DOI:
10.1039/D2RA00373B
(Paper)
RSC Adv., 2022,
12, 16779-16789
Computational identification of natural product inhibitors against EGFR double mutant (T790M/L858R) by integrating ADMET, machine learning, molecular docking and a dynamics approach†
Received
18th January 2022
, Accepted 13th May 2022
First published on 7th June 2022
Abstract
Double mutated epidermal growth factor receptor is a clinically important target for addressing drug resistance in lung cancer treatment. Therefore, discovering new inhibitors against the T790M/L858R (TMLR) resistant mutation is ongoing globally. In the present study, nearly 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules from various natural product libraries were screened by employing different ligand and structure-based techniques. Initially, the library was filtered to identify drug-like molecules, which were subjected to a machine learning based classification model to identify molecules with a higher probability of having anti-cancer activity. Simultaneously, rules for constrained docking were derived from three-dimensional protein–ligand complexes and thereafter, constrained docking was undertaken, followed by HYDE binding affinity assessment. As a result, three molecules that resemble interactions similar to the co-crystallized complex were selected and subjected to 100 ns molecular dynamics simulation for stability analysis. The interaction analysis for the 100 ns simulation period showed that the leads exhibit the conserved hydrogen bond interaction with Gln791 and Met793 as in the co-crystal ligand. Also, the study indicated that Y-shaped molecules are preferred in the binding pocket as it enables them to occupy both pockets. The MMGBSA binding energy calculations revealed that the molecules have comparable binding energy to the native ligand. The present study has enabled the identification of a few ADMET adherent leads from natural products that exhibit the potential to inhibit the double mutated drug-resistant EGFR.
000 molecules from various natural product libraries were screened by employing different ligand and structure-based techniques. Initially, the library was filtered to identify drug-like molecules, which were subjected to a machine learning based classification model to identify molecules with a higher probability of having anti-cancer activity. Simultaneously, rules for constrained docking were derived from three-dimensional protein–ligand complexes and thereafter, constrained docking was undertaken, followed by HYDE binding affinity assessment. As a result, three molecules that resemble interactions similar to the co-crystallized complex were selected and subjected to 100 ns molecular dynamics simulation for stability analysis. The interaction analysis for the 100 ns simulation period showed that the leads exhibit the conserved hydrogen bond interaction with Gln791 and Met793 as in the co-crystal ligand. Also, the study indicated that Y-shaped molecules are preferred in the binding pocket as it enables them to occupy both pockets. The MMGBSA binding energy calculations revealed that the molecules have comparable binding energy to the native ligand. The present study has enabled the identification of a few ADMET adherent leads from natural products that exhibit the potential to inhibit the double mutated drug-resistant EGFR.
Introduction
Natural products (NPs) have been used to treat various diseases for centuries. They are one of the most successful sources of potential leads as they have unique structural scaffolds making them the preferred choice over synthetic molecules.1 According to the latest update which analysed the data from Jan 1981 to Sep 2019, it was noted that in cancer 41% of drugs are inspired by natural products.2 Despite the several advantages, the development/conversion of NPs into drugs has always been challenging, due to which pharmaceutical companies had abandoned their natural product drug discovery programs in the early 1990s.3 This lack of interest resulted in a significant reduction in identifying new leads in the drug development pipeline and approval.4 Interestingly, this decline in identifying NCEs forced the pharmaceutical industry to refocus its attention on natural products, leading to natural product drug discovery renaissance. Thus, discovering and predicting new compounds from natural products as leads is essential to open new horizons for drug discovery.
The Epidermal Growth Factor Receptor (EGFR), a member of the ErbB family of tyrosine kinases, is among the most widely studied receptors in cancer biology. It is an important and well-established clinically used therapeutic target for non-small cell lung cancer (NSCLC).5–8 Hence, therapeutic leads that target EGFR were discovered, which led to the approval of gefitinib and erlotinib for the treatment of NSCLC.9 Although these first-generation inhibitors are highly effective, however during treatment within 6–12 months, these inhibitors become ineffective in 60% of NSCLC cases. It was shown that the ineffectiveness resulted from the acquisition of another mutation, wherein the gatekeeper residue threonine was replaced with methionine at position 790 (T790M).10,11 Therefore, the identification of new inhibitors against EGFR mutants, which inhibits both EGFR activating mutation (L858R) as well as secondary mutation (T790M), i.e. TMLR inhibitors, has emerged as an important clinical requirement.12–15
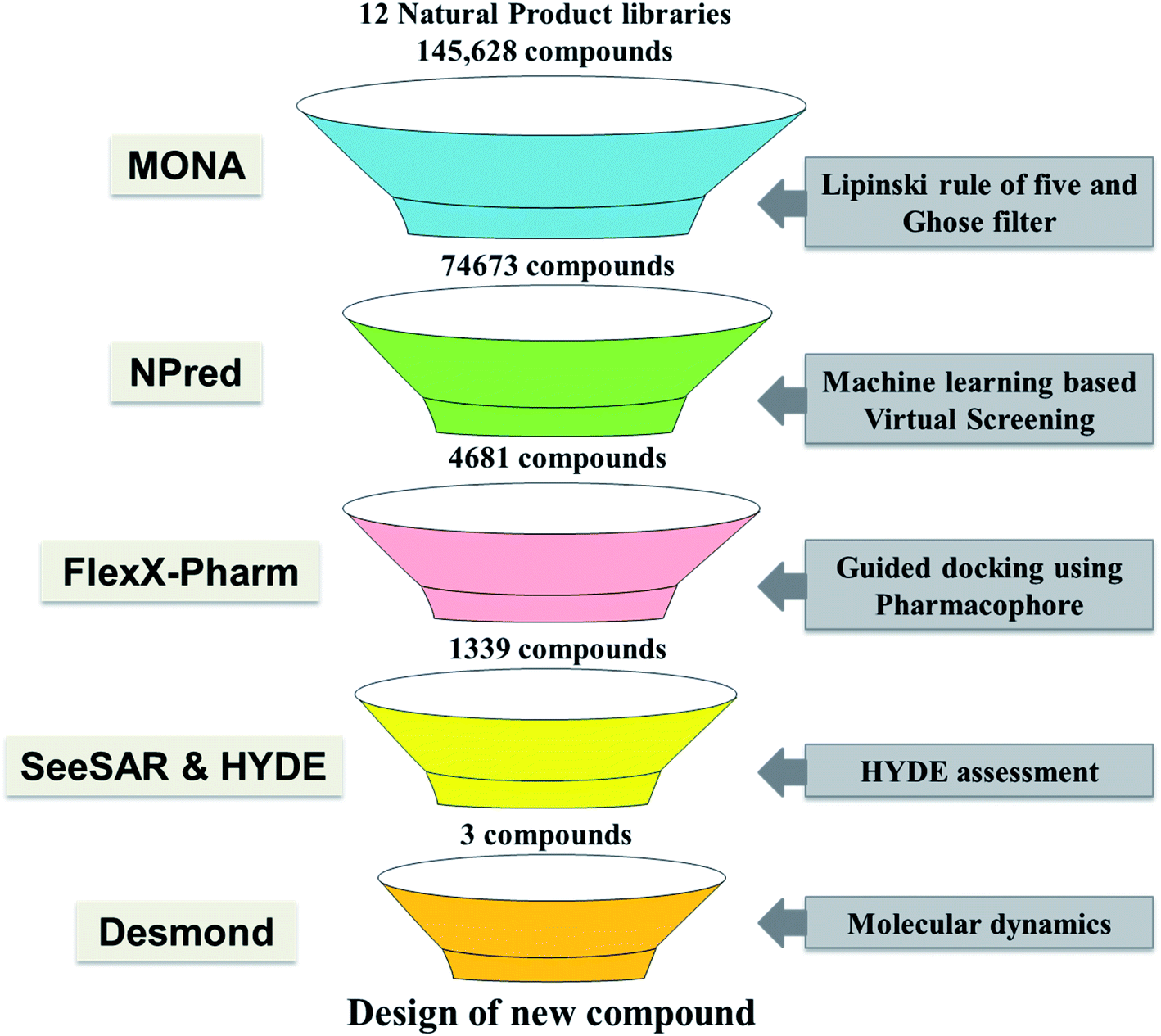
In recent years different structure and ligand-based computational methods have been regularly used to identify high binding affinity hits. However, it is essential to identify inhibitors that exhibit high affinity with the target binding site and have suitable ADMET properties to reduce the chances of attrition during the drug development phase.16,17 So, a novel computational approach has been employed wherein the rule for constrained-based docking was developed based on reported TMLR X-ray crystal structures so that the screened inhibitors mimic interactions similar to that of co-crystallized ligands. Additionally, prior to docking the NP library was screened using a machine learning based binary classification model (NPred) which categorizes molecules based on their anti-cancer potential. Also, the binding affinity and stability of ligands to the TMLR mutant protein was validated based on HYDE scoring function as well as molecular dynamics simulations. Overall, in the present work, twelve natural product databases were screened by an integrated workflow employing several techniques like ADMET, machine learning based prediction, constrained docking and molecular dynamics for identifying new ADMET adherent potential inhibitors against the mutant EGFR (Fig. 1). The current knowledge-based workflow can be useful for screening and identifying NP inhibitors against any cancer target.
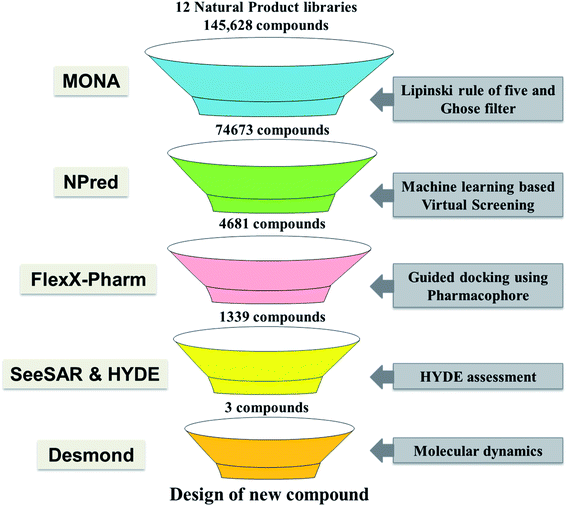 |
| | Fig. 1 Shows the workflow adopted in the present study. | |
Methods
Virtual library collection and drug likeliness filtering
Twelve different natural product databases (namely AfroDB Natural Products, AnalytiCon Discovery NP, Herbal Ingredients Target, Herbal Ingredients In Vivo Metabolism, IBScreen NP, Indofine Natural Products, NPACT Database, NubBe Natural Products, Princeton NP, Specs Natural Products, TCM Database@Taiwan, and UEFS Natural Products) containing a total of 152056 molecules available from the ZINC database (https://zinc.docking.org), were downloaded.18 MONA (https://www.zbh.uni-hamburg.de/mona), a cheminformatic application designed to process large small-molecule datasets was used to check the physico-chemical properties of the molecules.19 Firstly, the duplicate molecules present within these libraries were removed, leaving 145628 molecules for filtering. Thereafter, the drug-likeliness of these molecules was assessed using two-stage filtering criteria. The first filter operation was based on Rule of Five (Ro5), according to which the oral absorption is more likely when the compounds have ≤5 H-bond donors, ≤10 H-bond acceptors, molecular weight <500 Da, and logP of value <5.20 The second filter, which is even more stringent, was used to further reduce false-positive predictions about druggability/bioavailability. This screening was based on Ghose filter that defines the constraints for drug-likeness: logP should be between −0.4 and 5.0, molecular weight between 160 and 480 Da, and the number of atoms between 20 and 70.21
Anti-cancer activity prediction using binary QSAR model
To examine the anti-cancer potential of natural product compounds that were pre-screened for ADMET properties, machine learning based random forest binary classification model termed NPred was employed.22 NPred is a binary QSAR model developed to predict natural compounds probability as anticancer agents. The ones predicted active were subjected to pharmacophore-based virtual screening.
Generation of pharmacophore using protein–ligand interactions
As several EGFR mutant structures co-crystallized with different inhibitors are available in the PDB (https://www.rcsb.org/), twenty protein–ligand complexes of EGFR-TMLR mutant protein were retrieved. The hydrogen bonding interaction data in each of the ligand–receptor complexes were then identified and thereafter used to derive a pharmacophore pattern. As a result, a combination of “one essential” and “one optimal” interaction constituted the pharmacophore feature, which was then used for virtual screening.
Homology modeling
The crystal structure 5EDQ having a co-crystallized ligand (5N3) bound to it and exhibiting biological activity of 2 nM against the T790M/L858R mutant of EGFR was chosen for virtual screening and molecular dynamics study.23 However, it had a gap at the position 860–875 in its crystallographic structure. Therefore, homology modeling was undertaken to fill the gap at position 860–875 in the X-ray crystallographic structure of 5EDQ. Briefly, the sequence from 697 to 997 was considered as query and the 3D structure of double mutated (T790M/L858R) EGFR kinase (PDB: 5EDQ) was considered a template. The target and template sequences were aligned and ten homology models were built using MODELLER.24 The MODELLER pre-defined DOPE score (Discrete Optimized Protein Energy) was utilized to identify the best model. The model that exhibited the lowest DOPE score was selected for molecular docking.
Virtual screening using FlexX-Pharm
FlexX-Pharm is an advanced form of FlexX designed to utilize the information of pharmacophoric feature generated using protein–ligand binding data for docking.25 It works on the guiding principle that the defined pharmacophore constraints the docking process to find the solutions which exhibit the defined interactions. In this way, the probability of finding newer scaffolds showing interactions as in X-ray crystallographic complexes increases and the likelihood of false-positive decreases. Therefore, FlexX-Pharm was used to screen the library of molecules predicted to possess anti-cancer activity in the previous step. The binding pocket is defined as the location where the co-crystallized inhibitor molecule 5N3, is present in the protein structure 5EDQ and consists of amino acid residues: Leu718, Gly719, Phe723, Val726, Ala743, Lys745, Glu762, Leu788 Met790, Gln791, Leu792, Met793, Phe795, Gly796, Cys797, Asn842, Leu844 and Thr854.
Lead identification
The docking results were then imported in SeeSAR (SeeSAR; BioSolveIT GmbH, Sankt Augustin, Germany, https://www.biosolveit.de/SeeSAR) for visual analysis and binding free energy calculation using HYDE assessment method.26 HYDE implemented in SeeSAR relies on ligand's physico-chemical properties i.e. hydrogen bond and desolvation energy, to estimate ligand-binding affinity to the protein.
Molecular dynamics
The docked complexes were considered as the starting structures for the subsequent MD runs. The MD simulations were carried using Desmond simulation package included in Schrödinger Suite.27 Orthorhombic solvation box of TIP3P water model was used and the system was neutralized by adding chloride ions and an ionic strength of 0.15 M was set by adding NaCl. Prior to MD, Desmond's default system relaxation protocol was followed for equilibration. At the initial stage, 100 ps Brownian dynamics NVT simulation ensembled at 10 K employing constraint on protein's heavy atoms. The second stage consists of a 12 ps NVT simulation at 10 K and restricting the solute heavy atoms. The third stage corresponds to NPT simulation of 12 ps at 10 K, retaining the restrictions on the solute heavy atoms. The last relaxation stage was increasing temperature from 10 K to 300 K in 12 ps of NPT ensemble. Then the simulation was continued for the next 24 ps in NPT ensemble at 300 K with no restraint on any atom. The simulation length was 100 ns with a recording interval of 100 ps. The OPLS_2005 force field parameters were used in all simulations. As a control, MD using the same protocol described above was performed for the co-crystalized ligand 5N3 from 5EDQ structure.
After completion of the MD simulation, the trajectories were further examined to understand the ligand stability and protein conformational changes using parameters like root mean square deviation (RMSD), root mean square fluctuation (RMSF), solvent accessible surface area (SASA), radius of gyration (Rg) and molecular interactions between protein and ligand complexes. To estimate the relative free energy of ligand binding, Molecular Mechanics with Generalized Bonn Surface Area (MM-GBSA) was computed using the equation:
| ΔGbinding = ΔGcomplex − [ΔGprotein + ΔGligand] |
where, Δ
Gbinding is the free energy of the protein–ligand complex, and Δ
Gprotein and Δ
Gligand are energy values of protein and ligand, respectively. For the above purpose, Prime was utilized to calculate MM-GBSA using the last 5 ns simulation trajectories.
Results and discussion
Screening of natural product libraries for drug-likeness
In drug discovery, it has been suggested that the starting molecules should be drug/lead like28 i.e. it should have significant activity and favorable ADMET properties. As poor pharmacokinetics often results in failure in later stages of drug discovery, screening libraries for their ADMET properties before taking them for virtual screening studies is considered necessary.16,17,29 In the last few years, various in silico rules to evaluate the pharmacological properties and identify optimal molecules have been designed.30 The routinely used approach to screen for drug-like molecules is by estimating Lipinski's “Rule of Five” (Ro5). Therefore, twelve natural product libraries containing approximately 145000 unique molecules were screened for drug-like properties using the Lipinski filter. Application of Ro5 on the dataset resulted in the exclusion of ∼35% of the molecules, leaving ∼65%, which obeyed three out of four parameters of Lipinski (Fig. 2). Briefly, it was observed that 95758 followed three out of four parameters of Lipinski and are expected to have physico-chemical properties like drugs. Subsequently, the Ghose Rule of Three (Ro3), which is a variant of Ro5 was used to improve the drug likeliness prediction and reduce the false positives by narrowing the criteria's (−0.4 < logP < 5.6, 160 < MW < 480 Da, 20 < heavy atoms < 70). Implementation of the second filter based on Ro3 further eliminated compounds leaving behind ∼48% (∼75000 molecules) for further processing (Fig. 2). This filtered set of 74673 molecules that passed the Lipinski and Ghose filter are expected to have properties suitable for lead identification and therefore, only these were considered for anticancer activity prediction. It is also noted that the known inhibitor 5N3 i.e. (N-(7-chloro-1H-indazol-3-yl)-7,7-dimethyl-2-(1H-pyrazol-4-yl)-5H-furo[3,4-d]pyrimidin-4-amine), has molecular weight of 381.8, logP of 2.5, number of atoms is 27, number of H-bond donor and acceptor are 3 and 6, respectively. Similarly, the twenty reported co-crystallized ligands with TMLR EGFR protein from PDB were analyzed and found their physicochemical properties within the range of the Ghose filter. Thereby, it provided further confidence on the applicability of the Lipinski and Ghose filter for selecting dataset specifically for TMLR EGFR target.
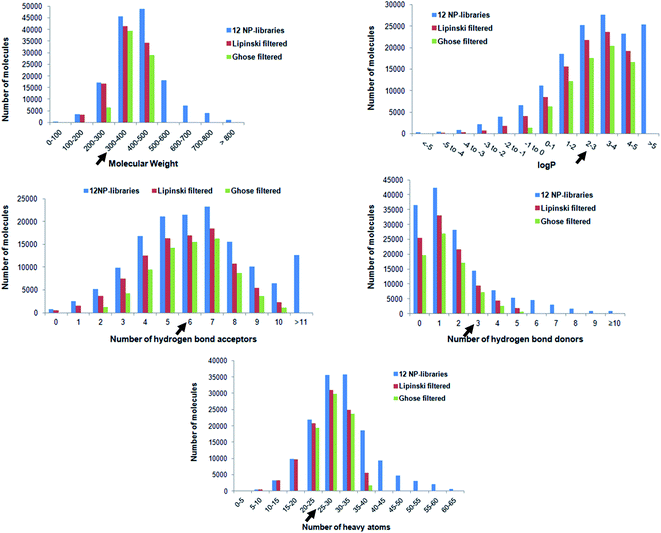 |
| | Fig. 2 Shows distribution of physicochemical properties of all molecules present in 12 natural product databases, namely AfroDB Natural Products, AnalytiCon Discovery NP, Herbal Ingredients Target, Herbal Ingredients In Vivo Metabolism, IBScreen NP, Indofine Natural Products, NPACT Database, NubBe Natural Products, Princeton NP, Specs Natural Products, TCM Database @ Taiwan, and UEFS Natural Products. The dark black arrow on each graph marks the position of known inhibitor (5N3). Lipinski's filtered compounds have ≤5 H-bond donors, ≤10 H-bond acceptors, molecular weight <500 Da, and logP value < 5.20 Ghose filtered molecules have logP between −0.4 and 5.0, molecular weight between 160 and 480 Da, and the number of heavy atoms between 20 and 70. | |
Virtual screening using a machine learning model
To speed up the drug discovery process, machine learning classification models are regularly employed to identify potential leads from a large set of molecules.31 Therefore, a random forest prediction model was utilized for screening and predicting their likeliness as anti-cancer inhibitors. The model predicts a score from 0 to 1 and a higher value signifies more likelihood for the compound to show anti-cancer activity. As a higher value reduces the probability of selecting false positives, only those compounds were selected that exhibited values >0.7. As a result, out of the 74673, nearly 6% of the compounds i.e. 4681 molecules, were predicted to have anti-cancer activity and therefore selected for molecular docking.
Pharmacophore based molecular docking
The receptor–ligand information derived from multiple co-crystal structures was used to define a pharmacophore pattern for constraint-based virtual screening to reduce false positives and improve docking results. Analysis of twenty double mutated TMLR co-crystal structures and their interactions with ligand–receptor complexes has demonstrated that Met793 acts as a donor and forms a hydrogen bond with the ligand in all the complexes. Also, it was shown that Gln791/Glu762 acts as a hydrogen bond acceptor while interacting with the ligand.32 Therefore, used the above structural features for defining the pharmacophore definition. As Met793 interaction showed an absolute 100% conservation, it was termed as “essential” constraint while interactions with either Gln791 or Glu762 was defined as “optimal”. The combination of “one essential” and “one optimal” interaction constituted the pharmacophore pattern which was used for constrained-based virtual screening using FlexX-Pharm. As a result of the pharmacophoric constraints during docking, only 1339 molecules out of 4681 could be docked in the active site as they demonstrated the ability to form the key defined interactions.
Lead identification and molecular interaction analysis
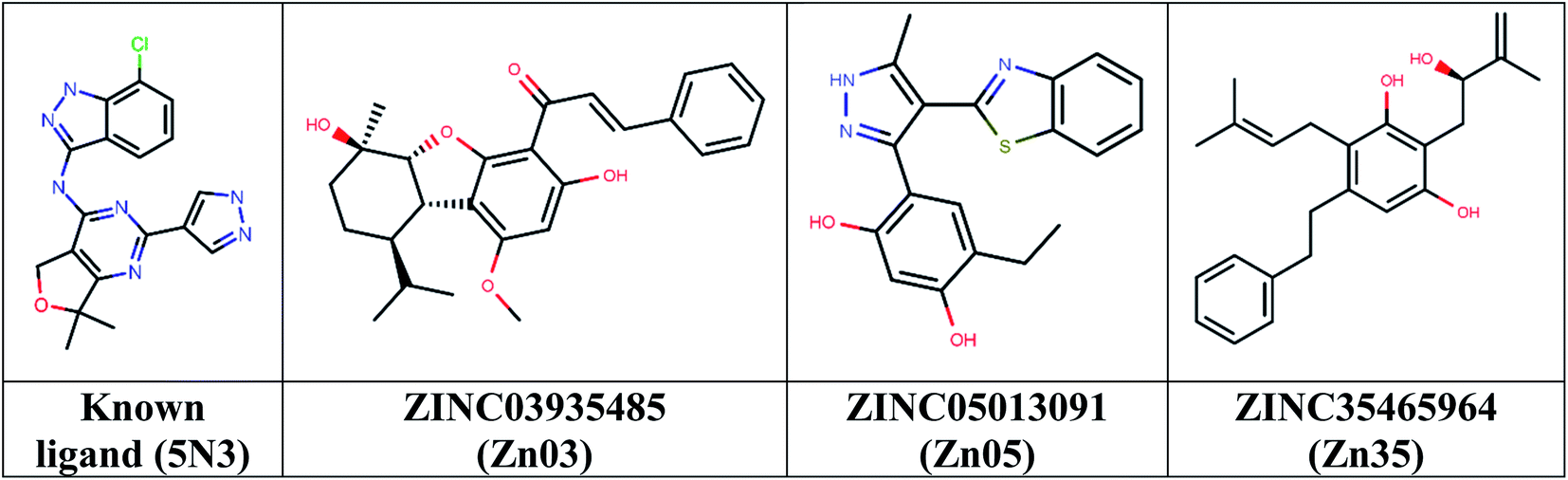
Then took 1339 docked molecules into SeeSAR for affinity assessment using HYDE. The top three ligands with the lowest HYDE affinity were chosen and their interactions in the protein's active site was analyzed (Fig. 3 and Table 1). It revealed that all the three ligands show the conserved H-bonding interaction with Gln791 and Met793, as in the case of co-crystallized inhibitors (Table 1). It confirmed that the defined pharmacophore during docking was constrained and only those docked poses were short-listed, which exhibited these binding characteristics in the active site of the TMLR mutant protein. Additionally, the three ligands showed similar hydrophobic interactions mainly with Leu718, Phe723, Val726, Ala743, Lys745, Leu788 and Leu844 (Table 1). As observed earlier, the binding pocket analysis shows that it prefers Y-shaped molecules to enable the ligand to occupy the pockets.33 Visual examination of the identified leads in the active site revealed that Zn05 and Zn35 have similar binding that is, they occupy both the pockets as in 5N3, the native ligand. However, Zn03 occupies only one of the two pockets showing hydrogen bond interaction with Gln791 and Met793. It instead orients towards the outer site and interacts with Leu718 and Phe795 to form hydrophobic interaction, thereby stabilizing the ligand in the binding pocket of TMLR.
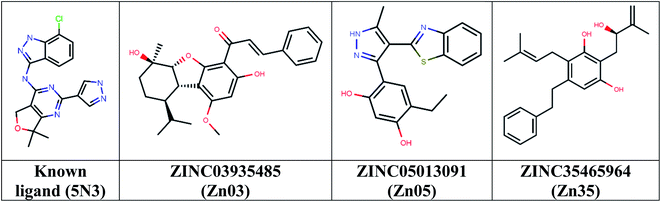 |
| | Fig. 3 Chemical structures of the three top-scoring molecules based on HYDE assessment and co-crystallized ligand. | |
Table 1 Hydrogen and hydrophobic interaction analysis of ligands with TMLR mutant
| Ligand |
Mol wt. |
logP |
H-bond interactions (AA residues) |
Hydrophobic interactions (AA residues) |
HYDE affinity |
| Co-crystallized ligand (5N3) |
382 |
2.5 |
Met793, Glu762 |
Leu 718, Gly 719, Phe 723, Val 726, Ala 743, Lys 745, Leu 788, Met 790, Leu 792, Leu 844 |
0.06–5.74 nM (experimental activity: 2.1 nM) |
| ZINC03935485 (Zn03) |
422 |
4.96 |
Gln 791, Met 793 |
Leu 718, Val 726, Ala 743, Met 790, Phe795, Leu 844 |
1–145 nM |
| ZINC05013091 (Zn05) |
351 |
4.64 |
Gln 791, Met 793 |
Leu 718, Phe 723, Val 726, Ala 743, Lys 745, Leu 788, Met 790, Leu 792, Leu 844 |
1–104 nM |
| ZINC35465964 (Zn35) |
366 |
4.87 |
Gln 791, Met 793 |
Leu 718, Phe 723, Val 726, Ala 743, Lys 745, Leu 788, Leu 844 |
0–18 nM |
Further, the three lead molecules were analyzed for their toxicological properties using DataWarrior.34 It relies on the registry of toxic effects of chemical substances (RTECS) database to predict the toxicity levels of a molecule and assess them for their mutagenicity, tumorogenicity (carcinogenicity), reproductive effect and irritant properties. Since the toxicity prediction capability of this tool is reliable with sensitivity and specificity levels of 87% and 77% respectively, thus this tool has been used in the current study. The results of the toxicity prediction analysis showed that none of the 3 compounds exhibited any adverse/toxic effects i.e. neither of them were predicted to be mutagenic, tumorogenic, have effect on reproductive ability or is an irritant.
As the three identified molecules follow drug like properties, are non-toxic and exhibit the key interactions similar to the three-dimensional protein–ligand complexes, therefore, to confirm the stability of these ligands in the binding pocket of TMLR mutant protein they were then subjected to 100 ns molecular dynamics simulation.
Molecular dynamics (MD) simulations
The use of molecular dynamics (MD) is an integral and well-established structure-based method for exploring the dynamic nature of protein–ligand complexes at an atomistic level.9,33 Therefore, to study the stability of the ligands (Zn03, Zn05 and Zn35) MD simulation was performed for 100 ns with 5N3 the co-crystallized ligand as the standard. The RMSD analysis of the protein backbone revealed that after 30 ns the system stabilized, for all three ligands. However, it required 50 ns to stabilize the protein with 5N3, the co-crystal ligand. Compared to the co-crystal ligand 5N3, Zn03, Zn05, and Zn35 showed better stability (lower RMSD) (Fig. 4A). To check for the flexibilities of the individual residues that may have contributed to the overall fluctuations in the system, RMSF was computed for all the protein–ligand complexes and it was observed to be similar (Fig. 4B). The radius of gyration was used to study the ligands compactness and stability. It was observed that Zn05 had the lowest Rg value, which was expected as it has benzothiazole and benzene linked to 2-pyrazoline in the center, thereby making the structure rigid and restricting the rotation (Fig. 4C). On the contrary, the Rg value was highest for Zn35, which has aliphatic structures, while the Rg of Zn03 was comparable with the standard ligand (Fig. 4C). The solvent accessible surface area (SASA) is an important measure of the surface area of a ligand that is available to its surrounding solvent. The SASA calculation for all the ligands revealed that the standard ligand has the lowest average SASA score indicating that a significant portion is buried in the receptor and only a smaller area is accessible to the solvent. The Zn05 ligand have higher SASA scores but are comparable to the standard ligand (5N3), revealing that only a few ligand atoms are exposed to the solvent (Fig. 4D). However, the SASA score of Zn03 and Zn35 was elevated, compared to the other ligands revealing that they are exposed to the solvent and thereby show an increased score.
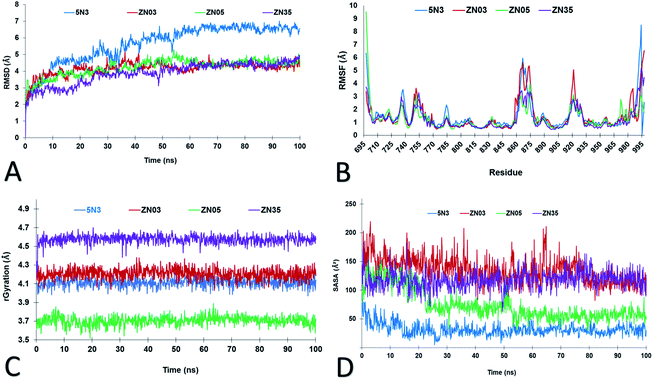 |
| | Fig. 4 Shows (A) RMSD of the protein backbone with respect to the first frame of MD simulation, (B) RMSF profile for Cα atoms of protein residues, (C) radius of gyration plot as a function of simulation time for ligands only, (D) solvent accessible surface area for three lead molecules and co-crystallized ligand. | |
Interaction analysis based on molecular dynamics
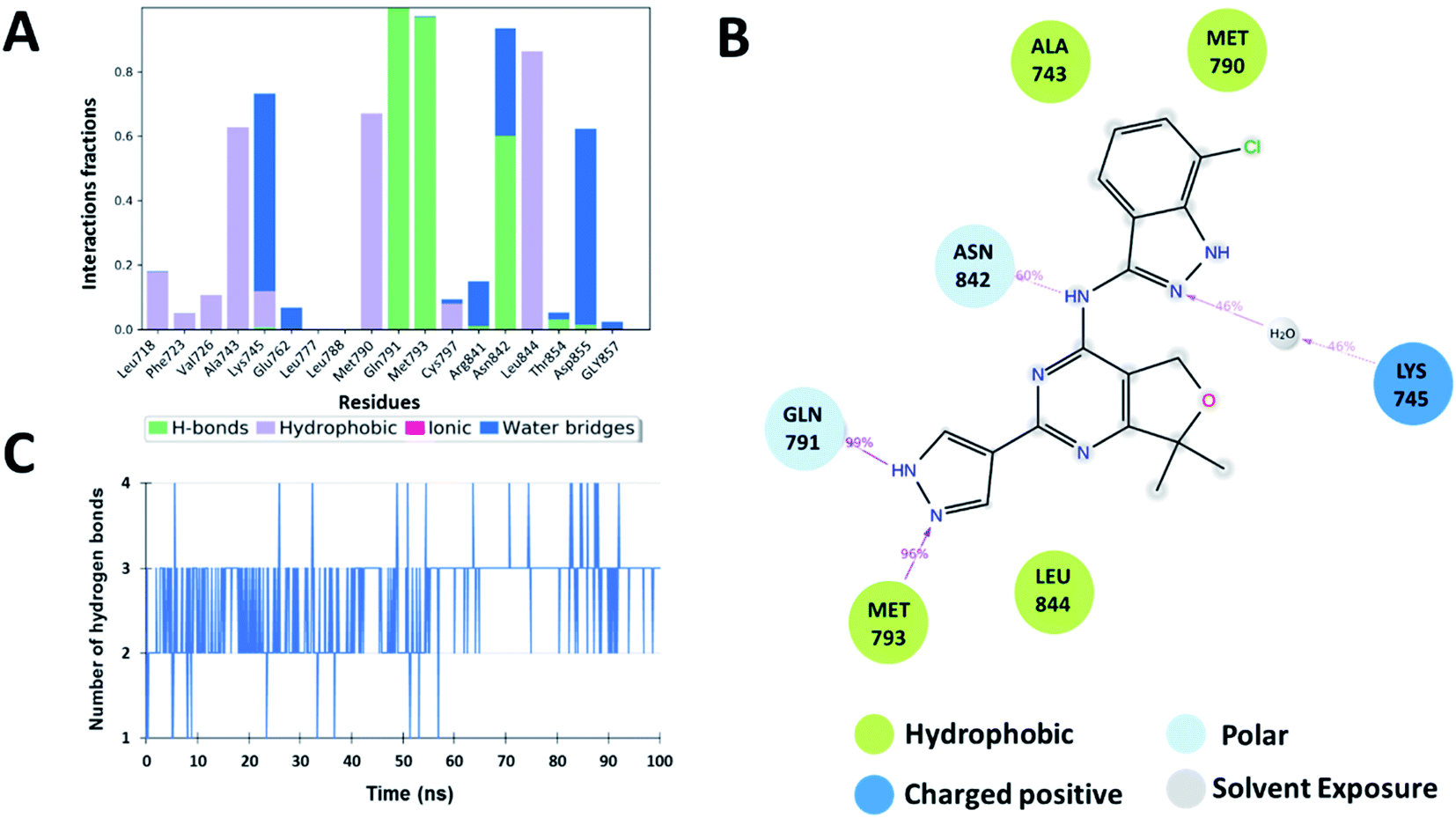
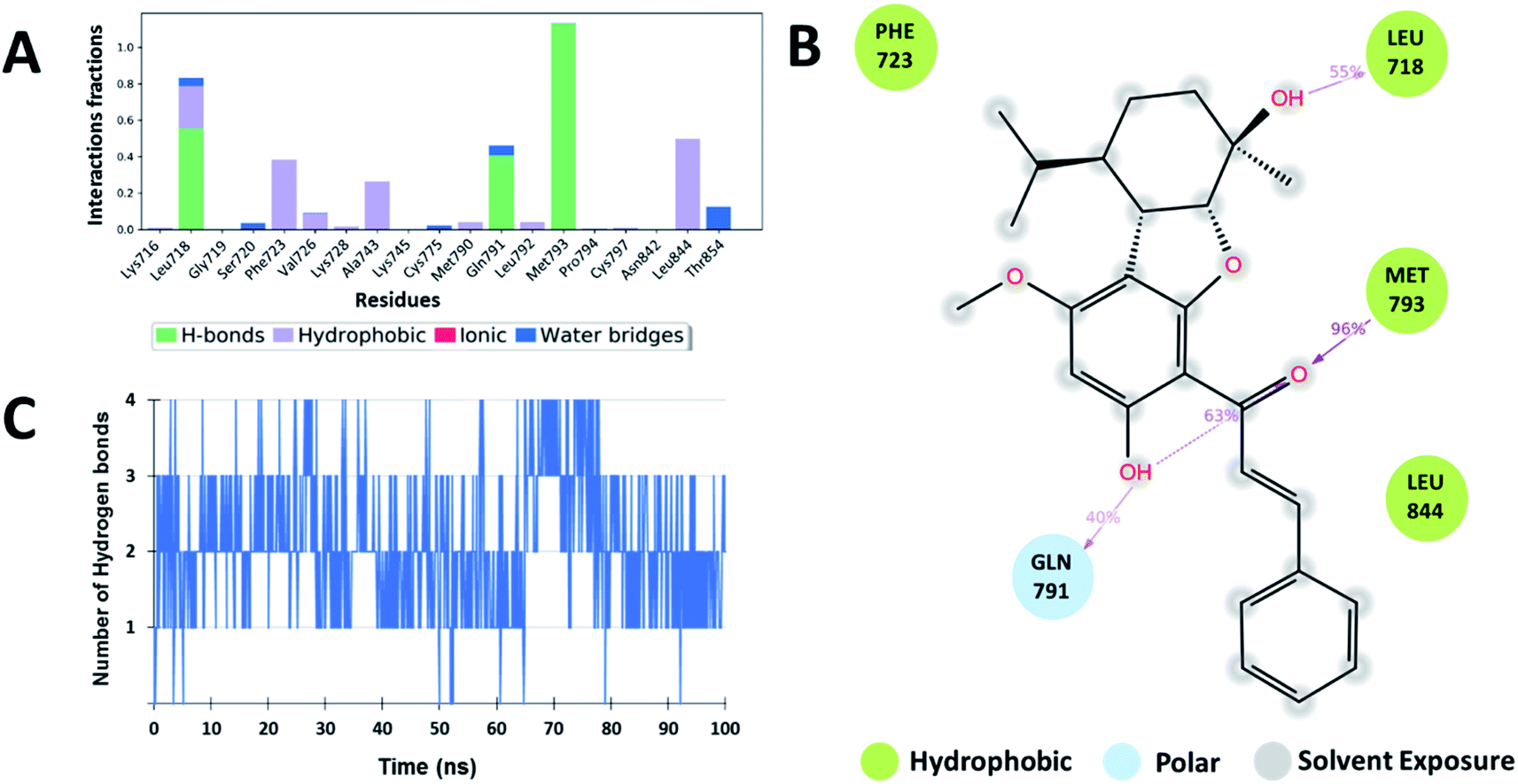
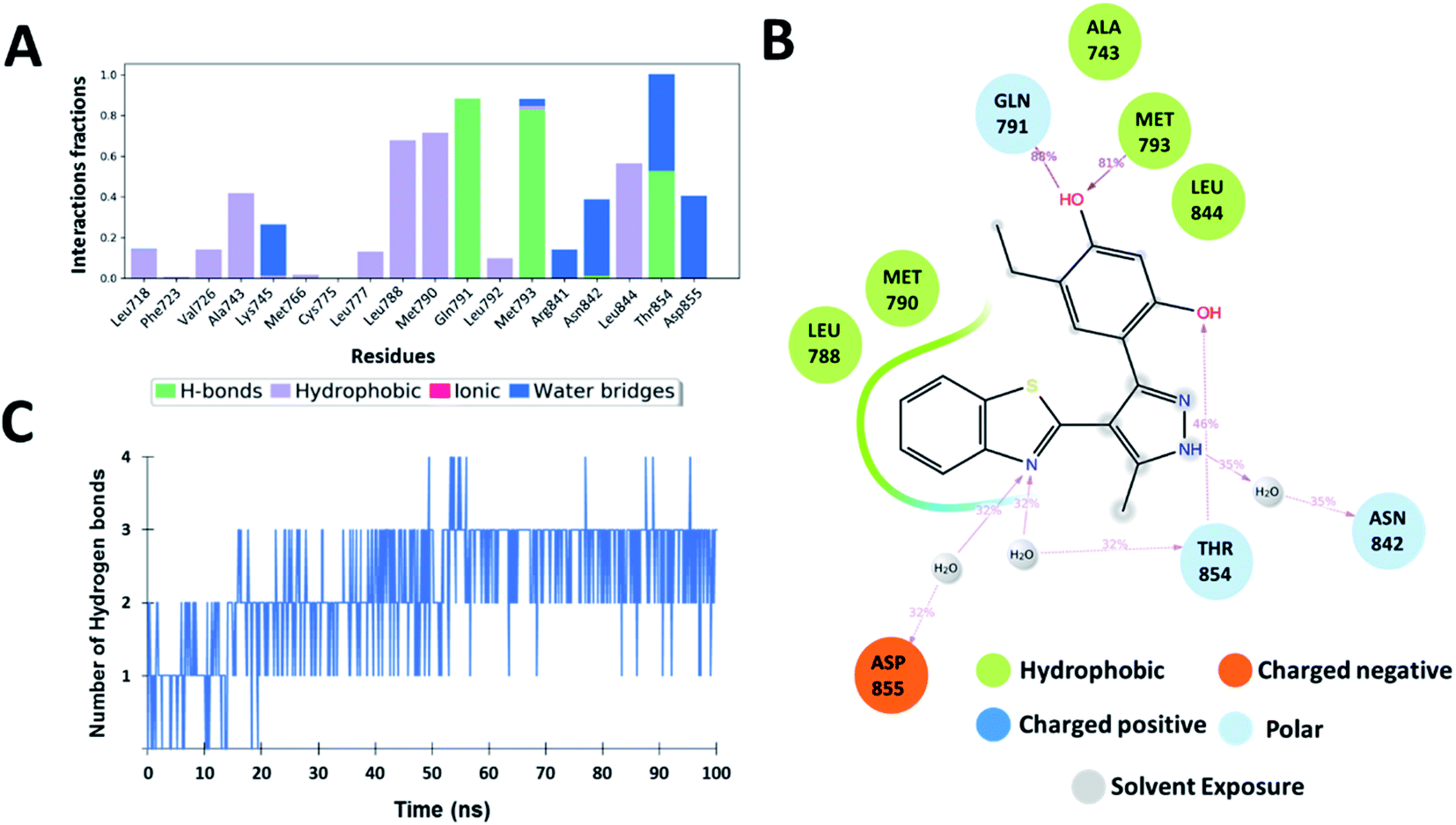
The interaction analysis of the ligand–receptor complexes for the 100 ns simulation time revealed that the leads Zn03 and Zn05 form hydrogen bonds with the protein backbone via Gln791 and Met793 same as in the case of co-crystal ligand (5N3) (Fig. 5–7 and ESI Fig. S1†). However, in Zn35, it shows hydrogen bond interaction prominently with the Met793 residue (Fig. 8 and ESI Fig. S1†), while with Gln791 the interaction probability was nearly 20%, so it is not captured in the Fig. 8. The three ligands Zn03, Zn05, and Zn35 interact with Met793 residue with connection probabilities of 96%, 81% and 82% respectively. On the other hand, two ligands (Zn03 and Zn05) interact with Gln791 residue with medium to strong connections of 40% and 88%, respectively.
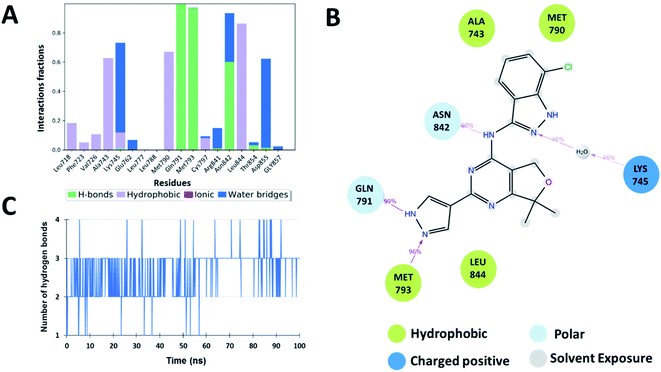 |
| | Fig. 5 Shows (A) residues involved in the interaction with 5N3 ligand and the interaction fraction during simulation. Different types of interactions are shown in different colors. (B) 2D ligand interaction diagram for the co-crystallized ligand 5N3 and surrounding residues with their percentage interactions and (C) number of H-bonds across the 50 ns MD simulation trajectory. The data and plots are generated using Schrödinger Suite. | |
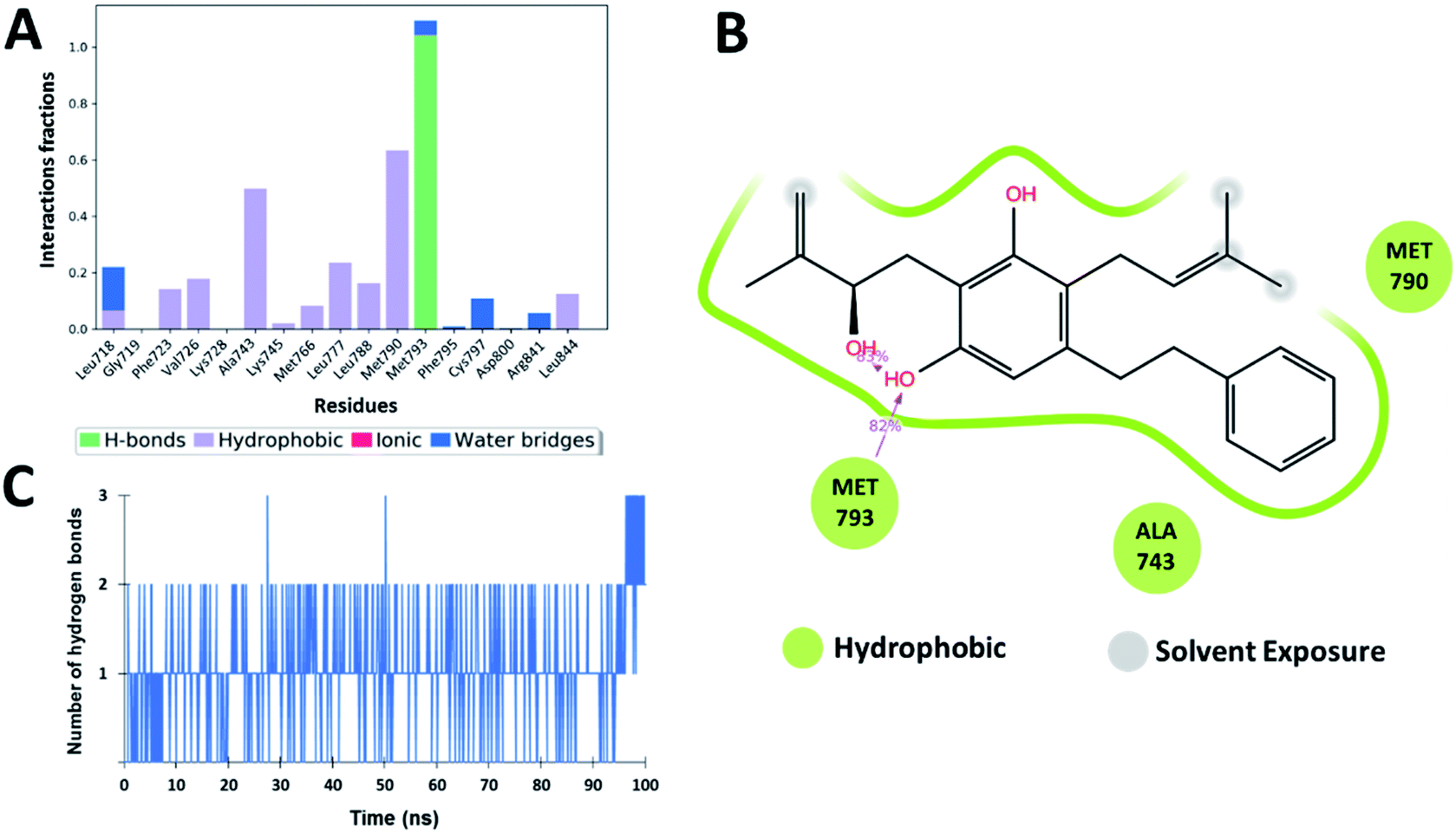
As hydrogen bonding is crucial for forming stable protein–ligand complexes, the number of hydrogen bonds throughout the simulation for all the ligands was also analyzed (Fig. 5–8). On average, the standard ligand (5N3) shows three hydrogen bonds, comparable to the Zn05 ligand (Fig. 7). In the case of Zn03 and Zn35, two hydrogen bonds are observed during the simulation period (Fig. 6 and 8).
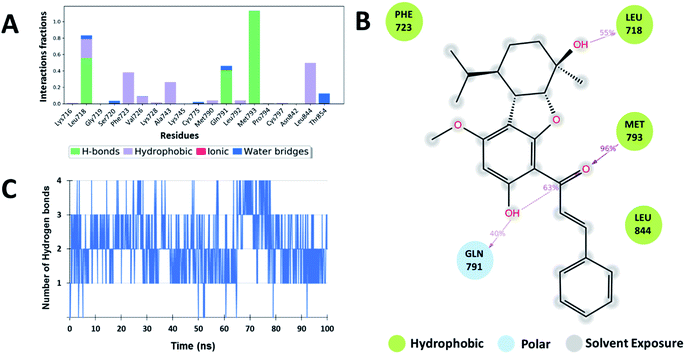 |
| | Fig. 6 Shows (A) residues involved in the interaction with ligand (Zn03) and the interaction fraction during simulation. Different types of interactions are shown in different colors. (B) 2D ligand interaction diagram for ligand Zn03 and surrounding residues with their percentage interactions, and (C) number of H-bonds across the 50 ns MD simulation trajectory. The data and plots are generated using Schrödinger Suite. | |
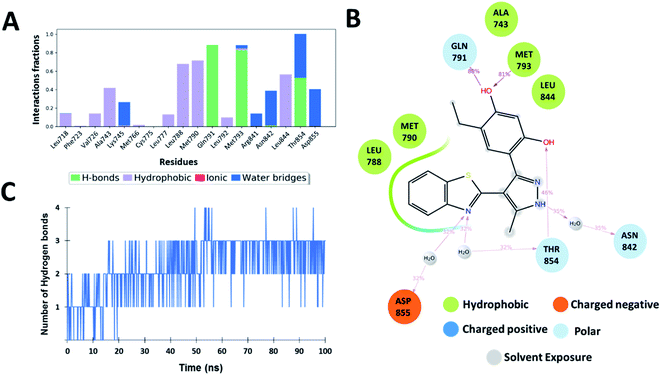 |
| | Fig. 7 Shows (A) residues involved in the interaction with ligand (Zn05) and the interaction fraction during simulation. Different types of interactions are shown in different colors. (B) 2D ligand interaction diagram for ligand Zn05 and surrounding residues with their percentage interactions, and (C) number of H-bonds across the 50 ns MD simulation trajectory. The data and plots are generated using Schrödinger Suite. | |
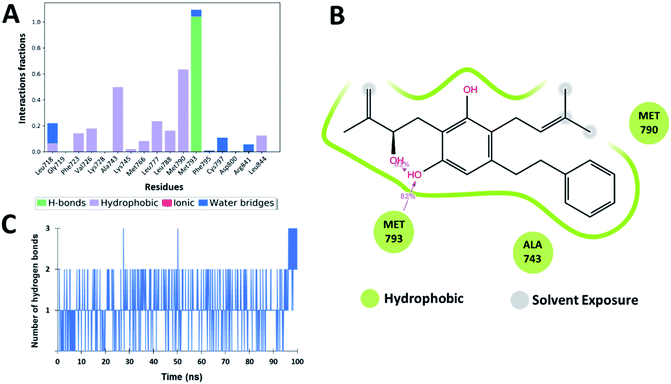 |
| | Fig. 8 Shows (A) residues involved in the interaction with ligand (Zn35) and the interaction fraction during simulation. Different types of interactions are shown in different colors. (B) 2D ligand interaction diagram for ligand Zn35 and surrounding residues with their percentage interactions, and (C) number of H-bonds across the 50 ns MD simulation trajectory. The data and plots are generated using Schrödinger Suite. | |
Apart from hydrogen bonds, hydrophobic interactions are also important for drug–target binding. The co-crystal ligand shows hydrophobic interactions predominantly with Ala743, Met790 and Leu844, which are also observed in the case of Zn05. In Zn03, additional H-bond interaction with Leu718 and hydrophobic interaction with Phe723 were observed, as phenyl ring orients outside the cavity and establishes interaction, enhancing the binding affinity.
Additionally, water-mediated interaction with Asn842 was observed in Zn05, similar to the co-crystal ligand. Also, additional water mediated H-bond interactions with Thr854 and Asp855 and hydrophobic interactions with Leu788 were observed, as benzothiazole ring orients inside the cavity thereby increasing the strength of interaction (Fig. 5 and 7).
Overall, the analysis indicated that the Y-shaped molecule (Zn05) is stabilized by the above interactions and is similar to the co-crystal ligand (5N3) interactions.
Optimization of Zn05
Based on the assessment of the interactions of ligands computed with the help of molecular dynamics calculations, the lead Zn05 was found to have the most interactions like the known ligand (5N3). It showed the RMSF and SASA were comparable to the co-crystal ligand. It even had RMSD and Rg values lower than the standard ligand suggesting the better stabilization of Zn05 in the active site of the TMLR mutant. Also, it showed conserved hydrogen bond interactions with Gln791, Met793, and Asn842 in comparison to co-crystal ligand (5N3). The analysis of co-crystallized ligand 5N3 bound in the active site of TMLR crystal structure (PDB: 5EDQ) revealed that in one of the pockets the nitrogen atom of pyrazole ring interacts via H-bond with Met793 while in the other pocket, the nitrogen of benzothiazole interacts with Glu762 to form hydrogen bond. From the co-crystallized ligand data, it was clear that interaction with Glu762 in addition to Gln791 and Met793 would further stabilize the ligand in the binding pocket of TMLR. The binding pattern analysis of Zn05 revealed an opportunity exists to make modifications that may allow interaction with Glu762. Therefore, a library of Zn05 analogues with several modifications were generated and selected the analog having a single –NH2 substitution on the benzothiazole group for molecular dynamics. The interaction analysis for the 100 ns simulation time revealed that the newly designed lead Zn05-NH2 forms hydrogen bonds with Gln791 and Met793 as well as additional interaction with Glu762 (Fig. 9 and ESI Fig. S1†). Also, the other H-bond interactions with Thr854 (water-mediated) are present but with a lower propensity (∼23%). Further, the number of hydrogen bonds throughout the simulation shows that nearly three hydrogen bonds are observed (Fig. 9). Overall, the modification allows the ligand to form additional interactions with TMLR protein and thereby increase binding affinity.
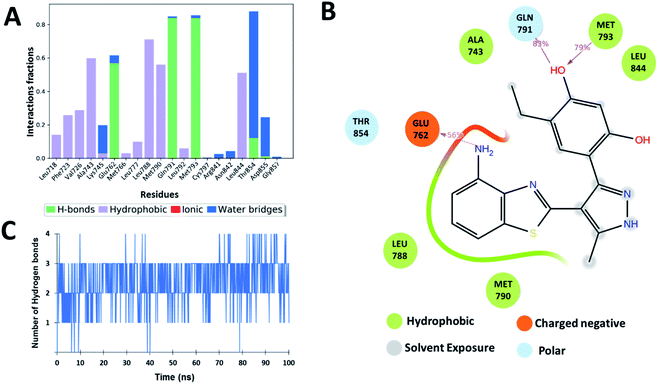 |
| | Fig. 9 Shows (A) residues involved in the interaction with ligand (Zn05-NH2) and the interaction fraction during simulation. Different types of interactions are shown in different colors. (B) 2D ligand interaction diagram for ligand Zn05-NH2 and surrounding residues with their percentage interactions (C) number of H-bonds across the 50 ns MD simulation trajectory. | |
Binding affinity calculations and per-residue-decomposition-analysis
To study the binding ability of inhibitors with the TMLR mutant EGFR protein, the binding free energies of complexes were computed using MM/GBSA method based on 50 snapshots taken from the last 5 ns MD simulation trajectories. It was observed that the binding free energy of Zn03, Zn05, Zn05-NH2 (analog of Zn05) and Zn35 are lesser but comparable to the reference ligand 5N3 (−72.45 kcal mol−1) with values −64.60, −61.07, −63.77 and −65.92 kcal mol−1, respectively. Further, the breakdown of energy terms reveals that in the case of Zn03, the contribution of coulomb energy is markedly lower while lipophilic and van der Waal and electrostatic solvation energy is nearly the same as in co-crystal ligand (Table 2). In Zn35, the contribution to the ΔGbind from lipophilic energy is comparatively higher than the native ligand (−23.86 vs. −19.56 kcal mol−1). This is in accordance with the ligand's fit in the binding pocket as it allows the benzyl group to have hydrophobic interactions in the pocket, thereby increasing the lipophilic energy. Similarly, in Zn05 there is a decrease in the contribution from lipophilic energy but a marked increase in coulomb energy compared to the crystal ligand (Table 2). Analysis of energy contributing terms of Zn05-NH2 ligand revealed an increase in lipophilic energy, thereby increasing the binding free energy in comparison to Zn05. Also, it is observed that van der Waals interactions are favorable for inhibitor binding in the active site of the mutant protein and all the three inhibitors Zn03, Zn05-NH2, Zn35 show similar van der Waals binding energy. Thus, this provides confidence that the identified molecules will have high affinity within the active site of the EGFR mutant protein.
Table 2 Binding free energy of studied protein–ligand complexes calculated using MM/GBSA method
| Ligand |
Binding free energy (ΔGbind) |
Coulomb energy (ΔEcoulomb) |
Lipophilic energy (ΔElipo) |
Van der Waals energy (ΔEvdw) |
| 5N3 |
−72.45 ± 3.45 |
−15.83 ± 1.68 |
−19.56 ± 0.95 |
−59.35 ± 2.11 |
| Zn03 |
−64.60 ± 3.91 |
−9.46 ± 3.60 |
−22.11 ± 1.90 |
−54.08 ± 2.55 |
| Zn05 |
−61.07 ± 4.24 |
−24.73 ± 3.12 |
−15.14 ± 0.81 |
−46.36 ± 2.23 |
| Zn35 |
−65.92 ± 4.00 |
−13.77 ± 3.46 |
−23.86 ± 1.26 |
−47.96 ± 2.62 |
| Zn05-NH2 |
−63.77 ± 5.67 |
−22.73 ± 3.85 |
−18.22 ± 1.17 |
−48.44 ± 2.47 |
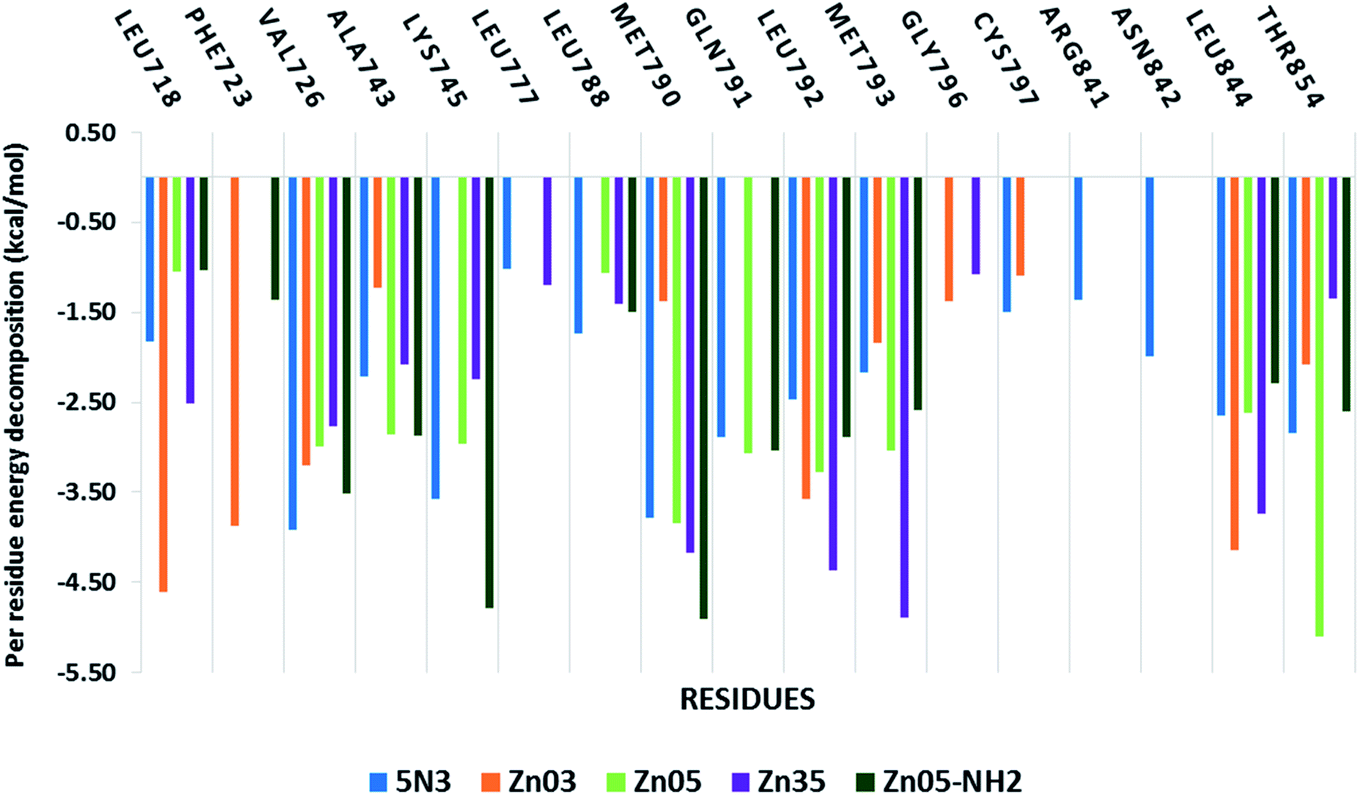
Further, to decipher the contribution of each amino acid involved in protein–ligand interactions per-residue decomposition analysis was undertaken (Fig. 10). The residues that exhibited binding free energy lesser than −1 kcal mol−1 are considered important and contribute towards the stability of the protein–ligand complex. It is observed that the amino acid residues Val718, Val726, Ala743, Lys745, Met790, Gln791, Leu792, Met793, Leu844 and Thr854 are significantly contributing towards the binding of all ligands including co-crystallized ligand 5N3. However, in the case of Zn03, the interaction with three residues Lys745, Leu788 and Gln791 was missing while it shows interaction with two other residues Gly796 and Cys797. On the other hand, in the case of Zn05-NH2, the residues Lys745 and Met790 are observed to have a remarkable contribution (<−4.5 kcal mol−1) towards the ligand binding.
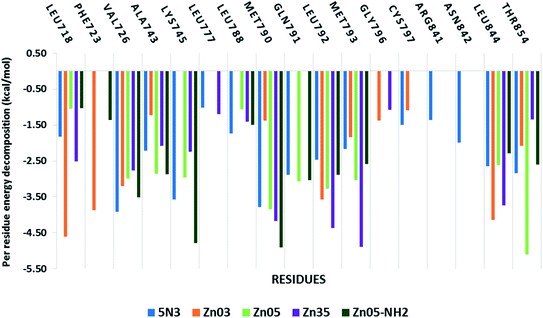 |
| | Fig. 10 Residue-wise energy decomposition analysis of the inhibitors against the TMLR mutant EGFR structure. | |
Conclusion
In this study, computational techniques including ligand and structure based were employed to shortlist a few leads from a myriad of natural compounds against EGFR double mutant for overcoming drug resistance in cancer. For this purpose, the drug-likeliness of the molecules was assessed using Lipinski and Ghose filter and then screened the library for its anticancer activity using a random forest based binary QSAR model. These potential anti-cancerous compounds were next examined for binding modes using pharmacophore constrained molecular docking against the EGFR-TMLR protein. Subsequent assessment of binding affinity allowed us to identify three lead molecules that occupy the critical sub-pockets and have the desired interaction critical for tight binding affinity with the receptor. The molecular dynamics simulations then confirmed the stability of the binding interactions predicted through molecular docking. Additionally, the MMGBSA binding affinity established that these molecules have comparable binding energy to the co-crystal ligand, which has low nM activity. Thus, we have integrated machine learning based virtual screening, molecular docking and molecular dynamics simulations to identify inhibitors against EGFR mutant protein from natural products libraries which may overcome resistance in cancer.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SMA would like to thank BioSolveIT (https://www.biosolveit.de/SeeSAR) for providing the software as part of the Scientific Challenge and Director, NICPR for the institutional support.
References
- M. Mangal, P. Sagar, H. Singh, G. P. S. Raghava and S. M. Agarwal, Nucleic Acids Res., 2013, 41, D1124–D1129 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- B. Shen, Cell, 2015, 163, 1297–1300 CrossRef CAS PubMed.
- J. W. H. Li and J. C. Vederas, Science, 2009, 35, 161–165 CrossRef PubMed.
- A. Díaz-Serrano, P. Gella, E. Jiménez, J. Zugazagoitia and L. Paz-Ares Rodríguez, Drugs, 2018, 78, 893–911 CrossRef PubMed.
- K. Gately, J. ÓFlaherty, F. Cappuzzo, R. Pirker, K. Kerr and K. ÓByrne, J. Clin. Pathol., 2012, 65, 1–7 CrossRef CAS PubMed.
- D. Raghav, V. Sharma and S. M. Agarwal, Interdiscip. Sci.: Comput. Life Sci., 2013, 5, 60–68 CrossRef CAS PubMed.
- I. Yadav, H. Singh, M. Khan, A. Chaudhury, G. P. S. Raghava and S. Agarwal, Adv. Anticancer Agents Med. Chem., 2014, 14, 928–935 CrossRef CAS PubMed.
- I. S. Yadav, P. P. Nandekar, S. Srivastava, A. Sangamwar, A. Chaudhury and S. M. Agarwal, Gene, 2014, 540, 131 CrossRef CAS.
- C. H. Yun, K. E. Mengwasser, A. V. Toms, M. S. Woo, H. Greulich, K. K. Wong, M. Meyerson and M. J. Eck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2070–2075 CrossRef CAS PubMed.
- G. M. ÓKane, T. A. Barnes and N. B. Leighl, Curr. Oncol., 2018, 25, S28–S37 CrossRef PubMed.
- R. Saini, S. Fatima and S. M. Agarwal, Chem. Biol. Drug Des., 2020, 96, 921–930 CrossRef CAS PubMed.
- S. Fatima, D. Pal and S. M. Agarwal, Chem. Biol. Drug Des., 2019, 94, 1306–1315 CrossRef CAS PubMed.
- S. Fatima and S. M. Agarwal, J. Recept. Signal Transduction, 2018, 38, 299–306 CrossRef CAS PubMed.
- S. Fatima and S. M. Agarwal, Med. Chem., 2019, 16, 52–62 CrossRef PubMed.
- S. Fatima, P. Gupta, S. Sharma, A. Sharma and S. M. Agarwal, Future Med. Chem., 2020, 12, 69–87 CrossRef CAS PubMed.
- A. Sharma, S. Sharma, M. Gupta, S. Fatima, R. Saini and S. M. Agarwal, Phytochem. Anal., 2018, 29, 559–568 CrossRef CAS PubMed.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- M. Hilbig, S. Urbaczek, I. Groth, S. Heuser and M. Rarey, J. Cheminf., 2013, 5, 1–10 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- K. Dhiman and S. M. Agarwal, RSC Adv., 2016, 6, 49395–49400 RSC.
- E. J. Hanan, M. Baumgardner, M. C. Bryan, Y. Chen, C. Eigenbrot, P. Fan, X. H. Gu, H. La, S. Malek, H. E. Purkey, G. Schaefer, S. Schmidt, S. Sideris, I. Yen, C. Yu and T. P. Heffron, Bioorg. Med. Chem. Lett., 2016, 26, 534–539 CrossRef CAS PubMed.
- M. A. Mart, A. C. Stuart, S. Roberto, F. Melo and S. Andrej, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef PubMed.
- S. A. Hindle, M. Rarey, C. Buning and T. Lengauer, J. Comput.-Aided Mol. Des., 2002, 16, 129–149 CrossRef CAS PubMed.
- N. Schneider, G. Lange, S. Hindle, R. Klein and M. Rarey, J. Comput.-Aided Mol. Des., 2013, 27, 15–29 CrossRef CAS PubMed.
- Schrödinger Release 2021: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021 Search PubMed; Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, p. 202 Search PubMed.
- V. Khanna and S. Ranganathan, J. Cheminf., 2011, 3, 30 CAS.
- S. Sharma, M. Gupta, A. Sharma and S. M. Agarwal, Lett. Drug Des. Discovery, 2018, 15, 1180–1188 CrossRef CAS.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- Y. Hu, Y. Lu, S. Wang, M. Zhang, X. Qu and B. Niu, Curr. Drug Targets, 2018, 20, 488–500 CrossRef PubMed.
- S. M. Agarwal, D. Pal, M. Gupta and R. Saini, Curr. Cancer Drug Targets, 2017, 17, 617–636 CAS.
- V. K. Sharma, P. P. Nandekar, A. Sangamwar, H. Pérez-Sánchez and S. M. Agarwal, RSC Adv., 2016, 6, 65725–65735 RSC.
- T. Sander, J. Freyss, M. Von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00373b |
| ‡ Equal contribution. |
| § Present address: Schrodinger Inc., Bengaluru, India, 560098. |
| ¶ Work done at ICMR-National Institute of Cancer Prevention and Research. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Prajwal Nandekar‡§
b and
Ravi Saini¶
c
*a,
Prajwal Nandekar‡§
b and
Ravi Saini¶
c