
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of thin solid electrolytes containing a small volume of an Li3OCl-type antiperovskite phase by RF magnetron sputtering†
Stephen J.
Turrell
 *ab,
Hyeon Jeong
Lee
abc,
Marco
Siniscalchi
ab,
Sudarshan
Narayanan
ab,
Mauro
Pasta
ab,
Susannah C.
Speller
a and
Chris R. M.
Grovenor
ab
*ab,
Hyeon Jeong
Lee
abc,
Marco
Siniscalchi
ab,
Sudarshan
Narayanan
ab,
Mauro
Pasta
ab,
Susannah C.
Speller
a and
Chris R. M.
Grovenor
ab
aDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: stephen.turrell@materials.ox.ac.uk
bThe Faraday Institution, Harwell Campus, Quad One, Becquerel Avenue, Didcot OX11 0RA, UK
cDivision of Chemical Engineering and Bioengineering, Kangwon National University, 1 Kangwondaehak-gil, Chuncheon 24341, Republic of Korea
First published on 31st October 2022
Abstract
Several attempts to synthesize Li3OCl – a lithium-rich antiperovskite compound envisaged as a potential solid electrolyte material for lithium metal batteries – have been reported, but few have yielded convincing results. There are two key challenges associated with this synthesis: the thermodynamic instability of Li3OCl at room temperature and its extreme hygroscopicity. Therefore, the likelihood of inadvertently forming the structurally similar thermodynamically stable hydroxide halide compound Li2OHCl is very high. In this report, we demonstrate the stabilization of a small volume fraction of antiperovskite phase with the characteristics expected for Li3OCl in ∼0.5 to ∼1 μm films fabricated from a Li2O + LiCl powder target by RF magnetron sputtering. Measures were taken to minimize the presence of moisture at all stages of synthesis and characterization. X-ray diffraction (XRD) experiments showed that reaction between the precursor phases occurred within the growing films to form a volume of antiperovskite phase with an identical lattice parameter to that predicted for cubic Li3OCl. This antiperovskite phase decomposed into Li2O and LiCl upon annealing at moderate temperatures. Characterization by Fourier transform infrared spectroscopy (FT-IR) confirmed the absence of O–H bonding in the films, providing further evidence that the antiperovskite phase was Li3OCl rather than Li2OHCl. Deposition of films with similar thicknesses from an Li2OHCl powder target was also performed for comparison. While FT-IR results showed that O–H bonding was present in these films, a small volume fraction of an antiperovskite phase with identical lattice parameter to Li2OHCl was only detected after heating the films to ∼100 °C. Owing to the low phase purities of films deposited from both target types, the Li+ conductivities were found to be on the order of 10−8 S cm−1. For Li2OHCl in particular, it is expected that further optimization of the processing conditions will lead to a significant increase in Li+ conductivity. This is the first reported attempt to synthesize lithium-rich antiperovskite compounds by RF magnetron sputtering.
1. Introduction
Significant efforts are underway to develop high-performance all-solid-state batteries (ASSBs) as successors to the commercially successful lithium-ion batteries ubiquitous in portable electronic devices and electric vehicles.1 Currently, lithium-ion cells suffer from safety and performance limitations due to the flammable liquid electrolyte, which has a narrow electrochemical stability window.2 Replacing the liquid electrolyte with a non-flammable solid lithium-ion conductor could enable the safe use of lithium metal anodes and high voltage cathodes, leading to improvements in energy density, charge capacity, cycle life and high temperature stability.3–7 Although some solid electrolytes possess Li+ conductivities on a par with – or in some cases higher than – those of liquid electrolytes,8,9 these high-conductivity electrolytes tend to be unstable in contact with the lithium metal anode and have narrow electrochemical stability windows.10–12 Furthermore, most solid electrolyte materials investigated to date are unable to block the growth of lithium metal filaments from the anode to the cathode during cycling at the rates required by applications, resulting in short-circuiting and hence cell failure at relatively low current densities.13–15 Solid electrolyte materials that can overcome these challenges must be found.Lithium-rich antiperovskite (LiRAP) compounds based on Li3OA oxyhalide, where A is a halogen or mixture of halogens, have attracted considerable attention since the first reports of promising lithium-ion conductivity in Li3OCl by Reckeweg et al.19 and Zhao and Daemen20 a decade ago. These compounds are predicted to have a cubic crystal structure (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group), as shown in Fig. 1(a) for Li3OCl.21 Computational investigations into Li3OCl have revealed that this material – and certain off-stoichiometric derivatives – may be well-suited to application as solid electrolytes.22–26 The closely related hydroxide halide antiperovskite Li2OHCl (Fig. 1(b)) has already shown promising performance in several experimental studies.7,16,17,27–31 Further, Braga and co-workers have published several studies on glassy electrolytes derived from Li3OCl which seem to show very high Li+ conductivity and some unusual, but advantageous, electrical properties.32–38 Here we are concerned with crystalline Li3OCl since it has been studied more widely as part of the growing research effort in the field of antiperovskite solid electrolytes.
m space group), as shown in Fig. 1(a) for Li3OCl.21 Computational investigations into Li3OCl have revealed that this material – and certain off-stoichiometric derivatives – may be well-suited to application as solid electrolytes.22–26 The closely related hydroxide halide antiperovskite Li2OHCl (Fig. 1(b)) has already shown promising performance in several experimental studies.7,16,17,27–31 Further, Braga and co-workers have published several studies on glassy electrolytes derived from Li3OCl which seem to show very high Li+ conductivity and some unusual, but advantageous, electrical properties.32–38 Here we are concerned with crystalline Li3OCl since it has been studied more widely as part of the growing research effort in the field of antiperovskite solid electrolytes.
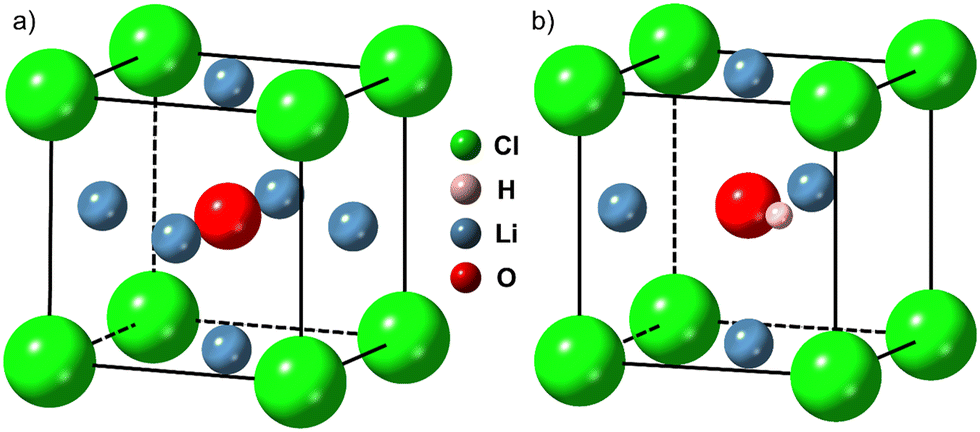 | ||
| Fig. 1 Crystallographic structures of (a) Li3OCl (Pmm) and (b) Li2OHCl (Pmc21 below ∼32 °C, Pmm above).7,16–18 Generated using CrystalMaker®: CrystalMaker Software Ltd, Oxford, England (https://www.crystalmaker.com). | ||
Among the relatively few experimental studies on Li3OCl, values of ionic conductivity between 1.26 × 10−7 S cm−1 and 8.5 × 10−4 S cm−1 have been reported.20,39–42 This wide range has been attributed to differences in composition and microstructure.43,44 Indeed, several computational studies have examined the roles of crystallographic defects – in particular Li+ vacancies and interstitials – in facilitating Li+ conduction in Li3OCl.15,21–24,45–50 These defects can be created by adjusting the ratio of Li2O and LiCl precursors used in synthesis or by adding aliovalent dopant ions such as Mg2+. A band gap of 3.7–4.4 eV44 has been predicted for grain boundaries in Li3OCl compared to the 1–3 eV band gap measured for 50% of grain boundaries in Li7La3Zr2O12;51 in both cases the grain boundary band gap is thought to be smaller than that of the bulk material, limiting the electrochemical stability of polycrystalline samples. The wider band gap predicted for Li3OCl may imply a lower electronic conductivity, which would help to limit self-discharge in ASSBs and could hinder the growth of lithium filaments during cycling,13 increasing the attainable power density and cycle life. Furthermore, several investigations have indicated that Li3OCl exhibits good stability in contact with lithium metal.25,40–42,52,53 This is readily understood by considering that, in contrast to most other solid electrolyte materials, cations reducible by lithium metal are absent.10 In addition to promising electrochemical properties, Li3OCl has a low raw materials cost, a low density and is non-toxic.32
Recently, growing scepticism18,27,54,55 has been directed towards the few published experimental studies on Li3OCl. This is a result of an increasing awareness of the synthesis challenges presented by this compound, with one recent review even raising the possibility that these challenges are insurmountable.54 The synthesis of Li3OCl is predicted to be difficult for two reasons. First, Li3OCl is thermodynamically unstable at room temperature, with a driving force for decomposition into Li2O and LiCl.15,22,23,46,47,56–58 Indeed, Mouta et al.23 calculated a negative Gibbs free energy change for this decomposition reaction in the 0 to 550 K temperature range, with values of −0.6785 eV at 0 K and −0.6740 eV at 550 K. Nevertheless, some studies propose that after synthesis at elevated temperatures it is possible to “freeze” the structure in a metastable state on cooling to room temperature owing to the kinetically limited decomposition reaction.26,59 The second issue is that Li3OCl is extremely hygroscopic: even if it is attained in a metastable state, it readily hydrates and reacts to form other materials viaeqn (1.1).57,58,60 Indeed, Dawson et al.60 explicitly calculated the hydration enthalpy of this compound using density functional theory and found it to be exothermic.
| Li3OCl + H2O → Li2(OH)Cl + LiOH | (1.1) |
| Li2O + LiCl → Li3OCl | (1.2) |
| 2LiOH + LiCl → Li3OCl + H2O | (1.3) |
| LiOH + LiCl → Li2OHCl | (1.4) |
Physical vapour deposition (PVD) techniques such as radio frequency (RF) magnetron sputtering and pulsed laser deposition (PLD) could be used to overcome the synthesis challenges encountered in the conventional ceramic processing routes. These processes create plasmas in which intimate mixing of the sputtered components occurs at the atomic level.64–68 This mixing could remove the diffusion limitation and significantly increase the extent of reaction between Li2O and LiCl. Furthermore, these deposition techniques are performed in vacuum chambers free of moisture, preventing hydration during synthesis.26 Although these techniques are limited to the fabrication of thin films (∼10 nm to ∼10 μm), thin Li3OCl layers could find application as the solid electrolyte in miniature thin-film cells and as electrode–electrolyte interlayers in bulk cells.42,61,69
The use of PLD to deposit Li3OCl thin films from a pellet target composed of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of Li2O and LiCl was reported previously,40 but no evidence for the absence of O–H bonding in the synthesized material was provided. Advantages of RF magnetron sputtering over PLD include the ability to use an unpressed and unsintered mixture of the precursor powders as the target, which simplifies processing and allows continual adjustment of the precursor ratio, and easier deployment of the process at an industrial scale.64,65,67,70,71 In this investigation we developed a radio frequency (RF) magnetron sputtering process to deposit thin film electrolytes containing a quantity of antiperovskite phase from mixtures of Li2O and LiCl powders, taking rigorous precautions to avoid exposure to moisture at all stages of synthesis and characterization. Deposition of films from Li2OHCl powder was also performed for comparison. Characterization was carried out using instruments which were either housed within Ar-filled gloveboxes or had air-free sample transfer systems. The precautions taken to avoid air exposure combined with careful and relevant characterization give us confidence that we have succeeded in fabricating thin films containing a quantity of oxyhalide (OH-free) LiRAP phase with a composition close to Li3OCl.
:1 molar mixture of Li2O and LiCl was reported previously,40 but no evidence for the absence of O–H bonding in the synthesized material was provided. Advantages of RF magnetron sputtering over PLD include the ability to use an unpressed and unsintered mixture of the precursor powders as the target, which simplifies processing and allows continual adjustment of the precursor ratio, and easier deployment of the process at an industrial scale.64,65,67,70,71 In this investigation we developed a radio frequency (RF) magnetron sputtering process to deposit thin film electrolytes containing a quantity of antiperovskite phase from mixtures of Li2O and LiCl powders, taking rigorous precautions to avoid exposure to moisture at all stages of synthesis and characterization. Deposition of films from Li2OHCl powder was also performed for comparison. Characterization was carried out using instruments which were either housed within Ar-filled gloveboxes or had air-free sample transfer systems. The precautions taken to avoid air exposure combined with careful and relevant characterization give us confidence that we have succeeded in fabricating thin films containing a quantity of oxyhalide (OH-free) LiRAP phase with a composition close to Li3OCl.
2. Experimental
2.1. Preparation of substrates and sputtering targets
Soda-lime glass, silicon, stainless steel (Fe–18Cr–8Ni [wt%], 304 stainless steel, Goodfellow Cambridge Ltd) and Waspaloy (58Ni–19Cr–14Co–Mo–Ti–Al–Fe [wt%], HAYNES® 282® alloy, Goodfellow Cambridge Ltd) substates were used for film deposition. The glass, silicon and alloys were cut from microscope slides, undoped wafers and 250 μm sheets, respectively, into approximately 10 mm × 10 mm square pieces. The alloy substrates were ground and polished, finishing on a 3 μm grade diamond suspension to achieve a surface free of visible scratches. All substrates were cleaned by sonication in distilled water and isopropanol prior to film deposition. Pt and Ni layers were sputter deposited on some of the glass substrates; these substrates were used to create layered glass/metal/electrolyte/metal samples for electrochemical impedance spectroscopy (EIS) measurements.The design of the sputtering target used to attempt Li3OCl synthesis was similar to that used by Lü et al.40 for the fabrication of “Li3OCl” by PLD, except that in the present investigation the powder was not pressed into a pellet. All stages of target preparation were performed inside an argon-filled glovebox (MBraun) with O2 and H2O concentrations below 0.1 ppm to avoid hydration of the reactants. To produce each target, 1.502 g of Li2O (99.5% purity, Sigma Aldrich) and 2.141 g of LiCl (≥99.0% purity, Sigma Aldrich) powders were mixed by grinding with an agate pestle and mortar for approximately 10 minutes. The powder mixture and the substrates and utensils required for preparing the target were dried in the heated antechamber of the glovebox under vacuum (∼1 mbar) at 80 °C for at least 2 hours to remove any residual moisture. The powder was then poured into a 2′′ diameter, ∼2 mm deep circular copper holder (Fig. S1a, ESI†) and lightly pressed and levelled using a glass microscope slide.
The target used to attempt Li2OHCl synthesis was prepared from Li2OHCl powder. To obtain Li2OHCl, 1.935 g of LiOH (≥99.0%, Sigma-Aldrich) and 3.426 g of LiCl (≥99.0% purity, Sigma Aldrich) were ground and heated at 350 °C for 30 minutes with a ramp rate of 5 °C min−1. The liquid state Li2OHCl was immediately cooled to room temperature and the resulting solid mass was ground with a mortar and pestle to produce Li2OHCl powder. A shallower (∼1 mm deep) holder was used for this target to improve thermal conduction and avoid melting of the Li2OHCl, which has a relatively low melting point (∼300 °C).16
2.2. Thin film deposition and post-deposition annealing
A PVD system (MB EVAP, MBraun) installed within a glovebox of the type described in Section 2.1 was used for the RF magnetron sputter deposition of metal and electrolyte thin films. This system contained a circular magnetron source (Gencoa Ltd), with a target-to-substrate distance of ∼12 cm as shown in Fig. S2 (ESI†). Substrate heating was not employed. The chamber was pumped down to achieve a base pressure below 5 × 10−5 mbar or 1 × 10−6 mbar for metal and electrolyte deposition, respectively.Pre-deposition sputtering was performed (with the substrates shielded) for approximately 30 minutes prior to the start of each deposition from an Li2O + LiCl target to remove contaminants from the surface. Pre-deposition sputtering was not performed for the Li2OHCl target due to uncertainty over the length of time the target would remain stable during sputtering. Post-deposition annealing at temperatures between 100 °C and 350 °C for 1 hour was performed on certain samples to see the effect on the film crystallinity and phase properties. A muffle furnace (KSL-1100X, MTI Corporation) located within one of the gloveboxes was used for this purpose. A controlled heating rate of 5 °C min−1 was used; after annealing, the samples were allowed to cool within the furnace at its natural rate to avoid the development of excessive stresses, which could lead to film cracking and delamination. The samples were held in an alumina crucible during annealing.
Due to the absence of prior reports on the deposition of LiRAP thin films by RF magnetron sputtering, a suitable set of processing conditions had to be developed from scratch. Initially the focus was on optimizing the sputtering process to achieve target stability, since even moderate values of RF power and process pressure led to target melting or excessive cracking. After stabilizing the Li2O + LiCl target, the fabrication conditions were optimized further to promote development of the desired antiperovskite structure within the films, as determined by XRD analysis. Similar conditions were also used with the Li2OHCl target. The conditions used with both target types are listed in Table 1. After processing, all samples were stored within the glovebox prior to characterization.
| Condition | Value | |
|---|---|---|
| Li2O + LiCl target | Li2OHCl target | |
| a The pressure fluctuated between these values. b BOC Pureshield argon (99.998% purity) and BOC N2.6 Zero Grade O2 (99.6%) purity were used. c Films deposited on alloy substrates were found to be very rough; the longer deposition time was used in an attempt to avoid pinholes. d The targeted duration was 12 hours, but the plasma became unstable leading to early cessation of the deposition. | ||
| Process pressure | 1–2 × 10−3 mbara | |
| Gases and flow rates | 8 sccm Ar, 8 sccm O2b | 5 sccm Arb |
| RF power | 20 W | 22 W |
| Deposition duration | 12 hours (glass and Si substrates); 24 hours (alloy substrates)c | ∼7 hours 15 mind |
2.3. Structural characterization
The crystallographic properties of the electrolyte films and powder from the sputtering targets were characterized using a compact X-ray diffractometer (Rigaku Miniflex with Cu K-alpha radiation and a θ–2θ geometry) located inside a glovebox with O2 and H2O concentrations below 1 ppm. Diffraction patterns were viewed in CrystalDiffract software (CrystalMaker Software Ltd) and indexed by comparison with reference powder patterns17,30,72–74 from the Inorganic Crystal Structure Database. Lattice parameter values were calculated from all clearly defined peaks for each phase.Scanning electron microscopy (SEM) was used to observe the electrolyte film morphologies in surface and cross-section views, measure film thicknesses and verify the dimensions of the electrical contacts on the samples used for impedance measurements. The samples were sputter-coated with ∼30 nm of Pt prior to examination to reduce charging. A plasma focused ion beam (PFIB) instrument (Thermo Scientific Helios G4 PFIB CXe DualBeam) with SEM column was used for secondary electron imaging and milling trenches to enable examination of the cross-sections of films deposited from Li2O + LiCl targets. Pt strap layers were deposited within the PFIB at locations selected for milling to protect the underlying film from damage by the xenon ion beam. A Zeiss Merlin SEM was used for secondary electron imaging of the films deposited from an Li2OHCl target; these samples were cross-sectioned by scoring their reverse side with a diamond scribe and fracturing them cleanly into two pieces.
2.4. Chemical characterization
The approximate chemical compositions of certain films deposited from Li2O + LiCl targets were determined by performing energy-dispersive X-ray spectroscopy (EDX) measurements on film cross-sections within the PFIB instrument at an accelerating voltage of 5 kV. An Oxford Instruments Ultim Max 170 X-ray detector was used for this purpose. The presence of lithium in these films was verified by X-ray photoelectron spectroscopy (XPS) using a PHI VersaProbe III instrument with an Al K-alpha source. Argon ion beam etching through the surface of each sample was carried out at 4 kV for 40 minutes prior to measuring the spectra to remove surface impurities. CasaXPS software (Casa Software Ltd) was used to perform energy calibration of the spectra using the adventitious carbon peak at 284.8 eV.Fourier transform infrared spectroscopy (FT-IR) was performed in a Thermo Scientific Nicolet iS10 instrument with a single reflection diamond attenuated total reflectance (ATR) module to detect the presence of O–H bonding in the films. The film side of each sample was placed facing the laser beam.
2.5. Electrical conductivity measurements
Electrochemical impedance spectroscopy (EIS) was used to measure the through-plane impedance of glass/metal/electrolyte/metal layered samples, where the electrolyte layer was a film deposited from an Li2O + LiCl or Li2OHCl target. Prior to the deposition of certain layers, shadow masks were applied to the samples to create the required layer shapes and areas. To reduce the risk of short circuiting, the overlap area between the top and bottom contacts was made as small as practically possible, minimizing the likelihood of encountering a pinhole in the electrolyte film. For the films deposited from Li2O + LiCl, this was achieved by depositing the metal layer over the full area of the glass substrate, masking off a narrow border with Kapton tape, depositing the electrolyte layer and then depositing ∼1 mm diameter circular metal top contacts spaced apart by several millimetres using a drilled stainless steel sheet as a shadow mask.A different technique inspired by Yu et al.75 was employed for the films deposited from Li2OHCl. Laser cut ‘single slot’ Kapton film masks (produced by Laser Micromachining Limited) were used to create 8 mm × 1 mm metal strips for the top and bottom contacts. These contacts were oriented at ∼90° to each other so that the area of overlap was ∼1 mm2. Hole-punched aluminium foil was applied after bottom contact deposition to mask an area for the electrolyte layer deposition, ensuring that the ends of the bottom contact remained accessible for electrical connection. Photographs of these masks and the stages of sample fabrication are shown in Fig. S3 (ESI†). This technique was also attempted for films deposited from Li2O + LiCl targets, but all of these samples failed (short-circuited or showed no conductivity).
To prevent air exposure during EIS measurements, each sample was inserted into a laminated aluminium pouch, connected to copper foil electrodes and sealed under vacuum within a glovebox. Standard materials and methods for laboratory scale pouch cell construction were used. Each sample was moved in turn from the glovebox to a temperature-controlled climatic chamber connected to an impedance analyser (ITS and MTZ-35, BioLogic Science Instruments) for measurements of impedance at temperatures between 25 °C and 90/100 °C using a voltage amplitude of 10 mV and a frequency range of 3.5 MHz to 0.1 Hz. Equivalent circuit models were fitted to the experimental impedance data using ZView software (Scribner Associates Inc.) to determine the total ionic resistance of the electrolyte layer.
3. Results and discussion
3.1. Stability of the sputtering targets
Sputtering the Li2O + LiCl target caused it to transform from a loose powder to a sintered mass that could be removed from the copper holder in one piece, as shown in Fig. S1 (ESI†). The onset of sintering was attributed to the low melting temperature of LiCl (605 °C), since even moderate target heating could achieve the homologous temperature required for an appreciable rate of sintering.Reaction between LiCl and Li2O powders was ruled out by performing XRD on powdered samples from the Li2O + LiCl target before and after sputtering, as shown in Fig. S1(g), ESI.† Detectable quantities of new phases were not formed during sputtering; the patterns differ only in terms of the relative peak intensities, which could be attributed to a combination of changes in grain orientations and chemical composition. The LiCl peaks from the top surface region of the target have higher intensities than those from the bottom surface region, while the opposite is true for the Li2O peaks. This indicates that the upper region of the target became enriched in LiCl during sputtering, possibly as a result of relatively low density molten LiCl rising to the target surface.76,77
The reasonable stability of the target during sputtering facilitated a steady, repeatable deposition process. Even though similar sputtering conditions were used for the Li2OHCl target, a combination of sintering and partial melting seemed to occur, which eventually caused holes to form (Fig. S4, ESI†). This was likely responsible for the plasma becoming unstable 7¼ hours into the 12 hour deposition. It was not possible to use milder sputtering conditions than those in Table 1 since the plasma extinguished shortly after ignition when this was tried.
3.2. Deposition from Li2O + LiCl targets
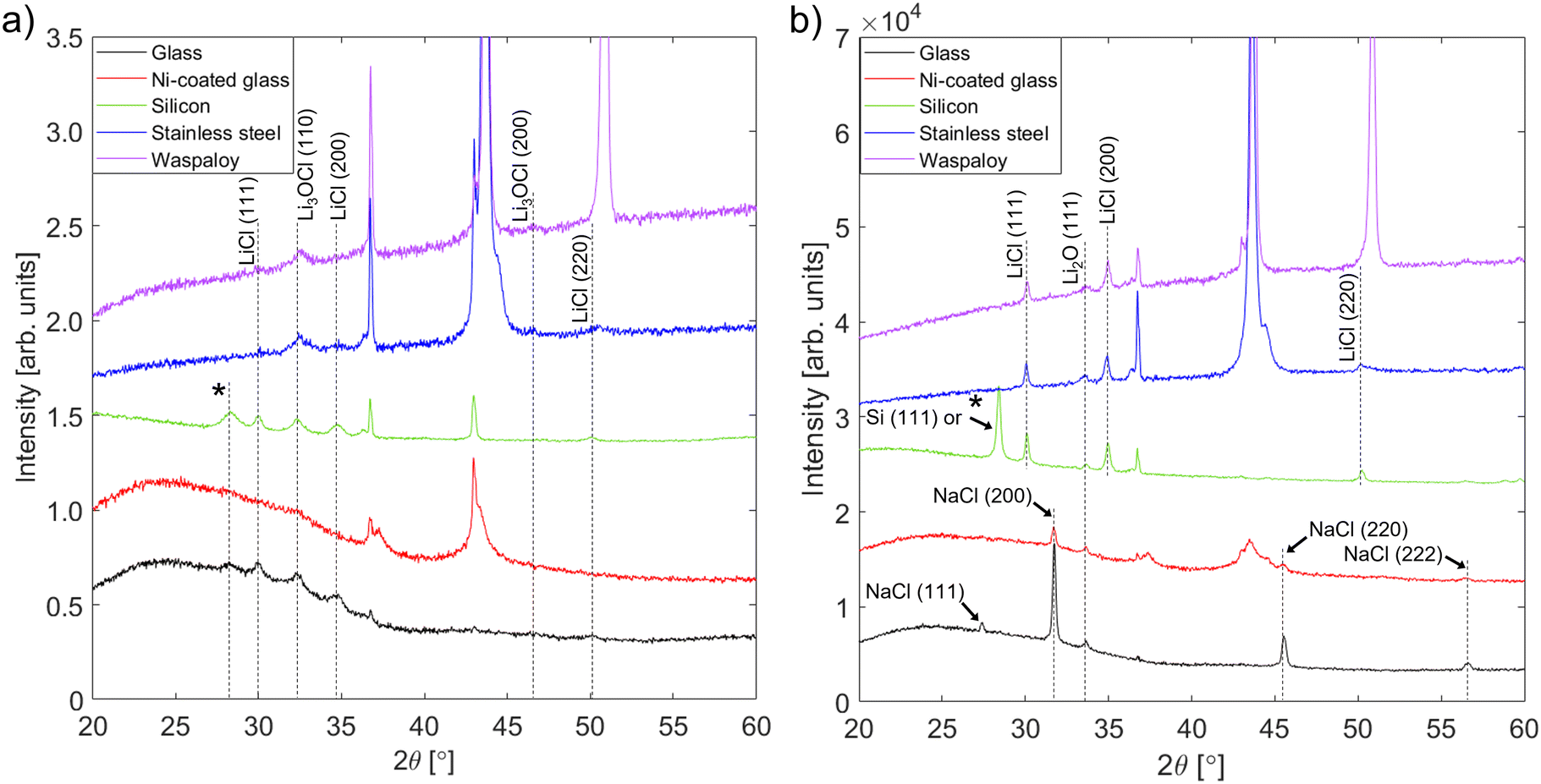 | ||
| Fig. 2 XRD patterns of films deposited from Li2O + LiCl targets onto several different substrate materials: (a) in the as-deposited condition and (b) after annealing at 250 °C for 1 hour. Peaks marked * correspond to impurity phases from the precursor mixture such as KCl. Unlabelled peaks are from the substrate or sample holder; the differing intensities of holder peaks are due to differences in the lateral positioning of samples within the holder. | ||
Fig. 2(a) shows that a phase with peaks matching the positions expected for Li3OCl (a = 3.91 Å)74 was present in all as-deposited films, regardless of the substrate type. These positions do not overlap with any of the peaks from the precursors or other possible phases in the Li–O–Cl system, which suggests that the in situ heating of the film by the impinging plasma ions was sufficient to stabilize an antiperovskite phase with composition close to Li3OCl. The presence of LiCl peaks could reflect incomplete reaction with Li2O, which may be present in amorphous form. However, EDX analysis (Section 3.2.3) revealed a significant Cl excess in the as-deposited films. This could be a result of LiCl enrichment in the upper regions of the target during sputtering (Fig. S1, ESI†). Further, if there is partial melting of the LiCl phase within the target, combined sputtering and evaporation may enhance the deposition rate of LiCl relative to that of Li2O.
The low intensities of the film peaks and their broadness compared to most of the substrate peaks is indicative of a semicrystalline film with a small grain size, which makes determination of the antiperovskite phase fraction very challenging. Post-deposition annealing was performed to encourage further reaction and crystallization into the antiperovskite phase. Since the melting temperature of Li3OCl is not known, an annealing temperature of 250 °C – which is close to the melting point of Li2OHCl16 – was chosen. However, after annealing at this temperature no peaks corresponding to Li3OCl were seen in the XRD patterns from any of the films (Fig. 2(b)). The only peaks from the films on silicon and alloy substrates correspond to Li2O and LiCl, which suggests that the antiperovskite phase decomposed during annealing. Since the results of prior experimental studies suggest that Li2OHCl is thermodynamically stable between room temperature and 250 °C,16,18,78,79 the fact that this decomposition occurred is good evidence that the antiperovskite phase attained here was indeed Li3OCl. Therefore, a quantity of Li3OCl was preserved in a metastable condition in the as-deposited films by a high rate of natural cooling from the deposition temperature (at which Li3OCl is thermodynamically stable) to room temperature (at which the decomposition kinetics are vanishingly slow). This demonstrates a key benefit of using RF magnetron sputtering to synthesize Li3OCl: relatively high cooling rates can be achieved due to the immediate quenching of the plasma at the end of deposition and the inherently high cooling rates of thin films compared with bulk samples such as pellets.80
A recent study57 estimated that Li3OCl is thermodynamically stable above 500 K (227 °C). While our results appear to contradict this, it is important to recognize that complete decomposition of the Li3OCl phase could have occurred before the annealing temperature was reached or during slow cooling back to room temperature. Evidence for this was provided by annealing films on glass substrates at 100 °C and 200 °C. As shown in the XRD patterns in Fig. S6 (ESI†), the main antiperovskite peak – (110) – is still present after annealing for 1 hour at 100 °C, but after annealing at 200 °C it is replaced by precursor peaks. Thus, the kinetics of decomposition become sufficiently fast at a temperature between 100 and 200 °C. No evidence for the presence of Li3OCl was seen in films deposited on silicon and annealed at 300 °C and 350 °C, also plotted in Fig. S6 (ESI†); a volume of Li3OCl may have re-formed at these temperatures and subsequently decomposed during slow cooling to room temperature.
The changes that occur within the films on glass and nickel-coated glass during annealing are different from those seen in the films on other substrates. Although Li3OCl peaks are absent from the XRD patterns and the Li2O peak (111) peak is present, there are no peaks corresponding to LiCl. Instead, the second set of film peaks matches exactly the positions expected for NaCl. EDX measurements performed before and after annealing (Section 3.2.3) indicate that a large volume of sodium (possibly in the form of Na2O) migrated into the films from the glass substrates during annealing. No Na2O peaks are visible in the XRD patterns, suggesting that the Na2O reacted with any LiCl present to form NaCl viaeqn (3.1).
| Na2O + 2LiCl → 2NaCl + Li2O | (3.1) |
Overall, on all substrate types the as-deposited films contain two principal crystalline phases: Li3OCl-type antiperovskite and LiCl. The XRD patterns indicate that the ratio of antiperovskite to LiCl volumes was greatest in the films on alloy substrates, while this ratio was very similar for films on glass and silicon substrates. Owing to the structural similarity between the films on glass and silicon substrates and the fact that metal coating layers were found to adhere poorly to silicon without the use of titanium or Al2O3 interlayers, the films on silicon were not subjected to further characterization.
 | ||
| Fig. 3 SEM micrographs of surfaces (left) and cross-sections made by PFIB sectioning (right) of films deposited from Li2O + LiCl targets onto (a) glass, (b) nickel-coated glass, (c) stainless steel and (d) Waspaloy substrates. The cross-sectional micrographs were taken at a 38° angle to the normal of the section plane, meaning that the scale bars are valid only for horizontal measurements. The electrolyte films are labelled ‘AP’ since they contain a quantity of antiperovskite phase. The material above the films is Pt that was deposited within the PFIB to protect the underlying films during sectioning. The site of a nickel top contact was chosen for imaging the cross-section in (b). Red circles on the surface view micrographs in (c) and (d) indicate holes in the electrolyte films. | ||
On all substrate types, the films have a “particulate” appearance, which reflects an “island-like” or Stranski–Krastanov (“layer plus island”) growth mode.81 Island-like growth occurs in situations where the surface energy of the substrate is low relative to that of the film and the film-substrate interfacial energy. Stranski–Krastanov (S–K) growth occurs if the film-substrate interfacial energy is relatively low. Layer growth initially prevails in this situation; however, a transition to island growth occurs after a few monolayers have formed to reduce the bulk strain energy. The films on alloy substrates have much coarser morphologies than those on glass and nickel-coated glass, shown quantitatively in Table 2 by the much greater thickness ranges and standard deviations of these films. Interestingly, although the deposition duration was twice that used for the other substrate types, the mean thicknesses of the films on the alloy substrates are lower than that of the film on uncoated glass. Along with the fact that the film on nickel-coated glass is ∼30% thinner than the film on uncoated glass, this indicates that the sticking coefficient is lower on metallic substrates than on uncoated glass. The red circles drawn on the surface view micrographs in Fig. 3(c) and (d) highlight areas where there are large holes in the films on the alloy substrates. Several similar areas can be seen at other locations in these micrographs and were found across the surface of each film. The presence of such features is particularly undesirable in electrolyte films since they could permit contact between the electrode layers.
| Substrate | Film thickness range [nm] | Mean [nm] | Standard deviation [nm] |
|---|---|---|---|
| Glass | 640–800 | 730 | 58 |
| Ni-coated glass | 410–600 | 520 | 59 |
| Stainless steel | 160–1390 | 620 | 360 |
| Waspaloy | 90–1410 | 670 | 404 |
The reasons for the morphological differences between the films on glass (uncoated and nickel-coated) and alloy substrates are not fully understood. However, one interesting observation is that the lattice parameters of 304 stainless steel and Waspaloy (both ∼3.6 Å82,83) are only about 8% smaller than that of Li3OCl (∼3.9 Å).74 Therefore, some degree of lattice matching between the alloy substrates and the Li3OCl phase fraction may occur during film nucleation. Growth would then follow the S–K mode, with the first few monolayers forming in a layer-by-layer fashion before transitioning to an island-like morphology.81 By contrast, the amorphous structure of glass favours purely island-like growth, with partial coalescence of the islands occurring as growth proceeds to minimize their surface area.
Although the lattice parameter of nickel (∼3.5 Å84) is also close to that of Li3OCl, the morphology of the film on nickel-coated glass is similar to that of the film on uncoated glass. A possible reason for this is the nanocrystallinity of the nickel layer, as evidenced by the lack of clear peaks corresponding to nickel in the XRD pattern in Fig. 2, which would preclude the lattice matching required for growth by the S–K mode. While the films on the glass-based substrates are much smoother than those on the alloy substrates, they are significantly rougher than the nickel bottom contact layer visible in the cross-sectional micrograph in Fig. 3(b). This can be attributed in part to the rather “soft” sputtering conditions used for the Li2O + LiCl target, since the use of low pressure and RF power limit two processes that promote the growth of smooth films: planarization (break-up of surface clusters) and the re-sputtering of loosely bound ions.81
The severe roughness and high density of holes displayed by the films on stainless steel and Waspaloy substrates renders them unsuitable for solid electrolyte applications. Therefore, subsequent characterization was performed using films on uncoated and metal-coated glass substrates.
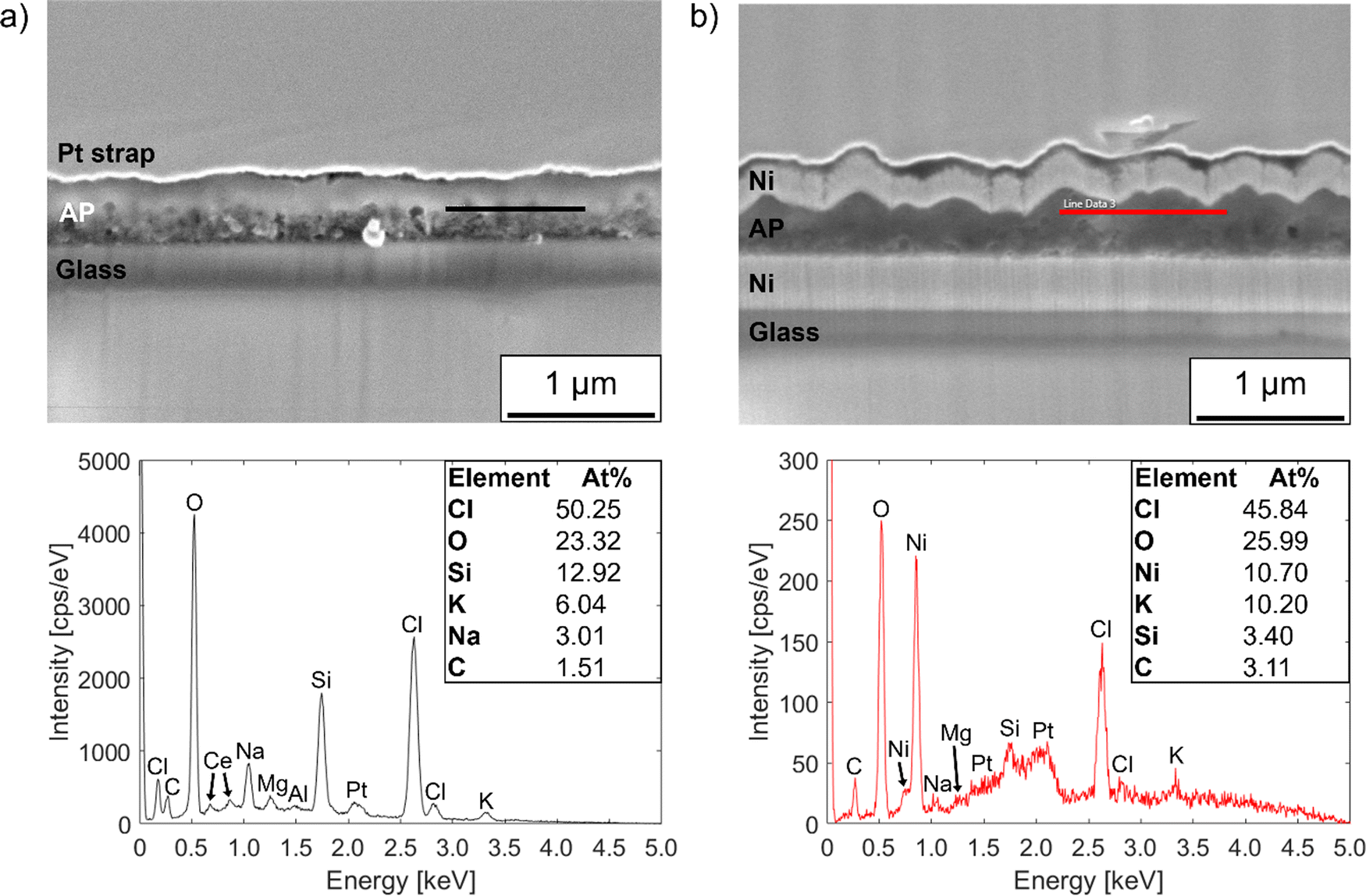 | ||
| Fig. 4 EDX spectra measured on the cross-sections of films (‘AP’) deposited from Li2O + LiCl targets onto (a) uncoated glass and (b) nickel-coated glass substrates. The black and red lines drawn on the SEM micrographs show the respective locations of the line scans. While the elemental concentrations are given in atomic percent, they do not account for the lithium content of the films or elements present at concentrations below 1 at% and are valid only for calculations of atomic ratio. | ||
The film on the uncoated glass substrate has a Cl/O atomic ratio of ∼2.2, while that of the film on nickel-coated glass is ∼1.8. This confirms the presence of an overall Cl excess in the films. However, the quantitative accuracy of the EDX measurements was compromised to some degree by the relatively low accelerating voltage used to avoid beam damage, which limited the intensities of the spectral lines.85 Furthermore, owing to the geometry of the area milled in the PFIB, measurements were performed with the cross-section plane normal tilted by 38° to the SEM beam, which may have increased the penetration of the beam into the substrate. This means that the relative concentrations of elements from the substrate such as Si, K and Na measured within the films may be higher than their true values.
EDX line scans were also performed on the cross-sections of annealed films to assess the degree of sodium migration from the glass substrates to the films during annealing. Fig. S7 (ESI†) shows composition profiles from line scans taken across films on uncoated and nickel-coated glass substrates after annealing for 1 hour at 250 °C. The results confirm observations from the XRD data in Fig. 2 that a significant amount of sodium migrates into the films from the glass substrates during annealing, but the nickel layer blocks the diffusion to a moderate degree. By inspection of the element concentration profiles, the film on uncoated glass has an Na/Cl atomic ratio of ∼0.8, whereas that of the film on nickel-coated glass is ∼0.4.
XPS was used to verify the presence of lithium and investigate its bonding environment. The spectrum measured from a film on an uncoated glass substrate is shown in Fig. 5.
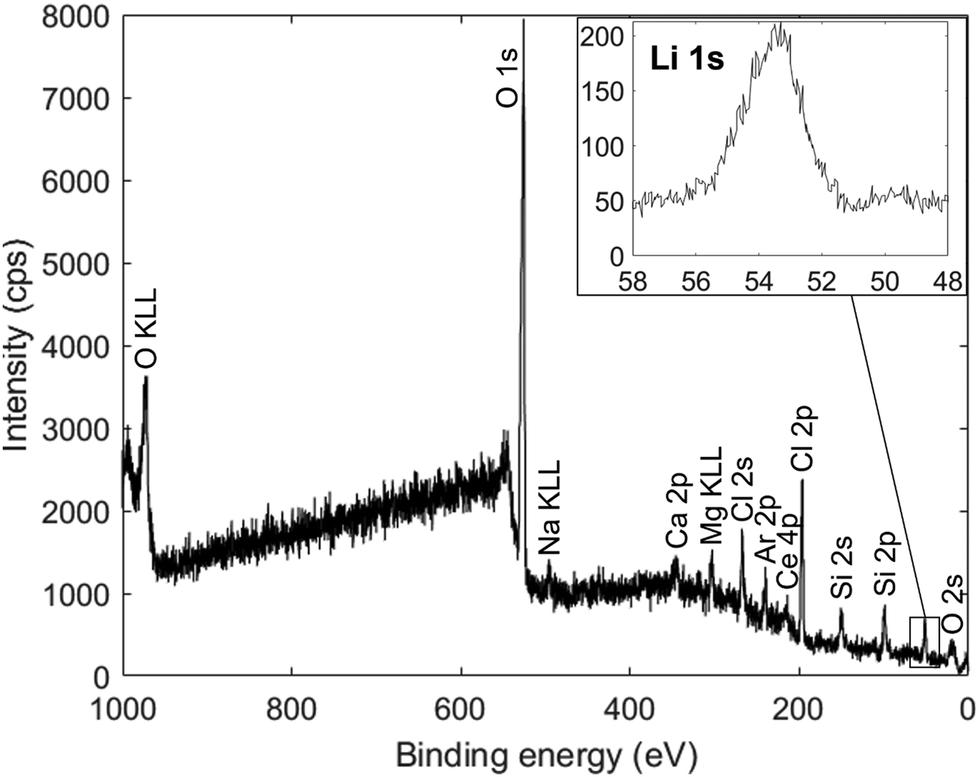 | ||
| Fig. 5 XPS spectrum of a film deposited from an Li2O + LiCl target onto a glass substrate, measured after etching the film surface using a 4 kV Ar-ion beam for 40 minutes to remove surface impurities and carbon-rich layers. The inset shows a high resolution plot of the Li 1s peak. Unlabelled peaks do not correspond to any probable film impurities and are thus attributed to contamination from the XPS chamber. | ||
The strong peaks corresponding to oxygen and chlorine, along with the sodium, calcium, magnesium, cerium and silicon peaks from the substrate, support the results from EDX measurements in Fig. 4. The Li 1s peak spans the energy range expected for the bonding in Li3OCl69 and likely contains components from Li–O (54.04 eV) and Li–Cl (54.87 eV). Overall, the XPS characterization confirms that lithium is present and in the expected bonding environments.
3.3. Comparison to films deposited from an Li2OHCl target
FT-IR experiments were performed to look for the presence of O–H bonding in the films deposited from Li2O + LiCl targets. To ensure correct interpretation of the results, reference films containing O–H bonding were deposited from an Li2OHCl powder target. In addition to FT-IR, XRD and SEM were used to investigate the structural properties of the new set of films. The FT-IR spectra and XRD patterns for both sets of films and a sample of Li2OHCl powder are shown in Fig. 6, while surface and cross-section view SEM micrographs taken after performing EIS are shown in Fig. S8 (ESI†).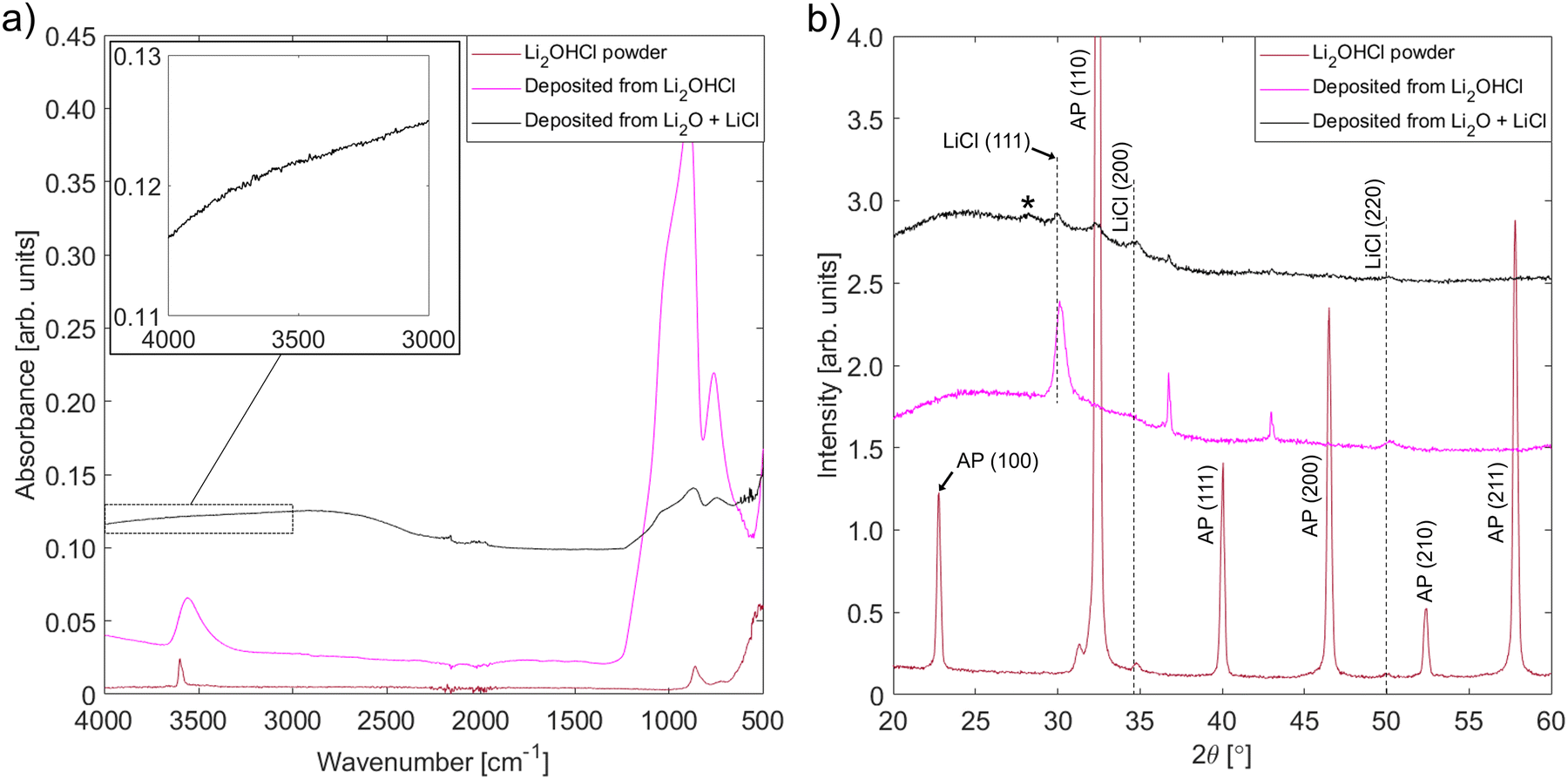 | ||
| Fig. 6 Characterization of films deposited from Li2O + LiCl and Li2OHCl targets onto uncoated glass substrates by (a) FT-IR and (b) XRD. A sample of Li2OHCl powder was also characterized for comparison. The enlarged IR spectral region covers the wavenumber range over which bands corresponding to O–H stretching vibrations are expected. On the XRD patterns, peaks marked * correspond to impurity phases from the precursor mixture such as KCl, while peaks marked ‘AP’ correspond to Li3OCl/Li2OHCl antiperovskite phases. Unlabelled peaks are from the sample holder; the differing intensities of holder peaks are due to differences in lateral positioning of the samples within the holder. | ||
A band centred at ∼3600 cm−1, which corresponds to the O–H stretching vibration, is present in the IR spectra corresponding to the Li2OHCl powder and film deposited from this powder. This shows that, like the powder, the film deposited from the powder contains O–H bonding. The absence of bands over the 3200–3600 cm−1 range in the spectrum corresponding to the film deposited from the Li2O + LiCl target shows that there is no detectable level of O–H bonding in this film. Therefore, the antiperovskite phase identified from the corresponding XRD pattern is likely to be Li3OCl or an off-stoichiometric variant rather than a hydroxide chloride compound such as Li2OHCl.
The XRD pattern for the film deposited from the Li2OHCl target does not contain any antiperovskite peaks: all detectable peaks correspond to LiCl and the sample holder. Amorphous Li2OHCl, LiOH or another phase containing OH such as Li4(OH)3Cl58 must also be present since the IR spectrum shows that the film contains O–H bonding. The reason that a quantity of antiperovskite phase is present in the film deposited from the Li2O + LiCl target but not in the film deposited from the Li2OHCl target is not clear. However, it could be due to the instability of Li2OHCl at the substrate temperature reached during deposition. A broader characterization of the properties of the films deposited from the Li2OHCl target, aside from characterization by EIS, was not performed since this is outside the scope of the present investigation.
The SEM micrographs in Fig. S8 (ESI†) show that the films deposited from the Li2OHCl target had a much smoother morphology than those deposited from Li2O + LiCl targets, which suggests that the film-substrate interfacial energy was lower. The thickness determined from 8 locations along the film cross section was (484 ± 4) nm, which is 2/3 of the thickness of the equivalent film deposited from Li2O + LiCl, as should be expected since the deposition duration was approximately 40% shorter.
The results of impedance measurements on films deposited from Li2O + LiCl and Li2OHCl targets are shown in Fig. 7, and the equivalent circuits used to fit the Nyquist plots are shown in Fig. S9 (ESI†). Ionic conductivity measurements on both sets of films proved to be extremely challenging: many films contained pinholes at the sites of the electrical contacts, which caused short circuits, and in some cases the interfaces between the contacts and the film were poor, giving purely capacitive behaviour.
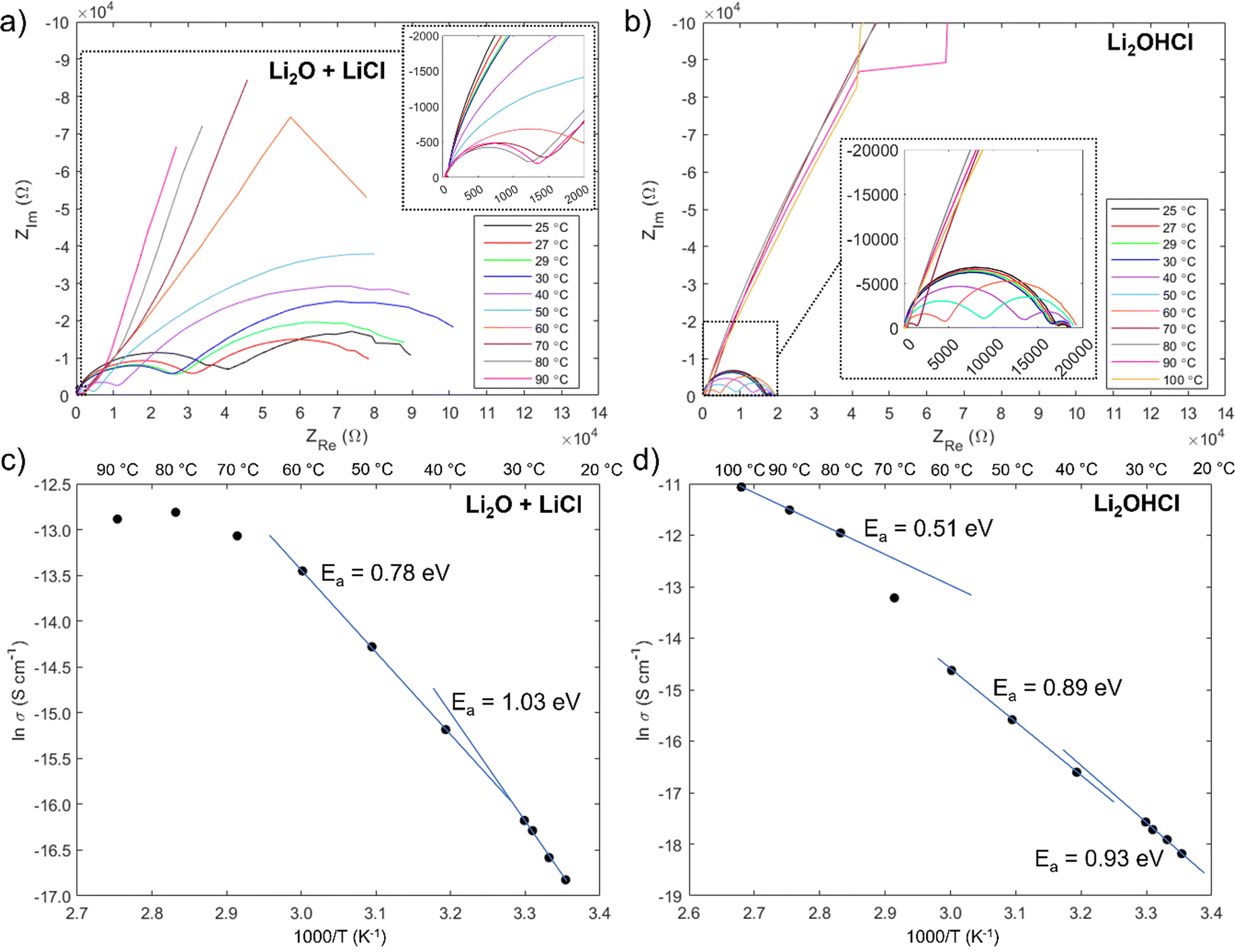 | ||
| Fig. 7 Results of through-plane impedance measurements performed at temperatures between 25 °C and 90/100 °C. Nyquist plots for the films deposited from Li2O + LiCl and Li2OHCl targets are shown in (a) and (b) respectively; (c) and (d) show the corresponding Arrhenius plots and activation energies calculated from the linear fit lines. | ||
It is noteworthy that a low frequency tail is absent from the Nyquist plots of both film samples up to 70 °C. The blocking behaviour of the electrical contacts is not seen in this temperature range because a “soft” electronic short circuit existed in both samples. Further evidence for this is provided by the semicircle at low frequency, which grows with increasing temperature as expected for the electronic component of the overall impedance. The electronic impedance was sufficiently high to enable measurement of the ionic impedance, which was determined from the high frequency semicircle by modelling the behaviour with an appropriate equivalent circuit (Fig. S9, ESI†). The resistance at the intercept of the high frequency semicircle with the real axis, Rint, is given by 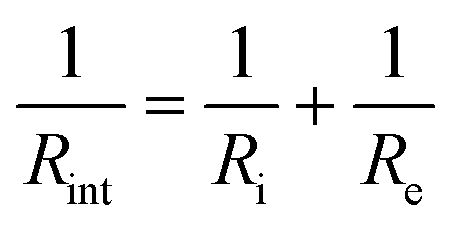 for the film deposited from Li2O + LiCl and
for the film deposited from Li2O + LiCl and 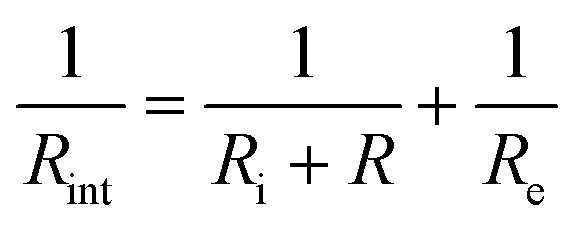 for the film deposited from Li2OHCl, where Ri, Re and R are the bulk ionic, electronic and additional (e.g. grain boundary or reaction layer) resistance components, respectively. Re corresponds to the intercept of the low frequency semicircle with the real axis. These intercepts are not actually reached by the curves in Fig. 7 due to interference between the high and low frequency behaviour, but the equivalent circuit modelling allows the points at which these intercepts would occur in the absence of interference to be determined. Thus, the value of Ri can be calculated for each temperature and converted to ionic conductivity using the film thickness (Table 2 and Fig. S8, ESI†) and the impedance contact overlap areas listed in Table S1 (ESI†). The values of ionic conductivity measured at 25 °C and 50 °C for both samples are shown in Table 3. On reaching 70 °C a low frequency tail appears in place of the low frequency semicircle in both Nyquist plots, indicating that the short circuits disappear, possibly due to the formation of insulating reaction layers.
for the film deposited from Li2OHCl, where Ri, Re and R are the bulk ionic, electronic and additional (e.g. grain boundary or reaction layer) resistance components, respectively. Re corresponds to the intercept of the low frequency semicircle with the real axis. These intercepts are not actually reached by the curves in Fig. 7 due to interference between the high and low frequency behaviour, but the equivalent circuit modelling allows the points at which these intercepts would occur in the absence of interference to be determined. Thus, the value of Ri can be calculated for each temperature and converted to ionic conductivity using the film thickness (Table 2 and Fig. S8, ESI†) and the impedance contact overlap areas listed in Table S1 (ESI†). The values of ionic conductivity measured at 25 °C and 50 °C for both samples are shown in Table 3. On reaching 70 °C a low frequency tail appears in place of the low frequency semicircle in both Nyquist plots, indicating that the short circuits disappear, possibly due to the formation of insulating reaction layers.
| Sputtering target | Li+ conductivity of film at 25 °C [S cm−1] | Li+ conductivity of film at 50 °C [S cm−1] |
|---|---|---|
| Li2O + LiCl | 4.93 × 10−8 | 6.29 × 10−7 |
| Li2OHCl | 1.27 × 10−8 | 1.71 × 10−7 |
The conductivity values are at the lower end of the ranges reported previously for Li2OHCl, and the low-temperature activation energies are somewhat higher than measured previously. However, it is important to bear in mind that both sets of films have low antiperovskite phase purity; in the case of the film deposited from Li2OHCl, no quantity of antiperovskite phase was detected (Fig. 6(b)), which may explain why its ionic conductivity is lower than that of the film deposited from Li2O + LiCl. Therefore, the overall ionic conductivity will have been supressed by the large volume fraction of low-conductivity secondary phases, including crystalline LiCl and amorphous phases undetectable by XRD. Furthermore, the broad antiperovskite peaks in the XRD pattern in Fig. 2 – and the lack of such peaks in Fig. 6 – suggest that the antiperovskite phases have a very small grain size. Since the grain boundaries of Li3OCl are predicted to have a significantly lower ionic conductivity than the grains,86 the large grain boundary areas of these samples should further suppress the overall ionic conductivity.
Nevertheless, the Arrhenius plots in Fig. 7(c) and (d) show the characteristic change in activation energy between 30 °C and 40 °C, which usually reflects the transition from an orthorhombic to a cubic crystal structure.7,16,17,79 The change is only slight for the film deposited from Li2OHCl, but it does suggest that some crystalline antiperovskite phase was present or formed during the low temperature heating. A much clearer change is seen for the film deposited from Li2O + LiCl. This is unexpected as an orthorhombic structure has not been predicted to be stable for Li3OCl. Nevertheless, it is quite possible that the orthorhombic phase is favoured below ∼32 °C for both Li2OHCl and Li3OCl, particularly if the composition of the Li3OCl-type phase is off-stoichiometric.
Unlike in previous studies on Li2OHCl, the activation energy seems to decrease further for both film types above 60 °C. Further structural or chemical changes must therefore occur in both films at the highest measurement temperatures. It was shown in Section 3.2.1 that only very minor structural changes occur in the films deposited from Li2O + LiCl targets onto uncoated glass substrates during annealing at 100 °C for 1 hour (Fig. S6, ESI†). However, the migration of sodium was not studied at this temperature; if it is significant, it may affect the ionic conductivity. In the case of the film deposited from Li2OHCl, the activation energy above 70 °C is 0.51 eV, which is slightly lower than the values reported previously for cubic Li2OHCl and may reflect an increase in the fraction of the cubic antiperovskite phase during heating between ∼60 °C and ∼80 °C.
To investigate the changes that occurred in the film deposited from Li2OHCl during the EIS measurements, XRD and FT-IR were performed after completing the experiment and cooling to room temperature. The results are shown in Fig. S10 (ESI†). Despite the continued presence of a strong LiCl(111) peak, the XRD patterns show that the main cubic antiperovskite peak – (110) – has appeared. While only a single peak corresponding to the antiperovskite phase can be seen clearly – since the other peaks overlap with those from the Pt electrical contacts – this is reasonable evidence that the antiperovskite phase fraction increased during the impedance measurements. The FT-IR spectrum shows that O–H bonding is still present after performing EIS, but interestingly the band intensity around 3600 cm−1 contains a sharp component (as seen in the spectrum from the Li2OHCl powder) and a broad component (as seen in the spectrum from the as-deposited film). The appearance of a sharp intensity component likely reflects the partial crystallization of the OH-containing phase (Li2OHCl) and suggests that annealing at a temperature of at least 100 °C will lead to improvements in the properties of the as-deposited films.
4. Conclusions
In this work, the formation of small volumes of LiRAP phases in thin film electrolytes deposited by RF magnetron sputtering was demonstrated for the first time. The XRD results showed that one of the structures present in films deposited from powdered Li2O + LiCl sputtering targets onto a variety of substrate materials matched that of Li3OCl. This phase was found to break down into Li2O and LiCl – the precursors of Li3OCl – on annealing at moderate temperatures, suggesting that rapid natural cooling from the deposition temperature and sluggish decomposition kinetics at room temperature locked a volume of this phase in a metastable state. By contrast, in films deposited from a powdered Li2OHCl sputtering target a quantity of thermodynamically stable antiperovskite phase with a structure matching that of Li2OHCl was partially crystallized on heating to 100 °C. Crucially, FT-IR experiments showed that the films deposited from Li2O + LiCl did not contain a detectable level of O–H bonding, whereas the thinner films deposited from Li2OHCl showed clear IR bands corresponding to O–H stretching vibrations. The results therefore suggest that quantities of Li3OCl and Li2OHCl type antiperovskite phases were formed in the films deposited from Li2O + LiCl and Li2OHCl targets, respectively.Due to the low phase purities of these films, it was not possible to isolate the antiperovskite phases and determine their precise chemical compositions. However, the fact that the measured lattice parameters were approximately the same as the expected values implies that the actual compositions were similar to the nominal chemical formulae. XRD and EDX analyses revealed a significant overall LiCl excess in the films deposited from Li2O + LiCl targets. Increasing the molar ratio of Li2O to LiCl in the target could reduce this excess and increase the volume fraction of the Li3OCl phase. In the case of the films deposited from Li2OHCl, the results show that the volume fraction of the Li2OHCl phase can be increased by annealing at a temperature of 100 °C, which is very low compared to most of the synthesis temperatures reported previously.7,16,17,27–29,31 For both film types, increasing the volume fraction of the antiperovskite phase should increase the overall Li+ conductivity, which may make the films viable as solid electrolytes.
RF magnetron sputtering is a promising fabrication technique for thin solid electrolytes containing LiRAP phases. Aside from further optimization of the processing conditions to maximize the antiperovskite phase fractions, future investigations should measure the film temperature during deposition from an Li2O + LiCl target and the rate of cooling to room temperature. This will increase understanding of the Li3OCl phase stability. Investigation of substrate materials that form low energy interfaces with films deposited from Li2O + LiCl targets and are free from highly mobile ions will also be required to improve electrolyte performance.
Data availability
Source data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the ISCF Faraday Challenge project SOLBAT [grant number FIRG026]. The authors acknowledge use of characterization facilities within the David Cockayne Centre for Electron Microscopy, Department of Materials, University of Oxford and capital equipment funded by the Henry Royce Institute (Grant ref. EP/R010145/1). SJT acknowledges support from an EPSRC PhD studentship [project reference 2114670 related to EP/R513295/1].References
- M. Pasta, D. Armstrong, Z. L. Brown, J. Bu, M. R. Castell, P. Chen, A. Cocks, S. A. Corr, E. J. Cussen, E. Darnbrough, V. Deshpande, C. Doerrer, M. S. Dyer, H. El-Shinawi, N. Fleck, P. Grant, G. L. Gregory, C. Grovenor, L. J. Hardwick, J. T. S. Irvine, H. J. Lee, G. Li, E. Liberti, I. McClelland, C. Monroe, P. D. Nellist, P. R. Shearing, E. Shoko, W. Song, D. S. Jolly, C. I. Thomas, S. J. Turrell, M. Vestli, C. K. Williams, Y. Zhou and P. G. Bruce, J. Phys. Energy, 2020, 2, 032008 CrossRef CAS.
- Y. Benabed, M. Rioux, S. Rousselot, G. Hautier and M. Dollé, Front. Energy Res., 2021, 9, 1–13 CrossRef.
- B. Wang, J. B. Bates, F. X. Hart, B. C. Sales, R. A. Zuhr and J. D. Robertson, J. Electrochem. Soc., 1996, 143, 3203–3213 CrossRef CAS.
- K. H. Park, K. Kaup, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, ACS Energy Lett., 2020, 533–539 CrossRef CAS.
- L. Zhou, C. Y. Kwok, A. Shyamsunder, Q. Zhang, X. Wu and L. F. Nazar, Energy Environ. Sci., 2020, 13, 2056–2063 RSC.
- V. Kumaravel, J. Bartlett and S. C. Pillai, Adv. Energy Mater., 2021, 11(3), 2002869 CrossRef CAS.
- Z. D. Hood, H. Wang, A. Samuthira Pandian, J. K. Keum and C. Liang, J. Am. Chem. Soc., 2016, 138, 1768–1771 CrossRef CAS PubMed.
- J. Lau, R. H. DeBlock, D. M. Butts, D. S. Ashby, C. S. Choi and B. S. Dunn, Adv. Energy Mater., 2018, 8, 1–24 CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- Y. Xiao, Y. Wang, S. H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- Z. Ning, D. S. Jolly, G. Li, R. De Meyere, S. D. Pu, Y. Chen, J. Kasemchainan, J. Ihli, C. Gong, B. Liu, D. L. R. Melvin, A. Bonnin, O. Magdysyuk, P. Adamson, G. O. Hartley, C. W. Monroe, T. J. Marrow and P. G. Bruce, Nat. Mater., 2021, 20, 1121–1129 CrossRef CAS PubMed.
- K. Kim and D. J. Siegel, J. Mater. Chem. A, 2019, 7, 3216–3227 RSC.
- H. J. Lee, B. Darminto, S. Narayanan, M. Diaz-Lopez, A. W. Xiao, Y. Chart, J. H. Lee, J. A. Dawson and M. Pasta, J. Mater. Chem. A, 2022, 10, 11574–11586 RSC.
- A. Koedtruad, M. A. Patino, N. Ichikawa, D. Kan and Y. Shimakawa, J. Solid State Chem., 2020, 286, 121263 CrossRef CAS.
- A. Y. Song, Y. Xiao, K. Turcheniuk, P. Upadhya, A. Ramanujapuram, J. Benson, A. Magasinski, M. Olguin, L. Meda, O. Borodin and G. Yushin, Adv. Energy Mater., 2018, 8, 1–11 CAS.
- O. Reckeweg, B. Blaschkowski and T. Schleid, Z. Anorg. Allg. Chem., 2012, 638, 2081–2086 CrossRef CAS.
- Y. Zhao and L. L. Daemen, J. Am. Chem. Soc., 2012, 134, 15042–15047 CrossRef CAS PubMed.
- M. J. Clarke, J. A. Dawson, T. J. Mays and M. S. Islam, ACS Appl. Energy Mater., 2021, 4, 5094–5100 CrossRef CAS.
- Y. Zhang, Y. Zhao and C. Chen, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 1–8 Search PubMed.
- R. Mouta, M. Á. B. Melo, E. M. Diniz and C. W. A. Paschoal, Chem. Mater., 2014, 26, 7137–7144 CrossRef CAS.
- Z. Lu, C. Chen, Z. M. Baiyee, X. Chen, C. Niu and F. Ciucci, Phys. Chem. Chem. Phys., 2015, 17, 32547–32555 RSC.
- M. S. Wu, B. Xu, W. W. Luo, B. Z. Sun and C. Y. Ouyang, Electrochim. Acta, 2020, 334, 135622 CrossRef CAS.
- J. A. S. Serejo, J. S. Pereira, R. Mouta and L. G. C. Rego, Phys. Chem. Chem. Phys., 2021, 23, 6964–6973 RSC.
- Y. Li, W. Zhou, S. Xin, S. Li, J. Zhu, X. Lü, Z. Cui, Q. Jia, J. Zhou, Y. Zhao and J. B. Goodenough, Angew. Chem., Int. Ed., 2016, 55, 9965–9968 CrossRef CAS PubMed.
- P. Hartwig and W. Weppner, Solid State Ionics, 1981, 3–4, 249–254 CrossRef CAS.
- Z. Lai, W. Feng, X. Dong, X. Zhou, Y. Wang and Y. Xia, J. Power Sources, 2021, 500, 229982 CrossRef CAS.
- T. Yamamoto, H. Shiba, N. Mitsukuchi, M. K. Sugumar, M. Motoyama and Y. Iriyama, Inorg. Chem., 2020, 59, 11901–11904 CrossRef CAS PubMed.
- Y.-S. Lee, S.-Y. Jung and K.-S. Ryu, J. Electrochem. Energy Convers. Storage, 2021, 18, 1–12 Search PubMed.
- M. H. Braga, J. A. Ferreira, V. Stockhausen, J. E. Oliveira and A. El-Azab, J. Mater. Chem. A, 2014, 2, 5470–5480 RSC.
- M. H. Braga, A. J. Murchison, J. E. Oliveira and J. B. Goodenough, ACS Appl. Energy Mater., 2019, 2, 4943–4953 CrossRef CAS.
- M. H. Braga, A. J. Murchison, J. A. Ferreira, P. Singh and J. B. Goodenough, Energy Environ. Sci., 2016, 9, 948–954 RSC.
- M. H. Braga, N. S. Grundish, A. J. Murchison and J. B. Goodenough, Energy Environ. Sci., 2017, 10, 331–336 RSC.
- M. H. Braga, C. M. Subramaniyam, A. J. Murchison and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 6343–6352 CrossRef CAS PubMed.
- M. H. Braga, J. E. Oliveira, T. Kai, A. J. Murchison, A. J. Bard and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 17968–17976 CrossRef CAS PubMed.
- M. H. Braga, J. E. Oliveira, A. J. Murchison and J. B. Goodenough, Appl. Phys. Rev., 2020, 7(1), 011406 CAS.
- X. Lü, G. Wu, J. W. Howard, A. Chen, Y. Zhao, L. L. Daemen and Q. Jia, Chem. Commun., 2014, 50, 11520–11522 RSC.
- X. Lü, J. W. Howard, A. Chen, J. Zhu, S. Li, G. Wu, P. Dowden, H. Xu, Y. Zhao and Q. Jia, Adv. Sci., 2016, 3, 1500359 CrossRef PubMed.
- M. Dondelinger, J. Swanson, G. Nasymov, C. Jahnke, Q. Qiao, J. Wu, C. Widener, A. M. Numan-Al-Mobin and A. Smirnova, Electrochim. Acta, 2019, 306, 498–505 CrossRef CAS.
- Y. Yang, J. Han, M. DeVita, S. S. Lee and J. C. Kim, Front. Chem., 2020, 8, 3–9 CrossRef PubMed.
- S. Li, J. Zhu, Y. Wang, J. W. Howard, X. Lü, Y. Li, R. S. Kumar, L. Wang, L. L. Daemen and Y. Zhao, Solid State Ionics, 2016, 284, 14–19 CrossRef CAS.
- B. Chen, C. Xu and J. Zhou, J. Electrochem. Soc., 2018, 165, A3946–A3951 CrossRef CAS.
- A. G. Squires, J. M. Dean and B. J. Morgan, ChemRxiv, 2021, preprint DOI:10.33774/chemrxiv-2021-hzrls.
- A. Emly, E. Kioupakis and A. Van Der Ven, Chem. Mater., 2013, 25, 4663–4670 CrossRef CAS.
- M. Wu, B. Xu, X. Lei, K. Huang and C. Ouyang, J. Mater. Chem. A, 2018, 6, 1150–1160 RSC.
- R. Mouta, E. M. Diniz and C. W. A. Paschoal, J. Mater. Chem. A, 2016, 4, 1586–1590 RSC.
- S. Stegmaier, J. Voss, K. Reuter and A. C. Luntz, Chem. Mater., 2017, 29, 4330–4340 CrossRef CAS.
- Z. Deng, B. Radhakrishnan and S. P. Ong, Chem. Mater., 2015, 27, 3749–3755 CrossRef CAS.
- X. Liu, R. Garcia-Mendez, A. R. Lupini, Y. Cheng, Z. D. Hood, F. Han, A. Sharafi, J. C. Idrobo, N. J. Dudney, C. Wang, C. Ma, J. Sakamoto and M. Chi, Nat. Mater., 2021, 20, 1485–1490 CrossRef CAS PubMed.
- T. K. Schwietert, A. Vasileiadis and M. Wagemaker, JACS Au, 2021, 1, 1488–1496 CrossRef CAS PubMed.
- K. Kim and D. J. Siegel, ACS Appl. Mater. Interfaces, 2019, 11, 39940–39950 CrossRef CAS PubMed.
- J. A. Dawson, T. Famprikis and K. E. Johnston, J. Mater. Chem. A, 2021, 9, 18746–18772 RSC.
- I. Hanghofer, G. J. Redhammer, S. Rohde, I. Hanzu, A. Senyshyn, H. M. R. Wilkening and D. Rettenwander, Chem. Mater., 2018, 30, 8134–8144 CrossRef CAS.
- M. H. Chen, A. Emly and A. Van Der Ven, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 1–8 Search PubMed.
- K. Kim, Y. Li, P.-C. Tsai, F. Wang, S.-B. Son, Y.-M. Chiang and D. J. Siegel, Chem. Mater., 2022, 34(3), 947–958 CrossRef CAS.
- M. B. Effat, J. Liu, Z. Lu, T. H. Wan, A. Curcio and F. Ciucci, ACS Appl. Mater. Interfaces, 2020, 12, 55011–55022 CrossRef CAS PubMed.
- J. Zhang, J. Han, J. Zhu, Z. Lin, M. H. Braga, L. L. Daemen, L. Wang and Y. Zhao, Inorg. Chem. Commun., 2014, 48, 140–143 CrossRef CAS.
- J. A. Dawson, T. S. Attari, H. Chen, S. P. Emge, K. E. Johnston and M. S. Islam, Energy Environ. Sci., 2018, 11, 2993–3002 RSC.
- B. Han, D. Feng, S. Li, Z. Zhang, Y. Zou, M. Gu, H. Meng, C. Wang, K. Xu, Y. Zhao, H. Zeng, C. Wang and Y. Deng, Nano Lett., 2020, 20, 4029–4037 CrossRef CAS PubMed.
- J. Howard, Z. D. Hood and N. A. W. Holzwarth, Phys. Rev. Mater., 2017, 1, 1–13 Search PubMed.
- F. Wang, H. A. Evans, K. Kim, L. Yin, Y. Li, P. C. Tsai, J. Liu, S. H. Lapidus, C. M. Brown, D. J. Siegel and Y. M. Chiang, Chem. Mater., 2020, 32, 8481–8491 CrossRef CAS.
- S. Swann, Phys. Technol., 1988, 19, 67–75 CrossRef CAS.
- G. Faraji, H. S. Kim and H. T. Kashi, Severe Plastic Deformation, Elsevier, 2018, pp. 1–17 Search PubMed.
- D. H. Lowndes, D. B. Geohegan, A. A. Puretzky, D. P. Norton and C. M. Rouleau, Science, 1996, 273, 898–903 CrossRef CAS PubMed.
- D. Benetti, R. Nouar, R. Nechache, H. Pepin, A. Sarkissian, F. Rosei and J. M. MacLeod, Sci. Rep., 2017, 7, 2–10 CrossRef PubMed.
- H. M. Christen and G. Eres, J. Phys.: Condens. Matter, 2008, 20(26), 264005 CrossRef CAS PubMed.
- C. Yan, R. Xu, J. Qin, H. Yuan, Y. Xiao, L. Xu and J. Huang, Angew. Chem., Int. Ed., 2019, 131, 15379–15382 CrossRef.
- J. Liimatainen, J. Piirto, J. Kaisto, A. Zolotukhin and S. Chaudhuri, J. Mater. Sci. Eng. B, 2015, 5, 196–205 CAS.
- Z. Vakulov, D. Khakhulin, E. Zamburg, A. Mikhaylichenko, V. A. Smirnov, R. Tominov, V. S. Klimin and O. A. Ageev, Materials, 2021, 14(17), 4854 CrossRef CAS PubMed.
- A. Watanabe, G. Kobayashi, N. Matsui, M. Yonemura, A. Kubota, K. Suzuki, M. Hirayama and R. Kanno, Electrochemistry, 2017, 85, 88–92 CrossRef CAS.
- G. I. Finch and S. Fordham, Proc. Phys. Soc., 1936, 48, 85–94 CrossRef CAS.
- K. Persson, Materials Data on Li3ClO (SG:221) by Materials Project, 2016.
- X. Yu, J. B. Bates, G. E. Jellison and F. X. Hart, J. Electrochem. Soc., 1997, 144, 524–532 CrossRef CAS.
- Lithium oxide, https://www.sigmaaldrich.com/GB/en/product/aldrich/374725, (accessed 9 June 2022).
- C. Zhang and M. F. Simpson, J. Nucl. Fuel Cycle Waste Technol., 2017, 15, 117–124 CrossRef.
- C. Eilbracht, W. Kockelmann, D. Hohlwein and H. Jacobs, Phys. B, 1997, 234–236, 48–50 CrossRef CAS.
- G. Schwering, A. Hönnerscheid, L. Van Wüllen and M. Jansen, ChemPhysChem, 2003, 4, 343–348 CrossRef CAS PubMed.
- G. Planini and M. Vollmer, Eur. J. Phys., 2008, 29, 369–384 CrossRef.
- M. Ohring, Materials Science of Thin Films, Academic Press, 2nd edn, 2001 Search PubMed.
- A. Boeuf, S. Crico, R. Caciuffo, F. Rustichelli, I. Pomot and G. Uny, Mater. Lett., 1985, 3, 115–118 CrossRef CAS.
- J. D. T. Allen, PhD thesis, University of Birmingham, 2019.
- M. Morinaga, Local Lattice Strains Around Alloying Elements in Metals, A Quantum Approach to Alloy Design: An Exploration of Material Design and Development Based Upon Alloy Design Theory and Atomization Energy Method, Elsevier, 2019 Search PubMed.
- D. Vaughan, R. Johnson, D. Seielstad, C. Meltzer, R. Woldseth, C. Ellwood, B. Fucci, J. Holm, A. Holzer, T. Stark, R. Vane, D. Wherry and J. Balser, Energy Dispersive X-ray Microanalysis An Introduction, 2008 Search PubMed.
- J. A. Dawson, P. Canepa, T. Famprikis, C. Masquelier and M. S. Islam, J. Am. Chem. Soc., 2018, 140, 362–368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00971d |
| This journal is © The Royal Society of Chemistry 2022 |