
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual CEA/CD44 targeting to colorectal cancer cells using nanobody-conjugated hyaluronic acid-modified mesoporous silica nanoparticles with pH- and redox-sensitivity
Xiying
Fan
a,
Tingting
Wang
a,
Min
Han
b,
Yi
Gu
ac,
Guochuan
Sun
a,
Xinying
Peng
ac,
Qinghui
Shou
a,
Haipeng
Song
d,
Wenshuai
Liu
*a and
Rui
Nian
 *a
*a
aCAS Key Laboratory of Biobased Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, No. 189 Songling Road, Qingdao 266101, China. E-mail: liuws@qibebt.ac.cn; nianrui@qibebt.ac.cn
bDepartment of Stomatology, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, No. 16766 Jingshi Road, Jinan 250101, China
cUniversity of Chinese Academy of Sciences, No. 19(A) Yuquan Road, Beijing 100049, China
dShenzhen Innova Nanobodi Co., Ltd, No. 1301 Guanguang Road, Shenzhen 518110, China
First published on 25th April 2022
Abstract
Colorectal cancer (CRC) is the second leading cause of cancer death among all malignancies. Drug delivery targeting tumor surface receptors improve therapeutic effects and lower side-effects since they can increase the drug accumulation in target tissues and cellular uptake. In this study, we established a pH- and redox-responsive drug delivery system based on mesoporous silica nanoparticles (MSNs) for the delivery of the anticancer drug doxorubicin hydrochloride (DOX). In this drug delivery system, MSNs-NH2 were modified with hyaluronic acid (HA) to form a highly loaded anticancer drug core; they were further coated with 11C12, a nanobody against carcinoembryonic antigen (CEA) to form a guiding outer layer (DOX@MSNs-HA-11C12). The accurate localization of nanoparticles would be mediated by two targeting molecules HA and nanobody 11C12, which target the CD44 receptor and the proximal membrane end of CEA on the surface of CRC cells, respectively. The release amount of DOX observed from in vitro drug release profiles increased as follows: pH 7.4 < pH 6.8 < pH 5.4, and (pH 7.4 + GSH) < (pH 6.8 + GSH) < (pH 5.4 + GSH), illustrating that the drug release could be accelerated in a weakly acidic environment as well as in the presence of reduced glutathione (GSH). In vitro cytotoxicity experiments showed that MSNs-HA-11C12 nanoparticles exhibited good biocompatibility and safety. Drug loaded nanoparticles modified by dual targeting molecules HA and 11C12 (DOX@MSNs-HA-11C12) led to higher LoVo CRC cell apoptosis in vitro, compared with DOX@MSNs-NH2 and DOX@MSNs-HA. Confocal laser scanning microscopy and fluorescence spectrophotometry further confirmed that the dual-targeting system significantly enhanced the uptake of DOX@MSNs-HA-11C12 by LoVo cells and the internalization of DOX was the highest, followed by DOX@MSNs-HA, DOX@MSNs-NH2, and DOX, subsequently. In this study, the nanobody targeting CEA was combined with the HA targeting CD44 receptor for the first time, which proved that the dual CEA/CD44 formulation can promote a more accurate drug delivery mode for CRC cells, and provide a basis for subsequent drug development for treating CRC and related cancers.
1. Introduction
Colorectal cancer (CRC) is a heterogeneous disease, which is mainly caused by precancerous polyps including traditional tubular adenomas and serrated polyps, and develops through the gradual accumulation of distinctive genetic and epigenetic alterations.1 Global cancer statistics for 2020 showed that CRC was the third most frequently diagnosed cancer and the second leading cause of cancer death among all malignancies.2 To date, the main treatments for CRC include surgery, chemotherapy, radiotherapy, biological immunity, and surgery combined with adjuvant chemotherapy. However, 86% of CRC patients die within 5 years after diagnosis due to the complex pathogenesis, poor solubility, and a large number of toxic side effects of chemotherapeutic drugs.3 Thus, considerable efforts have to be made to find effective treatments for CRC. To expand the therapeutic window, more efforts have been devoted to alternative therapies, including antibody-mediated targeted therapy, which has attracted great attention in the research of antitumor drugs because it can precisely clear cancer cells without damaging normal tissues.4 At the same time, the application of nanotechnology in a targeted drug delivery system (DDS) also plays a crucial role in improving cancer treatment.Due to the large surface area and pore volume, tunable pore structures, thermal stability, and high biocompatibility, mesoporous silica nanoparticles (MSNs) can serve as efficient carriers for various therapeutic agents.5 Moreover, surface modifications of MSNs with tumor-targeting ligands (e.g. antibodies, polysaccharides, peptides, nucleic acids, and small molecules) further realize the controllable and efficient delivery of MSNs as drug carriers.6,7 In addition, the controlled release of loaded drugs from MSNs can be further significantly boosted by using switchable gatekeeper modifications sensitive to certain external stimuli, including pH, temperature, oxygen, and enzymes, making MSNs exhibit great advantages as drug carriers.6,8,9
The preparation of DDS based on surface receptor recognition has attracted great attention. The common surface receptors of CRC include folate receptors, CD44 and carcinoembryonic antigen (CEA). CD44 is a common transmembrane glycoprotein in various cancer cells,10 and is particularly overexpressed in some types of colon cancers.11 Hyaluronic acid (HA), a disaccharide unit glycosaminoglycan composed of D-glucuronic acid and N-acetylglucosamine, exhibits a high-affinity for CD44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 and is reported as an excellent ligand for targeting CD44.13 Notably, although CD44 is also widely expressed in normal cells and tissues,14 partially weakening its specificity as a tumor target, CD44 on the surface of normal cells is in a resting state and cannot bind to HA, while CD44 on the surface of tumor cells is in an activated state and can bind to HA.15 Moreover, HA on the surfaces of MSNs enhances the dispersity of MSNs, which can be ascribed to two aspects: the electrostatic repulsions among the stretched hydrophilic HA chains and the steric hindrance among nanoparticles.16 Therefore, HA as a ligand attached to the surface of MSNs not only improves the dispersity and stability of MSNs, but also endows MSNs with the ability to target CD44 on CRC cells.
12 and is reported as an excellent ligand for targeting CD44.13 Notably, although CD44 is also widely expressed in normal cells and tissues,14 partially weakening its specificity as a tumor target, CD44 on the surface of normal cells is in a resting state and cannot bind to HA, while CD44 on the surface of tumor cells is in an activated state and can bind to HA.15 Moreover, HA on the surfaces of MSNs enhances the dispersity of MSNs, which can be ascribed to two aspects: the electrostatic repulsions among the stretched hydrophilic HA chains and the steric hindrance among nanoparticles.16 Therefore, HA as a ligand attached to the surface of MSNs not only improves the dispersity and stability of MSNs, but also endows MSNs with the ability to target CD44 on CRC cells.
Serum levels of CEA is one of the most meaningful tumor markers in the diagnosis of CRC and is of great value in post-operative follow-up, surveillance, and for evaluating the effectiveness of surgery.17 More importantly, CEA was reported to be anchored on the surface of tumor cells via glycosylphosphatidylinositol (GPI). Thus, CEA-targeted drugs, especially antibody drugs, have shown great potential in the treatment of CRC. Bacac et al. found that a novel IgG-based T-cell bispecific antibody strongly targets not only to the tumor cells with high CEA expression, avoiding damage to primary epithelial cells with low-CEA expressing tumor, but also to CD3 epsilon chains expressed on T cells.18 The Lacava group developed maghemite nanoparticles modified by anti-CEA antibody that successfully targeted CRC cells expressing the CEA.19
Monoclonal antibodies (mAbs) play a crucial role in targeted drug delivery. Benefiting from its specific binding ability toward cell surface receptors, targeted drug delivery to a variety of tumors is achieved by coupling antibodies to nanoparticles encapsulating small molecule chemotherapeutics, protein drugs, or nucleic acid based drugs.20 As a representative of a new generation of genetically engineered antibodies, nanobodies are engineered from the variable domain of the camelid (or shark) heavy chain only antibodies.21 With a molecular weight of only 12–15 kDa, they are regarded as the smallest antigen-binding entities.22,23 In addition to a pair of canonical disulfide bonds that maintain the basic structure, nanobodies contain two pairs of atypical disulfide bonds between their framework regions 2, 4 (FR2, 4) and complementarity determining region 3 (CDR3), forming tighter folds and greatly improving protein stability. Furthermore, the CDR3 region of the nanobody is longer than that of conventional antibodies and further enhances the recognition of otherwise cryptic epitopes.24 Meanwhile, the nanobody has a simple structure and is easily engineered into bivalent, multivalent as well as fusion forms by genetic engineering to exert more powerful biological functions. Compared to conventional antibodies, nanobodies can be manufactured to a high level in bacteria or yeast to significantly save on cost.25 In our previous study, we screened and produced a pair of nanobodies, 11C12 and 2D5, that specifically bind CEA (dissociation constants of 12 nM and 774 pM, respectively).26 They demonstrated superior specificity, with no binding to highly similar homologous proteins such as CEACAM1, 3, 6, 8 and fetoprotein, HE4 and CA125. They also exhibited exceptional stability. After storage in a high temperature and humidity environment (60 °C, 75% humidity) for 14 days, these nanobodies showed no degradation and no decrease in affinity. We have also developed a diagnosis kit for CEA based on these nanobodies, which can still accurately detect the content of CEA in clinical serum after storage at 37 °C and 50% relative humidity for 64 days.26 More interestingly, the epitope prediction analysis showed that the binding site of 11C12 was in the near membrane region of CEA. Therefore, 11C12 has the potential to be used as a targeting antibody to modify nanoparticles and achieve drug targeted delivery.
In this study, MSNs-NH2 were prepared using an organic template method. Doxorubicin hydrochloride (DOX) was loaded into the MSNs-NH2 (DOX@MSNs-NH2) via electrostatic adsorption (Scheme 1). Then, DOX@MSNs-NH2 were functionalized by HA. Subsequently, nanobody 11C12 was connected to the surface of MSNs-HA to construct nanoparticles (DOX@MSNs-HA-11C12) with dual active targeting. The successful construction of MSNs-HA-11C12 was characterized by different measurement methods, including scanning electron microscopy (SEM), dynamic laser light scattering (DLS), Fourier transform infrared (FTIR) and thermogravimetric analysis (TGA). Furthermore, the release of DOX from nanoparticles was evaluated in vitro. The in vitro cellular cytotoxicity and cellular uptake of nanoparticles were evaluated as well.
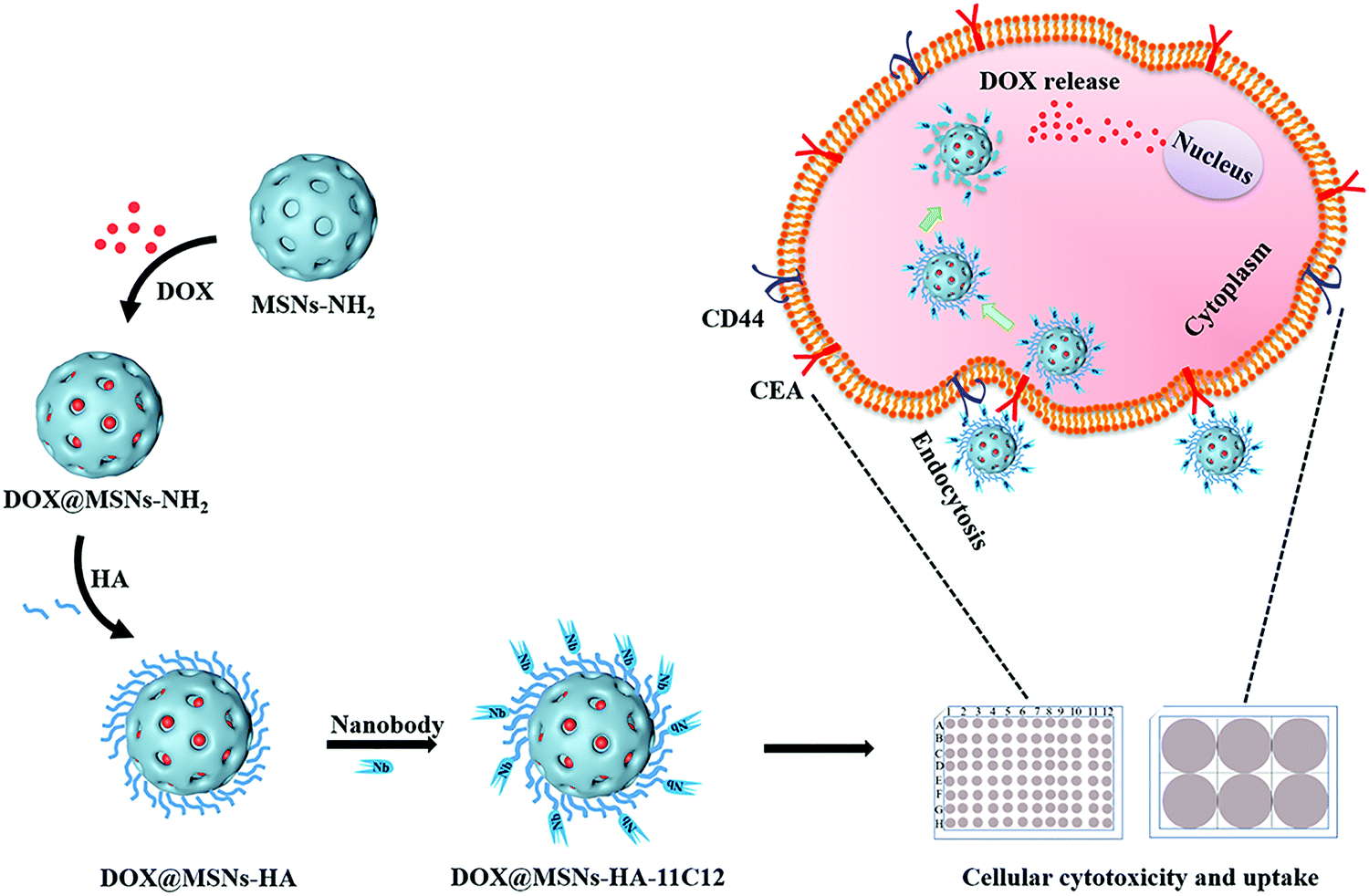 | ||
| Scheme 1 Schematic illustration of DOX@MSNs-HA-11C12. | ||
2. Materials and methods
2.1. Materials and cell lines
Cetyltrimethylammonium bromide (CTAB) and tetraethyl orthosilicate were obtained from Tianjin Bodi Chemical Holding Co., Ltd (Tianjin, China); polyethylene glycol, absolute ethanol and ethylene glycol (EG) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China); cell counting kit-8 (CCK-8), 4′,6-diamidino-2-phenylindole (DAPI), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Darmstadt, Germany). Phosphate-buffered saline (PBS) solution was purchased from Solarbio (Beijing, China). DOX and aminopropyltriethoxysilane (APTES) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd (Shanghai, China). HA was purchased from Beijing Mreda Technology Co., Ltd (Beijing, China). LoVo cells were obtained from Yuchi cell Biological Technology Co., Ltd (Shanghai, China). The BCA kit was purchased from Tiangen Biotech Co., Ltd (Beijing, China). All of the other chemicals were of analytical grade and were used as received.2.2. Preparation of MSNs-NH2
The MSNs-NH2 were synthesized according to the previously published methods27 and modified appropriately. 1.17 g CTAB was dissolved in a mixed solution containing 180 mL water, 30 mL EG, and 7.2 mL ammonia aqueous solution. After vigorous stirring at 600 rpm in a 50 °C water bath for 30 min, 4 mL TEOS and 0.74 mL APTES were rapidly added to the mixture. The final molar composition in the solution was 1 TEOS:0.18 APS:0.18 CTAB:4.7 NH3:10 EG:558.1 H2O. The resulting mixture was stirred for another 2 h at 50 °C and was then kept statically at the same temperature for 20 h. After reaction, sediment was collected by centrifugation at 14000 rpm for 20 min, and redispersed with deionized water and absolute ethanol three times alternately. The sediment was vacuum dried at 60 °C overnight to obtain MSNs-NH2.
In order to obtain the mesoporous structure, the organic template CTAB was removed using an ion exchange method.28 1 g MSN-NH2 was dispersed in ethanol (95%) containing 0.2 g NH4Cl, and the mixture was stirred at 60 °C for 15 min, then the sediment was collected by centrifugation and washed with cold ethanol three times. The sediment was vacuum dried at 60 °C overnight to obtain MSNs-NH2 with a mesoporous structure.
2.3. Conjugation of HA to MSNs-NH2
Hydrophilic HA modified MSNs-NH2 improve the dispersion of mesoporous silicon in PBS. HA (60 kDa) was first dissolved in water. The pH of the solution was adjusted by 0.1 M NaOH or 0.1 M HCl to 4.7–6.0, and then 1-(3-dimethylaminopropyl)-3-ethylcarbo-diimide/N-hydroxysuccinimide (EDC/NHS) was added for activation for 1 h. Ultrasonically dispersed MSNs-NH2 suspension was added to the mixture. Subsequently, the pH of the mixture was adjusted to 7–8, and the mixture was stirred overnight at room temperature; thus the carboxyl group of HA reacts with the amino group of mesoporous silicon to realize the covalent connection, resulting in the carboxyl group being modified on MSNs-NH2. After the reaction, the sample was centrifuged, and the precipitation was washed twice with deionized water, and finally centrifuged, precipitated, and lyophilized to obtain MSNs-HA.2.4. Conjugation of nanobody 11C12 to MSNs-HA
Pegylation nanobody 11C12 was obtained according to the following steps. Firstly, the nanobody 11C12 stored in our laboratory was exchanged into PBS (pH 8.0), then 2 mg mL−1 Traut's reagent was added and incubated at room temperature for 1 h (the molar ratio of Traut's reagent:nanobody was 40:1). The thiolated nanobody 11C12 was purified using an ultrafiltration tube and exchanged into PBS (pH 6.8). Then, 300 μL 20 mM NH2-PEG4-MAL was added, and the reaction was protected from light overnight under a nitrogen atmosphere. After the reaction, NH2-PEG4-11C12 was purified using an ultrafiltration tube and exchanged into PBS (pH 7.4).
16 mg MSNs-HA was dispersed uniformly in 4 mL PBS (pH 5.4) by sonication. EDC (1.6 mg)/NHS (1.6 mg) was added and then stirred at room temperature to activate the carboxyl group for 1 h. The pH of the mixture was adjusted to 7–8. Then, NH2-PEG-11C12 was added to the mixture. The mass ratio of the MSNs-HA to nanobody 11C12 was about 20:1. The reaction was processed as above at 4 °C overnight. After the reaction, the samples were separated by centrifugation, and the sediment was washed twice with PBS (pH 7.4) and the supernatant was retained. The initial concentration and the supernatant of 11C12 were determined using the BCA kit according to the manufacturer's method. The conjugation efficiency of 11C12 to MSNs-HA was determined indirectly.
2.5. Drug loading and stimuli-responsive drug release
The MSNs-NH2 were dispersed into 2 mL PBS (pH 7.4) and then 2 mL 2 mg mL−1 DOX solution was added with stirring at room temperature for 24 h without light. After the reaction, the samples were separated by centrifugation. The sediment was washed twice with PBS (pH 7.4) and the supernatant was retained. Above the sediment was DOX@MSNs-NH2, for subsequent modification with HA and 11C12. Drug loading on nanoparticles was measured using UV-visible spectroscopy at 480 nm. The DOX entrapment efficiency (EE) and drug loading (DL) were calculated according to the following formula.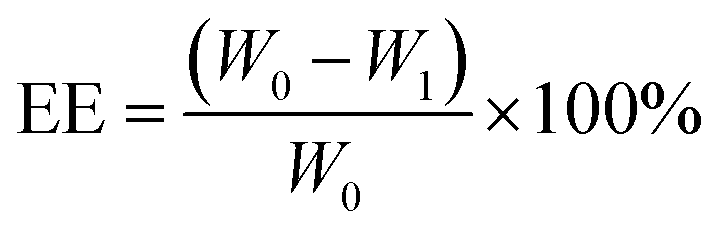 | (1) |
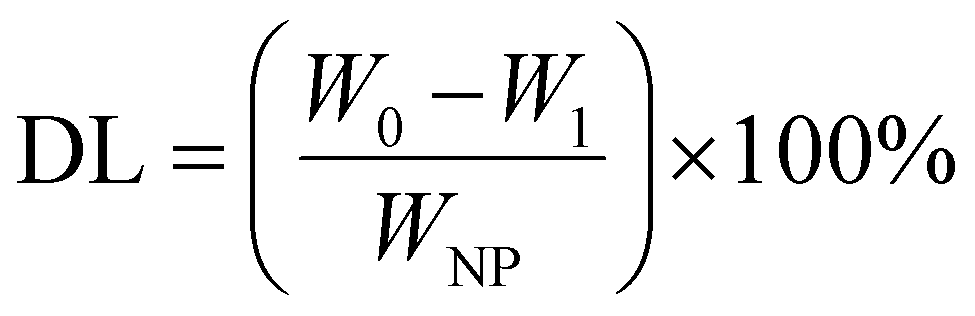 | (2) |
To investigate the release of DOX in response to pH and redox dual-stimuli, DOX@MSNs-HA-11C12 were dispersed in 2 mL PBS at different pH values of 5.4, 6.8 and 7.4 with or without 10 mM GSH, respectively. Meanwhile, DOX@MSNs-NH2 and DOX@MSNs-HA were incubated in 2 mL PBS (pH 7.4), respectively. The DOX released from DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSN-HA-11C12 was investigated at room temperature for 96 h. At a predetermined time, the supernatant was collected by centrifugation for quantitative analysis and replenished with the same volume of fresh medium. The absorbance of the supernatant was measured at 480 nm to calculate the cumulative DOX release.
2.6. Characterization of nanoparticles
2.7. In vitro cytotoxicity evaluation
In vitro cellular cytotoxicity of free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, DOX@MSNs-HA + free HA, and DOX@MSNs-HA-11C12 was evaluated by CCK8 assay. The LoVo cells in the logarithmic growth period were inoculated into 96-well plates with 10000 LoVo cells per well, and cultured in a 5% CO2 cell incubator. The culture medium in the well was discarded after 24 h. 100 μL of cell culture medium containing different concentrations of DOX loaded nanoparticles was added to each well. Free DOX was used as a positive control. After incubation for 48 h at 37 °C, the medium was removed, and fresh PBS was used to rinse the well three times. Then, 100 μL cell culture medium containing 10 μL CCK8 was added to each well. After incubation for 2 h at 37 °C, the absorbance was measured on a microplate reader at a double wavelength of 450 nm and 630 nm. Cell viability was calculated based on the reading.
2.8. In vitro cellular uptake
Confocal laser scanning microscopy (CLSM) assessed the internalization extent of DOX from free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 into LoVo cells. LoVo cells were seeded into 24-well plates at a density of 5 × 105 per well and incubated overnight. Then, the cells were treated with either 20 μg mL−1 free DOX or nanoparticles containing 20 μg mL−1 DOX. After incubation for 2 h in a 5% CO2 incubator at 37 °C, LoVo cells were washed with PBS three times and stained with 100 μL DAPI for 5–10 min. Subsequently, the cells were washed with cold PBS three times. Finally, the cells were soaked in PBS and stored away from light. The cells were visualized under CLSM with the following channels: DAPI is blue light, DOX is red light.The uptake of free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 was determined using a fluorescence spectrophotometer. LoVo cells were seeded in 6-well plates at a density of 5 × 105 per well and incubated overnight. Then, the cells were treated with either 20 μg mL−1 free DOX or nanoparticles containing 20 μg mL−1 DOX. The LoVo cells were cultured in a 5% CO2 incubator at 37 °C for 1 h, 2 h, and 4 h, the medium was discarded, and then the LoVo cells were washed with PBS three times. Trypsin was added to each well to digest the LoVo cells. After centrifugation at 1000 rpm for 5 min, the sediment was resuspended in PBS and washed two times. Then, cell lysate was added to lyse the cells. The fluorescence intensity at 480 nm excitation/590 nm emission wavelength was measured using a fluorescence spectrophotometer.
2.9. Statistical analysis
Each experiment was performed independently and in triplicate. All experimental data were presented as mean ± SD (standard deviation). Differences in means were tested using Duncan's tests, and P-values below 0.05 were considered significant.3. Results and discussion
3.1. Synthesis of DOX@MSNs-HA-11C12
The design and synthetic strategy of DOX@MSNs-HA-11C12 are illustrated in Scheme 1 and the procedure consists of four steps. Firstly, MSNs-NH2 were synthesized and modified according to the published methods.27 Under alkaline conditions, amino modified MSNs were synthesized in one-step with precursor tetraethoxysilane (TEOS) as raw materials. Then, DOX was loaded into the MSNs-NH2via electrostatic adsorption. Thereafter, HA functionalized DOX@MSNs-NH2 were prepared based on HA specific binding CD44 receptors which were abundantly expressed on CRC cells.11,15 Finally, owing to the specific binding of nanobody 11C12 to the near membrane end of CEA on the surface of CRC cells, nanobody 11C12 was connected to the surface of DOX@MSNs-HA to construct nanoparticles with dual active targeting.3.2. Characterization of nanoparticles
The average diameter of MSNs-NH2 shown in the TEM images was 144.9 ± 5.2 nm (Fig. 1A). After modification of HA and nanobody 11C12, the average particle sizes of MSNs-HA and MSNs-HA-11C12 had barely changed (Fig. 1C and D). Meanwhile, the surface morphologies of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 showed obvious differences (Fig. 1A–D). The surface of MSNs-NH2 was evenly distributed with small pits and pores, exhibiting a highly ordered state (Fig. 1A and B). After modification of HA, the outer layer of MSNs was uniformly encapsulated by HA, and the surface of the entire MSNs-HA presented a rough coat with unclear mesopores (Fig. 1C). After coupling with nanobody 11C12, the surface of MSNs-HA-11C12 became rougher and formed convex entanglement (Fig. 1D).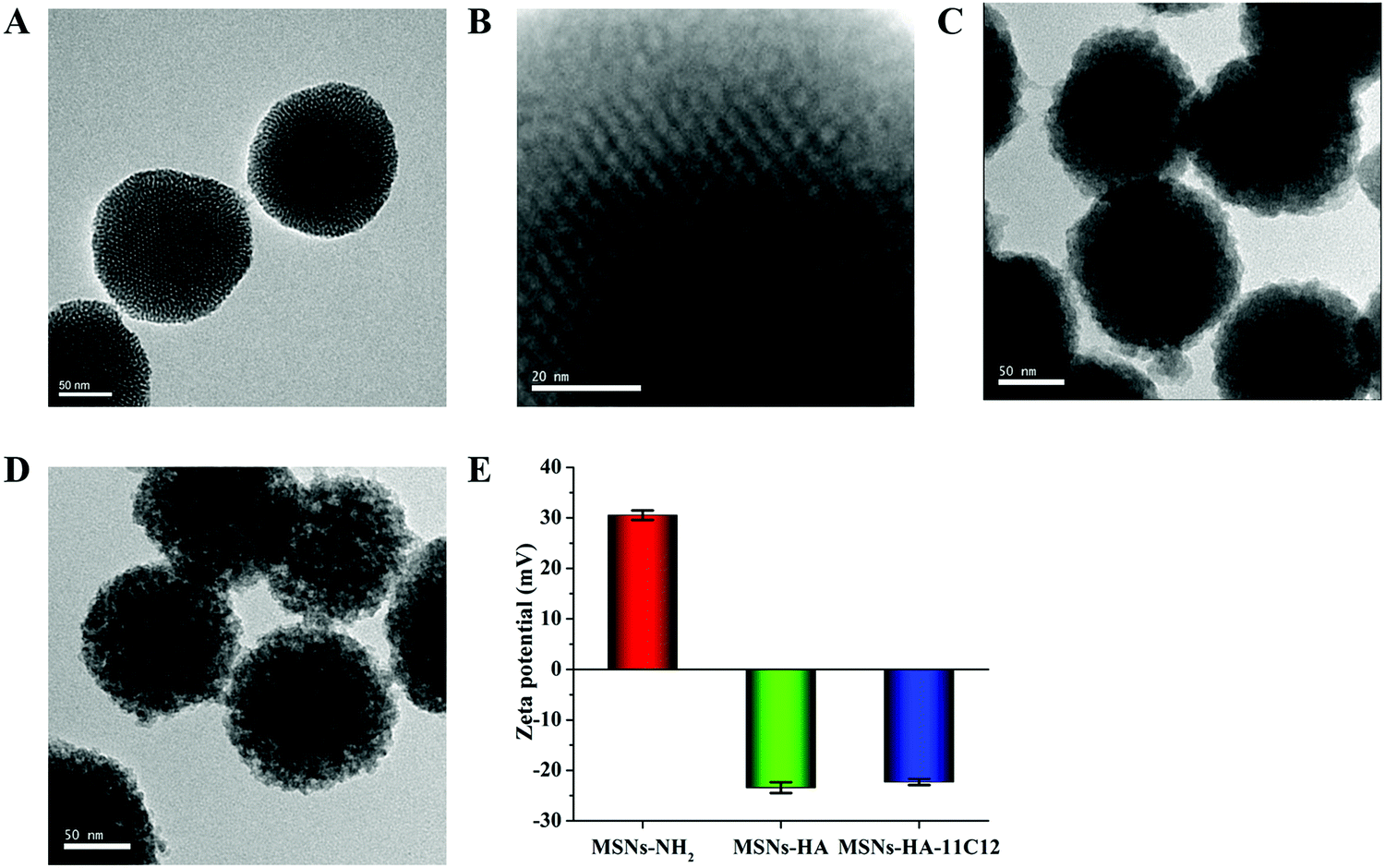 | ||
| Fig. 1 TEM images of (A) MSNs-NH2, (B) MSNs-NH2 with high resolution, (C) MSNs-HA, and (D) MSNs-HA-11C12. (E) Zeta potential of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12. Error bars and standard deviations from three independent experiments. | ||
Zeta potential is an important index used to characterize the stability of nanoparticles in a dispersion system through the electrostatic repulsion between the nanoparticles.30 Zeta potential values of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 were measured using DLS, and the data are displayed in Fig. 1E. The mean surface charge of MSNs-NH2 was +30.5 ± 1.0 mV. The positive zeta potential of MSNs-NH2 is attributed to the presence of a large amount of positively charged amino groups on MSNs.31 For HA functionalized MSNs, the zeta potential of MSNs-HA decreased to −23.4 ± 1.1 mV, which meant that the carboxyl group of HA on the surface of MSNs became dominant, and the negative charge carried by it completely sheltered the positive charge of the amino group on the surface of MSNs, making the whole nanoparticle negatively charged. The surface modification with nanobody 11C12 increased the charge value to −22.3 ± 0.6 mV due to the nanobody 11C12 with a slightly positive charge. The isoelectric point (pI) of nanobody 11C12 was 9.21, and nanobody 11C12 was connected to the surface of MSNs-HA under PBS (pH 8.0); thus nanobody 11C12 had a positive charge. The results revealed MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 were constructed successfully according to our design.
As reported by many researchers, MSNs and MSNs-NH2 agglomerated inevitably in saline and PBS. Therefore, they cannot be used directly for intravenous injection because the original charge balance between particles can be destroyed in blood circulation.16,32,33 In our research, MSNs-NH2 were uniformly dispersed in PBS at the initial stage and agglomerated largely within 5 min. In contrast, HA modified MSNs-NH2 and HA-11C12-dual modified MSNs-NH2 greatly alleviated the agglomeration (Fig. 2). The dispersity improvement likely results from the electrostatic repulsion and steric hindrance among stretched hydrophilic HA chains.34,35 In addition, CD44-positive cancer cells endocytose MSNs-HA more effectively via a receptor-mediated endocytosis mechanism.36 Besides, because of the high affinity and excellent solubility of nanobodies, nanobody-derived nanoparticles demonstrated a high target specificity, high stability, and good solubility. Therefore, HA and 11C12 capping not only provided the MSNs-NH2 with a more abundant biofunctionalized surface, but also effectively prevented the MSNs-NH2 from aggregating in saline solutions.
 | ||
| Fig. 2 Comparison of MSNs-NH2 (left), MSNs-HA (middle), and MSNs-HA-11C12 (right) suspended in PBS (pH 7.4) for 0 min, 5 min, 1 h, 5 h, 24 h, 36 h, 48 h, and 72 h. The same amount of both nanoparticles was weighed and the same volume of PBS was then added. The particles were fully suspended by shaking as the starting point. | ||
The isotherms and pore size distribution curves of MSNs-NH2, MSNs-HA, and DOX@MSNs-HA-11C12 were measured by N2 adsorption–desorption test analysis as shown in Fig. 3A and B. Meanwhile, Table 1 lists the Brunauer–Emmett–Teller (BET) surface area, total pore volume, and pore size. For MSNs-NH2, the BET surface area, pore volume, and pore size were 633 m2 g−1, 0.85 cm3 g−1, and 2.8 nm, respectively. After the modification of HA, the pore volume (0.7 cm3 g−1) and pore size (2.4 nm) of MSNs-HA were slightly decreased, which showed that the internal mesoporous network was not blocked by HA. After DOX loading (DOX@MSNs-HA-11C12), the BET surface area (70 m2 g−1), total pore volume (0.18 cm3 g−1), and pore size (about 0 nm) were sharply decreased, which indicated that DOX successfully entered the internal mesoporous network.
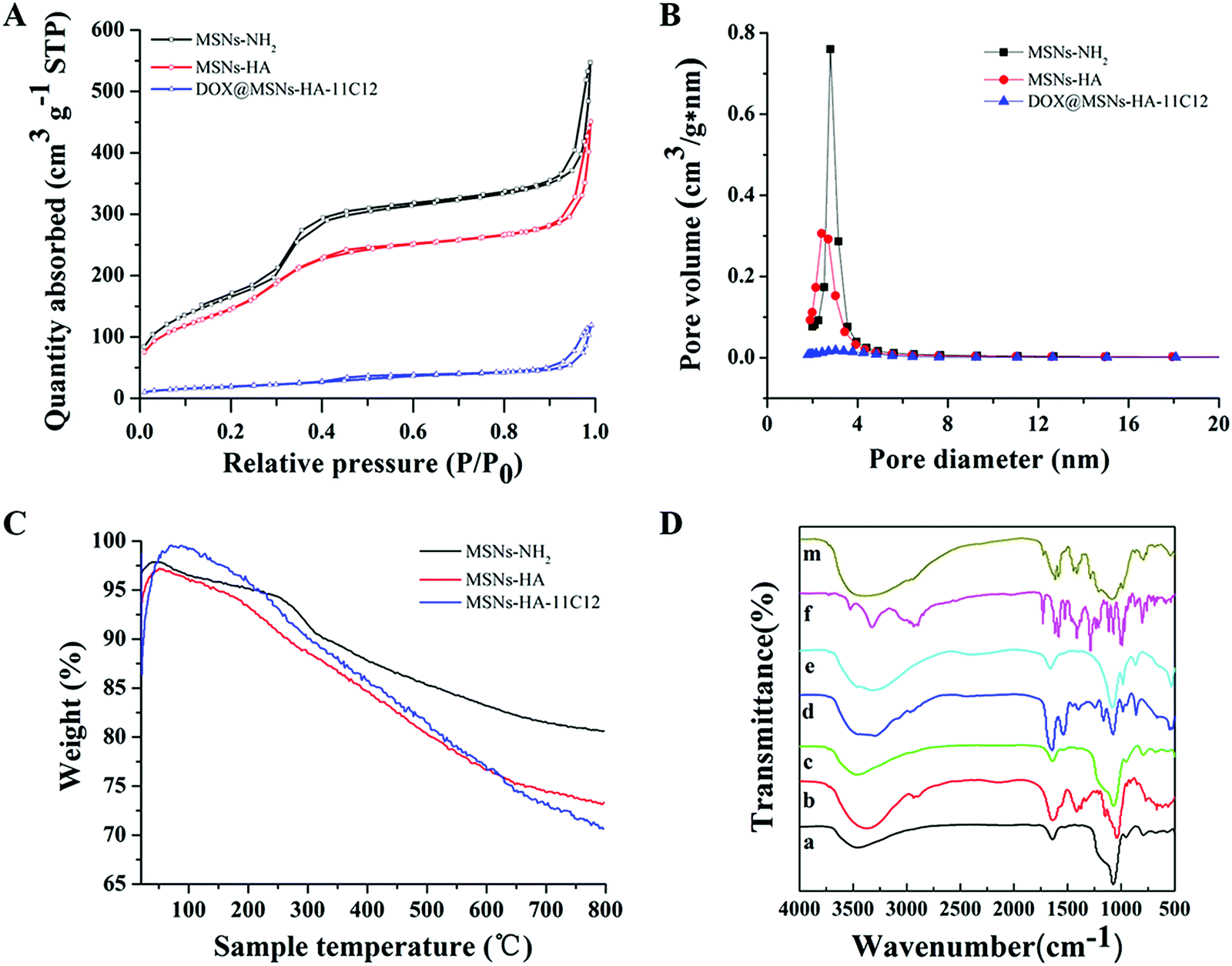 | ||
| Fig. 3 (A) N2 adsorption–desorption isotherms of MSNs-NH2, MSNs-HA, and DOX@MSNs-HA-11C12. (B) Pore size distributions of MSNs-NH2, MSNs-HA, and DOX@MSNs-HA-11C12. (C) TGA analysis of MSNs-NH2, MSNs-HA and MSNs-HA-11C12. (D) FTIR spectra of MSNs-NH2 (a), HA (b), MSNs-HA (c), 11C12 (d), and MSNs-HA-11C12 (e). | ||
| Sample | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| MSNs-NH2 | 633 | 0.85 | 2.8 |
| MSNs-HA | 567 | 0.70 | 2.4 |
| DOX@MSNs-HA-11C12 | 70 | 0.18 | 0 |
TGA curves of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 are shown in Fig. 3C. The weight loss of bare MSNs-NH2 was 19.38%. After surface functionalization of MSNs-NH2 with HA and 11C12, the weight loss of MSNs-HA and MSNs-HA-11C12 increased to 26.67% and 29.35%, respectively, which illustrated that about 7.29% of HA and 2.68% of 11C12 were introduced onto the surface of MSNs-NH2. According to the BCA assay, the amount of 11C12 was 24 μg mg−1 nanoparticles, which is consistent with the result of TGA. These results indicated that MSNs-HA and MSNs-HA-11C12 were successfully developed.
The surface conversion of the raw material and the prepared nanoparticles was evaluated using FTIR spectroscopy (Fig. 3D). In the spectrum of MSN-NH2, the corresponding absorption peaks of the silica structure were observed, the stretching vibration of Si–O at ∼1100 cm−1 and the bending vibration of Si–OH, Si–O–Si, and Si–O at ∼951 cm−1, ∼800 cm−1, and ∼464 cm−1, respectively. The stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O coming from HA appeared at ∼1406 cm−1 after the conjugation of the MSN-NH2 with HA. For MSNs-HA-11C12, new peaks of carbon associated stretching or bending vibration from 11C12 were shown from ∼500 cm−1 to 700 cm−1. These results indicated the successful conjugation of the MSNs-NH2 with HA and 11C12 in sequence.
O coming from HA appeared at ∼1406 cm−1 after the conjugation of the MSN-NH2 with HA. For MSNs-HA-11C12, new peaks of carbon associated stretching or bending vibration from 11C12 were shown from ∼500 cm−1 to 700 cm−1. These results indicated the successful conjugation of the MSNs-NH2 with HA and 11C12 in sequence.
3.3. DOX loading and in vitro DOX release kinetics
DOX, a cancer chemotherapeutic agent, was chosen as a guest model drug and loaded into the pores of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12. The EE of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 were 88.36%, 82.37%, and 73.12%, respectively. The DL of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 were 30.64%, 29.17%, and 26.77%, respectively. By comparison, it was found that the EE and DL of MSNs-HA-11C12 decreased slightly in that the loss of DOX occurred during HA and 11C12 modified MSNs-NH2. Although MSNs have made great progress as nanocarriers, their low DL, which is generally less than 20%,6,34,37,38 has limited their applications. This dual targeting MSNs based on HA and nanobody 11C12 achieved a relatively high drug loading efficiency up to 26.77%, which reduces the accumulation of foreign materials in vivo and enhances the safety of cancer treatment.To investigate the pH and redox dual-stimuli responsive release of MSNs-HA-11C12, the DOX release studies were taken under different pH (7.4, 6.8 and 5.4) with or without GSH (Fig. 4). A typically biphasic release pattern was observed in all of the nanoparticles: an initial burst release of DOX in the first 5 h owing to partial DOX adsorbed on the surface of nanoparticles was rapidly dissolved and diffused into the medium; then a sustained drug release up to 96 h in that DOX loaded into the pores of nanoparticles slowly released outward. In order to simulate the accumulative drug release experiments of MSNs-NH2, MSNs-HA, and MSNs-HA-11C12 under normal physiological conditions, the experiments were carried out in PBS (pH 7.4) at room temperature (Fig. 4A). The DOX release amount from MSNs-NH2 reached 13.1% within 5 h and then increased to 19.53% in 96 h; meanwhile, 13.09% and 8.88% of DOX were released within 96 h from MSNs-HA and MSNs-HA-11C12, respectively. All the profiles present the initial rapid DOX release, followed by a fairly plateau stage afterwards. The result indicates that the drug delivery systems release drug slowly and are good vehicles for cancer treatment.
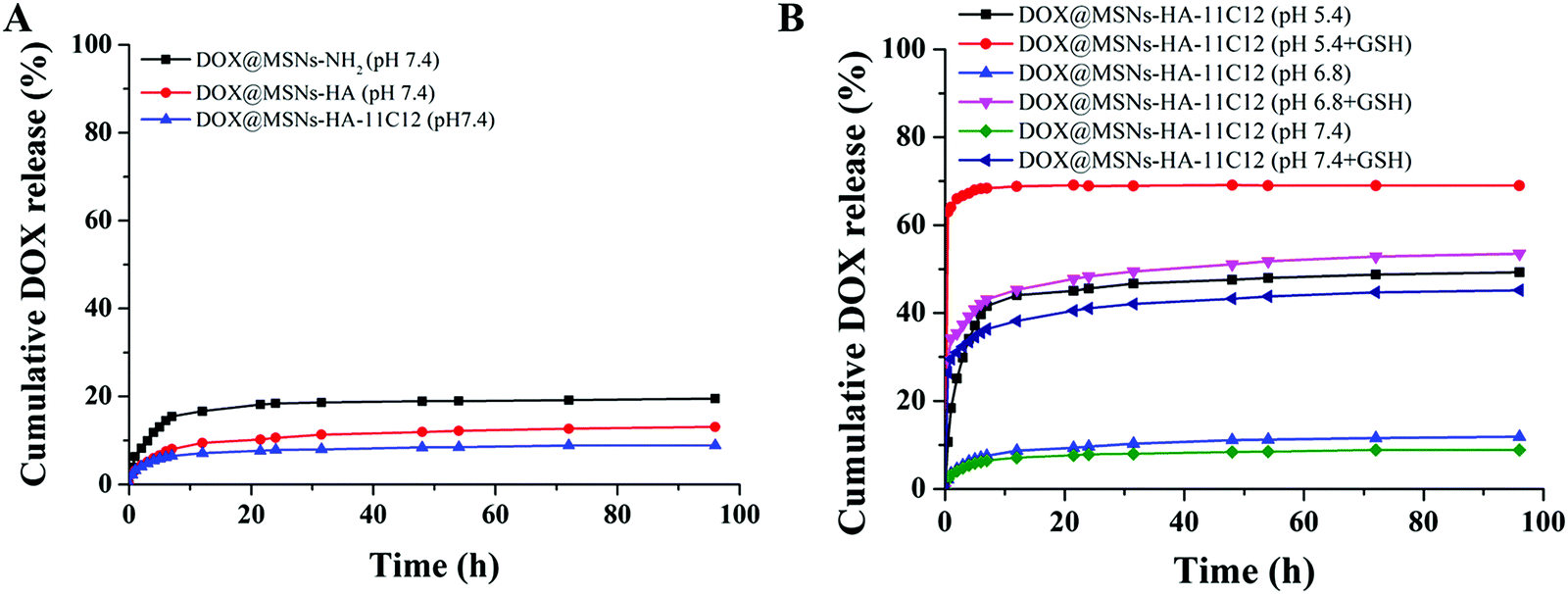 | ||
| Fig. 4 (A) In vitro drug release profiles of DOX@MSN-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 in media with physiological pH. (B) In vitro drug release profile of DOX@MSNs-HA-11C12 in media with different pH values (pH 7.4, 6.8 and 5.4) and with GSH or not. | ||
The cumulative DOX release was only 8.88% at pH 7.4 and 11.89% at pH 6.8, but reached as high as 49.27% at pH 5.4 as the DOX was rapidly released, indicating a pH responsive of the DOX release in this drug loaded system (Fig. 4B). This phenomenon mainly comes from the following two aspects: the DOX solubility was enhanced and electrostatic interaction between the DOX and nanoparticles was relatively weak under acidic conditions.14,39 When GSH was added into the releasing medium, the DOX release increased to 69% (pH 5.4 + GSH), 53.52% (pH 6.8 + GSH), and 45.21% (pH 7.4 + GSH). Therefore, the constructed MSNs-HA-11C12 drug loaded system has a dual stimulation response of pH and redox.
3.4. In vitro cellular cytotoxicity
The in vitro cytotoxicity of MSNs-HA-11C12 toward LoVo cells was analyzed by CCK8 assay (Fig. 5A). When the concentration of MSNs-HA-11C12 was between 1 and 50 μg mL−1, the cell viabilities were close to 100%, and when increasing the concentration of MSNs-HA-11C12 up to 75 μg mL−1 and 100 μg mL−1, the cell viability still reached 97% and 81%, respectively. The prepared MSNs-HA-11C12 without the loading of DOX did not show obvious apoptosis toward LoVo cells after treatment for 48 h, which illustrated the good biocompatibility and safety of MSNs-HA-11C12.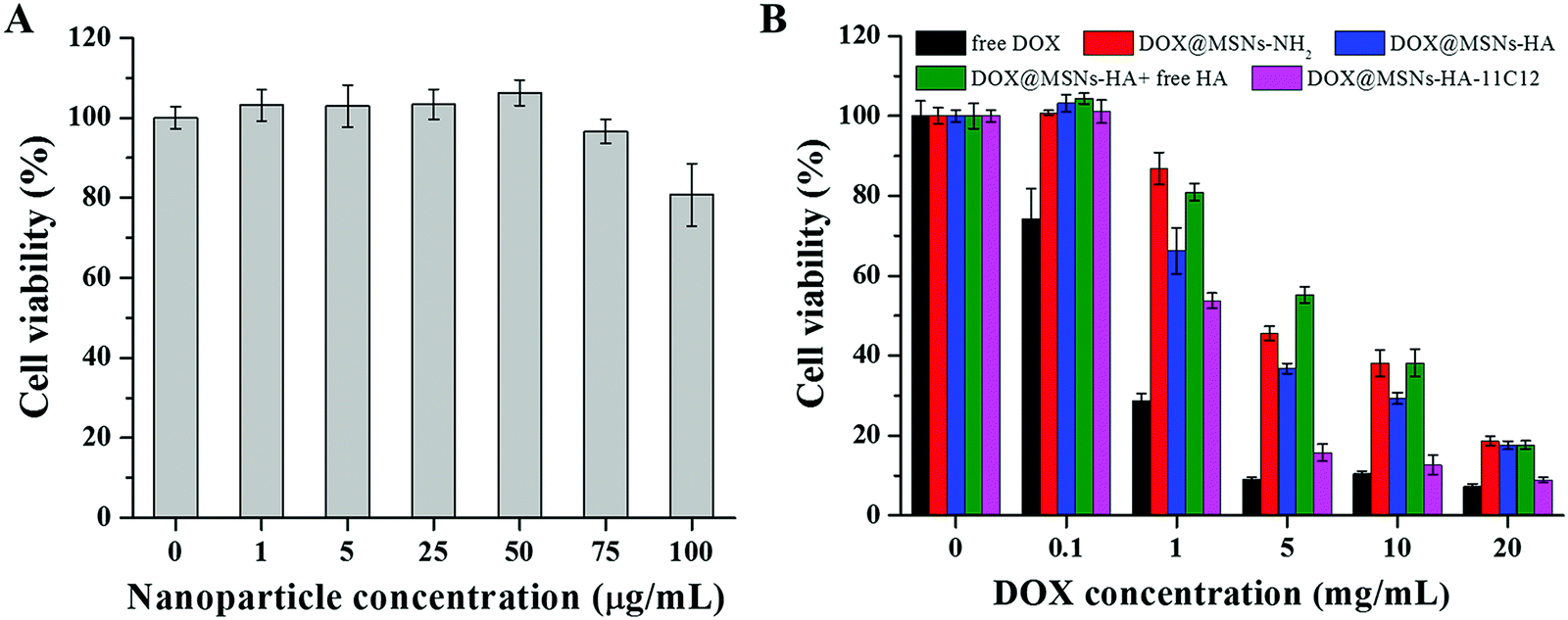 | ||
| Fig. 5 (A) In vitro cell viability of LoVo incubated with different concentrations of MSNs-HA-11C12 for 48 h. (B) In vitro cell viability of LoVo cells incubated with different concentrations of DOX@MSNs-NH2, DOX@MSNs-HA, DOX@MSNs-HA + free HA, DOX@MSNs-HA-11C12, and free DOX for 48 h. Error bars and standard deviations from three independent experiments. | ||
The cytotoxic effects of DOX@MSNs-NH2, DOX@MSNs-HA and DOX@MSNs-HA-11C12 at a tested concentration against LoVo cells for 48 h were also shown in Fig. 5B. The DOX loaded nanoparticles showed an obvious tendency to induce LoVo cell apoptosis in a dose-dependent manner. With free DOX as the control, it showed the highest cell killing effect compared with the drug loaded nanoparticles at all examined concentrations. Doses higher than 5 μg mL−1 of free DOX showed significantly pronounced killing of LoVo cells, whereas at 20 μg mL−1 free DOX for 48 h, the LoVo cell viability was only 7.3%. This might be due to the better diffusion effect of small molecules of DOX while increasing the probability of drug endocytosis in the in vitro cell experiment. More importantly, it was shown that DOX can likewise achieve highly efficient apoptosis toward CRC cells as a chemotherapeutic drug.40 The LoVo cellular viability was 86.9% (1 μg mL−1) and 45.6% (5 μg mL−1) for DOX@MSNs-NH2, and 66.2% (1 μg mL−1) and 36.8% (5 μg mL−1) for DOX@MSNs-HA after incubation with DOX@MSNs-NH2 or DOX@MSNs-HA for 48 h. The results illustrate that MSNs-NH2 modified by HA could significantly increase the apoptosis to LoVo cells resulting from the CD44 receptors of LoVo cell surface's targeting ability of the displayed HA on the MSNs-HA. When free HA was added to the DOX@MSNs-HA group as a competitive agent, the uptake of nanoparticles by cells was reduced due to free HA binding to the CD44 receptors of cell surfaces, which showed the reduction of LoVo cells apoptosis, and verified the targeting of HA in DOX@MSNs-HA. For example, the LoVo cellular viability (5 μg mL−1) was 55.2% for DOX@MSNs-HA + free HA and 36.8% for DOX@MSNs-HA. At the same concentration, DOX@MSNs-HA-11C12 which was modified by two targeting molecules HA and 11C12 had the strongest apoptosis toward LoVo cells owing to the displayed HA and 11C12 targeting to the CD44 receptors and the proximal membrane end of CEA on the surface of CRC cells, respectively. When the DOX concentration was 20 μg mL−1, the LoVo cellular viability (8.9%) was obtained for DOX@MSNs-HA-11C12. This phenomenon indicated that the modification of HA and 11C12 increased the targeting of the MSNs drug delivery system toward LoVo cells, improved the binding between the drug delivery system and LoVo cells, and enhanced the probability of LoVo cell apoptosis.
3.5. In vitro cellular uptake
The LoVo cells were incubated with DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 for 2 h, respectively. Then, the internalization of DOX was assessed qualitatively by CLSM. DOX was employed as the fluorescent probe as it emits red fluorescence under a specified wavelength of excitation light.41,42 The cell nuclei were stained in blue by DAPI since it is a fluorescent dye that binds strongly to DNA. As shown in Fig. 6, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 displayed obvious higher fluorescence intensity compared with free DOX, illustrating the amount of free DOX that was taken up by LoVo cells was minimal. The results were conflicted with the fact that free DOX led to the highest cell killing effect (Fig. 5), because the incubation time of free DOX and DOX loaded nanoparticles with LoVo cells were different in the cellular cytotoxicity and cellular uptake experiments. It was also suggested that the modified nanoparticles could transport more DOX to the cells in a shorter time. The fluorescence intensity trend was observed as follows: free DOX < DOX@MSNs-NH2 < DOX@MSNs-HA < DOX@MSNs-HA-11C12. The DOX fluorescence of the cells treated with free DOX appeared in the cell due to molecular free diffusion. While the DOX fluorescence of the cells treated with DOX@MSNs-NH2 was presented in the cell due to mainly enhanced permeability and the retention effect of the tumor. The increasing amount of DOX uptake from DOX@MSNs-HA-11C12 to DOX@MSNs-HA, higher than free DOX and DOX@MSNs-NH2, results from the targeting ability of functional molecules, as DOX@MSNs-HA-11C12 was modified by both HA and 11C12. This is the first study to modify nanoparticles with HA and 11C12 in order to enhance the dual target ability. The DOX@MSNs-HA-11C12 possesses a dual targeting property resulting from the CD44 receptor targeting ability of HA and the targeting ability of 11C12 toward the proximal membrane end of CEA on the surface of CRC cells.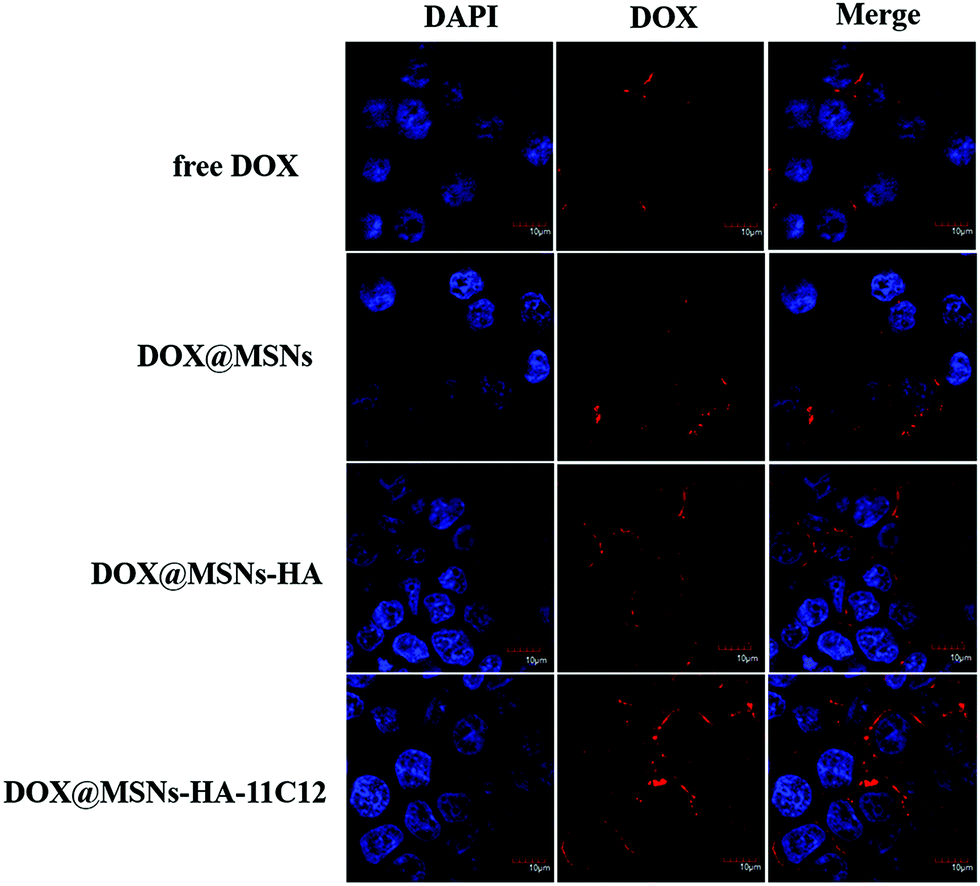 | ||
| Fig. 6 CLSM images of LoVo cells after incubation with free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 for 2 h. | ||
The fluorescence spectrophotometer quantitatively analysed the uptake of free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 by LoVo cells (Fig. 7). From the fluorescence intensity results, the uptake of DOX@MSNs-HA-11C12 by LoVo cells was the highest, followed by DOX@MSNs-HA, DOX@MSNs-NH2, DOX, and the blank, which was consistent with the results of CLSM. For example, after incubating for 4 h, the fluorescence intensity was 4546 for DOX@MSNs-HA-11C12, 4278 for DOX@MSNs-HA, 1411 for DOX@MSNs-NH2, and 570 for free DOX. Meanwhile, in a certain range, the uptake of free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 by LoVo cells increased gradually with the extension of incubation time.
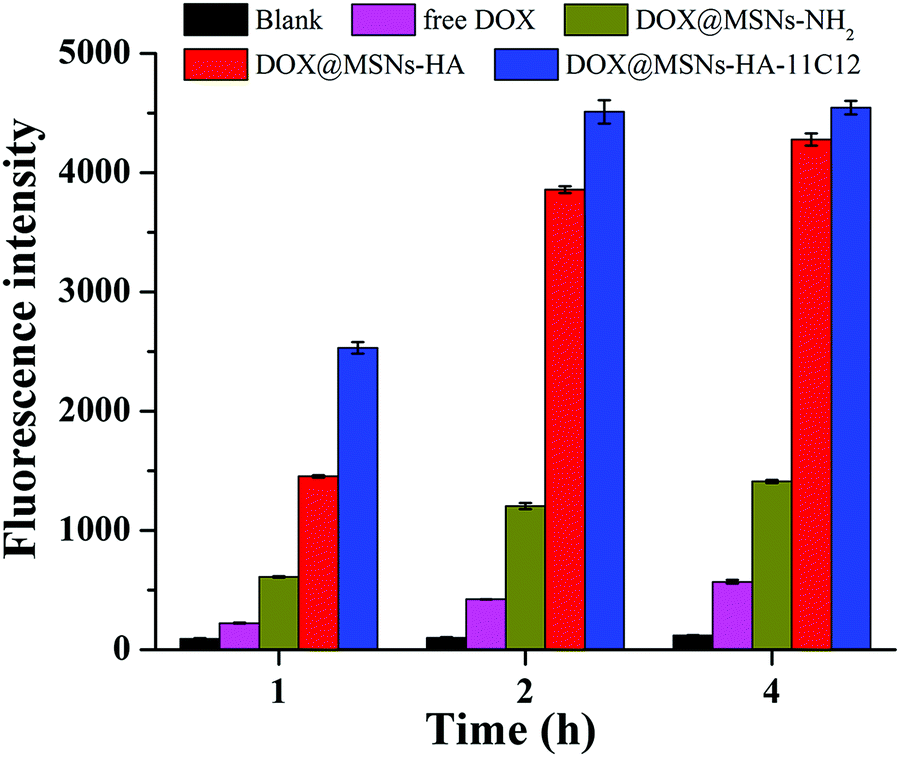 | ||
| Fig. 7 Fluorescence spectrophotometer results of LoVo cells incubated with free DOX, DOX@MSNs-NH2, DOX@MSNs-HA, and DOX@MSNs-HA-11C12 for 1 h, 2 h, and 4 h, respectively. Error bars and standard deviations from three independent experiments. | ||
4. Conclusions
In this study, we developed a dual targeted drug delivery system modified by HA and nanobody 11C12 (MSNs-HA-11C12) to sustain and control the delivery of DOX for targeting CRC. The results of TGA, DLS, and FTIR demonstrated that HA and 11C12 were successfully functionalized on the surface of MSNs. The DOX-EE and DOX-DL of MSNs-HA-11C12 were 73.12% and 26.77%, respectively. In vitro release profiles illustrated that DOX@MSNs-HA-11C12 was a pH- and redox– responsive drug delivery system for DOX. In vitro cytotoxicity experiments exhibited that MSNs-HA-11C12 without loading of DOX showed good biocompatibility and safety toward LoVo cells. CLSM and fluorescence spectrophotometry showed that the increasing amount of DOX uptake from DOX@MSNs-HA-11C12 to DOX@MSNs-HA, higher than free DOX and DOX@MSNs-NH2, results from the target ability of the functional molecule, as DOX@MSNs-HA-11C12 was modified by HA targeting for CD44 receptors and 11C12 targeting for the proximal membrane end of CEA on the surface of CRC cells. This is the first time that a drug delivery system has been combined with the targeting molecule HA and nanobody for CRC treatment, and this work provides new strategies for the clinical treatment of other major diseases.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by Shandong Energy Institute (SEI) [SEI I202144, SEI I202128].References
- L. H. Nguyen, A. Goel and D. C. Chung, Gastroenterology, 2020, 158, 291–302 CrossRef CAS PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- E. M. Stoffel and C. C. Murphy, Gastroenterology, 2020, 158, 341–353 CrossRef PubMed.
- Z. Zhai, X. Yu, B. Yang, Y. Zhang, L. Zhang, X. Li and H. Sun, Semin. Cell Dev. Biol., 2017, 64, 107–115 CrossRef PubMed.
- M. H. Chan and H. M. Lin, Biomaterials, 2015, 46, 149–158 CrossRef CAS PubMed.
- W. Cheng, J. Nie, L. Xu, C. Liang, Y. Peng, G. Liu, T. Wang, L. Mei, L. Huang and X. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 18462–18473 CrossRef CAS PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- E. Gullotti, J. Park and Y. Yeo, Pharm. Res., 2013, 30, 1956–1967 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- S. Wang, M. Cao, X. Deng, X. Xiao, Z. Yin, Q. Hu, Z. Zhou, F. Zhang, R. Zhang, Y. Wu, W. Sheng and Y. Zeng, Adv. Healthcare Mater., 2015, 4, 281–290 CrossRef CAS PubMed.
- C. W. Lin, K. Y. Lu, S. Y. Wang, H. W. Sung and F. L. Mi, Acta Biomater., 2016, 35, 280–292 CrossRef CAS PubMed.
- H. Ponta, L. Sherman and P. A. Herrlich, Nat. Rev. Mol. Cell Biol., 2003, 4, 33–45 CrossRef CAS PubMed.
- C. Chen, S. Zhao, A. Karnad and J. W. Freeman, J. Hematol. Oncol., 2018, 11, 64 CrossRef PubMed.
- B. Gupta, B. K. Poudel, H. B. Ruttala, S. Regmi, S. Pathak, M. Gautam, S. G. Jin, J. H. Jeong, H. G. Choi, S. K. Ku, C. S. Yong and J. O. Kim, Acta Biomater., 2018, 80, 364–377 CrossRef CAS PubMed.
- S. Ogino, N. Nishida, R. Umemoto, M. Suzuki, M. Takeda, H. Terasawa, J. Kitayama, M. Matsumoto, H. Hayasaka, M. Miyasaka and I. Shimada, Structure, 2010, 18, 649–656 CrossRef CAS PubMed.
- M. Ma, H. Chen, Y. Chen, K. Zhang, X. Wang, X. Cui and J. Shi, J. Mater. Chem., 2012, 22, 5615–5621 RSC.
- M. Campos-da-Paz, J. G. Dórea, A. S. Galdino, Z. G. M. Lacava and M. de Fatima Menezes Almeida Santos, Recent Pat. Biotechnol., 2018, 12, 269–279 CrossRef CAS PubMed.
- M. Bacac, T. Fauti, J. Sam, S. Colombetti, T. Weinzierl, D. Ouaret, W. Bodmer, S. Lehmann, T. Hofer, R. J. Hosse, E. Moessner, O. Ast, P. Bruenker, S. Grau-Richards, T. Schaller, A. Seidl, C. Gerdes, M. Perro, V. Nicolini, N. Steinhoff, S. Dudal, S. Neumann, T. von Hirschheydt, C. Jaeger, J. Saro, V. Karanikas, C. Klein and P. Umaña, Clin. Cancer Res., 2016, 22, 3286–3297 CrossRef CAS PubMed.
- M. C. da Paz, M. F. Santos and C. M. Santos, Int. J. Nanomed., 2012, 7, 5271–5282 Search PubMed.
- M. A. Fernandes, J. O. Eloy, M. T. Luiz, S. Junior and M. Chorilli, Colloids Surf., A, 2021, 611, 125806 CrossRef.
- C. Hamers-Casterman, T. Atarhouch, S. Muyldermans, G. Robinson, C. Hamers, E. B. Songa, N. Bendahman and R. Hamers, Nature, 1993, 363, 446–448 CrossRef CAS PubMed.
- S. Muyldermans, T. N. Baral, V. C. Retamozzo, P. De Baetselier, E. De Genst, J. Kinne, H. Leonhardt, S. Magez, V. K. Nguyen, H. Revets, U. Rothbauer, B. Stijlemans, S. Tillib, U. Wernery, L. Wyns, G. Hassanzadeh-Ghassabeh and D. Saerens, Vet. Immunol. Immunopathol., 2009, 128, 178–183 CrossRef CAS PubMed.
- M. Arbabi Ghahroudi, A. Desmyter, L. Wyns, R. Hamers and S. Muyldermans, FEBS Lett., 1997, 414, 521–526 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2017, 35, 1115–1117 CrossRef CAS PubMed.
- Q. Chen, Y. Zhou, J. Yu, W. Liu, F. Li, M. Xian, R. Nian, H. Song and D. Feng, Protein Expression Purif., 2019, 155, 43–47 CrossRef CAS PubMed.
- J. Lin, J. Yu, H. Wang, Y. Xu, F. Li, X. Chen, Y. Liang, J. Tang, L. Wu, Z. Zhou, C. Chen, M. Liu, X. Chun, R. Nian and H. Song, Anal. Bioanal. Chem., 2020, 412, 1723–1728 CrossRef CAS PubMed.
- J. Gu, W. Fan, A. Shimojima and T. Okubo, Small, 2007, 3, 1740–1744 CrossRef CAS PubMed.
- N. Lang and A. Tuel, Chem. Mater., 1966, 16, 1961–1966 CrossRef.
- Z. Bao, Y. Sun, K. Rai, X. Peng, S. Wang, R. Nian and M. Xian, Biomater. Sci., 2018, 6, 3042–3052 RSC.
- X. Zeng, W. Tao, L. Mei, L. Huang, C. Tan and S.-S. Feng, Biomaterials, 2013, 34, 6058–6067 CrossRef CAS PubMed.
- C. Tao, Y. Zhu, Y. Xu, M. Zhu, H. Morita and N. Hanagata, Dalton Trans., 2014, 43, 5142–5150 RSC.
- L. S. Wang, L. C. Wu, S. Y. Lu, L. L. Chang, I. T. Teng, C. M. Yang and J. Ho, ACS Nano, 2010, 4, 4371–4379 CrossRef CAS PubMed.
- J. Kim, J. E. Lee, J. Lee, Y. Jang, S. W. Kim, K. An, J. H. Yu and T. Hyeon, Angew. Chem., Int. Ed., 2006, 45, 4789–4793 CrossRef CAS PubMed.
- Q. Zhao, J. Liu, W. Zhu, C. Sun, D. Di, Y. Zhang, P. Wang, Z. Wang and S. Wang, Acta Biomater., 2015, 23, 147–156 CrossRef CAS PubMed.
- Z. Chen, Z. Li, Y. Lin, M. Yin, J. Ren and X. Qu, Chemistry, 2013, 19, 1778–1783 CrossRef CAS PubMed.
- Y. Zhou, G. Quan, Q. Wu, X. Zhang, B. Niu, B. Wu, Y. Huang, X. Pan and C. Wu, Acta Pharm. Sin. B, 2018, 8, 165–177 CrossRef PubMed.
- L. Cai, P. Zhu, F. Huan, J. Wang, L. Zhou, H. Jiang, M. Ji and J. Chen, Colloids Surf., B, 2021, 205, 111839 CrossRef CAS PubMed.
- M. Kim, D. Ki, Y. Na, H. Lee, J. Baek, J. Lee, H. Lee and C. Cho, Pharmaceutics, 2021, 13, 184 CrossRef CAS PubMed.
- L. Chen, L. Li, L. Zhang, S. Xing, T. Wang, Y. A. Wang, C. Wang and Z. Su, ACS Appl. Mater. Interfaces, 2013, 5, 7282–7290 CrossRef CAS PubMed.
- F. Muhammad, M. Guo, W. Qi, F. Sun, A. Wang, Y. Guo and G. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef CAS PubMed.
- R. Zhang, C. Wu, X. Wang, S. Qian, B. Chen, X. Li, S. Gutmann and L. Gang, Mater. Sci. Eng., C, 2009, 29, 1697–1701 CrossRef CAS.
- K. Dong, Z. Z. Zhao, J. Kang, L. R. Lin, W. T. Chen, J. X. Liu, X. L. Wu and T. L. Lu, Int. J. Nanomed., 2020, 15, 10285–10304 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |