
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation and selected catalytic properties of ruthenium, rhodium, osmium and iridium nanoparticles
Jan Krajczewski
*a,
Robert Ambroziak
 b and
Andrzej Kudelski
*a
b and
Andrzej Kudelski
*a
aFaculty of Chemistry, University of Warsaw, 1 Pasteur St., 02-093 Warsaw, Poland. E-mail: jkrajczewski@chem.uw.edu.pl; akudel@chem.uw.edu.pl
bInstitute of Physical Chemistry Polish Academy of Sciences, 44/52 Kasprzaka Str., 01-224 Warsaw, Poland
First published on 13th January 2022
Abstract
The synthesis and applications of nanoparticles formed from various platinum-group metals have been intensively investigated for many years because such systems have many important commercial applications, mainly as heterogeneous catalysts. Because of the very large number of data concerning nanoparticles of the platinum-group metals, in this review we focus only on nanoparticles formed from less frequently used members of the platinum family (i.e.: ruthenium, rhodium, osmium and iridium). Various methods of synthesis of unsupported and supported nanoparticles from these elements are described. Some example applications of such systems as catalysts have been also described, mainly those, for which there is a clear dependence of the catalytic activity on the size and shape of used nanoparticles.
 Jan Krajczewski | Jan Krajczewski is an associate professor in the Molecular Interactions and Spectroscopy group at the Faculty of Chemistry, University of Warsaw. His main research interest is the synthesis and characterization of metallic and plasmonic nanostructures and their application in optical spectroscopies. |
 Robert Ambroziak | Robert Ambroziak is pursuing his PhD at the University of Warsaw, Poland. His main research interest is SERS and the synthesis of nanostructured semiconductor substrates coated with silver and gold nanomaterials. |
 Andrzej Kudelski | Andrzej Kudelski is a professor at the Faculty of Chemistry, the University of Warsaw, Poland. His area of expertise is nanotechnology and applications of surface-enhanced Raman scattering (SERS) especially in biochemical and medical analysis. |
1. Introduction
The platinum-group metals (PGMs) – alternatively: platinum family or platinum metals – consist of six noble, precious metallic elements clustered together in the periodic table: iridium, osmium, palladium, platinum, rhodium, and ruthenium. All these metals have very useful catalytic properties and are widely used as catalysts in industry. In the last 3 decades, the automotive industry has emerged as the principal consumer of PGMs, although many other applications of these elements are still very important. To illustrate the very large interest in the investigations of nanoparticles of PGMs we carried out a search in the Scopus database using the combination of the keyword “nanoparticle” and the keyword being the name of the metal (iridium, osmium, palladium, platinum, rhodium, and ruthenium). The number of articles found in the Scopus database that contain in the title or abstract these two keywords are as follows: for platinum nanoparticles – 18.2 × 103 articles, for palladium – 13.9 × 103, for ruthenium – 4.1 × 103, for rhodium – 2.4 × 103, for iridium – 1.3 × 103, and for osmium – 1.8 × 102 (results from January 1st 2022). The results of this search also show that nanoparticles of ruthenium, rhodium, iridium and osmium are investigated significantly less often than those formed from platinum and palladium.In this review we describe the formation of unsupported and supported nanoparticles from less frequently used members of the platinum family (ruthenium, rhodium, osmium and iridium). The example applications of such nanoparticles as catalysts have been also described – in order to maintain a close correlation between the parts devoted to the synthesis of nanoparticles and their catalytic applications, we describe mainly catalytic processes for which there is a clear dependence of the catalytic activity of the platinum-group nanoparticles on the method of their synthesis (changing mainly their size and shape).
Due to much larger number of publications devoted to Pt and Pd nanoparticles than devoted to nanoparticles of other PGMs, Pt and Pd nanoparticles will not be described in this review, because in the case of a review containing a description of all PGMs, the part devoted to the Pt and Pd nanoparticles would dominate the entire manuscript and to obtain the description of Rh, Ru, Ir and Os nanoparticles as detailed, as in this contribution, the whole review would to be very long. The less frequently used members of the platinum family have also, however, very interesting properties. For example, ruthenium is the most active catalyst of the Fischer–Tropsch synthesis, and rhodium is preferable to the other platinum metals in the reduction of nitrogen oxides to nitrogen. Because of very widespread use of rhodium in catalytic converters, the nowadays worldwide consumption of this metal is much greater than the nowadays consumption of ruthenium, iridium and osmium.
2. Synthesis of unsupported ruthenium, rhodium, osmium and iridium nanoparticles
2.1. Ruthenium nanoparticles
Ruthenium is a metal from the platinum family with the smallest atomic number. There is a possibility of formation of ruthenium nanoparticles having various shapes and sizes, however, as in a case of nanoparticles of other noble metals, in a majority of cases spherical or almost spherical ruthenium nanoparticles are formed. A commonly used method of formation of ruthenium nanoparticles is so-called polyol synthesis. In high temperatures, many polyols are able to reduce ions of noble metals to the metallic form. In polyol synthesis, a hot solution of a polyol solvent, like: ethylene glycol (EG), diethylene glycol (DEG), triethylene glycol (TrEG), or other polyethers (in general, polyethylene glycol – PEG) is used both as a solvent and a reducing agent. Addition of some compounds can significantly effects on stability, size, shape and outermost crystal facets of formed nanoparticles.An interesting investigation of the formation of Ru nanoparticles in different polyols has been carried out by Yan and co-workers.1 Yan et al. heated to the boiling point solutions of RuCl3 and polyvinylpyrrolidone (PVP) in 3 various polyol solvents: EG, DEG and TrEG. Initially the color of all solutions was dark red and during heating changed to light yellow and then to dark brown. It suggests that the formation of Ru nanoparticles can be easily monitored by the UV-Vis spectrophotometry. Before heating, a polyol solution of RuCl3 and PVP exhibits absorption band at 347 nm. During heating, the intensity of this absorption band decreases indicating reduction of Ru3+ ions. TEM analysis showed that the size of formed Ru nanoparticles decreases in the order EG > DEG > TrEG, it means, the size decreases when the temperature of the boiling point of the solvent increases (the boiling points of EG, DEG, and TrEG are: 197 °C, 244 °C, and 285 °C, respectively). This effect can be explained by the fact that when the temperature is lower, the reaction rate of the formation of the nucleation centers is lower, the number of the formed nucleation centers is smaller, and hence the formation of a smaller number of bigger nanoparticles is observed. However, not always such a simple correlation between the temperature, at which the reaction in a polyol solvent is carried out, and the size of the formed nanoparticles is observed. For example, the heating to the boiling point of the solution of ruthenium(III) acetylacetonate (Ru(acac)3) and PVP in 1,4-butanediol (boiling point at 235 °C) leads to the formation of Ru nanoparticles with the diameter of 2.8 nm, whereas when using EG (boiling point at 197 °C), the average diameter of the formed nanoparticles is 2.1 nm.2 UV-Vis measurements showed that the kinetic of the reduction of Ru(acac)3 in both solvents is similar, which suggests that these polyols have a similar efficiency in the reducing of Ru3+ ions.
The size of formed Ru nanoparticles also depends on the molar ratio of PVP and the ruthenium precursor – lowering this ratio leads to the formation of a larger ruthenium nanoparticles with a narrow size distribution, however, when the PVP/Ru ratio is close to 1 or below, a precipitation of the formed Ru nanoparticles is often observed. Decreasing the amount of PVP in a solution allows for easier reduction of Ru3+ cations on the initially formed nucleation centers and hence the process of growth of nanoparticles occurs faster leading to formation of bigger nanoparticles (decreasing the time of the formation of nanoparticles also decreases the number of created nucleation centers).
Acetates are another stabilizers that can be used in an effective synthesis of ruthenium nanoparticles in a polyol environment. For example, Viau et al. formed ruthenium nanoparticles with very narrow particle diameter distribution by heating for 10 minutes of a polyol solution of RuCl3 and acetates.3 The reaction temperature was set in a range between 140 °C and 180 °C. The size of formed nanoparticles depends on the reaction temperature and on the concentration of acetate ions. Generally, higher reaction temperature leads to formation of smaller nanoparticles. As for PVP, a high concentration of acetates reduces the growth rate of nanoparticles and thus extends the duration of the process of formation of nanoparticles, allowing for creation of more nucleation centers. For this reason, formation of a larger amount of smaller nanoparticles is observed.
Ruthenium in the bulk form has hexagonal close-packed (hcp) crystallographic structure, and such a structure have also typical Ru nanoparticles. In 2013 Kitagawa et al. reported that by changing the Ru precursor and the polyol solvent it is possible to produce using a simple polyol method Ru nanoparticles having either hcp or fcc (face-centered cubic) crystallographic structures.4 Kitagawa et al. found that heating the Ru(acac)3 solution in TrEG to 200 °C leads to formation of Ru nanoparticles having fcc crystallographic structure, while heating the solution of RuCl3 in EG leads to formation of Ru nanoparticles having hcp crystallographic structure.4 Undoubtedly it is important result because Ru nanoparticles having different crystallographic structures exhibit significantly different catalytic activity. Example TEM images of fcc and hcp Ru nanoparticles and the corresponding XRD patterns are presented in Fig. 1. Alternative ways of formation of ruthenium nanostructures having the fcc crystallographic structure are significantly more complex, as, for example, the epitaxial growth of Ru shell on Pd–Cu alloy nanoparticles.5 In such synthesis, Pd–Cu nanoseeds were prepared by heating at 120 °C of the Pd(acac)2 and CuCl2 oleylamine ethanol solution with 1-octadecylene as a reducing agent. Subsequently, RuCl3 was added and the temperature of the reaction mixture was increased to 200 °C. TEM analysis revealed formation of the nanoparticles having homogeneous truncated octahedral shape and uniform size of 19.6 ± 0.8 nm. The epitaxial growth was initially induced by the galvanic replacement between the Ru precursor and PdCu3 seeds, during which the Pd–Cu seeds gradually adopts a Pd–Cu@Ru core–shell structure. Extending the heating time to 12 hours leads to formation of core–shell structures with the average core size of 14.9 nm and the shell thickness of 2.9 nm. Similar method to grow fcc ruthenium, which is thermodynamically unfavorable in a bulk form, was proposed by Ye et al.6 In this case, epitaxial growth of the Ru layer was realized on palladium truncated nanooctahedrals and nanocubes prepared using a polyol method. Subsequently, to the above sols of the Pd nanoparticles, a proper amount of RuCl3 and PVP was added. Then, ascorbic acid as a reducing agent was added and the obtained solution was heated to 200 °C under magnetic stirring. In order to obtain ruthenium nanoframes instead of the core–shell structures, formed Pd@Ru nanoparticles were placed in aqua regia which led to the selective removing of the palladium cores. Fig. 2a shows the general scheme of the formation of Ru nanoframes using this method. In a case of Ru nanostructures growth on Pd truncated octahedrals, detailed investigations proved that the formed Ru nanostructures actually exhibits fcc crystallographic structure and the dominant crystal facets are (100) at corners, (110) at edges and (111) at side faces, while, in a case of nanocubes, the dominant crystal facet is (100).6 Interestingly, Ru nanoparticles obtained in the same conditions but in an absence of Pd nanoseeds, have a shape of wavy nanowires and have a typical for ruthenium hcp crystallographic structure.6 TEM images of formed Ru nanoframes are presented on Fig. 2b and c.
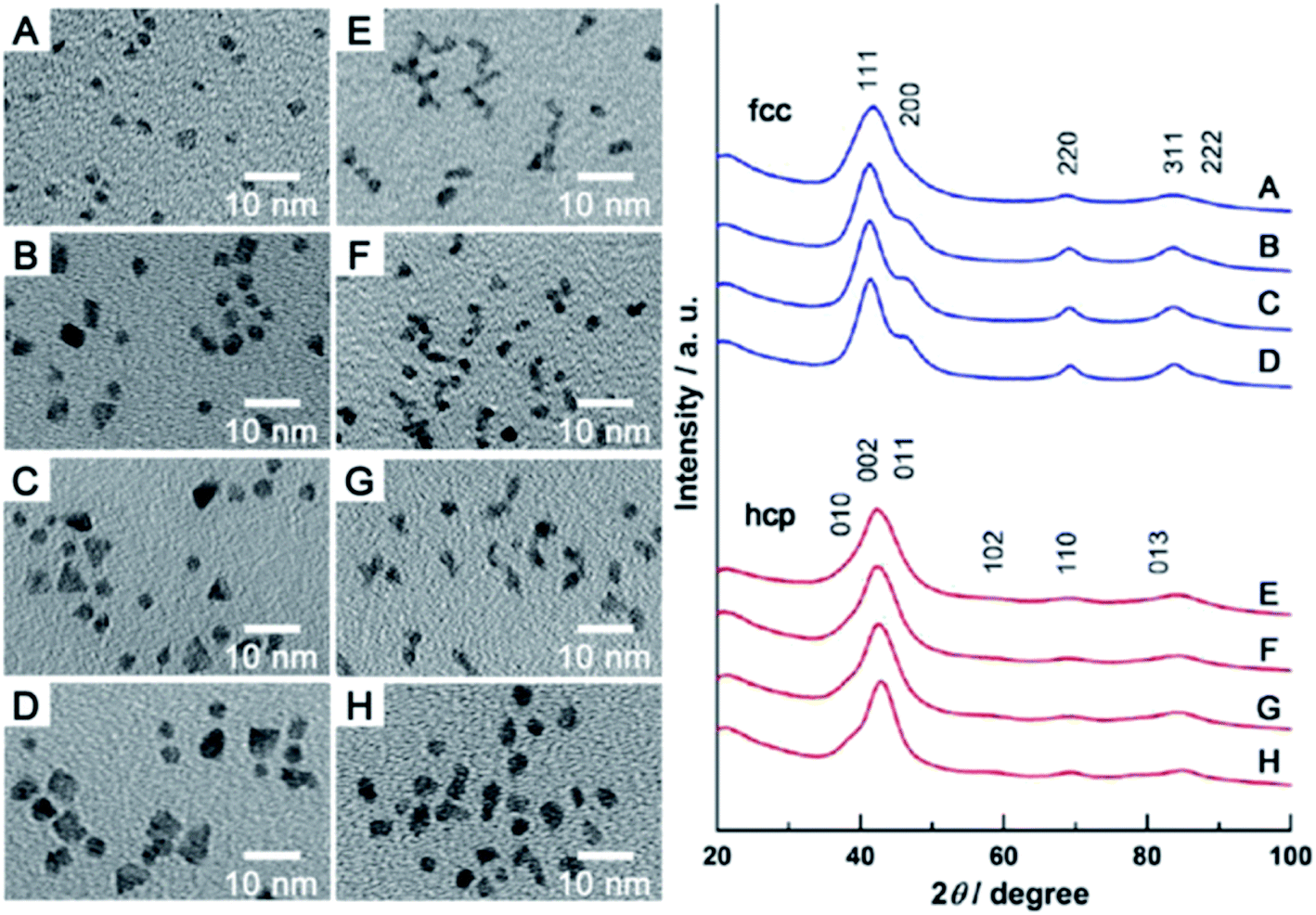 | ||
| Fig. 1 (A–D) TEM images of various Ru nanoparticles having fcc structure. (E–H) TEM images of various Ru nanoparticles having hcp structure. Right panel shows corresponding XRD patterns. Reprinted with permission from ref. 4. Copyright 2013 the American Chemical Society. | ||
 | ||
| Fig. 2 (a) General scheme showing the formation of Ru nanoframes: (1) nucleation of Ru on tops and edges of Pd nanoparticles, (2) etching of the Pd core. (b) and (c) TEM images (with lower and higher magnification, respectively) of Ru nanoframes obtained after Pd etching. Reprinted with permission from ref. 6. Copyright 2016 the American Chemical Society. | ||
Ruthenium nanoparticles have been also prepared in many other organic solvents. For example, Can and Metin prepared nearly monodisperse ruthenium nanoparticles by the reduction of Ru(acac)3 at 300 °C in a mixture of oleylamine and benzyl ether.7 Oleylamine plays here a dual role as surfactant and a reducing agent, while benzyl ether is used as a solvent boiling at a high temperature. TEM analysis showed very narrow size distribution of formed nanoparticles with the average diameter of 2.5 nm and with 12% standard deviation.7 Another strategy for the preparation of Ru nanoparticles with the tunable sizes ranging from 2.6 to 51.5 nm has been developed by Zhao et al.8 In this method, RuCl3 is used as a ruthenium precursor, PVP as a stabilizing agent and propanol is both a reducing agent and a solvent. A lower reaction temperature results in a slower formation of nucleation centers and thus larger Ru nanoparticles are generated. For example, the average diameter of nanoparticles formed at 98 °C is estimated as 2.6 ± 0.5 nm, nanoparticles formed at 70 °C have the average diameter of 4.5 ± 0.9 nm, whereas those formed at 30 °C have the average diameter of 51.5 ± 3.2 nm. Additionally, sizes of formed Ru nanoparticles decreases when pH of the solution is increasing because increasing in pH leads to increasing in the reduction ability of propanol.
Ruthenium nanoparticles of regular spherical shape (in some cases also having sponge-like structure) with various size can be obtained by the decomposition of (1,5-cyclooctadiene)(1,3,5-cyclooctatriene)ruthenium precursor under the H2 atmosphere (3 bar) in various alcohols or in an alcohol/THF mixture as a solvent.9 This reaction can be carried out without using further stabilizers and can be completed already after 45 minutes. The size of formed nanoparticles can be controlled in the range of 4–85 nm by the adjusting of the reaction temperature or by changing the composition of the solvent mixture. In line with previous reports, a higher reaction temperature leads to formation of smaller Ru nanoparticles.
Ru nanoparticles can be also formed at room temperature using a strong reducing agent. Such method of synthesis of Ru nanoparticles was realized, for example, by Abo-Hamed et al., who synthesized 1.2 nm ruthenium nanoparticles in an water–ethanol solution using sodium borohydride as a reducing agent.10 Ruthenium nanoparticles have been also prepared at 21 °C by the sonochemical reduction of a ruthenium chloride solution using ultrasound frequencies in the range of 20–1056 kHz.11 The proposed mechanism of this reaction is radical, sonolysis of water solution leads to formation of H˙ and OH˙ radicals which can reduce Ru3+ ions to the metallic form. The Ru nanoparticles produced by this ultrasound reduction have diameters between 10 and 20 nm as measured by the TEM imaging.
In many cases, using an organic solvent is undesirable due to various factors, like matrix effects or contamination of formed nanostructures by some organic compounds. Therefore, synthesis of metal nanoparticles in aqueous environment is often highly desirable and such methods of synthesis are continuously intensively developed. Ruthenium nanoparticles can be formed in aqueous solution, for example, by the reduction of RuCl3 by sodium borohydride.12 In the case of this synthesis, the pH of the reaction mixture should be lower than 4.9 – higher pH (too large increase in pH can be generated, for example, by the addition of too large amount of sodium borohydride) induces precipitation of Ru nanoparticles. The formed nanoparticles have narrow size distribution and their average size is 1.8 nm.12
2.2. Rhodium nanoparticles
In the platinum family, rhodium is the next element in terms of atomic number, after ruthenium. As in the case of ruthenium, also rhodium nanoparticles are often obtained by the polyol synthesis using RhCl3 as a source of Rh3+ ions, PVP as a stabilizing agent, and polyol (usually EG) as a solvent and a reducing agent.13 For example, Hoefelmeyer et al. formed small Rh nanocrystal seeds by the reduction of RhCl3 in EG in the presence of PVP and then, these seeds were further growth to form larger rhodium nanoparticles.13 It was found that the reaction temperature strongly effects on the process of growth of Rh nanoparticles. Growth of Rh nanocrystal seeds carried out at 190 °C leads to the formation of isotropic cubic nanoparticles with an average edge length of 12.7 ± 1.5 nm. Lowering the reaction temperature results in slightly different growth of Rh nanostructures, for example, at 140 °C Rh cubes with horns were formed, while at 90 °C multipods are synthetized. In temperatures lower than 90 °C, growth of Ru seeds does not occur. High resolution TEM and XRD analysis showed that the formed Rh cubes and multipods are single nanocrystals. Rhodium multipods have a central region of 3 nm in diameter and two to seven arms with length from 3 to 9 nm. It was found that also changing other (in addition to the reaction temperature) reaction parameters can strongly effect on the final geometry and size of produced nanoparticles. For example, Watson et al. synthetized monodisperse Rh planar tripods with the arm length of 8 nm and the width of 3.5 nm by heating at 140 °C of a solution of RhCl3 and PVP in EG (the process was carried out under a continuous nitrogen flow and the solution was stirred).14 Analogous planar tripod Rh structures were also obtained using Na3RhCl6 as a rhodium precursor.15,16 However, when trimethyl(tetradecyl)ammonium bromide (TTAB) is added to the RhCl3 and PVP solution in EG, heating of the obtained mixture leads to the formation of monodispersed Rh nanocubes.17 In a polyol synthesis one can also obtain starfish-like Rh nanocrystals with five wined arms.18 In the case of this synthesis, a solution of [(CF3COO)2Rh]2 and PVP in EG was heated to 180 °C for 6 hours. TEM analysis showed that the overall size of formed Rh nanoparticles was 30 nm, arms were 4–10 nm in width and 6–12 nm in length. TEM analysis also showed that the arms grew from the corners of a five-fold twinned decahedral cores.18 In a similar manner, the Rh decahedrons can be formed with 80–90% efficiency.19 In this case, Rh(acac)3 and PVP solution in tetraethylene glycol was heated at 250 °C for 1 hour. The average size of formed Rh decahedrons was 15 nm. Rhodium nanoparticles with the diameter in a range from 7 to 11 nm have been also formed at 220 °C in a solution of Rh(acac)3, sodium citrate and PVP in 1,4-butanediol.20In contrast to Ru nanoparticles, where hcp crystallographic structure is preferred (as for a bulk form), in a case of Rh nanoparticles the dominant crystallographic structure is thermodynamically stable for a bulk metal fcc structure. However, as in a case of ruthenium, it is possible to produce rhodium nanoparticles with different crystallographic structure than that of the bulk form. The first example of synthesis of Rh nanoparticles with the hcp crystallographic structure has been reported by Huang et al. in 2017.21 This group formed hcp Rh nanoparticles from Rh(acac)3 precursor dissolved in oleylamine (the process was carried out at 220 °C).21
Rh nanoparticles have been also prepared in anhydrous ethanol and anhydrous methanol, for example, by placing a solution of RhCl3 in these solvents in a polyfluoroethylene-lined stainless steel bomb and heating the sample at 175 °C – this process was carried out under high pressure auto-generated in such conditions.22 TEM analysis revealed that the size of formed Rh nanoparticles was in the range between 2 and 10 nm. The another organic solvent used for the synthesis of Rh nanoparticles is dimethylformamide. For example, Behr et al. synthesized Rh nanoparticles showing narrow size distribution from 2.7 to 4.8 nm by the reduction at 80 °C of Rh(acac)(CO)2 dissolved in dimethylformamide by the hydrogen under the pressure of 10 bar.23
Rhodium nanoparticles were also prepared in aqueous solution, for example, by the reduction of RhCl3 by sodium borohydride in a presence of PVP or N-alkyl-N-(2-hydroxyethyl)ammonium salts (with alkyl chain containing between 12 and 18 carbon atoms).24 This process, carried out at 20 °C under a nitrogen flow, is virtually instantaneous and induces very quick change of the color of the reaction mixture from intense red to black. The average diameter of formed Ru nanoparticles was 2.1 nm.24 A similar method of the synthesis of Rh nanoparticles in an aqueous environment (reduction of RhCl3 by sodium borohydride in a presence of PVP) was carried out by Ewers and co-workers.25 TEM analysis of the Rh nanoparticles formed using PVP as a stabilizing agent, showed that the produced nanoparticles consist of two fractions: smaller nanoparticles with the average diameter of 2.6 nm, and significantly larger nanoparticles with a diameter in a range between 12 and 70 nm. It was also found that the molecular weight of PVP does not effect on the shape of formed nanoparticles, however, when PVP with a lower molecular mass is used, the average diameter of formed nanoparticles is smaller. Ewers and co-workers introduced to the reaction mixture also the second surfactant: sodium dodecyl sulfate.25 In this case, the formed Rh nanoparticles have the average diameter of 35 ± 7 nm and their size distribution was significantly narrower.
Some groups synthesized Rh nanoparticles in a mixture of an organic solvent and water. For example, Harada et al. synthesized 2–3 nm rhodium nanoparticles stabilized by PVP in water–ethanol mixture by the reduction of Rh3+ ions at high-temperature and high-pressure.26 Rhodium nanoparticles can be also obtained in a mixture of water and methanol at the atmospheric pressure. For example, Baeza et al. formed ruthenium nanoparticles mostly pseudo – spherical in shape and with narrow size distribution in a range from 1.9 to 4.9 nm, by heating under reflux for 5 h at 90 °C (or at 95 °C, when methanol concentration was lower) a mixture composed from: RhCl3, aqueous solution of HCl, PVP, and methanol.27
For some applications microporous or mesoporous metallic nanoparticles seems to be significantly better than the standard solid nanostructures. Mesoporous Rh nanoparticles have been synthetized by Jiang et al. by the reduction of Na3RhCl6 by ascorbic acid in a presence of polymeric micelle templates from poly(ethylene oxide)-b-poly(methyl methacrylate).28 The schematic mechanism of the formation of mesoporous Rh nanoparticles is presented in Fig. 3a, whereas TEM images of formed nanoparticles are presented in Fig. 3b and c. Used polymeric micelle network plays a role of a pore-directing agent and template that directs the formation of uniformly sized mesoporous Rh nanoparticles. High surface area of the formed mesoporous Rh nanoparticles was thermally stable up to 400 °C.28
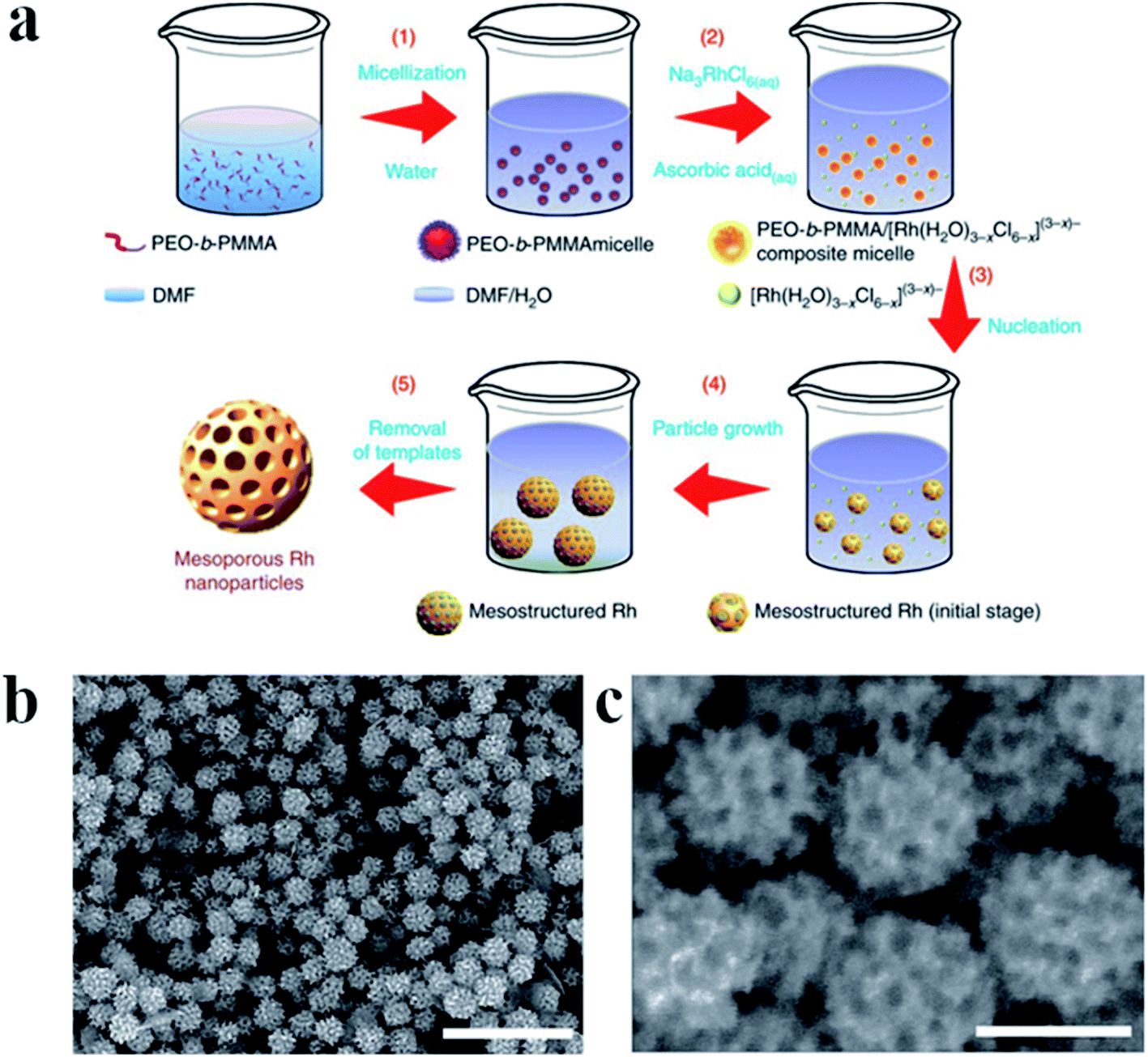 | ||
| Fig. 3 (a) Illustration showing the mechanism of the formation of mesoporous Rh nanoparticles. Poly(ethylene oxide)-b-poly(methyl methacrylate) was used to form soft micelle templates, Na3RhCl6 was used as Rh precursor, and ascorbic acid as a reducing agent. The reaction was carried out in a mixture of dimethylformamide and water. (b) and (c) TEM images of Rh mesoporous nanoparticles with lower and higher magnification, respectively. Scale bare is 500 nm for image (b) and 100 nm for image (c). Reprinted from ref. 28. Creative Commons Attribution 4.0 International License. | ||
Nanoparticles composed of two or more different metals are for some applications (for example catalytic ones) significantly better than those composed of only one element. One can find very large number of articles concerning synthesis of alloy or core–shell bimetallic nanostructures containing at least one metal from the platinum family. In some cases such nanoparticles are formed only from PGMs. Although, in these cases, nanoparticles are usually formed from alloys of platinum or palladium, there are also examples of synthesis of bimetallic nanoparticles composed only from less frequently used members of the platinum family (usually from alloys of: Ru–Rh, Ir–Rh, Ir–Ru and Os–Ru). For example, in 2015 Rakap described very simple synthesis of 3.4 ± 2.0 nm alloy Ru–Rh nanoparticles.29 The applied synthesis procedure involved: dissolution of RhCl3, RuCl3, and PVP in the absolute ethanol and long-term heating of the obtained solution.
2.3. Iridium nanoparticles
Iridium and osmium nanoparticles are synthesized not so often as Rh and Ru nanostructures. Nevertheless, many different approaches to synthetize the Ir and Os nanoparticles can be found in the literature.Monodisperse and uniform iridium nanoparticles are often synthesized in an aqueous solution. For example, Cui et al. synthesized 3.5 ± 0.5 nm Ir nanoparticles by the addition of aqueous solution of sodium borohydride to the aqueous solution containing IrCl3 and tannic acid.30 This reaction can be easily monitored by the UV-Vis spectrophotometry. Aqueous solution of IrCl3 is yellow and in the UV-Vis absorption spectrum of this solution one can observe two bands at 421 and 486 nm. The color of the sol of Ir nanoparticles is dark brown. Cui et al. developed another method of synthesis of iridium nanoparticles in an aqueous solution.31 In this case, iridium chloride, trisodium citrate and NaOH was dissolved in water and heated to boiling under reflux. During this heating the solution changes color from green to faint blue and then to faint yellow. After 20 minutes sodium borohydride was added, heater was switched off, and the solution was gradually cooled down to a room temperature. The final solution was yellow brown. The average diameter of formed Ir nanoparticles estimated from the TEM analysis is 2.5 ± 0.5 nm.31
As nanoparticles of all PGMs, iridium nanoparticles can be synthesized in organic solvents. For example, refluxing for 6 hours of the ethanolic solution of IrCl3 and PVP leads to the formation of pale yellow solution containing approximately spherical nanoparticles with an average size of about 1.5 nm.32
An interesting method of the synthesis of iridium nanoparticles is a photochemical synthesis developed by Kundu and Liang.33 This group irradiated for 4 hours using UV radiation of an aqueous solution containing: IrCl3, NaOH, hexadecyltrimethylammonium bromide (CTAB) and 2,7-dihydroxynapthalene (2,7-DHN). Illumination with UV radiation excites the photosensitizer molecules (2,7-DHN) what leads to the formation of radicals, that are able to reduce Ir3+ ions into the metallic form. The shape of formed nanoparticles depends on the concentration ratio between CTAB and IrCl3. It was observed that at lower CTAB concentrations, mostly approximately spherical nanoparticles were formed, while in a case of higher concentrations, nanoparticles with anisotropic shapes were formed. This is caused by the adsorption of CTAB molecules on the surface of nanoparticles which leads to the reduction of the growth rate of different crystal plates.33
Cubic iridium nanoparticles without any surfactants on the surface can be synthesized at room temperature by the galvanic replacement reaction.34 For this purpose, a drop of an aqueous solution containing a proper amount of IrCl3 and CuCl2 has been injected on a Cu foil. The size of the formed Ir nanocubes varies from 120 to 200 nm with the average edge length of 158 nm.34
Nanoparticles with well-defined iridium crystal facets can be obtained, for example, by the deposition of iridium layer on some regular palladium nanoparticles.35 Using cubic and octahedral Pd nanoparticles encased by {100} and {111} facets, respectively, as the seeds, it is possible to generate Pd@Ir cubic and octahedral nanoparticles with Ir{100} and Ir{111} facets. Cuboidal or octahedral palladium nanoparticles were prepared by the standard polyol synthesizes. For deposition of the iridium layer, EG solution containing PVP and ascorbic acid was heated at 100 °C for 1 h. Subsequently, a proper amount of palladium nanoparticles was added and the temperature was increased to the boiling point of EG. Immediately after the temperature of the boiling point of EG had been reached, a proper amount of Na3IrCl6 was introduced to the reaction mixture. TEM analysis showed that the formed Pd@Ir nanocubes had the average edge length of 7.5 nm, which is 1.5 nm greater than that of the initial Pd nanocubes. Therefore, the thickness of the deposited iridium layer was estimated on 0.75 nm. Crystallographic studies proved that the Ir surfaces formed on Pd@Ir nanocubes has {100} orientation, while in a case of octahedral Pd@Ir nanoparticles the dominant facet is {111}.35
2.4. Osmium nanoparticles
Osmium nanoparticles are often synthesized in aqueous solutions. For example, Chakrapani and Sampath prepared osmium nanostructures by the reduction of an aqueous solution of OsCl3 by ascorbic acid.36 This reaction was carried out for 90 minutes at 95 °C. SEM analysis showed that the synthetized nanomaterial contains nanochains formed by the connected together practically spherical 1–1.5 nm Os nanoparticles. The sol of such Os nanochains is violet (with the extinction band at 540 nm), and the formed nanostructures has plasmonic properties and can be used for surface-enhanced Raman scattering (SERS) measurements. Another method of synthesis of Os nanoparticles in an aqueous medium was developed by Ede et al.37 In this case, the formation of osmium nanoparticles was realized at room temperature by the reduction of OsO4 by 2,7-DHN in a presence of CTAB. This process takes place only after alkalization of the reaction mixture (realized by the addition of NaOH: the pale violet solution of OsO4 and 2,7-DHN becomes dark after addition of NaOH). Ede et al. assume that the formation of Os nanoparticles occurs in a several steps. At first, after alkalization of the reaction mixture, the perosmate anions ([OsO4(OH)2]2−) are formed. Such anions can be easily reduced in an alkaline solution to osmate anion ([OsO2(OH)4]4−). Finally, osmate anions are reduced by 2,7-DHN and osmium nanoparticles are generated. The degree of the agglomeration of formed Os nanoparticles depends on the concentration of CTAB in a solution. As in the case of osmium nanoparticles formed by the reduction with ascorbic acid,36 the osmium generated by the reduction with 2,7-DHN can be also used as an effective substrate in SERS measurements.37Although osmium in a bulk state adopts only hcp crystallographic structure, Wakisaka et al. showed that osmium nanoparticles with fcc crystallographic structure can be synthesized from osmium acetate in EG at 200 °C.38 This reduction was realized in a presence of PVP and the average size of formed Os nanoparticles was estimated as 1.2 ± 0.2 nm.38
2.5. Key parameters influencing the size, shape and structure of nanoparticles
To summarize the part of this article concerning synthesis of unsupported Ru, Rh, Ir and Os nanoparticles, the collected experimental details of some selected synthesis of such nanostructures are presented in Table 1. The presented data illustrates how the change of the experimental conditions influences on the shape, size and the crystallographic structure of the formed nanostructures (see Table 1). Moreover, below we briefly summarize how the change of the stabilizing agent, metal precursor, solvent and the reaction temperate influence on the size, shape and the structure of the formed nanostructures.| Metal precursor | Solvent | Temperature and the reducing agent, when different than a solvent | Stabilizing compound | Shape | Size/nm | Crystall-ographic structure | References |
|---|---|---|---|---|---|---|---|
| RuCl3 | Propanol | 30–98 °C | PVP | Spherical | 2.6–51.5 | fcc | 8 |
| RuCl3 | H2O | RT, NaBH4 | — | Spherical | 1.8 | fcc | 12 |
| RuCl3 | EG | 170 °C | PVP | Spherical | 3.5 | hcp | 4 |
| RuCl3 | EG | 160 °C – boiling point | PVP | Spherical | 2.9–7.4 | hcp | 1 |
| RuCl3 | DEG | 245 °C | PVP | Spherical | 1.8 | hcp | 1 |
| RuCl3 | TrEG | 285 °C | PVP | Spherical | 1.4 | hcp | 1 |
| RuCl3 | 1,2-Propanediol | 140–180 °C | Sodium acetate | Spherical | 1.5–6 | hcp | 3 |
| Ru(acac)3 | EG | 180 °C | PVP | Spherical | 2.1 | hcp | 2 |
| Ru(acac)3 | Butanediol | 215 °C | PVP | Spherical | 2.8 | hcp | 2 |
| Ru(acac)3 | TrEG | 200 °C | PVP | Spherical | 2.4 | fcc | 4 |
| Ru(acac)3 | Oleylamine, benzyl ether | 300 °C | Oleylamine | Spherical | 2.5 | fcc | 7 |
| RhCl3 | EG | 185 °C | TTAB, PVP | Cubic | 10 | fcc | 17 |
| RhCl3 | EG | 90–190 °C | PVP | Spherical | 3.6–5.5 | fcc | 13 |
| RhCl3 | EG | 140 °C | PVP | Tripods | 3.5–8 | fcc | 14 |
| Na3RhCl6 | EG | 140 °C | PVP | Tripods | — | fcc | 15 |
| RhCl3 | H2O | RT, NaBH4 | PVP | Spherical | Two fractions: 2.6, 12–70 | fcc | 25 |
| RhCl3 | H2O | RT, NaBH4 | PVP, SDS | Spherical | 35 | fcc | 25 |
| Rh(acac)3 | Oleylamine | 220 °C | Oleylamine | Spherical | 1–5 | fcc | 21 |
| IrCl3 | H2O | RT, NaBH4 | Tannic acid | Spherical | 3.5 | N/A | 30 |
| IrCl3 | Ethanol | 100 °C | PVP | Spherical | 1.5 | N/A | 32 |
| IrCl3 | H2O, NaOH | 130 °C, trisodium citrate | Trisodium citrate | Spherical | 2.5 | N/A | 31 |
| OsCl3 | H2O | 95 °C, ascorbic acid | — | Spherical | 1–1.5 | — | 36 |
| Os(acac)3 | EG | 200 °C | PVP | Spherical | 1.2 | fcc | 38 |
The other important factor influencing the synthesis of metallic nanoparticles is concentration of a stabilizing agent. Generally, increasing the concentration of a stabilizing agent leads to formation of smaller nanoparticles. This is caused by the fact that the formed metallic seeds are directly surrounded by a larger number of molecules of a stabilizing agent. In a case of using polymeric macromolecules as stabilizing agents, the length of the polymeric chain is also an important factor effecting on the size of formed nanoparticles. Additionally, the increasing concentration of a stabilizing agent often leads to an increase in the solution viscosity. The increased viscosity makes diffusion more difficult and hence limits the growth of nanoparticles.40 However, in many cases, it is a very small effect and therefore is often not considered.41
In a case of using polymeric macromolecules as stabilizing agents, the change of the molecular weight of the polymeric molecules usually also significantly influences on the stability of formed nanoparticles–nanoparticles stabilized by a polymeric stabilizing agent with a longer polymeric chain are usually more stable (longer polymeric molecules better keep metallic nanoparticles away from each other).42
Another important parameter is a heating rate. Successive decreasing of the heating rate leads to formation of larger nanoparticles with a broader size distribution.44 This can be connected with a less defined nucleation threshold. As a result, more nuclei can be formed during a longer time, what leads to a broader size distribution.
The rate of the addition of a metal precursor also effects on the final size of formed nanoparticles. The slower addition rate results in larger nanoparticles with a broader distribution of sizes. The slower addition rate induces a longer period of nucleation: immediately after injection a few nuclei are formed, while the precursor is continuously injected into the reaction mixture. Some of this precursor is converted into new nuclei and some is used for growing of pre-existed nuclei. Finally, the polydisperse particles with a large size are formed.45
When solvent plays also a role of a reducing agent, not only its boiling temperature but also its reducing potential is very important – reducing potential determines the kinetics of the reduction reaction that influences sizes and shapes of formed nanoparticles. For example, Kaneko et al. analyzed the reducing potential of various diols and showed that their reducing potential strongly depends on the number of carbon atoms between the –OH groups: the smaller distance between the hydroxyl groups results in higher reducing ability.46 The reactivity of polyols with different positions of the hydroxyl groups are almost similar as far as the number of carbon atoms between them is the same.
In a case of synthesis in aqueous solutions, application of a stronger reducing reagent produces smaller metallic nanoparticles with narrower size distribution.47 This effect can be explained by faster nucleation process when a stronger reducing agent is used.
3. Synthesis of ruthenium, rhodium, osmium and iridium nanoparticles attached to various supports
Nanoparticles of PGMs are often synthesized in a presence of another material. Such approach often leads to the formation of PGMs nanoparticles strongly attached to various supports. Attachment of nanoparticles of PGMs to the support can be also realized after their formation.There are many various reasons why nanoparticles of PGMs are attached to another materials. In many cases, the main reason of such attachment is modification of the electronic structure of the PGM nanoparticles, and hence, modification of some of their chemical (mainly catalytic) properties, induced by the PGM nanoparticles – support interactions. A significant modification of the catalytic properties of PGM nanoparticles is observed, for example, after their attachment to TiO2. The other very important reason of deposition of PGM nanoparticles on another materials is to facilitate their practical use, for example, as catalysts. When used as a heterogeneous catalyst, very expensive PGMs nanoparticles attached to some supports can be much easier and more efficiently removed from the reaction mixture after the completion of the reaction than the non-attached nanoparticles. For example, using a magnetic field easy manipulation of the PGM nanoparticles can be achieved when they are attached to a magnetic substrate. Below some examples of the synthesis of ruthenium, rhodium, osmium and iridium nanoparticles deposited on various supports are presented.
3.1. Supported ruthenium nanoparticles
Ruthenium nanoparticles can be deposited on various materials, for example, on graphene. To synthesize such a composite, Du et al. carried out co-reduction of RuCl3 and graphite oxide in EG using ascorbic acid as a reducing agent.47 Briefly, a solution of mentioned above compounds in EG was introduced to a stained steel autoclave and heated at 180 °C for 5 hours.47 TEM analysis of the material obtained after this process revealed that the Ru nanoparticles with the average diameter of 1.9 nm were well dispersed on the graphene surface. Fig. 4 presents TEM images of the obtained Ru nanoparticles attached to the surface of graphene. Ruthenium nanoparticles are also sometimes formed on magnetic substrates like magnetic silica-functionalized nanospheres or microspheres. To obtain such a material, Baig and Varma at first synthesized Fe3O4 magnetic nanoparticles functionalized by tetraethyl orthosilica, then added to this sol under stirring RuCl3, and after that increased pH to 10 using ammonia hydroxide and continued stirring for another 24 hours.48 TEM analysis confirmed formation of Fe3O4@SiO2Ru nanoparticles with a spherical morphology and a size in the range of 15–30 nm. | ||
| Fig. 4 TEM images of the Ru nanoparticles deposited on the surface of graphene: (a and b) before application as a catalyst and (c) after five cycles of the application of this material as a catalyst for the ammonia borane decomposition. Reprinted with permission from ref. 47. Copyright 2015 the Elsevier. | ||
It is also possible to attach already formed nanoparticles to the surface of the support material. For example, ruthenium nanoparticles can be prepared using previously described polyol method (heating a solution of RuCl3 in 1,2-propanediol at 150 °C)49 and then formed nanoparticles can be successfully immobilized on hydroxyapatite by heating a solution containing hydroxyapatite and Ru nanoparticles at 110 °C.
As examples of other interesting materials, on which ruthenium nanoparticles have been deposited, one can mention: carbon nanotubes,50,51 nitrogen-doped carbon nanofibers,52 or γ-Al2O3.4
3.2. Supported rhodium nanoparticles
Rhodium nanoparticles are often deposited on various carbon materials, and in such cases RhCl3 is usually used as a rhodium precursor. For example, Pan and Wai deposited Rh nanoparticles with an average diameter of 2.5 ± 0.7 nm on surfaces of carboxylate-functionalized multi-walled carbon nanotubes (MWCNTs) by a sonochemical reduction in an ethanol solution of RhCl3, MWCNTs and borane morpholine complex used as a reducing agent.53 It was found that when this synthesis is carried out without using of ultrasounds, formed Rh particles become very large and clumped.53 Reduction of RhCl3 by sodium borohydride in a presence of sodium citrate and carbon black allows to deposit Rh nanoparticles also on such carbon material.54 This reaction was carried out in an aqueous solution and leads to the formation of Rh nanoparticles with an average size of 4.1 nm and narrow size distribution well dispersed on the carbon surface. When reduction of RhCl3 is carried out without addition of sodium citrate, significantly bigger Rh nanoparticles were formed with the average size of 8.2 nm and with a broad size distribution from 3 to 16 nm. Rhodium nanoparticles with the average size of 5.7 ± 1.5 nm have been also deposited on reduced graphene oxide by the reduction at elevated temperature of RhCl3 by H2 in a presence of this carbon material.55 In some cases also other than RhCl3 rhodium compounds are used as rhodium precursors during deposition of Rh nanoparticles on carbon materials. For example, rhodium nanoparticles have been deposited on carbon nanofibers by the thermal decomposition of Rh4(CO)12 in boiling toluene in a presence of carbon nanofibers.56Rh nanoparticles are often deposited on various metal oxides like silica, alumina, titania, zirconia, hafnia or ceria.57 For example, to deposit rhodium nanoparticles on ceria, Akbayrak et al. mixed a proper amount of ceria with an aqueous solution of RhCl3. Subsequently, to the above mixture, aqueous solution of sodium borohydride was added dropwise. After 30 minutes of stirring, formed Rh/CeO2 was isolated by centrifugation. TEM analysis showed that the average diameter of formed Ru nanoparticles was 3.2 ± 0.8 nm.57
A very important parameter in a case of nanocatalysts is catalyst recovery. Catalyst recovery allows for isolation of nanocatalyst from a reaction solution, purification and application of nanocatalyst for the next reaction cycle. One of the options of separation of nanostructures is centrifugation. However, in a case of small nanoparticles centrifugation can take a long time or even can be impossible. Therefore, one of a promising alternative is formation of conjugates of metallic nanoparticles of a catalyst and a magnetic matrix. An example of such a catalyst are rhodium nanoparticles on magnetic silica-coated Fe3O4 nanoparticles synthesized by Jacinto et al.58 In this case, RhCl3 was added to a solution of amino functionalized silica coated magnetic nanoparticles and injected for reduction at 75 °C in H2 at 6 bar. TEM analysis showed that the formed catalyst is composed of silica spheres that contains magnetic cores and Rh nanoparticles of about 3–5 nm decorating the silica shell.
3.3. Supported iridium nanoparticles
Iridium nanoparticles have been also deposited on many different materials, for example, on carbon nanotubes.59 To form such a composite, a solution of (acetylacetonato)(1,5-cyclooctadiene)iridium(I) and carbon nanotubes in dichloromethane was placed in a stainless steel autoclave. Then, the introduced mixture was stirred and the solvent was slowly evaporated. Subsequently, autoclave was filled with hydrogen at 150 bar and was kept in these conditions for 12.5 h, which led to the formation of Ir nanoparticles. It was found that the reaction temperature effects on the size and catalytic activity of formed Ir nanoparticles. At low temperature nucleation is slow and the nucleation occurs only after collision with an already existing nucleation seed, what leads to formation of large microparticles or microfilms, which are catalytically less active than the smaller nanoobjects. At higher temperature nucleation is significantly faster and hence formation of smaller nanoparticles with higher catalytic activity is observed. Du et al. proposed a slightly different method of deposition of Ir nanoparticles on carbon nanotubes: mixing carbon nanotubes with an aqueous solution of H2IrCl6, evaporation of water at 100 °C, and then the final reduction of H2IrCl6 at 300 °C in H2/Ar atmosphere.60 The average size of formed Ir nanoparticles was about 1.9 nm.Another carbon material which had been applied as a support for Ir nanoparticles is graphene. Wu et al. formed Ir nanoparticles supported on graphene by the following three steps process: (1) sonication of a graphene oxide functionalized with dopamine (graphene oxide was synthesized by the Hummers' method) and IrCl3, (2) product collection by the centrifugation and (3) heating of the obtained mixture under Ar atmosphere to 750 °C and keeping the sample in this temperature for 3 h.61 TEM measurements showed that the size of formed Ir nanoparticles was 2–3 nm. Similar approach for formation of Ir nanoparticles on graphene (however with using Ir(acac)3, as a iridium precursor) has been applied by Cunci et al.62 This group also found that the final size of Ir nanoparticles formed in such synthesis depends on the gas, in which the heating was realized. Samples heated in air exhibit a wide size dispersion of formed Ir nanoparticles in a range of 2–30 nm, while heating in N2 atmosphere leads to the formation of small (1–4 nm) Ir nanoparticles.
Iridium nanoparticles have been also supported on nitrogen-functionalized carbon (NC) matrix.63,64 For this purpose, NC powder, ascorbic acid and hexachloroiridium acid was dispersed in EG. Obtained reaction mixture was transferred into a teflon-lined stainless steel autoclave and heat treated at 180 °C for 5 h. TEM analysis of the obtained product showed that the formed spherical Ir nanoparticles with a diameter of 1.4 nm are homogeneously dispersed on NC matrix without any significant signs of aggregation.64
In many cases iridium nanoparticles has been deposited on various oxides supports. For example, Fan et al. synthetized iridium nanoparticles supported on hydrous zirconia.65 At first, a solution containing: NH3, ZrOCl2 and H2IrCl6 has been aged for 12 h at 50 °C, then the obtained product was filtered and washed with distilled water until no chlorine was found in an elution, and subsequently the obtained sample was reduced for 2 h by a pure hydrogen at the pressure of 40 bar and the temperature of 180 °C. TEM analysis showed formation of 2.5–5.0 nm Ir nanoparticles uniformly distributed over the substrate. Another oxide substrate, on which Ir nanoparticles has been deposited, were MoO3 nanorods.66 The MoO3 nanorods were obtained by the calcination of a Mo3O10(C6H8N)2·2H2O precursor at 400 °C for 2 h under air flow. Then, the obtained MoO3 support was impregnated by an aqueous solution of H2IrCl6. After drying, the sample was reduced at 300 °C for 2 h using a stream of 5 vol% H2/Ar. TEM analysis showed that the formed Ir nanoparticles have a diameter of 2.5 ± 0.1 nm. A TiO2 nanopowder has been also used as a support for Ir nanoparticles. In this case, a TiO2 nanopowder was mixed with an aqueous solution of IrCl4 hydrate and urea, the obtained suspension was heated to 80 °C and stirred at this temperature for 16 hours, then, the product was filtered, dried under vacuum and calcined at 400 °C for 2 h.67 The average size of Ir nanoparticles formed on the surface of TiO2 nanostructures was about 1.5 nm.
3.4. Supported osmium nanoparticles
As another PGM nanostructures, osmium nanoparticles are often supported on various carbon nanomaterials. For example, Kavitha et al. deposited Os nanoparticles on the surface of reduced graphene oxide68 carrying out a reduction process with the phase transfer of the osmium precursor (briefly: OsCl3 transferred to the toluene phase using tetraoctylammonium bromide was mixed with an aqueous suspension of graphene oxide prepared by the Hummers' method, then hydrazine was added and the system was heated to 90 °C for 90 minutes). TEM analysis showed that the size of formed Os nanoparticles was 2–3 nm.68 The Os nanoparticles was also deposited on carbon nanotubes.69 At first, the metalorganic Os precursor [Os3(CO)10(NCCH3)2] was dissolved in acetone and sonicated. Then, the solvent was evaporated under N2 stream in a pyrolysis tube. Subsequently, the tube was sealed and heated to 400 °C under vacuum (10−6 bar) in furnace for 12 hours. The as synthesized Os nanoparticles were supported on carbon nanotubes by the chemical vapor deposition. TEM analysis revealed formation of 2 nm Os nanoparticles attached to the surface of carbon nanotubes. Electron diffraction measurements showed, that the formed Os nanoparticles has a polycrystalline nature.Low et al. synthesized osmium nanoparticles dispersed on silica.70 For this purpose, pentane was added to a flask containing dehydroxylated silica and subsequently osmium(cyclooctadiene)(cyclooctatetraene) was introduced to the flask. Then pentane was removed under reduced pressure. The addition and removal of pentane was repeated three times to achieve a good dispersion of osmium precursor on the surface of silica. After that, the silica with the adsorbed Os precursor was dried under vacuum and subsequently loaded to a glass reactor filled with H2 at the pressure of 0.66 bar, heated to 300 °C and kept at this temperature for 16 hours.
Using silica structures as sacrificial templates Molefe and co-workers formed Os nanoparticles trapped into a hollow carbon spheres.71 At first, the SiO2 microspheres were synthesized by a modified Stober method. Subsequently, obtained SiO2 structures was mixed with ethanol, urea and (NH4)2OsCl6 (as a osmium precursor) and sonicated until formation of uniform suspension. After homogenization, the obtained material was heated to 95 °C and kept at this temperature for 12 hours. This step leads to the formation of Os nanoparticles supported on the surface of SiO2 microspheres. Then, the carbon layer was formed by a chemical vapor deposition at 950 °C for 2 hours using toluene as a carbon source. The product formed at this stage can be described as SiO2@Os/C nanoparticles. Finally, the SiO2 cores were etched by HF to form Os nanoparticles encapsulated inside hollow carbon nanospheres.
Osmium nanoparticles have been also deposited on the surface of zeolite Y.72 For this purpose, zeolite Y was mixed with an aqueous solution of OsCl3. After 72 h, the initially opaque-brown supernatant turned colorless due to the complete ion exchange – osmium ions were introduced into the surface of zeolite Y by the ions exchange reaction. Then Os3+ ions were reduced by sodium borohydride. TEM analysis showed that the average diameter of formed Os nanoparticles was 3.2 ± 0.9 nm.72
4. Selected applications of Ru, Rh, Ir and Os nanoparticles in catalysis
PGMs have many very useful properties and therefore they have many various important applications (for example, in production of: electrical contacts, electronic elements, thermocouples, alloys with special properties – e.g. for dentistry, elements of jewelry, filters in some X-rays devices, anticancer drugs, etc.). However, without any doubts, the most important group of applications of nanoparticles of PGMs is heterogeneous catalysis. Below we present some selected examples of applications of Ru, Rh, Ir and Os nanoparticles in catalysis. Due to very large number of publications on the catalytic applications of Ru, Rh, Ir, and Os nanoparticles, we discuss only a small part of the available literature, mainly those works that show a change in the catalytic activity of such nanoparticles caused by a change in the method of their synthesis.4.1. CO oxidation
Very important use of PGMs is in catalytic converters in automobiles, which are used for transferring harmful carbon monoxide, nitrogen oxides and unburned hydrocarbons exhaust emissions into less noxious gases. Therefore, the description of the catalytic applications of nanoparticles formed from less commonly used members of the platinum family will be started from the presentation of some their applications as catalyst for CO oxidation.In our opinion, for PGMs, the most interesting mechanism of the catalytic oxidation of CO to CO2 is observed on ruthenium. Moreover, the catalytic activity of Ru nanoparticles strongly depends on their size and structure, and therefore depends on the method of their preparation. Hence, as an example of the application of Ru/Rh/Ir/Os nanoparticles in CO oxidation, we will describe some results concerning CO oxidation over Ru nanoparticles. Since the catalytic oxidation of CO over PGMs is very widely studied and is described in numerous publications, interested readers are referred to the more specialized review articles.73–75
Oxidation of CO over Ru nanoparticles has been studied, for example, by Joo et al.2 In this case, Ru nanoparticles were supported on silica wafers and the catalytic reaction was carried out using a chamber filled with a CO/O2/He mixture within a temperature range from 180 to 240 °C. Fig. 5a shows the turnover frequency (TOF) for the CO to CO2 oxidation at various temperatures over Ru nanoparticles having different size, and Fig. 5b shows the turnover frequency and the activation energy (Ea) of this process at 240 °C over Ru nanoparticles having different size.2 These experiments showed that at 240 °C, the catalyst based on 6 nm Ru nanoparticles exhibits 8-fold higher catalytic activity than that based on 2.1 nm Ru nanoparticles.2 The potential reason of this effect is connected with the fact, that in a case of oxidizing conditions, the surface of metallic ruthenium is at first converted to a catalytically active thin ruthenium oxide layer (Ru2O) which subsequently transforms into an inactive oxide phase.76 Interactions of the ruthenium nanoparticles with a substrate significantly decrease the stability of this catalytically active thin ruthenium oxide layer, whereas increase in the size of the ruthenium nanoparticles increases stability of this film.
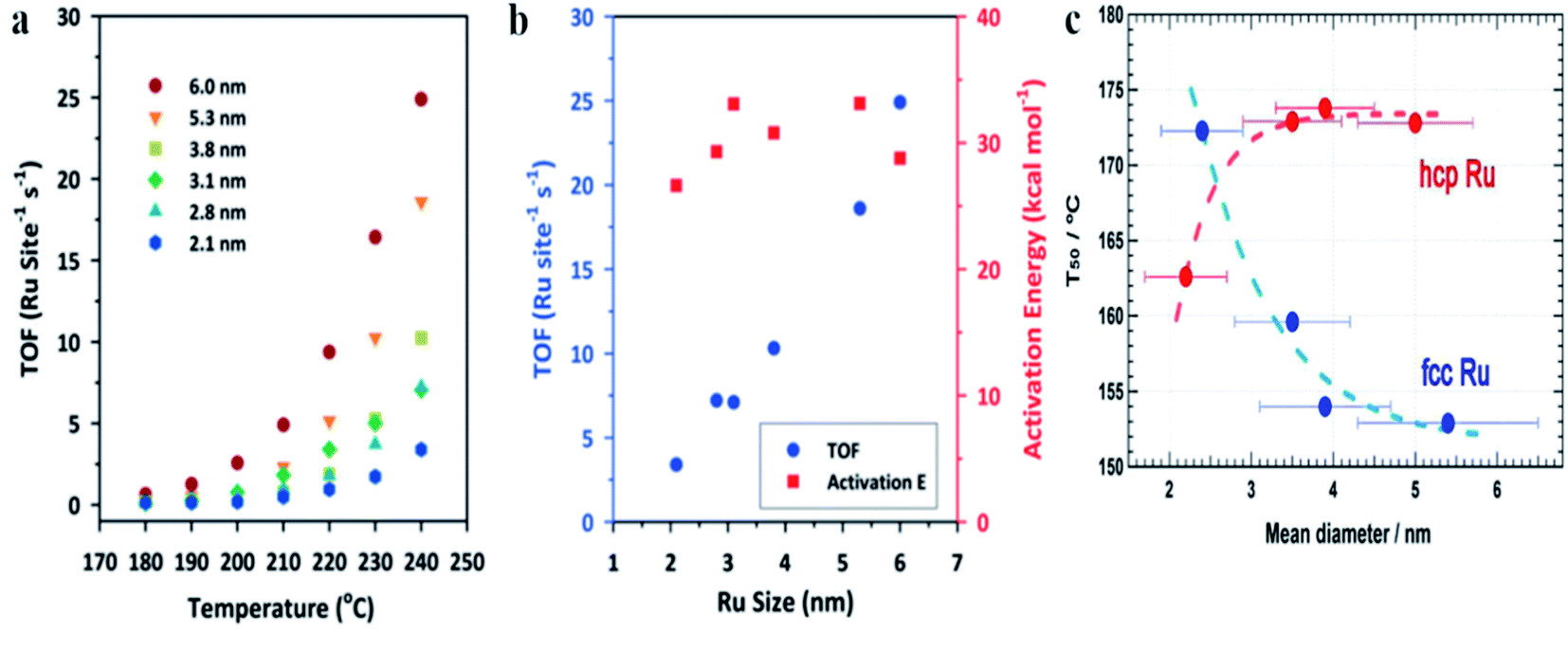 | ||
| Fig. 5 (a) The turnover frequency (TOF) for the CO to CO2 oxidation at various temperatures over Ru nanoparticles having different size. (b) The turnover frequency (TOF) for the CO to CO2 oxidation and the activation energy (Ea) of this process at 240 °C over Ru nanoparticles having different size. (c) The temperature for 50% conversion of CO to CO2 (T50) over fcc and hcp Ru nanoparticles having different size. (a) and (b) Reprinted with permission from ref. 2. Copyright 2010 the American Chemical Society. (c) Reprinted with permission from ref. 4. Copyright 2013 the American Chemical Society. | ||
Kusada et al. synthesized Ru nanoparticles with hcp (a standard structure of a bulk ruthenium) and fcc crystallographic structures and compared their catalytic activity towards oxidation of CO (see Fig. 5c).4 The CO oxidation was carried out in a tubular quartz reactor containing a gas mixture composed of CO/O2/He. The catalyst was heated to 100 °C and after 15 minutes, the eluent gas was collected and was analyzed by gas chromatography. It was found, that for hcp Ru nanoparticles, the temperature for 50% conversion of CO to CO2 (T50) increases with increasing the particle size (see Fig. 5c), a trend similar to that observed for Au or Rh nanoparticles.20,77 However, different situation is observed for fcc Ru nanoparticles, when T50 decreases with increasing the particle size (see Fig. 5c), a similar to the trend observed for Pt nanoparticles.78 As mentioned above, some reports revealed that CO oxidation on hcp Ru nanoparticles begins from oxidation of Ru(001) to a RuO2(110) layer, and a subsequent CO oxidation occurs on RuO2(110). In a case of fcc Ru nanoparticles, the dominant facet is (111), which has the lowest surface energy. It means that the surface of fcc Ru should exhibit higher reactivity than the surface of hcp Ru. Additionally, the higher catalytic activity of fcc Ru nanoparticles (it means having another structure than a bulk metal) may be attributed in part to a higher number of defects in such structures, such as twin boundaries.
4.2. Fischer–Tropsch synthesis
Another example of an economically important process which can be carried out using PGMs nanoparticles (ruthenium catalysts seem to be the most active ones) and which shows strong dependence on the structure of used catalytic nanostructures is a Fischer–Tropsch synthesis (FTS). The FTS is a class of chemical reactions that converts a mixture of CO and H2 into hydrocarbons. The FTS occurs at 150–350 °C through polymerization of CHx building blocks generated from syngas by the CO dissociation followed by the C hydrogenation. The main products of FTS are paraffin,79 waxes80 and small amounts of oxygenates like alcohols, acids, esters, ketones and aldehydes.81It was found that the size of used ruthenium nanoparticles significantly effects the composition of the product of the FTS process.82 The larger Ru nanoparticles supported on TiO2 have improved reduction properties which leads to the preference of methanation, while smaller Ru nanoparticles are less effective in reduction which promotes the chain growth.
Another important factor affecting on the composition of the products of FTS is a pore size of the support material.83 It was found that the CO conversion and the selectivity towards C5+ hydrocarbons increase with increasing pore size, whereas the ethane selectivity shows an opposite trend.
Using osmium nanoparticles (instead of ruthenium) on various supports (e.g. SiO2,71 Al2O3,84 and carbon spheres71) leads to formation in the FTS of practically only C1 and C2 hydrocarbons and no long chain hydrocarbons are formed.
4.3. Decomposition of nitrogen oxides
Nitrogen oxides (NOx) generated by industry and by the internal combustion engines in various vehicles are very important air pollutants. Decomposition of NOx is effectively catalyzed by many types of materials, such as: zeolites, metal oxides and PGMs.85 The generally accepted mechanism of NO decomposition in a presence of PGMs catalysts involves adsorption of molecules of NO on the metal surface, NO dissociation leading to N and O atoms adsorbed on the metal surface, and recombination of the adsorbed atoms leading to the formation of molecules of N2 and O2. N2 desorbs easily from the surface of PGMs even in relatively low temperatures (e.g. 190 °C),86 but desorption of O2 is significantly more difficult and low efficiency of this process may even lead to a deactivation of the catalyst.87,88 Hence, many groups are working on increasing the effectiveness of O2 desorption from PGMs. Rhodium is preferable to other PGMs in the nitrogen oxides decomposition and therefore nowadays, the majority of rhodium consumed worldwide is used in automobiles in catalytic converters.As an example of the rhodium catalyst for NO conversion, one can mention rhodium nanoparticles deposited on carbon nanotubes which, at 400 °C, convert NO into N2, O2 and N2O with almost 100% efficiency – decrease in the temperature leads to decrease in the efficiency of the NO conversion.89 Interestingly, this catalyst allows for the production of a significant amounts of N2O – at 300 °C the efficiency of the transformation to N2O reaches 55%.89 Beyer et al. also investigated this system (rhodium nanoparticles on carbon nanotubes) and reported that the conversion rate of NO reaches a maximum (99%) at 400 °C, with the selectivity towards N2 equal to about 94%.90 Vermisoglou et al. showed that for Rh nanoparticles supported on single-wall carbon nanotubes functionalized with PEG, an efficient oxygen desorption is observed at a temperature as low as 110 °C.91 What more, in a presence of CO as a reducing agent, the catalyst retained high activity for more than 24 h on stream with intermediate steps of regeneration in helium atmosphere every 4.5 h. This catalyst exhibits good selectivity towards N2 formation (about 76–96%).91
In many cases catalysts containing mixtures of rhodium and another metal (or metals) from the platinum group are used. For example, Salker and Desai investigated M0.05Co2.95O4 structures containing Rh, Ru and Pd (where M is used to denote Rh and Ru and Pd).92 In the temperature range between 300 and 400 °C, the efficiency of the NO conversion over pure Co3O4 is about 40%.92 Above this temperature the yield drops.92 The addition of PGMs increases the efficiency of the catalytic reaction even in the temperature range between 200 and 300 °C. The rhodium-doped catalyst allows for even 100% conversion of NO. In a case of admixtures with ruthenium, the conversion rate was lower (about 70%), however, the stability of the catalyst and the selectivity of reduction to N2 instead of N2O is increased.92
Not only rhodium and its alloys has been used as PGMs catalysts for NO decomposition. For example, Baudin et al. synthetized Ir/Ce0.6Zr0.4O2 catalyst and showed that at about 250 °C, the 1-propanol assisted-reduction of NOx reaches over this catalyst almost 100% efficiency.93
It was found that the catalytic activity of bimetallic nanoparticles can be significantly higher than those of standard one element structures. For example, the bimetallic Ru–Ir/TiO2 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) catalyst exhibits more than two times higher activity towards removal of NOx than the one element Ru/TiO2 and Ir/TiO2 catalysts.94 Ir–Ru nanoparticles have been also supported on Al2O3.95 This catalyst exhibits high NOx conversion rate (91%) which could be achieved at 175 °C, while the monometallic Ru and Ir catalysts have no activity in NOx-decomposition at this temperature.
:1) catalyst exhibits more than two times higher activity towards removal of NOx than the one element Ru/TiO2 and Ir/TiO2 catalysts.94 Ir–Ru nanoparticles have been also supported on Al2O3.95 This catalyst exhibits high NOx conversion rate (91%) which could be achieved at 175 °C, while the monometallic Ru and Ir catalysts have no activity in NOx-decomposition at this temperature.
Another example of alloy nanoparticles from PGMs that have a significant activity in NO decomposition are Pd0.5Ru0.5 nanostructures. Sato et al. showed that the degree of NOx to N2 conversion over these nanostructures reaches almost 100% already at a temperature of about 200 °C (the tests were carried out using a gas derived from gasoline combustion).96 For comparison, platinum nanoparticles achieves a similar degree of conversion in a significantly higher temperature (at about 350 °C). It should be noted that the obtained Pd0.5Ru0.5 structures reveal a similar efficiency of the catalytic NO decomposition as rhodium nanoparticles (the most efficient PGMs catalyst of this process).
4.4. Hydrogenation processes
PGMs nanoparticles are often used as catalysts in various hydrogenation processes. For example, Esteban et al. carried out hydrogenation of benzene and cyclohexene using iridium nanoparticles supported on a reduced graphene oxide.97 With this catalyst benzene hydrogenation was possible at 100 °C and under 10 bar of H2. Esteban et al. found that the iridium nanoparticles with a diameter of 3.6 ± 1.0 nm have the highest catalytic activity, whereas bigger and smaller Ir nanoparticles are significantly less active.97 Hydrogenation of various substituted benzenes can be also carried out using as a catalyst rhodium nanoparticles supported on carbon nanofibers.56 Motoyama et al. found that for such systems, the catalytic activity strongly depends on the structure of the support, it means, on the arrangement of the graphite layers in carbon nanofibers: the highest activity was observed in a case of the tubular arrangement, lower for the platelet arrangement and the lowest activity for the herringbone arrangement.56 Interestingly, when the same group studied the process of hydrogenation of nitroarenes on Pt nanoparticles deposited on carbon nanofibers, they observed reverse influence of the arrangement of the graphite layers on the activity of the catalyst.98Pan and Wai studied influence of the size of rhodium nanoparticles supported on carbon nanotubes on their catalytic activity in the hydrogenation of o-, m- and p-xylenes.99 Hydrogenation of xylenes was carried out in liquid arenes without addition of any other solvent to avoid solvent effects. Pan and Wai found that the hydrogenation rate reveals a negative particle size effect.99 Hydrogenation of xylenes can occur in two different ways leading to trans or cis dimethylcyclohexane. Formation of trans isomer is thermodynamically preferable. Pan and Wai also found that even the cis–trans ratio in the obtained product depends on the size of rhodium nanoparticles that are used as a catalyst – decreasing the size of Rh nanoparticles leads to increase in the cis–trans ratio.99 Another investigations showed that not only size, but also the shape of rhodium nanoparticles could affected on their catalytic activity in the hydrogenation of arenes.100 For example, the 4.9 ± 0.4 nm tetrahedral rhodium nanoparticles supported on charcoal exhibit 109 times higher catalytic activity than the 4.8 ± 0.4 nm spherical rhodium nanoparticles supported on charcoal and 5.8 times higher catalytic activity than the commercial Rh/C catalyst.100 Jacinto et al. showed that one can easily improve reusability of the catalyst Rh nanoparticles by immobilization of such nanostructures on amino-modified silica-coated magnetic nanoparticles.58 Rh nanoparticles immobilized on magnetic supports can be easily efficiently recovered from the reaction medium by applying an external magnetic field – Jacinto et al. showed that such very simple method of separation allows for using this catalyst up to 20 time without a significant loss in its catalytic activity.
Cyclohexene and phenylacetylene can be hydrogenated in a methanolic solution under mild condition in a presence of iridium nanoparticles deposited on microporous sodalite-like zeolitic imidazole framework as a catalyst.101 Zahmakiran showed that also in this case the catalytic activity of Ir nanoparticles depends on their average size – the highest activity was observed for the smallest nanoparticles.101
4.5. Conversion of CO2
It is well known that increasing CO2 amount in the atmosphere leads to a climate change and a global warming. Therefore, in recent years, conversion of CO2 into hydrocarbons or other organic compounds is a major research focus of many groups. Reduction of CO2 by H2 has been analyzed, for example, by Kwak et al.102 This group performed their studies using a series of Ru/Al2O3 catalysts with ruthenium content in the 0.1–5% range, and they found that with increasing metal loading, the selectivity toward formation of CH4 increases, while that for the CO production decreases (see Fig. 6).102 Kwak et al. explained observed change of the reaction pathway by the clustering of a highly dispersed metal phase and formation of active centers of a different nature.102 On the other hand, Aitbekova et al. found that more active for CO2 methanation are smaller Ru nanoparticles.103 This group also showed that a type of a support strongly effects on the selectivity of the CO2 catalytic reduction.103 The highest selectivity for the CO production is observed when Ru nanoparticles are supported on CeO2, slightly lower selectivity occurs in a case of using TiO2 support, while the lowest is observed for Al2O3 support.103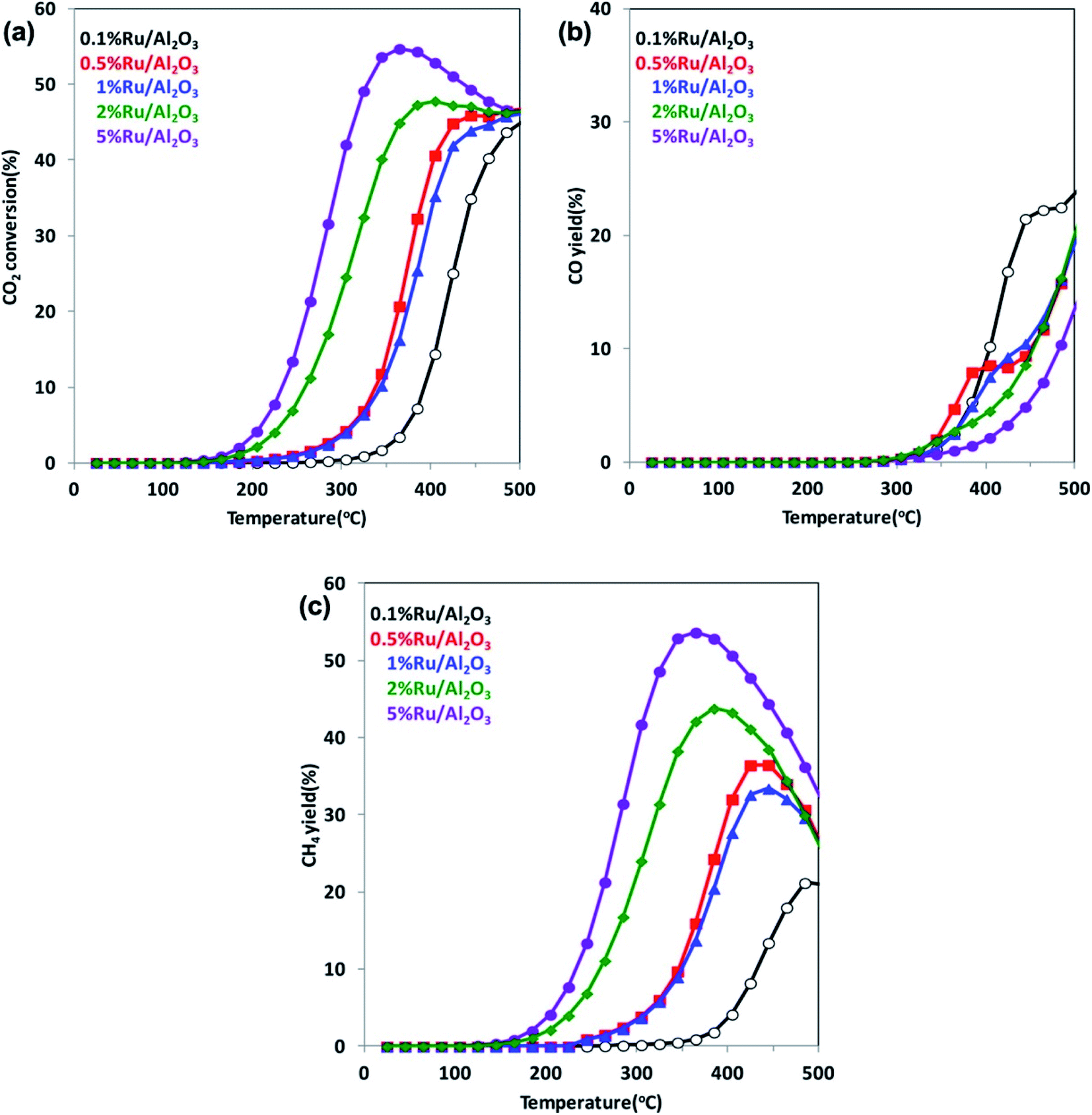 | ||
| Fig. 6 (a) Temperature dependences of the CO2 conversion rate over Ru/Al2O3 catalysts for various Ru loadings. (b) and (c) Temperature dependences of the yields of the formation of CO and CH4, respectively, over Ru/Al2O3 catalysts for various Ru loadings. Reprinted with permission from ref. 102. Copyright 2013 the American Chemical Society. | ||
The crystal phase effects of the substrate was also studied by Lin and co-workers for CO2 methanation catalyzed by Ru nanoparticles deposited on TiO2.104 In these studies, ruthenium nanoparticles were formed by a wet impregnation (impregnation of a support with a precursor-containing solution and drying) on two types of TiO2 surface: the rutile-type TiO2 (r-TiO2) and the anatase-type TiO2 (a-TiO2).104 TEM analysis showed, that the average size of Ru nanoparticles formed on the surface of r-TiO2 is 1.1 ± 0.2 nm, while in a case of formation of Ru nanoparticles on the surface of r-TiO2, the obtained nanoparticles were significantly larger and had the average size of 4.0 ± 2.4 nm. Results of the catalytic tests showed, that the Ru/r-TiO2 nanoparticles exhibit a much higher activity and thermal stability in CO2 methanation than the Ru/a-TiO2 nanostructures.104 A significant substrate effect on the conversion of CO2 on the supported Ru nanoparticles was also observed by Wang and co-workers.105 This group analyzed conversion of CO2 catalyzed by the Ru nanoparticles supported on CeO2 and Al2O3. It was found that the reaction route depends on a type of a support. In a case of a CeO2 support, the main product is CH4, while in a case of a Al2O3 support the dominant product is CO.105
Reduction of CO2 was also carried out using as a catalyst iridium nanoparticles supported on CeO2.106 It was found that also in this case a size of metal nanoparticles plays a crucial role in the reduction process. For example, when the size of Ir nanoparticles dropped from 2.5 to 1 nm, there was no methane in the products and the selectivity toward formation of CO was 100%. The potential reason of this effect is a fact that with the decreasing size of Ir nanoparticles more oxygen atoms is incorporated into the metal what changes the chemical properties of Ir nanoparticles and therefore their catalytic performance.
4.6. Reduction reactions
Aromatic nitro compounds are widely applied in industry. The reduction of nitro compounds is an important method for preparation of aniline and its derivatives. Traditionally, the reduction of nitro compounds is carried out in a presence of Fe/HCl. However, this method leads to formation of a large amount of waste what is not environmentally friendly. Therefore, a development of novel highly efficient and selective catalysts towards reduction of nitro compounds is highly desirable.Very promising catalysts for this process are rhodium nanoparticles (often supported).107–111 It is worth mentioning that the reduction of nitro compounds can be often easily monitored by simple UV-Vis absorption measurements. For example, in a case of reduction of 4-nitrophenol, an initial solution of 4-nitrophenol exhibits pale yellow color (the absorption band at 316 nm). After reduction, the solution fades to colorless, due to the formation of 4-aminophenol with the absorption band at 303 nm.107 The catalytic activity and selectivity of some PGMs nanoparticles towards reduction of various aromatic compounds are shown in Table 2.
| Nanocatalyst | Substrate | Selectivity | Turnover frequency (TOF) | Catalytic activity in time | References |
|---|---|---|---|---|---|
| Rh | Aromatic ketones | High, ketone group is untouched | 94 for acetophenone | — | 122 |
| Rh on carbon nanofibers | Compounds with aromatic rings and epoxide groups | High | 1000 for benzene | 100% after 5 cycles | 56 |
| Rh on carbon nanotubes | Polycyclic aromatic hydrocarbons | 80% | 2414 for benzene, 1949 for toluene, 468 for anisole | 100% after 6 cycles | 53 |
| Rh on hydroxyapatite | Benzene, toluene | >99% | 1770 for benzene, 980 for toluene | 92% after 5 cycles | 123 |
| Rh on carbon nanotubes | o-, m-, p-xylene | >90% | 761 for o-xylene, 866 for m-xylene, 918 for p-xylene | — | 99 |
| Ru | Aromatic ketones | High aromatic ring is untouched | 74 | — | 122 |
| Ru on magnesia | Heteroaromatic compounds with nitrogen and sulfur | High | 3700 for pyrrole, 4400 for pyridine, 0.2 for thiophene | 100% after several cycles | 124 |
| Ir on hydrous zirconia | Haloaromatic nitrocompounds | 99.9% dehalogenation product not observed | 253 | 100% after 6 cycles | 65 |
| Ir on microporous sodalite ZIF 8 | Styrene, phenylacetylene | 90% for styrene | 195 for styrene | 92–98% after 5 cycles | 101 |
| Ir on reduced graphene oxide | Benzene | High | 10000 |
— | 97 |
4.7. Hydrogen generation from various compounds used for hydrogen storage
Currently due to increase in the CO2 emission and hence global warming and climatic changes, new compounds used to “storage energy” that do not generate CO2 during their conversion into an “energy” are developed. One of them is hydrogen. Important problem in a case of hydrogen is its storage. A very promising method of hydrogen storage is its storage in various chemical compounds, for example, ammonia borane (NH3BH3). The hydrogen stored in NH3BH3 could be released in various ways, but the hydrolytic dehydrogenation of NH3BH3 seems to be the most promising one: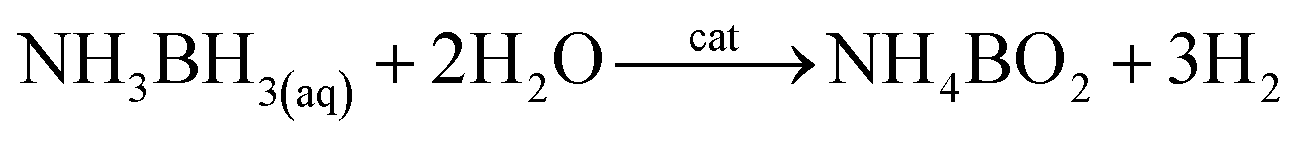
It was found that nanoparticles of PGMs can be used as highly efficient catalysts towards hydrolysis of NH3BH3 (see Table 3). For example, a very promising catalyst of this reaction is ruthenium in a form of either unsupported nanoparticles10 or nanoparticles supported on various substrates: graphene,47 γ-Al2O3,7 or silica nanospheres.112 Using Ru catalysts hydrolysis of NH3BH3 be carried out at room temperature and in some cases it takes only a few minutes to finalize this process (for more details how various parameters influence this process see Fig. 7).
| Nanocatalyst | Ea [kJ mol−1] | TOF | Catalytic activity in time | References |
|---|---|---|---|---|
| Rh nanoclusters in zeolite | 66.9 ± 1.3 | 92 | Total turnover number: 47200 |
125 |
| Rh on carbon black | 37.1 | 60 | 50% after 5 runs | 54 |
| Rh on nanodiamonds | 25.6 | 729 | 42.2% after 5 runs | 126 |
| Rh on WO3−x | 45.2 | 115 | 19.1% after 10 cycles | 127 |
| Rh on WO3 | 39 ± 2 | 749 | 34% after 5 cycles | 128 |
| Rh on graphene | 19.7 | 146 | 58% after 5 cycles | 129 |
| Rh on γ-C3N4 | 24.2 | 969 | 44.1% after 4 cycles | 115 |
| Rh on CeO2 | 43 ± 2 | 217 | 67% after 5 cycles | 57 |
| Rh on Co3O4 | 63 | 1880 | — | 130 |
| Rh on Fe3O4 | 57.5 | 272 | 100% after 5 cycles | 131 |
| Rh on silica-coated Fe3O4 | 54 ± 2 | 17 | 5 cycles | 132 |
| Rh on TiO2 | 47 | 643 | 57% after 5 cycles | 133 |
| Rh on carbon | 37.1 | 336 | 50% after 5 cycles | 54 |
| Rh on carbon nanotubes | 32 ± 1 | 706 | 61% after 5 cycles | 114 |
| Rh on CeO2 | 43 ± 2 | 2010 | 67% after 5 cycles | 57 |
| Rh on nanoTiO2 | 65.5 ± 2 | 260 | 134 | |
| Rh on graphene | 19.7 | 325 | ∼50% after 5 cycles | 129 |
| Rh on γ-Al2O3 | 21 | 178 | — | 135 |
| Ru–Rh | 47.4 ± 2 | 386 | 68% after 5 cycles | 29 |
| Ru | 27.5 | 21.8 | — | 10 |
| Ru on nanodiamonds | 50.7 | 229 | 40% after 4 cycles | 136 |
| Ru on MWCNTs | 33 ± 2 | 329 | 50% after 5 cycles | 137 |
| Ru on MOF MIL53 | 28.9–33.7 | 260.8–266.9 | 71–75% after 5 cycles | 138 |
| Ru on zeolite ZK-4 | 139 | |||
| Ru on TiO2 | 70 ± 2 | 241 | 90% after 3 cycles | 140 |
| Ru on cubic mesoporous silica | 24.13 | 202 | — | 141 |
| Ru on carbon black | 34.8 | 429.5 | 43.1% after 5 cycles | 142 |
| Ru on ZrO2 | 58 ± 2 | 173 | 67% after 5 run | 143 |
| Ru on HfO2 | 65 | 170 | 75% after 5 run | 144 |
| Ru on MOF MIL96 | 48 | 231 | 65% after 5 run | 145 |
| Ru on CeO2 | 51 | 361 | 60% after 5 run | 146 |
| Ru on hydroxyapatite | 58 | 137 | 92% after 5 run | 147 |
| Ru on zeolite MCM41 | 28–47 | 288 | 73–61% after 5 run | 148 |
| Ru on Fe3O4 | 77 | 127 | — | 149 |
| Ru on graphene | 12.7 | 600 | 80% after 5 cycles | 47 |
| Ru on SiO2 | 38.2 | 200 | — | 112 |
| Ru on carbon | 76 | 113 | — | 150 |
| Ru on graphene | 11.7 | 100 | — | 151 |
| Ru on γ-Al2O3 | 48 ± 2l | 83.3 | 90% after 10 cycles | 7 |
| Ru on SiO2 | 38.2 | 200 | — | 112 |
| Ru on MWCNTs | 33 | 329 | Total turnover number: 26400 |
137 |
| Ru on carbon | 76 | 113 | — | 150 |
| Ru on γ-Al2O3 | 23 | 77 | — | 135 |
| Ru on γ-Al2O3 | 67 | 23.05 | — | 152 |
| Ru on γ-C3N4 | 35.6 | 241 | 67.4% after 4 run | 153 |
| Ru on cotton fibers | 32.41 | 322 | 80% after 5 runs | 154 |
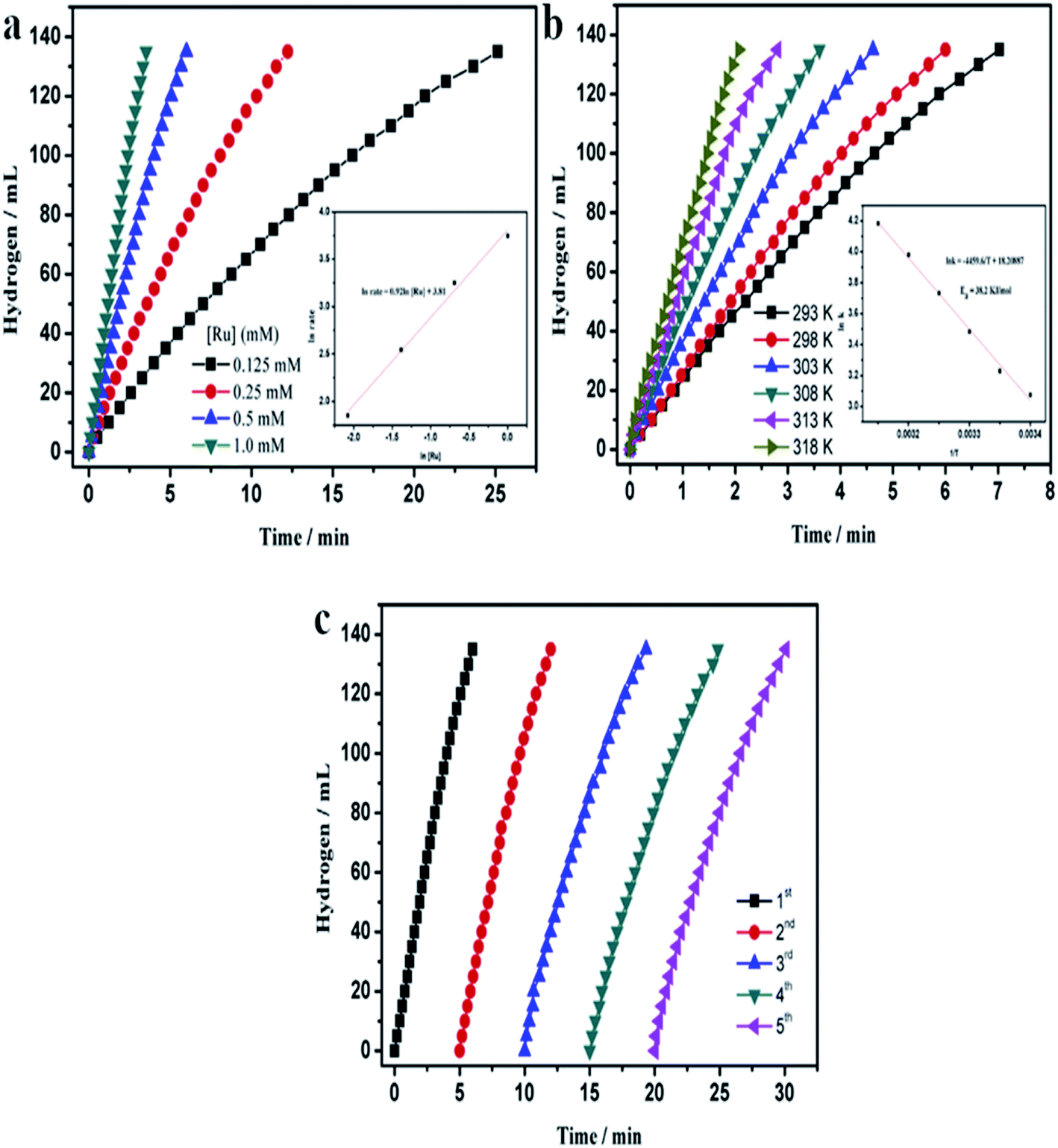 | ||
| Fig. 7 Temporal dependences of the amount of hydrogen generated by the hydrolysis of a solution of ammonia borane (10 ml, 200 mM) – reaction catalysed by Ru on silica nanospheres. (a) Dependences obtained for various concentrations of the Ru catalyst for the process carried out at 25 °C (inset shows the hydrogen generation rate vs. Ru concentrations – both values are presented in a logarithmic scale). (b) Dependences obtained at various temperatures – inset shows the Arrhenius plot (lnk vs. 1/T). (c) Dependences obtained for the subsequent runs of the process after addition of the same amount of ammonia borane. Reprinted with permission from ref. 112. Copyright 2014 the Elsevier. | ||
Hydrolysis of NH3BH3 can be also carried using both unsupported rhodium nanoparticles113 and rhodium nanoparticles deposited on carbon black,54 carbon nanotubes,114 graphitic carbon nitride,115 and various oxides, such as: Al2O3, SiO2, TiO2, HfO2 and CeO2.57 Chen and co-workers found that smaller Rh nanoparticles exhibit better catalytic activity than the bigger ones.54 Also the material of the support influences on the catalytic activity of rhodium nanoparticles, for example, the activity of Rh nanoparticles deposited on CeO2 is higher than the activity of Rh nanoparticles deposited on Al2O3, SiO2, TiO2, and HfO2.57
Hydrogen can be also produced from ammonia borane by the following reaction with methanol:
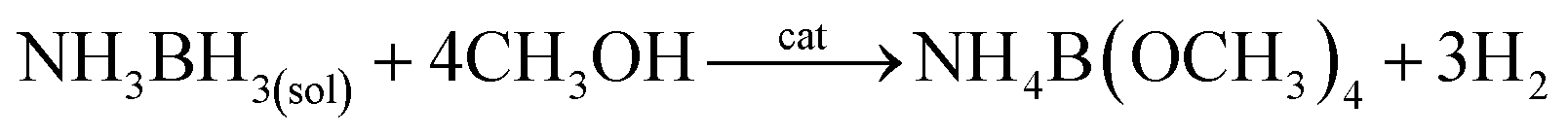
This process is also effectively catalyzed by rhodium nanoparticles supported on carbon116 or on nanosilica117 – when unsupported Rh nanoparticles are used, the turnover frequency is very low due to the aggregation of nanoparticles.
Another compound appearing to be a promising candidate for the chemical hydrogen storage is hydrazine borane. Similarly like in a case of ammonia borane, during hydrolysis of hydrazine borane a significant amount of hydrogen is formed. This reaction is effectively catalyzed by rhodium nanoparticles supported, for example, on hydroxyapatite.118 Moreover, the hydrogen evolution at room temperature starts immediately after addition of the catalyst, without induction period, even when the concentration of Rh nanoparticles is low.
4.8. Other reactions
The catalytic applications of PGMs are numerous and in addition to the examples mentioned above, there are many other chemical reactions that can be catalyzed by PGMs. For example, osmium nanoparticles supported on the surface of zeolite Y exhibit high catalytic activity and selectivity towards aerobic oxidation of alcohols to carbonyl compounds.72 The same catalyst can be applied towards cis-dihydroxylation of olefins and acetylation of 1,2-cis-diols.119 Ruthenium nanoparticles supported on active carbon can be also used as a catalyst in aerobic oxidative cross-coupling between alcohols (e.g. benzyl alcohol) and amines (both aromatic, like aniline, and various acyclic and cyclic aliphatic ones) what leads to the formation of proper imines.120 Specific flat-shaped pristine fcc ruthenium nanoparticles having a large fraction of {111} facets on their surfaces can be applied as a highly selective and reusable heterogeneous catalyst for the production of various primary amines by the reductive amination of carbonyl compounds.121 Using this catalyst, amination reactions are realized with extremely high reaction rates at low temperatures.1215. Conclusion and future outlook
In this review article we described various methods of synthesis of nanoparticles from some platinum groups metal (PGMs). Due to very numerous literature concerning synthesis of Pt and Pd nanoparticles, in this work we focused only on synthesis of nanoparticles from less frequently used members of the platinum family (ruthenium, rhodium, osmium and iridium). Such nanoparticles can be obtained by many various processes using different metal precursors, solvents, reducing and stabilizing agents (for examples see Table 1). We showed that changing various factors like the temperature of the process, type of the metal precursor, type of the solvent, or concentration of the reagents, it is possible to control the size, geometry or even the crystallographic structure of the formed nanoparticles. In many cases nanoparticles of PGMs are supported on various substrates, that can also influence on their properties, mainly their catalytic activity. Obtained nanoparticles of PGMs are often used commercially as catalysts of many important reactions.In the last part of this review article, we briefly present selected applications of nanoparticles of PGMs as catalysts – we mainly selected examples when the catalytic activity of such nanostructures depends on their size, shape and the crystallographic structure – it means examples, when the catalytic activity of nanoparticles clearly depends on the method of their synthesis. It shows how important is to improve methods of synthesis of PGMs nanoparticles to increase in the control of their size, shape, crystallographic structure and the homogeneity. We believe that unsupported and supported PGMs nanoparticles obtained according to recently developed procedures will replace many of the currently used commercial catalysts due to their improved catalytic activity and selectivity. Although this issue is not addressed in this review article, some future efforts should also focus on increasing the stability of unsupported and supported PGMs nanoparticles. Another problem is sometimes low recoverability of PGMs nanoparticles – an interesting possibility to increase reusability is the formation of composites from PGMs nanoparticles and magnetic nanostructures, and the concentration of PGMs from the reaction mixture using magnetic separation. PGMs are very expensive and any significant reduction in their consumption is very important from an economic point of view. Therefore, another likely modification of standard procedures for the preparation of commercial catalysts is the wider use of core–shell nanostructures, in which the precious PGMs only form the outermost layer of the system.
We hope that our work will convince readers that PGMs nanoparticles are fascinated subjects of research.
Abbreviations
| 2,7-DHN | 2,7-Dihydroxynapthalene |
| acac | Acetylacetonate |
| CTAB | Hexadecyltrimethylammonium bromide |
| DEG | Diethylene glycol |
| EG | Ethylene glycol |
| FTS | Fischer–Tropsch synthesis |
| MOF | Metal–organic framework |
| MWCNTs | Multi-walled carbon nanotubes |
| NC | Nitrogen-functionalized carbon |
| PEG | Polyethylene glycol |
| PGMs | Platinum-group metals |
| PVA | Polyvinyl acetate |
| PVP | Polyvinylpyrrolidone |
| SDS | Sodium dodecyl sulfate |
| TEM | Transmission electron microscopy |
| THF | Tetrahydrofuran |
| TrEG | Triethylene glycol |
| TTAB | Trimethyl(tetradecyl)ammonium bromide |
| UV | Ultraviolet |
| UV-Vis | Ultraviolet-visible |
| XRD | X-ray diffraction |
Conflicts of interest
Authors declared that there is no conflict of interest regarding the publication of this paper.Acknowledgements
This work has been financed by the University of Warsaw.References
- X. Yan, H. Liu and K. Y. Liew, J. Mater. Chem., 2001, 11, 3387–3391 RSC.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709–2713 CrossRef CAS PubMed.
- G. Viau, R. Brayner, L. Poul, N. Chakroune, E. Lacaze, F. Fiévet-Vincent and F. Fiévet, Chem. Mater., 2003, 15, 486–494 CrossRef CAS.
- K. Kusada, H. Kobayashi, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 5493–5496 CrossRef CAS PubMed.
- Y. Yao, D. S. He, Y. Lin, X. Feng, X. Wang, P. Yin, X. Hong, G. Zhou, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5501–5505 CrossRef CAS PubMed.
- H. Ye, Q. Wang, M. Catalano, N. Lu, J. Vermeylen, M. J. Kim, Y. Liu, Y. Sun and X. Xia, Nano Lett., 2016, 16, 2812–2817 CrossRef CAS PubMed.
- H. Can and Ö. Metin, Appl. Catal., B, 2012, 125, 304–310 CrossRef CAS.
- Y. Zhao, Y. Luo, X. Yang, Y. Yang and Q. Song, J. Hazard. Mater., 2017, 332, 124–131 CrossRef CAS PubMed.
- K. Pelzer, O. Vidoni, K. Philippot, B. Chaudret and V. Collière, Adv. Funct. Mater., 2003, 13, 118–126 CrossRef CAS.
- E. K. Abo-Hamed, T. Pennycook, Y. Vaynzof, C. Toprakcioglu, A. Koutsioubas and O. A. Scherman, Small, 2014, 10, 3145–3152 CrossRef CAS PubMed.
- Y. He, K. Vinodgopal, M. Ashokkumar and F. Grieser, Res. Chem. Intermed., 2006, 32, 709–715 CrossRef CAS.
- J. Yang, J. Y. Lee, T. Deivaraj and H.-P. Too, J. Colloid Interface Sci., 2004, 271, 308–312 CrossRef CAS PubMed.
- J. D. Hoefelmeyer, K. Niesz, G. A. Somorjai and T. D. Tilley, Nano Lett., 2005, 5, 435–438 CrossRef CAS PubMed.
- A. M. Watson, X. Zhang, R. Alcaraz de la Osa, J. M. Sanz, F. González, F. Moreno, G. Finkelstein, J. Liu and H. O. Everitt, Nano Lett., 2015, 15, 1095–1100 CrossRef CAS PubMed.
- N. Zettsu, J. M. McLellan, B. Wiley, Y. Yin, Z.-Y. Li and Y. Xia, Angew. Chem., Int. Ed., 2006, 45, 1288–1292 CrossRef CAS PubMed.
- R. Das and R. K. Soni, Analyst, 2018, 143, 2310–2322 RSC.
- Y. Zhang, M. E. Grass, J. N. Kuhn, F. Tao, S. E. Habas, W. Huang, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 5868–5869 CrossRef CAS PubMed.
- H. Zhang, X. Xia, W. Li, J. Zeng, Y. Dai, D. Yang and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 5296–5300 CrossRef CAS PubMed.
- S. R. Lee, M. Vara, Z. D. Hood, M. Zhao, K. D. Gilroy, M. Chi and Y. Xia, ChemNanoMat, 2018, 4, 66–70 CrossRef CAS.
- M. E. Grass, Y. Zhang, D. R. Butcher, J. Y. Park, Y. Li, H. Bluhm, K. M. Bratlie, T. Zhang and G. A. Somorjai, Angew. Chem., Int. Ed., 2008, 47, 8893–8896 CrossRef CAS PubMed.
- J. L. Huang, Z. Li, H. H. Duan, Z. Y. Cheng, Y. D. Li, J. Zhu and R. Yu, J. Am. Chem. Soc., 2017, 139, 575–578 CrossRef CAS PubMed.
- S. Gao, J. Zhang, Y.-F. Zhu and C.-M. Che, New J. Chem., 2000, 24, 739–740 RSC.
- A. Behr, Y. Brunsch and A. Lux, Tetrahedron Lett., 2012, 53, 2680–2683 CrossRef CAS.
- J. Schulz, A. Roucoux and H. Patin, Chem.–Eur. J., 2000, 6, 618–624 CrossRef CAS.
- T. D. Ewers, A. K. Sra, B. C. Norris, R. E. Cable, C.-H. Cheng, D. F. Shantz and R. E. Schaak, Chem. Mater., 2005, 17, 514–520 CrossRef CAS.
- M. Harada, D. Abe and Y. Kimura, J. Colloid Interface Sci., 2005, 292, 113–121 CrossRef CAS PubMed.
- J. A. Baeza, L. Calvo, M. A. Gilarranz and J. J. Rodriguez, Chem. Eng. J., 2014, 240, 271–280 CrossRef CAS.
- B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood, J. Henzie and Y. Yamauchi, Nat. Commun., 2017, 8, 15581 CrossRef CAS PubMed.
- M. Rakap, J. Alloys Compd., 2015, 649, 1025–1030 CrossRef CAS.
- M. Cui, J. Zhou, Y. Zhao and Q. Song, Sens. Actuators, B, 2017, 243, 203–210 CrossRef CAS.
- M. Cui, Y. Zhao, C. Wang and Q. Song, Microchim. Acta, 2016, 183, 2047–2053 CrossRef CAS.
- H. Su, D.-D. Liu, M. Zhao, W.-L. Hu, S.-S. Xue, Q. Cao, X.-Y. Le, L.-N. Ji and Z.-W. Mao, ACS Appl. Mater. Interfaces, 2015, 7, 8233–8242 CrossRef CAS PubMed.
- S. Kundu and H. Liang, J. Colloid Interface Sci., 2011, 354, 597–606 CrossRef CAS PubMed.
- R. Zhang, X. Liu, L. Shi, X. Jin, Y. Dong, K. Li, X. Zhao, Q. Li and Y. Deng, Nanomaterials, 2019, 9, 76 CrossRef PubMed.
- X. Xia, L. Figueroa-Cosme, J. Tao, H.-C. Peng, G. Niu, Y. Zhu and Y. Xia, J. Am. Chem. Soc., 2014, 136, 10878–10881 CrossRef CAS PubMed.
- K. Chakrapani and S. Sampath, Chem. Commun., 2013, 49, 6173–6175 RSC.
- S. R. Ede, U. Nithiyanantham and S. Kundu, Phys. Chem. Chem. Phys., 2014, 16, 22723–22734 RSC.
- T. Wakisaka, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, G. Ibrahima, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Commun., 2020, 56, 372–374 RSC.
- Z. Fereshteh, R. Rojaee and A. Sharifnabi, Superlattices Microstruct., 2016, 98, 267–275 CrossRef CAS.
- T. Zhao, R. Sun, S. Yu, Z. Zhang, L. Zhou, H. Huang and R. Du, Colloids Surf., A, 2010, 366, 197–202 CrossRef CAS.
- R. Harpeness, Z. Peng, X. Liu, V. G. Pol, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2005, 287, 678–684 CrossRef CAS PubMed.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Langmuir, 2002, 18, 2888–2894 CrossRef CAS.
- H. Liu, H. Zhang, J. Wang and J. Wei, Arab. J. Chem., 2020, 13, 1011–1019 CrossRef CAS.
- J. L. Hernández-Pinero, M. Terrón-Rebolledo, R. Foroughbakhch, S. Moreno-Limón, M. F. Melendrez, F. Solís-Pomar and E. Pérez-Tijerina, Appl. Nanosci., 2016, 6, 1183–1190 CrossRef.
- D. Kim, S. Jeong and J. Moon, Nanotechnology, 2006, 17, 4019 CrossRef CAS PubMed.
- H. Kaneko, T. Matsumoto, J. L. C. Huaman, M. Ishijima, K. Suzuki, H. Miyamura and J. Balachandran, Inorg. Chem., 2021, 60, 3025–3036 CrossRef CAS PubMed.
- C. Du, Q. Ao, N. Cao, L. Yang, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2015, 40, 6180–6187 CrossRef CAS.
- R. B. N. Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2013, 1, 805–809 CrossRef.
- C.-M. Ho, W.-Y. Yu and C.-M. Che, Angew. Chem., Int. Ed., 2004, 43, 3303–3307 CrossRef CAS PubMed.
- J. Wang, S. Shen, B. Li, H. Lin and Y. Yuan, Chem. Lett., 2009, 38, 572–573 CrossRef CAS.
- J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2009, 48, 2565–2568 CrossRef CAS PubMed.
- A. B. Ayusheev, O. P. Taran, I. A. Seryak, O. Y. Podyacheva, C. Descorme, M. Besson, L. S. Kibis, A. I. Boronin, A. I. Romanenko, Z. R. Ismagilov and V. Parmon, Appl. Catal., B, 2014, 146, 177–185 CrossRef CAS.
- H.-B. Pan and C. M. Wai, J. Phys. Chem. C, 2009, 113, 19782–19788 CrossRef CAS.
- J. Chen, M. Hu, M. Ming, C. Xu, Y. Wang, Y. Zhang, J. Wu, D. Gao, J. Bi and G. Fan, Int. J. Hydrogen Energy, 2018, 43, 2718–2725 CrossRef CAS.
- N. Karachi, M. Hosseini, Z. Parsaee and R. Razavi, J. Photochem. Photobiol., A, 2018, 364, 344–354 CrossRef CAS.
- Y. Motoyama, M. Takasaki, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2009, 11, 5042–5045 CrossRef CAS PubMed.
- S. Akbayrak, Y. Tonbul and S. Özkar, Appl. Catal., B, 2016, 198, 162–170 CrossRef CAS.
- M. J. Jacinto, P. K. Kiyohara, S. H. Masunaga, R. F. Jardim and L. M. Rossi, Appl. Catal., A, 2008, 338, 52–57 CrossRef CAS.
- M. Rueping, R. M. Koenigs, R. Borrmann, J. Zoller, T. E. Weirich and J. Mayer, Chem. Mater., 2011, 23, 2008–2010 CrossRef CAS.
- X. Du, Y. Liu, J. Wang, Y. Cao and K. Fan, Chin. J. Catal., 2013, 34, 993–1001 CrossRef CAS.
- X. Wu, B. Feng, W. Li, Y. Niu, Y. Yu, S. Lu, C. Zhong, P. Liu, Z. Tian, L. Chen, W. Hu and C. M. Li, Nano Energy, 2019, 62, 117–126 CrossRef CAS.
- L. Cunci, C. V. Rao, C. Velez, Y. Ishikawa and C. R. Cabrera, Electrocatalysis, 2013, 4, 61–69 CrossRef CAS.
- H. Wang, M. Ming, M. Hu, C. Xu, Y. Wang, Y. Zhang, D. Gao, J. Bi, G. Fan and J.-S. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 22340–22347 CrossRef CAS PubMed.
- M. Huang, H. Wang, D. He, P. Jiang and Y. Zhang, Chem. Commun., 2019, 55, 3634–3637 RSC.
- G.-Y. Fan, L. Zhang, H.-Y. Fu, M.-L. Yuan, R.-X. Li, H. Chen and X.-J. Li, Catal. Commun., 2010, 11, 451–455 CrossRef CAS.
- S. He, L. Xie, M. Che, H. C. Chan, L. Yang, Z. Shi, Y. Tang and Q. Gao, J. Mol. Catal. A: Chem., 2016, 425, 248–254 CrossRef CAS.
- O. Hernández-Cristóbal, G. Díaz and A. Gómez-Cortés, Ind. Eng. Chem. Res., 2014, 53, 10097–10104 CrossRef.
- C. Kavitha, K. Bramhaiah, N. S. John and S. Aggarwal, R. Soc. Open Sci., 2017, 4, 170353 CrossRef CAS PubMed.
- K.-F. Yung and W.-T. Wong, J. Cluster Sci., 2007, 18, 51–65 CrossRef CAS.
- J. E. Low, A. Foelske-Schmitz, F. Krumeich, M. Wörle, D. Baudouin, F. Rascón and C. Copéret, Dalton Trans., 2013, 42, 12620–12625 RSC.
- T. Molefe, R. P. Forbes and N. J. Coville, Catal. Lett., 2021, 151, 875–887 CrossRef CAS.
- M. Zahmakıran, S. Akbayrak, T. Kodaira and S. Özkar, Dalton Trans., 2010, 39, 7521–7527 RSC.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24–65 CrossRef CAS.
- A. K. Santra and D. W. Goodman, Electrochim. Acta, 2002, 47, 3595–3609 CrossRef CAS.
- T. Engel and G. Ertl, Adv. Catal., 1979, 28, 1–78 CAS.
- J. Assmann, V. Narkhede, L. Khodeir, E. Löffler, O. Hinrichsen, A. Birkner, H. Over and M. Muhler, J. Phys. Chem. B, 2004, 108, 14634–14642 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- E. McCarthy, J. Zahradnik, G. C. Kuczynski and J. J. Carberry, J. Catal., 1975, 39, 29–35 CrossRef CAS.
- X. Dupain, R. A. Krul, C. J. Schaverien, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2006, 63, 277–295 CrossRef CAS.
- Q. Zhang, X. Li, K. Asami, S. Asaoka and K. Fujimoto, Catal. Today, 2005, 104, 30–36 CrossRef CAS.
- G. Henrici-Olivé and S. Olivé, Angew. Chem., Int. Ed. Engl., 1976, 15, 136–141 CrossRef.
- Y. Zhang, X. Su, L. Li, H. Qi, C. Yang, W. Liu, X. Pan, X. Liu, X. Yang, Y. Huang and T. Zhang, ACS Catal., 2020, 10, 12967–12975 CrossRef CAS.
- K. Xiong, Y. Zhang, J. Li and K. Liew, J. Energy Chem., 2013, 22, 560–566 CrossRef CAS.
- M. Leconte, A. Theolier and J. M. Basset, J. Mol. Catal., 1985, 28, 217–231 CrossRef CAS.
- A. Fritz and V. Pitchon, Appl. Catal., B, 1997, 13, 1–25 CrossRef CAS.
- M. Aryafar and F. Zaera, J. Catal., 1998, 175, 316–327 CrossRef CAS.
- T. W. Root, L. D. Schmidt and G. B. Fisher, Surf. Sci., 1983, 134, 30–45 CrossRef CAS.
- V. P. Zhdanov and B. Kasemo, Surf. Sci. Rep., 1997, 29, 31–90 CrossRef.
- J. Z. Luo, L. Z. Gao, Y. L. Leung and C. T. Au, Catal. Lett., 2000, 66, 91–97 CrossRef CAS.
- H. Beyer, K. Chatziapostolou and K. Köhler, Top. Catal., 2009, 52, 1752–1756 CrossRef CAS.
- E. C. Vermisoglou, G. E. Romanos, G. N. Karanikolos and N. K. Kanellopoulos, J. Hazard. Mater., 2011, 194, 144–155 CrossRef CAS PubMed.
- A. V. Salker and M. S. F. Desai, Appl. Surf. Sci., 2016, 389, 344–353 CrossRef CAS.
- F. Baudin, P. Da Costa, C. Thomas, S. Calvo, Y. Lendresse, S. Schneider, F. Delacroix, G. Plassat and G. Djéga-Mariadassou, Top. Catal., 2004, 30/31, 97–101 CrossRef CAS.
- R. Zhu, Q. Yan, J. He, G. Cao and F. Ouyang, Appl. Catal., A, 2017, 541, 42–49 CrossRef CAS.
- Y.-W. You, Y. J. Kim, J. H. Lee, M. W. Arshad, S. K. Kim, S. M. Kim, H. Lee, L. T. Thompson and I. Heo, Appl. Catal., B, 2021, 280, 119374 CrossRef CAS.
- K. Sato, H. Tomonaga, T. Yamamoto, S. Matsumura, N. D. B. Zulkifli, T. Ishimoto, M. Koyama, K. Kusada, H. Kobayashi, H. Kitagawa and K. Nagaoka, Sci. Rep., 2016, 6, 28265 CrossRef CAS PubMed.
- R. M. Esteban, K. Schütte, P. Brandt, D. Marquardt, H. Meyer, F. Beckert, R. Mülhaupt, H. Kölling and C. Janiak, Nano-Struct. Nano-Objects, 2015, 2, 11–18 CrossRef CAS.
- M. Takasaki, Y. Motoyama, K. Higashi, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2008, 10, 1601–1604 CrossRef CAS PubMed.
- H.-B. Pan and C. M. Wai, J. Phys. Chem. C, 2010, 114, 11364–11369 CrossRef CAS.
- K. H. Park, K. Jang, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2007, 46, 1152–1155 CrossRef CAS PubMed.
- M. Zahmakiran, Dalton Trans., 2012, 41, 12690–12696 RSC.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 13736–13745 CrossRef CAS PubMed.
- Q. Lin, X. Y. Liu, Y. Jiang, Y. Wang, Y. Huang and T. Zhang, Catal. Sci. Technol., 2014, 4, 2058–2063 RSC.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- J. Krajczewski, K. Kołątaj and A. Kudelski, Appl. Surf. Sci., 2016, 388, 624–630 CrossRef CAS.
- P. Luo, K. Xu, R. Zhang, L. Huang, J. Wang, W. Xing and J. Huang, Catal. Sci. Technol., 2012, 2, 301–304 RSC.
- Y. Lee, S. Jang, C.-W. Cho, J.-S. Bae, S. Park and K. H. Park, J. Nanosci. Nanotechnol., 2013, 13, 7477–7481 CrossRef CAS PubMed.
- A. George, D. Selvan and S. Mandal, ChemistrySelect, 2017, 2, 9718–9721 CrossRef CAS.
- N. R. Guha, D. Bhattacherjee and P. Das, Tetrahedron Lett., 2014, 55, 2912–2916 CrossRef CAS.
- Q. Yao, W. Shi, G. Feng, Z.-H. Lu, X. Zhang, D. Tao, D. Kong and X. Chen, J. Power Sources, 2014, 257, 293–299 CrossRef CAS.
- T. Ayvalı, M. Zahmakıran and S. Özkar, Dalton Trans., 2011, 40, 3584–3591 RSC.
- Q. Yao, Z.-H. Lu, Y. Jia, X. Chen and X. Liu, Int. J. Hydrogen Energy, 2015, 40, 2207–2215 CrossRef CAS.
- R. Lu, M. Hu, C. Xu, Y. Wang, Y. Zhang, B. Xu, D. Gao, J. Bi and G. Fan, Int. J. Hydrogen Energy, 2018, 43, 7038–7045 CrossRef CAS.
- W. Luo, W. Cheng, M. Hu, Q. Wang, X. Cheng, Y. Zhang, Y. Wang, D. Gao, J. Bi and G. Fan, ChemSusChem, 2019, 12, 535–541 CrossRef CAS PubMed.
- D. Özhava and S. Özkar, Appl. Catal., B, 2016, 181, 716–726 CrossRef.
- D. Çelik, S. Karahan, M. Zahmakıran and S. Özkar, Int. J. Hydrogen Energy, 2012, 37, 5143–5151 CrossRef.
- Ö. Metin, N. A. Alp, S. Akbayrak, A. Biçer, M. S. Gültekin, S. Özkar and U. Bozkaya, Green Chem., 2012, 14, 1488–1492 RSC.
- Y. Zhang, F. Lu, H.-Y. Zhang and J. Zhao, Catal. Lett., 2017, 147, 20–28 CrossRef CAS.
- D. Chandra, Y. Inoue, M. Sasase, M. Kitano, A. Bhaumik, K. Kamata, H. Hosono and M. Hara, Chem. Sci., 2018, 9, 5949–5956 RSC.
- J. Llop Castelbou, E. Bresó-Femenia, P. Blondeau, B. Chaudret, S. Castillón, C. Claver and C. Godard, ChemCatChem, 2014, 6, 3160–3168 CrossRef CAS.
- M. Zahmakıran, Y. Román-Leshkov and Y. Zhang, Langmuir, 2012, 28, 60–64 CrossRef PubMed.
- M. Fang and R. A. Sánchez-Delgado, J. Catal., 2014, 311, 357–368 CrossRef CAS.
- M. Zahmakıran and S. Özkar, Appl. Catal., B, 2009, 89, 104–110 CrossRef.
- Z. Wen, J. Wu and G. Fan, Catalysts, 2020, 10, 1037 CrossRef CAS.
- X. Li, Y. Yan, Y. Jiang, X. Wu, S. Li, J. Huang, J. Li, Y. Lin, D. Yang and H. Zhang, Nanoscale Adv., 2019, 1, 3941–3947 RSC.
- S. Akbayrak, Y. Tonbul and S. Özkar, Int. J. Hydrogen Energy, 2021, 46, 14259–14269 CrossRef CAS.
- J. Shen, L. Yang, K. Hu, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2015, 40, 1062–1070 CrossRef CAS.
- S. Akbayrak, Y. Tonbul and S. Özkar, ACS Sustainable Chem. Eng., 2020, 8, 4216–4224 CrossRef CAS.
- Y. Tonbul, S. Akbayrak and S. Özkar, J. Colloid Interface Sci., 2019, 553, 581–587 CrossRef CAS PubMed.
- E. Taşçı, S. Akbayrak and S. Özkar, Int. J. Hydrogen Energy, 2018, 43, 15124–15134 CrossRef.
- Y. Tonbul, S. Akbayrak and S. Özkar, Int. J. Hydrogen Energy, 2019, 44, 14164–14174 CrossRef CAS.
- S. Akbayrak, S. Gençtürk, İ. Morkan and S. Özkar, RSC Adv., 2014, 4, 13742–13748 RSC.
- M. Chandra and Q. Xu, J. Power Sources, 2007, 168, 135–142 CrossRef CAS.
- G. Fan, Q. Liu, D. Tang, X. Li, J. Bi and D. Gao, Int. J. Hydrogen Energy, 2016, 41, 1542–1549 CrossRef CAS.
- S. Akbayrak and S. Özkar, ACS Appl. Mater. Interfaces, 2012, 4, 6302–6310 CrossRef CAS PubMed.
- K. Yang, L. Zhou, G. Yu, X. Xiong, M. Ye, Y. Li, D. Lu, Y. Pan, M. Chen, L. Zhang, D. Gao, Z. Wang, H. Liu and Q. Xia, Int. J. Hydrogen Energy, 2016, 41, 6300–6309 CrossRef CAS.
- M. Zahmakiran, Mater. Sci. Eng. B, 2012, 177, 606–613 CrossRef CAS.
- S. Akbayrak, S. Tanyıldızı, İ. Morkan and S. Özkar, Int. J. Hydrogen Energy, 2014, 39, 9628–9637 CrossRef CAS.
- J. R. Deka, D. Saikia, K.-S. Hsia, H.-M. Kao, Y.-C. Yang and C.-S. Chen, Catalysts, 2020, 10, 267 CrossRef CAS.
- H. Liang, G. Chen, S. Desinan, R. Rosei, F. Rosei and D. Ma, Int. J. Hydrogen Energy, 2012, 37, 17921–17927 CrossRef CAS.
- Y. Tonbul, S. Akbayrak and S. Özkar, J. Colloid Interface Sci., 2018, 513, 287–294 CrossRef CAS PubMed.
- E. B. Kalkan, S. Akbayrak and S. Özkar, Mol. Catal., 2017, 430, 29–35 CrossRef CAS.
- L. Wen, J. Su, X. Wu, P. Cai, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 17129–17135 CrossRef CAS.
- S. Akbayrak, Y. Tonbul and S. Özkar, Dalton Trans., 2016, 45, 10969–10978 RSC.
- S. Akbayrak, P. Erdek and S. Özkar, Appl. Catal., B, 2013, 142–143, 187–195 CrossRef CAS.
- L. Wen, Z. Zheng, W. Luo, P. Cai and G.-Z. Cheng, Chin. Chem. Lett., 2015, 26, 1345–1350 CrossRef CAS.
- M. Sait Izgi, M. Ş. Ece, H. Ç. Kazici, Ö. Şahin and E. Onat, Int. J. Hydrogen Energy, 2020, 45, 30415–30430 CrossRef CAS.
- S. Basu, A. Brockman, P. Gagare, Y. Zheng, P. V. Ramachandran, W. N. Delgass and J. P. Gore, J. Power Sources, 2009, 188, 238–243 CrossRef CAS.
- N. Cao, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2013, 38, 11964–11972 CrossRef CAS.
- G. P. Rachiero, U. B. Demirci and P. Miele, Catal. Today, 2011, 170, 85–92 CrossRef CAS.
- Y.-T. Li, S.-H. Zhang, G.-P. Zheng, P. Liu, Z.-K. Peng and X.-C. Zheng, Appl. Catal., A, 2020, 595, 117511 CrossRef CAS.
- L. Fu and L. Cai, Int. J. Hydrogen Energy, 2021, 46, 10749–10762 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |