
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence9-Nitrobenzo[b]quinolizinium as a fluorogenic probe for the detection of nitroreductase in vitro and in Escherichia coli†
Peter Jonas
Wickhorst
,
Heiko
Ihmels
 *,
Melanie Marianne
Lammert-Baumgartner
,
Mareike
Müller
and
Holger
Schönherr
*,
Melanie Marianne
Lammert-Baumgartner
,
Mareike
Müller
and
Holger
Schönherr
Department of Chemistry – Biology, University of Siegen, Center of Micro- and Nanochemistry and (Bio-)Technology (Cμ), Adolf-Reichwein-Str. 2, Siegen 57068, Germany. E-mail: ihmels@chemie.uni-siegen.de
First published on 30th November 2021
Abstract
The non-fluorescent 9-nitrobenzo[b]quinolizinium is readily reduced by nitroreductase to a fluorescent reaction product. The resulting light-up effect enables the selective detection of nitroreductase activity in vitro and in Escherichia coli bacterial cultures.
Hypoxia is a condition in cells with insufficient oxygen supply, which is related to several diseases such as inflammations,1 strokes2 or infections with the COVID-19 virus.3 Furthermore, hypoxia is an indicator of tumors, because it is observed in cancerous tissue that is disconnected from blood supply.4,5 Therefore, the detection and determination of the hypoxia status of cells is an important task in analytical biology and medicine.5 For this purpose, fluorescent probes based on organic dyes have been developed that are able to detect hypoxia-related phenomena, such as a low pH, low oxygen levels or activity of reducing enzymes, such as azoreductase or nitroreductase, by a strong increase of their emission intensity or a distinct shift of their emission maximum in the presence of the target analyte.5 Specifically, nitroreductase is a useful biomarker for the fluorimetric detection of hypoxic cells and bacteria with a high nitroreductase activity, e.g. Escherichia coli (E. coli) and Mycobacterium tuberculosis,6,7 as this enzyme reduces non-fluorescent substrates, such as nitroarenes, to fluorescent products in the presence of NAD(P)H as cofactor.4,5,8 However, several reaction products may be formed in this reaction along with the desired hydroxylamino- and amino-substituted fluorophores, for example in secondary reactions such as dimerization of intermediates or reduction of alkenes.8 Therefore, there is still a high demand for novel fluorogenic probes that are able to detect nitroreductase based on a clean and predictable reaction. As a result, fluorescent and luminescent light-up probes that specifically target nitroreductase in living organisms have been developed based on, e.g., lanthanoid complexes,9 BODIPY,10 coumarin,11 naphthalimide,12 xanthene,13 benzothiazole,14,15 and amino-silapyronine.16 Another useful fluorophore that may be employed for this purpose is the benzo[b]quinolizinium ion (1a), which has been shown to be a versatile building block for the development of fluorescent probes for bioanalytes due to its high fluorescence quantum yield (Φfl = 0.52 in H2O) and good water solubility, but it has not been utilized for enzyme detection, so far.17 Therefore, we explored the potential of quinolizinium derivatives to act as fluorescent probes for the detection of nitroreductase. To this end, we selected the 9-nitrobenzo[b]quinolizinium (1b, Fig. 1) as starting point, because the anticipated reduction product 9-aminobenzo[b]quinolizinium (1c) exhibits the highest fluorescence quantum yield (Φfl = 0.41 in H2O) among the known aminobenzo[b]quinolizinium derivatives.18 For comparison, the nitrostyryl-substituted quinolizinium 2 and the nitrophenyl-substituted benzo[b]quinolizinium 1d were selected also as substrates to investigate whether structure variations have an influence on the reactivity towards nitroreductase. Herein, we demonstrate that, indeed, the 9-nitrobenzo[b]quinolizinium (1b) may be used as novel fluorogenic probe for the detection of nitroreductase in vitro and in E. coli bacteria and that, strikingly, two different emitting species are formed on reduction of 1b depending on the reaction conditions.
 | ||
| Fig. 1 Structures of quinolizinium derivatives 1 and 2. | ||
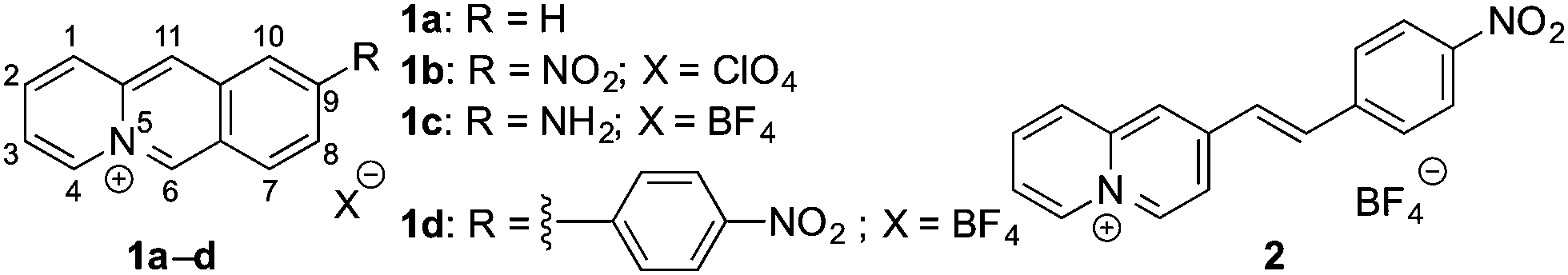
In first experiments, the absorption and emission properties of probes 1b, 1d and 2 were investigated in aqueous buffer solution. These derivatives exhibit long-wavelength absorption maxima at 422 nm (1b), 371 nm (1d) and 413 nm (2) (Fig. 2 and Fig. S2, ESI†) and are essentially non-fluorescent in aqueous buffer solution. The low fluorescence intensity of these quinolizinium derivatives, especially as compared with the high emission quantum yield of the parent compound 1a, is likely caused by the nitro substituent, because nitroarenes are known to undergo fast relaxation by intersystem crossing (ISC) in the excited state leading to fluorescence quenching.19 Notably, upon addition of nitroreductase and NADH all derivatives developed red-shifted absorption bands with long-wavelength absorption maxima at 480 nm (1b), 385 nm (1d), and 415 nm (2) (Fig. 2 and Fig. S2, ESI†), indicating their reductive conversion under these reaction conditions. Specifically, the red shift is explained by the formation of an amino functionality and the resulting formation of a donor–acceptor chromophore.17 Furthermore, the reaction of compound 1b with nitroreductase led to the transformation of 1b into a fluorescent product with a fluorescence light-up effect (Φfl = 0.05), that reached its maximum intensity approx. 20 min after addition of the enzyme under the employed conditions (Fig. 2 and 3). In contrast, no such light-up effect was observed for derivatives 1d and 2 under the same reaction conditions, presumably because the styryl and biaryl functionalities still cause emission quenching in the resulting products. Therefore, only derivative 1b was selected for further investigations. In order to determine the optimal reaction conditions, fluorescence spectra were recorded at different temperature and pH under otherwise identical conditions (Fig. S3, ESI†). Thus, a continuous increase of the fluorescence intensity was observed with increasing pH and temperature up to pH 7 and 37 °C, whereas at larger values the fluorescence intensity started to decrease. The effect of the temperature on the reaction of 1b with nitroreductase is in good agreement with the reported activities of the enzyme towards nitroarene derivatives9–11,15 but the optimal pH value lies usually slightly higher at pH 7.4.9–11,15 The lower fluorescence intensity at higher pH values is presumably caused by a slow degradation of the substrate by nucleophilic addition of the hydroxide ion to the benzo[b]quinolizinium at C6 position and subsequent ring opening reaction, as usually observed for this class of compounds.20 Furthermore, excitation spectra indicated that the new long-wavelength absorption maxima at 447 nm and 480 nm correspond to the newly formed emitting species (Fig. 2 and Fig. S4A, ESI†).
 | ||
| Fig. 2 (A) Absorption before (red) and after (black) incubation of probe 1b (c = 10 μM) with nitroreductase (5 μg mL−1) and NADH (50 μM) in PBS buffer (pH = 7, T = 37 °C) for 40 min. (B) Change of the emission after addition of nitroreductase (5 μg mL−1) and NADH (50 μM) to probe 1b (c = 10 μM) in PBS buffer (pH = 7, T = 37 °C); λex = 415 nm. The arrows indicate the changes of absorption and emission with increasing reaction time. | ||
 | ||
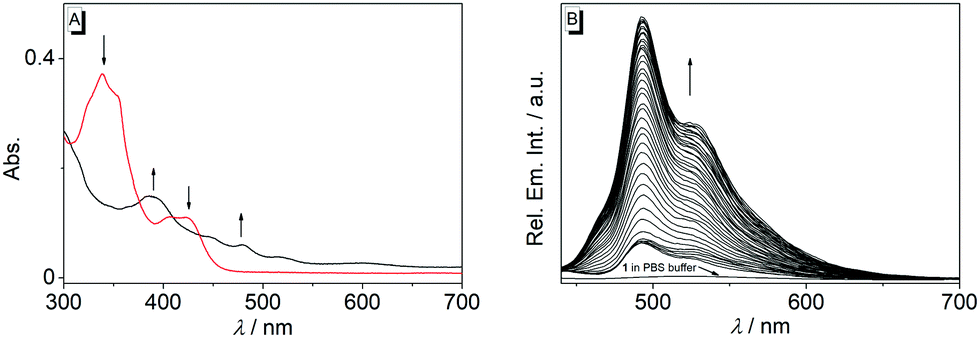
| Fig. 3 (A) Change of the emission intensity during the reaction of 1b with nitroreductase at different concentrations (black: 0 μg mL−1, red: 1.25 μg mL−1, blue: 1.75 μg mL−1, green: 2.5 μg mL−1, magenta: 5 μg mL−1) in the presence of NADH (50 μM) in PBS buffer (pH = 7, T = 37 °C); λex = 415 nm. Inset (A) Picture of the emission color before (i) and after (ii) incubation of 1b with nitroreductase (5 μg mL−1) and NADH (50 μM) in PBS buffer (pH = 7, T = 37 °C) for 40 min; λex = 366 nm. (B) Relative emission intensity of solutions of NaCl (5 mM), KCl (5 mM), CaCl2 (2.5 mM), MgCl2 (2.5 mM), cystein (Cys, 1 mM), glutamic acid (Glu, 1 mM), vitamin C (1 mM), glucose (1 mM), BSA (10 μM), NaSH (10 μM), H2O2 (10 μM), NaClO (10 μM), nitroreductase (NTR, 5 mg mL−1) and NADH (50 μM) after treatment with 1b for 40 min in PBS buffer (pH = 7, T = 37 °C); λex = 415 nm. | ||
The reaction of substrate 1b with nitroreductase was followed at different concentrations of both components. Thus, the fluorescence intensity increased strongly with increasing concentration of compound 1b (Fig. 3A and Fig. S7, ESI†). Furthermore, the conversion of substrate 1b to the reaction product was accelerated when the nitroreductase concentration was increased from 1.25 μg mL−1 to 2.5 μg mL−1; however, no further increase of the reaction rate was observed at higher concentrations, i.e. 5.0 μg mL−1, of the enzyme (Fig. 3A). Only at very low nitroreductase concentrations (c < 0.2 μg mL−1), an essentially linear relationship between signal intensity and nitroreductase concentration was observed, that was further utilized to determine a detection limit for the enzyme of 6 ng mL−1 (r2 = 0.996; Fig. S4B, ESI†),21 which lies within the range of most reported nitroreductase probes (0.79–48 ng mL−1).5,9–11
To exclude chemical conditions that can cause false positive signals, the fluorimetric response of probe 1b was examined towards components that may be encountered at physiological conditions, namely NaCl, KCl, CaCl2, MgCl2, L-cystein, L-glutamic acid, ascorbic acid, glucose, BSA, NaSH, H2O2, NaClO and NADH. Notably, other than with nitroreductase, no significant changes of the emission intensity of solutions of substrate 1b were observed in the presence of these tested substances (Fig. 3B), which points to an undisturbed fluorescence light-up effect induced by the enzyme.
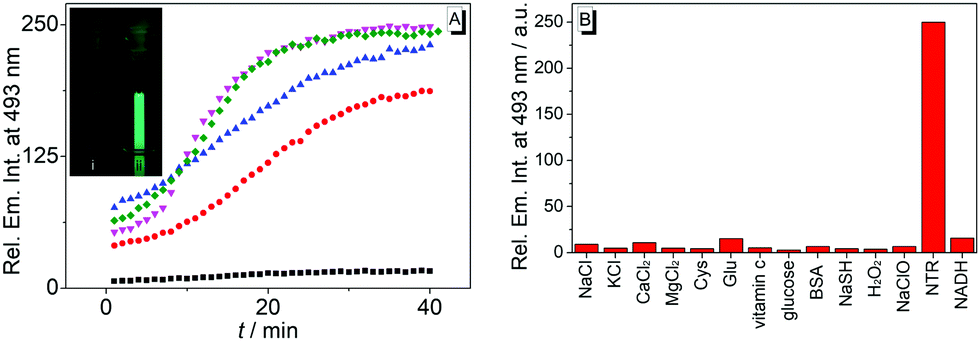
Strikingly, the absorption and emission spectra obtained after reaction of substrate 1b with nitroreductase/NADH did not resemble the ones of the expected product 1c. Instead, the main product exhibited a blue-shifted emission band and a red-shifted absorption as compared with the spectra of 1c.18 But extraction of the aqueous reaction mixture with organic solvent and exposure of the extract to aerobic conditions for 24 h revealed the subsequent formation of the aminobenzoquinolizinium 1c under these conditions, as unequivocally shown by the emission band of this compound and comparison with the one of an authentic sample (Fig. S6A, ESI†). These observations indicate that not only the nitro group, but also the benzoquinolizinium unit is reduced by nitroreductase. Indeed, it is well known that benzo[b]quinolizinium derivatives can be reduced with hydride- or hydrogen-donating reagents, such as LiAlH4, NaBH4, H2/PtO2 or H2/Pd to the dihydro- and tetrahydroquinolizinium derivatives;22–24 however, these products have been reported as colorless compounds. Yet, the deprotonation of 6,11-dihydrobenzoquinolizinium has been shown to give indirectly the reduction product 6H-benzo[b]quinolizine as red to purple-red compound.23 Unfortunately, spectroscopic data are not available from resembling chromophores/fluorophores in the literature. But at least preliminary TD-DFT calculations revealed absorption bands, without fine structure, for compound 4 that lie in the same range as the ones observed for the primary reduction product of 1b (Fig. S6B, ESI†). Based on the above-mentioned observations, we propose that the nitro-substituted substrate 1b is reduced to the 9-amino-6H-benzo[b]quinolizine (4) by nitroreductase and NADH (Scheme 1).
 | ||
| Scheme 1 Nitroreductase-mediated reaction of 9-nitrobenzo[b]quinolizinium (1b) to the proposed product 4 and subsequent oxidation to 9-aminobenzo[b]quinolizinium (1c) upon release of 4 from the binding site. | ||
The initially obtained organic-phase extract from the reaction mixture still showed the emission band of the primary product 4 directly after extraction and no traces of 1c. In another control experiment, it was also shown that the absorption and emission properties of the primary product did not change when it was stored in solutions that still contained nitroreductase and NADH for 24 h. Additionally, the amino-substituted derivative 1c was not reduced by nitroreductase/NADH, which indicates that 1c is not sensitive towards reduction under these conditions and that it is not formed as intermediate in the reaction of 1b with nitroreductase. In contrast, treatment of the nitro- or amino-substituted derivatives 1b and 1c with chemical reducing agents NaBH4 or H2/Pd,C and photometric and fluorimetric monitoring of the reaction indicated the reduction to hexahydro- and tetrahydrobenzo[b]quinolizine derivatives as already reported for the parent benzo[b]quinolizinium.22 Only in the case of the reduction of 1b with NaBH4 a similar emission as the one observed on treatment with nitroreductase was temporarily detected in the course of the reaction (Fig. S6C, ESI†), but it was rather weak and slightly red-shifted, presumably due to the different employed solvent systems.
Altogether, the experimental observations point to a specific transformation of the nitrobenzoquinolizinium 1b to 9-amino-6H-benzo[b]quinolizine (4) by nitroreductase, because it is not formed and fairly unstable under different conditions. Therefore, it may be assumed that the primary product 4 still binds to the enzyme and is protected within the binding site against further reduction within the reaction mixture (Scheme 1). This assumption is supported by CD-spectroscopic monitoring of the reaction, because the CD spectrum of the enzyme in the reaction mixture changed slightly during the reaction (Fig. S5A, ESI†), which may be caused by the association with the reaction product.25 Furthermore, the addition of acetamide as competing nitroreductase-binding ligand26 to the reaction mixture led to a slow decrease of the emission maximum at 493 nm of intermediate 4 in favor of the red-shifted band of the amine 1c at 510 nm (Fig. S5B, ESI†). This change of the emission spectrum indicates the formation of compound 1c by displacement of 4 from the enzyme and subsequent oxidation. Overall, these observations lead to a plausible mechanism, that starts with the initial reduction of the substrate 1b to the 9-amino-6H-benzo[b]quinolizine within the active site of the enzyme without formation of the aminobenzoquinolizinium 1c (Scheme 1). Presumably, the reduction of the heteroaromatic core takes place prior to or during the reduction of the nitro group already. Only upon release from the binding site the reactive intermediate 4 is oxidized to the final product 1c. To the best of our knowledge, such a sequence of overreduction and re-oxidation that is governed by association and dissociation of the nitroarene substrate has not been reported for nitroreductase reductions.
Lastly, the potential of probe 1b to detect nitroreductase activity in living cells was investigated by fluorimetric analysis of E. coli bacteria, which are known to exhibit a high nitroreductase activity.7 The E. coli cultures were treated with solutions of probe 1b and exhibited a strong green emission after incubation for 4 h (Fig. 4 and Fig. S9, ESI†) and did not show a bactericidal effect, even after 24 h, as shown by a high viability of E. coli suspensions (Fig. S10, ESI†), while previous reports have indicated a moderate growth inhibition on eukaryotic cells.27 Fluorimetric analysis showed that compound 1c was formed as the main emitting species in E. coli, as clearly indicated by a complete match of the emission and excitation spectra with the ones of an authentic sample (Fig. S9C, ESI†). Based on the observation in vitro, the fluorimetric response results from the initial formation of intermediate 4, which is subsequently displaced from the enzyme by competing ligands in the cells, such as acetate.26 Finally, the intermediate 4 is transformed into 1c within the bacteria, as already observed in vitro upon displacement of the nitroreductase bound compound 4 by acetamide (Fig. S5B, ESI†). Since the conversion of 4 into compound 1c is irreversible under these conditions, even small amounts of oxygen or other oxidants like NAD+ or NADP+, which are also present in E. coli or hypoxic cells in low concentration4 are apparently sufficient to convert intermediate 4 into derivative 1c.
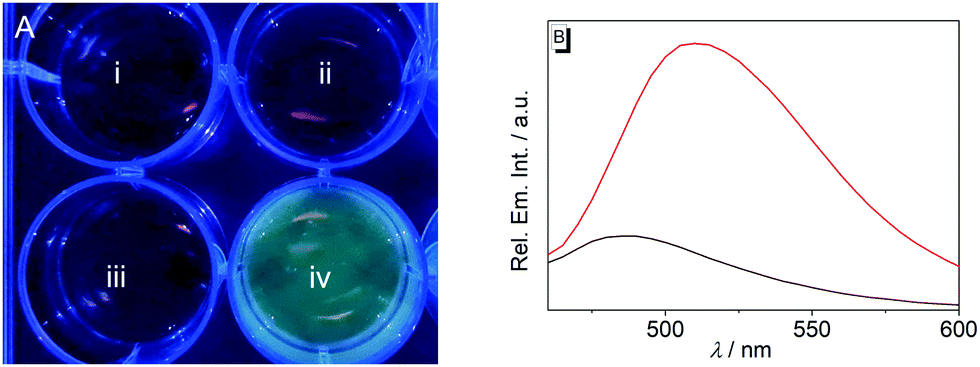 | ||
| Fig. 4 (A) Photograph (acquired under illumination with λ = 365 nm) of the emission color of Tris-HCl buffer (i), E. coli culture in Tris-HCl buffer (ii), 20 μM probe 1b in Tris-HCl buffer (iii) and E. coli culture in Tris-HCL buffer after incubation with 20 μM probe 1b (iv) for 4 h. (B) Relative emission intensity of E. coli culture with an optical density of OD600 = 0.2 in the absence of probe 1b (black) and after incubation with 20 μM probe 1b for 4 h (red) in Tris-HCl buffer; λex = 389 nm. | ||
Conclusions
In summary, it was demonstrated that the nitrobenzoquinolizinium 1b has the potential to act as a probe for the fluorimetric detection of nitroreductase with a comparatively good sensing performance.5,9–11 The fluorogenic reaction in the enzyme binding site proceeds on formation of an “over-reduced” intermediate 4 that is eventually transformed to the fluorescent 9-aminobenzo[b]quinolizinium (1c) upon release from the binding site. These results shall constitute the starting point for the development of a novel class of water-soluble, nitroreductase-targeting fluorescent probes based on the benzo[b]quinolizinium fluorophore, which can be utilized for the fluorimetric detection of the hypoxia status of cells. Specifically, the formation of two different emitting species at different stages of the incubation, i.e.4 and 1c, constitutes a special and so far unparalleled feature of this system that may be used to monitor nitroreductase activity at different emission wavelengths.Author contributions
Experiments and writing (P. J. Wickhorst); experiments (M. M. Lammert-Baumgartner); supervision and writing (M. Müller, H. Schönherr, H. Ihmels).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the Deutsche Forschungsgemeinschaft and the University of Siegen is gratefully acknowledged. We thank Bernd Meyer, Marcus Raabe and Christoph Dohmen for technical assistance.Notes and references
- T. McGarry, M. Biniecka, D. J. Veale and U. Fearon, Free Radical Biol. Med., 2018, 125, 15 CrossRef CAS PubMed
.
- P. Ferdinand and C. Roffe, Exp. Transl. Stroke Med., 2016, 8, 1 CrossRef PubMed
- A. Solhpour, A. Jafari, M. A. Pourhoseingholi and F. Soltani, Trends Anaesth. Crit. Care, 2020, 34, 63 CrossRef
- A. Sharma, J. F. Arambula, S. Koo, R. Kumar, H. Singh, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2019, 48, 771 RSC
- J.-N. Liu, W. Bu and J. Shi, Chem. Rev., 2017, 117, 6160 CrossRef CAS PubMed
- S. E. Cellitti, J. Shaffer, D. H. Jones, T. Mukherjee, M. Gurumurthy, B. Bursulaya, H. I. Boshoff, I. Choi, A. Nayyar, Y. S. Lee, J. Cherian, P. Niyomrattanakit, T. Dick, U. H. Manjunatha, C. E. Barry, G. Spraggon and B. H. Geierstanger, Structure, 2012, 20, 101 CrossRef CAS PubMed
- M. D. Roldán, E. Pérez-Reinado, F. Castillo and C. Moreno-Vivián, FEMS Microbiol. Rev., 2008, 32, 474 CrossRef PubMed
- K. Durchschein, M. Hall and K. Faber, Green Chem., 2013, 15, 1764 RSC
- B. Brennecke, Q. Wang, Q. Zhang, H.-Y. Hu and M. Nazaré, Angew. Chem., Int. Ed., 2020, 132, 8512 CrossRef PubMed
- S. Karan, M. Y. Cho, H. Lee, H. Lee, H. S. Park, M. Sundararajan, J. L. Sessler and K. S. Hong, J. Med. Chem., 2021, 64, 2971 CrossRef CAS PubMed
- R. Peng, J. Yuan, D. Cheng, T. Ren, F. Jin, R. Yang, L. Yuan and X. Zhang, Anal. Chem., 2019, 91, 15974 CrossRef CAS
-
(a) Z. He, Y. Chou, H. Zhou, H. Zhang, T. Cheng and G. Liu, Org. Biomol. Chem., 2018, 16, 3266 RSC
- K. N. More, T.-H. Lim, J. Kang, H. Yun, S.-T. Yee and D.-J. Chang, Molecules, 2019, 24, 3206 CrossRef CAS PubMed
- Q. Yang, S. Wang, D. Li, J. Yuan, J. Xu and S. Shao, Anal. Chim. Acta, 2020, 1103, 202 CrossRef CAS PubMed
- L. Fan, Q. Zan, B. Lin, X. Wang, X. Gong, Z. Zhao, S. Shuang, C. Dong and M. S. Wong, Analyst, 2020, 145, 5657 RSC
- S. Sarkar, H. Lee, H. G. Ryu, S. Singha, Y. M. Lee, Y. J. Reo, Y. W. Jun, K. H. Kim, W. J. Kim and K. H. Ahn, ACS Sens., 2021, 6, 148 CrossRef CAS PubMed
-
(a) S. Schramm and D. Weiß, Adv. Heterocycl. Chem., 2019, 128, 103 CrossRef CAS
- K. Faulhaber, A. Granzhan, H. Ihmels, D. Otto, L. Thomas and S. Wells, Photochem. Photobiol. Sci., 2011, 10, 1535 CrossRef CAS PubMed
- G. J. S. Zugazagoitia, C. X. Almora-Diaz and J. Peon, J. Phys. Chem. A, 2008, 112, 358 CrossRef PubMed
-
(a) G. E. Gomez Pinheiro, H. Ihmels and C. Dohmen, J. Org. Chem., 2019, 84, 3011 CrossRef CAS PubMed
- Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem., Int. Ed., 2013, 52, 1688 CrossRef CAS PubMed
- T. Miyadera and R. Tachikawa, Tetrahedron, 1969, 25, 5189 CrossRef CAS
- L. L. Braun and C. K. Bradsher, J. Org. Chem., 1968, 33, 1296 CrossRef CAS
- C. Bradsher and N. Yarrington, J. Org. Chem., 1960, 25, 294 CrossRef CAS
-
A. Rodger, R. Marrington, D. Roper and S. Windsor, in Protein-Ligand Interactions, ed. G. U. Nienhaus, Humana Press, New Jersey, 2005, pp. 343–364 Search PubMed
- P. R. Race, A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton and E. I. Hyde, J. Biol. Chem., 2005, 280, 13256 CrossRef CAS PubMed
- For cytotoxicity of the reduction product 1c, see: A. K. Ernst, M. Liebscher, S. Mathea, A. Granzhan, J. Schmid, M. R. Popoff, H. Ihmels, H. Barth and C. Schiene-Fischer, Sci. Rep., 2016, 6, 20301 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional spectroscopic data, NMR spectra of 1b and 1d, bacterial viability test. See DOI: 10.1039/d1nj05230f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |