
Size and isotopic ratio measurements of individual nanoparticles by a continuous ion-monitoring method using Faraday detectors equipped on a multi-collector-ICP-mass spectrometer
Shuji
Yamashita
 a,
Kota
Yamamoto
a,
Hiroaki
Takahashi
b and
Takafumi
Hirata
*a
a,
Kota
Yamamoto
a,
Hiroaki
Takahashi
b and
Takafumi
Hirata
*a
aGeochemical Research Center, Graduate School of Science, The University of Tokyo, Hongo 7-3-1, Bunkyo-ku, Tokyo, 113-0033, Japan
bRegulatory Standard and Research Department, Secretariat of Nuclear Regulation Authority (S/NRA/R), Roppongi 1-9-9, Minato-ku, Tokyo, 106-8450, Japan
First published on 3rd December 2021
Abstract
We investigated the capability of high-gain Faraday detectors equipped on a multi-collector-ICP-mass spectrometer (MFC-ICP-MS) in performing both particle size and Ag isotopic ratio determination for individual silver nanoparticles (Ag NPs). Two high-gain Faraday detectors equipped with 1013 Ω resistors were used to detect transient signals emanating from individual NPs (particle events). A continuous ion-monitoring method (CIM) was adopted to obtain reliable isotopic ratio data from the particle events. The CIM measurements of the signal intensities obtained from Ag NPs (40, 60, 80, 100, and 200 nm) exhibited a good linearity with the sizes of the Ag NPs. The variations of the measured 109Ag/107Ag ratio for the particles of each size in the isotopic ratio measurements (about 100 particles sampled per size) were about: 12% for 40 nm, 4.9% for 60 nm, 1.9% for 80 nm, 1.2% for 100 nm, and 0.26% for 200 nm. The resulting precision of the 109Ag/107Ag ratio measurements can be basically attributed to both shot noise (counting statistics) and background fluctuation on amplifiers. The data obtained here demonstrated that the MFC-ICP-MS system combined with the CIM can be employed to determine both the NP size and isotopic ratio of constituent elements.
Introduction
Isotopic ratio data for individual particles are beneficial for environmental studies dealing with source identification of artificial airborne particles. The production, transfer, and fate of these airborne particles (e.g., PM0.1 and PM2.5) can affect human health and influence climate, and need to be investigated.1 Pb isotopic signatures have been widely used for provenance studies on airborne particles in many countries (e.g., Europe,2,3 America,4,5 and Japan6,7). Moreover, several reports have revealed that nuclear fuel materials such as plutonium have been released into the environment in trace amounts.8–12 Submicron-sized colloid-facilitated transport is probably the principal means of long-distance transport of actinides in groundwater.13–15 The chemical fingerprints of radioactive particles based on the isotopic ratio of radioactive elements (Pb, Th, U or Pu) can provide key information on the identity of the reactors, the source of the particles or the processes used to produce them.16,17 Thus, the isotopic signature of individual colloids and particles can be used for the identification of actinides on colloids in groundwater and surface water, as well as nuclear safeguards.18,19A major problem associated with the isotopic ratio determination for individual particles (e.g., colloids and nanoparticles) is the long analysis time stemming from complicated sample preparation and analytical procedures. Kober et al. reported precise Pb isotopic ratio data from single zircon (ZrSiO4) crystals using thermal ionisation-mass spectrometry (TIMS).20 Although this technique allows Pb isotopic analyses for single particles, complicated sample preparation and time-consuming analytical procedures are required. Besides TIMS, sensitive and rapid isotopic analysis can be performed by multi-collector-inductively coupled plasma-mass spectrometry (MC-ICP-MS).21,22 The multi-collector array using Faraday detectors is still the principal system setup for MC-ICP-MS systems (MFC-ICP-MS). As such, this technique can be useful to determine the isotopic ratio of individual nanoparticles (NPs).
Recently, ICP-MS has been used for sensitive and rapid detection of individual particles (nano- and submicron level) to characterise properties such as size, particle number concentrations, and elemental composition.23–25 NPs are detected as transient signals (0.2–0.6 ms) in ICP-MS. To obtain reliable signal intensity data within the duration of each particle (particle event), data acquisition with a short dwell time (<100 μs) is generally used. However, the rate of coincidence (i.e., overlap of two or more particle events) increases with increased particle concentration even with a short dwell time. To avoid errors due to the overlapping of the particle events, the NP suspension has to be diluted.
We recently reported on Pt and Os isotopic ratio measurements from single NPs using an MC-ICP-MS utilising four high-gain ion detectors combined with a high-time resolution data acquisition system (HTR-MC-ICP-MS).26,27 A time resolution of up to 10 μs was possible with this system. However, the major drawback of the HTR-MC-ICP-MS system is that careful calibration of detector gains and correction of counting loss due to dead time are essential to obtain reliable elemental and isotopic data, making the system not user-friendly.
On the other hand, the MFC-ICP-MS system has the following unique features: (a) a basic system setup for many MC-ICP-MS instruments, (b) a simpler system configuration compared to multiple ion counters, and (c) a wide dynamic range in ion monitoring. Moreover, the Faraday detector is capable of monitoring high-count rate signals greater than 108 cps (counts per second). This is a very important capability for the monitoring of large-sized NPs (>80 nm) and submicron particles. For the HTR-MC-ICP-MS system, the monitoring of particles > 100 nm is hindered by the large contribution of the counting loss caused by the detector dead time. With the Faraday detector equipped with a 1011 Ω resistor, the system can monitor an ion count rate of 3 × 109 cps, which suggests high capability for submicron particle detection. Another feature of the Faraday detector is its higher amplification gain for small signals. Several pioneering studies revealed that Faraday detectors equipped with a 1013 Ω resistor instead of a 1011 Ω resistor can provide a better signal-to-noise ratio (S/N).28–30 The use of the Faraday detector equipped with a 1013 Ω resistor, however, leads to a slow response. The response of the Faraday detector takes about 100 ms, a time duration widely believed to disallow the use of Faraday detectors to keep up with the rapid changes in the signal intensities produced by the particles.
Due to the slow response of the Faraday detector, the measured isotopic ratios deviate from the true ratios when rapidly changing signal intensities were monitored.31 Several pioneering studies have developed practical correction methods to minimise the contribution of the slow response of the Faraday detector, including tau correction32 and linear correlation.31 Despite the ability to obtain better precision and accuracy in the measured isotopic ratio, systematic errors remained when transient signals were monitored.28,29 This is a critical problem in isotopic ratio determination for transient signals of particles. To obtain reliable isotopic ratio data from transient signals, a new correction method for the slow response of the Faraday detector has been described (i.e., a continuous ion-monitoring: CIM method).33 Reproducible 234U/238U and 235U/238U ratio determination is possible even for transient signals produced by laser ablation with a short time duration (<3 s) with the CIM. The resulting precision of the isotopic ratio measurement was approximately 2–10 times better than that obtained by the conventional tau correction method.
The objective of this research is to extend the analytical capability of conventional MFC-ICP-MS systems to NP analysis. The application of MFC-ICP-MS systems to the isotopic analysis of NPs can open new doors to environmental, medical, and biological studies. Hence, the goal of this study is to determine both the size and isotopic ratio of individual Ag NPs using the MFC-ICP-MS system with the CIM correction method. Here, Ag NPs were used to demonstrate the capability of our method because of several reasons: (a) the size standards of Ag NPs are commercially available, (b) naturally occurring Ag has two stable isotopes, 107Ag (51.8%) and 109Ag (48.2%), which exist in an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, (c) variation of the Ag isotopic ratio is limited (< 0.2%),34 and (d) Ag NPs are conventionally used for performance evaluation in spICP-MS analyses.
:1 ratio, (c) variation of the Ag isotopic ratio is limited (< 0.2%),34 and (d) Ag NPs are conventionally used for performance evaluation in spICP-MS analyses.
Experimental
Instrumentation
A double focusing MC-ICP-MS (Neptune XT, Thermo Fisher Scientific, Germany) equipped with nine Faraday detectors utilising 1013 Ω resistors was used for particle size and 109Ag/107Ag isotopic ratio measurements. The instrument was operated in low-resolution mode throughout this study to obtain maximum sensitivity. Moreover, a Jet sampling cone and X skimmer cone were used to enhance the signal intensities of the Ag isotopes.35 A desolvating nebuliser (APEX-Ω, Element Scientific Inc., USA) was used for introducing the sample into the ICP to improve the ionisation efficiency of the analytes by minimising the water load into the ICP.36 In fact, several studies have revealed that a desolvating nebuliser can effectively allow the detection of smaller-sized NPs (<10 nm).37–39 The resulting sensitivity was about 820 V per μg g−1 for Ag, which was about 4 times higher than that obtained with the conventional setting. The details of the instrumentation and operational settings are listed in Table 1.| ICP-MS instrument | |
|---|---|
| ICP-MS | Neptune XT, multiple collector-ICP-MS (Thermo Fisher Scientific, Germany) |
| RF power | 1200 W |
| Guard electrode | Enabled |
| Flow rate of cooling gas | 15.0 L min−1 |
| Flow rate of auxiliary gas | 0.75 L min−1 |
| Flow rate of sample gas | 0.8 L min−1 |
| Type of nebuliser | MicroMist U-series nebuliser (Glass Expansion, Australia) |
| Sample uptake rate | 0.1 mL min−1 |
| Sampling cone | Ni JET cone |
| Skimmer cone | Ni X cone |
| Integration time | 0.131 s |
|
|
| Faraday cup detector | |
| 107Ag | C (1013 Ω amplifier) |
| 109Ag | H1 (1013 Ω amplifier) |
|
|
| Sample introduction system | |
| Instrument | APEX-Ω (Element Scientific Inc., USA) |
| Spray chamber temperature | 110 °C |
| Peltier cooler temperature | 3 °C |
| Desolvator temperature | 160 °C |
| Sweep gas (Ar) | 4.2 L min−1 |
| Flow rate of N2 | 3.5 mL min−1 |
Materials
Commercially available Ag NPs purchased from nanoComposix (USA) were used in this study. These Ag NPs were citrate-stabilized particles with nominal diameters of 41 ± 5 nm, 59 ± 6 nm, 77 ± 9 nm, 97 ± 13 nm, and 205 ± 21 nm. All of the Ag NP suspensions were diluted with high-purity water at a resistivity of 18.2 MΩ cm (Merck Millipore, USA). Due to the slow response of the high-gain Faraday amplifiers, the typical time duration of a particle event is about 2.5–4 s. To avoid the introduction of two or more particles, the number concentrations of the Ag NP suspensions were adjusted to about 70 particles per mL. In addition, calibration solutions in a concentration range of 0.02–0.5 ng mL−1 were prepared from Ag+ standard solution (Kanto Chemical Co., Inc., Japan), by diluting them with high-purity water without acid.Data processing
Data based on TRA (time resolved analysis) mode for about 30 min were acquired. An integration time, corresponding to the “dwell time” defined on conventional scanning mass spectrometers, of 0.131 s (time slice) was adopted to monitor the output voltages for the Ag isotope signals. The resulting TRA data were used to identify particle events and calculate the signal intensities of the Ag isotopes for each single particle event.For the separation of the particle event from the background signal, a discrimination setting for the ion currents was defined. The limit of detection (LOD) is defined as
| LOD = μ + 5σ |
Multiple collector arrays using Faraday detectors have been widely used for high-precision isotopic ratio measurements with both the MC-ICP-MS and TIMS instruments.28,40 The response of the Faraday detector is widely recognized to lack the speed necessary for the rapid changes of the signal intensity produced by single NPs.31 The CIM was adopted to overcome this limitation.33
Size and 109Ag/107Ag isotopic ratio measurements
The resulting signal intensity data were used for both NP size and isotopic ratio determination using NanoQuant software developed in-house.41 The total counts of 107Ag+ and 109Ag+ for individual particle events were calculated by the integration of the total time range of the particle events (from the starting to terminal channels). The total ion counts of a particle event are proportional to the analyte mass, and hence, to the NP size. Based on this, the information of the NP size in the sample was obtained by calibration with Ag NP standards, assuming that the number of atoms correlates with the measured ion counts.41,42 This implies that the measured ion counts from individual Ag NPs would be linearly correlated with the cube of diameter (or radius) of the monitored Ag NPs. The particle sizes were also determined by using a protocol developed by Pace et al.24 using external calibration with dissolved standards.Several causes, such as (a) the mass discrimination effect, (b) a difference in the gains of the amplification, and (c) ion collection efficiencies of Faraday cups, can cause systematic errors in the measured isotopic ratios. To minimise the contributions of these causes, the 109Ag/107Ag ratio for the 0.5 ng mL−1 Ag+ solution was used for the normalisation. The systematic error in the 109Ag/107Ag ratio measurements was corrected by normalising the measured 109Ag/107Ag ratio to 0.929.43 The typical magnitude of the bias factors was about 2% μ−1, which conforms well with our previous reports.44
Results and discussion
Detection and size analysis of individual particles
Fig. 1(a) shows the signal intensity profiles of both 107Ag+ (black) and 109Ag+ (blue) obtained from 200 nm Ag particles. Similar to particle events detected with high-gain ion detectors,26,27 the particle events were transient signals. With a solution containing 70 particles per mL of Ag NPs, a total of about 100 particle events were observed over an analysis period of 30 min. Fig. 1(b) shows enlarged-scale particle events obtained with the MFC-ICP-MS system. The time durations of the particle events ranged from about 2.5 to 4 s. NPs which are supposed to be detected within <1 ms were detected as signals extending to a few seconds. This was due to the slow response of the Faraday detectors.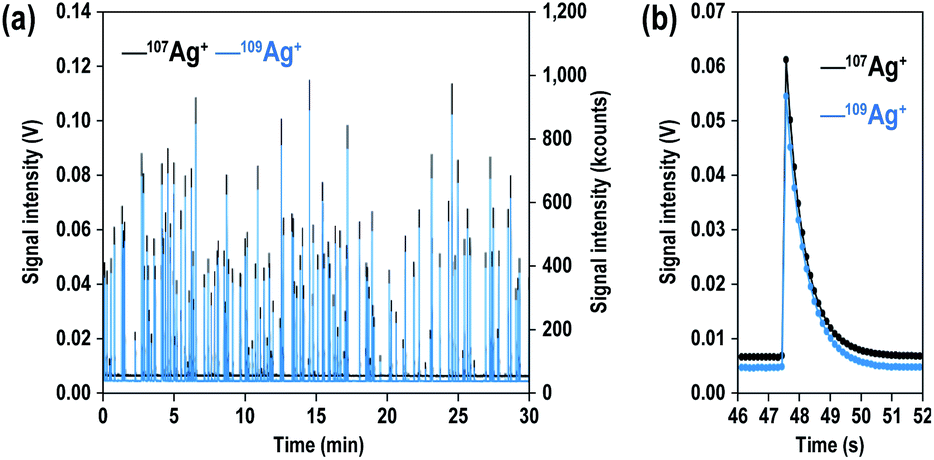 | ||
| Fig. 1 (a) Signal time profile for 107Ag+ (black) and 109Ag+ (blue) obtained from 200 nm Ag particles (70 particles per mL) with integration times of 0.131 s. (b) A 200 nm particle event as measured by MFC-ICP-MS. | ||
Next, we evaluated the capability of MFC-ICP-MS for the determination of Ag NP size and its correlation with signal intensity, to demonstrate that the size analysis of NPs is possible even with the slow response of the Faraday detectors. The signal intensities of 107Ag+ and 109Ag+ for single particle events were calculated by averaging the signal intensities of all 100 particles sampled. Fig. 2 illustrates the correlation of the measured signal intensities with the size of the NPs (40, 60, 80, 100, and 200 nm). The calculated signal intensity data were plotted against the size of the NPs. As shown in Fig. 2, the measured ion counts obtained from particles of various sizes give a straight line with a slope of 3.00 ± 0.13 (2SD). This linear correlation between the signal intensities and Ag NP size implies that the measured ion counts correlate with the number of atoms in single particles, suggesting that quantitative ion counts can be obtained even from transient signals emanating from the Ag NPs.
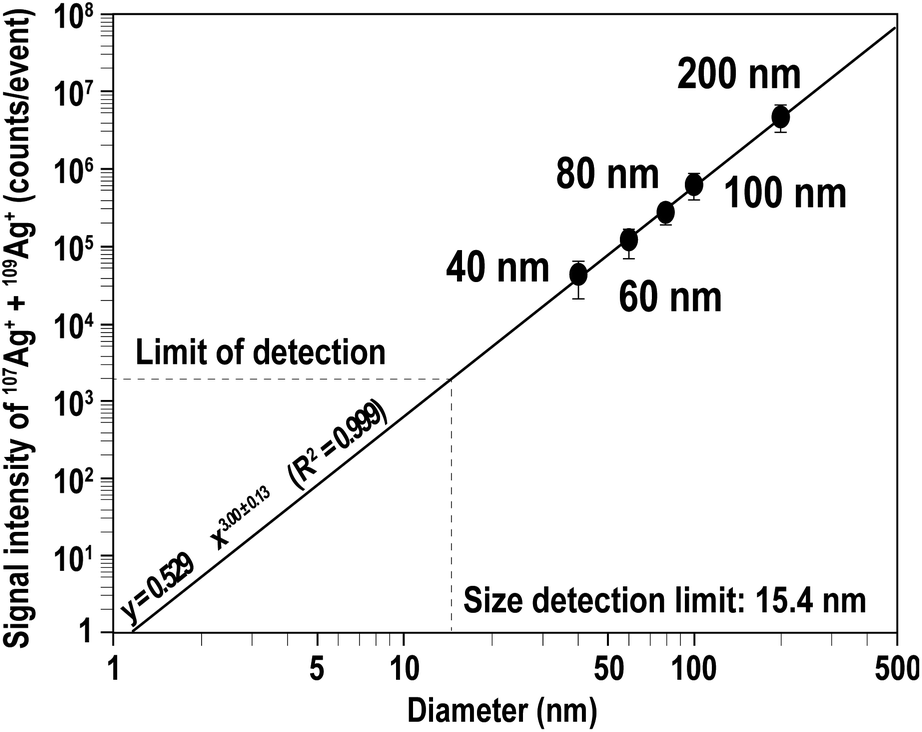 | ||
| Fig. 2 Signal intensity (counts per event) plotted against the diameters of the Ag NPs. Mean ion counts were calculated from samples containing about 100 Ag particles. The horizontal dashed line shows the LOD (1932 counts for total Ag+ calculated from the baseline of the time scan recorded for the Ag NP sample). Based on the LOD, the size detection limit was 15.4 nm. | ||
To demonstrate the potential application of MFC-ICP-MS on a wide variety of samples, the size analysis of Ag NPs was performed using an external calibration method24 with Ag+ standard solutions, under the assumption that it is difficult to obtain particle standards.
In this study, the transport efficiency was determined by the particle size method, which utilizes the ratio of ionic solution sensitivity and NP sensitivity.24 To obtain ionic solution sensitivity, Ag+ standard solutions in a concentration range of 0.02–0.5 ng mL−1 were used to construct an Ag+ standard calibration curve. The total mass entering the sample introduction system for each event was calculated by multiplying each Ag+ standard concentration by the sample uptake rate and the dwell time. On the other hand, NP sensitivity was determined by constructing an Ag NP calibration curve, by using three sizes of monodisperse Ag NP suspensions (i.e., 40, 80, and 200 nm) as reference materials. The transport efficiency, determined by the particle size method, was about 15% based on the quotient of the signal-to-mass ratios of both the Ag+ standard solution and the Ag NP standard. Finally, the mass flux calibration curve was created by multiplying each Ag+ standard concentration by the sample uptake rate, the dwell time, and the transport efficiency (data not shown). Then, the size analysis of 60 and 100 nm Ag NPs was performed using the mass flux calibration curve.
Fig. 3 shows the size distributions obtained from 60 and 100 nm Ag NPs calibrated using the mass flux calibration curve obtained using the protocol mentioned above (striped columns). The mean sizes and variations (defined as one standard deviation: 1SD) were 56 ± 7 nm for 60 nm Ag NPs and 96 ± 14 nm for 100 nm Ag NPs. The size distribution provided by the manufacturer using TEM analysis is also shown in Fig. 3 (grey columns). The mean size and variations defined by 1SD of the measurements were 59 ± 6 nm for 60 nm Ag NPs and 97 ± 13 nm for 100 nm Ag NPs. The results demonstrated that the mean sizes and variations obtained by MFC-ICP-MS were in good agreement with the TEM data within analytical uncertainty. Moreover, the distribution pattern obtained by MFC-ICP-MS did not vary significantly from the distribution data provided by the manufacturer using TEM analysis. The results demonstrated the validity of the size analysis using the present MFC-ICP-MS.
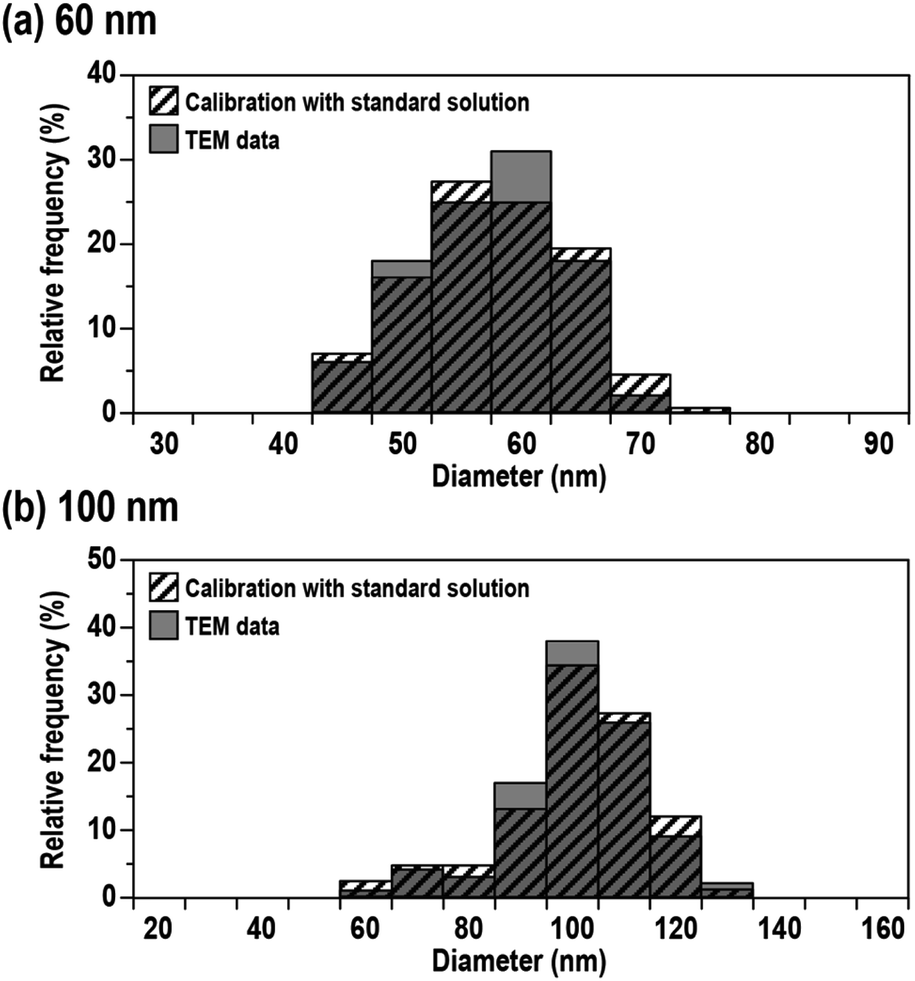 | ||
| Fig. 3 Size distributions obtained from (a) 60 nm and (b) 100 nm Ag NPs. The size of Ag NPs was calibrated with Ag+ standard solution (striped columns) and TEM data provided by the manufacturer (grey columns). | ||
109Ag/107Ag isotopic ratio for individual particles
With the MFC-ICP-MS system combined with CIM, 109Ag/107Ag ratio measurements for the analysed Ag NPs were conducted. In this study, suspensions containing 70 particles per mL Ag NPs of each sample were analysed to avoid the overlapping of particle events. Fig. 4 shows the measured 109Ag/107Ag ratios as a function of the Ag NP diameter. The particle diameters were calculated based on the total ion counts from 107Ag+ and 109Ag+ isotopes. The accuracy of the measured 109Ag/107Ag ratio (i.e., deviation from the natural value of the 109Ag/107Ag ratio: 0.929)38 was 1.76% for 40 nm, 1.08% for 60 nm, 1.03% for 80 nm, 0.33% for 100 nm, and 0.89% for 200 nm. The relative deviations became smaller as the particle size increased, except in the case of 200 nm. The reason why the 109Ag/107Ag ratio for 200 nm Ag particles was poorer than that for 100 nm Ag NPs will be stated later. From our results, we can see that the relative deviations of the measured 109Ag/107Ag ratio were smaller than 2% for all sizes. This is significantly better than the relative deviation observed for the 195Pt/194Pt ratio (5% for 70 nm Pt NPs) measured by HTR-MC-ICP-MS.27 This larger deviation found for the high-gain ion detectors can be attributed to the insufficient correction of the detector dead time.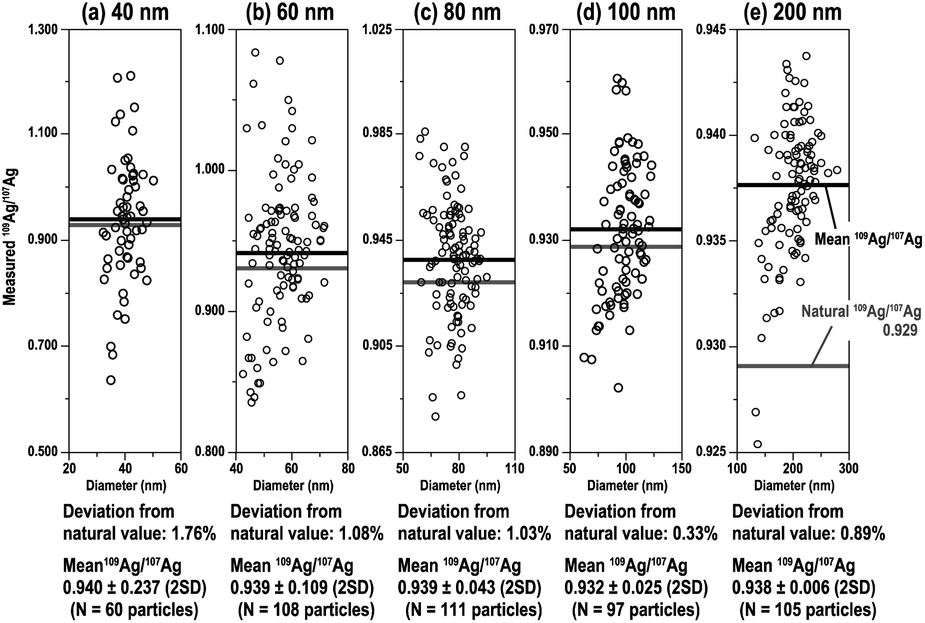 | ||
| Fig. 4 109Ag/107Ag ratio for Ag NPs of various sizes ((a) 40 nm, (b) 60 nm, (c) 80 nm, (d) 100 nm, and (e) 200 nm) plotted against the diameter of individual particles. | ||
As for why the relative deviation of the 109Ag/107Ag ratio for 200 nm Ag particles (0.89%) was poorer than that of 100 nm Ag NPs (0.33%), neither background fluctuation nor shot noise can be a major source of the accuracy deterioration for 200 nm Ag NPs. The poorer accuracy can be explained either by the lacking signal-output linearity of the amplifier systems or by the leakage of ion currents through high-impedance amplifiers. In fact, the measured 109Ag/107Ag ratio for 200 nm Ag particles approached unity, suggesting that the output voltages for 109Ag+ signals are similar to those of 107Ag+ signals. Another possibility is that polarisation affects the high-impedance resistors. This can be serious when high voltages (>10 V) are applied onto high-impedance resistors (e.g., 1013 Ω). The polarisation effect acts as a negative feedback circuit onto the high-gain amplifiers, and thus, the measured isotopic ratio could be biased. For the analysis of large-sized particles using MFC-ICP-MS, the resulting isotopic ratio would deviate from the true ratio due to the lack of signal-output linearity in the Faraday amplifier, possibly caused by leakage of ion currents through high-impedance amplifiers, or a polarisation effect. Hence, we do not have any further evidence to investigate this, so this should remain a possibility.
The mean and standard deviation (2SD) of 109Ag/107Ag ratios were 0.940 ± 0.237 for 40 nm, 0.939 ± 0.109 for 60 nm, 0.939 ± 0.043 for 80 nm, 0.932 ± 0.025 for 100 nm, and 0.938 ± 0.006 for 200 nm. The precision of the 109Ag/107Ag ratio measurements improved as the Ag NP size increased (i.e., number of atoms per particle increased). The low precision in smaller-sized Ag NPs can be attributed to a larger contribution of shot noise (counting statistics).
To investigate the effect of the signal intensity on the precision of the 109Ag/107Ag isotopic ratio measurements, the % RSD of the measured 109Ag/107Ag ratio was plotted against the signal intensity of 107Ag+ for individual Ag NPs (Fig. 5). The major source of errors, 109Ag/107Ag ratio data originating from shot noise (counting statistics), background fluctuations, and the combination of both sources (propagated with both the shot noise and background fluctuation) are also plotted on the same figure. The lines of errors were defined at the 95% confidence level. The % RSD of the 109Ag/107Ag ratio for NPs was 11.5% for 40 nm, 4.88% for 60 nm, 1.87% for 80 nm, 1.20% for 100 nm, and 0.26% for 200 nm, suggesting that the precision of the 109Ag/107Ag ratio measurements improved as the signal intensities increased. The variation of the 109Ag/107Ag ratio for lower signal intensity (i.e., <2 × 106 counts per events) mainly originated from background fluctuation, whereas the variation of the 109Ag/107Ag ratio for higher signal intensity (i.e., >2 × 106 counts per events) was mainly due to shot noise. The proportions of the data points plotted underneath the lines defined by the combined source of errors were about 90.5% for 40 nm, 92.9% for 60 nm, 96.4% for 80 nm, 94.3% for 100 nm, and 84.8% for 200 nm. This suggests that the precision of 109Ag/107Ag ratio measurements can be well explained by the combined influences of shot noise and background fluctuation.
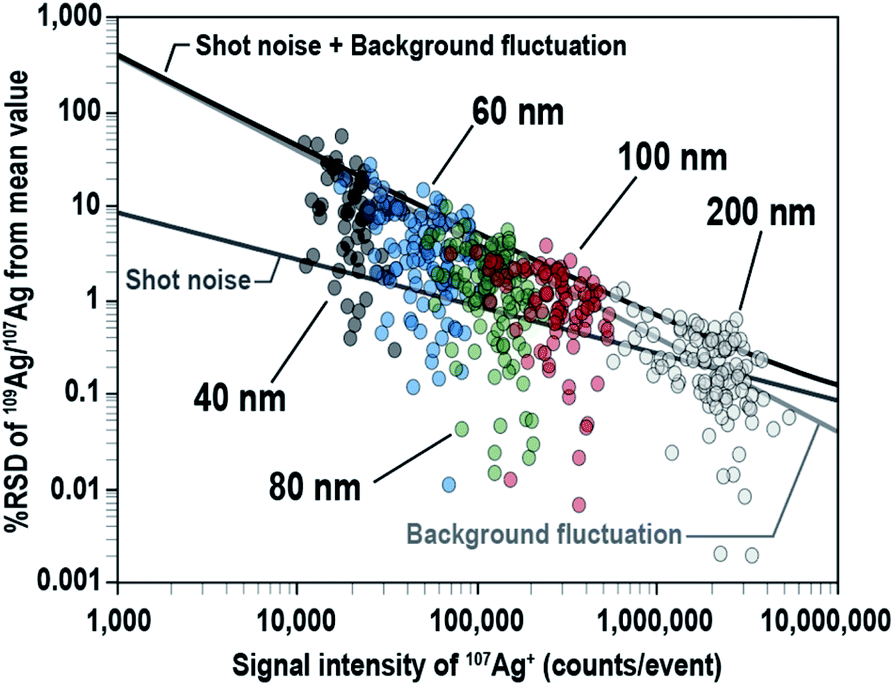 | ||
| Fig. 5 Logarithmic-measured 109Ag/107Ag ratios for Ag NPs (40 nm, 60 nm, 80 nm, 100 nm, and 200 nm) plotted against the signal intensity of 107Ag+ for the individual NPs. Uncertainty (95% confidence level) of the 109Ag/107Ag ratio originating from the combined shot noise and background fluctuation is shown by the black line. | ||
From our above results, precise and accurate isotopic ratios can be measured even in the presence of high-flux ion currents emanating from larger NPs. The advantage of using Faraday detectors is that these detectors are free from counting loss due to detector dead time. This characteristic of the Faraday detector can lead to a simpler calibration and correction procedure for isotopic analysis of NPs, in contrast to using high-gain ion detectors used in our previous study.
Conclusions
In this study, we demonstrated the capability of MFC-ICP-MS for particle analysis by determining the size and isotopic ratio for individual Ag NPs. The resulting mean sizes and variations for samples agreed well with TEM results provided by the manufacturer. The % RSD of the 109Ag/107Ag ratio for Ag NPs of each size was about: 12% for 40 nm, 4.9% for 60 nm, 1.9% for 80 nm, 1.2% for 100 nm, and 0.26% for 200 nm, showing the MFC-ICP-MS provides more accurate isotopic ratio data than HTR-MC-ICP-MS (i.e., % RSD of the 195Pt/194Pt ratio for 70 nm Pt NPs was about 5%). Although HTR-MC-ICP-MS used in our previous study was capable of isotopic ratio measurements from smaller NPs (e.g., 10 nm Pt NPs) with a high-throughput of 100 grains per second, careful calibrations and considerations such as detector gains, backgrounds, and dead time were needed to obtain reliable elemental/isotopic ratio data. This is especially true in correction methods (i.e., whether extendable or non-extendable types) for the counting loss due to detector dead time. In contrast, the conventional MC-ICP-MS utilising multiple Faraday detectors used in this study offers a wide dynamic analytical range, long-term stabilities in amplification gain and background, and no contribution of counting loss due to detector dead time. Combining with the CIM correction method, a simple calibration and correction procedure can be achieved with the system. However, high-throughput particle analysis using MFC-ICP-MS still remains a challenge as the analysis time can be prolonged by the slow response of the Faraday detectors.We hope that our manuscript can provide preliminary insights into the application of MFC-ICP-MS to size and isotopic analysis of single particles in the geochemistry and environmental chemistry research community where multiple collector instruments are widely used. MFC-ICP-MS can become a powerful tool for the identification, production, transfer, and determination of the status of airborne particles and radioactive particles, which will provide important information to various scientists dealing with isotope geochemistry and environmental chemistry.
Author contributions
S. Y. and H. T. designed the study. S. Y. wrote the manuscript. S. Y. and K. Y. carried out the experiments with MFC-ICP-MS and CIM. H. T. and T. H. reviewed and edited the manuscript. T. H. supervised the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Hisashi Asanuma (The University of Tokyo) for scientific advice. We are also grateful to Hui Hsin Khoo (The University of Tokyo) for scientific advice and critical reading of the manuscript. This work was financially supported by a Grant-in-Aid for Scientific Research (A26247094 and JP19H01081) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.References
- T. Kyotani and M. Iwatsuki, Atmos. Environ., 2002, 36, 639–649 CrossRef CAS.
- A. M. Veysseyre, A. F. Bollhöfer, K. J. R. Rosman, C. P. Ferrari and C. F. Boutron, Environ. Sci. Technol., 2001, 35, 4463–4469 CrossRef CAS PubMed.
- R. Kurkjian, C. Dunlap and A. R. Flegal, Atmos. Environ., 2002, 36, 1421–1429 CrossRef CAS.
- K. J. R. Rosman, W. Chisholm, C. F. Boutron, J. P. Candelone and U. Gorlach, Nature, 1993, 362, 333–334 CrossRef CAS PubMed.
- D. Woolard, R. Franks and D. R. Smith, J. Anal. At. Spectrom., 1998, 13, 1015–1019 RSC.
- H. Mukai, N. Furuta, T. Fujii, Y. Ambe, K. Sakamoto and Y. Hashimoto, Environ. Sci. Technol., 1993, 27, 1347–1356 CrossRef CAS.
- H. Mukai, A. Tanaka, T. Fujii, Y. Zeng, Y. Hong, J. Tang, S. Guo, H. Xue, Z. Sun, J. Zhou, D. Xue, J. Zhao, G. Zhai, J. Gu and P. Zhai, Environ. Sci. Technol., 2001, 35, 1064–1071 CrossRef CAS PubMed.
- J. Zheng, K. Tagami, Y. Watanabe, S. Uchida, T. Aono, N. Ishii, S. Yoshida, Y. Kubot, S. Fuma and S. Ihara, Sci. Rep., 2012, 2, 14 Search PubMed.
- M. Yamamoto, A. Sakaguchi, S. Ochiai, T. Takada, K. Hamataka, T. Murakami and S. Nagao, J. Environ. Radioact., 2014, 132, 31 CrossRef CAS PubMed.
- P. J. Kershaw, D. S. Woodhead, M. B. Lovett and K. S. Leonard, Appl. Radiat. Isot., 1995, 46, 1121–1134 CrossRef CAS.
- M. H. Dai, J. M. Kelley and K. O. Buesseler, Environ. Sci. Technol., 2002, 36, 3690–3699 CrossRef CAS PubMed.
- K. Hirose and Y. Sugimura, J. Radioanal. Nucl. Chem., 1990, 138, 127–138 CrossRef CAS.
- W. R. Penrose, W. L. Polzer, E. H. Essington, D. M. Nelson and K. A. Orlandini, Environ. Sci. Technol., 1990, 24, 228 CrossRef CAS.
- D. I. Kaplan, P. M. Bertsch, D. C. Adriano and K. A. Orlandini, Radiochim. Acta, 1994, 66/67, 181 CrossRef CAS.
- A. B. Kersting, D. W. Efurd, D. L. Finnegan, D. J. Rokop, D. K. Smith and J. L. Thompson, Nature, 1999, 396, 56–59 CrossRef.
- J. Igarashi, J. Zheng, Z. Zhang, K. Ninomiya and Y. Satou, Sci. Rep., 2019, 9, 1–10 CAS.
- Y. Kurihara, N. Takahata, T. D. Yokoyama, H. Miura, Y. Kon, T. Takagi, H. Shogo, N. Yamaguchi, Y. Sano and Y. Takahashi, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- D. L. Donohue, Anal. Chem., 2002, 74, 28A–35A CrossRef CAS.
- A. Axelsson, D. M. Fischer and M. V. Pénkin, J. Radioanal. Nucl. Chem., 2009, 282, 725–729 CrossRef CAS.
- B. Kober, R. T. Pidgeon and H. J. Lippolt, Earth Planet. Sci. Lett., 1989, 91, 286–296 CrossRef CAS.
- K. Hattori, S. Sakata, M. Tanaka, Y. Orihashi and T. Hirata, J. Anal. At. Spectrom., 2017, 32, 88–95 RSC.
- H. Obayashi, M. Tanaka, K. Hattori, S. Sakata and T. Hirata, J. Anal. At. Spectrom., 2017, 32, 686–691 RSC.
- D. M. Mitrano, A. Barber, A. Bednar, P. Westerhoff, P. Higgins and J. F. Ranville, J. Anal. At. Spectrom., 2012, 27, 1131–1142 RSC.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jimenez-Lamana, Anal. Chem., 2014, 86, 2270–2278 CrossRef CAS PubMed.
- T. Hirata, S. Yamashita, M. Ishida and T. Suzuki, Mass Spectrom., 2020, 9, 1–8 Search PubMed.
- S. Yamashita, M. Ishida, T. Suzuki, M. Nakazato and T. Hirata, Spectrochim. Acta, Part B, 2020, 169, 1058812 CrossRef.
- J. M. Koornneef, C. Bouman, J. B. Schwieters and G. R. Davies, J. Anal. At. Spectrom., 2013, 28, 749–754 RSC.
- J. M. Koornneef, C. Bouman, J. B. Schwieters and G. R. Davies, Anal. Chim. Acta, 2014, 819, 49–55 CrossRef CAS PubMed.
- J.-I. Kimura, Q. Chang, N. Kanazawa, S. Sasaki and B. S. Vaglarov, J. Anal. At. Spectrom., 2016, 31, 790–800 RSC.
- T. Hirata, Y. Hayano and T. Ohno, J. Anal. At. Spectrom., 2003, 18, 1283–1288 RSC.
- T. Pettke, F. Oberli, A. Audétat, U. Wiechert, C. R. Harris and C. A. Heinrich, J. Anal. At. Spectrom., 2011, 26, 475–492 RSC.
- K. Yamamoto, H. Asanuma, H. Takahashi and T. Hirata, J. Anal. At. Spectrom., 2021, 36, 668–675 RSC.
- R. Mathur, A. Arribas, P. Megaw, M. Wilson, S. Stroup, D. Meyer-Arrivillaga and I. Arribas, Geochim. Cosmochim. Acta, 2018, 224, 313–326 CrossRef CAS.
- L. Gou, Z. Jin, L. Deng, M. He and C. Liu, Spectrochim. Acta, Part B, 2018, 146, 1–8 CrossRef CAS.
- J. Zheng, J. Nucl. Radiochem. Sci., 2015, 15, 7–13 CAS.
- M. Hadioui, G. Knapp, A. Azimzada, I. Jreije, L. Frechette-Viens and K. J. Wilkinson, Anal. Chem., 2019, 91, 13275–13284 CrossRef CAS PubMed.
- J. Kocic, D. Günther and B. Hattendorf, J. Anal. At. Spectrom., 2021, 36, 233–242 RSC.
- S. Yamashita, M. Nakazato and T. Hirata, Anal. Sci., 2021, 37, 1637–1640 CrossRef CAS PubMed.
- M. Pfeifer, N. S. Lloyd, S. T. M. Peters, F. Wombacher, B.-M. Elfers, T. Schulz and C. Münker, J. Anal. At. Spectrom., 2017, 32, 130–143 RSC.
- T. Suzuki, S. Yamashita, Y. Yoshikuni and T. Hirata, J. Mass Spectrom. Soc. Jpn., 2019, 67, 147–153 CrossRef CAS.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- G. Audi, O. Bersillon, J. Blachot and A. H. Wapstra, Nucl. Phys. A, 2003, 729, 3–128 CrossRef.
- T. Hirata, Analyst, 1996, 121, 1407–1411 RSC.
| This journal is © The Royal Society of Chemistry 2022 |