DOI:
10.1039/D2MA00206J
(Paper)
Mater. Adv., 2022,
3, 5027-5033
Selective and sensitive determination of capsaicin using polymelamine formaldehyde decorated over carbon nanotubes†
Received
22nd February 2022
, Accepted 20th April 2022
First published on 26th April 2022
Abstract
An electrochemical biosensor based on polymelamine formaldehyde (PMF) supported over oxygen functionalised carbon nanotubes (OCNTs) was designed for selective and sensitive determination of capsaicin. The synergy between the highly porous PMF and conductive OCNTs enhanced the electron transport at the electrode/electrolyte interface and eliminated the interference of common organic and inorganic ions at pH 1. The PMF/OCNT composite exhibited superior selectivity and sensitivity towards capsaicin with a low detection limit of 71.5 nM, a response time of ∼1 minute and a wider detection range of 0.1–500 μM with two linear ranges of 100 nM–240 μM and 240–500 μM and high sensitivities of 960 μA μM−1 cm−2 and 2900 μA μM−1 cm−2, respectively. The proposed sensor was successfully applied to the determination of capsaicin in different chilli samples, with the recoveries between 99.2–114%, demonstrating the practical applicability of the sensor.
1. Introduction
Capsaicin is an important phytochemical present in peppers which constitutes 90% of total alkaloids present in chilli.1,2 The hotness of the peppers is due to the capsaicin content, which ranges from 0.1 to 1% w/w.3 Along with this, capsaicin is also known for possessing various human health benefits, including antioxidant,4 anti-tumorigenic,5 antimutagenic,6 anti-bacterial,7 anti-inflammatory,8 and anti-carcinogenic,9 as well as protective properties against cholesterol and obesity.10–12 Besides this, capsaicin has also been used as a topical analgesic for post herpetic neuralgia as well as the mouth sores caused due to chemotherapy or radiation, osteoarthritis and rheumatoid arthritis.13 Despite the numerous benefits associated with capsaicin, excess consumption can lead to disastrous effects on human health. Hence, determining the amount of capsaicin with high accuracy and precision remains one of the hottest topics in the field of medicine, the food industry and pharmacology.14 The heat level of pepper and hot sauces has been conventionally measured using “Scoville's organoleptic method” which is rather subjective and relies on the sensations received by trained personnel.15 Other existing techniques include thin layer chromatography (TLC), gas phase chromatography, high-performance liquid phase chromatography (HPLC), UV-Vis spectroscopy, fluorescence spectroscopy capillary electrophoresis and the HPLC technique associated with UV-Vis and mass spectrometry16–22 commonly employed for selective determination of capsaicin in a complex mixture of various capsaicinoids. Among the various techniques used for the determination of capsaicin, electrochemical methods are in vogue and have drawn considerable attention because of their quick response, high sensitivity, and ease of operation.23 Electrochemical determination of capsaicin was firstly introduced by Kachoosangi et al., who developed an adsorptive stripping voltammetry-based method for determining capsaicin accurately within commercially available hot-pepper sauces using carbon nanotubes (CNTs).24 Later on, continuous research attempts have been made to improve the sensitivity towards capsaicin by developing efficient catalysts.25 Until now, various catalytic materials, including boron-doped diamond,26 carbon-based disposable electrodes,27,28 metal nanoparticles (Ag/Ag2O, CeO2, Ru, Sn etc.),29–32 mesoporous cellular foams33 and recently, N-doped atom graphene nanoplates34 have been explored for selective and sensitive electrochemical determination of capsaicin. However, the synthesis of these catalysts is a time consuming, complicated procedure that requires harsh conditions and high temperature, and low stability and high cost are major concerns.
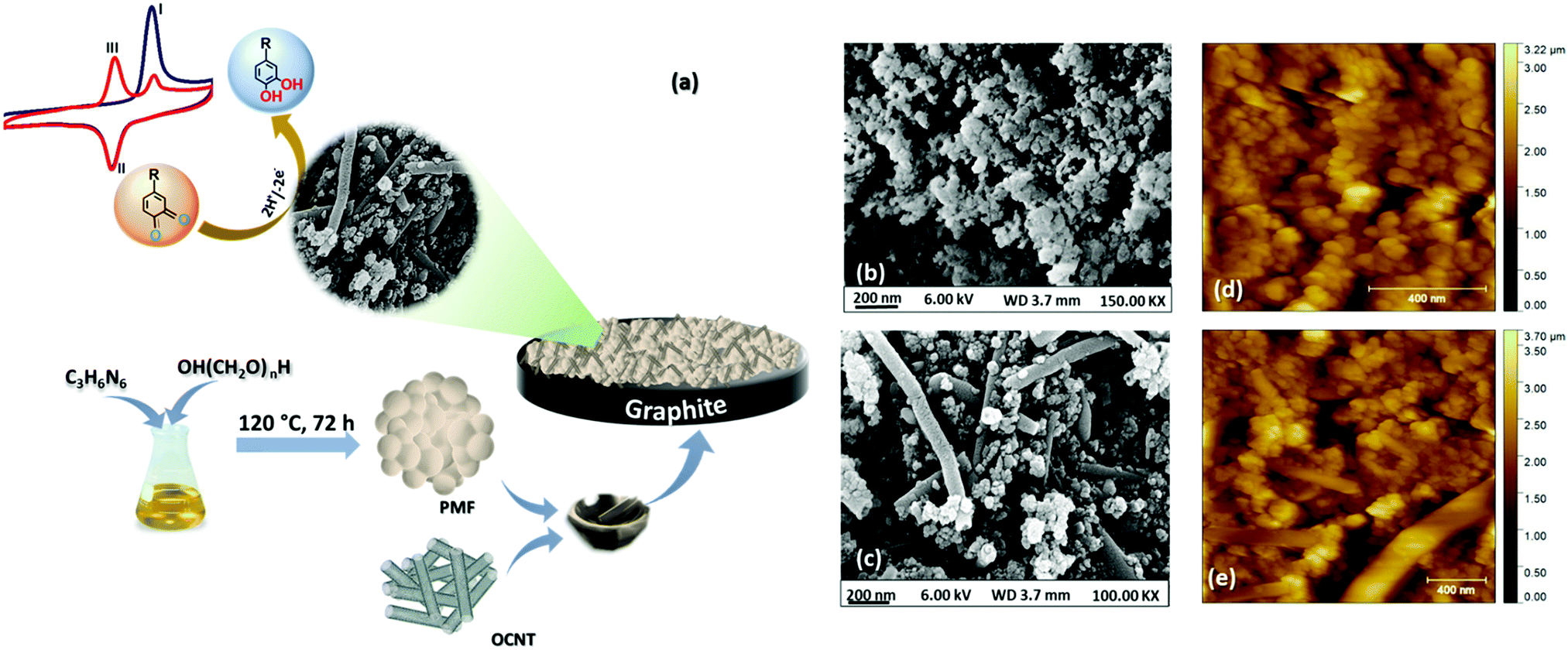
In the present report, we have explored polymelamine formaldehyde (PMF) along with oxygen functionalized carbon nanotubes (OCNTs) as an efficient catalyst for selective electrochemical detection of capsaicin. Recent past porous organic polymers by virtue of their unique properties like porosity, high surface area, adjustable functionality, biocompatibility, good chemical and thermal resistivity and physicochemical stability, have been utilized for various applications, including drug delivery, adsorbents, filters and electrochemical sensors.35–37 Nevertheless, no work has been reported on employing PMF as an electrode material to construct an electrochemical sensor for capsaicin. Herein, we demonstrated PMF in combination with OCNTs as a PMF/OCNT composite for selective determination of capsaicin. The natural π–π affinity between PMF and OCNTs facilitates the electron transfer at the electrode/electrolyte interface. Moreover, the huge number of amine functional groups of PMF in hybridization with OCNTs enhances the sorption capacity of PMF. In addition, the polymers enhance the electrical conductivity, mechanical strength, and adhesive property towards the substrates.38 The proposed sensor has shown outstanding physicochemical stability in highly acidic media (pH 1) along with high selectivity towards capsaicin with detection of a wide detection range of 0.1–500 μM and lowest detection limit (LOD) of 71.5 nM. A schematic representation (Fig. 1(a)) shows the fabrication procedure of the PMF/OCNT electrochemical sensor for capsaicin.
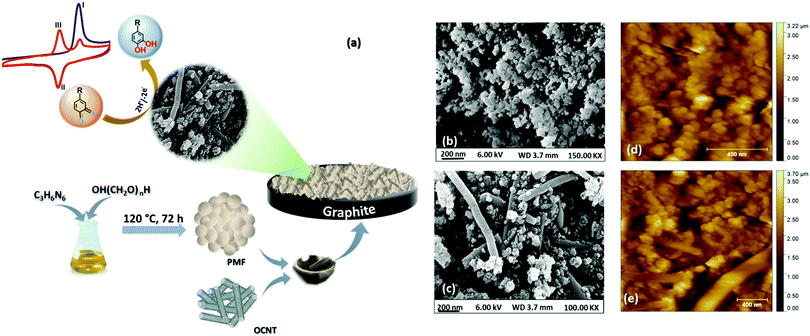 |
| | Fig. 1 (a) Schematic representation of the PMF/OCNT composite preparation process and electrochemical measurement of capsaicin; (b) and (c) FE-SEM images of PMF and the PMF/OCNT composite; (d) and (e) AFM images of PMF and the PMF/OCNT composite, respectively. | |
2. Experimental section
2.1. Materials and reagents
All chemicals were of analytical reagent grade and used without any further purification. Paraformaldehyde and melamine were purchased from Loba Chemie. Capsaicin was commercially purchased from Sigma Aldrich. A stock solution of capsaicin was prepared in anhydrous ethanol and stored at 4 °C. Briton–Robinson (BR) buffer solution (50 mM) was prepared by mixing boric acid, acetic acid and phosphoric acid and the pH was adjusted from 1 to 5 by using 0.1 M NaOH solution. All the aqueous solutions were prepared by using deionized water obtained from a Millipore system with resistivity of >12 MΩ cm−1.
2.2. Synthesis of polymelamine formaldehyde
Polymelamine formaldehyde was synthesized via one-pot synthesis; briefly, both the monomers, melamine (0.378 g, 3 mmol) and paraformaldehyde (1.8 equiv., 0.162 g, 5.4 mmol), were mixed with 3.36 mL (overall concentration of 2.5 M) of dimethyl sulfoxide (DMSO) in a Teflon container protected in a steel reactor. The reaction mixture was heated to 120 °C in an oven for 1 h. The reactor was kept stirring for 15 minutes to obtain a homogeneous solution. The solution was then heated to 170 °C for 72 h.39 The reaction was allowed to cool at room temperature, then crushed, filtered, and washed with different solvents, DMSO, acetone, tetrahydrofuran (THF) and CH2Cl2. The resulting filtered solid was dried in oven 80 °C for 24 h.
2.3. Preparation of the PMF/OCNT/GCE electrochemical sensor
The PMF/OCNT composite was prepared by physically grinding a mixture of PMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OCNT (70:30) using a mortar and pestle. The homogenous slurry was prepared by dispersing 1.3 mg of composite in 500 μL containing isopropyl alcohol (IPA 100 μL) and Millipore water (400 μL, 12 MΩ) by ultrasonication for 30 min. Afterwards, 20 μL (52 μg) of the as prepared slurry was drop cast on a graphite electrode and dried at room temperature.
:OCNT (70:30) using a mortar and pestle. The homogenous slurry was prepared by dispersing 1.3 mg of composite in 500 μL containing isopropyl alcohol (IPA 100 μL) and Millipore water (400 μL, 12 MΩ) by ultrasonication for 30 min. Afterwards, 20 μL (52 μg) of the as prepared slurry was drop cast on a graphite electrode and dried at room temperature.
2.4. Physical characterization
The synthesized material was characterized by X-ray diffractometer (XRD) using a PAN analytical X’Pert-Pro diffraction system with CuKα radiation of 1.54 Å operated at 45 kV and 40 mA at a scanning of 2° min−1 ranging from 5° to 80°. Morphological analysis was performed by field emission scanning electron microscopy (FESEM, JEOL, JSM-6610l) at 20 kV. FT-IR spectra were recorded using a BRUKER TENSOR-27 spectrometer in the range of 600–4000 cm−1 with a spectral resolution of 4 cm−1 and number of scans of 100. FT-IR data were collected and analysed by OPUS data collection and analysis software. AFM analysis was carried out using a Bruker multimedia 8 instrument operating in non-contact mode during AFM analysis.
2.5. Electrochemical measurements
Electrochemical experiments were carried out using a three-electrode assembly in a single compartment electrochemical workstation (Bio-logic (VSP)) consisting of a graphite electrode (∅ 2 mm) used as a working electrode (WE), platinum wire as an auxiliary/counter electrode (CE) and a Ag/AgCl/3 M KCl electrode as a reference electrode. All the analyses were done by cyclic voltammetry (CV), square wave voltammetry (SWV) and electrochemical impedance spectroscopy (EIS) techniques. CV measurements were made between 0.0 to 1.0 V vs. Ag/AgCl/3M KCl at a scan rate of 25 mV s−1 in a three-electrode assembly containing 10 mL BR buffer. SWV measurements were done with a step potential of 5 mV at a scan rate of 25 mV s−1 between 0.0 V to 1.0 V. All the experiments were performed at pH 1.0 with the optimized catalyst (70:30) and the accumulation time of one minute.
3. Results and discussion
The surface morphology of the synthesized PMF and PMF/OCNT composite were analysed by FE-SEM. The FE-SEM image (Fig. 1(b)) showed the spherical morphology of the polymer material and the particles became aggregated among themselves to form clusters. After physical mixing of the polymer with OCNTs, a homogenous distribution of polymer particles over the OCNT surface could be seen without any aggregation of the carbon nanotubes (Fig. 1(c)). This was further supported by AFM images showing that the spherical clusters of PMF fully covered the surface of the carbon nanotubes without altering the original structure of the CNTs (Fig. 1(d) and (e)). The energy dispersive spectroscopy (EDS) studies confirm the coexistence of N, C, and O elements in the PMF/OCNT composite (Fig. S1, ESI†). This was further supported by the XPS survey spectrum, which confirms the presence of all N, C, and O elements without any additional peak signifying the purity of the composite (Fig. S2a, ESI†). The XPS spectrum of C1s was deconvoluted into five peaks (Fig. S2b, ESI†). The peaks centred at 284.6 eV, 285.7 eV and 287.6 eV were assigned to C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N bonds, respectively. Whereas other peaks at 286.4 eV and 288.5 eV were attributed to carbon–oxygen functional groups (labelled as C–OH and –COOH). The N 1s XP spectra were fitted into three peaks at 398.7 eV, 399.7 eV, and 400.7 demonstrating the presence of pyridinic N (C–NC), pyrrolic N (C–N–C) and N–H bonds (Fig. S2c, ESI†). Also, the O 1s spectrum shows three oxygen peaks arising from CO (530.3 eV), C–O (531.3 eV) and O–CO (532.9 eV) groups ascribed to OCNTs (Fig. S2d, ESI†).30,42 The microstructural attributes of the PMF/OCNT composite were further characterized by powdered XRD. The presence of a sharp intense peak (Fig. S3a, ESI†) at 26.07° with a feeble hump at 44.02° corresponded to graphitic (002) and (001) planes, respectively, in the OCNTs. As expected, a broadened peak of PMF at 2θ of 21.08° signified the amorphous nature of the polymer.43 Furthermore, in the FT-IR spectrum (Fig. S3b, ESI†), the appearance of a broad peak at 3390 cm−1 together with absorption bands at 2946 cm−1 was attributed to the stretching of free –NH & –CH2 groups and the band at 1189 cm−1 was due to C–N stretching of secondary amines, confirming the existence of numerous –NH2 and –NH–CH2–NH groups in PMF. Moreover, the absorption bands located at 1568 and 1471 cm−1 indicated successful polymerization. The thermal stability of the polymer was further examined by TGA (Fig. S4, ESI†). The obtained curve showed a weight loss of ca. 53% up to 200 °C upon heating in air. The plausible reason could be the outgassing of moisture and entrapment of the solvent as well as CO2 adsorption in the pores of the polymer matrix.40 Furthermore, the weight loss above temperature 300 °C was due to decomposition of the polymer network and was completely decomposed beyond 500 °C. The obtained results indicated the excellent thermal stability of PMF up to 300 °C in air.41
N and C–N bonds, respectively. Whereas other peaks at 286.4 eV and 288.5 eV were attributed to carbon–oxygen functional groups (labelled as C–OH and –COOH). The N 1s XP spectra were fitted into three peaks at 398.7 eV, 399.7 eV, and 400.7 demonstrating the presence of pyridinic N (C–NC), pyrrolic N (C–N–C) and N–H bonds (Fig. S2c, ESI†). Also, the O 1s spectrum shows three oxygen peaks arising from CO (530.3 eV), C–O (531.3 eV) and O–CO (532.9 eV) groups ascribed to OCNTs (Fig. S2d, ESI†).30,42 The microstructural attributes of the PMF/OCNT composite were further characterized by powdered XRD. The presence of a sharp intense peak (Fig. S3a, ESI†) at 26.07° with a feeble hump at 44.02° corresponded to graphitic (002) and (001) planes, respectively, in the OCNTs. As expected, a broadened peak of PMF at 2θ of 21.08° signified the amorphous nature of the polymer.43 Furthermore, in the FT-IR spectrum (Fig. S3b, ESI†), the appearance of a broad peak at 3390 cm−1 together with absorption bands at 2946 cm−1 was attributed to the stretching of free –NH & –CH2 groups and the band at 1189 cm−1 was due to C–N stretching of secondary amines, confirming the existence of numerous –NH2 and –NH–CH2–NH groups in PMF. Moreover, the absorption bands located at 1568 and 1471 cm−1 indicated successful polymerization. The thermal stability of the polymer was further examined by TGA (Fig. S4, ESI†). The obtained curve showed a weight loss of ca. 53% up to 200 °C upon heating in air. The plausible reason could be the outgassing of moisture and entrapment of the solvent as well as CO2 adsorption in the pores of the polymer matrix.40 Furthermore, the weight loss above temperature 300 °C was due to decomposition of the polymer network and was completely decomposed beyond 500 °C. The obtained results indicated the excellent thermal stability of PMF up to 300 °C in air.41
Electrochemical behavior of capsaicin
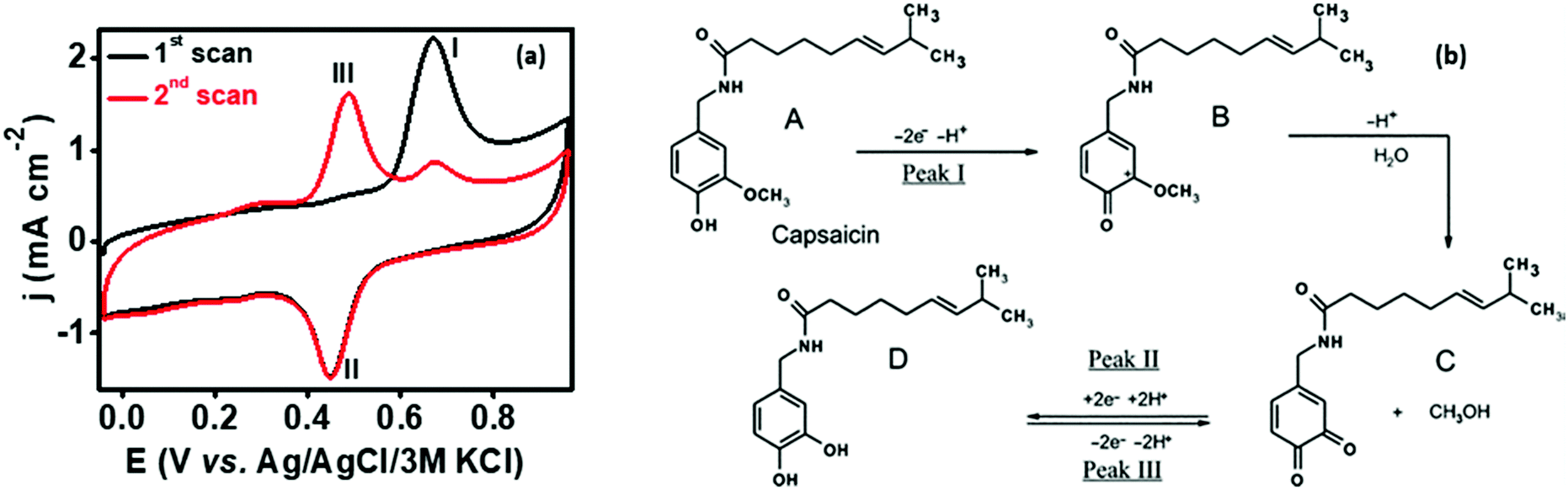
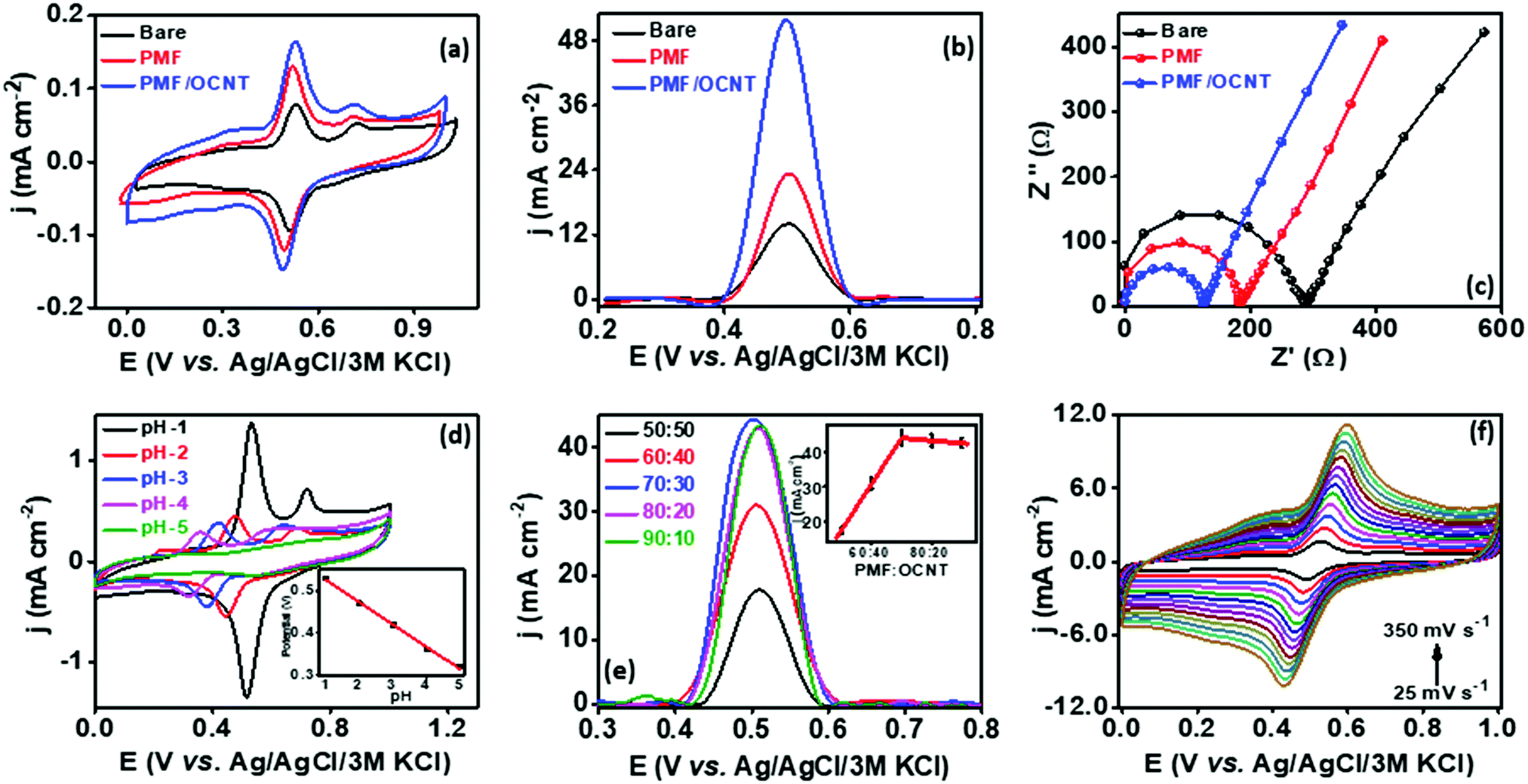
In order to analyze the electrocatalytic behavior of the proposed PMF/OCNT composite towards capsaicin, preliminary experiments were performed using a 3 electrode assembly in 50 mM BR buffer electrolyte in the presence of 100 μM of capsaicin by sweeping two successive scans of cyclic voltammogram between a voltage range of 0.0 to 1.0 V at 25 mV s−1 (Fig. 2(a)). In the first scan, a sharp oxidation peak (peak I) at 0.67 V was observed, which corresponds to the electrooxidation of capsaicin, followed by an irreversible hydrolysis (Cirr) step ascribed to conversion of the 2-methoxy group into o-benzoquinone and giving rise to a well-known catechol redox couple shown by peak II and peak III at 0.49 V and 0.45 V, respectively. Furthermore, the redox cycle takes place between peak II & peak III as shown in the mechanism (Fig. 2(b)).1,24 To further authenticate the capability of PMF/OCNT towards electrocatalytic oxidation of capsaicin, preliminary studies were conducted with the PMF/OCNT composite. Fig. 3(a) and (b) display the well-defined redox peak in the second cycle of CV with an oxidation peak current density (0.16 mA cm−2) and peak separation (ΔEp) of 0.04 V. Comparatively the PMF and bare graphite showed an inferior current density of 0.13 mA cm−2 and 0.078 mA cm−2 fortifying that the superior activity of PMF/OCNT towards electrochemical oxidation of capsaicin was due to the synergic effect of the porous PMF and highly conductive OCNTs. The above results were further supported by electrochemical impedance spectroscopy (EIS), wherein the lowest Rct (Fig. 3(c) and Table S1, ESI†) in PMF/OCNT as compared to the bare graphite and PMF signifies the dominance of faster kinetics towards the electrochemical oxidation of capsaicin. This governs the effective charge transfer at the electrode–electrolyte interface and was fitted to an equivalent circuit shown in Fig. S5, ESI,† which was further confirmed by calculating the double layer capacitance in the capacitive region (Fig. S6 and Table S2 detailed in the ESI†).
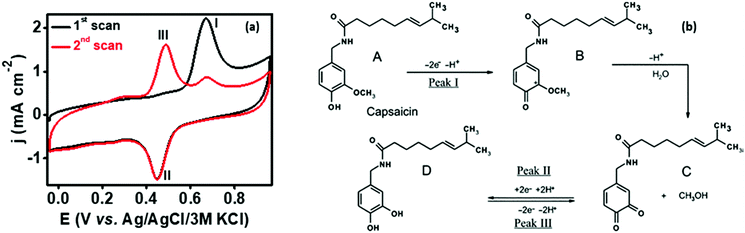 |
| | Fig. 2 (a) Two successive cyclic voltammetric scans in the presence of 100 μM capsaicin at a scan rate of 25 mV s−1 showing electrochemical oxidation of capsaicin; (b) mechanism showing the reaction pathway of electrochemical oxidation of capsaicin.40,41 | |
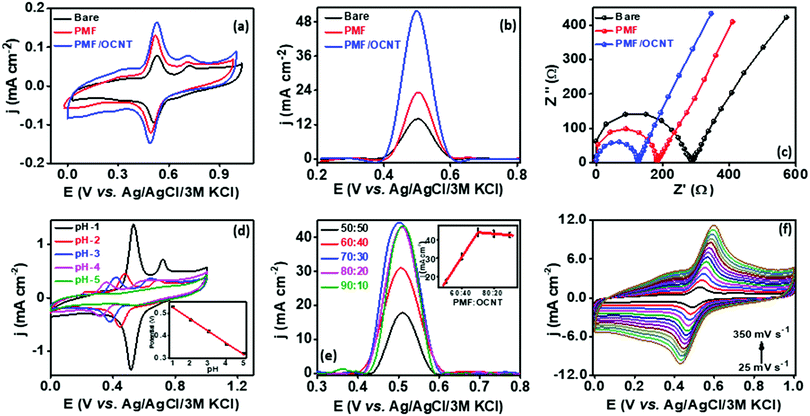 |
| | Fig. 3 (a) CV showing the response of various catalysts towards capsaicin, (b) corresponding SWV and (c) EIS, (d) CV showing the effect of different pHs on the PMF/OCNT modified electrode in 100 μM capsaicin (inset: corresponding linearity graph), (e) SWV showing the optimization of the catalyst ratio for PMF/OCNT (inset: corresponding linearity graph) and (f) CV showing the PMF/OCNT modified electrode at various scan rates in 50 mM BR buffer (pH 1.0) solution containing 100 μM capsaicin; CE: Pt wire, RE: Ag/AgCl/3 M KCl. | |
Optimization of the sensing parameters
pH optimization.
The effect of pH on the peak potential and the current response was investigated by varying the pH from 1.0 to 5.0 in BR buffer containing 100 μM capsaicin (Fig. 3(d)). The decrease in the peak current density (jp) with the increase in pH from 1.00 to 5.00, was apparently due to the partial deprotonation of the phenolic moiety in capsaicin at higher pH.24 Moreover, the peak potential varied linearly per unit increase in pH (Fig. 3(d) inset) and shifted to negative by 0.053 V, which was close to the −0.059 V as predicted by the Nernst equation. This suggested that electrooxidation of capsaicin involves an equal number of electrons and protons and the overall electrochemical as well as chemical step involves two electrons and protons. Therefore, BR buffer of pH 1.0 was taken as the optimum in subsequent experiments for better sensitivity and better signal to noise ratio.
Capsaicin accumulation time and composite optimization
In order to understand the effect of accumulation time of capsaicin on the fabricated sensor, SWV analysis was performed in BR buffer solution having pH 1.0 at 100 μM of capsaicin. The current density started decreasing from 55.7 to 8.4 mA cm−2 when the accumulation time increased from 1 to 4 min, and became constant (8.4 mA cm−2) with a further increase in time up to 6 minutes, indicating the saturation of the electrode surface (Fig. S7a and b, ESI†). Therefore, one minute was selected for operational direction of capsaicin.
The electrocatalytic activity of PMF is strongly influenced by the conductive support, which was analysed by taking different ratios of PMF:OCNT. As displayed in Fig. 3(e), different ratios of polymer and OCNT (PMF:OCNT) viz. 50:50, 60:40, 70:30, 80:20 and 90:10 showed particular responses towards the oxidation of capsaicin. The response current density increased with the increase in the amount of OCNTs, reaching the maximum value (44.20 mA cm−2) at 70:30, and then started decreasing. Therefore, 70:30 ratio was selected for subsequent experiments. The kinetic behaviour of capsaicin was further examined by studying the influence of the scan rate on the peak potential and peak current for 100 μM capsaicin on the PMF/OCNT modified electrode. CV was performed by varying the scan rate from 25 mV s−1 to 300 mV s−1 (Fig. 3(e)). The linear increase in the oxidation and reduction peak currents with scan rate indicated that both oxidation and reduction were adsorption-controlled processes (Fig. S8a, ESI†). In addition, the slope of the plot between the log of peak current and the log of scan rate was found to be 0.743, which is higher than 0.5 confirming that the process is adsorption controlled. The relationship between redox peak potential Ep and logarithm of scan rate was also investigated and it was found that Ep also changes linearly with the logarithm of the scan rate in the range of 25–350 mV s−1. Based on the slope between the peak potential and scan rate, the number of electrons involved in the oxidation and reduction of capsaicin was calculated via Laviron's equation (Fig. S8b, ESI†).44
The total number of electrons involved in the oxidation and reduction process was found to be two. The value of the charge-transfer rate constant (ks) for the composite in 100 μM capsaicin was found to be 7.15 × 102 s−1. Moreover, the concentration of capsaicin adsorbed on the PMF/OCNT modified electrode was analysed according to the formula using the slope of the relationship between the peak current and scan rate (Ipavs. ν) (Fig. S7c, ESI†). Furthermore, the electrocatalytic efficiency of the composite for oxidation of capsaicin was evaluated by calculating the catalytic rate constant using the Galus equation.45
| Icat/IL = π1/2(kcatC0t)1/2 |
wherein,
Icat,
IL,
t, and
C0 are the catalytic current for capsaicin, the limited current in the absence of capsaicin, the time elapsed, and the bulk concentration of capsaicin, respectively. Based on the slope of
Icat/
ILversus t1/2, the value of
Kcat was found to be 32.294 M
−1 s
−1 (Fig. S9a and b, ESI
†). This high value could be attributed to the increased efficiency of PMF/OCNT towards electrochemical oxidation of capsaicin.
Analytical detection of capsaicin
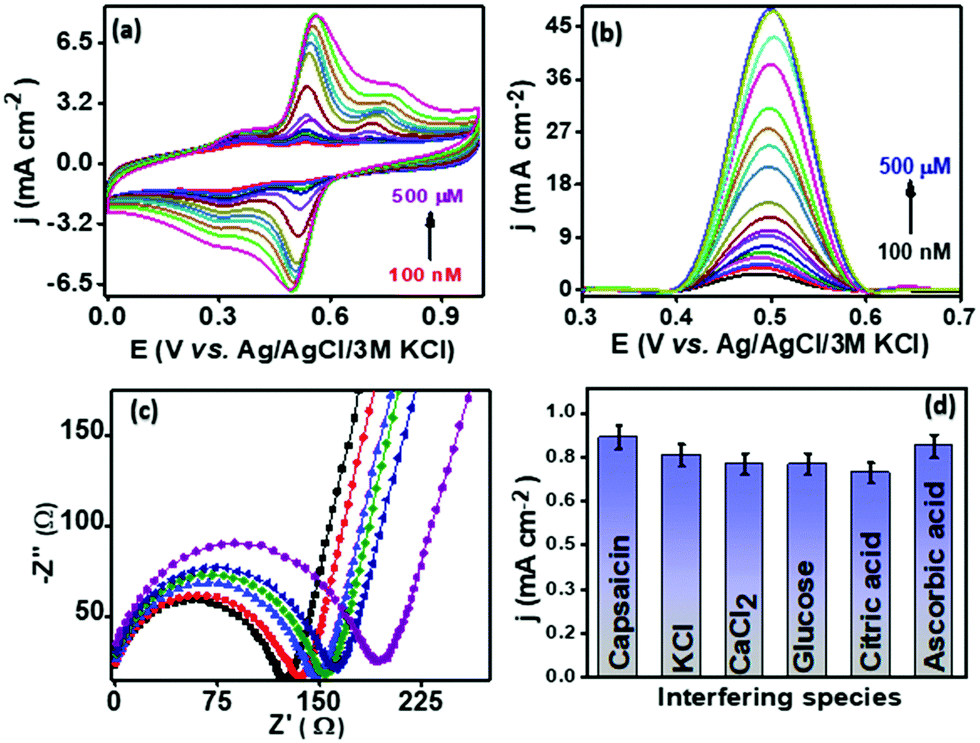
The analytical performance of the proposed sensor PMF/OCNT was investigated using CV and SWV under optimal conditions by varying the concentration of capsaicin from 100 nM–500 μM with an accumulation time of 1 minute for each concentration. Fig. 4 shows that with an increase in the concentration of capsaicin, the oxidation peak current increased and gets saturated at a current density of 48.14 mA cm−2 when the concentration reaches 500 μM, which indicates that all of the active sites that are present became saturated resulting in no further adsorption of capsaicin (Fig. 4(b)). Furthermore, the corresponding calibration curve was plotted using the oxidation peak current density (jpa) vs. capsaicin concentration. The proposed sensor exhibited two linear ranges, that is, 100 nM–240 μM and 240–500 μM with a superior sensitivity of 960 μA μM−1 cm−2 and 2900 μA μM−1 cm−2, respectively (Fig. S8d, ESI†). The limit of detection (LOD) was calculated using the LOD = 3Sa/b formula, where Sa is the standard deviation of the intercept and b is the slope of the calibration curve and was found to be 71.5 nM, respectively. The presence of two linear ranges with different sensitivities was attributed to the adsorptive nature of capsaicin over the electrode surface. At low concentration capsaicin directly adsorbs as a monolayer on the electrode surface and at higher concentration it absorbs in the form of multilayers and passivating all available active sites on the electrode surface provides a second range with lower sensitivity.46 In addition, the effect of interferences of some common organic and inorganic compounds (i.e., KCl, MgCl2, CaCl2, citric acid, glucose and ascorbic acid) present in the chilli samples along with capsaicin was also evaluated using SWV. The results depicted that no significant increase in peak current was observed for addition of 10 mM of each interferent in the presence of 100 μM capsaicin suggesting good anti-interference properties of the proposed sensor and that it can be used for real sample analysis (Fig. 4(d)). The operational stability of PMF/OCNT was investigated by cyclic voltammogram studies over 100 consecutive cycles with the scan rate of 25 mV s−1 (Fig. S10a, ESI†). The negligible decay in either current or potential demonstrated the excellent stability of the composite. Furthermore, the sensor was stored for twelve days and the current response was measured on alternate days. Fig. S10b, ESI† shows that the sensor retained its initial response after being stored for 12 days. This result indicated the long-term storage stability of the proposed sensor. Moreover, the PMF/OCNT based electrochemical sensor displays a wider linear range as well as the shortest accumulation time compared to the previously reported sensors for capsaicin detection (Table S3, ESI†).
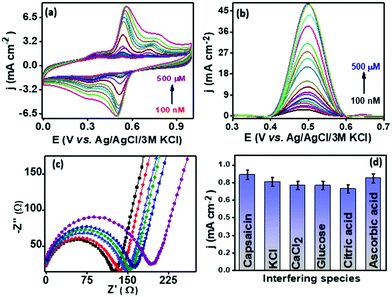 |
| | Fig. 4 (a) CV, (b) SWV and (c) EIS response of the PMF/OCNT modified electrode in 50 mM BR buffer at various conc. of capsaicin; (d) bar diagram representing the current density w.r.t. interferants at a 100 μM concentration of capsaicin. | |
Real sample analysis
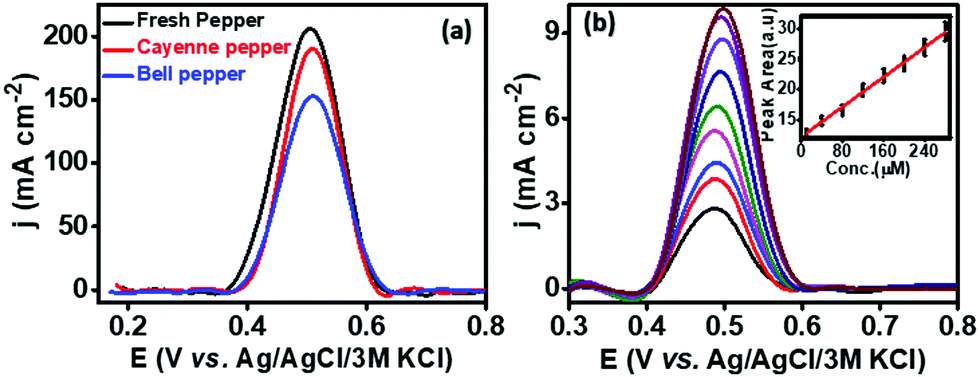
The practical applicability of the developed PMF/OCNT sensor was carried out by choosing fresh green pepper, bell pepper and cayenne pepper available in a local market. The chillies were crushed, and filtered to remove any solid waste. The extraction of capsaicin from the real samples was carried out by dissolving 10 mg of each sample in 2 mL of 30% ethanol containing BR buffer (pH 1.0). The diluted samples were then ultrasonicated for 30 minutes to obtain a uniform dispersion and 100 μL of the above-diluted sample was then added to 10 mL of BR buffer. To ascertain the authentication of the proposed sensor, certain known amounts of capsaicin were added to the diluted sample and then detected using the standard addition method (Fig. 5). The results obtained are tabulated in Table S4, ESI†. Furthermore, the recovery was found from 99.2–114%. The obtained results were in good agreement (Fig. S11 and Table S5, ESI†) with HPLC, signifying that the sensor possesses good accuracy and can be promising for practical applications.
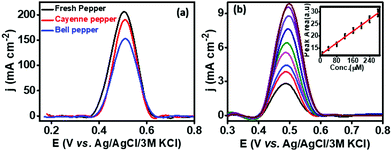 |
| | Fig. 5 (a) SWV response of PMF/OCNT modified electrodes for different pepper samples and (b) SWV responses for the addition of 10, 40, 60, 80, 120, 140, 180, 200, 240, and 280 μM capsaicin in diluted samples of different chillies after 1 min accumulation time in 50 mM BR buffer (pH 1) (inset: corresponding calibration curve between concentration and peak area for additions of capsaicin). | |
4. Conclusion
Herein, we have developed a PMF/OCNT based electrochemical sensor for direct capsaicin sensing, wherein the PMF/OCNT exhibits high surface area and good electrochemical properties. The proposed sensor exhibited high sensitivities of 0.96 mA μM−1 cm−2 and 2.90 mA μM−1 cm−2, with LOD of 71.5 nM. The interference study demonstrated good selectivity among various interferents with high adsorbing power of capsaicin over PMF/OCNT due to strong interactions. The heterogeneous electron transfer rate constant (ks) was calculated to be 7.15 × 102 s−1 which is higher than those so far reported. Additionally, the proposed sensor was also tested for the detection of capsaicin in real samples and showed excellent activity, which suggests that the proposed electrochemical sensor can be used for the trace detection of capsaicin in a wide range of water and vegetable samples with good accuracy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
T. C. Nagaiah thanks Science and Engineering Research Board (SERB, CRG/2018/004478 & SR/NM/NS-1376/2014) for funding.
References
- A. da Silva Antonio, L. S. M. Wiedemann and V. F. da Veiga Junior, Food Anal. Methods, 2019, 12, 1327–1345 CrossRef.
- A. Laskaridou-Monnerville, J. Chromatogr. A, 1999, 838, 293–302 CrossRef CAS.
- I. Perucka and W. Oleszek, Food Chem., 2000, 71, 287–291 CrossRef CAS.
- D. E. Henderson, A. M. Slickman and S. K. Henderson, J. Agric. Food Chem., 1999, 47, 2563–2570 CrossRef CAS PubMed.
- A. M. Sánchez, S. Malagarie-Cazenave, N. Olea, D. Vara, A. Chiloeches and I. Díaz-Laviada, Apoptosis, 2007, 12, 2013–2024 CrossRef PubMed.
- Y.-J. Surh and S. Sup Lee, Life Sci., 1995, 56, 1845–1855 CrossRef CAS PubMed.
- C.-S. Kim, T. Kawada, B.-S. Kim, I.-S. Han, S.-Y. Choe, T. Kurata and R. Yu, Cell Signalling, 2003, 15, 299–306 CrossRef CAS.
- E. Marini, G. Magi, M. Mingoia, A. Pugnaloni and B. Facinelli, Front. Microbiol., 2015, 6, 1281 Search PubMed.
- R. Clark and S.-H. Lee, Anticancer Res., 2016, 36, 837–843 CAS.
- J. Zheng, S. Zheng, Q. Feng, Q. Zhang and X. Xiao, Biosci. Rep., 2017, 37 CAS , BSR20170286.
- A. Tremblay, H. Arguin and S. Panahi, Int. J. Obes., 2016, 40, 1198–1204 CrossRef CAS PubMed.
- R. Kempaiah, H. Manjunatha and K. Srinivasan, Mol. Cell. Biochem., 2005, 275, 7–13 CrossRef CAS PubMed.
- R. Mohammad, M. Ahmad and L. Y. Heng, Sens. Actuators, B, 2017, 241, 174–181 CrossRef CAS.
- M. D. Reyes-Escogido, E. G. Gonzalez-Mondragon and E. Vazquez-Tzompantzi, Molecules, 2011, 16, 1253–1270 CrossRef CAS PubMed.
- W. L. Scoville, J. Am. Pharm. Assoc., 1912, 1, 453–454 CAS.
- M. Monforte-González, F. Medina-Lara, G. Gutiérrez-Carbajal and F. Vázquez-Flota, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 1697–1704 CrossRef.
- S. Saha, S. Walia, A. Kundu, C. Kaur, J. Singh and R. Sisodia, Int. J. Food Prop., 2015, 18, 1535–1545 CrossRef CAS.
- C. J. Welch, E. L. Regalado, E. C. Welch, I. M. Eckert and C. Kraml, Anal. Methods, 2014, 6, 857–862 RSC.
- N. M. Maula, Muhaimin and Millasari, AIP Conf. Proc., 2020, 2229, 030015, DOI:10.1063/5.0002779.
- G. F. Barbero, M. Palma and C. G. Barroso, J. Agric. Food Chem., 2006, 54, 3231–3236 CrossRef CAS PubMed.
- L. Liu, X. Chen, J. Liu, X. Deng, W. Duan and S. Tan, Food Chem., 2010, 119, 1228–1232 CrossRef CAS.
- Q. Zhang, J. Hu, L. Sheng and Y. Li, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2292–2297 CrossRef CAS PubMed.
- I. N. Jovanović, L. Čižmek and Š. Komorsky-Lovrić, Electrochim. Acta, 2016, 208, 273–281 CrossRef.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Analyst, 2008, 133, 888–895 RSC.
- R. D. Crapnell and C. E. Banks, Analyst, 2021, 146, 2769–2783 RSC.
- Y. Yardım, Electroanalysis, 2011, 23, 2491–2497 CrossRef.
- Y. Yardım and Z. Şentürk, Talanta, 2013, 112, 11–19 CrossRef PubMed.
- P. B. Deroco, O. Fatibello-Filho, F. Arduini and D. Moscone, Electrochim. Acta, 2020, 354, 136628 CrossRef CAS.
- Y. Wang, B. Huang, W. Dai, J. Ye and B. Xu, J. Electroanal. Chem., 2016, 776, 93–100 CrossRef CAS.
- M. A. Salam and R. Burk, Arabian J. Chem., 2017, 10, S921–S927 CrossRef CAS.
- A. K. Baytak and M. Aslanoglu, Food Chem., 2017, 228, 152–157 CrossRef CAS PubMed.
- W. Numphud, O. Chienthavorn and W. Siriwatcharapiboon, Sci. Asia, 2020, 46, 586–594 CAS.
- Z. Xue, C. Hu, H. Rao, X. Wang, X. Zhou, X. Liu and X. Lu, Anal. Methods, 2015, 7, 1167–1174 RSC.
- A. Soleh, K. Saisahas, K. Promsuwan, P. Thavarungkul, P. Kanatharana and W. Limbut, ACS Appl. Nano Mater., 2020, 3, 10094–10104 CrossRef CAS.
- S. Das, I. H. Chowdhury, A. Chakraborty, M. K. Naskar, M. Sarkar and S. M. Islam, Mater. Adv., 2022, 3, 3165–3173 RSC.
- R. Yuan, Z. Yan, A. Shaga and H. He, Sens. Actuators, B, 2021, 327, 128949 CrossRef CAS.
- L. Huang, R. Liu, J. Yang, Q. Shuai, B. Yuliarto, Y. V. Kaneti and Y. Yamauchi, Chem. Eng. J., 2021, 408, 127991 CrossRef CAS.
- M. X. Tan, L. Gu, N. Li, J. Y. Ying and Y. Zhang, Green Chem., 2013, 15, 1127–1132 RSC.
- S. Zhang, Q. Yang, X. Zhou, Z. Li, W. Wang, X. Zang, C. Wang, M. J. Shiddiky, A. C. Murugulla and N.-T. Nguyen, Analyst, 2019, 144, 342–348 RSC.
- S. Søpstad, K. Imenes and E. A. Johannessen, Biosens. Bioelectron., 2019, 130, 374–381 CrossRef PubMed.
- P. Supchocksoonthorn, N. Thongsai, W. Wei, P. Gopalan and P. Paoprasert, Sens. Actuators, B, 2021, 329, 129160 CrossRef CAS.
- J. Yin, T. Zhang, E. Schulman, D. Liu and J. Meng, J. Mater. Chem., 2018, 6, 8441–8448 RSC.
- A. A. Ensafi, H. A. Alinajafi and B. Rezaei, J. Mater. Chem., 2018, 6, 6045–6053 RSC.
- N. Hui, R.-f Gao, X.-q Li, W. Sun and K. Jiao, J. Braz. Chem. Soc., 2009, 20, 252–258 CrossRef CAS.
- S. Tajik, H. Beitollahi, R. Hosseinzadeh, A. Aghaei Afshar, R. S. Varma, H. W. Jang and M. Shokouhimehr, ACS Omega, 2021, 6, 4641–4648 CrossRef CAS PubMed.
- A. Soleh, P. Kanatharana, P. Thavarungkul and W. Limbut, Microchem. J., 2020, 153, 104379 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*