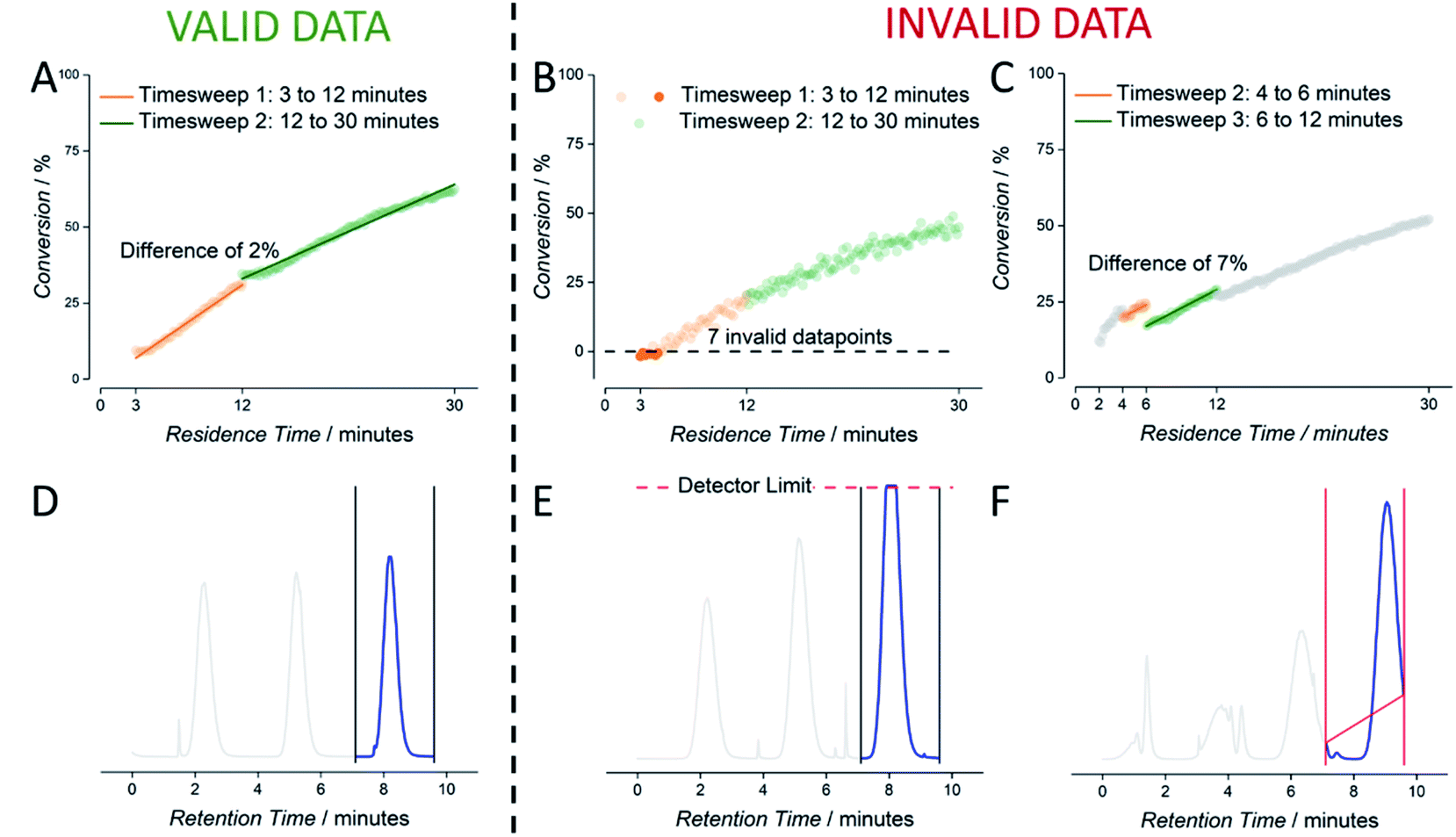 | ||
| Fig. 4 Visualization of the employed data cleaning algorithm. NMR data is checked for negative conversion (B) and timesweep jumps; valid data is shown in (A). SEC data is checked for detector saturation (E) and incorrect baseline parameters (F); valid data is shown in (D). | ||
Also, SEC data is prone to minor irregularities, mostly stemming from incorrect setting of automatic integration limits. Oftentimes, manual correction in SEC analysis can be performed to obtain better data. Predefined baseline borders on the raw elugram obviously lead to misinterpretation of the polymer sample. Such data are flagged and need to be reviewed by the operator. In future we will seek to make this correction fully automatic, yet this requires full integration of the commercial SEC software used in data acquisition. At this stage, SEC deviations are only automatically flagged, and trigger a correction by the operator. Adjusting the integration region prevents the loss of valid data. Only data where measured elugram intensities exceeded detector saturation are removed from the dataset entirely (Fig. 4D).
Data aggregation
After all data had been cleaned and checked for internal consistency, the next issue to address was to how to present such a large dataset coherently. Individual plots are shown in the ESI† for all screenings for all eight acrylic monomers, each at 4, 2 and 1 M concentration (0.005 M AIBN, DPtarget of 50, 80 °C, butyl acetate as solvent). It is obvious, that a form of data aggregation, hence presentation of the data in a reduced form is required for meaningful interpretation of data. Such aggregation (again, this can be obtained automatically via feeding the csv file into python code) is shown in Fig. 5. The simplest form of aggregation is to carry out the same test as given in Fig. 3. Since experiments were designed to keep the target degree of polymerisation constant, ideally all polymerizations should yield identical DP/conversion plots. Fig. 5C shows such an overlap of data. As can be seen, the data exhibits quite some scatter, and a linear regression reports a slope of 56 versus the theoretically expected 50. While this deviation (merely 10% from theory) is in principle satisfying, it must be noted that no individual calibrations of all monomers are available, and hence deviations between experiment and theory most likely stem from SEC calibration errors that have no root in the automation or the carried out automated experiment and analysis. If anything, individual datasets all show much lower internal scatter (as exemplified in Fig. 3), and the overall good average proves, if anything, the power of statistics over individual systematic errors (here absence of correct calibration).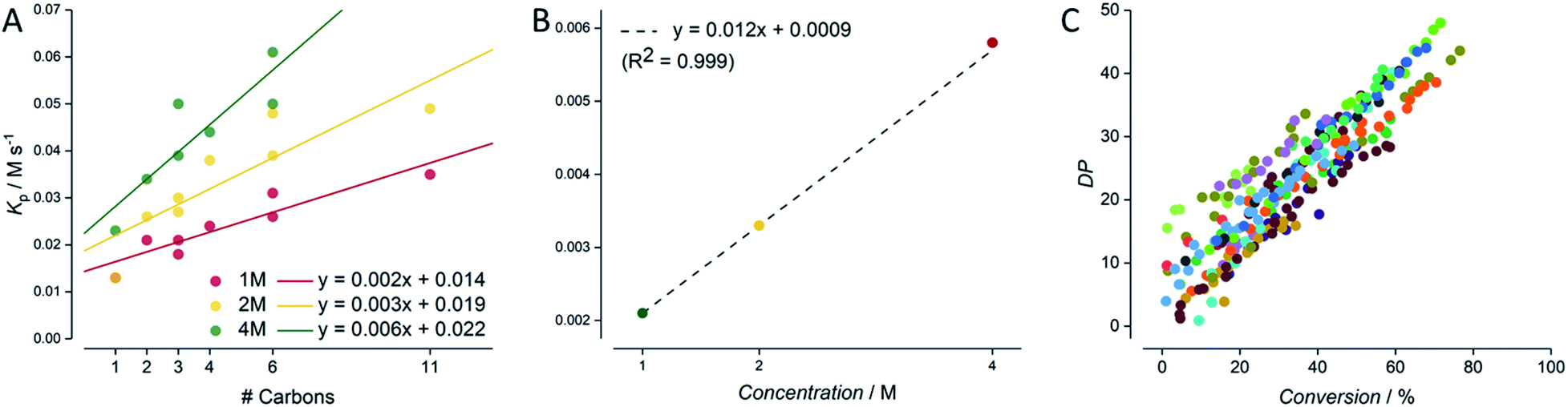 | ||
| Fig. 5 Analysis of the lab-wide monomer screening carried out by 8 individual operators. The ester side-chain length of the acrylic monomer correlates with the rate of polymerisation (slope tres − ln([M]0/[M]t)) (A). Moreover, a strong concentration dependency is observed (B). A combined conversion–DP plot confirms the RAFT kinetics and highlights the consistent data acquisition (C). | ||
More interesting, and at the same time also more robust since not dependent on individual calibrations, is the meta-analysis of the conversion data obtained from NMR. Each individual time/conversion plot can be reduced to an overall polymerization rate coefficient, obtained as the slope of a first order kinetic plot of data. While such rate coefficient KP has no direct meaning (true rate coefficients can only be expressed for each individual chain reaction), this method provides nonetheless a good quantification of data, and makes the various experiments comparable. Fig. 5A shows the apparent rate of polymerization RP for each experiment as a function of number of carbon atoms in the side chain of the monomer. This presentation is chosen since the propagation rate coefficient of monomers is known to scale with the length of the ester side chain, and RP in turn correlates typically directly with the propagation rate coefficient times the monomer concentration.36–38 Indeed, Fig. 5A shows nicely that this trend is also observed across all experiments. The rate of reaction increases with the number of carbon atoms. Also, as expected, the incline becomes steeper with increasing monomer concentration. While this correlation is crude (monomers with branched side chains are known to behave differently than linear side chains), it offers a further data reduction. If data for each individual monomer concentration is fitted linearly, all rates can be combined into a single correlation for monomer concentration. Since the overall rate of polymerization necessitates a linear correlation with monomer concentration (doubling of monomer concentration leads directly to doubling of the rate, since RP = KPcM) this correlation is directly identified, showing a remarkably high precision with an R2 of 0.999, as seen in Fig. 5B (underpinning the power of statistics to reduce experimental scatter). With this data plot, it is now possible to predict the rate for any acrylate RAFT polymerization that is carried out with the same RAFT agent and initiator concentration, interpolating over the whole monomer concentration and monomer space. One can only imagine the predictivity of polymerization rates if future experiments will also target initiator concentration and other RAFT agents as the next screening dimensions. It is obvious that provision of such data will then allow to model the reactions of practically any RAFT polymerization. As mentioned in the introduction, such modelling is to date not possible by using detailed kinetic modelling due to a lack of precise data and missing details in mechanistic understanding. We are certain, that our approach will in future not only fill this gap in predictivity, but that it will also provide the data basis to refine the mechanistic and kinetic understanding of the RAFT process. At first glance it may seem contradictory to use overall rate of polymerization data to determine mechanistic details of polymerization, as this usually involves complex and detailed kinetic studies into distinct chain growth reaction steps. However, with the present precision, once enough data is available, deep learning methods will become feasible that can bridge the current knowledge gap in mechanistic and kinetic understanding of radical polymerizations, especially when combined with deterministic modelling of reactions.
Having said that, we are confident though that the present work outlines the method nicely, and demonstrates its potential. Obviously, there are no limits in terms of data interpretation, and connection of this experimental data with kinetic data already available in literature. It is, however, sure that provision of big data, and aggregation of data will be a key in the future to exploit the full potential unfolded here.
Conclusions
We have demonstrated how an online-monitoring flow polymerization setup can be fully automated to yield very consistent and statistically robust collections of data in a high-throughput fashion. The operation of the setup is shown to be independent of the operator, including undergraduate students, PhD without RAFT or flow chemistry background, and the principle investigator of the research group, who has no daily lab practice. While this seems to be an incremental software improvement “only” on first glance, this is a significant development towards creation of big chemical data for use in machine learning advances.Next to automation, we have shown how datasets are automatically cleaned by algorithms to provide fully consistent data treatment and determination of statistical outliers. After data cleaning, all results are aggregated in simple plots, which in turn then allow to interpolate rate information in the full experimental space covered. As such, our method does not only mark the crossing of the high-throughput line in polymerization monitoring, it also demonstrates the full digitalization of the process, providing machine-readable outputs ready to be transferred into databases. We envisage for the future that such databases can be expanded – via interlaboratory collaboration – using open data and FAIR (findability, accessibility, interoperability and reusability) data use principles. Such approach would lead to true generation of ‘big data’ and deep learning approaches towards a better understanding of reaction kinetics. At the same time, this approach would eliminate systematic errors that might be present in single reaction setups, and increase not only the precision, but also accuracy of rate determinations.
Data availability
The code for the software described in the manuscript is available via the GitHub platform. All acquired data is collated in the ESI† section of the manuscript.Author contributions
Joren Van Herck: conceptualization, setup optimization, software development, data cleaning algorithm, final analysis, writing and editing. Iyomali Abeysekera, Axel-Laurenz Buckinx, Kewei Cai, Jordan Hooker, Kirti Thakur, Emma Van de Reydt, Pieter-Jan Voorter, Dries Wyers: experimental work. Tanja Junkers: experimental work, conceptualization, writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Monash University for general funding.Notes and references
- C. Guerrero-Sanchez, R. Yañez-Macias, M. Rosales-Guzmán, M. A. De Jesus-Tellez, C. Piñon-Balderrama, J. J. Haven, G. Moad, T. Junkers and U. S. Schubert, High-Throughput/High-Output Experimentation in RAFT Polymer Synthesis, RAFT Polymerization, Wiley-VCH, Weinheim, 2021 Search PubMed.
- F. Soheilmoghaddam, M. Rumble and J. Cooper-White, Chem. Rev., 2021, 121, 10792–10864 CrossRef CAS.
- R. Potyrailo, K. Rajan, K. Stoewe, I. Takeuchi, B. Chisholm and H. Lam, ACS Comb. Sci., 2011, 13, 579–633 CrossRef CAS PubMed.
- Y. Zheng, Y. Luo, K. Feng, W. Zhang and G. Chen, ACS Macro Lett., 2019, 8, 326–330 CrossRef CAS PubMed.
- J. J. Haven, E. Baeten, J. Claes, J. Vandenbergh and T. Junkers, Polym. Chem., 2017, 8, 2972–2978 RSC.
- I. Perevyazko, A. Lezov, A. S. Gubarev, E. Lebedeva, G. Festag, C. Guerrero-Sanchez, N. Tsvetkov and U. S. Schubert, Polymer, 2019, 182, 121828 CrossRef.
- M. Rubens, J. Van Herck and T. Junkers, ACS Macro Lett., 2019, 1437–1441 CrossRef CAS PubMed.
- S. T. Knox, S. Parkinson, R. Stone and N. J. Warren, Polym. Chem., 2019, 10, 4774–4778 RSC.
- M. Rosales-Guzmán, O. Pérez-Camacho, C. Guerrero-Sánchez, S. Harrisson, R. Torres-Lubián, J. Vitz, U. S. Schubert and E. Saldívar-Guerra, ACS Comb. Sci., 2019, 21, 771–781 CrossRef PubMed.
- D. J. Keddie, C. Guerrero-Sanchez, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2011, 44, 6738–6745 CrossRef CAS.
- H. Zhang, V. Marin, M. W. M. Fijten and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1876–1885 CrossRef CAS.
- R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2425–2434 CrossRef CAS.
- S.-J. Richards, A. Jones, R. M. F. Tomás and M. I. Gibson, Chem.–Eur. J., 2018, 24, 13758–13761 CrossRef CAS PubMed.
- S. Oliver, L. Zhao, A. J. Gormley, R. Chapman and C. Boyer, Macromolecules, 2019, 52, 3–23 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS.
- J. Van Herck and T. Junkers, Chemistry–Methods, 2022, 2, e202100090 Search PubMed.
- J. Niu, Z. A. Page, N. D. Dolinski, A. Anastasaki, A. T. Hsueh, H. T. Soh and C. J. Hawker, ACS Macro Lett., 2017, 6, 1109–1113 CrossRef CAS PubMed.
- L. L. Fillbrook, M. D. Nothling, M. H. Stenzel, W. S. Price and J. E. Beves, ACS Macro Lett., 2022, 166–172 CrossRef CAS PubMed.
- J. J. Haven and T. Junkers, Eur. J. Org. Chem., 2017, 2017, 6474–6482 CrossRef CAS.
- C. Botha, J. Höpfner, B. Mayerhöfer and M. Wilhelm, Polym. Chem., 2019, 10, 2230–2246 RSC.
- C. F. Carter, H. Lange, S. V Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393–404 CrossRef CAS.
- J. J. Haven, J. Vandenbergh and T. Junkers, Chem. Commun., 2015, 51, 4611–4614 RSC.
- M. Rubens, J. H. Vrijsen, J. Laun and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 3183–3187 CrossRef CAS PubMed.
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086–1094 RSC.
- C. Rosenfeld, C. Serra, S. O'Donohue and G. Hadziioannou, Macromol. React. Eng., 2007, 1, 547–552 CrossRef CAS.
- C. A. Shukla and A. A. Kulkarni, Beilstein J. Org. Chem., 2017, 13, 960–987 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- D. J. G. Devlaminck, P. H. M. Van Steenberge, M.-F. Reyniers and D. R. D’hooge, Polymers, 2019, 11, 320 CrossRef PubMed.
- D. Matioszek, S. Mazières, O. Brusylovets, C. Y. Lin, M. L. Coote, M. Destarac and S. Harrisson, Macromolecules, 2019, 52, 3376–3386 CrossRef CAS.
- J. J. Haven, C. Guerrero-Sanchez, D. J. Keddie, G. Moad, S. H. Thang and U. S. Schubert, Polym. Chem., 2014, 5, 5236–5246 RSC.
- J. Vandenbergh and T. Junkers, Macromolecules, 2014, 47, 5051–5059 CrossRef CAS.
- J. J. Haven, N. Zaquen, M. Rubens and T. Junkers, Macromol. React. Eng., 2017, 11, 1700016 CrossRef.
- S. T. Knox, S. J. Parkinson, C. Y. P. Wilding, R. A. Bourne and N. J. Warren, Polym. Chem., 2022, 13, 1576–1585 RSC.
- M. Grootveld, B. Percival, M. Gibson, Y. Osman, M. Edgar, M. Molinari, M. L. Mather, F. Casanova and P. Wilson, Progress in Low Field Benchtop NMR Spectroscopy in Chemical and Biochemical Analysis, Anal. Chim. Acta, 2019, 1067, 11–30 CrossRef CAS PubMed.
- A. P. Haehnel, M. Schneider-Baumann, L. Arens, A. M. Misske, F. Fleischhaker and C. Barner-Kowollik, Macromolecules, 2014, 47, 3483–3496 CrossRef CAS.
- A. P. Haehnel, M. Schneider-Baumann, K. U. Hiltebrandt, A. M. Misske and C. Barner-Kowollik, Macromolecules, 2013, 46, 15–28 CrossRef CAS.
- S. Beuermann, S. Harrisson, R. A. Hutchinson, T. Junkers and G. T. Russell, Polym. Chem., 2022, 13, 1891–1900 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2dd00035k |
| This journal is © The Royal Society of Chemistry 2022 |