DOI:
10.1039/D2MA00209D
(Paper)
Mater. Adv., 2022,
3, 5778-5785
Surface coating and characteristics of ester-free poly(trimethylene carbonate) bearing an aromatic urea moiety for biomaterials use†
Received
23rd February 2022
, Accepted 2nd May 2022
First published on 7th May 2022
Abstract
As a contribution to future developments in biomaterials surface science, this study investigated the polymer properties and surface coating performance of urea-functionalized ester-free poly(trimethylene carbonate), 1-(2-mercaptoethyl)urea poly(trimethylene carbonate) (PTMCM-SU). An aromatic PTMC derivative bearing a vinyl moiety (PTMCM-VB) as a precursor was modified to PTMCM-SU via UV irradiation. In polymer design, hydrogen interaction may be induced in the PTMCM-SU by the presence of 1-(2-mercaptoethyl)urea (SU). Here, the glass transition temperature (Tg) of PTMCM-SU was increased to −2 °C compared to that of non-functionalized PTMC at −29 °C. The wettability of the polymer coatings on glass, stainless steel, and polyethylene (PE) was also studied. PTMC and PTMCM-VB had similar hydrophobic surfaces but the PTMCM-SU surface was hydrophilic. Interestingly, the protein adsorption on PTMCM-SU showed a higher affinity to the coated materials, thus enhancing their bioactivity. We demonstrated that, by modifying PTMC with a urea pendant, we could tune the polymer functionality and promote biological responsiveness for its potential use as a coating surface for biomaterials.
Introduction
With the advent of biotechnology, biodegradable polymeric coating materials have attracted attention in clinical applications due to their flexible functionalization. Biodegradable polymers possess good bioresorbability, biocompatibility and non-toxicity.1 The polymeric coating on biomaterials can be controlled and surface properties such as wettability, bioactivity, adherence, and scratch and abrasion resistance can be modified for specific requirements and advanced functionalization in a wide range of biomedical applications.1 Diversity of functionalization is required to increase their applicability for cell proliferation,2 tissue growth and repair,3 and delivery of biomolecules such as drugs,4 growth factors,5 and antimicrobial agents.6 To date, aliphatic polyesters and polycarbonates that have been used in medical products have been mainly derived from polylactic acid (PLA), polyglycolide (PGA) and polycaprolactone (PCL) for the development of sutures, bone screws, tissue scaffolds, etc.7 However, due to the limited chemical structure of polyesters, we focused on trimethylene carbonate bearing various functional groups, while avoiding ester bonding, which produces organic acidic compounds after hydrolysis.8–10
Poly(trimethylene carbonate) (PTMC) is a well-known biodegradable candidate due to its biocompatibility, flexibility, uniform surface erosion characteristics and non-acidic by-products during the degradation process. Therefore, synthetic PTMC can be regarded as a promising surface coating for biomedical applications.11 Many efforts to functionalize PTMC with various moieties, including alkyl,12 aryl,13 alkene,14 alkyne,15 halogen,16 azide,17 and amino15 have been reported. However, most of the currently published studies have used ester-based side chain modification,18 which may induce acidic by-products upon degradation.19 Hence, our group has focused on an ester-free based structure of functionalized PTMC. We have previously reported the successful functionalization of ester-free PTMC with improved properties using our own unique designs and syntheses. For instance, PTMC with oligo(ethylene glycol) shows thermal responsivity at near body temperature with minimal acidity of the generated degradation product.20,21 Also, ester-free aromatic22 and alicyclic23 PTMC have improved the thermal properties of commercial PTMC.
In addition to expanding the applicability of ester-free PTMC derivatives, introduction of a functional group that is beneficial for biomaterials and tissue interactions by tailoring the wettability,24 surface charge,25 and intermolecular interactions26 would be desirable. For instance, Wei et al. precisely designed grafted poly(hexamethyldisiloxane) (PHMDSO) brushes with different surface wettability by manipulating the duration of oxygen plasma treatment, to study the effect of cell adherence performance.27 Ding et al. demonstrated the modulation of protein adsorption, vascular cell selectivity and platelet adhesion by multifunctionality of the poly(dopamine) (PDA) coating for vascular stent applications.28
Recently, urea derivatives have been shown to be excellent functional groups that facilitate interesting performance in terms of biomimetics to biomolecules,29 tailorable hydrophilicity and hydrogen bonding,30etc. These derivatives impart a structure similar to peptide bonds that could have a better chance of binding to protein molecules and enhancing bioactivity. Urea is notably associated with hydrogen bonding as a strategy for improving physical properties and desired intermolecular interaction such as drug release control.31 Kim et al. reported that introducing a urea-bearing functional group into a copolymer lowered the critical micelle concentration, enhanced the kinetic stability of the micelles and, with the non-covalent interaction with doxorubicin (a model anticancer drug), facilitated drug delivery control.4 Ying et al. explored the hindered urea bond, a urea structure with a bulky substituent on one of the nitrogen atoms. The hydrolysis kinetics were tuned by manipulating the substituent's bulkiness, which has potential in tissue engineering applications for controlling the release kinetics of cells or proteins to wound sites.32 Inspired by these findings, it is proposed that the addition of a urea group into the side chain of PTMC with an ester free design would enhance its biocompatibility. Since the presence of a urea moiety could make polymerization difficult, post-modification provides an easy synthetic route and the possibility for incorporating a urea pendant group through a reactive site such as a double bond. In our previous study, we successfully synthesized cross-linked PTMC bearing an aromatic pendant and 1-(2-mercaptoethyl)urea (SU). The PTMCM-SU in the form of a film incorporating a 50% crosslinker of 2,2′-bis(trimethylene carbonate-5-ly)-butyl ether (BTB) showed a greater tensile strength of 9.94 MPa than non-functionalized PTMC (0.89 MPa).33 However, the polymer materials could not be coated on any substrate, and the post-modification behaviour was unclear due to the crosslinked gel material. We were motivated to apply the polymer materials for the surface coating of biomaterials using soluble PTMC derivatives without crosslinking. For specific use in the medical field, the study of biological responses is essential to understand the behaviour and interaction with the surrounding biological microenvironment. The requirements for biological interaction or effects of the biomaterials differ depending on the therapeutic application.34 This study aimed to evaluate the bioactivity of SU for potential use in biomaterials using linear polymers of PTMC derivatives, for tissue engineering, and drug release.
In this study, an aromatic bearing vinyl moiety was introduced into a PTMC side chain followed by post-modification of PTMC with a thiol bearing a urea group through UV irradiation. The polymers were coated on glass, stainless steel (SS) and polyethylene (PE) to study their characteristics. For use as a polymeric coating material in clinical applications, the thermal properties, contact angles, roughness and protein adsorption are discussed along with an investigation of the existence of hydrogen bonding between urea-functional PTMC.
Experimental
Materials and procedures
Trimethylolethane, ethyl chloroformate, hexafluoro-2-propanol, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU) and 2,2-dimethyl-1-propanol were purchased from Tokyo Chemical Industry (TCI), Japan. Potassium hydroxide pellets (KOH), potassium cyanate (KOCN) and 1 M hydrochloric acid (HCl) were purchased from Nacalai Tesque. 2,2-Dimethoxy-2-phenylacetophenone (DMPA) and 4-vinylbenzyl chloride were purchased from Sigma-Aldrich. Trimethylamine was purchased from Fujifilm Wako Pure Chemical (WAKO), Japan. Anhydrous dimethyl sulfoxide (DMSO), dimethylformamide (DMF), tetrahydrofuran (THF), ethyl acetate, hexane, isopropanol, sodium chloride, and dichloromethane (DCM) were used without further purification.
Monomer syntheses
2-Methyl-2-[{(4-vinylbenzyl)methoxy}methyl]-1,3-propanediol (diol-styrene), 5-methyl-5-[{(4-vinylbenzyl)methoxy}methyl]-1,3-dioxanone (TMCM-VB) and SU were synthesized using the same procedure as reported from our previous paper.33
Polymer syntheses.
Synthesis of PTMCM-VB and PTMC.
Under a N2 atmosphere, TMCM-VB (1.0 g, 3.81 mmol) was dissolved in anhydrous DCM solution with MS4A and stirred for 6 hr. The solution was then transferred to another flask and the solvent was removed by vacuum evaporated at room temperature overnight. The polymerization was carried out by using 2,2-dimethyl-1-propanol and DBU as an initiator and catalyst. Briefly, the dried monomer was dissolved in anhydrous DCM (1.91 ml) with 2 M concentration. Subsequently, DBU (56.9 μl, 0.381 mmol) and 1 M 2,2-dimethyl-1-propanol (38.1 μl, 0.0381 mmol) were added. The polymerization was carried out for 24 hr under room temperature. The polymerization was stopped by dissolving a small amount of DCM. After precipitation of the mixture against a large excess of cold methanol, the polymer was recovered by decantation and centrifugation and finally dried in vacuo at room temperature (88% yield). PTMC was polymerized from monomer 1,3-dioxane-2-one with the same procedure as described above (91% yield).
Post modification of the thiol-ene of PTMCM-VB with SU.
Post-modification was performed under a nitrogen atmosphere. The PTMCM-VB (0.8 g, 3 mmol, ∼25 vinyl aromatic units per chain) was dissolved in 31 ml of DMF until a homogenous solution was formed. SU (3.67 g, 31 mmol) and DMPA as a photo-initiator (0.16 g, 0.62 mmol) were added into the mixture. The mixture was stirred at room temperature under UV light (365 nm). The reaction was monitored at specific time intervals by NMR. After 4 h, the UV light was turned off and the mixture was vacuum evaporated to remove the solvent. The obtained compound was re-dissolved in HFIP/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) and then placed into a dialysis bag (cut off Mn 2 kDa) and dialyzed against water for 1 day to remove unreactive thiol–urea. The solution outside the mixture of the dialysis bag was replaced with fresh water every 12 h. After that, the mixture in the dialysis bag was vacuum dried at 50 °C overnight to obtain post-modified polymer (PTMCM-SU) (22% yield).
:10) and then placed into a dialysis bag (cut off Mn 2 kDa) and dialyzed against water for 1 day to remove unreactive thiol–urea. The solution outside the mixture of the dialysis bag was replaced with fresh water every 12 h. After that, the mixture in the dialysis bag was vacuum dried at 50 °C overnight to obtain post-modified polymer (PTMCM-SU) (22% yield).
Coating procedure
Spin coating of synthetic polymers was applied on the glass, stainless steel (SS), and polyethylene (PE). A solution of the synthetic polymer in HFIP/DCM (1:1) at 10 mg ml−1 was made and 40 μl of the polymeric solution was dropped onto the spinning substrate at 1000 rpm for 30 s for two time-repeated coatings (Spincoater 1H-D7, Mikasa Co., Ltd). The coated substrates were then dried under vacuum before testing.
Characterizations
1H NMR (400 MHz) spectra was recorded on a JEOL JNM-ECX400 instrument with tetramethylsilane (TMS) as an internal standard. Chemical shifts were measured in CDCl3 or DMSO-d6 at 25 °C depending on the polymer solubility. PTMCM-VB was measured using CDCl3 while PTMCM-SU was measured using DMSO-d6. Fourier transform infrared spectrometer (FT-IR) spectra were obtained with an IR Affinity-1S Shimadzu. Size Exclusion Chromatography (SEC) measurement was conducted to determine the average molecular weight (Mn) and dispersity distribution (PDI) of the synthetic polymers. The system was equipped with an RI-2031 Plus Intelligent RI detector, PU-2080 Plus Intelligent HPLC pump, AS-2055 Plus Intelligent Sampler, CO-2065 Plus Intelligent Column oven (JASCO), and commercial columns (TSKgel SuperH3000 and TSKgel GMHXL) connected in series. Polystyrene standards (Shodex) (1 mg ml−1, 0.6 ml min−1) were employed and tetrahydrofuran for PTMCM-VB while dimethylformamide for PTMCM-SU was used as an eluent at 40 °C.
Contact angles
The static water contact angle of the coated substrates was measured by a Flow Design CAM-004 (Tokai Hit TPX-S). Image were taken after 40 μl DI water was dropped on the film surface with three replicates at different locations to obtain the mean value of the contact angle.
Laser scanning microscope
The morphology and roughness of the coated substrates were measured using an Olympus LEXT OLS4100 3D laser scanning microscope (horizontal: X–Y direction resolution of 0.12 μm; light source: 405 nm semiconductor laser; detector: photomultiplier).
Protein adsorption
Protein adsorption of the coated samples was examined using bovine serum albumin (BSA) (Sigma, St. Louis, USA) with a concentration of 4.5 mg ml−1. The samples were soaked in 900 μm protein solution and incubated for 4 h at 37 °C. After rinsing with PBS, the adsorbed protein was detached by 1 wt% n-sodium dodecyl sulfate (SDS) for 4 h. Bicinchoninic acid (BCA) protein assay reagent was used as an indicator for protein adsorption on the samples. UV measurement at 562 nm for the protein on the sample surface was interpreted by an absorbance microplate reader (MTP-310lab, Corona Electric) based on the standard control and calibration curve of the protein.
Thermal resistance
The thermal stability of the polymer was determined by a TGA-50 thermo-gravimetric analyser (Shimadzu) under a nitrogen atmosphere with 10 °C min−1 flow rate at 500 °C. Differential scanning calorimetry (DSC) spectra were also analysed by a Hitachi DSC6200 in the atmosphere with temperature ramp rate 10 °C min−1 in the range 70–200 °C.
Cell adhesion
The cell adhesion study was carried out by using L929 fibroblasts with SS as a substrate for all the polymer coatings. Fluorescent staining with cell tracker green was used. Cells were plated onto the sample surface at an initial density 4000 cells per cm2 and cultured under a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 7 h. The samples were washed with PBS and nutrient medium and the cells were observed using a fluorescence microscope. The number of adherent cells was counted using the fluorescent images obtained.
Results and discussion
Polymerization and post-modification
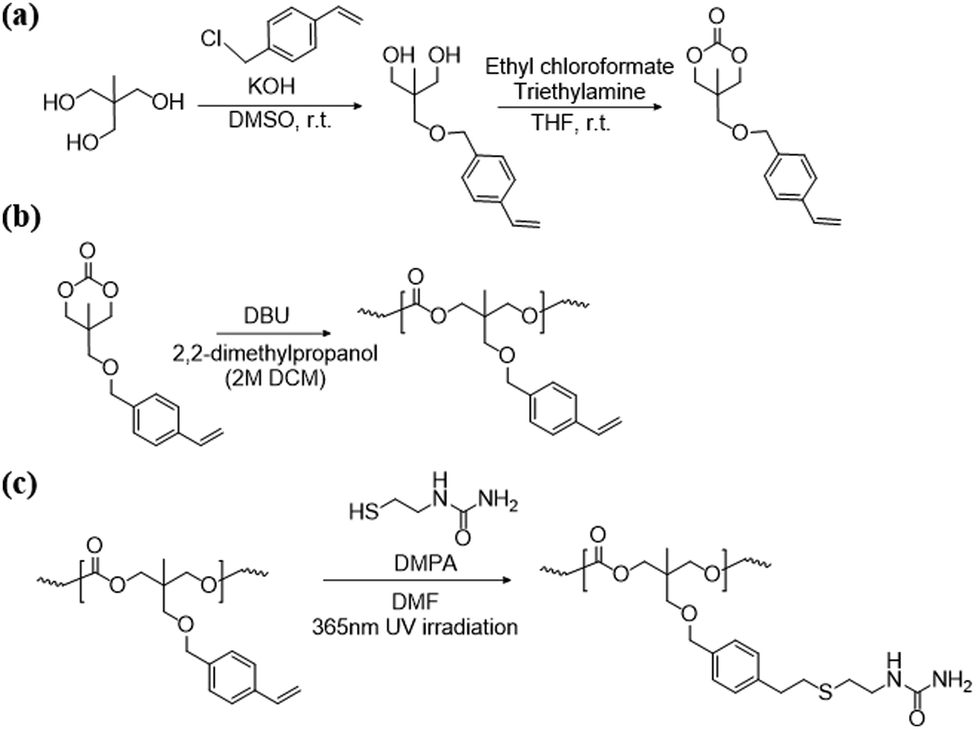
The synthetic route for vinyl aromatic TMC monomers is depicted in Scheme 1 and the functionalized ester-free design was conducted as previously reported.33 SU comprising a urea moiety was selected for introduction into the PTMCM-VB side chain to enhance the intermolecular interaction and bioactive properties. In this study, post-modification by UV light (365 nm) was used for irradiation with the aid of a DMPA photo-initiator. The reaction was performed at room temperature, thus preventing any side reaction.
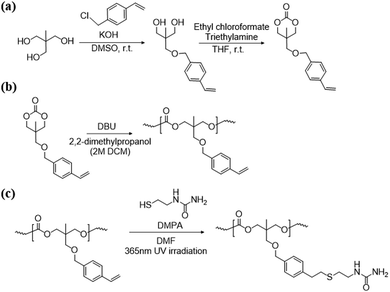 |
| | Scheme 1 Synthesis of the TMCM-VB monomer (a), polymerization of TMC-VB (b), and post polymer-modification of PTMCM-SU (c). | |
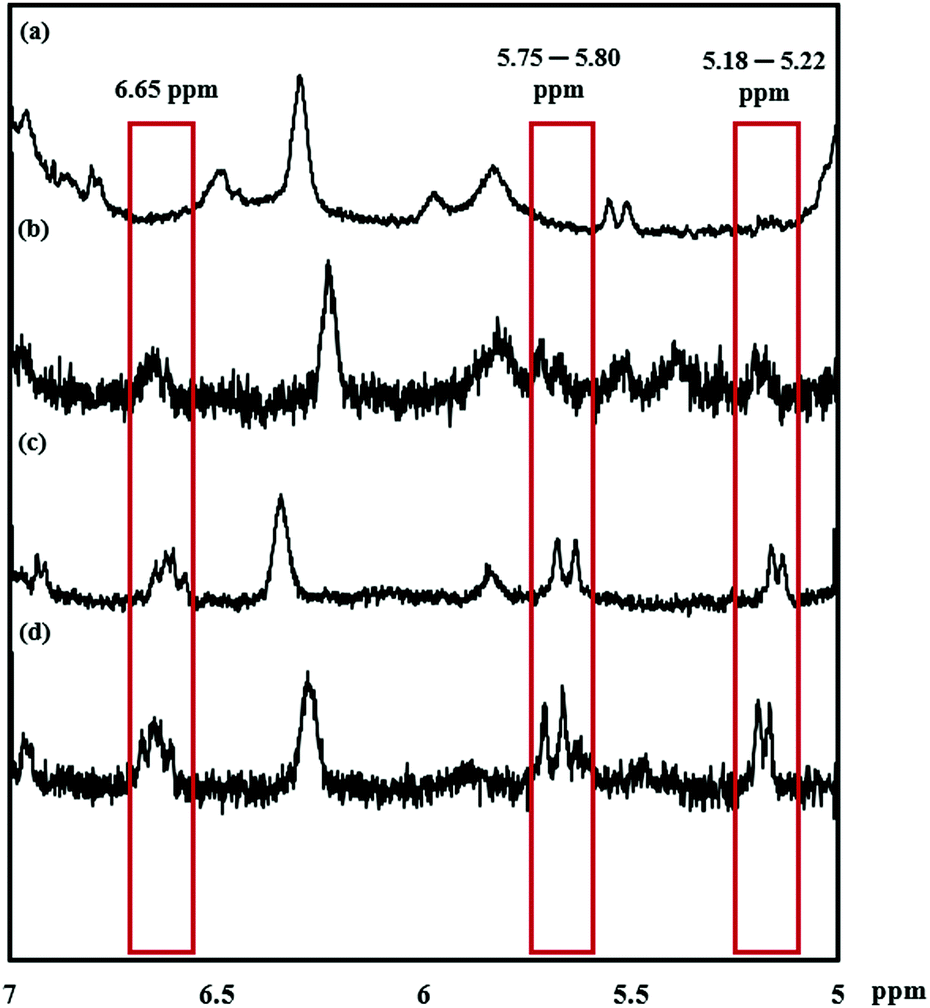
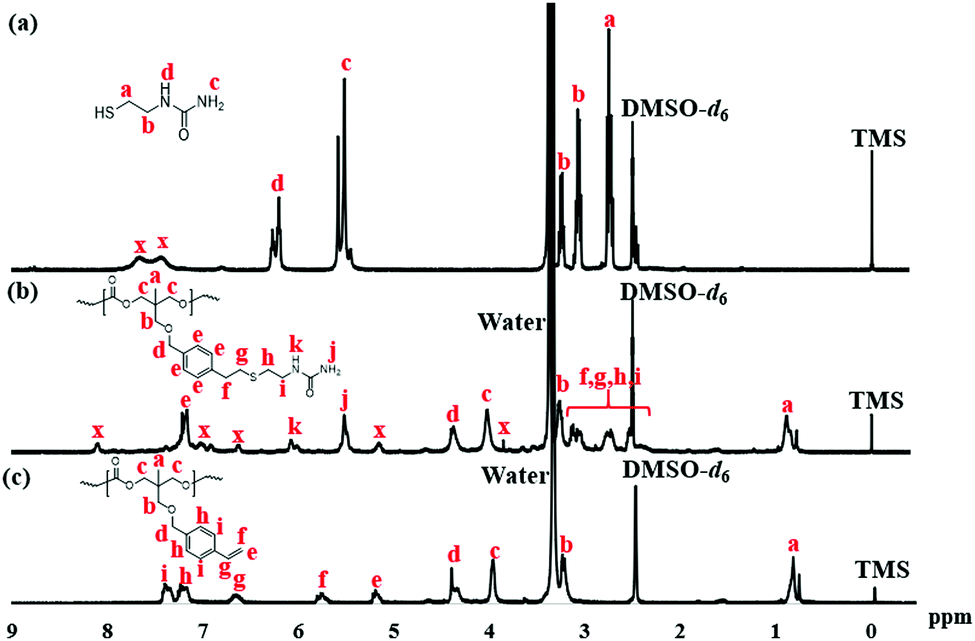
The reaction was monitored by 1H NMR at time intervals of 30, 60, 120, and 240 min, as shown in Fig. 1a–d, respectively. Resonance signals at 5.18–5.22, 5.75–5.80, and 6.65 ppm (Fig. 1) were attributed to the vinyl group and slightly decreased in intensity upon reaction time. Also, after SU modification, the aromatic proton signal at 7.17–7.22 ppm, and 7.35–7.40 ppm in Fig. 2c shifted to 7.17 ppm in Fig. 2b, indicating successful modification of PTMCM-SU. The appearance of resonances at 2.53 ppm, 2.73 ppm, 3.12 ppm, 3.26 ppm, 5.52 ppm, and 6.02 ppm, which corresponded to the SU group, is also shown in Fig. 2b. To complete the conversion of the vinyl double bond of PTMCM-VB and prevent consumption of active thiols as a possible side reaction, a thiol–ene molar ratio of 10:1 was used.35 A modification percentage of almost 100% was achieved according to the 1H NMR spectra. Excess of SU at resonances of 2.53 ppm, 2.73 ppm, 3.12 ppm, 3.26 ppm, 5.52 ppm, and 6.02 ppm was removed by dialysis after reaction, as shown in Fig. 2. The polymers PTMC, PTMCM-VB and PTMCM-SU were synthesized for comparison of their properties. The molecular weights of PTMC, PTMCM-VB and PTMCM-SU are detailed in Table 1. The molecular weight of non-functionalized PTMC was 6400 g mol−1 with a PDI of 1.5 (Table 1, entry 1). The molecular weights and PDI of PTMCM-VB (Table 1, entry 2) and after urea post-modification with SU (Table 1, entry 3) were 5100 g mol−1 and 1.2, and 7900 g mol−1 and 2.0, respectively. The increase in molecular weight of the post-modified polymer demonstrates that the SU had been incorporated into the side chain of PTMC whereas the broadening of the polydispersity was possibly due to either a side reaction for attachment of SU at different positions on the vinyl or chain scission that occurred during UV irradiation.
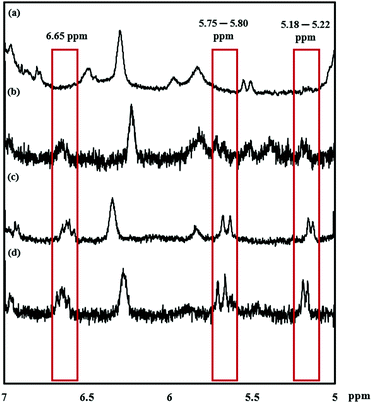 |
| | Fig. 1
1H NMR spectra of PTMCM-VB modified with SU at specific time points. (a) 240 min. (b) 120 min. (c) 60 min. (d) 30 min. | |
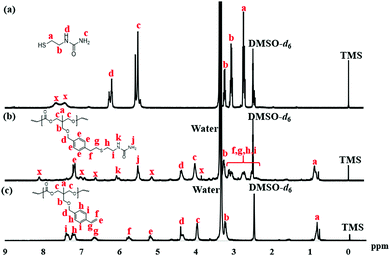 |
| | Fig. 2
Fig. 2
1H NMR spectra of SU (a), PTMCM-SU (b), and PTMCM-VB (c). | |
Table 1 Summary of polymer PTMC, PTMCM-VB, and PTMCM-SU properties
| Entry |
Polymer |
Yield (%) |
M
n
|
PDIc |
T
g (°C) |
|
[M]:[C]:[I] = 100:10:1. Initiator = 2,2-Dimethyl-1-propanol. Catalyst = DBU. Solvent = THF. Methanol insoluble part. Mn determined by SEC in THF with PS standards.
Water insoluble part. Mn determined by SEC in DMF with PS standards.
PDI was defined as Mw/Mn.
|
| 1 |
PTMCa |
91 |
6400 |
1.5 |
−29 |
| 2 |
PTMCM-VBa |
88 |
5100 |
1.2 |
−11 |
| 3 |
PTMCM-SUb |
22 |
7900 |
2.1 |
−2 |
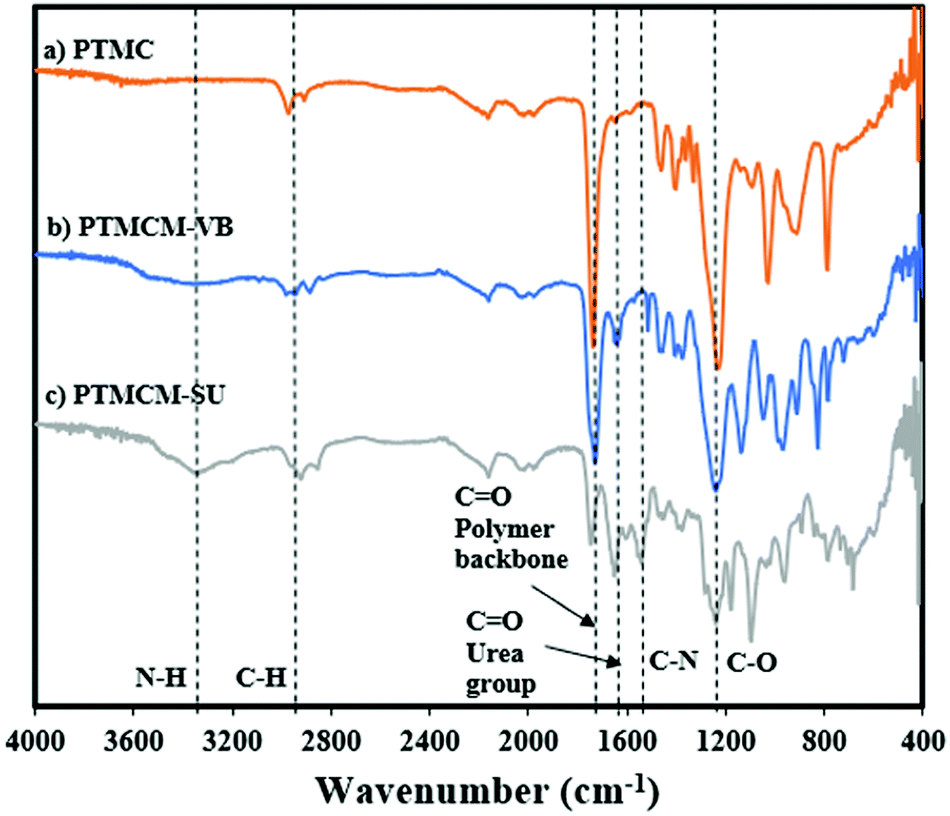
The chemical structures of PTMC, PTMCM-VB and PTMCM-SU were also determined by FT-IR (Fig. 3a–c), respectively. The significant wavenumbers for PTMC were at 2970 cm−1, 1744 cm−1 and 1233 cm−1 which were attributed to the C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching at the polymer backbone. After introduction of the vinyl aromatic as PTMCM-VB, there was a new peak at 1,645 cm−1 represented as a C = C double bond stretching vibration of the vinyl aromatic group. However, after modification of PTMCM-VB with the thiol–urea, a new peak appeared at 3337 cm−1 showing the frequencies of N–H stretching. Also, the CO of the urea group appeared at 1651 cm−1 while the C–N stretching frequency was seen at 1557 cm−1. These observations lead to the conclusion that the vinyl aromatic and thiol-urea groups were successfully introduced. To investigate the presence of hydrogen bonding, a thiol–urea precursor was used to measure 1H NMR (Fig. 2). Splitting peaks were observed between 5.52–5.58 and 6.21–6.27 ppm, which could be due to the hydrogen bonding between the amide and carbonyl groups of SU. In the case of the modified polymer, although the peaks were displayed in a broad range making it difficult to observe clear splitting, they still showed an analogous pattern.
O and C–O stretching at the polymer backbone. After introduction of the vinyl aromatic as PTMCM-VB, there was a new peak at 1,645 cm−1 represented as a C = C double bond stretching vibration of the vinyl aromatic group. However, after modification of PTMCM-VB with the thiol–urea, a new peak appeared at 3337 cm−1 showing the frequencies of N–H stretching. Also, the CO of the urea group appeared at 1651 cm−1 while the C–N stretching frequency was seen at 1557 cm−1. These observations lead to the conclusion that the vinyl aromatic and thiol-urea groups were successfully introduced. To investigate the presence of hydrogen bonding, a thiol–urea precursor was used to measure 1H NMR (Fig. 2). Splitting peaks were observed between 5.52–5.58 and 6.21–6.27 ppm, which could be due to the hydrogen bonding between the amide and carbonyl groups of SU. In the case of the modified polymer, although the peaks were displayed in a broad range making it difficult to observe clear splitting, they still showed an analogous pattern.
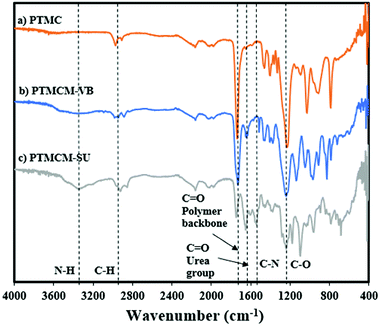 |
| | Fig. 3 FT-IR spectra of PTMC (a), PTMCM-VB (b), and PTMCM-SU (c). | |
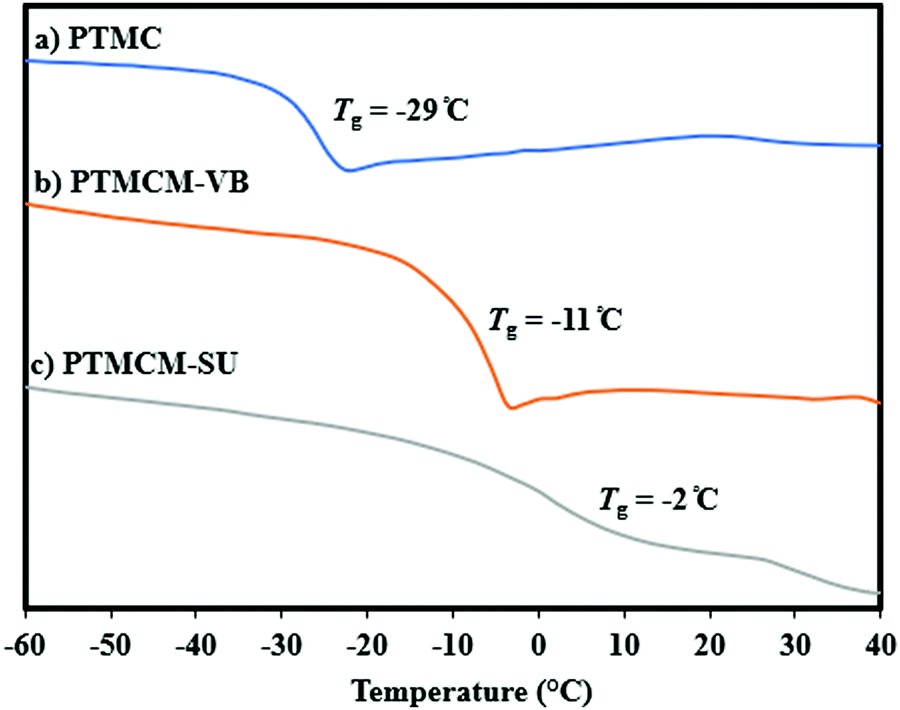
Thermal stability
The thermal stability of non-functionalized PTMC usually has a lower Tg so its performance has been restricted even though it has good biodegradability. For this reason, introduction of a rigid functional group or using intermolecular bonding allows us to overcome these limitations. Fig. 4 shows a comparison of Tg for PTMC, PTMCM-VB and PTMCM-SU, which were −29 °C, −11 °C and −2 °C, respectively. Lizundia et al. reported that ring-opening polymerization of a novel monomer, 2,3-dihydro-5H-1,4-benzodioxepin-5-one (2,3-DHB), yielded a biodegradable aliphatic polyester and that introducing an aromatic into the polymer backbone resulted in exceptional thermal properties with a Tg of nearly 27 °C.36 The results were similar to those of Nobuoka et al. where the introduction of aromatic derivatives into the side chain of PTMC yielded a Tg of up to 1 °C.37 The presence of an SU group in the side chain of PTMC further improved Tg, suggesting the presence of self-association hydrogen bonding of the polymer chains.
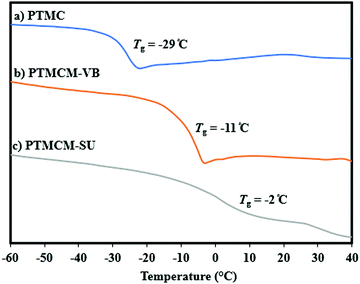 |
| | Fig. 4 DSC trace of PTMC (a), PTMCM-VB (b), and PTMCM-SU (c). | |
This is supported by the findings of Shieh et al. who showed that incorporating acrylic acid units into poly(N-isopropylacrylamide) (PNIPAAm) tended to increase the Tg due to strong hydrogen bonding between the amide groups present in the PNIPAAm.38 Based on the results, the presence of aromatic as well as thiol-urea groups was asserted to form π–π interaction and hydrogen bonding that restricted the polymer chain, thus increasing Tg.
Surface morphology
The synthetic polymers PTMC, PTMCM-VB and PTMCM-SU were spin-coated on a substrate to assess their suitability as coating materials. Glass (12 mm diameter, 0.1 mm thickness), SS (8 mm diameter, 0.1 mm thickness), and PE (8 mm diameter, 1 mm thickness) were used as coating substrates. All the polymers were dissolved in HFIP/DCM at a 1:1 ratio. The roughness and surface morphologies were investigated by laser scanning microscopy, as shown in Fig. 5. Based on the surface morphologies, the surface on the substrate would not be fully covered by the polymer using droplet solution. This could possibly be due to polymer solutions behaving as viscous solutions that are more resistant to deformation from the shear forces of the spin coating process. Both PTMC and PTMCM-VB had a homogeneous surface while PTMCM-SU showed slight agglomeration on the substrate surface. This could be due to solvent evaporation driving the concentration above the solubility limit, leading to interaction of SU within the polymer chains. The roughnesses of bare glass, SS and PE were 0.01 μm, 0.055 μm and 0.169 μm, respectively. After coating, the roughness increased to 0.188 μm, 0.093 μm and 0.2 μm for PTMC, 0.134 μm, 0.079 μm and 0.133 μm for PTMCM-VB, and 0.127 μm, 0.054 μm and 0.182 μm for PTMCM-SU on the glass, SS and PE, respectively.
 |
| | Fig. 5 Surface morphologies of glass (a), PTMC/glass (b), PTMCM-VB/glass (c), PTMCM-SU/glass (d), SS (e), PTMC/SS (f), PTCM-VB/SS (g), PTMCM-SU/SS (h), PE (i), PTMC/PE (j), PTMCM-VB/SS (k), and PTMCM-SU/PE (l). | |
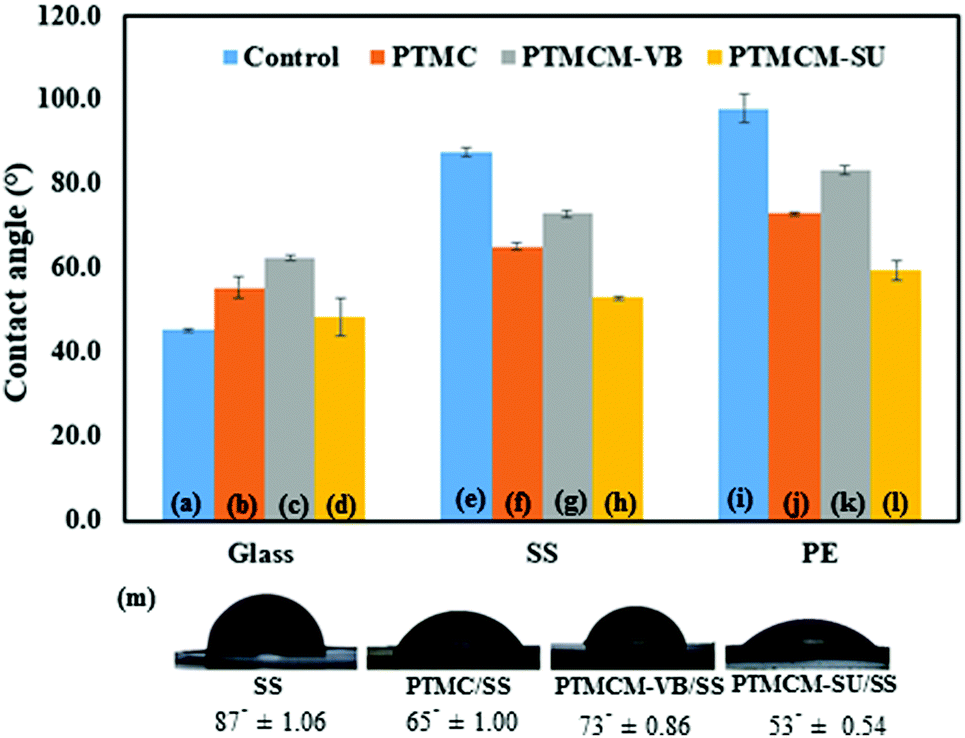
Contact angle
Surface hydrophilicity may play an important role in the biocompatibility of a material by causing quantitative protein adsorption. The water contact angles of spin-coated polymers on the glass, SS and PE respectively were determined, as shown in Fig. 6. All the contact angles of the spin-coated polymers were influenced by the substrate surface. For the control groups, which were non-coated substrates of glass, SS and PE, the contact angles were 45.3°± 0.40, 87.4° ± 1.06 and 98.0° ± 3.45, respectively. The bare glass possessed hydrophilic attributes, while bare SS and PE gave hydrophobic surfaces. After coating with the PTMC, the angles changed to 55.4° ± 2.74 for glass, 65.20° ± 1.00 for SS, and 73.0° ± 0.44 for PE. Compared to bare SS and PE, the PTMC coating was slightly hydrophilic. This could possibly be attributed to the low molecular weight and presence of a hydroxyl group in the end chain of the polymer. However, the contact angle of the aromatic PTMCM-VB coating was hydrophobic compared to PTMC, 62.4° ± 0.65 for glass, 73.1° ± 0.86 for SS and 83.3° ± 1.00 for PE due to the presence of the aromatic group. Conversely, after introducing the SU coating, the contact angle changed to 48.3° ± 4.44 for glass, 52.9° ± 0.54 for SS, and 59.5° ± 2.25 for PE. In comparison to bare SS and PE, the PTMCM-SU coating was hydrophilic. Amino groups could have improved the hydrophilicity by interacting with water molecules through hydrogen bonding.
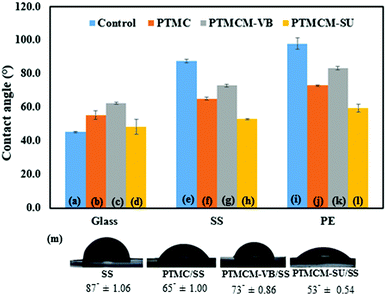 |
| | Fig. 6 Contact angles of glass (a), PTMC/glass (b), PTMCM-VB/glass (c), PTMCM-SU/glass (d), SS (e), PTMC/SS (f), PTCM-VB/SS (g), PTMCM-SU/SS (h), PE (i), PTMC/PE (j), PTMCM-VB/SS (k), and PTMCM-SU/PE (l). (m) Images of contact. | |
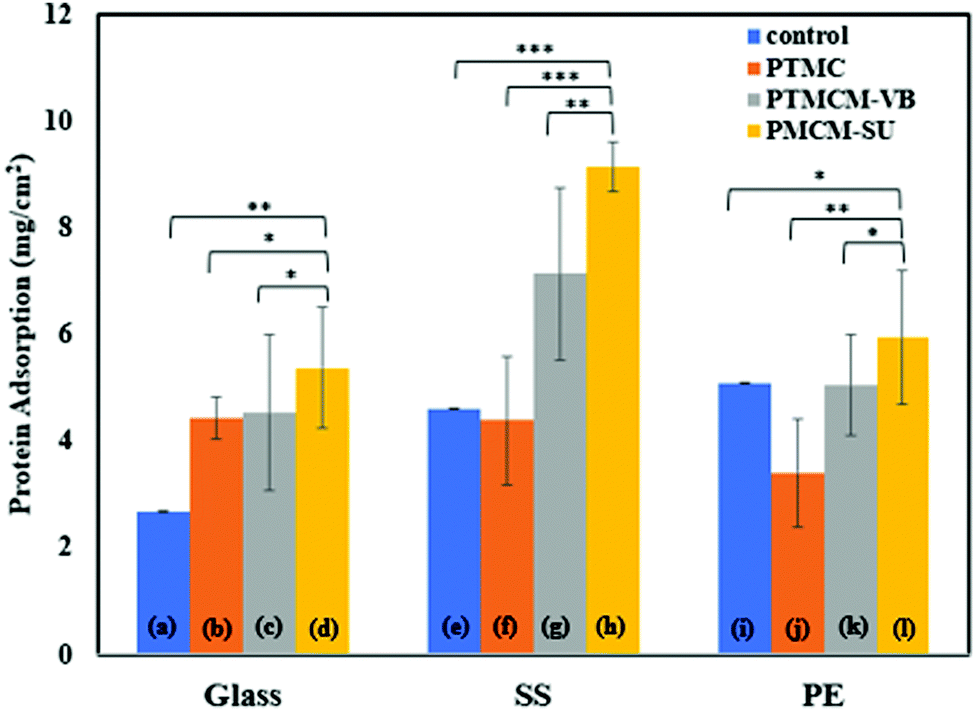
Protein adsorption
Protein adsorption of a coating biomaterial is important for determining its biocompatibility when exposed to the physiological environment. It is believed that interaction between the proteins and biomaterials may mediate cellular adhesion that affects the long-term performance of the medical device. Thus, investigating the surface modification of a coating is a matter of concern. Fig. 7 shows the protein behaviours in response to PTMC, PTMCM-VB and PTMCM-SU on glass, SS and PE substrates. Albumin, the major protein of human blood plasma, was used in this study. The protein adsorptions of the non-coated substrates of glass, SS and PE, which acted as control groups, were 2.68 μg cm−2 ± 0.002, 4.61 μg cm−2 ± 0.002 and 5.10 μg cm−2 ± 0.01, respectively. After coating with PTMC, PTMCM-VB and PTMCM-SU, the protein adsorption was 4.43 μg cm−2 ± 0.38, 4.53 μg cm−2 ± 1.46 and 5.38 μg cm−2 ± 1.13 for the glass substrate, 4.39 μg cm−2 ± 1.20, 7.13 μg cm−2 ± 1.63 and 9.14 μg cm−2 ± 0.46 for the SS substrate and 3.41 μg cm−2 ± 1.01, 5.05 μg cm−2 ± 0.94 and 5.94 μg cm−2 ± 1.24 for the PE substrate. The results for protein adherence on each polymer were influenced by the substrate type. This suggests the presence of areas not covered by the polymer coating as seen in the morphologies shown in Fig. 5, leading to exposure of the bare substrate with protein exposure. Based on this result, there is a marked difference in protein adsorption for PTMCM-SU, which is attributed to its high affinity to adsorb protein compared to non-functionalized and other PTMC derivatives used in this study. According to Arima et al., the effects of surface functional groups such as amines showed higher protein adsorption compared to CH3, OH and COOH.39 The aggregation on the substrate could be a factor to consider in a large, adsorbed amount of protein due to more of the surface area of PTMCM-SU being accessible. Furthermore, the effect of wettability could contribute to inducing favourable conditions for protein adsorbed on the surface. Cells effectively adhere onto a polymer surface with moderate wettability with water contact angles of 40–70°. This is in line with our result as the contact angles of PTMCM-SU were 53– 59°, and thus greater protein interaction with the polymer surface is expected as there is a correlation between cell adherence and protein adsorption.
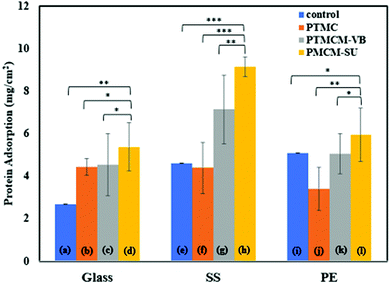 |
| | Fig. 7 Albumin (BSA) protein adsorption on glass (a), PTMC/glass (b), PTMCM-VB/glass (c), PTMCM-SU/glass (d), SS (e), PTMC/SS (f), PTCM-VB/SS (g), PTMCM-SU/SS (h), PE (i), PTMC/PE (j), PTMCM-VB/SS (k), and PTMCM-SU/PE (l). (n = 4, *p < 0.5, **p < 0.1, ***p < 0.01). | |
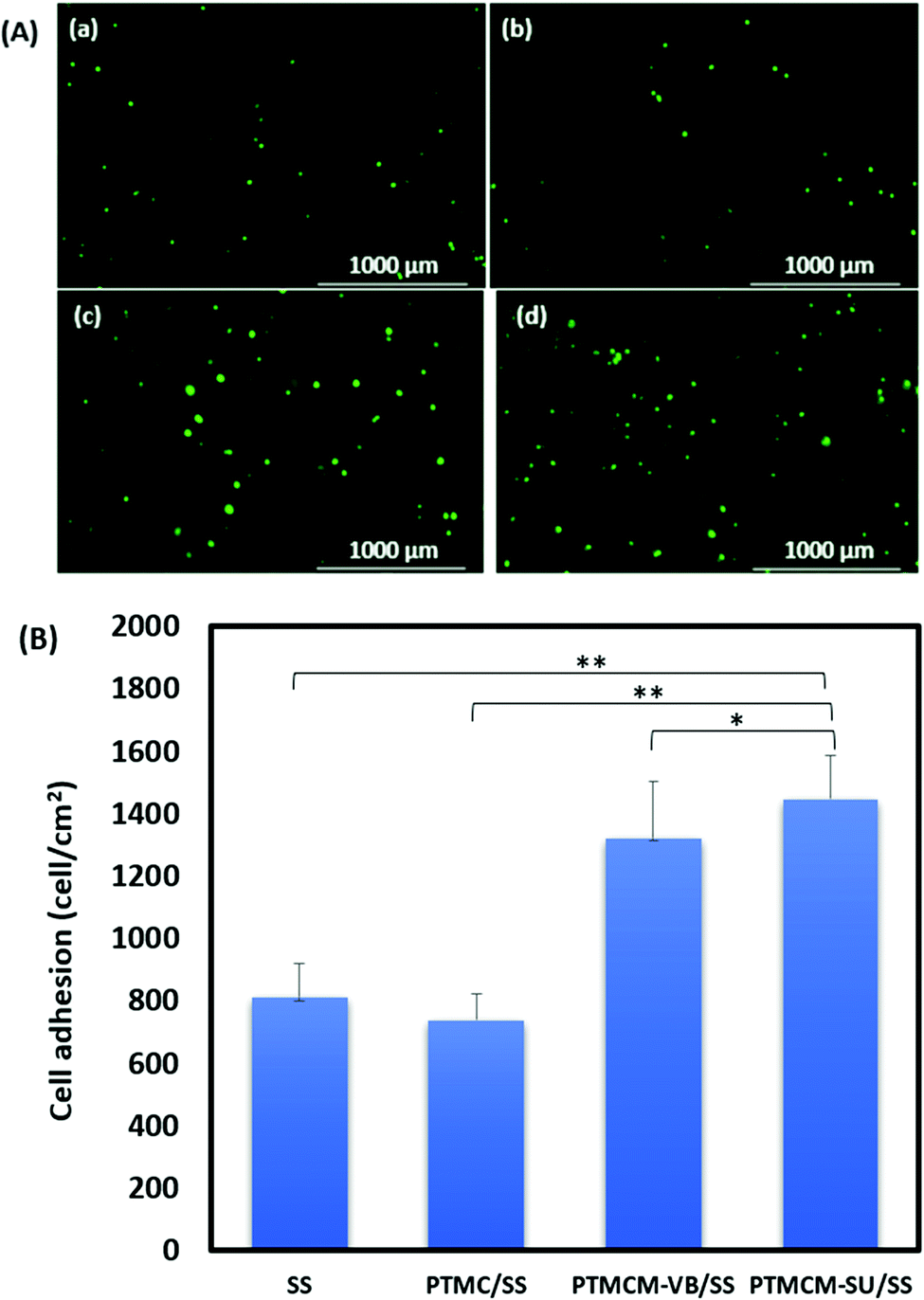
Cell adhesion
The L929 fibroblasts were cultured for up to 7 h on the polymer-coated stainless steel (SS) substrate for the adhesion study (Fig. 8). The cells were more adherent on the functionalized PTMC-coated compared to non-functionalized PTMC and bare SS. The cell adhesions for bare SS, PTMC/SS, PTMCM-VB/SS, and PTMCM-SU were 812 ± 106, 738 ± 85, 1320 ± 181, 1450 ± 138 per cm2, respectively. There was a significant effect on cell adherence after coating with PTMCM-VB and PTMCM-SU. Surface chemistry, wettability, roughness, rigidity, etc., are prone for better adhesion.40 The possible dominant factors could be due to the rigidity of the aromatic ring, and specific hydrogen bonding between the urea group of the polymer and the polar groups of the cell surfaces.
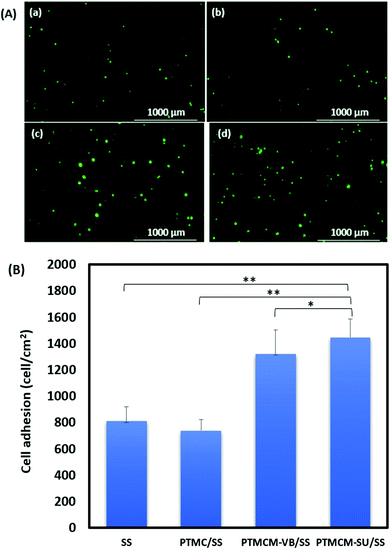 |
| | Fig. 8 (A) Images of L929 cell adhesion on bare stainless steel (SS) (a), PTMC/SS (b), PTMCM-VB/SS (c) and PTMCM-SU/SS (d), and (B) the cell adhesion percentage of SS, PTMC/SS and PTMCM-VB/SS and PTMCM-SU/SS (n = 5, *p < 0.5, **p < 0.1). | |
Conclusions
In this study we report on the successful design and performance of an ester-free PTMC derivative with an aromatic urea moiety, PTMCM-SU, for biomaterials coating. The PTMCM-SU was effectively synthesized via post-modification of the vinyl aromatic under UV irradiation. Induction of hydrogen bonding between the polymer chains was investigated as well as its effect on the thermal properties with an increase of Tg to −2 °C. The presence of amines on the urea pendant tended to increase the hydrophilicity of the modified polymer with contact angles from 48° to 59°. Besides, the protein adsorption of PTMCM-SU showed high affinity to the albumin on the surface in the range of 5.38 μg cm−2 to 9.14 μg cm−2. The cell adherence of PTMCM-SU showed better cell adhesion compared to non-functionalized PTMC. As a result, the synthetic polymer showed promise for use as a biomaterial in tissue engineering and scaffold applications by promoting the bioactivity of the material.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is partly supported by AMED under Grant Number JP20lm0203014. This work is also supported by KAKENHI (JP19KK0277, JP20H02799). The author is grateful for the MEXT scholarship, NAIST competitive research support and partially NAIST Foreigner and woman incentive fund.
Notes and references
- A. J. Nathanael and T. H. Oh, Polymers, 2020, 12, 3061 CrossRef CAS PubMed.
- A. Joy, D. M. Cohen, A. Luk, E. Anim-Danso, C. Chen and J. Kohn, Langmuir, 2011, 27, 1891–1899 CrossRef CAS PubMed.
- M. Tallawi, E. Rosellini, N. Barbani, M. Grazia Cascone, R. Rai, G. Saint-Pierre and A. R. Boccaccini, J. R. Soc., Interface, 2015, 12 Search PubMed.
- S. H. Kim, J. P.-K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y. Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, W. W. Lu, W. Zhen, D. Yang and S. Peng, NPG Asia Mater., 2017, 9, e435–e435 CrossRef CAS.
- Y. Qin, J. Yang and J. Xue, J. Mater. Sci., 2014, 50, 1150–1158 CrossRef.
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef CAS PubMed.
- R. Hou, L. Wu, J. Wang, Z. Yang, Q. Tu, X. Zhang and N. Huang, Biomolecules, 2019, 9, 1–15 CrossRef PubMed.
- C. G. Pitt and G. Zhong-wei, J. Controlled Release, 1987, 4, 283–292 CrossRef CAS.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741–1748 CrossRef CAS PubMed.
- A.-C. Albertsson and M. Eklund, J. Appl. Polym. Sci., 1995, 57, 87–103 CrossRef CAS.
- C. Bartolini, L. Mespouille, I. Verbruggen, R. Willem and P. Dubois, Soft Matter, 2011, 20, 9628–9637 RSC.
- N. Nemoto, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1305–1317 CrossRef CAS.
- F. He, C.-F. Wang, T. Jiang, B. Han and R. X. Zhuo, Biomacromolecules, 2010, 11, 3028–3035 CrossRef CAS PubMed.
- R. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS PubMed.
- X. Zhang, Z. Zhong and R. Zhuo, Macromolecules, 2011, 44, 1755–1759 CrossRef CAS.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 3 Search PubMed.
- N. Chanthaset and H. Ajiro, Materialia, 2019, 5, 100178 CrossRef CAS.
- H. Ajiro, Y. Takahashi and M. Akashi, Macromolecules, 2012, 45, 2668–2674 CrossRef CAS.
- Y. Haramiishi, N. Chanthaset, K. Kan, M. Akashi and H. Ajiro, Polym. Degrad. Stab., 2016, 130, 78–82 CrossRef CAS.
- H. Nobuoka and H. Ajiro, Tetrahedron Lett., 2019, 60, 164–170 CrossRef CAS.
- H. Nobuoka, R. Miyake, J. Choi, H. Yoshida, N. Chanthaset and H. Ajiro, Eur. Polym. J., 2021, 160, 110782 CrossRef CAS.
- S. C. Luo, S. S. Liour and H. H. Yu, Chem. Commun., 2010, 46, 4731–4733 RSC.
- J. M. Schakenraad and H. J. Busscher, Colloids Surf., 1989, 42, 331–343 CrossRef CAS.
- S. Yuan, G. Xiong and A. Roguin, Biointerphases, 2012, 7, 30 CrossRef CAS PubMed.
- J. Wei, M. Yoshinari, S. Takemoto, M. Hattori, E. Kawada, B. Liu and Y. Oda, J. Biomed. Mater. Res., Part B, 2007, 81, 66–75 CrossRef PubMed.
- Y. Ding, Z. Yang, C. W.-C. Bi, M. Yang, J. Zhang, S. L. Xu, X. Lu, N. Huang, P. Huang and Y. Leng, J. Mater. Chem. B, 2014, 2, 3819–3829 RSC.
- N. Shimada, W. Song and A. Maruyama, Biomater. Sci., 2014, 2, 1480–1485 RSC.
- M. Yokoya, S. Kimura and M. Yamanaka, Chem. – Eur. J., 2021, 27, 5601–5614 CrossRef CAS PubMed.
- S. H. Kim, J. P.-K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y. Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CrossRef CAS PubMed.
- H. Ying, J. Yen, R. Wang, Y. Lai, J. L.-A. Hsu, Y. Hu and J. Cheng, Biomater. Sci., 2017, 5, 2398–2402 RSC.
- L. Y. Tan, N. Chanthaset, S. Nanto, R. Soba, M. Nagasawa, H. Ohno and H. Ajiro, Macromolecules, 2021, 54, 5518–5525 CrossRef CAS.
- H. Ang, Y. Huang, S. Lim, P. Wong, M. Joner and N. Foin, J. Thorac. Dis., 2017, 9, S923–S934 CrossRef PubMed.
- J. Yue, X. Li, G. Mo, R. Wang, Y. Huang and X. Jing, Macromolecules, 2010, 43, 9645–9654 CrossRef CAS.
- E. Lizundia, V. A. Makwana, A. Larrañaga, J. L. Vilas and M. P. Shaver, Polym. Chem., 2017, 8, 3530–3538 RSC.
- H. Nobuoka and H. Ajiro, Macromol. Chem. Phys., 2019, 220, 1900051 Search PubMed.
- Y. T. Shieh, P. Y. Lin, T. Chen and S. W. Kuo, Polymers, 2016, 12, 434 CrossRef PubMed.
- Y. Arima and H. Iwata, J. Mater. Chem., 2007, 17, 4079–4087 RSC.
- J. H. Lee, H. W. Jung, I. K. Kang and H. B. Lee, Biomaterials, 1994, 15, 705–711 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab