
Efficient diffusion of superdense lithium via atomic channels for dendrite-free lithium–metal batteries†
Shiyuan
Zhou‡
 a,
Weixin
Chen‡
a,
Jie
Shi‡
b,
Gen
Li
a,
Fei
Pei
a,
Sangui
Liu
a,
Weibin
Ye
c,
Liangping
Xiao
a,
Ming-Sheng
Wang
c,
Dan
Wang
b,
Yu
Qiao
ad,
Ling
Huang
a,
Gui-Liang
Xu
e,
Hong-Gang
Liao
*ad,
Jian-Feng
Chen
b,
Khalil
Amine
efg and
Shi-Gang
Sun
a
a,
Weixin
Chen‡
a,
Jie
Shi‡
b,
Gen
Li
a,
Fei
Pei
a,
Sangui
Liu
a,
Weibin
Ye
c,
Liangping
Xiao
a,
Ming-Sheng
Wang
c,
Dan
Wang
b,
Yu
Qiao
ad,
Ling
Huang
a,
Gui-Liang
Xu
e,
Hong-Gang
Liao
*ad,
Jian-Feng
Chen
b,
Khalil
Amine
efg and
Shi-Gang
Sun
a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: hgliao@xmu.edu.cn
bState Key Laboratory of Organic Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P. R. China
cState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Materials, Pen-Tung Sah Institute of Micro-Nano Science and Technology, Xiamen University, Xiamen 361005, P. R. China
dInnovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen 361005, P. R. China
eChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, IL 60439, USA
fMaterials Science and Engineering, Stanford University, Stanford, CA, USA
gInstitute for Research & Medical Consultations, Imam Abdulrahman Bin Faisal University (IAU), Dammam, Saudi Arabia
First published on 11th November 2021
Abstract
The non-uniform aggregation of fast-diffused Li on an anode surface would aggravate its tip-effect-induced nucleation/growth, leading to the notorious dendrite growth in Li metal batteries (LMBs). Tuning the Li diffusion on the anode surface has been regarded previously as a mainstream method to induce its uniform deposition, while the diffusion of Li in the anode bulk is usually ignored. Here, conceptually different from the classic surface modification, we propose a molecular tunnelling strategy to construct atomic channels in graphite bulk, which enables the fast diffusion of superdense Li. Density functional theory calculations and ab initio molecular dynamics simulations prove that the bulk diffusion through atomic channels could become a new and dominating path. Its reversible and efficient diffusion has been further visualized by in situ transmission electron microscopy. As a result, when coupled with high-loading LiFePO4 cathodes (20 mg cm−2), a high areal capacity and 100% capacity retention are achieved over 370 cycles. Through this work a new strategy is developed based on the bulk-diffusion of superdense Li for dendrite-free LMBs, which can be pervasive in other high-performance energy storage systems.
Broader contextThe uncontrollable aggregation of Li at the anode surface would always lead to notorious dendrite growth, which extremely limits the practical application of Li metal batteries (LMBs) in terms of electrochemical performance and safety. Since the surface diffusion of Li in an anode is much faster than bulk diffusion, tuning its surface diffusion/deposition has been regarded as a mainstream method. Prior to the surface deposition of Li, the intercalation of Li+ would lead to the formation of a typical C6LiC6 state, sacrificing its diffusibility in the bulk phase. Therefore, the diffusion of Li in graphite layers has been largely prohibited. Here, different from the traditional surface modification of the anode, we built the interlayer and intralayer atomic channels in graphite by pre-tunnelling the graphite layers. Benefitting from its unique configuration, the obtained atomic channels allow the free and fast diffusion of Li with much enhanced kinetics. As a result, high-performance LMB full cells have been achieved with no capacity fading after long-term cycling. The use of atomic channels to diffuse compact Li provides a new path for the inhibition of Li dendrites. |
Introduction
As an alternative to commercial Li ion batteries, energy storage systems using Li metal as an anode have been considered as an effective solution to the next-generation high-energy-density batteries, mainly owing to its extremely high theoretical capacity (3860 mA h g−1) and low redox potential (−3.040 V vs. the standard hydrogen electrode).1,2 Despite the great prospects, the practical application of Li metal batteries (LMBs) is still hindered by the severe safety issues resulting from Li dendrite growth.3,4 Compared with electron transfer, the diffusion of highly reactive Li in the anode side, which proceeds mainly through the surface, is much more sluggish, and is consequently a dominating factor.5 The inhomogeneous and uncontrollable aggregation of Li at the electrode/electrolyte interface would always lead to notorious dendrite growth and limit their further application due to the unsatisfying electrochemical performance and severe safety issues. Since the surface diffusion of Li in the anode is much faster than bulk diffusion,6–9 tuning the diffusion/deposition of Li on the anode surface has been regarded as a mainstream method to induce its uniform deposition.Previous studies have focussed on constructing three-dimensional open-structure carbon skeletons and/or introducing guidable seeds, such as Au and Ag metal nanoparticles and Co and Ni single atoms.10–13 Prior to its surface deposition, Li+ would have to overcome a large energy barrier to intercalate into graphite layers, leading to a layer expansion of ∼0.2 Å,14,15 and it would be restrained in a typical C6LiC6 state in the bulk, sacrificing its diffusibility.16 Consequently, graphite bulk has rarely been considered for carrying dense and rapid Li flux (Fig. 1a), and the potential for diffusion of multilayer Li through graphite layers has not been fully utilized. By using density functional theory (DFT) calculations and in situ transmission electron microscopy (TEM), pioneering work reported by Kuhne et al. has demonstrated the feasibility of multilayer compact Li existing between two graphene layers, which far exceeds the typical C6LiC6 structure.17 However, the intercalation and diffusion properties of the reported bi-layer graphite sheets are inequivalent to those of the bulk carbon they exfoliated from. Additionally, their non-scalable material preparation makes them far from practical application in high-performance LMBs. Inspired by this work, a new Li diffusion pathway through bulk carbon can be constructed by pre-tunnelling graphite layers. The obtained atomic channels can allow the free and fast diffusion of superdense Li with much enhanced kinetics and safety (Fig. 1b).
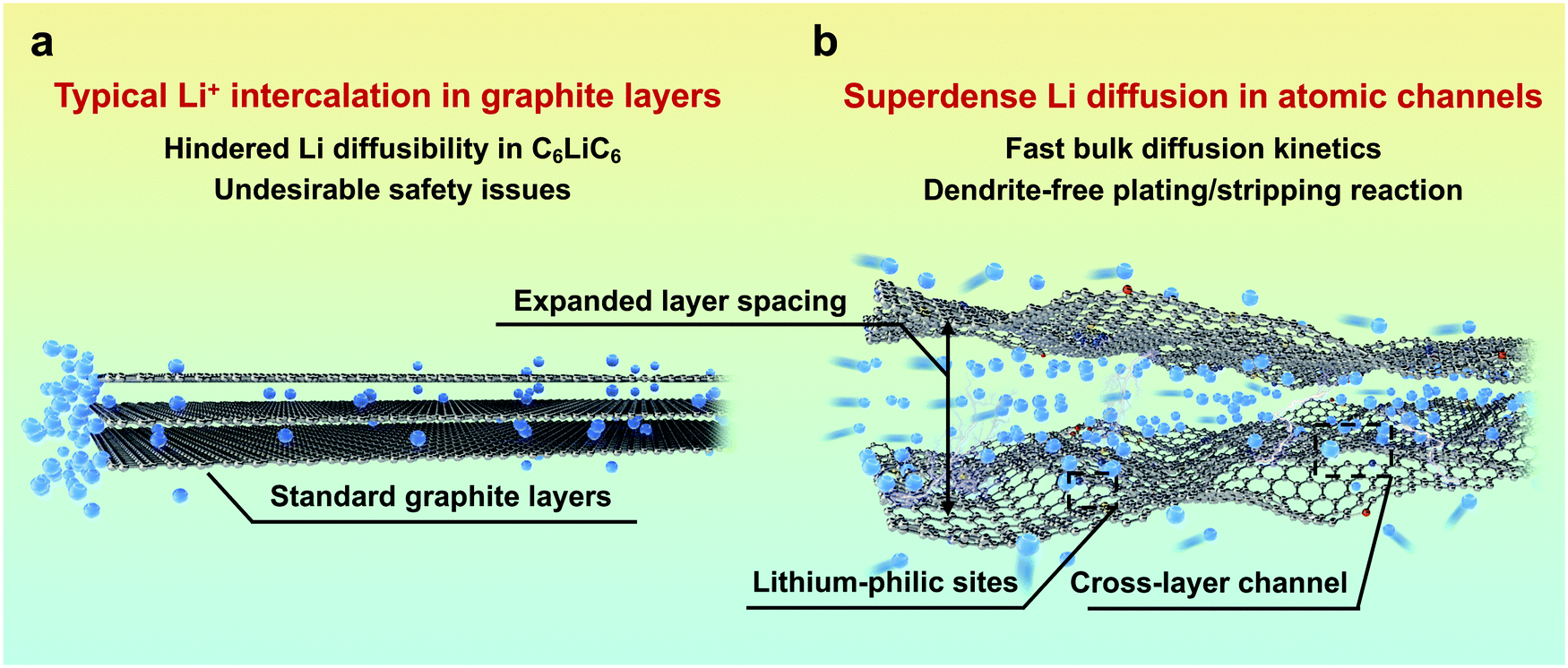 | ||
| Fig. 1 Comparative illustration of graphite layers and atomic channels. Schematic illustration of (a) typical Li+ intercalation in graphite layers and (b) superdense Li diffusion in atomic channels. | ||
In this contribution, by adopting a molecular tunnelling strategy, we construct a bulk diffusion Li conductor (BDLC) with abundant atomic channels for superdense Li transportation. Via pre-tunnelling graphite layers (layer spacing large as ∼7 Å), introducing voids and lithium-philic sites simultaneously, the interlayer and intralayer channels for Li diffusion are thus built. Different from the conventional surface diffusion/deposition mechanism, the atomic channel can effectively alleviate the dendrite issues caused by nonuniform surface deposition, and achieve rapid bulk diffusion. DFT calculations illustrate that due to the high affinity and reduced migration barrier of the BDLC, bulk diffusion through atomic channels could become a new and dominating path for Li transportation with high diffusion kinetics compared with its surface diffusion. Climbing image nudged elastic band (CI-NEB) and ab initio molecular dynamics (AIMD) simulations validate the diffusion behavior of superdense Li within interlayer and intralayer of atomic channels. Furthermore, a highly reversible and dendrite-free plating/stripping process of superdense Li in the BDLC bulk is visualized by in situ TEM. As a result, the Li@BDLC||Li@BDLC symmetric cell can operate for more than 2000 hours with a low hysteresis of 27 mV. When paired with high-loading LiFePO4 (LFP) cathodes over 20 mg cm−2, a high areal capacity of 3.9 mA h cm−2 (1.1× excess of Li) is reached, and 100% capacity retention over 370 cycles is achieved (1.3× excess of Li), respectively. The bulk diffusion strategy would provide a new perspective that is different from conventional surface diffusion, and would expand the knowledge about superdense Li diffusion and redefine the research of Li dendrite inhibition as well.
Results and discussion
Molecular dynamics simulations of interlayer/intralayer diffusion
Previous studies have evaluated whether superdense Li can form in two graphene layers using DFT calculations, but the dynamic diffusion process has not been studied.17 Here, we perform AIMD simulations to investigate the diffusion kinetics of different Li layers. Constrained by the hexagonal symmetry of graphene, AA-stacking shows a higher structural stability and smaller layer spacing than that of AB-stacking in both C6LiC6 and C3LiC3 structures (Fig. S1, ESI†). In order to avoid the structure distortion, the AA-stacking is mainly considered for Li diffusion in the BDLC. AIMD simulations are performed to calculate the layer spacing required for different amounts of Li in between graphite layers. At a high interlayer Li density, Li would quickly redistribute and shrink to a denser state after relaxation, and the final state suggests a stable C2LiC2 structure (Fig. 2a and b, and Fig. S2, ESI†). The increment in the layer spacing to 6.79 Å when 3 times more Li compared to C6LiC6 is inserted reveals that the widened layer spacing is a prerequisite for superdense Li diffusion (Fig. 2c). The energy barrier of Li migration is compared between the BDLC and graphite layers under different layer spacings (3.4 Å/6.8 Å) to evaluate the diffusion kinetics (Fig. 2d, and Fig. S3, ESI†). Under the same atomic arrangement, the expansion of layer spacing can lead to a decreased energy barrier and a lower final-state energy in the BDLC. The energy advantage of migration reflects the reduced barrier of crossing the bottleneck and boosted the kinetics of interlayer diffusion. Besides, benefitting from a high degree of electron localization, the N active sites exhibit a much better lithium-philicity than typical C sites (Fig. S4 and S5 and Table S1, ESI†). The high affinity can make Li more inclined to diffuse through the bulk channel of the BDLC than the surface of the graphite layer.18–20 Therefore, taking into account the N active sites, we find that the BDLC would allow a faster and tighter stacking of Li, which should be attributed to the strong ionization and high affinity to Li (Fig. S6 and S7, ESI†). What is more, the energy barrier is also reduced with the presence of N active sites (Fig. S8–S10, ESI†).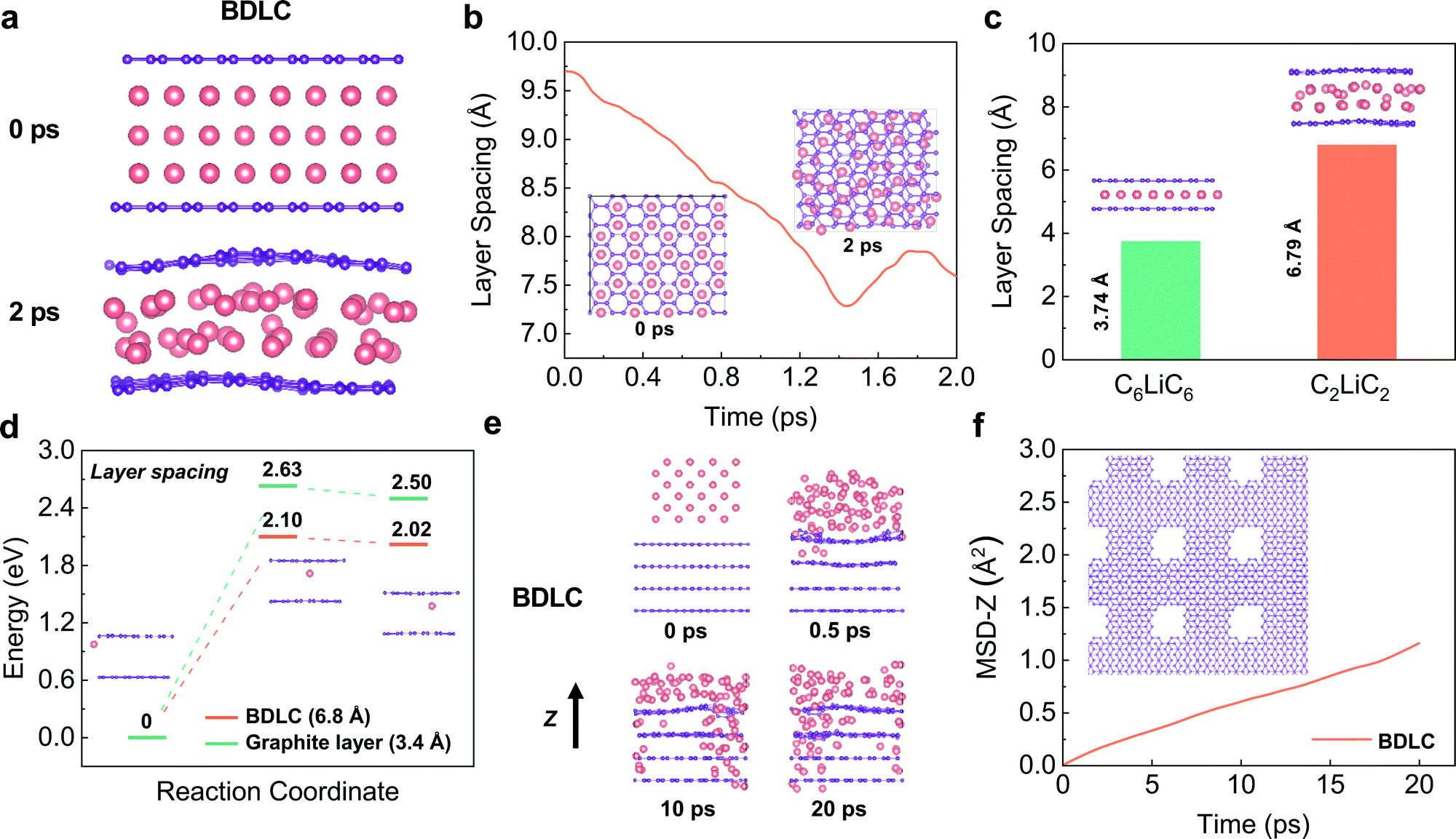 | ||
| Fig. 2 DFT calculations and AIMD simulations of Li diffusion through the atomic channels of BDLC. (a) Snapshots of AIMD simulations for interlayer diffusion in BDLC. (b) Variation of layer spacing over time for interlayer diffusion according to AIMD simulations. The images in the inset of 2b correspond to the top view of snapshots. (c) Layer spacing of C6LiC6 and C2LiC2. The images in the inset of 2c correspond to the structure of C6LiC6 and C2LiC2. (d) Migration energy profiles of Li diffusion with respect to different layer spacings. The images in the inset of 2d are the structures of BDLC (6.8 Å) corresponding to the initial, transition and final state of Li diffusion. (e) Snapshots of AIMD simulations for cross-layer Li diffusion in BDLC. (f) MSD curves of Li diffusion along the z-axis direction. The images in the inset of 2f correspond to the top view of the typical model of BDLC for Li intralayer diffusion. The pink and violet balls represent Li and C atoms, respectively. | ||
The typical cross-layer diffusion of high-dense Li is studied by AIMD simulations. In Fig. 2e and f, Li mainly diffuses through the voids of the BDLC, which is quantitatively evidenced by the time evolution of the mean square displacement along the z axis (MSD-z). Besides, N active sites in BDLC could provide an additional intralayer channel to mediate Li transportation, and realize a faster and more uniform intralayer diffusion (Fig. S11, ESI†): (i) the N–Li interaction allows N active sites to continuously capture the diffused Li; (ii) owing to the potential energy fluctuation, Li is inclined to achieve cross-layer diffusion in BDLC. From static and dynamic aspects, CINEB calculations and AIMD results validate the efficient diffusion and enhanced kinetics of superdense Li in the atomic channels of the BDLC, which may lead to a different plating/stripping behavior.
Construction of the BDLC with atomic channels
Based on the thermally activated NH3 molecules, we introduce a molecular tunnelling strategy to penetrate the standard graphite layers for the construction of atomic channels (Fig. S12, ESI†).21 In scanning electron microscopy (SEM) images, compared with the smooth interface of graphite carbon (GC), the BDLC reveals a hierarchical and rough surface structure (Fig. 3a and b). By comparing the intensity of the D-band and G-band (i.e. ID/IG) located at around 1330 and 1570 cm−1 of Raman spectra (Fig. 3c), a higher ratio of BDLC (0.99) indicates a larger proportion of the disordered structure,22 which is further confirmed from the aberration corrected high resolution TEM images (HRTEM). In Fig. 3d and e, in contrast to the well-defined graphite layers of GC, the BDLC presents a twisty interlayer structure and expanded layer spacings, while at the same time, the long-range order of graphite layers would not be sacrificed. Three typical interlayer channels of BDLC can be summarized as (I) bulging channel, (II) single-layer-graphene supported channel and (III) multi-layer-graphene supported channel (Fig. 3f). We believe that it is the undulating interlayer structure and small graphene nanosheets that stabilize the significantly expanded graphite layers. The average layer spacing demonstrates an expansion of 170%, from 3.33 Å of GC to 5.65 Å of BDLC (Fig. S13, ESI†). It should be noted that the layer spacing can even reach ∼7 Å in some areas. Besides, the thermal treatment at 450 °C leads to the most optimized interlayer structure, avoiding insufficient expansion and structure collapse (Fig. S14, ESI†). The intralayer structure of BDLC also experience an evident variation, with the generation of abundant cross-layer voids (Fig. 3g and h). In addition, the energy dispersive spectrum mappings and N 1s X-ray photoelectron spectroscopy results confirm the rich N active sites uniformly dispersed in the BDLC (Fig. S15, ESI†). The BDLC also presents a good structural flexibility when tension is applied or released (Fig. S16, ESI†). Therefore, the BDLC is constructed with stable atomic channels, which is consistent with the calculation model. | ||
| Fig. 3 Structure characterization of BDLC and GC. SEM images of (a) GC and (b) BDLC. (c) Raman spectra of GC and BDLC. Aberration corrected HRTEM images of the interlayer structure of (d) GC and (e) BDLC. The expanded layer spacings are marked with red squares. (f) Schematic illustration along with the HRTEM images of a typical expanded interlayer structure of BDLC, including the (I) bulging channel, (II) single-layer-graphene supported channel and (III) multi-layer-graphene supported channel. Aberration corrected HRTEM images of the intralayer structure of (g) GC and (h) BDLC. The cross-layer voids are marked with red circles. | ||
Visualization of plating/stripping of superdense Li
In order to investigate the Li diffusion behavior in the BDLC, we conduct in situ TEM to study the plating/stripping of superdense Li (Movie S1, ESI†), and its reversibility over 10 cycles (Movie S2, ESI†) and in situ selected-area electron diffraction (SAED) to distinguish Li in atomic channels (Movie S3, ESI†). In an open-cell nanobattery setup, Li and Li2O are used as the counter electrode and solid electrolyte, respectively (Fig. 4a). The diffusive direction of Li is driven by applying an opposite bias. Sequential TEM images show the typical morphologies of the BDLC during the plating/stripping process (Fig. 4b, c and Movie S1, ESI†). With the continuous plating of Li, the contrast of the areas outlined with orange dashed lines would increase gradually. The plated Li of the BDLC reveals an irregular shape and undergoes a continuous reshaping, unlike the typical Li crystal, which usually has a preferred crystal facet due to the rule of lowest surface energy. The as-plated Li metal would not be fixed in specific sites, but present a liquid-like diffusibility in the BDLC. Even so, the plated Li should not be attributed to the liquid phase since it is proved to be nanocrystalline by the SAED pattern. A reciprocal lattice rod, attributed to the (110) plane of Li metal, is observed in an in situ SAED investigation of BDLC (Fig. 4g and Movie S3, ESI†). The shape effect of the specimen, especially for an ultra-thin two-dimensional crystal, would cause the electron diffraction to deviate from the Bragg condition (2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ). The reciprocal lattice is no longer a geometric point, but a reciprocal lattice rod that expands along the thickness of the crystal. And the length of the reciprocal lattice is inversely proportional to the thickness of the crystal, that is, the thinner the crystal, the longer the reciprocal lattice rod. It should be noted that only when the Li metal appears in the atomic channels of the BDLC, it is possible to be thin enough to induce the intensity distribution of the diffracted beam with a certain width range. The crystal lattice of a few layered Li would deform along with the distortion and expansion of BDLC graphite layers, which would eventually elongate a diffraction spot into a reciprocal lattice rod.23 Therefore, the SAED pattern verifies the few layered Li in the atomic channels of the BDLC. These channels are in good contact with the flowable superdense Li metal, so they can diffuse freely in a Coble creep state,24 and their inhomogeneous aggregation and the formation of Li dendrites on the surface can be effectively avoided. During Li stripping, the contrast of the BDLC decreases rapidly. The plated Li outlined in Fig. 4b2 begins to shrink in a similar flowable state until totally stripped (Fig. 4c). Noticeably, even though the outlined areas appear to be isolated and not directly connected to the Li2O electrolyte on the surface of the BDLC, the stripping of metallic Li is not affected (Fig. 4c1). In contrast, the discontinuously plated areas and its creep-like stripping exemplify that the diffusion of Li occurs efficiently within the atomic channels of the BDLC rather than on the surface. It is commonly accepted in all solid-state in situ TEM that inefficient ion diffusion would always lead to the direct deposition of alkali metal on the surface, and it is often difficult to achieve a complete stripping process due to the Li concentration gradient.25,26 However, in atomic-channel-rich BDLC, either on the side close to the Li2O electrolyte or the side far from it we do not observe the dendrite growth (Fig. 4c2 and c3). All the plated Li metal could be completely stripped from the BDLC, ensuring a good reversibility on the electrode (Fig. 4c4). Such a dendrite-free and fast bulk diffusion behavior of Li highlights the role of atomic channels.
sinθ = nλ). The reciprocal lattice is no longer a geometric point, but a reciprocal lattice rod that expands along the thickness of the crystal. And the length of the reciprocal lattice is inversely proportional to the thickness of the crystal, that is, the thinner the crystal, the longer the reciprocal lattice rod. It should be noted that only when the Li metal appears in the atomic channels of the BDLC, it is possible to be thin enough to induce the intensity distribution of the diffracted beam with a certain width range. The crystal lattice of a few layered Li would deform along with the distortion and expansion of BDLC graphite layers, which would eventually elongate a diffraction spot into a reciprocal lattice rod.23 Therefore, the SAED pattern verifies the few layered Li in the atomic channels of the BDLC. These channels are in good contact with the flowable superdense Li metal, so they can diffuse freely in a Coble creep state,24 and their inhomogeneous aggregation and the formation of Li dendrites on the surface can be effectively avoided. During Li stripping, the contrast of the BDLC decreases rapidly. The plated Li outlined in Fig. 4b2 begins to shrink in a similar flowable state until totally stripped (Fig. 4c). Noticeably, even though the outlined areas appear to be isolated and not directly connected to the Li2O electrolyte on the surface of the BDLC, the stripping of metallic Li is not affected (Fig. 4c1). In contrast, the discontinuously plated areas and its creep-like stripping exemplify that the diffusion of Li occurs efficiently within the atomic channels of the BDLC rather than on the surface. It is commonly accepted in all solid-state in situ TEM that inefficient ion diffusion would always lead to the direct deposition of alkali metal on the surface, and it is often difficult to achieve a complete stripping process due to the Li concentration gradient.25,26 However, in atomic-channel-rich BDLC, either on the side close to the Li2O electrolyte or the side far from it we do not observe the dendrite growth (Fig. 4c2 and c3). All the plated Li metal could be completely stripped from the BDLC, ensuring a good reversibility on the electrode (Fig. 4c4). Such a dendrite-free and fast bulk diffusion behavior of Li highlights the role of atomic channels.
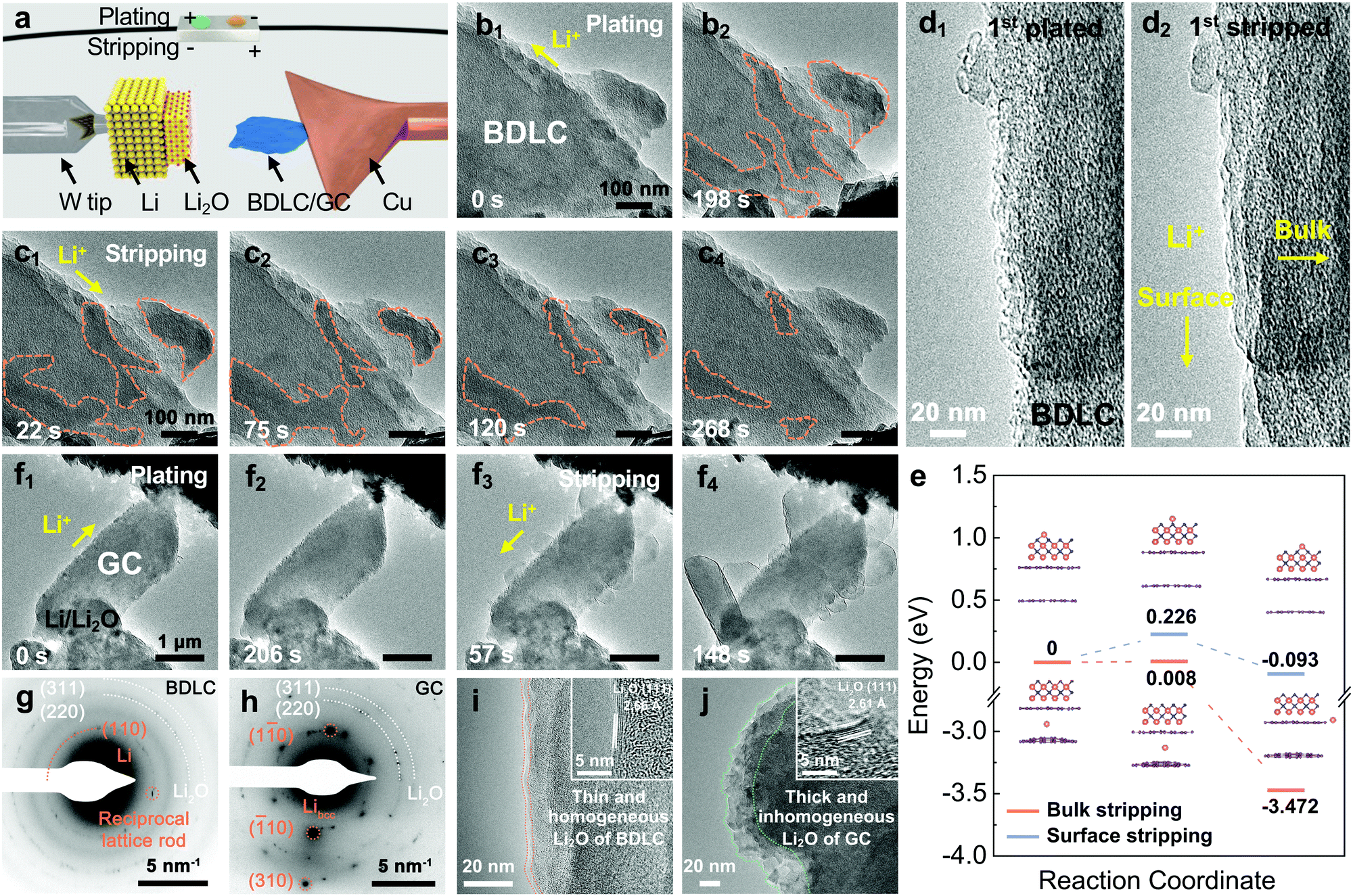 | ||
| Fig. 4 In situ TEM characterization of the Li plating/stripping process. (a) Schematic illustration of the in situ TEM device. (b and c) In situ TEM images of BDLC plated for (b1) 0 s, (b2) 198 s and then stripped for (c1) 22 s, (c2) 75 s, (c3) 120 s, and (c4) 268 s. (d) Surface details of the 1st Li (d1) plated/(d2) stripped BDLC selected from a 10 cycle reversible Li plating/stripping process under a higher magnification. (e) Migration energy profiles of Li diffusion regarding bulk stripping and surface stripping. The models of BDLC have a layer spacing of 6.8 Å to imitate bulk stripping, and a single layer of Li2O is fixed on it to imitate surface stripping. The images in the inset of 4e are the structures of bulk stripping and surface stripping corresponding to the initial, transition and final state. (f) In situ TEM images of GC plated for (f1) 0 s, (f2) 206 s and then stripped for (f3) 57 s, (f4) 148 s. (g) SAED images of BDLC obtained after a 10 cycle reversible Li plating/stripping process in Fig. S19 (ESI†), (h) GC obtained after an irreversible Li plating/stripping process in Fig. S20 (ESI†). (i and j) Comparison of surface structures between BDLC and GC after in situ TEM plating/stripping under a higher magnification. The HRTEM images in the inset of 4i and j are the layer spacings of Li2O corresponding to BDLC and GC, respectively. The diffusive directions of Li+ are marked in 4b1, c1, d2, f1 and f3. The pink, violet and blue balls represent Li, C and O atoms, respectively. | ||
To quantitatively distinguish the density and distribution of plated Li, we have estimated the content ratio of Li and C atoms. For the in situ TEM images in Fig. 4b, c and Movie S1 (ESI†), the mass-thickness contrast contributed by the scattering of C and Li atoms is the dominant factor. Among them, the ratio of scattered electrons (N) to the total number of electrons (N0) from specimens (1, 2,…) arriving at the detector could be expressed as:27–29
 | (1) |
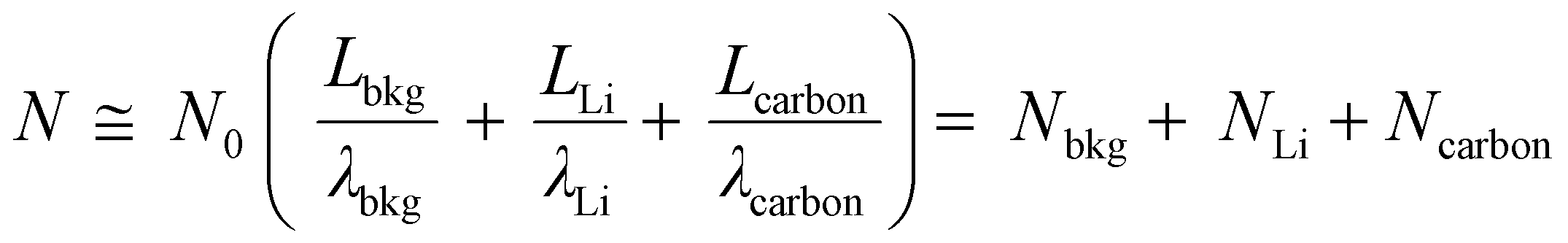 | (2) |
Therefore, by analyzing the relative contrast before and after Li plating in specific areas (detailed calculation is presented in Fig. S17 and Eqn S4–S7, ESI†), we could calculate the overall specimen thickness of Li (LLi = 250.5 Å) and C (Lcarbon = 89.3 Å), respectively. In the fully plated BDLC, the highest overall Li/C atomic ratio is around 2.8. By comparing the fully plated and fully stripped BDLC (Fig. S18, ESI†), most areas have limited Li plated on the surface, which is further confirmed from HRTEM images to be ultra-small nanograins (Fig. 4d). Then, by measuring the surface areas, the average thickness of surface Li (Lsurface Li) reaches 95.2 Å. Even considering an ideal surface-plated Li on both sides in the TEM imaging mode, the thickness of bulk Li (Lbulk Li) is measured to be 60.1 Å, and the bulk Li/C atomic ratio can be correspondingly estimated to be at least 0.67 (Lbulk Li/Lcarbon), 4 times of the conventional C6LiC6 structure (0.167). This indicates the superdense Li within the atomic channels of the BDLC. More surface details of the BDLC can be found from a 10 cycle reversible Li plating/stripping process under higher magnification (Fig. 4d, Fig. S19 and Movie S2, ESI†). After the 1st Li plating, numerous tiny nanograins are observed on the surface of the BDLC (Fig. 4d1), which then can be immediately and completely stripped within a short time, leaving a smooth surface without Li dendrite and dead Li (Fig. 4d2). Even after the 10th plating/stripping, the surface of the BDLC still remains smooth and intact (Fig. S19, ESI†). This not only verifies a small amount of Li on the surface of the BDLC, but also demonstrates a highly reversible Li plating/stripping behavior. More importantly, both the surface and the bulk of BDLC provide fast Li diffusion channels. Therefore, it motivates us to further comparatively study the Li stripping efficiency through these channels, and the DFT-based calculation shows that both bulk and surface stripping of Li have extremely low energy barriers (<0.5 eV), as shown in Fig. 4e.
In comparison, in situ TEM plating/stripping of GC nanosheets with a standard graphite layer is studied under the same conditions (Fig. 4f and Movie S4, ESI†). During Li plating, the edges of GC would become rough gradually, but seldom can we observe the distinct contrast change on GC (Fig. 4f1 and f2). On comparing with the plating behaviors of superdense Li in atomic channels of BDLC, it is found that the plated Li is mainly on the surface rather than in the bulk of GC. However, when the stripping begins, explosive growth of Li takes place on the surface of GC directly (Fig. 4f3 and f4), and the as-formed Li metal cannot disappear during the subsequent plating process, remaining as Li dendrite or dead Li on the surface of GC (Fig. S20, ESI†). In the in situ TEM plating/stripping of GC under a higher magnification, the sluggish Li diffusion and rapid dendrite growth during stripping have been further verified (Fig. S19 and Movie S5, ESI†). As shown in the SAED pattern of GC (Fig. 4h), unlike the polycrystalline diffraction rings of Li in the BDLC, GC presents a superposition of multiple separate metallic Li diffraction spots, which is achieved through irreversible growth of Li dendrites.25 The stripped Li would grow along a preferred crystal facet on the surface of GC, and this contributes to the typical reciprocal lattice of metallic Libcc (body-centered cubic).30 After stripping of Li, a thin and homogenous Li2O layer is observed on the surface of the BDLC (Fig. 4i), in sharp contrast to the thick and inhomogeneous Li2O layer of GC (Fig. 4j). Their lattice fringes reach 2.66 and 2.61 Å, respectively, belonging to the (111) plane of Li2O. Two sets of SAED patterns and HRTEM images comparatively reflect the difference in Li morphology and SEI layers of BDLC and GC. The in situ TEM observation of GC intuitively reflects that Li diffusion has a higher energy barrier and lower spontaneity, so Li metal tends to be plated on its surface instead of in the bulk. During stripping, the sluggish diffusion kinetics and local concentration of Li would lead to rapid dendrite growth. If it happens in a real battery, the plating/stripping behaviors of GC would probably cause unbearable problems such as low CE, poor cycling performance, sever safety issues, etc.
To verify the performance of the BDLC in a real coin-type cell, ex situ SEM characterization is conducted to observe the plated/stripped Li morphologies of the BDLC and GC at the 1st, 5th and 10th cycles (0.5 mA cm−2, 5 mA h cm−2, ester-based electrolyte), accompanied by the corresponding electrochemical profiles (Fig. S21, ESI†). After the 1st plating, the BDLC presents a compact and smooth surface morphology, while some gaps are observed in GC. These unfilled areas would increase after the 5th plating in GC with its decreased CE (91.4%), leading to the formation of blocky Li metal. The non-uniformly plated Li is hard to be fully stripped. An irreversible and uneven Li stripping is observed in GC after the 5th stripping, in comparison with the fully stripped surface of the BDLC. After the 9th plating/stripping, BDLC||Li maintains a high CE of 96.6%, while GC||Li quickly declines to 65.8%. Therefore, in sharp contrast to the smooth surface of the BDLC, filamentous Li dendrites are observed in the most area of GC after the 10th plating. This would increase the irreversible reaction in GC and finally lead to battery failure and safety issues.31,32 In comparison, the BDLC demonstrates a uniform and stable plating/stripping reaction after several cycles, which is also in accordance with in situ TEM results. Generally, bulk diffusion of superdense Li predicted by DFT and AIMD has been visualized by in situ TEM and ex situ SEM studies. The distinct difference of the Li plating/stripping behavior between the BDLC and GC confirms that atomic channels are constructed in the entire self-supported BDLC electrode, which enable the fast diffusion of massive Li and facilitate the highly reversible reaction via a dendrite-free way.
Electrochemical performance of BDLC
To study the Li plating/stripping reversibility of BDLC in LMBs, half cells are employed to investigate the Coulombic efficiency (CE) with different current densities and Li areal capacities, using an ester-based electrolyte (1 M LiPF6 in EMC/EC + 5 wt% FEC). At a current density of 1 mA cm−2 and an areal capacity of 2 mA h cm−2, the CE of BDLC||Li reaches 98.5% after 150 cycles, while the CE of GC||Li would decline to 91% (Fig. 5a). What is worse, the CE of Cu||Li would quickly get exhausted to 40.9% around 60 cycles. To evaluate the diffusion kinetics, the CE is tested at 2 and 5 mA cm−2 with an areal capacity of 1 mA h cm−2, BDLC||Li delivers a stable CE at the increasing current densities, maintaining at 99.6% and 98.6% after 100 cycles (Fig. 5b). Even at a fast-charging current density of 8 mA cm−2, BDLC||Li can still operate stably after 100 cycles (85.9%), while GC||Li and Cu||Li would experience an obvious fluctuation of CE and decrease rapidly. Benefitting from the atomic channels, Li in the BDLC achieves a rapid diffusion and the unsatisfying cyclability decay caused by Li dendritic growth has also been effectively inhibited. Compared with the previous studies, BDLC||Li reveals an appealing stability of CE in the ester-based electrolyte (Table S2, ESI†). In electrochemical impedance spectroscopy (Fig. S22 and Table S3, ESI†), BDLC presents a much smaller charge-transfer resistance (Rct) of 53.8 Ω compared with GC (86.3 Ω) and Cu (337.9 Ω). The above investigation should be attributed to the high electrode conductivity and enhanced diffusion kinetics of the BDLC. To estimate the stability of the plating/stripping process, symmetric cells are assembled by pairing two identical electrodes with the same Li areal deposition of 3 mA h cm−2. After 2000 h at 1 mA cm−2 for 1 mA h cm−2, the hysteresis voltage of Li@BDLC||Li@BDLC is maintained at 27 mV, showing a highly stable Li plating/stripping (Fig. 5c and Fig. S22, ESI†). No obvious increase in overpotential can be found in the voltage curves at the selected cycles of Li@BDLC (inserted image of Fig. 5c). In contrast, the hysteresis of Li@GC||Li@GC reaches 281.9 mV and tends to increase continuously while the hysteresis of Li@Cu||Li@Cu quickly soars up in just 46 cycles. Moreover, even at a higher current density of 3 mA cm−2, the Li@BDLC||Li@BDLC still exhibits a high stability with a hysteresis of 28.5 mV after 800 cycles (Fig. S23, ESI†). The stable cycling performance of the BDLC in symmetric cells should be ascribed to the uniform and reversible Li plating/stripping process endowed by its abundant atomic channels. | ||
| Fig. 5 Electrochemical performance of BDLC and GC. CE at a current density of (a) 1 mA cm−2 for 2 mA h cm−2, (b) 2, 5 and 8 mA cm−2 for 1 mA h cm−2. For the half-cell tests, a pre-cycle is performed at 0.2 mA cm−2 between 0 and 1.0 V for 5 cycles to form a stable SEI layer. (c) Constant charging/discharging voltage profiles of symmetric cells at a current density of 1 mA cm−2 for 1 mA h cm−2. After a pre-cycle at 0.2 mA cm−2 between 0 and 1.0 V for 5 cycles, 3 mA h cm−2 of Li is pre-electrodeposited at 0.2 mA cm−2 to obtain Li@BDLC, Li@GC, Li@Cu. The image in the inset of Fig. 6c shows the details of representative voltage-time profiles. (d) Charge/discharge profiles of LFP cathodes against Li@BDLC at the 4th, 100th, 200th and 300th cycles (after activation at 0.2 C for 3 cycles). (e) Comparison of cycling performance between Li@BDLC||LFP and Li@GC||LFP with high LFP areal loading of 20 mg cm−2, 1.3 × limited excess of Li, at 0.5 C. (f) Comparison of electrochemical performance against the Li@BDLC||LFP full cell regarding LFP areal loading, capacity retention rate and specific capacity (details are provided in Table S4, ESI†). All the coin-type cells are tested in the ester-based electrolyte. | ||
To investigate the application potential of the BDLC, the full cells are assembled by pairing Li@BDLC anodes with LFP cathodes under the harsh conditions of high LFP areal loading, limited Li excess, long cycles and fast charge/discharge rates. For high areal loading of LFP (Fig. S24, ESI†), in order to better utilize its capacity, the full cells are activated for a few cycles between 2.5 and 4.0 V, for instance, 0.2 C for cycling performance and 0.05 C for rate performance (170 mA h g−1 = 1 C). In Fig. 5d and e, after activation (20 mg cm−2 LFP, 1.3× excess of Li, 0.5 C), the full cell of Li@BDLC||LFP delivers a higher discharge capacity and more stable cyclability (156 mA h g−1versus 147 mA h g−1, 1st cycle at 0.5 C), while the Li@GC||LFP experiences a rapid capacity decline with an obvious fluctuation of CE only after 60 cycles and finally retains 27% of the initial capacity after 220 cycles (Fig. S25, ESI†). Noticeably, the Li@BDLC||LFP achieves an ultra-high cycling stability, delivering a discharge capacity of 157 mA h g−1 over 370 cycles with 100% capacity retention. When we further increase the areal loading of LFP and decrease the excess of Li (25 mg cm−2 LFP, 1.1× excess of Li, 0.5 C), the highest areal capacity of Li@BDLC||LFP could reach 3.9 mA h cm−2, and it can still operate stably over 100 cycles after activation (Fig. S26, ESI†). Compared with the most reported LFP-based LMBs, the Li@BDLC||LFP reveals a superior cycling performance especially when considering the high areal loading and limited excess of Li (Fig. 5f and Table S4, ESI†). When we increase the charge/discharge rate to 1 C (10 mg cm−2 LFP, 1.3× excess of Li), Li@BDLC||LFP can still achieve a stable cycling performance, delivering 138 mA h g−1 after 500 cycles and 97% capacity retention (Fig. S27, ESI†). In comparison, Li@GC||LFP would experience a much faster capacity fading, retaining 36% after 110 cycles (49 mA h g−1). This should be attributed to the sluggish Li+ diffusion kinetics and the irreversible Li plating/stripping process, as this difference would exacerbate in the rate performance (10 mg cm−2 LFP, 1.3× excess of Li). Li@BDLC||LFP demonstrates a better rate capability especially with the increase of charge/discharge rates, delivering discharge capacities of 157, 154, 151, 146, 135, 122 and 105 mA h g−1 at 0.1, 0.2, 0.3, 0.5, 1, 1.5 and 2 C, respectively (Fig. S28, ESI†). However, Li@GC||LFP can only provide 151, 148, 143, 132, 109, 87 and 69 mA h g−1. Even when operating back to 0.1 C, the capacity of Li@GC||LFP would still decrease rapidly. To further understand the reasons for the distinct difference in the electrochemical performance, ex situ TEM investigation is conducted for both anodes and cathodes of Li@BDLC||LFP and Li@GC||LFP after rate performance at a discharge state (Fig. 6). It can be observed that the carbon-coated LFP cathodes of both Li@BDLC||LFP and Li@GC||LFP remain well after rate performance compared with the pristine LFP, which can be consequently excluded for the capacity decay in LFP electrochemical performance (Fig. 6a–d). In sharp contrast, Li@BDLC and Li@GC anodes demonstrate a distinct difference on the surface. Compared with the homogeneous surface structure of Li@BDLC (Fig. 6e and f), Li@GC has a thick and rough surface (Fig. 6g). Investigation at a higher magnification reveals an irregular mosaic and inhomogeneous SEI layer of Li@GC (Fig. 6h), which is in accordance with the in situ TEM study. This illustrates that the atomic channels of the BDLC can facilitate uniform Li plating/stripping at various rates, diminish focused Li nucleation on the surface, and enable a dendrite-free electrochemical performance.
 | ||
| Fig. 6 HRTEM investigation of Li@BDLC||LFP and Li@GC||LFP after rate performance at a discharge state. HRTEM images of the pristine LFP cathode at (a) low magnification, (b) high magnification, showing a typical carbon-coated structure. HRTEM images of cycled LFP cathodes from (c) Li@BDLC||LFP and (d) Li@GC||LFP. HRTEM images of cycled (e and f) Li@BDLC and (g and h) Li@GC anodes at different magnifications. | ||
Considering the excellent electrochemical performance of pre-electrodeposited Li@BDLC, we further explore its potential through a simpler fabrication (molten Li adsorption in an argon-filled glovebox) and a practical application (directly using as an interlayer without pre-electrodeposited Li).33 Specifically, for molten Li adsorption in an argon-filled glovebox, the BDLC is placed on the top of molten Li made by heating fresh Li foil on a hotplate to 330 °C (Fig. S29, ESI†). Benefitting from its lithium-philicity, it is easy to wet the BDLC and it filled with the molten Li in a short time. The SEM images reveal the uniform surfaces of Li deposited areas. By further regulating the heating area and the amount of Li, it is relatively easy to prepare a large-scale Li@BDLC electrode with a specific areal capacity. For the BDLC interlayer, the coin-type cell is assembled with a high-loading LFP cathode (10 mg cm−2), BDLC interlayer without pre-electrodeposition and 100 μm Li foil, and is operated at 1 C (Fig. S30, ESI†). During the initial charging, Li+ would be deintercalated from the LFP cathode and intercalated/deposited in the BDLC. It is worth noting that the simple and practical battery assembly method would not sacrifice the excellent cycling performance of the BDLC, and a discharge capacity of 141 mA h g−1 is achieved with 100% capacity retention after 100 cycles, compared with 27 mA h g−1 of GC (19% capacity retention after 100 cycles). The above preparation method and battery applications further verify the significant role of atomic channels of the BDLC, and also reveal its large-scale prospect for dendrite-free and high-performance LMBs.
Conclusions
In conclusion, different from the traditional surface modification of the current collector, we construct atomic channels inside graphite to enable the bulk diffusion of superdense Li, for realizing dendrite-free LMBs. By using DFT calculations and AIMD simulations, the dynamic diffusion of multilayer dense Li in atomic channels is predicted. We further visualize the fast and reversible diffusion of superdense Li through the atomic channels of the BDLC by in situ TEM. This demonstrates the ideal application potential under limited Li excess when paired with a high areal loading of LFP cathodes, delivering the highest areal capacity of 3.9 mA h cm−2 and 100% capacity retention for 370 cycles. This work validates the possibility of bulk diffusion through atomic channels instead of surface diffusion, which points out a new avenue in guiding reversible Li plating/stripping for dendrite-free LMBs.Author contributions
H.-G. L., S.-G. S., K. A. and G.-L. X. conceived and designed this work. S. Z. and W. C. conducted the experiments and wrote the paper. J. S., D. W. and J.-F. C. designed and conducted the DFT calculations and AIMD simulations. G. L., W. Y. and L. X. conducted the TEM characterization. F. P. and S. L. measured the electrochemical performance. Y. Q. and L. H. analysed the characterization. All authors participated in the analysis of experimental data and discussion of the results, as well as in the writing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants no. 91934303, 21673198, 21673194, 21621091 and 21620102007) and the National Key Research and Development Program of China (2016YFB0100202). Research at the Argonne National Laboratory was funded by the US Department of Energy (DOE), Vehicle Technologies Office under contract no. DE-AC02-06CH11357. K. A. and G. X. also thank the support from Clean Vehicles, US-China Clean Energy Research Centre (CERC-CVC2).Notes and references
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, Chem. Rev., 2020, 120, 6783–6819 CrossRef CAS PubMed.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- R. Rojaee and R. Shahbazian-Yassar, ACS Nano, 2020, 14, 2628–2658 CrossRef CAS PubMed.
- J. Zheng, M. S. Kim, Z. Tu, S. Choudhury, T. Tang and L. A. Archer, Chem. Soc. Rev., 2020, 49, 2701–2750 RSC.
- J. Shen, G. Liu, Y. Han and W. Jin, Nat. Rev. Mater., 2021, 6, 294–312 CrossRef CAS.
- Z. Hong, Y. Zhen, Y. Ruan, M. Kang, K. Zhou, J. M. Zhang, Z. Huang and M. Wei, Adv. Mater., 2018, 14, e1802035 CrossRef PubMed.
- J. Chen, B. Yang, H. Hou, H. Li, L. Liu, L. Zhang and X. Yan, Adv. Energy Mater., 2019, 9, 201803894 Search PubMed.
- X. Sun, C. Wang, Y. Gong, L. Gu, Q. Chen and Y. Yu, Small, 2018, 14, e1802218 CrossRef PubMed.
- Q. Jin, K. Wang, H. Li, W. Li, P. Feng, Z. Zhang, W. Wang, M. Zhou and K. Jiang, Chem. Eng. J., 2021, 417, 128104 CrossRef CAS.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 1–7 CrossRef.
- R. Zhang, X. Chen, X. Shen, X.-Q. Zhang, X.-R. Chen, X.-B. Cheng, C. Yan, C.-Z. Zhao and Q. Zhang, Joule, 2018, 2, 764–777 CrossRef CAS.
- K. Xu, M. Zhu, X. Wu, J. Liang, Y. Liu, T. Zhang, Y. Zhu and Y. Qian, Energy Storage Mater., 2019, 23, 587–593 CrossRef.
- P. Zhai, T. Wang, W. Yang, S. Cui, P. Zhang, A. Nie, Q. Zhang and Y. Gong, Adv. Energy Mater., 2019, 9, 1804019 CrossRef.
- W. Cai, C. Yan, Y.-X. Yao, L. Xu, R. Xu, L.-L. Jiang, J.-Q. Huang and Q. Zhang, Small Struct., 2020, 1, 2000010 CrossRef.
- G. Yang, Z. Liu, S. Weng, Q. Zhang, X. Wang, Z. Wang, L. Gu and L. Chen, Energy Storage Mater., 2021, 36, 459–465 CrossRef.
- Y. Zhu, P. Simon and J. Ye, Natl. Sci. Rev., 2020, 7, 191–201 CrossRef.
- M. Kuhne, F. Borrnert, S. Fecher, M. Ghorbani-Asl, J. Biskupek, D. Samuelis, A. V. Krasheninnikov, U. Kaiser and J. H. Smet, Nature, 2018, 564, 234–239 CrossRef.
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, e1805121 CrossRef.
- X.-R. C. Xiang Chen, T.-Z. Hou, B.-Q. Li, X.-B. Cheng, R. Zhang and Q. Zhang, Sci. Adv., 2019, eaau7728 CrossRef PubMed.
- R. Zhang, X. R. Chen, X. Chen, X. B. Cheng, X. Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 7764–7768 CrossRef CAS PubMed.
- W. Chen, F. S. Cannon and J. R. Rangel-Mendez, Carbon, 2005, 43, 573–580 CrossRef CAS.
- X. Zhou, W. Huang, C. Shi, K. Wang, R. Zhang, J. Guo, Y. Wen, S. Zhang, Q. Wang, L. Huang, J. Li, X. Zhou and S. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 35296–35305 CrossRef CAS PubMed.
- R. D. Diehl, J. Ledieu, N. Ferralis, A. W. Szmodis and R. McGrath, J. Phys.: Condens. Matter, 2003, 15, 63–81 CrossRef.
- Y. Chen, Z. Wang, X. Li, X. Yao, C. Wang, Y. Li, W. Xue, D. Yu, S. Y. Kim, F. Yang, A. Kushima, G. Zhang, H. Huang, N. Wu, Y. W. Mai, J. B. Goodenough and J. Li, Nature, 2020, 578, 251–255 CrossRef CAS PubMed.
- X. H. Liu, Y. Liu, A. Kushima, S. Zhang, T. Zhu, J. Li and J. Y. Huang, Adv. Energy Mater., 2012, 2, 722–741 CrossRef CAS.
- W. Ye, F. Pei, X. Lan, Y. Cheng, X. Fang, Q. Zhang, N. Zheng, D. L. Peng and M. S. Wang, Adv. Energy Mater., 2020, 10, 1902956 CrossRef CAS.
- A. Wank, B. Wielage, H. Podlesak and T. Grund, J. Therm. Spray Technol., 2006, 15, 280–283 CrossRef CAS.
- J. Lu, Z. Aabdin, N. D. Loh, D. Bhattacharya and U. Mirsaidov, Nano Lett., 2014, 14, 2111–2115 CrossRef CAS.
- N. D. Loh, S. Sen, M. Bosman, S. F. Tan, J. Zhong, C. A. Nijhuis, P. Král, P. Matsudaira and U. Mirsaidov, Nat. Chem., 2017, 9, 77–82 CrossRef CAS PubMed.
- P. Wei, Y. Cheng, X. Yan, W. Ye, X. Lan, L. Wang, J. Sun, Z. Yu, G. Luo, Y. Yang, M. H. Rummeli and M.-S. Wang, Adv. Mater., 2021, 2105228, DOI:10.1002/adma.202105228.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef.
- Z. Xu, Z. Guo, R. Madhu, F. Xie, R. Chen, J. Wang, M. Tebyetekerwa, Y.-S. Hu and M.-M. Titirici, Energy Environ. Sci., 2021 10.1039/D1EE01346G.
- J. Wang, G. Huang and X.-B. Zhang, Batteries Supercaps, 2020, 3, 1006–1015 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02205a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |