
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the phase change material properties by an energy-efficient one-step preparation method using organogelator–polyolefin composites†
Felix
Leven
 *ab,
Johannes
Limberg
a,
Jessica
Noll
a,
Mathias
Ulbricht
b and
Rainer
Ostermann
a
*ab,
Johannes
Limberg
a,
Jessica
Noll
a,
Mathias
Ulbricht
b and
Rainer
Ostermann
a
aTechnische & Makromolekulare Chemie, Westfälische Hochschule, August-Schmidt-Ring 10, 45665, Recklinghausen, Germany. E-mail: FelixLeven@gmx.de
bLehrstuhl für Technische Chemie II, Universität Duisburg-Essen, Universitätsstraße 7, 45141, Essen, Germany
First published on 9th August 2022
Abstract
The synergistic combination of various sorbitol-based organogelators with polyolefins allows the preparation of porous support structures for immobilized phase change materials (PCMs). Using a PCM as a solvent for the preparation leads to dimensionally stable composite materials with extremely high loading rates and low leakage of PCMs. Detailed investigations were performed on the kind of polyolefin support and its mass fraction concentration in the PCM, the temperature-dependent softening and failure under superimposed load, the efficiency of heat transport and the retention capacity over several melting/solidification cycles in various measurement setups. In particular, paraffin wax in combination with 1,2,3-trideoxy-4,6:5,7-bis-O-[(4-propylphenyl)methylene]-nonitol (TBPMN) and ultrahigh molecular weight polyethylene (UHMWPE) showed the best results in terms of high dimensional stability, low leakage, excellent processability and competitive heat capacity. The herein-established one-step preparation method saves time and energy compared to the loading of pre-formed porous supports and improves application-related properties at the same time.
1. Introduction
Climate change and the resulting urgent need to change the entire energy sector impose challenges for all of us to use the available energy more responsibly. Worldwide one-third of the whole energy consumption is related to air conditioning; in Germany, this proportion is about 27%.1,2 Typical insulation materials are organic polymers like polystyrene (PS) or polyurethane (PU) foams or glass and rock wool.3 In terms of performance, aerogels are superior to conventional insulators, but their worse price/performance ratio turns them into niche products for special applications. Optimized insulators and latent heat storage systems are a highly relevant starting point for developments towards a more sustainable energy balance.Phase change materials (PCMs) are a group of materials with high melting energy. This property makes them interesting for heat transport in the fluid state and heat absorption and storage in the immobilized solid state. Typical applications are solar energy storage,4–7 heat recovery in industrial processes,8 temperature control in electrical applications9 and energy storage in buildings.10 Both inorganic materials like hydrated salts11 and organic components like fatty acids,12 waxes13 and polyethyleneglycol14–16 are used. On the one hand, typical organic materials show a lower melting point and fewer problems with a phase separation over the melting and solidification cycles and are less corrosive. On the other hand, leakages can occur in the molten state connected with a potential material loss. In order to prevent leakage, such materials are often encapsulated and consequently immobilized using porous polymers,17 polymer blends,13,18 as well as aerogels based on graphene12,19 or silicon dioxide,16,20 graphite21,22 and diatomaceous earth.23,24 Gelators (cf. below) can also be used,25,26 but they have shown a much worse retention capability.26,27 Recently, Fan et al. described the preparation of a form-stable PCM using polynorbornene-based bottlebrush polymers as gelators and sulphur-treated nickel foams with interesting properties for application in solar technology. To avoid material loss, the PCM needs to show a high phase compatibility to the support material when molten, but should not dissolve the support.28 Looking at the production costs of an immobilized PCM, both the synthesis of the support material and the subsequent loading of PCM, by “flooding” the support, have a high contribution.
Aerogels are nanostructured open porous solids, which can also function as support materials for PCMs. Aerogels can be made via a sol–gel process from many inorganic materials such as silicon dioxide or titanium dioxide, which were the first documented ones.29 Furthermore, the classical synthetic polymers (PU, PS),30 biopolymers like cellulose15,31 and starch32 or carbon frameworks33 can also be used as the solid phase.
Organogelators are molecules which, in a suited liquid, can build up three-dimensional networks by chemical or physical interactions. These are, for example, hydrogen bonds of OH- or NH-functionalities, π–π stacking, ionic interactions and van der Waals forces.34–38 The specific network structure depends on the solvent or the molten porogen. Typical gelator material classes are amino acids,38,39 isosorbide derivatives35,37,40–43 or amines.43–45 Despite their micro-scale structure, gelator networks are not applicable for aerogels because of their missing covalent bonds and the resulting low mechanical stability, which leads to a collapse of the structure after extraction of the solvent. Finally, compact bundles of associated gelator aggregate strings result. However, in 2016, Jamart-Grégoire et al. described an amino acid-based organogel that was supercritically dried to obtain small samples of monolithic aerogels.46 However, these aerogels were very brittle and powdery and would therefore not be suitable as support for PCM.
In this work, an alternative and new approach for aerogel-immobilized PCM synthesis by using organogelators and cheap polyolefins in the PCM as a solvent is introduced. Recently, we demonstrated that by combining organogelators with suitable high molecular weight polymers, for example polyolefins, mechanically robust composite aerogels can be obtained, because a thin polymer film on the gelator fibers reinforces the network.47 Here, we use such composites as support for PCMs such as paraffin or polyethyleneglycol. Importantly, we show that instead of subsequent loading of the support with PCMs, also a one-step fabrication directly in the PCM as solvent is possible and that robust materials at very high PCM/carrier ratios of up to 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 can be obtained. The main advantage of the composites obtained here compared to flooded PCM supports and gelator-stabilised PCMs is that they can be produced fast and easily while at the same time retaining very good material properties. The method is not limited to the system described and can also be adapted for various applications.
:1 can be obtained. The main advantage of the composites obtained here compared to flooded PCM supports and gelator-stabilised PCMs is that they can be produced fast and easily while at the same time retaining very good material properties. The method is not limited to the system described and can also be adapted for various applications.
2. Experimental
2.1. Materials
The following support polymers were used in this work: Stamylan P, isotactic polypropylene (iPP), Mw ∼ 300 kg mol−1, from Sabic; high-density polyethylene (HDPE), Mw ∼ 500 kg mol−1, from Sabic; Stamylan UH, ultrahigh molecular weight polyethylene (UHMWPE), Mw ∼ 6.000 kg mol−1, from DSM. Millad NX8000, 1,2,3-trideoxy-4,6:5,7-bis-O-[(4-propylphenyl)methylene]-nonitol (TBPMN) from Milliken, di-(4-ethylbenzylidene)sorbitol (DEBS), di-(2,4-dimethylbenzylidene)sorbitol (D2,4-DMBS) and di-(3,4-dimethylbenzylidene)sorbitol (D3,4-DMBS) (own synthesis, see the ESI† 1) were used as gelators. The solvents used were 1,2,4-trichlorobenzene (TCB) ≥98% from AppliChem; benzene ≥99.7%, AnalaR NORMAPUR, from VWR; cyclohexane ≥99%, GPR RECTAPUR, from VWR. The PCMs used were 1-tetradecanole (TD), p.a., from Merck, paraffin wax from uncolored candles, and three polyethyleneglycoles (PEG), Polyglykol 2000S, 8000S, and 20000S from Clariant.2.2. Synthesis of organogelator-based aerogels and PCM-composite materials
Herein, the synthesis of PCM composites by flooding a separate aerogel support and the one-step synthesis are described.For the preparation of a polymer-based aerogel, defined amounts of polymer and gelator TBPMN (e.g. mass ratio 25:75 for iPP) are heated in a suitable high boiling solvent, e.g. TCB, under stirring, until a homogeneous solution is obtained. The stirring is stopped and the solution is cooled down to room temperature. During cooling, the gelator solidifies as fibrils first, followed by the crystallization of the polymer. Low concentrations of the polymer (≤0.5%) lead to a blueish shimmer caused by the gelator. At higher concentrations, it is covered by the white turbidity of the crystallizing polymer. Since freeze-drying from a high-boiling solvent is hardly possible, the solvent is exchanged to benzene. Therefore, the gel body is stored in five to ten times the amount of benzene for 16 to 20 hours on a shaker and then the solvent is refreshed. This is repeated at least three times. The freeze drying is done at −10 °C under vacuum (≤3 μbar). During this process, the gel body slightly shrinks (0–5 vol%). The obtained aerogel is then inserted into a molten PCM (at 90 °C) until mass constancy is reached. The PCM composite is freed from exceeding material and can now be used. Fig. 1 shows the structures of the preferred gelator used, an SEM image of the intermediate aerogel structure and a sketch of the cross-section of a support fibril.
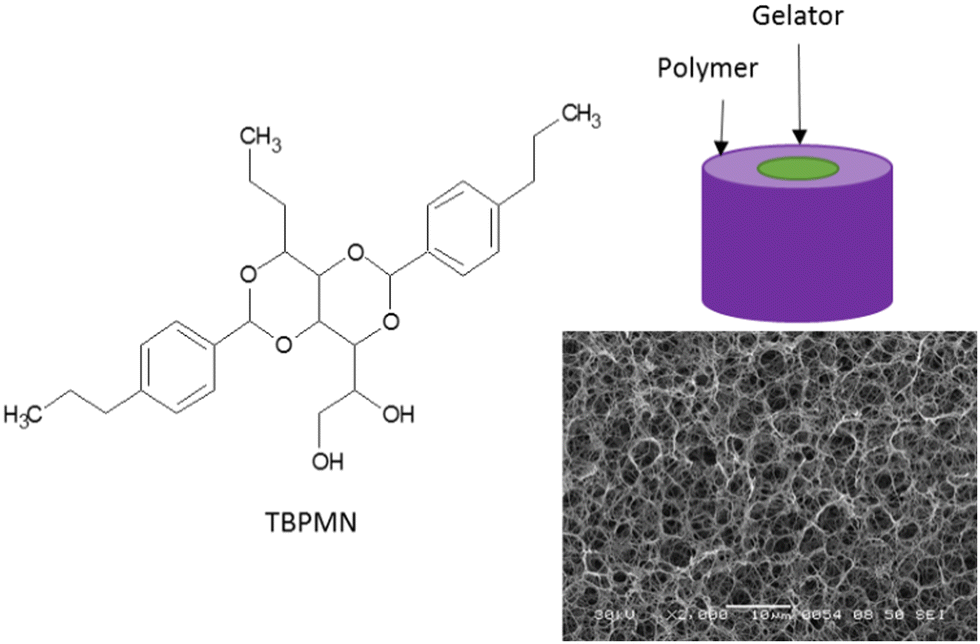 | ||
| Fig. 1 Gelator structure, schematic drawing of the cross-section of a polymer-stabilized gelator fibril and SEM image of a freeze-dried TBPMN/polyolefin sample. | ||
For the one-step synthesis, the polymer and the gelator are directly solved in the hot PCM. Compared to aerogel formulations, it is necessary to increase the ratio of polymer to gelator (e.g. mass ratio 40:60 for iPP:TBPMN). After cooling, the PCM composite is taken out of the synthesis pot under slight heating of the pot at the walls. The excess of PCMs is removed analogously to the process described above. Both manufacturing methods for aerogel-immobilized PCMs are illustrated in Fig. 2.
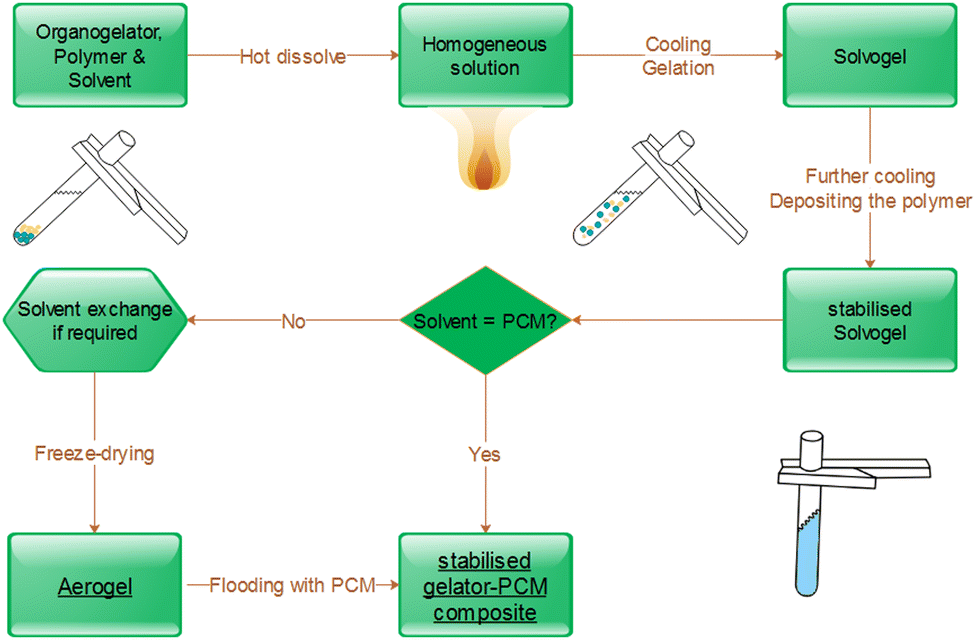 | ||
| Fig. 2 Manufacturing scheme for PCM composites. | ||
The direct path with PCM as a solvent is generally to be preferred, as this saves time, energy and material. The final samples can be cut into the desired shape and used according to their intended purpose. Furthermore, composites can be heated above the melting point of the PCM and their shape can easily be adjusted.
2.3. Characterization
The thermal behavior was analyzed with a Mettler Toledo DSC 822 and Netzsch DSC 214 Polyma (see the ESI† for Temperature program). The morphological structure of the aerogels was investigated with a JEOL JSM-6460LV scanning electron microscope (SEM) (30 kV; high vacuum mode). All samples were cryogenically fractured in liquid nitrogen and gold coated. The mass loss of the samples was determined in an oven at 95 °C. Fig. 3 outlines the experimental setup.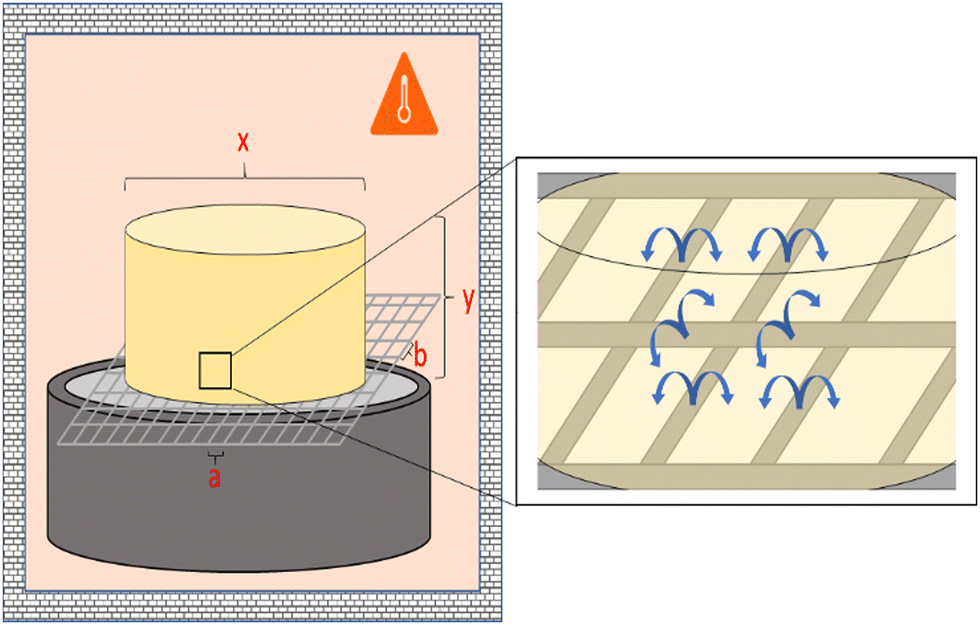 | ||
| Fig. 3 Sketch of the measurement setup for determining the mass loss over several melting/solidification cycles with the sample on a grid. | ||
The samples were placed on a grid. Different grids were used (for more details see the ESI† 2). All samples were heated for ≥1 hour per cycle, cooled to room temperature (RT) for another hour, and then weighed on an analytical balance (Nimbus NBL 164i). Cylindrical specimens with a diameter of 21 mm and an initial weight of 7.5 g were used.
To measure the rate of temperature increase in the material, samples were fabricated with a centered temperature sensor and placed in a heating block designed for this purpose. The temperature sensor was kept centered even when the sample was melted, using a 3D-printed holder installed above the setup. During the measurement, the temperatures of the heating block, the sample's cores and the room were recorded simultaneously via an Arduino microcontroller. The samples used were 20 g cylindrical specimens with a diameter of 27 mm.
3. Results and discussion
Besides the intrinsic latent heat storage capability of a PCM, a high loading capacity of the support and its retention capacity over multiple melting and freezing cycles are central requirements of immobilized PCM composites. Therefore, the compatibility of the PCM with the support material and an open-pore structure with very high porosity at sufficient mechanical stability are necessary. Additionally, a low density is ideal to make high mass-based loading ratios possible. It is feasible to synthesize aerogels with TBPMN as a gelator and iPP or UHMWPE in TCB with densities of 0.03 to 0.25 g cm−3.47 Paraffin as a PCM was infiltrated as described in Section 2.2 and very well retained in the composite material. Depending on the support density, mass ratios PCM:support of up to 25 could be obtained.Based on these results, the synthesis was done directly in the PCM in a single-step process. Although the structure of the support gets slightly rougher by the worse solvent (compared to TCB), PCM:support ratios of 39:1 with good retention capability are obtained. This can be attributed to the complete filling of the support structure, whereas when the aerogel is flooded, empty areas remain.
To prove that the network structure described in the previous work47 is also formed in paraffin as the solvent, different TBPMN/polyolefin samples were synthesized and then exchanged against cyclohexane via Soxhlet extraction.47 After subsequent freeze drying, the samples were analyzed by SEM. Fig. 4 shows the obtained aerogel structure of an extracted paraffin-derived sample compared to the structure obtained from TCB.
 | ||
| Fig. 4 SEM images of the support structure – left: 5% TBPMN/PP sample (60/40) obtained in paraffin after solvent exchange for cyclohexane and subsequent freeze-drying – right: 1.5% TBPMN/PP (75/25) aerogel (analogous to ref. 47). | ||
The structures are very similar. Because paraffin is not the ideal solvent for the gelator-polyolefin system, the structures are slightly coarser, but still mesoporous. This observation is in agreement with the findings in previous work,47 that using cyclohexane instead of aromatic solvents (like TCB) yielded coarser aerogels.
For the measurement of PCM loss from the composite material, the samples were placed on different grids in an oven and the weight loss over 20 melting/freezing cycles (RT to 95 °C to RT) was observed (cf. Section 2.3). It should be noted that no relevant change in mass could be detected when heating the sample just above the melting point, which is why the thermal stress on the specimens was increased by overheating to 95 °C. On the one hand, the higher temperature far above the melting point ensures a higher creeping capability of the PCM; on the other hand, this leads to a further volume increase of the PCM. If the support is too rigid, PCMs will be lost inevitably. Because there is no standardized method for the measurement of PCM loss, it was tested using different setups (see the ESI† 2). Consequently, only values from the same setup are comparable. The mass losses on the PTFE grid are higher than those on, for example, a PE grid. The more demanding experimental setup was chosen at this point to show the difference between the samples more clearly. Under milder testing conditions (other grid than PTFE, lower overheating), the mass loss is nearly zero for many samples. However, the fact that overheating of the specimens is necessary in order to observe significant changes in the material at an early stage of the test sequence with a good measurement setup underlines the good cycling stability and thus performance of the composites established.
UHMWPE and TBPMN individually show a stabilizing effect for the PCM; hence, a comparative analysis against the polymer/gelator composite material was performed. Here, it was possible to show that the mixture shows significantly better PCM retention behavior than the single components. In particular after long thermal stress, it was observed that the composites creep less and show a better dimensional stability. In Fig. 5, the mass loss of polymer, gelator and polymer/gelator composite PCM samples on a PTFE grid is shown. The composite (UHMWPE/TBPMN) shows significantly better retention properties than the individual components (UHMWPE or TBPMN).
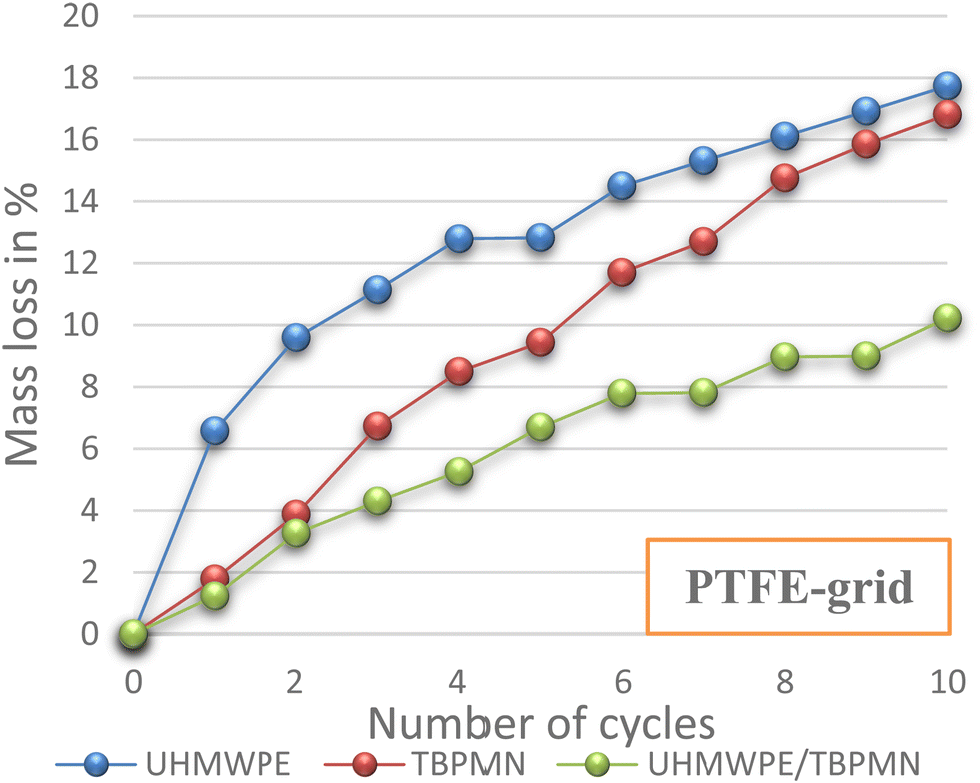 | ||
| Fig. 5 Mass loss of three different PCM samples (4 wt% support; composite 1:1 vs. 4 wt% individual components; PTFE grid). | ||
If HDPE or PP alone is used instead of the highly swellable UHMWPE, the samples already fail while the first melting. In contrast, gelator composites with PP or HDPE perform similarly to the UHMWPE-TBPMN composites.
Fig. 6 shows the mass loss of samples with different support fractions. The retention capability, presented as residual weight, initially increases with increasing weight fraction. Interestingly, an optimum is obtained at about 4 wt% of support material. Samples with more support show similar performance. This can possibly be explained by the structure of the support obtained in the PCM or by its lower flexibility when volume increases upon heating. The mass loss increments decrease with increasing cycle numbers; almost constant values are observed after 20 cycles. Exemplary measurements with 50 cycles and 24 h of thermal stress showed no further mass loss.
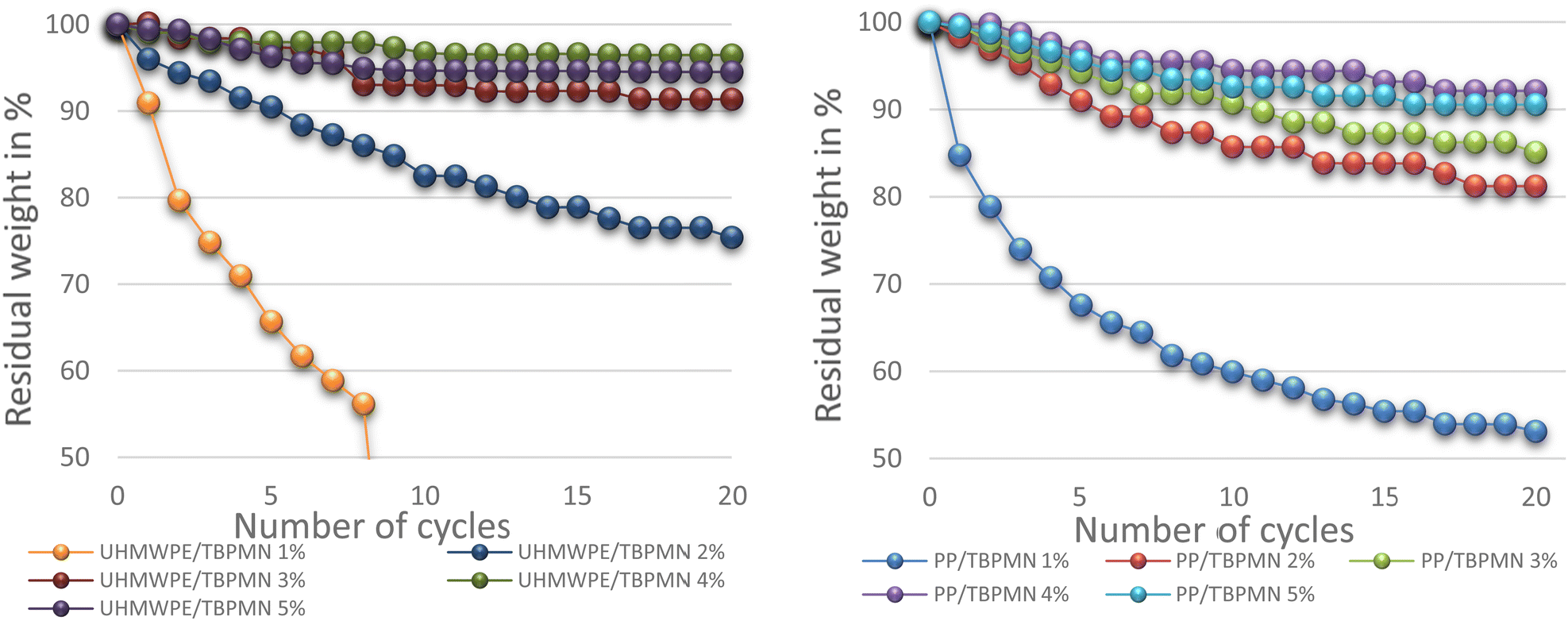 | ||
| Fig. 6 Influence of the support mass fraction on the retention capacity (polymer/gelator 1:1) (PE grid). | ||
The composition of the aerogels described in the previous paper47 does not necessarily represent the ideal one as PCM support; therefore, different formulations were evaluated. The requirement here is to obtain a stable and PCM-retaining network and not the finest porous structure possible, as it is required for thermal insulation applications. Experiments with different polymer/gelator ratios revealed that a significantly higher polymer content is advantageous compared to the aerogel mixtures; optima as PCM support were found for UHMWPE/TBPMN at 1:1 and PP/TBPMN at 2:3.
These interesting results have been verified with other systems. For the synthesis of the PCM composites, different sorbitol based gelators (themselves and UHMWPE stabilized) in the PCM materials paraffin, PEG (2 kg mol−1, 8 kg mol−1, 20 kg mol−1) and 1-tetradecanole were evaluated. The best and most interesting results were observed with the paraffin/TBPMN/polyolefin system already described above. We propose that the optimum support material results from several synergistic contributions: TBPMN finely distributes the material, the crystalline fraction of the polyolefin stabilizes the gelator network especially at high temperatures and the amorphous and swellable part of the polyolefin improves the retention of the PCM. Finally, the specific properties (molecular weight, surface tension, polarity, viscosity) of the PCM also influence the retention capability.
An overview of all results obtained with various gelators, polyolefins and PCM is provided in the ESI† 3. Already in the preparation step, it is obvious that nonpolar gelators, exactly as polyolefins, show a better phase compatibility to PEG with increasing molecular weight. TBPMN can stabilize PEG even in low concentrations without any losses. The higher creeping tendency of samples with a lower support content and without polymer is optically visible. Long thermal stress increases this effect. While in paraffin as the PCM, the composite of gelator and polymer brings enhanced properties, in PEG, the amount of the gelator has the dominating effect on stability. The shape stability can be viewed as exemplary material in Fig. 7.
 | ||
| Fig. 7 Shape stability after 20 melting/solidification cycles of PEG 8K samples with different gelators (2% composites; gelator/UHMWPE 70/30). | ||
While the PCM composites with PEG show very good retention properties (mass loss after 20 cycles is close to zero for many of the PEG-containing specimens), their mechanics in the molten state are less good compared to the paraffin-containing materials. Most of the PEG-containing composites tend to creep and will fail in the long term. The solvogel “smears” easily on surfaces and does not transfer forces throughout the specimen. This may be due to the relatively hard crystallization of the PEG, destroying potentially cellular parts of the gel network. In the solid state, PEG samples behave in a brittle, fragile manner. The opposite is observed with paraffin. In the solid state, the composite can be easily cut and shaped. When the wax melts, the space-filling solvogel network provides high flexibility without losing material. Fig. 8 shows a paraffin composite before and after melting.
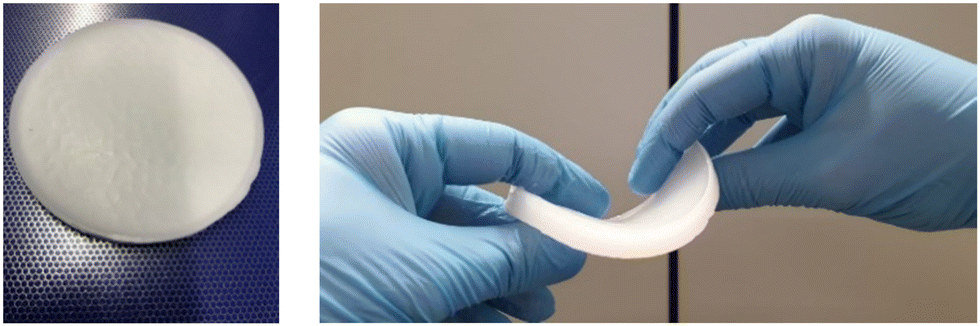 | ||
| Fig. 8 Paraffin gelator/polyolefin composite before and after melting. | ||
All the gelators show good retention properties for PEG with increasing molar mass, whereby the composites with higher form stability are preferred because of their longer lifetime. TBPMN is more suitable for paraffin than the other gelators. Tetradecanol, which can also be used as a PCM, has much lower performance in form stability tests, which was quite expectable. The gelators build up their network by π–π stacking and hydrogen bonds, which are disturbed by the hydroxyl groups of the long chain alcohol and the retention property is weakened. The stabilizing effect of the sorbitol derivatives for 1-tetradecanol reported in another study26 could not be demonstrated in connection with the here used overheating of samples.
The gel stability was further tested temperature dependent with a modified ball drop test. For this, different samples of 2.5 g weight were loaded by a 5 g heavy stamp and successively heated. Samples with TBPMN and UHMWPE or PP were analyzed. The region of softening was defined as the temperature range from the first penetration of the stamp into the material and the point where the stamp fell through the gel sample. In Table 1, these softening regions are shown.
| Sample | Softening temperature | Sample | Softening temperature |
|---|---|---|---|
| 0.075% UHMWPE | Melting of the wax (60 °C) | 0.375% PP | Melting of the wax (60 °C) |
| 1.5% TBPMN/UHMWPE (95/5) | 140–180 °C | 1.5% TBPMN/PP 3:1 |
140–180 °C |
| 3% TBPMN/UHMWPE (95/5) | 160–200 °C | 3% TBPMN/PP 3:1 |
160–190 °C |
| 3% TBPMN/UHMWPE (50/50) | 150–180 °C | 2% TBPMN/PP 3:2 |
150–190 °C |
| 1.425% TBPMN | 170–190 °C | 4% TBPMN/PP 3:2 |
180–190 °C |
The experiments confirm that higher gelator fractions have a positive effect on the temperature stability. The polymer has a smaller influence, at least until the weight is increased. When the stamp weight is six times, the sample mass material failure appears earlier. In this case, the polymer concentration gets a higher influence, because the failure happens near the polymer melting temperature in the case of UHMWPE. For the sample with 1.5% TBPMN/UHMWPE (95/5), the region of failure shifts to 90–120 °C. Increasing the polymer proportion to 75/25, the region of failure widens to 90–140 °C. The same effect is visible in PP-containing samples. It is important that the measurements were all performed with paraffin as the PCM. If, for example, a solvogel with TCB is measured, the values decrease by 30–40 °C caused by the better solvent properties of TCB.
Since the PCM is immobilized in the PCM composites, this affects the heat transport in the material. While non-stabilized PCMs quickly reach a uniform temperature after melting by convection in the liquid, this is not the case with the materials described here. To illustrate this effect, cylindrical samples were heated from the outside by using a heating block, and the temperature was measured in their center. The influence of the support structure was determined using TBPMN in paraffin at various concentrations and plotted in Fig. 9.
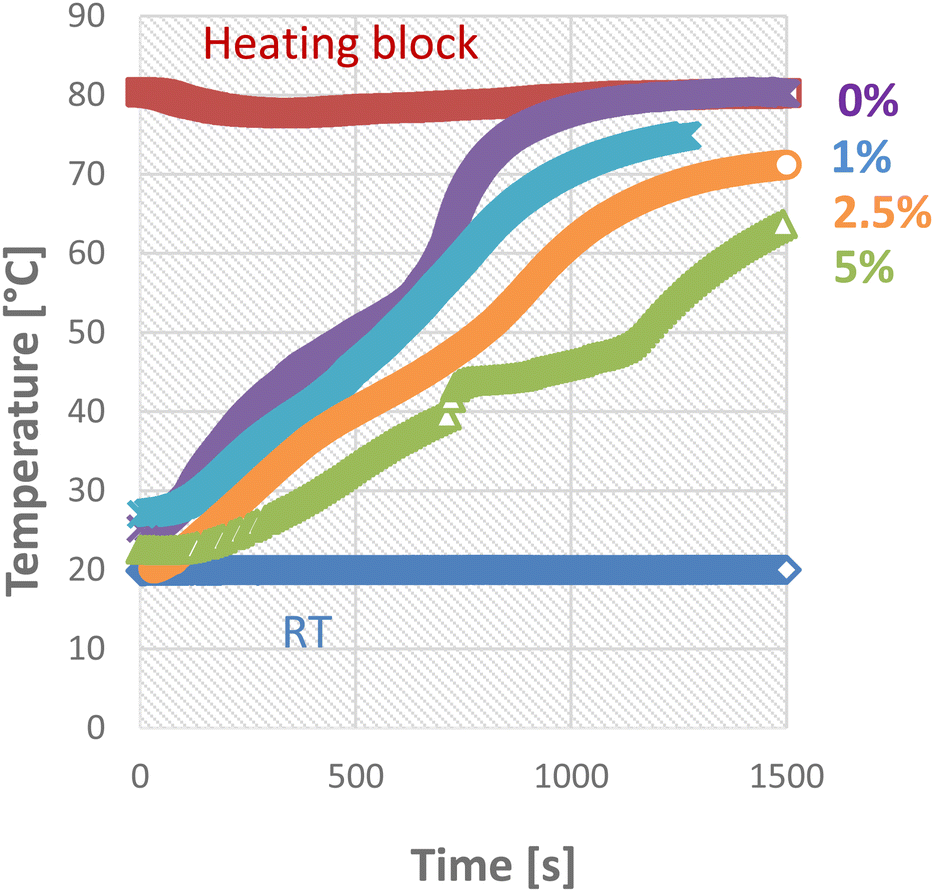 | ||
| Fig. 9 Melting behaviour of paraffin waxes stabilized with different amounts of TBPMN. | ||
In the case of pure paraffin, the change from solid to liquid state can be observed in the bending of the t (time) curve. Due to the gel network of the TBPMN, this effect disappears and the curve becomes largely linear. With an increasing fraction of the support structure, the heat transport slows down. This slower heat transport can possibly be a problem in applications where a lot of heat has to be dissipated in a short time. To counteract this, additives with high thermal conductivity such as carbon black or silver nanoparticles can be added.48–51
The same behavior can also be observed using DSC (see the ESI† 4). In addition to the broadening of the temperature range for melting and solidification processes, the DSC curves show the associated enthalpies. In Table 2, the obtained data of some selected materials are presented.
| Sample | Solidification process | Melting process | ||
|---|---|---|---|---|
| T s [°C] | ΔHs [J g−1] | T m [°C] | ΔHm [J g−1] | |
| Pure | 36.4 | 186.3 | 59.7 | 180.3 |
| 4% TBPMN | 36.7 | 184.1 | 66.3 | 182.0 |
| 4% TBPMN/UHMWPE (50/50) | 36.4 | 177.5 | 66.9 | 172.6 |
The PCM composites obtained have a high melting enthalpy, with PEG 8K composites between 156.9 and 166.7 J g−1, PEG 20K composites between 152.6 and 154.3 J g−1 and paraffin composites between 172.6 and 182.0 J g−1, in case of 4% support samples.
In Table 3, the loading rates achieved in this work are compared with references from the literature.
| Classical PCM support composites | ||
|---|---|---|
| Composite | Ref. | w% PCM (%) |
| a The PP aerogel/paraffin system is chemically and structurally the most comparable to the composites shown in this work. Despite the lower loading rate and more moderate melting conditions, a PCM loss of 9.7% after 10 cycles and 14.9% after 20 cycles was observed in that study. The gelator/polyolefin composite established in the present work, with <5% PCM loss after 20 cycles, clearly shows better performance (or comparable performance in the suboptimal test setup (PTFE grid)). b For uncoated expanded perlite (EPW), a mass loss of 48% was found, for hydrophobic coated expanded perlite (EPO) 0%. c In these works, the loss of mass after 24 h in the molten state was observed. Depending on the PCM, the mass loss varied widely. Samples with 3% 3,4-DMDBS lost 13.6% of their mass with n-octadecane and 36.3% with 1-octadecanol. Xu et al.27 describe a lower mass loss with 3,4-DMDBS and 1-octadecanol of ≤20% with 3% gelator. By adding 6% expanded graphite, the loss is reduced to 15.6%. This is also significantly worse than the results obtained in the present work. Long-term shape stability is not sufficiently given in all comparable works using organogelators. | ||
| EG/MA–PA–SA | 52 | 93 |
| EG/paraffin | 22, 53 | 92 |
| PP-aerogel/paraffin | 17 | 90–91 |
| Gypsum/C18–C24 | 54 | 18 |
| C-SiO2-PA | 20 | 61 |
| C-SiO2-OD | 20 | 73 |
| SiO2/PEG | 14 | 80 |
| HDPE/paraffin | 18 | 77 |
| EPO/paraffin | 55 | 50 |
| Diatomite/capric–lauric acid | 24 | 62 |
| Carbon/lauric acid | 56 | 33 |
| PCM-gelator-composit | ||
| G18/paraffin | 25 | 97 |
| 3,4-DMDBS/C18;C18-1ol | 26 | 97 |
| DMDBS/EG-OD | 27 | 83–97 |
| G18/C18 | 45 | 94 |
| This work | ||
| Isosorbide derivatives + UHMWPE/PEG | 95–98 | |
| Isosorbide derivatives + UHMWPE/paraffin | ||
The literature comparison shows that the composites prepared in this work are in the range of the loading rates of gelator-stabilized composites, but show significantly better properties, especially in the case of the TBPMN + UHMWPE/paraffin composites. Of course, there are alternative PCM composites such as PP aerogel and EG stabilized materials, but these are much more complex and cost-intensive to produce. The stabilization of the gel network, which was shown in a previous work,47 has a significant positive effect. This innovation gives the material a fundamental advantage over the established systems.
4. Conclusions
The most important accomplishment of this work is the massive simplification of the preparation step with parallel optimization of the product properties for the supported PCM composites. In particular, the separate production of porous support materials is time- and energy-consuming. For aerogels, the drying process with supercritical CO2 or freeze drying is the limiting step; therefore, these materials only have limited applications. Because with the one-step method established in the present work, the fine structuring of the support is realized already in the PCM itself, the tedious procedures needed in traditional fabrication can be completely omitted. We also propose a rationale for the synergistic behavior of gelators and polymers in the composite structure. In other studies, composites with gelators and polymers have already been used, but without the synergetic effect of these two, which leads to problems with retention property, loading capacity, shape and thermal stability. In particular with paraffin as PCM, the composite of gelator and polyolefin shows a particularly strong improvement. In contrast, PCM immobilized in either only a gelator or only polyolefin showed a strong tendency to creep during prolonged overheating, resulting in shape and material loss.In principle, all gelators that were studied are suitable for composite synthesis. However, the best results were obtained with the industrially established gelator TBPMN. The PCM loading rate of the supports can be set between 19:1 and 39:1, with a rate of 24:1 giving the best results, which in any case is extremely high. In this range, the amount of energy that can potentially be absorbed per mass of material is only marginally smaller compared with the plain PCM. In the case of a commercial application, the cost of the gelator is the decisive factor. The best composites obtained show a mass loss of 0–5% after 20 cycles. However, this usually occurs during the first few cycles, after which a mass constancy is observed; tests over 50 cycles and thermal stress over 24 h have confirmed the stability of the composites.
It can be assumed that such appropriately stabilized PCM composites have great potential for long-term and environmentally friendly energy savings or recycling. The composites themselves can be renewed by melting and can also be produced with recycled polyolefins. It is hoped that the knowledge gained in this work will help to provide solutions to mitigate problems caused by the climate and energy crisis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Milliken company for providing the gelator. We acknowledge support from the TC II lab at UDE and TMC lab at WHS. F. L. also gratefully acknowledges the Villigst foundation for a scholarship.References
- D. Ürge-Vorsatz, L. F. Cabeza, S. Serrano, C. Barreneche and K. Petrichenko, Heating and cooling energy trends and drivers in buildings, Renewable Sustainable Energy Rev., 2015, 41, 85–98 CrossRef.
- U. Bigalke, A. Armbruster, F. Lukas, O. Krieger, C. Schuch and J. Kunde, Der dena-Gebäudereport 2016, Statistiken und Analysen zur Energieeffizienz im Gebäudebestand, Berlin, 2016 Search PubMed.
- A. Kumar and B. M. Suman, Experimental evaluation of insulation materials for walls and roofs and their impact on indoor thermal comfort under composite climate, Build. Environ., 2013, 59, 635–643 CrossRef.
- M. Kenisarin and K. Mahkamov, Solar energy storage using phase change materials, Renewable Sustainable Energy Rev., 2007, 11, 1913–1965 CrossRef CAS.
- Q. Mao, H. Chen and Y. Yang, Energy Storage Performance of a PCM in the Solar Storage Tank, J. Therm. Sci., 2019, 28, 195–203 CrossRef.
- Y. Wang, B. Tang and S. Zhang, Single-Walled Carbon Nanotube/Phase Change Material Composites: Sunlight-Driven, Reversible, Form-Stable Phase Transitions for Solar Thermal Energy Storage, Adv. Funct. Mater., 2013, 23, 4354–4360 CrossRef CAS.
- J. Yang, G.-Q. Qi, L.-S. Tang, R.-Y. Bao, L. Bai, Z.-Y. Liu, W. Yang, B.-H. Xie and M.-B. Yang, Novel photodriven composite phase change materials with bioinspired modification of BN for solar-thermal energy conversion and storage, J. Mater. Chem. A, 2016, 4, 9625–9634 RSC.
- T. Inagaki and T. Ishida, Computational Design of Non-natural Sugar Alcohols to Increase Thermal Storage Density: Beyond Existing Organic Phase Change Materials, J. Am. Chem. Soc., 2016, 138, 11810–11819 CrossRef CAS PubMed.
- R. Kandasamy, X.-Q. Wang and A. S. Mujumdar, Application of phase change materials in thermal management of electronics, Appl. Therm. Eng., 2007, 27, 2822–2832 CrossRef.
- A. M. Khudhair and M. M. Farid, A review on energy conservation in building applications with thermal storage by latent heat using phase change materials, Energy Convers. Manage., 2004, 45, 263–275 CrossRef CAS.
- Y. Liu, Y. Yang and S. Li, Graphene oxide modified hydrate salt hydrogels: form-stable phase change materials for smart thermal management, J. Mater. Chem. A, 2016, 4, 18134–18143 RSC.
- A. R. Akhiani, M. Mehrali, S. Tahan Latibari, M. Mehrali, T. M. I. Mahlia, E. Sadeghinezhad and H. S. C. Metselaar, One-Step Preparation of Form-Stable Phase Change Material through Self-Assembly of Fatty Acid and Graphene, J. Phys. Chem. C, 2015, 119, 22787–22796 CrossRef CAS.
- X. Zhang, P. Deng, R. Feng and J. Song, Novel gelatinous shape-stabilized phase change materials with high heat storage density, Sol. Energy Mater. Sol. Cells, 2011, 95, 1213–1218 CrossRef CAS.
- H. Yang, L. Feng, C. Wang, W. Zhao and X. Li, Confinement effect of SiO2 framework on phase change of PEG in shape-stabilized PEG/SiO2 composites, Eur. Polym. J., 2012, 48, 803–810 CrossRef CAS.
- J. Yang, E. Zhang, X. Li, Y. Zhang, J. Qu and Z.-Z. Yu, Cellulose/graphene aerogel supported phase change composites with high thermal conductivity and good shape stability for thermal energy storage, Carbon, 2016, 98, 50–57 CrossRef CAS.
- Q. Guo and T. Wang, Influence of SiO2 pore structure on phase change enthalpy of shape-stabilized polyethylene glycol/silica composites, J. Mater. Sci., 2013, 48, 3716–3721 CrossRef CAS.
- H. Hong, Y. Pan, H. Sun, Z. Zhu, C. Ma, B. Wang, W. Liang, B. Yang and A. Li, Superwetting polypropylene aerogel supported form-stable phase change materials with extremely high organics loading and enhanced thermal conductivity, Sol. Energy Mater. Sol. Cells, 2018, 174, 307–313 CrossRef CAS.
- A. Sarı, Form-stable paraffin/high density polyethylene composites as solid–liquid phase change material for thermal energy storage: preparation and thermal properties, Energy Convers. Manage., 2004, 45, 2033–2042 CrossRef.
- J. Yang, X. Li, S. Han, Y. Zhang, P. Min, N. Koratkar and Z.-Z. Yu, Air-dried, high-density graphene hybrid aerogels for phase change composites with exceptional thermal conductivity and shape stability, J. Mater. Chem. A, 2016, 4, 18067–18074 RSC.
- X. Huang, Z. Liu, W. Xia, R. Zou and R. P. S. Han, Alkylated phase change composites for thermal energy storage based on surface-modified silica aerogels, J. Mater. Chem. A, 2015, 3, 1935–1940 RSC.
- L. Xia, P. Zhang and R. Z. Wang, Preparation and thermal characterization of expanded graphite/paraffin composite phase change material, Carbon, 2010, 48, 2538–2548 CrossRef CAS.
- Z. Zhang, N. Zhang, J. Peng, X. Fang, X. Gao and Y. Fang, Preparation and thermal energy storage properties of paraffin/expanded graphite composite phase change material, Appl. Energy, 2012, 91, 426–431 CrossRef CAS.
- B. Xu and Z. Li, Paraffin/diatomite composite phase change material incorporated cement-based composite for thermal energy storage, Appl. Energy, 2013, 105, 229–237 CrossRef CAS.
- M. Li, Z. Wu and H. Kao, Study on preparation and thermal properties of binary fatty acid/diatomite shape-stabilized phase change materials, Sol. Energy Mater. Sol. Cells, 2011, 95, 2412–2416 CrossRef CAS.
- D. Wu, W. Wen, S. Chen and H. Zhang, Preparation and properties of a novel form-stable phase change material based on a gelator, J. Mater. Chem. A, 2015, 3, 2589–2600 RSC.
- L. Niu, G. Bai and J. Song, 1,3:2,4-di-(3,4-dimethyl)benzylidene sorbitol organogels used as phase change materials: solvent effects on structure, leakage and thermal performance, RSC Adv., 2015, 5, 21733–21739 RSC.
- J. Xu, X. Cheng, Y. Li and G. Yu, Preparation and Properties of l-octadecanol/1,3:2,4-di-(3,4-dimethyl) Benzylidene Sorbitol/Expanded Graphite Form-stable Composite Phase Change Material, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2019, 34, 728–735 CrossRef CAS.
- D. Fan, Y. Cao, J. Liu, D. Xiong and T. Qian, Polynorbornene-based bottlebrush polymers confining phase change materials for ultra-stable latent heat storage derived from solar irradiation, Sol. Energy Mater. Sol. Cells, 2022, 236, 111547 CrossRef CAS.
- S. S. Kistler, Coherent Expanded-Aerogels, J. Phys. Chem., 1932, 36, 52–64 CrossRef CAS.
- S. Jiang, S. Agarwal and A. Greiner, Low-Density Open Cellular Sponges as Functional Materials, Angew. Chem., Int. Ed., 2017, 56, 15520–15538 CrossRef CAS PubMed.
- S. T. Nguyen, J. Feng, N. T. Le, A. T. T. Le, N. Hoang, V. B. C. Tan and H. M. Duong, Cellulose Aerogel from Paper Waste for Crude Oil Spill Cleaning, Ind. Eng. Chem. Res., 2013, 52, 18386–18391 CrossRef CAS.
- N. Guerrero-Alburquerque, S. Zhao, N. Adilien, M. M. Koebel, M. Lattuada and W. J. Malfait, Strong, Machinable, and Insulating Chitosan-Urea Aerogels: Toward Ambient Pressure Drying of Biopolymer Aerogel Monoliths, ACS Appl. Mater. Interfaces, 2020, 12, 22037–22049 CrossRef CAS PubMed.
- R. Brandt and S. Katalysierte, Unterkritisch getrocknete Resorcin-Formaldehyd-Aerogele und daraus abgeleitete Kohlenstoff-Aerogele, Bayerische Julius-Maximilians-Universität Würzburg, Würzburg, 2004 Search PubMed.
- M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Effects of Hydrogen Bonding and van der Waals Interactions on Organogelation Using Designed Low-Molecular-Weight Gelators and Gel Formation at Room Temperature, Langmuir, 2003, 19, 8622–8624 CrossRef CAS.
- S. Yamasaki, Y. Ohashi, H. Tsutsumi and K. Tsujii, The Aggregated Higher-Structure of 1,3:2,4-Di-O-benzylidene-D-sorbitol in Organic Gels, Bull. Chem. Soc. Jpn., 1995, 68, 146–151 CrossRef CAS.
- D. R. Trivedi and P. Dastidar, Instant Gelation of Various Organic Fluids Including Petrol at Room Temperature by a New Class of Supramolecular Gelators, Chem. Mater., 2006, 18, 1470–1478 CrossRef CAS.
- W.-C. Lai and Y.-C. Lee, Self-assembly behavior of gels composed of dibenzylidene sorbitol derivatives and poly(ethylene glycol), RSC Adv., 2016, 6, 98042–98051 RSC.
- K. Hanabusa and M. Suzuki, Development of low-molecular-weight gelators and polymer-based gelators, Polym. J., 2014, 46, 776–782 CrossRef CAS.
- M. Suzuki, H. Saito and K. Hanabusa, Two-component organogelators based on two L-amino acids: effect of combination of L-lysine with various L-amino acids on organogelation behavior, Langmuir, 2009, 25, 8579–8585 CrossRef CAS PubMed.
- K. K. Diehn, H. Oh, R. Hashemipour, R. G. Weiss and S. R. Raghavan, Insights into organogelation and its kinetics from Hansen solubility parameters. Toward a priori predictions of molecular gelation, Soft Matter, 2014, 10, 2632–2640 RSC.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, 1,3:2,4-Dibenzylidene-D-sorbitol (DBS) and its derivatives--efficient, versatile and industrially-relevant low-molecular-weight gelators with over 100 years of history and a bright future, Soft Matter, 2015, 11, 4768–4787 RSC.
- K. Bernland, T. Tervoort and P. Smith, Phase behavior and optical- and mechanical properties of the binary system isotactic polypropylene and the nucleating/clarifying agent 1,2,3-trideoxy-4,6:5,7-bis-O-[(4-propylphenyl) methylene]-nonitol, Polymer, 2009, 50, 2460–2464 CrossRef CAS.
- C. Lindqvist, J. Bergqvist, C.-C. Feng, S. Gustafsson, O. Bäcke, N. D. Treat, C. Bounioux, P. Henriksson, R. Kroon, E. Wang, A. Sanz-Velasco, P. M. Kristiansen, N. Stingelin, E. Olsson, O. Inganäs, M. R. Andersson and C. Müller, Fullerene Nucleating Agents: A Route Towards Thermally Stable Photovoltaic Blends, Adv. Energy Mater., 2014, 4, 1301437 CrossRef.
- F. Abraham, R. Kress, P. Smith and H.-W. Schmidt, A New Class of Ultra-Efficient Supramolecular Nucleating Agents for Isotactic Polypropylene, Macromol. Chem. Phys., 2013, 214, 17–24 CrossRef CAS.
- Z. Liu, L. Jiang, X. Fu, J. Zhang and J. Lei, Preparation and characterization of n-octadecane-based reversible gel as form-stable phase change materials for thermal energy storage, J. Therm. Anal. Calorim., 2020, 140, 2159–2170 CrossRef CAS.
- B. Jamart-Grégoire, S. Son, F. Allix, V. Felix, D. Barth, Y. Jannot, G. Pickaert and A. Degiovanni, Monolithic organic aerogels derived from single amino-acid based supramolecular gels: physical and thermal properties, RSC Adv., 2016, 6, 102198–102205 RSC.
- F. Leven, M. Ulbricht, J. Limberg and R. Ostermann, Novel finely structured polymer aerogels using organogelators as a structure-directing component, J. Mater. Chem. A, 2021, 9, 20695–20702 RSC.
- W. Liang, L. Wang, Z. Zhu, C. Qian, H. Sun, B. Yang and A. Li, In Situ Preparation of Polyethylene Glycol/Silver Nanoparticles Composite Phase Change Materials with Enhanced Thermal Conductivity, ChemistrySelect, 2017, 2, 3428–3436 CrossRef CAS.
- H. Zhang, Q. Sun, Y. Yuan, Z. Zhang and X. Cao, A novel form-stable phase change composite with excellent thermal and electrical conductivities, Chem. Eng. J., 2018, 336, 342–351 CrossRef CAS.
- L. Xia, P. Zhang and R. Z. Wang, Preparation and thermal characterization of expanded graphite/paraffin composite phase change material, Carbon, 2010, 48, 2538–2548 CrossRef CAS.
- M. Xiao, B. Feng and K. Gong, Thermal performance of a high conductive shape-stabilized thermal storage material, Sol. Energy Mater. Sol. Cells, 2001, 69, 293–296 CrossRef CAS.
- X. Yang, Y. Yuan, N. Zhang, X. Cao and C. Liu, Preparation and properties of myristic–palmitic–stearic acid/expanded graphite composites as phase change materials for energy storage, Sol. Energy, 2014, 99, 259–266 CrossRef CAS.
- A. Sarı and A. Karaipekli, Thermal conductivity and latent heat thermal energy storage characteristics of paraffin/expanded graphite composite as phase change material, Appl. Therm. Eng., 2007, 27, 1271–1277 CrossRef.
- A. Karaipekli, A. Sarı and A. Biçer, Thermal regulating performance of gypsum/(C18–C24) composite phase change material (CPCM) for building energy storage applications, Appl. Therm. Eng., 2016, 107, 55–62 CrossRef CAS.
- S. Ramakrishnan, J. Sanjayan, X. Wang, M. Alam and J. Wilson, A novel paraffin/expanded perlite composite phase change material for prevention of PCM leakage in cementitious composites, Appl. Energy, 2015, 157, 85–94 CrossRef CAS.
- Z. Chen, F. Shan, L. Cao and G. Fang, Synthesis and thermal properties of shape-stabilized lauric acid/activated carbon composites as phase change materials for thermal energy storage, Sol. Energy Mater. Sol. Cells, 2012, 102, 131–136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00578f |
| This journal is © The Royal Society of Chemistry 2022 |