
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransparent, self-recoverable, highly tough, puncture and tear resistant polyurethane supramolecular elastomer with fast self-healing capacity via “hard–soft” hard domain design†
Kangming Xu,
Guoqing Chen,
Mingjie Zhao,
Weiyi He,
Qiaoman Hu * and
Yong Pu
* and
Yong Pu
College of Materials Science and Engineering, Chongqing University of Arts and Sciences, Chongqing 402160, China. E-mail: qmhuwit@163.com
First published on 20th January 2022
Abstract
The integration of superior mechanical properties and fast healing efficiency for self-healing polyurethane supramolecular elastomers is challenging due to the confliction between high chain mobility for healing and high chain rigidity for mechanical properties. Herein, a strategy to design a “hard–soft” hard domain by the cooperation of quadruple hydrogen bonds (HBs) in the mainchain as restriction units and single HBs in the side chain as diffusion units is reported. The resulting transparent supramolecular elastomer exhibited fast self-recoverability, good puncture resistance and superior mechanical properties with a tensile strength of 20.5 MPa, an extensibility of 2043.7%, a toughness of 146.1 MJ m−3 and a tear resistance of 13.8 kJ m−2. Moreover, the fast self-healing capacity (healing efficiency > 82% within 3 h under moderate condition) was realized due to the soft effects of weak HBs in the side chain on the strong HBs in the mainchain. Taking advantage of the merits of the supramolecular elastomer, a flexible sensor was simply fabricated, which showed good self-repairable and stable sensing properties. Thus, the elastomer has great potential in the field of flexible electronics and wearable devices.
Introduction
Self-healing polymeric materials with the ability to repair cracks and restore original functions, have been widely developed to improve service reliability, extend service lifetime and reduce material consumption and environmental pollution.1–4 As such, these promising materials show great value in numerous fields, including flexible electronics,5–9 soft robotics,10,11 protective coatings12,13 and healthcare devices.14–17 To put self-healing polymeric materials into use in the above fields, the key factor lies in the contradiction between fast healing efficiency and good mechanical properties of the materials.18–20 The high diffusibility and exchange of the healing agent for fast healing conflict with the good chain rigidity, entanglement and crystallinity of materials for good mechanical properties.21–23 The integration of good mechanical properties and fast healing efficiency for self-healing polymeric materials is a challenging task, which attracts great interest in both academia and industry.Among numerous self-healing polymeric materials, elastomer especially polyurethane (PU) is the most suitable candidate to meet the aforementioned requirements.24 PU usually exhibits two phases: soft matrix and hard domains. The soft matrix has segments with low glass transition temperature (Tg), which endows the material with deformability. The hard domains have segments with strong interactions, which provides the material with good shape fixity.25–27 At the same time, lots of macromolecular diols and vast amounts of diisocyanates and chain extenders can be used as soft and hard segments, respectively. Thus, the desired mechanical and healing properties may be obtained by adjusting the composition or ratio of soft and hard segments.
Basically, two kinds of interactions are often introduced into the hard segments of PU to regulate the self-healing property. One is the non-covalent interaction, including hydrogen bonds (HBs),18,19,22,24,28–32 metal–ligand coordination,33,34 ionic interactions,35 π–π stacking,36 host–guest interactions37 and van der Waals forces.38 The other is the dynamic covalent interaction such as Diels–Alder reaction,39 disulfide bond,40 transesterification,41 siloxane exchange,42 olefin metathesis,43 hindered urea bond,44 boronic ester45 and so on. Among these, HBs have attracted significant attention in terms of binding energy and self-healing condition. Numerous efforts have been done to fabricate fascinating PU with good mechanical and self-healing property through HBs.19,32,46–54 For example, Kang et al. introduced multistrength (weak and strong) HBs into poly(urethane urea), which resulted in the elastomer with high stretchability, high toughness, notch-insensitive and water-insensitive self-healing.32 Xu et al. proposed a “multiphase active HBs” strategy to create a loosely packed hard segment of poly(urethane urea), which also resulted in a ultra-stretchable, notch-insensitive and fast self-healing elastomer.46 Based on this, Wang et al. further putted forward the “dynamic hard domains” concept. A poly(urethane urea) with decent mechanical property, outstanding self-recoverability and self-healing property was fabricated.47 However, the tensile strength of the above elastomers was somewhat not good enough because of the relatively low binding energy of the hard segments. By adopting quadruple HBs with high binding energy as side chain in the hard segments, Song et al. reported a healable PU with remarkable tensile strength. However, the healing efficiency of the elastomer was relatively low.19 To the best of our knowledge, the simultaneous improvement of the tensile strength, stretchability and self-healing property is difficult to achieve by the reported hard segments regulation, because of the natural limitation of the single type of the hard segments.
Herein, based on the different demand of the mechanical and self-healing properties to the segments, we propose a strategy to improve the two properties simultaneously through the “hard–soft” hard domain design. The quadruple HBs in the mainchain with high binding energy form “hard” hard domain and act as strong physical crosslinking points, which provide not only high tensile strength, but also good shape fixity. The single HBs in the side chain with low binding energy form “soft” hard domain, which diffuse and exchange easily and result in the fast healing efficiency at a moderate condition.55,56 Moreover, by the cooperation of the two different hard segments, the elastomer with high toughness, puncture and tear resistance is fabricated. Besides, the self-healing property of the elastomer in the applied flexible electronic is also simply examined.
Experimental section
Materials
Guanidine carbonate, ethyl acetoacetate, alpha-acetyl-gamma-butyrolactone, triethylamine, ethylene glycol monoallyl ether, 1-thioglycerol, 2,2-dimethoxy-2-phenylacetophenone, hexamethylene diisocyanate, polytetramethylene ether glycol (PTMEG, Mn = ∼1000 g mol−1), ditinbutyl dilaurate (DBTDL) were purchased from Aladdin Chemistry Regent Co., Ltd (China). Triethylene glycol bis(3-tert-butyl-4-hydroxy-5-methylphenyl)propionate (AO-70) was obtained from Beijing Additive Research Institute (China). Hydroxyl groups modified multi-walled carbon nanotubes (MWCNTs-OH) with hydroxyl content of 1.85 wt% was purchased from Chengdu Organic Chemicals Co. Ltd (China). 4,4′-Dicyclohexylmethane diisocyanate (HMDI) was kindly provided by Wanhua Chemical Group Co., Ltd (China). Absolute alcohol, sodium hydroxide, dichloromethane, hydrochloric acid, p-toluene sulfonic acid, cyclohexane, sodium bicarbonate, tetrahydrofuran, anhydrous sodium sulfate, acetone, petroleum ether and dimethyl formamide (DMF) were purchased from Chengdu Kelong Chemical Reagent Co. Ltd (China). DMF was dehydrated with activated 4A molecular sieves for at least one week before use, the other reagents were used as received.Characterizations
Nuclear magnetic resonance spectroscopy (1H-NMR, 400 Hz) was recorded on a Bruker Avance III 400 spectrometer (Bruker Daltonics Inc., Germany). Fourier transform infrared spectroscopy (FT-IR) was collected from 4000 to 400 cm−1 with a resolution of 2 cm−1 using a PerkinElmer Spectrum Two instrument (PerkinElmer, USA) fitted with KBr Pellets or the attenuated total reflectance (ATR) accessory at room temperature. Molecular weight (Mn and Mw) and polydispersity index (PDI) were determined by a LC20 gel permeation chromatography (GPC) instrument (Shimadzu, Japan) with DMF or chloroform as mobile phase. The sample concentration was approximately 10 mg ml−1, with an injection volume of 10 μl. The X-ray diffraction (XRD) was measured from 5° to 50° on a Tongda TD-3500X X-ray diffractometer (Dandong Tongda Instrument Co., China). Thermo gravimetric analysis (TGA) was performed on the STA 449 F3 instrument (Netzsch, Germany). PU and MWCNTs samples of about 5–10 mg were heated from 30 to 800 °C and 30 to 1000 °C at a heating rate of 20 °C min−1 in nitrogen atmosphere, respectively. Differential scanning calorimetry (DSC) was performed on the Q200 instrument (TA, USA). Samples of about 5–10 mg were first heated from room temperature to 200 °C, then cooled to −80 °C, finally heated to 220 °C at a heating and cooling rate of 10 °C min−1 in nitrogen atmosphere. The glass transition temperature (Tg) was obtained from the second heating scan. The transmittance of PU films was measured by the Cary series UV-VIS-NIR spectrophotometer (Agilent Technologies, USA) over a spectral range from 200 nm to 800 nm. The scratch recovery was observed under an DMM-200C optical microscope (Caikon, China) equipped with a high precision heating stage device. The responses of the PU based sensor against multiple stimuli were measured by the CHI 760E electrochemical workstation (CH Instruments, China).The tensile tests were measured at room temperature using a tensile tester (Teststar, Wance, China). Rectangle samples were prepared with a gauge region of 8 mm × 6.5 mm × 0.7 mm. At least three individual tensile tests were measured for each sample. The strain rate was 100 mm min−1. Tensile toughness of the samples can be defined by integrating the area under the engineering stress–strain curves. For the cyclic tensile test at a constant strain of 500%, five cyclic were performed with a strain rate of 100 mm min−1 at room temperature. Besides a relaxation sample was performed after the second cycle and relaxed at room temperature for 1 h. In addition, five cyclic tensile tests with a variable strain from 100% to 500% were also performed. The fracture energy test was conducted by the tensile test using the single-edge notched sample (length of the slit is 1 mm). The notched and unnotched specimens (gauge region of 10 mm × 5 mm × 0.7 mm) were both tested at a strain rate of 100 mm min−1. To assess the healing efficiency, the rectangle samples were cut off with a knife in the middle and then brought into contact immediately, followed by keeping the samples at different temperatures for different times. The healed samples were further measured by the tensile test with a strain rate of 100 mm min−1 at room temperature.
Synthesis of PU elastomers
Taking PU-HM4-HP6 as an example, the synthesis process (Fig. S14†) is as follows: PTMEG (20 g, 20 mmol) in a dried flask equipped with a mechanical stirrer was heated at 120 °C under vacuum of 60 Pa for 2 h to remove residual moisture. After cooled to 85 °C, HMDI (9.81 ml, 40 mmol) was injected into the flask and stirred for 1 h with a stirring rate of 220 rpm under nitrogen atmosphere. After cooled to 80 °C, DBTDL (2 drop) was injected into the flask and stirred for another 3 h. Then, 5-(2-hydroxyethyl)-6-methyl-2-aminouracil (HMIC, 1.352 g, 8 mmol, Fig. S1 and S2†) was added into the flask and stirred for 0.5 h at a stirring rate of 250 rpm. After that, a mixture of 3-(2-((2,3-dihydroxypropyl)thio)ethoxy)propyl 3-(3-(tert-butyl)-4-hydroxy-5-methylphenyl)propanoate (GL, 5.139 g, 12 mmol, heated at 100 °C under vacuum of 60 Pa for 2 h to remove residual moisture, Fig. S3–S8†) and DMF (20 ml) was injected into the flask and stirred for another 3 h. The resulting solution was poured into a Teflon mold and transferred into an oven, which was kept at 80 °C for 24 h. Then the mold was transferred into a vacuum and kept at 70 °C for 24 h to remove residual solvent, resulting in PU-HM4-HP6. The molar ratio of the raw materials of all PU-HM-HP was summarized in Table 1.| Sample name | PTMEG (mmol) | HMDI (mmol) | HMIC (mmol) | GL (mmol) |
|---|---|---|---|---|
| PU-HM5-HP5 | 20 | 40 | 10 | 10 |
| PU-HM4-HP6 | 20 | 40 | 8 | 12 |
| PU-HM3-HP7 | 20 | 40 | 6 | 14 |
| PU-HM2-HP8 | 20 | 40 | 4 | 16 |
| PU-HM1-HP9 | 20 | 40 | 2 | 18 |
| PU-HM0-HP10 | 20 | 40 | 0 | 20 |
Fabrication of films for mechanical properties testing
A mixture of PU-HM-HP (12 g) and DMF (200 ml) was added into a dried flask and stirred with a stirring rate of 400 rpm at room temperature for 12 h. The resulting solution was poured into a Teflon mold with dimensions of 130 mm × 130 mm × 60 mm. The mold was transferred into an oven and kept at 80 °C for 24 h, then was transferred into a vacuum and kept at 70 °C for 48 h, which resulted a PU-HM-HP film for mechanical properties testing with a thickness of about 0.7 mm.Fabrication of self-healing sensor
To a glass plate the MWCNTs-UPy (Fig. S9–S13†) solution was coated for two times and drying with an infrared lamp. Then the solution of PU-HM4-HP6 in DMF (30 wt%) was poured onto the MWCNTs-UPy coated glass plate and dried at room temperature for 6 h. After that, the glass plate was transferred into an oven and kept at 80 °C for 24 h, then was transferred into a vacuum and kept at 70 °C for 24 h. Finally, the sensor was peeled off from the glass plate.Results and discussion
To synchronously regulate the mechanical and self-healing properties, HMIC and hindered phenol (HP) units were introduced into the mainchain and side chain of PU-HM-HP elastomer, respectively (Fig. 1a). The chemical structures of the elastomer were confirmed by the 1H-NMR and FT-IR analysis (Fig. S15 and Table S1†). HMIC can form quadruple HB interactions with an AADD–DDAA (A-proton acceptor, D-proton donor) type, which exhibits a high dimerization constant (>107 L mol−1) and excellent stability.57,58 Thus, HMIC acted as strong physical crosslinking points in the elastomer with the combination of rigid HMDI (Fig. 1b). HP units can form single HB interactions, which shows a weak binding energy and strong exchangeability.59 By attaching HP units to the end of a long side chain, the easy diffusion of HP units was achieved in the elastomer. Besides, the excessive entanglement of chains in the elastomer was also weaken due to the long side chain (Fig. 1b). Further, the influence of the two components on the microstructure of the elastomer was studied by FT-IR. The stretching vibration region of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of PU-HM-HP elastomers can be divided into four bands by using Gauss Lorzne peak fitting (Fig. S16†). The bands appeared at about 1720 cm−1, 1695 cm−1, 1665 cm−1 and 1635 cm−1 are attributed to free urethane CO groups, H-bonded urethane CO groups (and free urea CO groups), disordered H-bonded urea CO groups and ordered H-bonded urea CO groups, respectively.19 PU-HM-HP samples with high HP content showed a high intensity of the free and H-bonded urethane CO groups and an extremely low intensity of the ordered and disordered H-bonded urea CO groups (Fig. S17†), which indicated a “soft and weak” hard domain. The situation of samples with high HMIC content was opposite, which indicated a “hard and strong” hard domain. By the cooperation of the “soft and weak” hard domain and the “hard and strong” hard domain, PU-HM-HP elastomers with desired properties were achieved. For example, the average transmittance of the colorless PU-HM-HP films under visible range from 400 to 800 nm was about 90% (Fig. 1c). Moreover, PU-HM-HP exhibited excellent repeatable deformation recovery and superior puncture resistance. The shape of the 0.7 mm thick sample was almost completely recovered within 30 s from an elongation of 650% (Fig. 1d and S18†) and the 0.7 mm thick sample was not pierced even punctured up to an elongation of 500% (Fig. 1e and S19†).
O group of PU-HM-HP elastomers can be divided into four bands by using Gauss Lorzne peak fitting (Fig. S16†). The bands appeared at about 1720 cm−1, 1695 cm−1, 1665 cm−1 and 1635 cm−1 are attributed to free urethane CO groups, H-bonded urethane CO groups (and free urea CO groups), disordered H-bonded urea CO groups and ordered H-bonded urea CO groups, respectively.19 PU-HM-HP samples with high HP content showed a high intensity of the free and H-bonded urethane CO groups and an extremely low intensity of the ordered and disordered H-bonded urea CO groups (Fig. S17†), which indicated a “soft and weak” hard domain. The situation of samples with high HMIC content was opposite, which indicated a “hard and strong” hard domain. By the cooperation of the “soft and weak” hard domain and the “hard and strong” hard domain, PU-HM-HP elastomers with desired properties were achieved. For example, the average transmittance of the colorless PU-HM-HP films under visible range from 400 to 800 nm was about 90% (Fig. 1c). Moreover, PU-HM-HP exhibited excellent repeatable deformation recovery and superior puncture resistance. The shape of the 0.7 mm thick sample was almost completely recovered within 30 s from an elongation of 650% (Fig. 1d and S18†) and the 0.7 mm thick sample was not pierced even punctured up to an elongation of 500% (Fig. 1e and S19†).
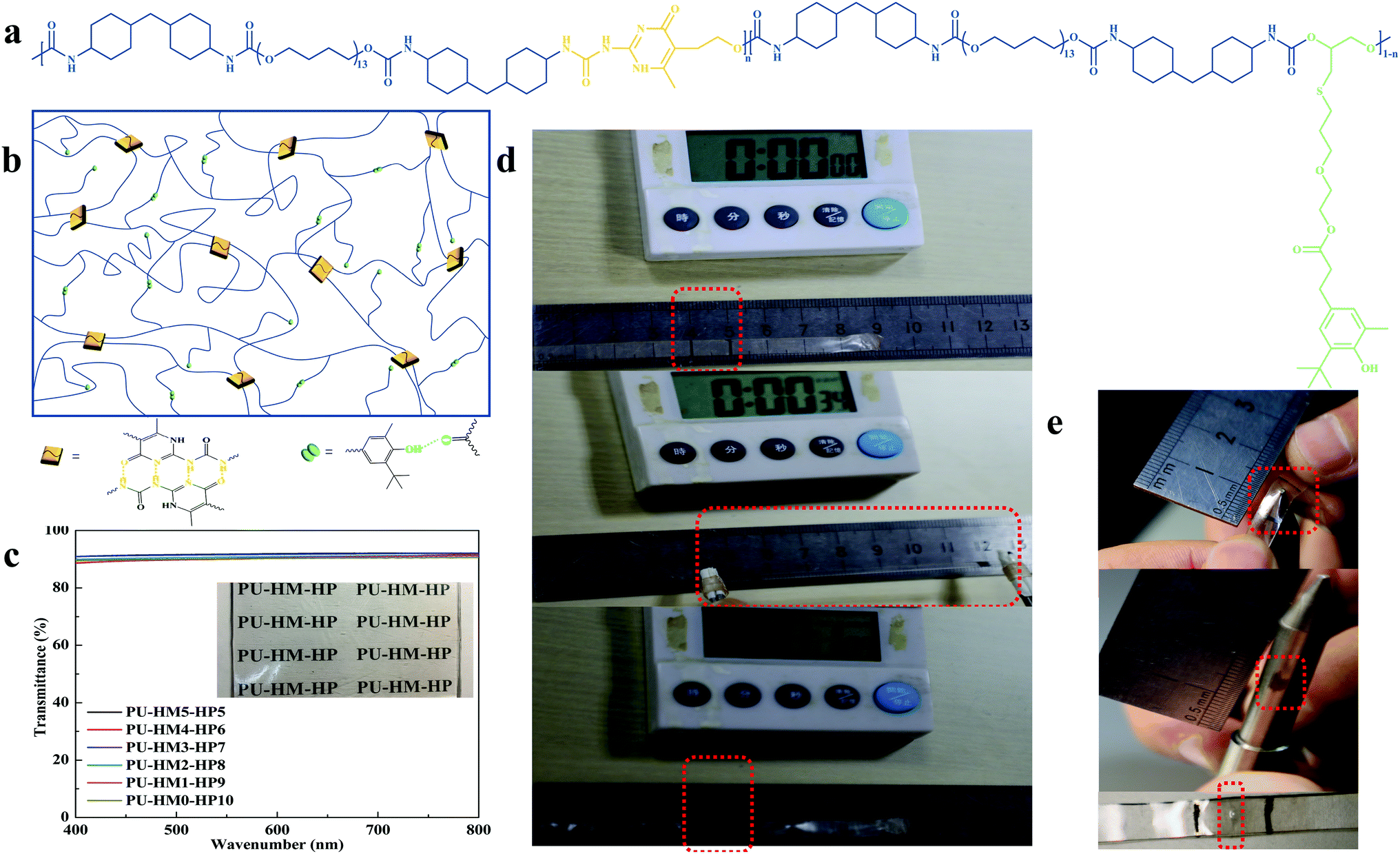 | ||
| Fig. 1 Schematic and performance of PU-HM-HP elastomer. (a) Chemical structure of PU-HM-HP. (b) Schematic illustration of microphase-separated structure of PU-HM-HP. (c) Transmittance spectrum of PU-HM-HP film with a thickness of 180 μm. Inset photograph of the film is transparent and colorless. (d) Optical images of PU-HM4-HP6 that can recover its original length after being stretched to 650% strain and (e) optical images of PU-HM4-HP6 that can tolerate puncture. | ||
In addition, basic characterization experiments of PU-HM-HP were shown in the ESI.† The number-average molecular weight of the elastomers ranged from 61 to 91 kDa with polydispersity indexes of about 1.98 (Fig. S20 and Table S2†). The Tg of the elastomers was detected at about −45 °C and neither exothermic nor endothermic peaks were observed in the DSC curve, which indicated the noncrystalline hard domains in PU-HM-HP (Fig. S21†). The XRD results further confirmed the amorphous structure of PU-HM-HP (Fig. S22†). The initial decomposition temperature was about 300 °C, which indicated excellent thermal stability of PU-HM-HP (Fig. S23†).
The mechanical properties of PU-HM-HP elastomers with different HM/HP molar ratio were quantitatively examined via uniaxial tensile testing. Typical strain–stress curves of the elastomers were shown in Fig. 2a and all PU-HM-HP samples showed rubber-like properties. PU-HM0-HP10 exhibited a young's modulus of 0.283 ± 0.016 MPa, a tensile stress of 1.383 ± 0.134 MPa and an elongation of 1536.246 ± 114.104% (Fig. 2b and c), which was attributed to the weak restraint of the “soft” HP-type side chain dominated hard domains to the elastomer. With HM/HP molar ratio increasing, the young's modulus and tensile stress gradually increased, which was attributed to the strong crosslinking effects of the “hard” HMIC dominated hard domains. In particular, the young's modulus showed a significant improvement from 1.896 ± 0.082 MPa of PU-HM4-HP6 to 2.670 ± 0.143 MPa of PU-HM5-HP5 (Fig. 2b). Associated with the strain-induced crystallization phenomenon only observed in PU-HM5-HP5 system during tensile testing (not presented in this paper) and the significant increase of the intensity of the ordered H-bonded urea CO groups from PU-HM4-HP6 to PU-HM5-HP5 as well as the highest intensity of disordered H-bonded urea CO groups of PU-HM4-HP6 (Fig. S17†), the relatively regular alinement of chains induced by the strong restriction of HMIC hard domains was generated in PU-HM5-HP5 system, which resulted a tensile stress of 27.237 ± 1.051 MPa.60 Although the elongation at break showed a fluctuation change along with HM/HP molar ratio variation, the elongation of all PU-HM-HP samples was higher than 1450%, which was mainly attributed to the weakened entanglement of the chains and the “sacrificial bond” effects of the weak HBs. Typically, PU-HM4-HP6 exhibited the highest elongation of 2043.749 ± 136.098% with a relatively high tensile strength of 20.531 ± 0.528 MPa, which resulted in a toughness of 146.097 ± 8.241 MJ m−3. PU-HM5-HP5 exhibited the elongation of 1831.124 ± 95.457% and resulted in the highest toughness of 211.935 ± 12.957 MJ m−3 (Fig. 2d). It is noteworthy that the toughness of our elastomers was higher than that of man-made fibers such as Kevlar 49 (50 MJ M−3), Nylon 66 (80 MJ M−3) and spider dragline silk (180 MJ m−3). Moreover, the toughness was comparable or even exceeded the record of some materials reported in the literature.24,28,31,37,55,61–66 Such superordinary toughness is beneficial for avoiding crack formation and prolonging service lifetime.
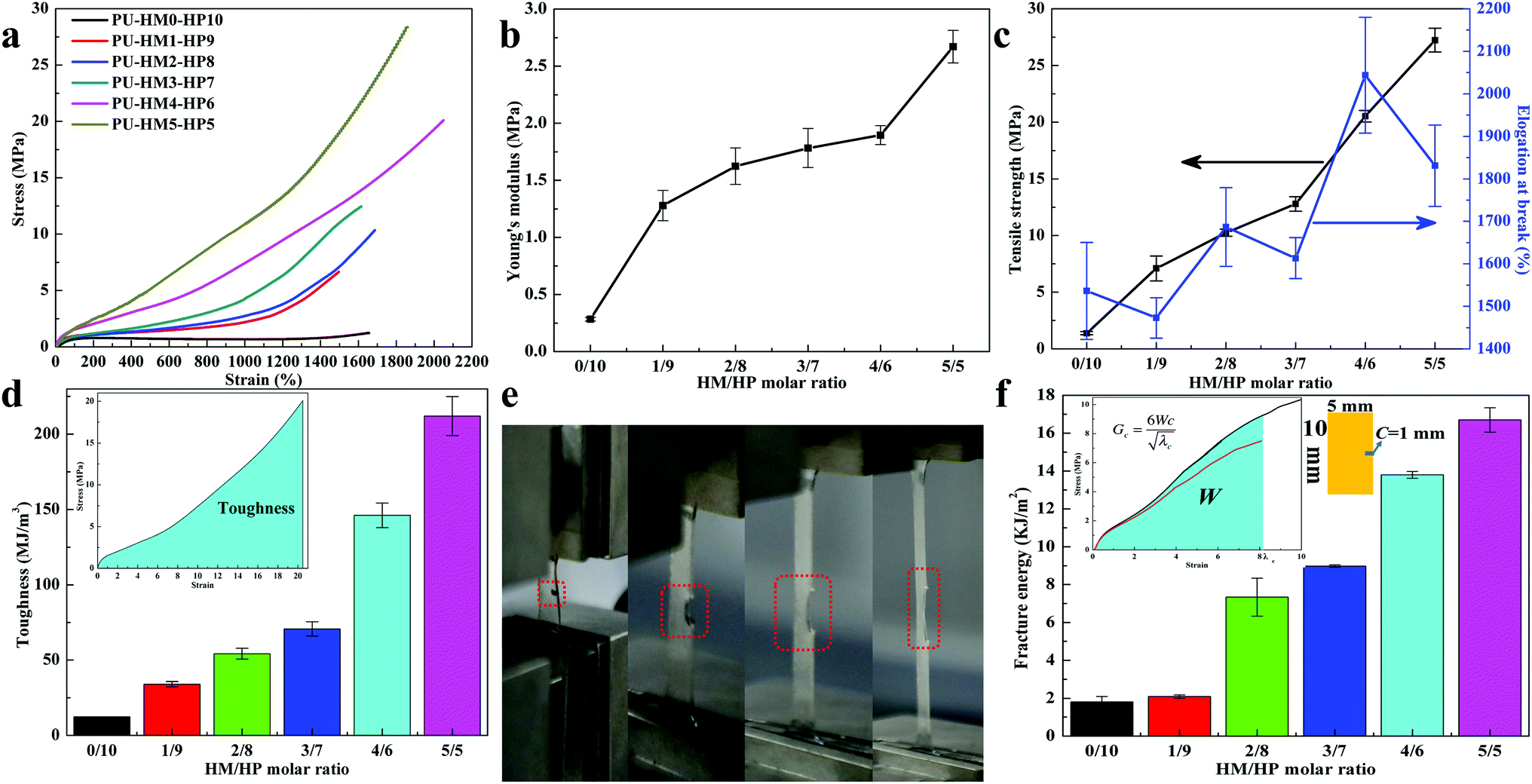 | ||
| Fig. 2 The mechanical properties of PU-HM-HP elastomers. (a) Strain–stress curves of PU-HM-HP with a loading velocity of 100 mm min−1. (b) Young's modulus variation of PU-HM-HP. (c) Tensile strength and elongation at break variation of PU-HM-HP. (d) Toughness variation of PU-HM-HP. Inset graph illustrates the calculation method of toughness. (e) Optical images of the single-edge-notched PU-HM4-HP6 sample from 0% to 800% strain and (f) fracture energy variation of PU-HM-HP. Inset graph illustrates the calculation method of fracture energy. | ||
Tear resistance of elastomers is also important for the protection of the relevant devices. As Fig. 2e and S24† shows, PU-HM4-HP6 with a cut notch could be stretched up to 8 times without fracture, which was also attributed to the weakened entanglement of the chains and the strong–weak HB network effects.19,32 With HM/HP molar ratio increasing, the fracture energy of PU-HM-HP system gradually increased (Fig. 2f). PU-HM4-HP6 and PU-HM5-HP5 showed a fracture energy of 13.798 ± 0.184 kJ m−2 and 16.698 ± 0.641 kJ m−2, respectively, which revealed the good tear resistance of the elastomers by comparing with natural rubber (fracture energy is about 10 kJ m−2).
The “sacrificial bond” effects of the weak HBs at small deformation and the crosslinking effects of the strong HBs at large deformation of PU-HM-HP allows energy dissipation at multiple scales. To reveal the role of different HBs in energy dissipation, five successive loading-unloading process with a strain of 500% in every cycle were performed (Fig. S25†). All PU-HM-HP exhibited large hysteresis loops in the first loading–unloading process and sharply decreased in the second loading–unloading process, which was attributed to the massive breaking of the multiple HBs. Then the hysteresis showed slight decrease in the following three loading–unloading process, which was mainly assigned to the recombination of the weak HBs. In each cycle, the higher HM/HP molar ratio, the larger the hysteresis area (Fig. 3a). The hysteresis difference between the first and second successive cyclic tensile testing (ΔW) represents the ability of chain rearrangement, a larger value represents the harder rearrangement because of the molecular interactions. The ratio between the integrated area in the hysteresis loop and that under the loading curve of the first cyclic tensile testing (W1/W) represents the efficiency of energy dissipation, a higher value represents the better efficiency. With HM/HP molar ratio increasing, the ΔW gradually increased due to the increasing restriction of the strong HBs. The W1/W first decreased then increased and PU-HM0-HP10 showed the highest value among the elastomers, which indicated that the efficiency of energy dissipation of the weak HBs was better than that of the strong HBs and the high density of the strong HBs was benefit for the efficiency (Fig. 3b). Besides, with more strong HBs, the stress difference between the first and second successive cyclic tensile testing at 500% strain and residual strain of the first cyclic tensile testing gradually increased and decreased, respectively (Fig. 3c). Dissipated energy during loading-unloading process under 100% to 500% strains was also examined (Fig. S26† and 3d). At low strain (100%), the hysteresis areas of all PU-HM-HP showed little difference, which was attributed to the preferentially breaking of weak HBs. With strain increasing, the difference of the hysteresis areas of PU-HM-HP became larger and larger, which was attributed to the different amounts of strong HBs dissociation at high strain. Thus, the effective energy dissipation process was achieved due to the combination of the weak sacrificial HBs and the strong restrictive HBs, which resulted in a tough PU-HM-HP elastomer.
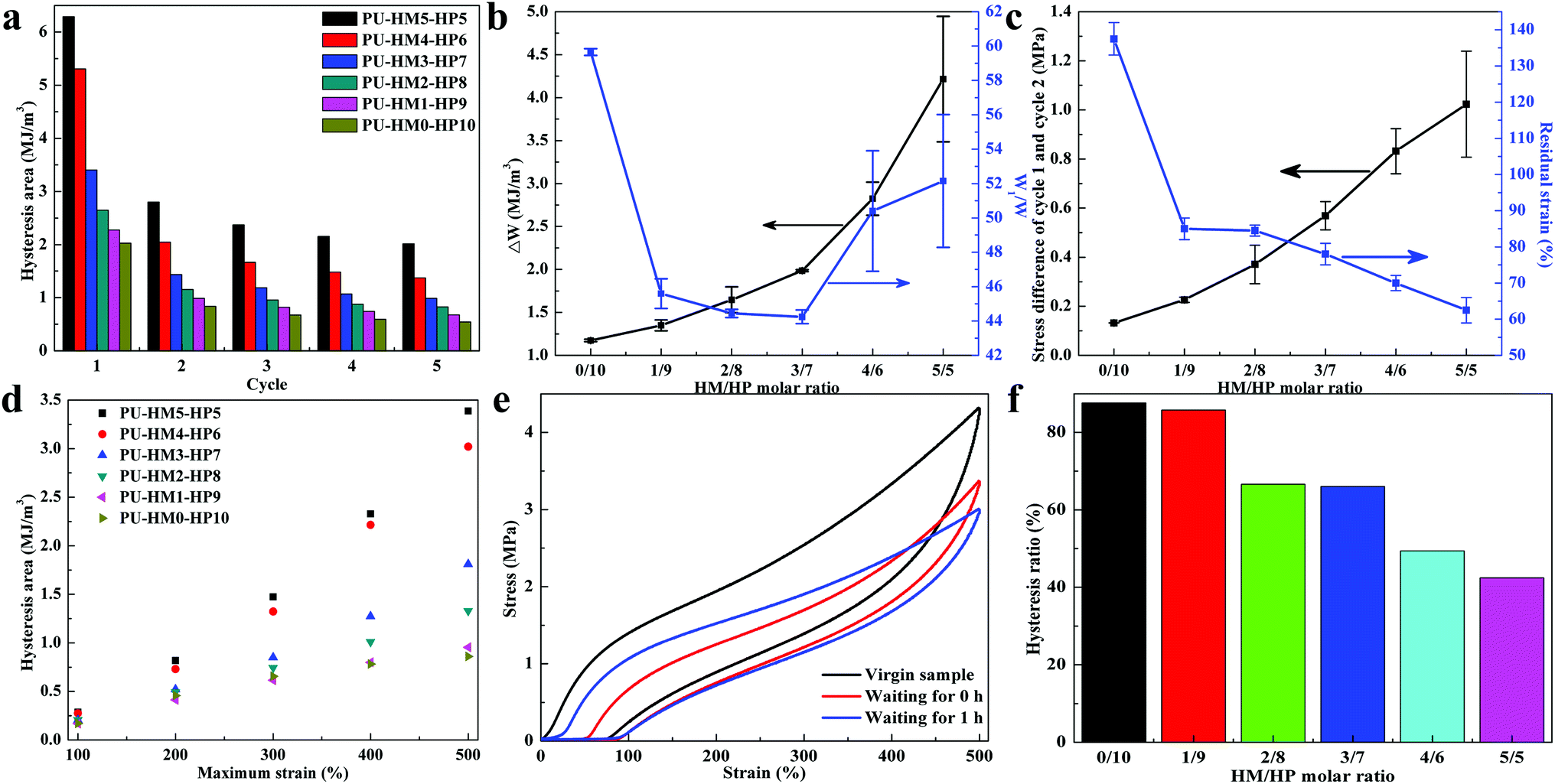 | ||
| Fig. 3 The energy dissipation of PU-HM-HP elastomers. (a) Dissipated energy during five successive loading-unloading process with a strain of 500% in every cycle. (b) Variation of the hysteresis difference between the first and second successive cyclic tensile testing (ΔW) and the hysteresis ratio of the first cyclic tensile testing (W1/W). (c) Variation of the stress difference between the first and second successive cyclic tensile testing at 500% strain and residual strain of the first cyclic tensile testing. (d) Dissipated energy during loading–unloading process under 100% to 500% strains. (e) Cyclic tensile testing curves in successive cycles of PU-HM4-HP6 with a strain of 500% in each cycle and a delay time of 0 h and 1 h, and (f) the hysteresis ratio between the 1 h cyclic tensile testing and the initial cyclic tensile testing. | ||
To elucidate the recovery property of PU-HM-HP elastomers, cyclic tensile testing curves in successive cycles were conducted after 1 h relaxation (Fig. 3e). As Fig. 3f shows, the hysteresis ratio between the 1 h cyclic tensile testing and the initial cyclic tensile testing gradually decreased with HM/HP molar ratio increasing. PU-HM0-HP10 had a hysteresis ratio of about 88% after 1 h relaxation while that of PU-HM5-HP5 was about 42%. The reconnection process arising from weak HBs dominates the faster elastic recovery, while the strong HBs showed a weak dynamic, which needed more time or more energy to completely reformed.
The introduction of single HBs at the end of a long side chain makes the easy exchange and diffusion of the HBs, which is expected to improve the self-healing efficiency. To verify our assumption, PU-HM-HP elastomers healed at different temperature for different time were conducted. For quadruple HBs contained elastomers such as PU-HM4-HP6 (Fig. 4a), the tensile strength healed at room temperature (25 °C) for 6 h could reach about 1.3 MPa due to the easily exchange and diffusion of single HBs at side chain. However, the tensile strength showed little change when the healing time was extended, as the quadruple HBs were stable at room temperature. When healed at 80 °C, the tensile strength exhibited a noticeable increase within 3 h, while the elongation at break possessed a large value even healed for only 1 h. Therefore, the healing efficiency in this paper is defined as the ratio between the healed and virgin tensile strength for accurate assessment. As Fig. 4b shows, only PU-HM0-HP10 system had an excellent healing efficiency at room temperature (about 100% for 6 h healing). For other PU-HM-HP elastomers, though the healed tensile strength could reach about 1.3 MPa, the increased virgin tensile strength with HM/HP molar ratio increasing resulted in the rapid decrease of the healing efficiency at room temperature. For high temperature self-healing (80 °C), PU-HM0-HP10 to PU-HM3-HP7 samples exhibited a high healing efficiency after 2 h healing (>92%). PU-HM4-HP6 showed a healing efficiency of above 82% after 3 h healing. However, the healing efficiency for PU-HM5-HP5 was only about 36% even after 3 h healing. As is well known, the healing process goes through five steps: (a) surface rearrangement, (b) surface approach, (c) wetting, (d) diffusion and (e) randomization, during which the diffusion is the most critical step for healing.76,77 For PU-HM-HP system, the introduction of weak HBs in the side chain not only weakened chain entanglement of soft domain, but also had soft effects on the strong restraint hard domain, which promoted the chain diffusion and resulted in the fast self-healing of the elastomer. As the scratch recovery of PU-HM1-HP9 and PU-HM4-HP6 showed (Fig. 4c), the scratches of about 100 μm shrink to about 17 μm and 32 μm for PU-HM1-HP9 and PU-HM4-HP6 within 5 min, which was attributed to the fast diffusion of chains. Then the scratches showed little change in the following 1 h. The size of the scratch of PU-HM4-HP6 after 1 h healing was about 2 times than that of PU-HM1-HP9, which was attributed to the restriction of the quadruple HBs. The non-desirable healing efficiency of PU-HM5-HP5 was mainly attributed to the regular arrangement of chains caused by the restriction of the quadruple HBs as mentioned above. Therefore, PU-HM4-HP6 exhibited the balanced mechanical and self-healing properties. As Fig. 4d shows, though the incision of PU-HM4-HP6 system was not fully repaired, the sample could lift a 5 kg object without changes of incision after healing at 80 °C for only 1 h. Such fast healing efficiency had obvious advantages over other reported UPy-type self-healing elastomers (Fig. 4e).
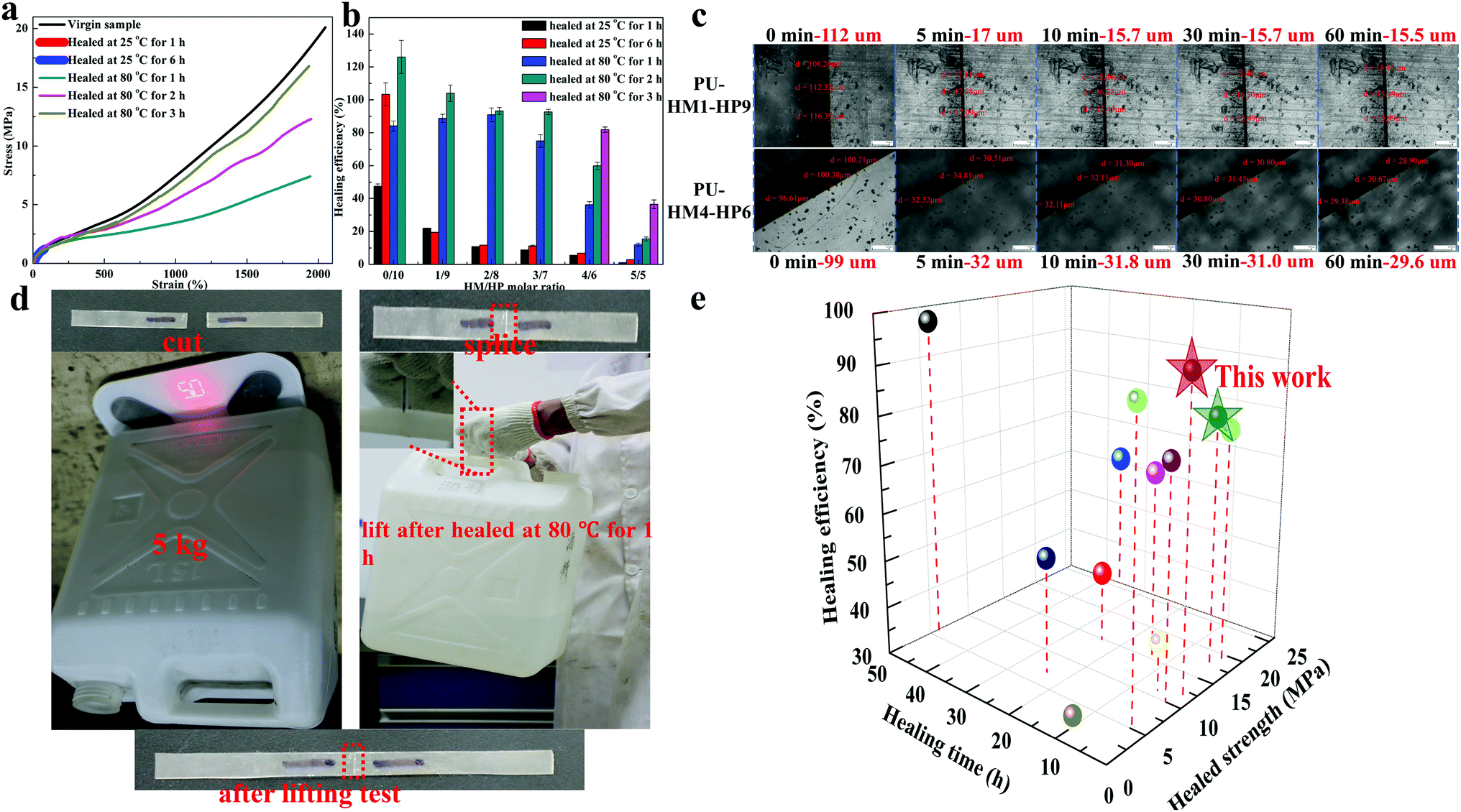 | ||
| Fig. 4 The self-healing properties of PU-HM-HP elastomers. (a) Representative stress–strain curves of virgin and self-healed PU-HM4-HP6 sample. (b) The healing efficiency of PU-HM-HP elastomers at different temperature for different time. (c) Optical microscopy images of scratch recovery of PU-HM1-HP9 and PU-HM4-HP6 films. (d) Load-bearing test of the self-healed PU-HM4-HP6 sample and (e) graphic comparison of self-healed tensile strength, healing time and healing efficiency at 80 °C for UPy-type self-healing elastomers.19,67–75 | ||
Taking advantage of PU-HM-HP elastomer, a flexible sensor composed of MWCNTs and PU-HM4-HP6 was fabricated. The conductivity and sensing property in response to vocalization, finger and wrist bending were simply examined. The healed sensor film, which was cut in half with a razor blade and healed at 80 °C for 1 h after splicing, was used for the conductivity test (Fig. 5a). When power on, the LED lamp connected in series with the healed film was lighted up, which indicated the conductive network of the film was reconstructed after healing. When the film was stretched to 65%, the brightness of LED lamp remained unchanged and no crackle was observed, which indicated that the conductive film exhibited good flexibility and stability. When the film was stretched further to 130%, the LED lamp dimed, which was attributed to the obvious crackle of the MWCNTs layer on the surface of PU-HM-HP layer. With this conductive and flexible sensor film, the sensing property of the virgin and healed sample (cut in half with a razor blade and healed at 80 °C for 1 h after splicing) was compared. The current signal of the virgin and healed sensors in response to vocalization could both be detected, which indicated the high sensitivity of the sensors (Fig. 5b). Both of the virgin and healed sensors showed a repeatable and wide response peak, while the sensing sensitivity of the healed sensor showed a little decline compared with that of the virgin sensor. For the current signal in response to finger and wrist bending, both of the virgin and healed sensors showed a sharp double splitting peak (Fig. 5c and d). Despite fluctuations, both the peaks of the virgin and healed sensors showed good stability and repeatability. Though the sensing sensitivity of the healed sensors showed a certain degree of decline for both actions, PU-HM-HP elastomer still showed great potential in the field of flexible electronics and wearable devices associate to the superior puncture and tear resistance of the elastomer.
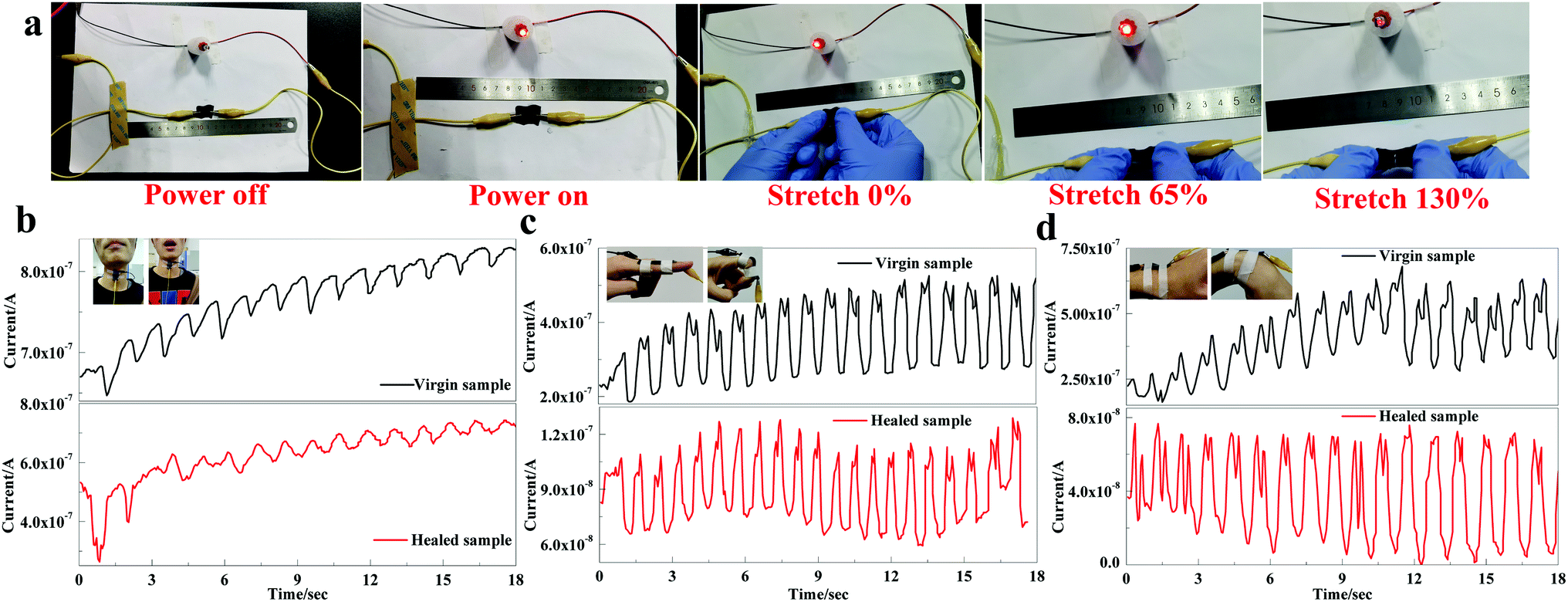 | ||
| Fig. 5 The conductivity and sensing properties of PU-HM-HP elastomers. (a) Optical images of conductive behavior of the healed sensor under stretching. (b) Optical images and the current signal of the virgin and healed sensors in response to vocalization. (c) Optical images and the current signal of the virgin and healed sensors in response to finger bending and (d) optical images and the current signal of the virgin and healed sensors in response to wrist bending. | ||
Conclusion
In this study, a design concept based on “hard–soft” hard domain by the cooperation of quadruple HBs in the mainchain as restriction units and single HBs in the side chain as diffusion units is proposed to synthesize a novel polyurethane supramolecular elastomer. The elastomer exhibited colorless transparency, high strength, extensibility and toughness, superior puncture and tear resistance, fast self-recoverability and self-healing capacity. The synchronous improvement of the mechanical and self-healing properties was based on the corporation of the two different hard domains and related strong and weak HB networks. Thereinto, the strength of the weak HBs at side chain is the key factor, which not only had direct effects on the exchange and diffusion performance, but also had decisive effects on the self-healing capacity at room temperature. The regulation of the strength between strong HBs in the mainchain and weak HBs in the side chain provides a promising construction strategy for tuning the conflict between mechanical property and self-healing property, which facilitates to find use in the field of flexible electronics and wearable devices.Author contributions
K. X. is in charge of the investigation, data curation and writing; Q. H. is in charge of the conceptualization; G. C., M. Z. and W. H. are in charge of the formal analysis and methodology; Y. P. is in charge of the funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (51703016) and the Science and Technology Research Program of Chongqing Municipal Education Commission of China (KJQN202001345 and KJQN201801332) is gratefully acknowledged.References
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- Z. Liu, Z. Zhang and R. O. Ritchie, Adv. Mater., 2018, 30, 1705220–1705235 CrossRef PubMed.
- N. Chen, L. Qin and Q. Pan, J. Mater. Chem. A, 2018, 6, 6667–6674 RSC.
- G. F. Cai, J. X. Wang, K. Qian, J. W. Chen, S. H. Li and P. S. Lee, Adv. Mater., 2017, 4, 1600190 Search PubMed.
- J. Kang, J. B. H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- J. Y. Oh, D. Son, T. Katsumata, Y. Lee and Y. Kim, Sci. Adv., 2019, 5, eaav3097 CrossRef CAS PubMed.
- S. H. Shin, W. Lee, S. M. Kim, M. Lee, J. M. Koo, S. Y. Hwang, D. X. Oh and J. Park, Chem. Eng. J., 2019, 371, 452–460 CrossRef CAS.
- Z. Zou, C. Zhu, Y. Li, X. F. Lei, W. Zhang and J. L. Xiao, Sci. Adv., 2018, 4, eaaq0508 CrossRef PubMed.
- E. Acome, S. K. Mitchell, T. G. Morrissey, M. B. Emmett, C. Benjamin, M. King, M. Radakovitz and C. Keplinger, Science, 2018, 359, 61–65 CrossRef CAS PubMed.
- S. Terryn, J. Brancart, D. Lefeber, G. Van Assche and B. Vanderborght, Sci. Robot., 2017, 2, eaan4268 CrossRef PubMed.
- H. Wei, Y. Wang, J. Guo, S. Y. Wei and Z. H. Guo, J. Mater. Chem. A, 2015, 3, 469–480 RSC.
- S. Nevejans, N. Ballard, M. Fernández, B. Reck and J. M. Asua, Polymer, 2019, 166, 229–238 CrossRef CAS.
- S. Choi, Y. Eom, S. M. Kim, S. Y. Hwang, J. Park and D. X. Oh, Adv. Mater., 2020, 32, 1907064 CrossRef CAS PubMed.
- M. Diba, S. Spaans, K. Ning and S. C. G. Leeuwenburgh, Adv. Mater. Interfac., 2018, 5, 1800118 CrossRef.
- D. L. Taylor and M. I. H. Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- J. H. Yoon, S. M. Kim, Y. Eom, S. Y. Hwang, J. Park and B. G. Choi, ACS Appl. Mater. Interfaces, 2019, 11, 46165–46175 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- Y. Song, Y. Liu, T. Qi and G. L. Li, Angew. Chem., Int. Ed., 2018, 57, 13838–13842 CrossRef CAS PubMed.
- J. C. Lai, X. Y. Jia, D. P. Wang, Y. B. Deng, P. Zheng, C. H. Li, J. L. Zuo and Z. Bao, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- J. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 1702616 CrossRef PubMed.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef PubMed.
- S. M. Kim, H. Jeon, S. H. Shin, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- W. B. Ying, Z. Yu, D. H. Kim, K. J. Lee, J. Shang and R. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 11072–11083 CrossRef CAS PubMed.
- S. Wang, J. Y. Oh, J. Xu, H. Tran and Z. Bao, Acc. Chem. Res., 2018, 51, 10331045 Search PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Adv. Funct. Mater., 2018, 28, 1706050 CrossRef.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561–10565 CrossRef CAS PubMed.
- X. Wu, J. Wang, J. Huang and S. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 7387–7396 CrossRef CAS PubMed.
- P. Cao, B. Li, T. Hong, J. Townsend, Z. Qiang, K. Xing, K. D. Vogiatzis, Y. Wang, J. W. Mays, A. P. Sokolov and T. Saito, Adv. Funct. Mater., 2018, 28, 1800741–1800749 CrossRef.
- J. Kang, D. Son, G. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. H. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846–1706853 CrossRef PubMed.
- Y. Peng, L. Zhao, C. Yang, Y. Yang, C. Song, Q. Wu, G. Huang and J. Wu, J. Mater. Chem. A, 2018, 6, 19066–19074 RSC.
- X. Wang, S. Zhan, Z. Lu, J. Li, X. Yang, Y. Qiao, Y. Men and J. Sun, Adv. Mater., 2020, 32, 2005759 CrossRef CAS PubMed.
- A. Das, A. Sallat, F. Böhme, M. Suckow, D. Basu, S. Wießner, K. W. Stöckelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS PubMed.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325–1605331 CrossRef PubMed.
- A. Susa, R. K. Bose, A. M. Grande, S. van der Zwaag and S. J. Garcia, ACS Appl. Mater. Interfaces, 2016, 8, 34068–34079 CrossRef CAS PubMed.
- W. Pu, D. Fu, Z. Wang, X. Gan, X. Lu, L. Yang and H. Xia, Adv. Sci., 2018, 5, 1800101 CrossRef PubMed.
- Y. Zhang, L. Yuan, G. Liang and A. Gu, Ind. Eng. Chem. Res., 2018, 57, 12397–12406 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- H. Liu, A. Z. Nelson, Y. Ren, K. Yang, R. H. Ewoldt and J. S. Moore, ACS Macro Lett., 2018, 7, 933–937 CrossRef CAS.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- K. Song, W. Ye, X. Gao, H. Fang, Y. Zhang, Q. Zhang, X. Li, S. Yang, H. Wei and Y. Ding, Mater. Horiz., 2021, 8, 216–223 RSC.
- J. H. Xu, P. Chen, J. W. Wu, P. Hu, Y. S. Fu, W. Jiang and J. J. Fu, Chem. Mater., 2019, 231, 7951–7961 CrossRef.
- D. Wang, J. H. Xu, J. Y. Chen, P. Hu, Y. Wang, W. Jiang and J. J. Fu, Adv. Funct. Mater., 2020, 30, 1907109 CrossRef CAS.
- B. Wu, Z. Liu, Y. Lei, Y. Wang, Q. Liu, A. Yuan, Y. Zhao, X. Zhang and J. Lei, Polymer, 2020, 186, 122003 CrossRef CAS.
- X. Yan, Z. Liu, Q. Zhang, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- J. Wu, L. H. Cai and D. A. Weitz, Adv. Mater., 2017, 29, 1702616 CrossRef PubMed.
- X. Dai, L. B. Huang, Y. Du, J. Han, Q. Zheng, J. Kong and J. Hao, Adv. Funct. Mater., 2020, 30, 1910723 CrossRef CAS.
- S. Chen, L. Sun, X. Zhou, Y. Guo, J. Song, S. Qian and Z. You, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- X. Chen, Q. Zhong, C. Cui, L. Ma, S. Liu, Q. Zhang and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 30847–30855 CrossRef CAS PubMed.
- P. Song and H. Wang, Adv. Mater., 2020, 32, 1901244 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- H. Wang, H. Liu, Z. Cao, G. Huang and J. Wu, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 11299–11305 CrossRef CAS PubMed.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef.
- G. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 810–811 CrossRef CAS PubMed.
- K. Xu, F. Zhang, X. Zhang, Q. Hu, H. Wu and S. Guo, J. Mater. Chem. A, 2014, 2, 8545–8556 RSC.
- Y. Li, W. Li, A. Sun, M. Jing, X. Liu, L. Wei, K. Wu and Q. Fu, Mater. Horiz., 2021, 8, 267–275 RSC.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- X. An, R. H. Aguirresarobe, L. Irusta, F. Ruipérez, J. M. Matxain, X. Pan, N. Aramburu, D. Mecerreyes, H. Sardon and J. Zhu, Polym. Chem., 2017, 8, 3641–3646 RSC.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- T. Wang, Y. Zhang, Q. Liu, W. Cheng, X. Wang, L. Pan, B. Xu and H. Xu, Adv. Funct. Mater., 2018, 28, 1705551 CrossRef.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, F. Qing, X. Bao and Z. You, Adv. Mater., 2019, 31, 1901402 CrossRef PubMed.
- J. Li, H. Ejima and N. Yoshie, ACS Appl. Mater. Interfaces, 2016, 8, 19047–19053 CrossRef CAS PubMed.
- X. Lin, Q. Xie, C. Ma and G. Zhang, J. Mater. Chem. B, 2021, 9, 1384–1394 RSC.
- Z. Yang, F. Wang, C. Zhang, J. Li, R. Zhang, Q. Wu, T. Chen and P. Sun, Polym. Chem., 2019, 10, 3362–3370 RSC.
- Z. Yang and D. Sun, J. Appl. Polym. Sci., 2020, 137, 49413 CrossRef CAS.
- J. Hu, R. Mo, X. Jiang, X. Sheng and X. Zhang, Polymer, 2019, 183, 121912 CrossRef CAS.
- C. J. Fan, Z. C. Huang, B. Li, W. Xiao and E. Zheng, Sci. China Mater., 2019, 62, 1188–1198 CrossRef CAS.
- H. Xie, D. Sheng, Y. Zhou, S. Xu, H. Wu, X. Tian, Y. Sun, X. Liu and Y. Yang, New J. Chem., 2020, 44, 13584–13590 RSC.
- L. Xiao, J. Shi, K. Wu and M. Lu, React. Funct. Polym., 2020, 148, 104482 CrossRef CAS.
- Y. Yang, Z. Ye, X. Liu and J. Su, J. Mater. Chem. C, 2020, 8, 5280–5292 RSC.
- J. Rong, J. Zhong, W. Yan, M. Liu, Y. Zhang, Y. Qiao, C. Fu, F. Gao, L. Shen and H. He, Polymer, 2021, 221, 123625 CrossRef CAS.
- R. P. Wool and K. M. O’connor, J. Appl. Physiol., 1981, 52, 5953–5963 CrossRef CAS.
- Y. H. Kim and R. P. Wool, Macromolecules, 1983, 16, 1115–1120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07083e |
| This journal is © The Royal Society of Chemistry 2022 |