
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis and microstructure analysis of Fe-doped NiS2 for efficient oxygen evolution electrocatalysis†
Haofei
Zhao‡
,
Wenrui
Li‡
and
Rongming
Wang
 *
*
Beijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory for Magneto-Photoelectrical Composite and Interface Science, School of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China. E-mail: rmwang@ustb.edu.cn
First published on 4th August 2022
Abstract
The development of low-cost and effective electrocatalysts for water splitting is important for the production and application of green hydrogen as a renewable energy source. In this work, Fe-doped NiS2 with different doping contents was successfully prepared through a one-pot solvothermal process utilizing a PEGylated deep eutectic solvent. The morphology, microstructure, electronic structure and oxygen evolution reaction (OER) catalytic activity of the synthesized samples are systematically investigated. The sample with an Fe content of ∼16% possesses the best electrocatalytic performance with a low overpotential of 257 mV at 10 mA cm−2, a small Tafel slope of 41 mV dec−1 and a superior stability for 60 h at ∼20 mA cm−2. Stable and homogenous Ni(Fe) oxide porous nanostructures with residual S, including pores of several nanometers, were found after the long-term OER test. X-Ray photoelectron spectroscopy and electron energy loss spectroscopy mapping indicate that the enhanced OER property may be attributed to the local composition variation of Fe at the nanoscale and the synergic effect of Fe and Ni. This study provides insights into the rational design and synthesis of superior transition-metal based water splitting electrocatalysts.
Introduction
With the imminent threat of global warming and the rapid depletion of fossil fuels, it is urgent to develop green and renewable energy systems. Due to its high energy density, hydrogen is a promising candidate as a carbon-free energy carrier and may play a role in reducing industrial carbon emissions.1 Water electrolysis using renewable electricity is an attractive strategy to produce hydrogen. However, the oxygen evolution reaction (OER) at the anode is a sluggish kinetic process involving four electrons, thus limiting the overall efficiency. The state-of-the-art electrocatalysts are based on noble metals, and their high cost and insufficient reserves restrict their wide application.2 Therefore, it is important to develop low-cost and effective OER catalysts for water splitting.In the past few decades, earth-abundant-transition-metal based compounds have been widely explored as OER electrocatalysts, including oxides, chalcogenides, nitrides, phosphides et al.3,4 Recent studies have suggested that transition metal sulfides may exhibit superior OER performance.5–8 Previous studies have revealed that the oxyhydroxides in situ formed at the surface of sulfides during the OER catalysis in alkaline media are the real active species.9–12 And a systematic study of 3d transition-metal oxyhydroxides reveals that the OER activity of these samples follows a trend of Ni > Co > Fe > Mn.13 Thus, nickel sulfides are promising electrocatalysts for water splitting.14,15
Heteroatom doping is an effective strategy to improve the electrocatalytic performance. Yan et al. revealed that Fe3+ doping in the NiS2(002) facet lowers the activation energy of H2 generation of active sites, resulting in a good electrocatalytic hydrogen evolution activity.16 Han et al. reported that confined Fe overdoping in Ni3S2 induces a synergic effect of size decrease and electronic structure modulation, thus resulting in a 70 mV decrease in the overpotential at 10 mA cm−2 for the OER.17 Ding et al. suggested that the Fe incorporation in NiS2 regulates the electronic structure of pyrite NiS2, increasing its conductivity, and the conductive Fe-doped NiS2 with an in situ electrochemical formed surface layer of oxyhydroxides during the OER exhibits a low overpotential of 260 mV at 10 mA cm−2.18 These doped nickel sulfides are usually synthesized through a two-step precursor sulfuration route with a sulfur source under high temperature. Combing the two steps into a one-step synthesis should be cost-effective.
As quasi-ionic liquids, deep eutectic solvents (DES) are promising for applications in nanomaterial synthesis due to their unique physicochemical properties, such as low cost, easy preparation, environmental friendliness etc.19,20 Jiang et al. developed a PEGylated DES by simply mixing polyethylene glycol 200 (PEG-200) with thiourea in the molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and demonstrated its prospective application in the one-pot solvothermal synthesis of metal sulfides with tunable composition.21 PEG 200 in the solvent could act as a shape-control agent, while thiourea could work as the sulfur source and help to stabilize the M–S bonds in metal sulfides.21,22
:1, and demonstrated its prospective application in the one-pot solvothermal synthesis of metal sulfides with tunable composition.21 PEG 200 in the solvent could act as a shape-control agent, while thiourea could work as the sulfur source and help to stabilize the M–S bonds in metal sulfides.21,22
In this work, we synthesized a series of Fe-doped NiS2 with different composition through a one-pot solvothermal process using PEGylated DES, and performed a detailed characterization of the prepared samples. The doped samples exhibit enhanced OER activity compared to pure NiS2, with the best found in (NiFe)S2-3 (∼16% Fe). (NiFe)S2-3 needs a low overpotential of 257 mV to deliver a current density of 10 mA cm−2 and a good stability (> 60 h at ∼20 mA cm−2) in 1 M KOH.
Experimental
Synthesis
The Fe-doped NiS2 samples were synthesized by a simple solvothermal method. In a typical procedure, a PEGylated DES was firstly prepared by mixing PEG 200 and thiourea in the molar ratio of 2:1.21 Then, 0.5 mmol nickel acetate and 0.5 mmol iron(II) acetate were dissolved in 40 ml PEGylated DES and stirred and sonicated for about 1 h to form a transparent homogeneous solution. The solution was subsequently transferred to a 50 ml Teflon-line autoclave, maintained at 200 °C for 10 h, and then cooled down to room temperature naturally. The product was washed with ethanol and water several times. Afterward, the sample was collected by centrifugation (3000 rpm, 3 min) after a low-speed centrifugation (200 rpm, 1 min) to remove large particles (FeS2 and some aggregations), and dried in an oven at 80 °C for 4 h. This sample was denoted as (NiFe)S2-3. Following a similar procedure, samples with different Fe doping content were prepared by adding different amounts of acetates while the total amount of Ni and Fe acetates was kept as 1.0 mmol, and labeled as NiS2 (No Fe doping), (NiFe)S2-1 (0.2 mmol Fe acetate), (NiFe)S2-2 (0.4 mmol Fe acetate) and (NiFe)S2-4 (0.6 mmol Fe acetate).
Characterizations
Powder X-ray diffraction (XRD) patterns were acquired on a Rigaku Ultima IV using Cu Kα radiation at a scan rate of 0.02 °C s−1. Transmission electron microscopy (TEM) investigations were performed with a JEOL JEM 2200FS electron microscope. X-Ray photoelectron spectra (XPS) were collected on a PHI5000 VersaProbe III spectrometer with a microfocus and monochromatic Al Kα (1486.6 eV) X-ray excitation source. The core-level XPS profiles were fitted using Gaussian–Lorentzian lineshapes with a Shirley-type background.Electrochemical measurements
Electrochemical tests were performed with a CHI760E electrochemical workstation in a standard three-electrode system in 1 M KOH, using a Pt foil as the counter electrode and a Hg/HgO electrode as the reference electrode. To prepare the working electrode, 3 mg of the prepared samples and 30 μL 5 wt% Nafion solution were dispersed in 470 μL ethanol by 30 min sonication to form a homogeneous ink, and then 50 μL ink was drop-cast on a piece of carbon paper and dried naturally with a catalyst loading of ∼0.3 mg cm−2. The electrolyte was bubbled with O2 for 30 min before the electrochemical measurements. The prepared working electrodes were activated via consecutive cyclic voltammetry (CV) scans for 20 cycles at a scan rate of 10 mV s−1. 100% iR-compensated cyclic voltammograms with a scan rate of 2 mV s−1 were used to characterize the OER activity of the samples. The Tafel plots were extracted from the backward sweep of the CV curves to avoid the effect of the redox peaks of Ni. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range from 1 Hz to 100 kHz with an amplitude of 5 mV at the potential of 10 mA cm−2 for each sample. The electrochemical active surface area (ECSA) was evaluated by carrying out a series of CV measurements in a non-faradaic potential range (0.1 V–0.2 V vs. Hg/HgO) at various scan rates (5, 10, 20, 30 and 40 mV s−1). The overpotentials were calculated using the equation: η = EHg/HgO + 0.098 V + 0.059*pH − 1.23 V.Results and discussion
Structure and morphology
TEM investigations reveal that the as-prepared NiS2 sample exhibits porous nanostructures (Fig. S1, ESI†), while the Ni–Fe sulfides show similar morphology and are composed of irregular nanoparticles of tens of nanometers, as shown in Fig. 1(a–e). Apparent diffraction contrast in the bright-field TEM images indicates the crystal nature of the nanoparticles. Fig. 1(b–e) suggests that the size of the crystallites decreases with the increase of the Fe content, which should have resulted from the decrease of the Ni content in the raw materials and the lattice instability due to Fe overdoping.17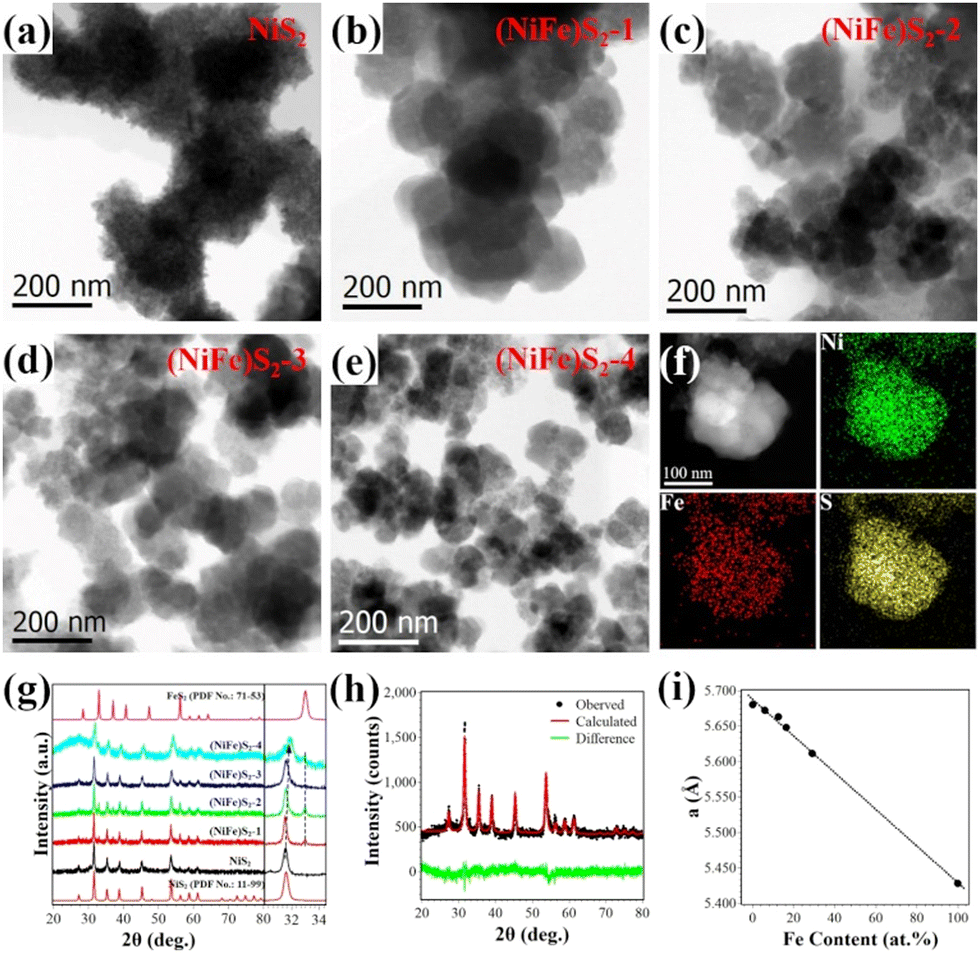 | ||
| Fig. 1 (a–e) Bright-field TEM images of NiS2, (NiFe)S2-1, (NiFe)S2-2, (NiFe)S2-3 and (NiFe)S2-4. (f) High-angle annular dark field image of a nanoparticle composed of several crystallites and corresponding EDX elemental maps of (NiFe)S2-3. (g) XRD patterns (left) and enlarged (200) main peak (right) of the as-prepared samples. (h) Graphic results of the Rietveld refinement fitting of the XRD data of (NiFe)S2-3 using MAUD software. (i) Variation of lattice parameters with Fe content in the prepared samples. | ||
Fig. 1(f) presents typical energy dispersive X-ray spectroscopy (EDX) elemental maps of the nanostructures consisting of several nanocrystals. Homogenous distribution of Ni, Fe and S in the crystallites could be observed in the maps.
The crystal structures of the samples were characterized by XRD. As shown in Fig. 1(g), major peak positions in the XRD patterns matched well with those of NiS2 (PDF No. 11-99). Minor peaks related to FeS2 (PDF No. 71-53) can also be distinguished in the spectra of the samples, suggesting the existence of tiny FeS2 nanoparticles. Detailed investigations indicate that the major diffraction peaks shift to higher angles slightly. The right panel of Fig. 1(g) depicts enlarged (200) peaks of NiS2. The observed gradually increased positions from 31.58° (NiS2) to 31.88° ((NiFe)S2-4) indicate that the lattice parameters of the samples decrease with Fe doping. The cations in disulfides are coordinated by 6 sulfur anions, and the ionic radius of Fe2+ (75 pm) is smaller than that of Ni2+ (83 pm) under octahedral coordination. The lattice contraction should be attributed to the replacement of Ni2+ by smaller Fe2+. The broadening of the peaks maybe due to the size effect of the nanocrystals, which is consistent with the TEM observations. Quantitative analysis of the XRD data was performed by Rietveld fitting (Fig. 1(h)) using MAUD software.23 The lattice parameters and average crystal sizes of the samples were retrieved and summarized in Table S1 (ESI†). The Fe contents determined by EDX quantitative analysis are also shown in Table S1 (ESI†). Fig. 1(i) depicts the variation of the lattice parameters with the Fe content. The almost linear relationship between them as previously reported18,24 suggests successful doping of Fe in NiS2.
High resolution TEM (HRTEM) investigations provide further insights into the nanostructures. Fig. 2(a) shows a representative HRTEM image of (NiFe)S2-3, exhibiting two well-resolved perpendicular lattice fringes with interplanar spacings of 0.40 nm and 0.34 nm, which could be indexed to the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (11) planes of cubic disulfide, respectively. The observed amorphous structure on the surface with 1∼3 atomic layers in thickness may have resulted from the residual carbonaceous species or surface oxidation of the sample. The inset shows the fast Fourier transform (FFT) pattern of the marked square in Fig. 2(a). The inhomogeneous contrast in the HRTEM image and the diffusing of the diffraction spots in the FFT pattern suggest lattice variations in the sample. Fig. 2(b) shows the inverse FFT (IFFT) image of the marked area. Contrast with stripes of several nanometers can be distinguished in the IFFT image, which may come from the local variation of the lattice parameters with different doping of Fe at the nanometer scale. The local distributions of Ni and Fe at the nanometer scale were obtained with electron energy loss spectroscopy (EELS) mapping in scanning TEM mode with a nanometer-size electron beam. As shown in Fig. 2(c), Fe distributes throughout the nanocrystal inhomogeneously at the nanometer scale and is rich in the surfaces/interfaces. The composition variation could induce strains and electronic structure modulation, which may benefit the electrocatalytic performance.17 The selected-area electron diffraction (SAED) pattern of (NiFe)S2-3 in Fig. 2(d) exhibits a pronounced ring structure, which can be indexed with NiS2, indicating the polycrystalline nature of the Fe-doped NiS2.
0) and (11) planes of cubic disulfide, respectively. The observed amorphous structure on the surface with 1∼3 atomic layers in thickness may have resulted from the residual carbonaceous species or surface oxidation of the sample. The inset shows the fast Fourier transform (FFT) pattern of the marked square in Fig. 2(a). The inhomogeneous contrast in the HRTEM image and the diffusing of the diffraction spots in the FFT pattern suggest lattice variations in the sample. Fig. 2(b) shows the inverse FFT (IFFT) image of the marked area. Contrast with stripes of several nanometers can be distinguished in the IFFT image, which may come from the local variation of the lattice parameters with different doping of Fe at the nanometer scale. The local distributions of Ni and Fe at the nanometer scale were obtained with electron energy loss spectroscopy (EELS) mapping in scanning TEM mode with a nanometer-size electron beam. As shown in Fig. 2(c), Fe distributes throughout the nanocrystal inhomogeneously at the nanometer scale and is rich in the surfaces/interfaces. The composition variation could induce strains and electronic structure modulation, which may benefit the electrocatalytic performance.17 The selected-area electron diffraction (SAED) pattern of (NiFe)S2-3 in Fig. 2(d) exhibits a pronounced ring structure, which can be indexed with NiS2, indicating the polycrystalline nature of the Fe-doped NiS2.
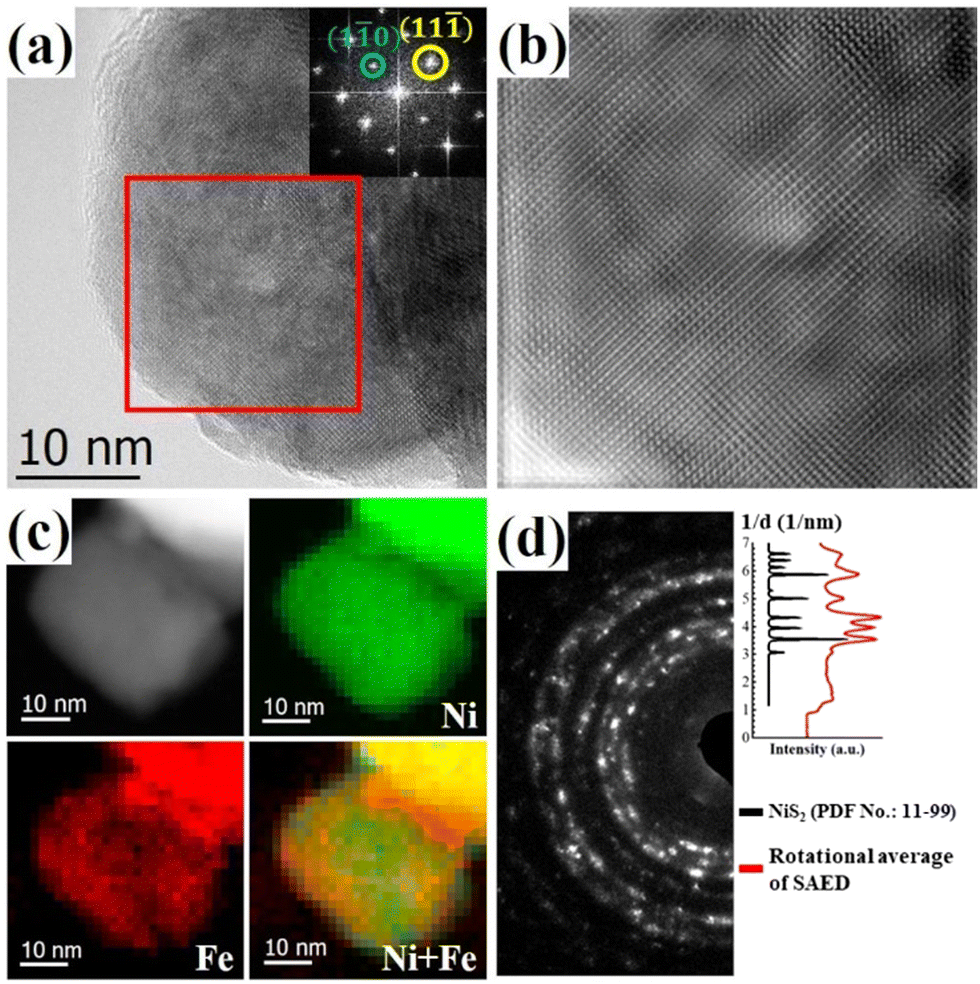 | ||
| Fig. 2 Representative TEM images of (NiFe)S2-3. (a) High-resolution image and corresponding FFT pattern (inset). (b) Inverse FFT image of the area marked by the red square in (a). (c) EELS elemental maps of a crystallite with a pixel size of 1.6 nm showing the local distributions of Ni and Fe. (d) SAED pattern (left) and the comparison of the peak positions between its rotational average and the reference pattern of NiS2 (right). | ||
XPS analysis
The surface chemical states of Ni, Fe and S in (NiFe)S2-3 were investigated with XPS. The survey spectrum clearly proves the presence of Ni, Fe and S (Fig. 3(a)), while the C and O should be due to the surface adsorbed contaminants and/or oxidation. The Ni 2p3/2 region exhibits a strong peak at 853.4 eV along with a satellite peak at 860.3 eV, corresponding to Ni2+ in disulfides,25–27 as shown in Fig. 3(b). The shoulder peak at 855.7 eV might be due to the presence of Ni3+.11,28,29 The Fe 2p3/2 spectrum overlaps with two broad Ni LMM Auger peaks.30–32 The peak positions, widths, area ratio etc. of the two Auger peaks are obtained by fitting the XPS spectrum of NiS2 ranging from 701 eV and 717 eV (Fig. S2, ESI†), and used in the fitting of the Fe 2p3/2 spectrum. The fitted narrow peaks at 707.3 eV and 709.2 eV could be attributed to Fe2+ in disulfides and Fe3+, respectively.11,26,33 The S 2p profile could be deconvoluted to 6 peaks, as demonstrated in Fig. 3(d). The peaks at 162.5 eV and 163.5 eV unequivocally originate from 2p spin–orbital splitting of S22−,33–35 and the peaks at 164.2 eV and 166.1 eV are ascribed to polysulfide Sn or Sn2− often presented at the disulfide surfaces.33,36 The fitting peaks at 161.4 eV and 168.6 eV could be assigned to S2− and SO42− species, respectively.26,29,37 The XPS spectra of other samples exhibit similar features (Fig. S3, ESI†) and could be well fitted with the same components, the fitting parameters are listed in Table S2 (ESI†) (Ni 2p3/2) and Table S3 (ESI†) (S 2p). Thus, these analysis results further confirm the successful doping of Fe in NiS2.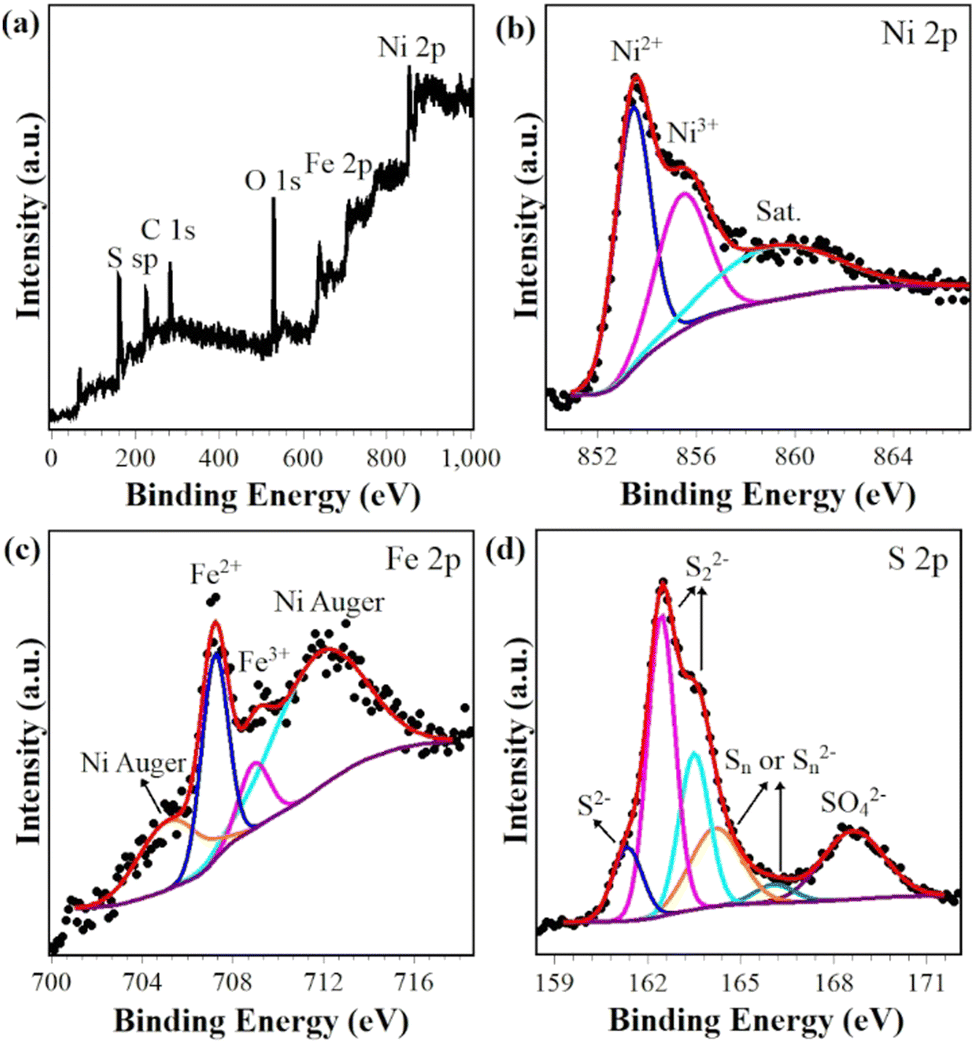 | ||
| Fig. 3 XPS survey spectrum (a) and core-level spectra with deconvolution (b) Ni 2p, (c) Fe 2p and (d) S 2p of (NiFe)S2-3. | ||
Electrochemical oxygen evolution activity
The OER activities of Fe-doped NiS2 were investigated in O2-saturated 1.0 M KOH using a standard three-electrode system. Blank CFP was also tested under the same conditions, and exhibited negligible activity. The 100% iR-compensated cyclic voltammograms with a scan rate of 2 mV s−1 were shown in Fig. 4(a). The undoped sample (NiS2) needs a high overpotential (η10) of 411 mV at 10 mA cm−2. With Fe doping, the η10 values are significantly reduced, and (NiFe)S2-3 exhibits the best activity with a low overpotential of 257 mV, which is comparable or even superior to that of other reports, such as Fe0.1Ni0.9S2 (260 mV),18 V0.1Ni0.9S2 (290 mV),34 (NiFe)S2 (230 mV),11 Fe–NiS2/C (256 mV),38 (NiFe)S2@Graphene (320 mV),26 (NiCo)S2 (270 mV),27 Fe0.2Co0.8S2 (290 mV)39etc. A detailed comparison with some reported transition-metal based OER catalysts (sulfides, phosphides, hydroxides etc.) can be found in Table S5 (ESI†). The Fe content of (NiFe)S2-3 is ∼16%, close to the optimal Fe content of Fe-doped Ni based OER electrocatalysts in previous reports.25,26,40,41 For better evaluation of the electrocatalytic performance, the Tafel plots were derived from the backward sweep of the voltammograms.42 As depicted in Fig. 4(b), (NiFe)S2-3 also shows the smallest Tafel slope of 41 mV dec−1, indicating a fast reaction kinetics. For all samples that have similar ECSA (Fig. S4, ESI†), it is reasonable to conclude that (NiFe)S2-3 should possess a high intrinsic OER activity.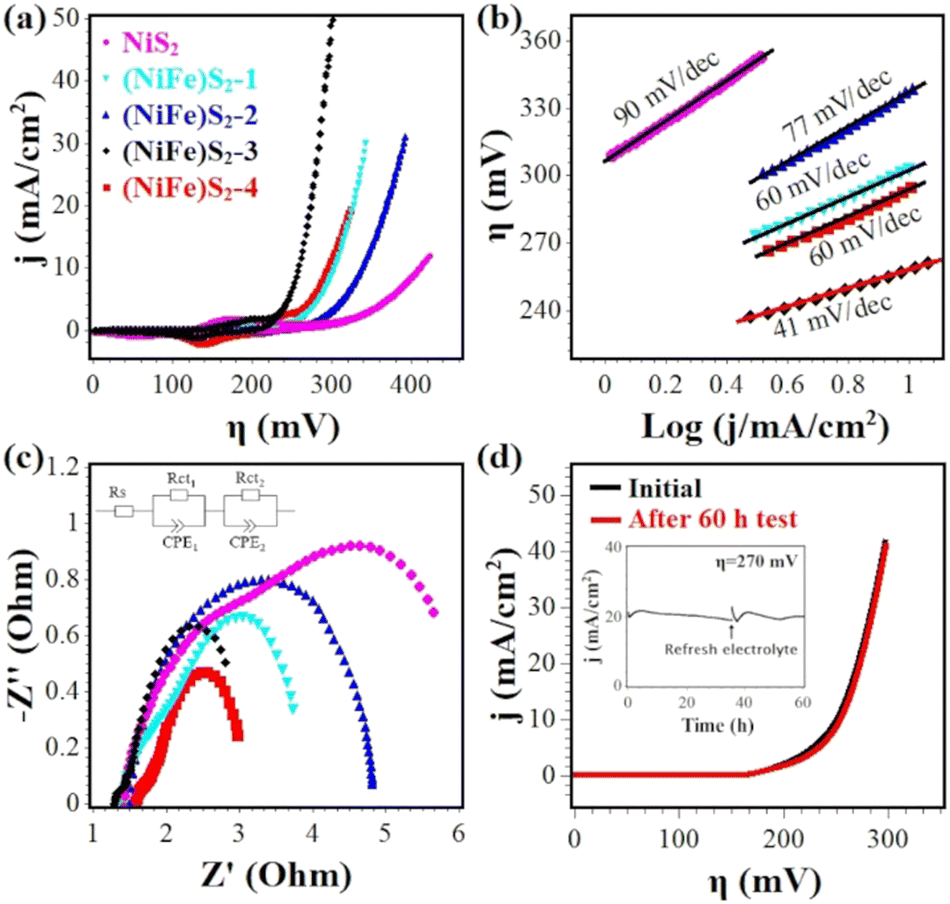 | ||
| Fig. 4 (a) Cyclic voltammograms of NiS2, (NiFe)S2-1, (NiFe)S2-2, (NiFe)S2-3 and (NiFe)S2-4 with 100%-iR correction at a scan rate of 2 mV s−1 in 1 M KOH. (b) Tafel plots extracted from the backward sweep of the corresponding voltammograms. (c) Nyquist plots of the EIS data in the frequency range of 100 kHz to 1 Hz; the inset is the equivalent circuit model used to fit the data. (d) Linear sweep voltammograms of (NiFe)S2-3 before and after 60 h test at ∼20 mA cm−2; the inset is the measured i–t curve. | ||
EIS measurements were carried out to characterize the charge-transfer resistance during the OER catalysis. The Nyquist plots of the EIS data are shown in Fig. 4(c) and fitted with a simplified Randles equivalent circuit (inset), where RS is the solution resistance (∼1.5 Ω), Rct1 and Rct2 represent the internal resistance of the electrode and the interfacial resistance between the electrode and the electrolyte, respectively.43 As listed in Table S4 (ESI†), (NiFe)S2-3 displays a low total resistance (Rct1 + Rct2) of ∼2.3 Ω, suggesting a fast charge transfer during the OER process.
Fig. 4(d) shows the stability test results of (NiFe)S2-3. After being tested for 60 h at ∼20 mA cm−2, the activity loss is negligible and the overpotential to achieve 10 mA cm−2 increased slightly.
Sample after the OER durability test
As shown in Fig. 5(a), (NiFe)S2-3 transformed to porous nanostructures after a long-time durability test (∼20 mA cm−2, 60 h), and holes of several nanometers presented in the tested sample, which may help to expose reaction sites and provide pathways of mass transport, thus contributing to the superior electroactivity.10,44 The inset in Fig. 5(a) shows an HRTEM image, suggesting that the nanostructures are composed of many poorly-crystallized nanocrystals. Compared with the EDX spectrum of the as-prepared sample (Fig. 5(c)), the intensity of the S peak in that of the tested sample is significantly reduced, while the O peak increased, indicating the desulfurization and oxidation of the sample in the strongly oxidative environment during OER catalysis. Elemental maps of Ni, Fe, S, and O suggest that these elements are distributed uniformly (Fig. 5(d)). The polycrystalline rings in the SAED pattern could be well indexed with NiO (PDF No.: 78-643). The Ni 2p3/2 XPS profile is well fitted with a peak at 854.5 eV along with a satellite peak at 859.1 eV (Fig. 5(e)), which could be attributed to Ni2+ in NiO.28,45 The O 1s spectrum is composed of two components with the binding energies of 529.9 eV and 531.7 eV (Fig. 5(f)), which could be assigned to metal-O and metal-OH bands, respectively.45–47 These characterization results suggest the formation of nanoporous Ni(Fe) oxides after the stability test.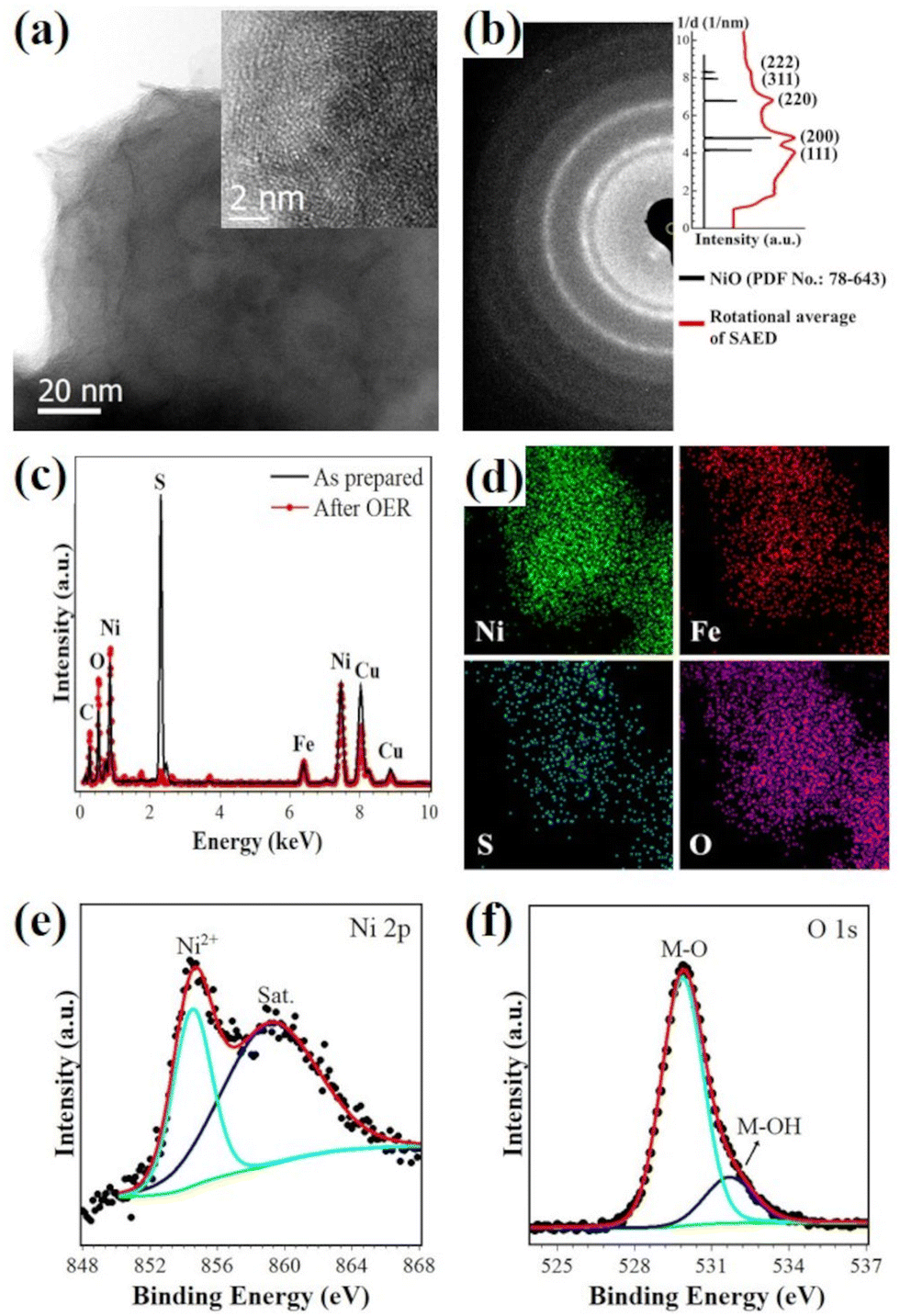 | ||
| Fig. 5 Morphology, composition and surface chemical states of (NiFe)S2-3 after the durability test (∼20 mA cm−2, 60 h). (a) Bright-field and high-resolution TEM images (inset). (b) SAED pattern (left panel) and indexing (right panel), (c) comparison of the EDX spectra before and after the OER test, (d) EDX elemental maps of Ni, Fe, S and O, and the (e) Ni 2p and (f) O 1s XPS profiles. | ||
It has been shown that sulfides will be oxidized (at least on the surface) to oxyhydroxides in the strong oxidative environment during OER catalysis in an alkaline solution, which should be the real active species.9,11,18,32,48 Based on the reports on the redox behavior of Ni–Fe oxyhydroxides,49–51 it is reasonable to conclude that Ni(Fe) oxides result from the electrochemical reduction of in situ formed oxyhydroxides with the gradual decrease of the applied potential, and would be oxidized to oxyhydroxides when the potential increases. It should be noted that the sample is not fully desulfurized even after 60 h electrolysis at ∼20 mA cm−2, and the residual S may play a role in the high OER activity.25
Previous studies suggest that the stable electrocatalytic performance of sulfides during OER catalysis may be ascribed to the in situ electrochemical formation of a stable core–shell heterostructure consisting of an active oxyhydroxide layer and conductive disulfide core,11,12,18 stable oxyhydroxides,10,25,52etc. We investigated the composition change of (NiFe)S2-3 during the durability test with TEM EDX. As shown in Fig. S5 (ESI†), the significant change in the EDX spectra is the reduction of the S peak, indicating the desulfurization of the prepared sample. After being tested for 1 h at ∼20 mA cm−2, the S peak is still relatively strong, and is significantly reduced after being tested for 5 h, which is close to that of the sample being tested for 60 h. This result indicates the gradual transformation of (NiFe)S2-3 during the long-term electrocatalysis. Considering the negligible activity loss after a 60 h test (Fig. 4(d)), it is reasonable to conclude that the long-term stable catalytic performance may be due to the in situ electrochemically-formed stable oxyhydroxides.
Conclusions
In summary, we have successfully synthesized and systematically investigated Fe doped NiS2via a one-pot solvothermal process in PEGylated DES. When used as OER catalysts, the Fe-doped samples exhibit enhanced activity compared to the undoped one. The sample containing ∼16% Fe ((NiFe)S2-3) shows the best electrocatalytic activity, and only needs a low overpotential of 257 mV to achieve a current density of 10 mA cm−2 and displays a small Tafel slope of 41 mV dec−1. Furthermore, (NiFe)S2-3 maintains its activity even after a stability test of 60 h at ∼20 mA cm−2. The post-OER (NiFe)S2-3 is found to be nanoporous Ni(Fe) oxide with residual S, which should be in situ electrochemically transformed to active oxyhydroxides during the OER catalysis. Detailed investigations reveal the presence of local lattice distortions and nanoscale variation of Fe doping content, which may contribute to the superior performance of (NiFe)S2-3. This work may contribute to the development of a low-cost and effective non-noble electrocatalyst for water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Beijing Natural Science Foundation (Grant No. 2212034), the National Natural Science Foundation of China (No. 51971025 and 12034002) and the Fundamental Research Funds for the Central Universities (FRF-IDRY-19-028).Notes and references
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- J. Mohammed-Ibrahim, J. Power Sources, 2020, 448, 227375 CrossRef CAS.
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1–20 Search PubMed.
- S. Shit, S. Chhetri, W. Jang, N. C. Murmu, H. Koo, P. Samanta and T. Kuila, ACS Appl. Mater. Interfaces, 2018, 10, 27712–27722 CrossRef CAS PubMed.
- Y. Wu, X. Liu, D. Han, X. Song, L. Shi, Y. Song, S. Niu, Y. Xie, J. Cai, S. Wu, J. Kang, J. Zhou, Z. Chen, X. Zheng, X. Xiao and G. Wang, Nat. Commun., 2018, 9, 1425 CrossRef PubMed.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, Q. Zhang, X. Jia, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- X. Feng, Q. Jiao, H. Cui, M. Yin, Q. Li, Y. Zhao, H. Li, W. Zhou and C. Feng, ACS Appl. Mater. Interfaces, 2018, 10, 29521–29531 CrossRef CAS PubMed.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- X. Zou, Y. Wu, Y. Liu, D. Liu, W. Li, L. Gu, H. Liu, P. Wang, L. Sun and Y. Zhang, Chem, 2018, 4, 1139–1152 CAS.
- M. Zhou, Q. Weng, X. Zhang, X. Wang, Y. Xue, X. Zeng, Y. Bando and D. Golberg, J. Mater. Chem. A, 2017, 5, 4335–4342 RSC.
- Y. Liu, S. Ju, C. Wang, M. Li, W. Zhu, D. Chen, A. Yuan and G. Zhu, Ind. Eng. Chem. Res., 2019, 58, 18976–18985 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- P. Luo, H. Zhang, L. Liu, Y. Zhang, J. Deng, C. Xu, N. Hu and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 2500–2508 CrossRef CAS PubMed.
- M. Yao, B. Sun, L. He, N. Wang, W. Hu and S. Komarneni, ACS Sustainable Chem. Eng., 2019, 7, 5430–5439 CrossRef CAS.
- J. Yan, H. Wu, P. Li, H. Chen, R. Jiang and S. Liu, J. Mater. Chem. A, 2017, 5, 10173–10181 RSC.
- C. Han, W. Li, C. Shu, H. Guo, H. Liu, S. Dou and J. Wang, ACS Appl. Energy Mater., 2019, 2, 5363–5372 CrossRef CAS.
- X. Ding, W. Li, H. Kuang, M. Qu, M. Cui, C. Zhao, D. C. Qi, F. E. Oropeza and K. H. L. Zhang, Nanoscale, 2019, 11, 23217–23225 RSC.
- F. M. Perna, P. Vitale and V. Capriati, Curr. Opin. Green Sustainable Chem., 2020, 21, 27–33 CrossRef.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- J. Jiang, C. Yan, X. Zhao, H. Luo, Z. Xue and T. Mu, Green Chem., 2017, 19, 3023–3031 RSC.
- C. Lamiel, V. H. Nguyen, M. Baynosa, D. C. Huynh and J. J. Shim, J. Electroanal. Chem., 2016, 771, 106–113 CrossRef CAS.
- L. Lutterotti, S. Matthies, H. R. Wenk, A. S. Schultz and J. W. Richardson, J. Appl. Phys., 1997, 81, 594–600 CrossRef CAS.
- L. Wang, T. Y. Chen, C. L. Chien, J. G. Checkelsky, J. C. Eckert, E. D. Dahlberg, K. Umemoto, R. M. Wentzcovitch and C. Leighton, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 144402 CrossRef.
- T. Wang, G. Nam, Y. Jin, X. Wang, P. Ren, M. G. Kim, J. Liang, X. Wen, H. Jang, J. Han, Y. Huang, Q. Li and J. Cho, Adv. Mater., 2018, 30, 1–9 Search PubMed.
- C. Liu, H. Ma, M. Yuan, Z. Yu, J. Li, K. Shi, Z. Liang, Y. Yang, T. Zhu, G. Sun, H. Li and S. Ma, Electrochim. Acta, 2018, 286, 195–204 CrossRef CAS.
- J. Zhang, X. Bai, T. Wang, W. Xiao, P. Xi, J. Wang, D. Gao and J. Wang, Nano-Micro Lett., 2019, 11, 2 CrossRef CAS PubMed.
- P. Dubey, N. Kaurav, R. S. Devan, G. S. Okram and Y. K. Kuo, RSC Adv., 2018, 8, 5882–5890 RSC.
- P. Xu, J. Li, J. Luo, L. Wei, D. Zhang, D. Zhou, W. Xu and D. Yuan, Sci. Rep., 2018, 8, 1–9 Search PubMed.
- G. Zhang, Y. S. Feng, W. T. Lu, D. He, C. Y. Wang, Y. K. Li, X. Y. Wang and F. F. Cao, ACS Catal., 2018, 8, 5431–5441 CrossRef CAS.
- D. L. Legrand, G. M. Bancroft and H. W. Nesbitt, Int. J. Miner. Process., 1997, 51, 217–228 CrossRef CAS.
- W. Li, H. Zhao, H. Li and R. Wang, Nanoscale Adv., 2022, 4, 1220–1226 RSC.
- M. V. Morales-Gallardo, A. M. Ayala, M. Pal, M. A. Cortes Jacome, J. A. Toledo Antonio and N. R. Mathews, Chem. Phys. Lett., 2016, 660, 93–98 CrossRef CAS.
- H. Liu, Q. He, H. Jiang, Y. Lin, Y. Zhang, M. Habib, S. Chen and L. Song, ACS Nano, 2017, 11, 11574–11583 CrossRef CAS PubMed.
- L. Gao, C. Guo, X. Liu, X. Ma, M. Zhao, X. Kuang, H. Yang, X. Zhu, X. Sun and Q. Wei, New J. Chem., 2020, 44, 1711–1718 RSC.
- V. Toniazzo, C. Mustin, J. M. Portal, B. Humbert, R. Benoit and R. Erre, Appl. Surf. Sci., 1999, 143, 229–237 CrossRef CAS.
- X. Han, X. Wu, C. Zhong, Y. Deng, N. Zhao and W. Hu, Nano Energy, 2017, 31, 541–550 CrossRef CAS.
- L. Xie, D. Zhao, J. Dai, Z. Wu and L. Li, Catalysts, 2019, 9, 458 CrossRef CAS.
- Y. Zhou, M. Luo, Z. Zhang, W. Li, X. Shen, W. Xia, M. Zhou and X. Zeng, Appl. Surf. Sci., 2018, 448, 9–15 CrossRef CAS.
- J. Landon, E. Demeter, N. Inoǧlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS.
- N. Cheng, Q. Liu, A. M. Asiri, W. Xing and X. Sun, J. Mater. Chem. A, 2015, 3, 23207–23212 RSC.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed.
- X. Y. Yu and X. W. David Lou, Adv. Energy Mater., 2018, 8, 1–37 Search PubMed.
- R. A. Patil, C. P. Chang, R. S. Devan, Y. Liou and Y. R. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 9872–9880 CrossRef CAS PubMed.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 CrossRef.
- Q. Liu, J. Huang, Y. Zhao, L. Cao, K. Li, N. Zhang, D. Yang, L. Feng and L. Feng, Nanoscale, 2019, 11, 8855–8863 RSC.
- R. He, X. Huang and L. Feng, Energy Fuels, 2022, 36, 6675–6694 CrossRef CAS.
- A. G. Marrani, V. Novelli, S. Sheehan, D. P. Dowling and D. Dini, ACS Appl. Mater. Interfaces, 2014, 6, 143–152 CrossRef CAS PubMed.
- M. Gorlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- D. Di Girolamo, M. Piccinni, F. Matteocci, A. G. Marrani, R. Zanoni and D. Dini, Electrochim. Acta, 2019, 319, 175–184 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00447j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |