
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLewis base sites of non-oxide supports boost oxygen absorption and activation over supported Pt catalysts†
Jianye Liua,
Wenbin Chena,
Taihe Hea,
Yiwen Fanga,
ZiYi Zhong bc,
Xiaoming Wanga,
Zhen Li*a and
Yibing Song*a
bc,
Xiaoming Wanga,
Zhen Li*a and
Yibing Song*a
aDepartment of Chemistry, Shantou University, Guangdong 515063, China. E-mail: ybsong@stu.edu.cn
bDepartment of Chemistry Engineering, Guangdong Technion-Israel Institute of Technology (GTIIT), Guangdong 515063, China
cTechnion-Israel Institute of Technology (IIT), Haifa 32000, Israel
First published on 26th April 2022
Abstract
Formaldehyde (HCHO) oxidation to improve indoor air quality has attracted extensive attention. Designing efficient catalysts for HCHO removal at room temperature still remains challenging. Herein, we report a novel strategy to boost HCHO oxidation by the synergistic effect of Pt nanoparticles and C3N4. The pyridine nitrogen of C3N4 can create Lewis base sites, which function in adsorbing and activating O2 molecules. As the preparation temperature increased, the pyridine nitrogen content increased on the C3N4 surface, leading to a more significant synergistic effect. The mechanism study by in situ DRIFTS indicated that the adsorbed O2 molecules were activated by Pt/C3N4. As a result, the Pt/C3N4-650 has the most outstanding performance for HCHO oxidation at room temperature. HCHO can be completely eliminated with a concentration of 80 ppm at room temperature at a GHSV of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ml g−1 h−1. This study will provide a new perspective to design efficient HCHO oxidation catalysts.
000 ml g−1 h−1. This study will provide a new perspective to design efficient HCHO oxidation catalysts.
1 Introduction
Formaldehyde (HCHO) as a typical indoor air pollutant seriously threatens human health, because prolonged exposure to HCHO can lead to skin irritation, cancer and other diseases.1–3 In order to meet environment regulations and human health needs, lots of approaches have been studied to remove indoor air HCHO in the past few decades. Among them, physical adsorption, photocatalysis and catalytic oxidation were considered as effective methods for indoor air purification.4–6 Catalytic oxidation technology utilizes catalyst activated oxygen to thoroughly purify gaseous pollutants, it can completely convert HCHO into harmless CO2 and H2O without generating secondary pollutants, and has been widely used in industrial fields.Generally, catalytic oxidation of HCHO follows the Mars–Van Krevelen mechanism, in which the oxygen species (O2−, O−, etc.) on the surface of the catalyst play a significant role.7–10 Platinum-based materials are efficient and widely used catalysts for HCHO oxidation because Pt0 active sites can efficiently activate O–O bonds to produce surface oxygen species for promoting HCHO oxidation.11–13 Unfortunately, as a precious metal, platinum's high cost limits its widely used, so improving catalytic performance and reducing usage are effective means to promote its industrial application process. The application of carrier maybe one of the best methods to resolve these problems, due to carrier can increase the Pt dispersion to further advance its utilization.14 In the past few decades, a large numbers of carriers supported Pt catalyst have been studied for HCHO oxidation. For example, metal oxides (MnO2, Co3O4, CeO2, Al2O3, etc.), compound oxide (MnOx–CeO2) and non-metallic oxide (SiO2) as carriers can not only increase the Pt dispersion, but also provide a space for forming reactive oxygen species to enhance the catalytic performance.11,15–19 In addition, it has been confirmed that the hydroxyl-rich carrier (TiO2, AlOOH, FeOOH, etc.) can resulted in a new reaction path over Pt for HCHO oxidation.20–24 In the presence of hydroxyls near the platinum, HCHO can proceed as follow steps: HCHO → HCOO− + OH− → H2O + CO2, which has lower energy barriers compared to the direct HCHO oxidation route (HCHO → HCOO− → CO → CO2).22,25–27 However, when oxides used as supports, the use of catalysts may be limited by the environment, such as acidic environment. Therefore, non-oxide supported noble metal catalysts are still a highly importance research topic.
Carbon nitride (especially g-C3N4) as a star material is a conjugated polymer composed of carbon and nitrogen.28 The structure characteristics endow carbon nitride with special physical and chemical properties, such as excellent hardness, resistance to acids and alkalis and so on.29 It has been proved that using urea as raw material, polymerized carbon nitride is formed when the synthesis temperature is higher than 390 °C, and polymerized C3N4 crystallizes into g-C3N4 when the synthesis temperature is higher than 520 °C.30,31 Recently, Gan et al. reported carbon nitride supported Pt nanoparticles possess extraordinary capability for catalytic toluene oxidation because it can boost O2 activation.32 Similarly, activation of molecular oxygen is also a crucial step for HCHO oxidation.
In this work, we report a novel strategy to boost HCHO oxidation over Pt/C3N4: the C species neared pyridinic N of carbon nitride could effectively adsorb and activate O2 molecular to assist Pt catalytic HCHO oxidation. Due to the increase of carbon nitride preparation temperature, it's crystal structure would change, resulting in the increase of pyridine nitrogen content on carbon nitride. Pyridine nitrogen is suggested to create the Lewis based site and the oxygen molecules can be adsorbed on Lewis based sites.33 The increased pyridinic N of carbon nitride was demonstrated as an efficient strategy to improve the ability to activate oxygen.34 The result shows that the Pt NPs supported on C3N4-650 exhibited superior catalytic performance for HCHO oxidation and the catalyst with Pt loading as low as 0.48 wt%. In situ DRIFTS was used to confirm the promotion effect of using C3N4 as Pt catalyst support. This study provides guidance on the non-oxide and metal-free material as catalyst carrier for HCHO oxidation.
2 Experimental
2.1 Preparation of different Pt/CN catalysts
A certain amount of urea was sealed in a ceramic crucible and placed into muffle furnace. It was heated up to corresponding temperatures (450, 550 and 650 °C) with a heating rate of 5 °C min−1, and kept at this temperature for 2 h. The product was cooled down to room temperature and ground to powder. The obtained samples were denoted as CN-450, CN-550, CN-650, respectively.35,36Taking Pt/CN-450 as an example, 200 mg of CN-450 and 1.65 mL K2PtCl4 (5 mmol L−1) were added to a three-necked flask with 300 mL deionized water and ultrasonic dispersion it for 30 min. Continuously pour 10% H2/Ar into the three-necked flask for 4 h at room temperature while magnetic stirring. The solid products were collected by centrifugation and washed four times with deionized water and dried in vacuum overnight. The Pt/CN-550, Pt/CN-650, were prepared with similar procedure except for using the different carbon nitride supports.
2.2 Characterization
X-ray diffraction (XRD) patterns were obtained from a Bruker's D8 ADVANCE powder X-ray diffractometer with Cu Kα radiation (λ = 0.15418 nm; the scanning rate was 4° min−1 in the range of 2θ = 10–80°). The contents of Pt were measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES, Agilent 720, USA). Fourier transform infrared (FTIR) spectra were recorded with a Nicolet iS10 FTIR spectrometer ex situ in air. Field Emission Scanning Electron Microscope (FE-SEM) images were performed by using ZEISS Gemini 300 (Germany). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images were obtained using a JEM-F200. X-ray photoelectron spectroscopy (XPS) was measured on Thermo SCIENTIFIC ESCALAB 250Xi. Nitrogen, carbon and hydrogen content in catalyst was measured by a Vario El cube elemental analyzer (Elementar, Germany). H2-temperature programmed reduction (H2-TPR) was carried out in AMI-300 with a TCD. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) analysis of the nitrogen adsorption and desorption isotherms at 77 K recorded on micromeritics ASAP 2020. Diffused reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were performed on a Nicolet iS50 spectrometer, equipped with a liquid N2 cooled MCT detector. The turnover frequency (TOF) calculation formula of the samples were shown in the ESI.† The calculation of pyridine nitrogen distribution is as follows,| D = m × nnitrogen × npyridinic N × S |
2.3 Catalytic activity evaluation
A fixed bed reactor is used to evaluate the performance of HCHO removing. The 80 ppm HCHO vapour was generated by bubbling the aqueous formaldehyde with a flow of dry air (20 mL min−1) and then diluted with dry air (30 mL min−1). HCHO vapor was passed through a quartz tube (d = 7 mm) with 0.06 g catalyst loaded. The total flow rate was controlled at 50 mL min−1 by mass-flow meters, affording a gas hourly space velocity (GHSV) of 50000 mL g−1 h−1. The HCHO concentration in the inlet ([HCHO]inlet) and outlet ([HCHO]outlet) gas stream was analyzed by the phenol spectrophotometric method (the details of method are shown in the ESI†).23 The HCHO conversion was calculated as: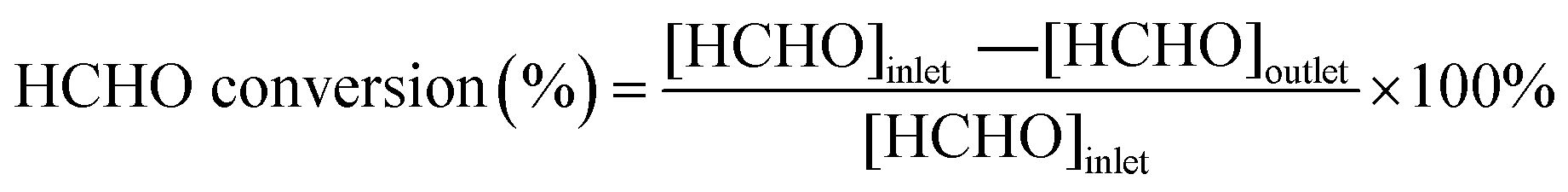
3 Results and discussion
3.1 Structural properties of the synthesized Pt/CN-x
In order to study the effect of CN supports on HCHO oxidation performance, the XRD and FT-IR were utilized to study the structure of the series supports and the results are shown in Fig. 1. In Fig. 1a, the result demonstrates that crystalline C3N4 polymer materials were synthesized at different temperatures. The characteristic peaks at about 12.7° and 27.5° are attributed to (100) and (002) facets of g-C3N4. The weaker peak is related to an in-plane structural packing motif, which represents an about 0.680 nm repeat distance.37 While the stronger peak is related to polymeric or graphitic sheets, which corresponds to an about 0.324 nm repeat distance.38 With the synthesis temperature increased, the observed changes can be attributed to distortions of crystal lattice of the synthesized g-C3N4.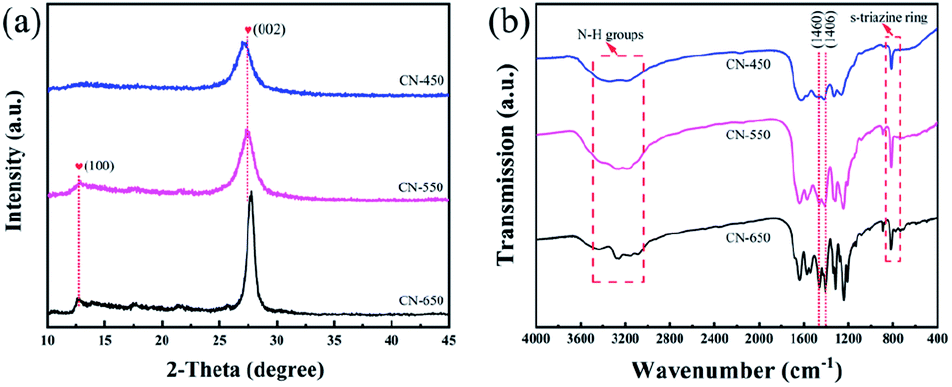 | ||
| Fig. 1 Structure properties of different carbon nitride supports (a) X-ray diffraction (XRD) patterns. (b) Fourier transform infrared (FT-IR) spectra. | ||
In Fig. 1b, the results of FT-IR also confirm the formation of g-C3N4 under different prepared temperature.39,40 Broad band near 3200 cm−1 contains absorption peaks represent NH groups. Peaks at 1683 and 1638 cm−1 correspond to double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond stretching mode and NH2 groups overlapping deformation mode, respectively. For these three carriers, the more obvious difference is the peaks in 1460 and 1406 cm−1 which are a tri-s-triazine aromatic rings double bond CN stretching modes. Group of adsorption lines near 1200–1350 cm−1 (1324, 1246 and 1213 cm−1) can be assigned to the C–N bonds stretching. And 1324 cm−1 is related to the C–N stretch in the threefold N-bridge linking the tri-s-triazine rings. Adsorption in 812 cm−1 is a s-triazine ring. These results show that the structure of carbon nitride changes as the preparation temperature increases.
N bond stretching mode and NH2 groups overlapping deformation mode, respectively. For these three carriers, the more obvious difference is the peaks in 1460 and 1406 cm−1 which are a tri-s-triazine aromatic rings double bond CN stretching modes. Group of adsorption lines near 1200–1350 cm−1 (1324, 1246 and 1213 cm−1) can be assigned to the C–N bonds stretching. And 1324 cm−1 is related to the C–N stretch in the threefold N-bridge linking the tri-s-triazine rings. Adsorption in 812 cm−1 is a s-triazine ring. These results show that the structure of carbon nitride changes as the preparation temperature increases.
The structure and morphology of catalysts were observed by scanning electron microscope. It can be seen from SEM images (Fig. 2) that carbon nitride is formed by stacking, and the higher the preparation temperature, the greater the degree of polymerization. Therefore, the specific surface area of carbon nitride will increase with the increase of calcination temperature.
 | ||
| Fig. 2 SEM images of different supported Pt samples. (a and b) Pt/CN-450, (c and d) Pt/CN-550, (e and f) Pt/CN-650. | ||
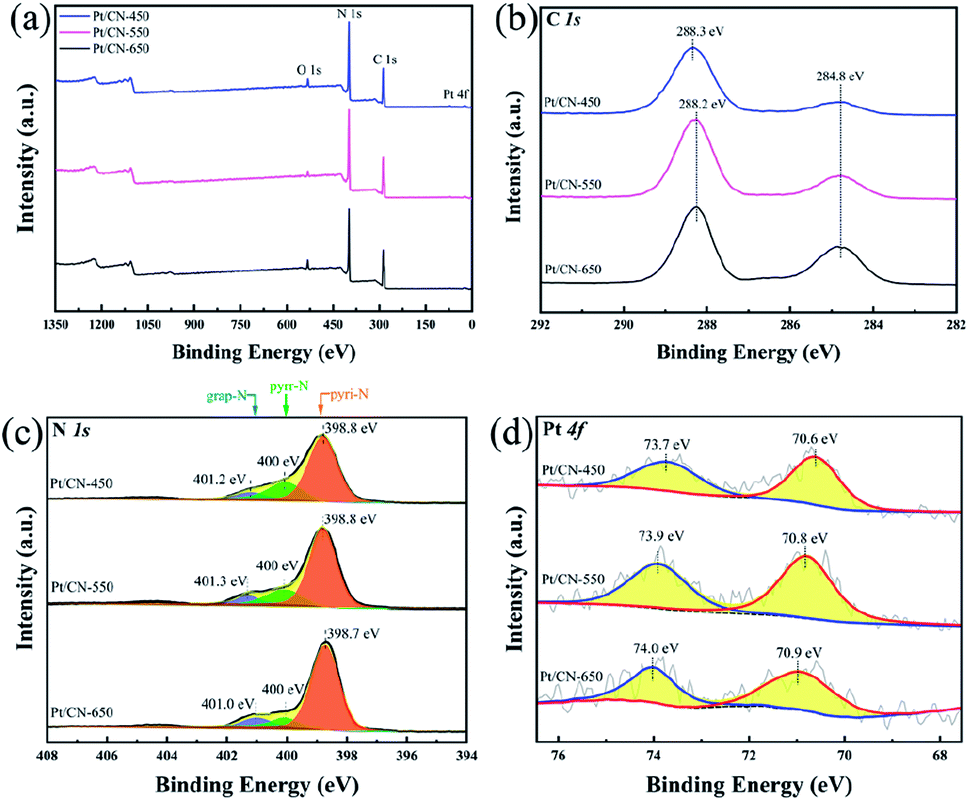
The surface chemical states of the series Pt/CN catalysts were characterized by XPS to further study the effect of CN supports on HCHO oxidation performance as shown in Fig. 3. As displayed in Fig. 3a, the survey scan XPS spectrum reveals the existence of C, N, O and Pt elements in the Pt/CN sample. In Fig. 3b, the C 1s spectra were fitted to two peaks, belonging to C(N)3 (288.2 eV) and adventitious carbon (284.8 eV).32 In the Fig. 3c, the sharp N 1s XPS signal with a shoulder can be assigned to pyridinic N (398.8 eV), pyrrolic N (400.0 eV) and graphitic N (401.1 eV), respectively.34,41 The quantify of N species are analyzed by elemental analyzer and XPS as shown in Table 1. As we can see, with the increase of roasting temperature, the content of N species increased from 58.1% to 61.6% in C3N4 supports. Among them, the graphitic N remains almost constant, while the pyrrolic N decreases from 25% to 13%. As for pyridinic N, its content increases with the increase of roasting temperature and achieves more than about 70%. It is generally known that pyridinic N can create Lewis base sites, which can effectively adsorb and activate oxygen molecules, it may be one of the key factors for the high activity of HCHO removing over Pt/CN-650, due to the synergistic effect of activated oxygen.34,42–44 Calculating the distribution of pyridine nitrogen on the surface of catalysts, the result increased from 0.48 to 1.39 (Table 1). It indicates that the pyridinic N can be more widely distributed on the catalysts at a higher preparation temperature of carbon nitrogen. The chemical states of Pt were investigated using Pt 4f XPS spectra as shown in Fig. 3d. As we can see, the dominant peaks located at 73.7° and 70.6° can be assigned to Pt0, indicating the Pt species exist in metal.45 However, with the increase of roasting temperature, the binding energy of Pt are significantly shift from 73.7 to 74.0 eV, suggesting the interaction between Pt and CN support are enhanced. In addition, it can be seen from the literature review, Pt2+ belongs to soft acids and pyridine nitrogen is borderline bases. According to the HSAB theory, acids and bases interact is strong interactions, which are turn to form stable Pt–N bonds.46 As a result, the activated oxygen over CN-650 can migrate faster to Pt, leading to the highest activity of HCHO oxidation over Pt/CN-650.
 | ||
| Fig. 3 Surface chemical property of the samples (a) XPS survey spectra of samples. (b) C 1s XPS spectra. (c) N 1s XPS spectra. (d) Pt 4f XPS spectra. | ||
| Samples | Nitrogen concentrationa (%) | Graphitic Nb | Pyrrolic N | Pyridinic N | Distribution of pyridine nitrogenc | |||
|---|---|---|---|---|---|---|---|---|
| eV | at% | eV | at% | eV | at% | |||
| a The actual content of N obtained from elemental analyzer.b Surface functionalities calculated based on peaks fit to XPS spectra.c The calculation method of pyridine nitrogen distribution was shown in characterization. | ||||||||
| Pt/CN-450 | 58.8 | 401.2 | 6 | 400 | 25 | 398.8 | 69 | 0.48 |
| Pt/CN-550 | 60.8 | 401.3 | 9 | 400 | 18 | 398.8 | 73 | 1.09 |
| Pt/CN-650 | 61.6 | 401.0 | 9 | 400 | 13 | 398.7 | 78 | 1.39 |
In order to determine whether the size and morphology of Pt species is affected by CN supports, which may result in different performance of HCHO oxidation. The TEM was used to investigate the Pt dispersion on the surface of different CN supports and the results are shown in Fig. 4. As we can see, the Pt nanoparticles (NPs) are uniformly dispersed on the surface of CN in Fig. 4a, c and e, and corresponding to the size of 4.49, 4.22 and 4.70 nm respectively as shown in insert graphs. It indicates the size and morphology of Pt NPs maintain a similar state on the surface of different CN supports. In addition, the lattice with a d spacing of 0.23 nm was observed in the HRTEM images as shown in Fig. 4b, d and f, which could be ascribed to the (111) facet of metal Pt.32 The loading of Pt NPs was also test by ICP-AES as shown in Table 2 and the Pt loading is almost the same in the series Pt/CN catalyst. These results suggest that the different performance of the series Pt/CN must be associated with the CN supports rather than the size, morphology, chemical state and loading of Pt NPs.
 | ||
| Fig. 4 TEM and HRTEM images of different supported Pt samples. (a and b) Pt/CN-450, (c and d) Pt/CN-550, (e and f) Pt/CN-650. | ||
3.2 Catalytic oxidation performance of different Pt/CN-x catalysts
The performances of HCHO oxidation over Pt/CN-450, Pt/CN-550, and Pt/CN-650 were investigated in a fixed bed reactor and the results were shown in Fig. 5. Fig. 5a and b illustrates the profile of HCHO conversions over the series catalysts. As we can see, the conversion of HCHO increased with the increase of CN supports preparation temperature, the Pt/CN-650 exhibits the best catalytic ability, the HCHO conversion stays at 100% in the temperature range of 25–105 °C and the gas hourly space velocity (GHSV) of 50000 mL g−1 h−1 is achieved, while T100 (complete conversion temperature of HCHO) were achieved at 75 °C for Pt/CN-550 catalyst and only 70% of HCHO is oxidized at 25 °C. The Pt/CN-450 catalyst shows the lowest activity, about 47.5% of HCHO is removed at 25 °C and it cannot be completely removed within the test temperature range. It can be seen from Table S3† that compared with other non-oxide supported Pt catalysts, the Pt/C3N4 shows excellent catalytic oxidation performance of HCHO. In addition, the TOF values of the catalysts were calculated and summarized in Table 2. The Pt/CN-450, Pt/CN-550 and Pt/CN-650 showed a TOF of 15.28, 19.55 and 30.72 h−1, respectively. It is clear that the increase of preparation temperature of carbon nitride significantly enhanced the activity of the Pt/C3N4 catalysts. The HCHO oxidation stability over Pt/CN-650 was test as presented in Fig. 5c and d. The result shows it still maintained a high conversion (>90%) even after 40 hours, indicating that the Pt/CN-650 catalyst have excellent stability. Meantime, the recycling runs of HCHO removal was also performed to evaluate the stability of samples. Fig. 5d showed the removal efficiency of the Pt/CN-650 remained stable after 3 cycles. The results above suggest the different performance of the series Pt/CN must be associated with the CN supports or Pt species.
 | ||
| Fig. 5 (a) HCHO conversion rate over samples at room temperature. (b) HCHO conversions over Pt/C3N4 catalysts at various temperature. (c) HCHO removal efficiency as a function of test time over Pt/CN-650 at room temperature. (d) HCHO removal efficiency as a function of time over Pt/CN-650 in three recycling runs. Reaction conditions: 80 ppm HCHO/21%O2/N2, RH = 35%, space velocity (SV) = 50000 mL g−1 h−1. | ||
3.3 Mechanism study
In Fig. 6, H2-TPR measurement was used to study the ability to activate oxygen over the series Pt/CN catalysts, which was an important factor for the HCHO removal as mentioned above.47 Before reducing by 10% H2/Ar, the samples were treated with oxygen at 30 °C for 90 minutes to produce surface O species. As shown in Fig. 6, the reduction temperature was shift from 57 to 50 °C with the increase of CN preparation temperature. The lowest reduction temperature indicates oxygen was most easily activated on the surface of Pt/CN-650. With the synergistic effect of this activated O species, the performance of HCHO oxidation is greatly improved.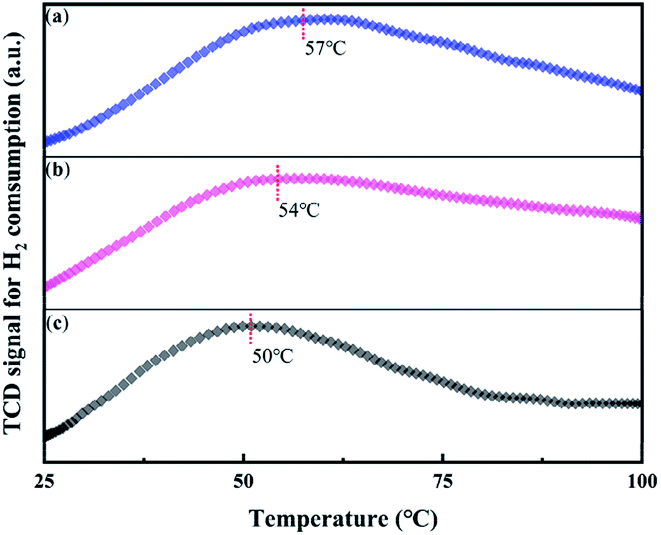 | ||
| Fig. 6 H2-TPR patterns of the samples (a) Pt/CN-450, (b) Pt/CN-550, (c) Pt/CN-650. | ||
In situ DRIFTS were performed to study the mechanism of HCHO oxidation over the Pt/CN-450 and Pt/CN-650 catalysts and the results are shown in Fig. 7. The characteristic peaks of intermediates could be observed in the spectrum by differential spectroscopy. Several weak peaks originated from the gradual accumulation were detected in the range of 800–3000 cm−1 over Pt/CN-450. The peaks at about 1010 and 1540 cm−1 are attributed to the dioxymethylene (DOM) species and νas(COO−) of the formate species on the surface of Pt catalyst, respectively.47,48 While the peak at about 1700 cm−1 is ascribed to the bridge adsorbed νas(CO). Compared to Pt/CN-450 catalyst, the peaks on the surface of Pt/CN-650 are significantly enhanced and two new peaks at 1347 and 2919 cm−1, assigned to νs(COO−) and the C–H stretching vibration of HCOOH*, are observed as shown in Fig. 7b, indicating the performance of HCHO oxidation has a substantial improvement with the synergistic effect of activated oxygen.49
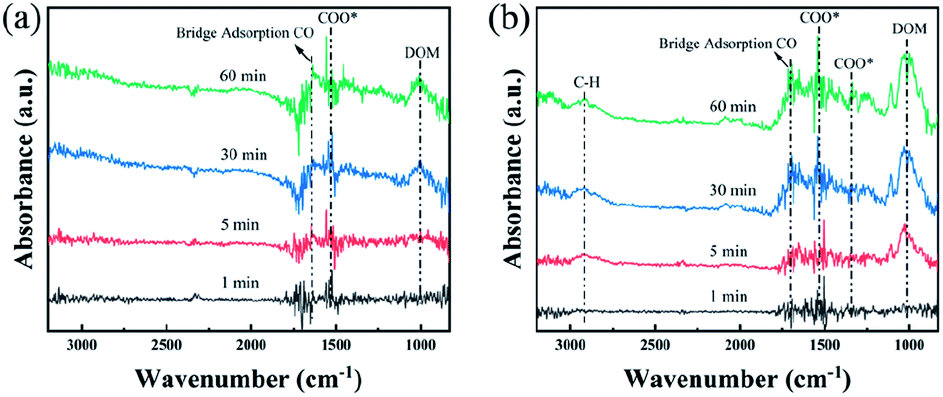 | ||
| Fig. 7 In situ DRIFTS spectra of O2 + N2 + HCHO gas mixture exposure for up to 60 min at room temperature over (a) Pt/CN-450, (b) Pt/CN-650. | ||
Combined with the above, we speculate a possible reaction process for HCHO oxidation over the Pt/CN-650 catalyst as illustrated in Scheme 1. Firstly, HCHO and O2 molecules are adsorbed on the surface of Pt/CN-650 catalyst, then the CN-650 promotes the dissociation of oxygen to produce oxygen species (O*, O−, O2−, etc.), which could oxidize HCHO to DOM or formate species. Then, the adsorbed formate species will react with H ions to form H2O and CO, which was further oxidized to CO2.
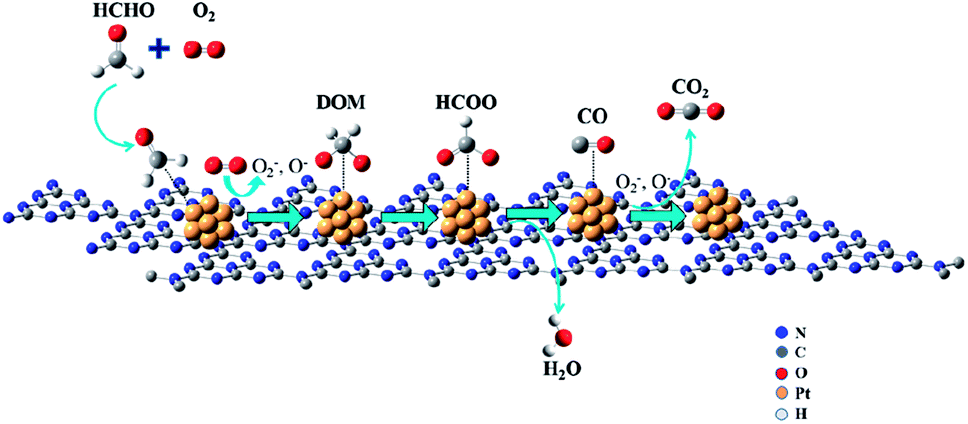 | ||
| Scheme 1 Reaction pathway of catalytic-oxidation of HCHO on the Pt/C3N4 catalysts at room temperature. | ||
4 Conclusions
In the present work, we report a new synergistic effect between C3N4 and Pt NPs for high efficiency HCHO oxidation at room temperature. The pyridine nitrogen of C3N4 can create Lewis base sites, functioning in adsorbing and activating O2 molecules. Under this synergistic effect, the activated O species greatly promote HCHO oxidation over Pt NPs. We found the pyridine nitrogen content increased at a higher preparation temperature, such that the Pt/C3N4-650 has the most outstanding performance for HCHO oxidation at room temperature. This work provides a new strategy to boost HCHO oxidation by the synergistic effect between activated O species and Pt NPs, it will open a new vision on designing high-performance catalysts for HCHO oxidation at room temperature.Author contributions
Jianye Liu: methodology, data curation, writing – original draft, writing – review & editing. Wenbin Chen: formal analysis. Taihe He: formal analysis. Yiwen Fang: supervision. ZiYi Zhong: supervision, resources. Xiaoming Wang: supervision. Zhen Li: supervision, writing – review & editing. Yibing Song: supervision, resources, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21043006 and 21902121) and the Education Department of Guangdong Province (2018KTSCX063) and the Science and Technology Planning Project of Guangdong Province (2014A020216045) and the 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG09A) and Science and Technology Project of China Huaneng Group (HNKJ19-G011).Notes and references
- E. L. Hult, H. Willem, P. N. Price, T. Hotchi, M. L. Russell and B. C. Singer, Indoor Air, 2015, 25, 523–535 CrossRef CAS PubMed.
- K. H. Kim, S. A. Jahan and J.-T. Lee, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2011, 29, 277–299 CrossRef CAS PubMed.
- C. J. Jiang, D. D. Li, P. Y. Zhang, J. G. Li, J. Wang and G. Yu, Build. Environ., 2017, 117, 118–126 CrossRef.
- C. J. Na, M. J. Yoo, D. C. W. Tsang, H. W. Kim and K. H. Kim, J. Hazard. Mater., 2019, 366, 452–465 CrossRef CAS PubMed.
- X. Yue, N. L. Ma, C. Sonne, R. Guan, Su S. Lam, Q. V. Le, X. Chen, Y. Yang, H. Gu, J. Rinklebe and W. Peng, J. Hazard. Mater., 2021, 405, 124138 CrossRef CAS PubMed.
- J. Li, W. Cui, P. Chen, X. Dong, Y. Chu, J. Sheng, Y. Zhang, Z. Wang and P. Dong, Appl. Catal., B, 2020, 260, 118130 CrossRef CAS.
- B. Chen, B. Wu, L. Yu, M. Crocker and C. Shi, ACS Catal., 2020, 10, 6176–6187 CrossRef CAS.
- D. Chen, G. Zhang, M. Wang, N. Li, Q. Xu, H. Li, J. He and J. M. Lu, Angew. Chem., Int. Ed., 2021, 60, 6377–6381 CrossRef CAS PubMed.
- C. Ma, G. Pang, G. He, Y. Li, Y. He and Z. Hao, J. Environ. Sci., 2016, 39, 77–85 CrossRef CAS PubMed.
- X. Sun, J. Lin, Y. Wang, L. Li, X. Pan, Y. Su and X. Wang, Appl. Catal., B, 2020, 268, 118741 CrossRef CAS.
- G. Huang, Z. Yan, S. Liu, T. Luo, L. An and Z. Xu, J. Environ. Sci., 2020, 87, 173–183 CrossRef PubMed.
- J. Ye, M. Zhou, Y. Le, B. Cheng and J. Yu, Appl. Catal., B, 2020, 267, 118689 CrossRef CAS.
- Z. Yan, Z. Xu, J. Yu and M. Jaroniec, Environ. Sci. Technol., 2015, 49, 6637–6644 CrossRef PubMed.
- J. Guo, C. Lin, C. Jiang and P. Zhang, Appl. Surf. Sci., 2019, 475, 237–255 CrossRef CAS.
- Z. X. Yan, Z. H. Xu, B. Cheng and C. J. Jiang, Appl. Surf. Sci., 2017, 404, 426–434 CrossRef CAS.
- G. Li and L. Li, RSC Adv., 2015, 5, 36428–36433 RSC.
- L. Nie, A. Meng, J. Yu and M. Jaroniec, Sci. Rep., 2013, 3, 3215 CrossRef PubMed.
- X. Tang, J. Chen, X. Huang, Y. Xu and W. Shen, Appl. Catal., B, 2008, 81, 115–121 CrossRef CAS.
- N. H. An, W. L. Zhang, X. L. Yuan, B. Pan, G. Liu, M. J. Jia, W. F. Yan and W. X. Zhang, Chem. Eng. J., 2013, 215–216, 1–6 CrossRef CAS.
- C. Zhang, H. He and K. i. Tanaka, Catal. Commun., 2005, 6, 211–214 CrossRef CAS.
- C. Zhang, H. He and K. i. Tanaka, Appl. Catal., B, 2006, 65, 37–43 CrossRef CAS.
- Z. Xu, J. Yu and M. Jaroniec, Appl. Catal., B, 2015, 163, 306–312 CrossRef CAS.
- J. Chen, J. Ding, H. Li, J. Sun, Z. Rui and H. Ji, Catal. Sci. Technol., 2019, 9, 3287–3294 RSC.
- C. Zhang, F. Liu, Y. Zhai, H. Ariga, N. Yi, Y. Liu, K. Asakura, M. Flytzani-Stephanopoulos and H. He, Angew. Chem., Int. Ed., 2012, 51, 9628–9632 CrossRef CAS PubMed.
- L. Miao, J. Wang and P. Zhang, Appl. Surf. Sci., 2019, 466, 441–453 CrossRef CAS.
- D. W. Kwon, P. W. Seo, G. J. Kim and S. C. Hong, Appl. Catal., B, 2015, 163, 436–443 CrossRef CAS.
- Y. Huo, X. Wang, Z. Rui, X. Yang and H. Ji, Ind. Eng. Chem. Res., 2018, 57, 8183–8189 CrossRef CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- D. Feng, Y. Cheng, J. He, L. Zheng, D. Shao, W. Wang, W. Wang, F. Lu, H. Dong and H. Liu, Carbon, 2017, 125, 454–463 CrossRef CAS.
- Y. Zheng, Z. Zhang and C. Li, J. Photochem. Photobiol., A, 2017, 332, 32–44 CrossRef.
- T. Gan, J. Yang, D. Morris, X. Chu, P. Zhang, W. Zhang, Y. Zou, W. Yan, S. H. Wei and G. Liu, Nat. Commun., 2021, 12, 2741 CrossRef CAS PubMed.
- H. Metiu, S. Chrétien, Z. Hu, B. Li and X. Y. Sun, J. Phys. Chem. C, 2012, 116, 10439–10450 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- J. Xu, Y. Li, S. Peng, G. Lub and S. Li, Phys. Chem. Chem. Phys., 2013, 15, 7657–7665 RSC.
- A. Mishra, A. Mehta, S. Basu, N. P. Shetti and K. R. Reddy, Carbon, 2019, 149, 693–721 CrossRef CAS.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS.
- E. B. Chubenko, N. M. Denisov, A. V. Baglov, V. P. Bondarenko, V. V. Uglov and V. E. Borisenko, Cryst. Res. Technol., 2020, 55, 1900163 CrossRef CAS.
- Y. Li, H. Xu, S. Ouyang, D. Lu, X. Wang, D. Wang and J. Ye, J. Mater. Chem. A, 2016, 4, 2943–2950 RSC.
- S. Maldonado, S. Morin and K. J. Stevenson, Carbon, 2006, 44, 1429–1437 CrossRef CAS.
- H. W. Liang, X. Zhuang, S. Brüller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- B. Li, X. Y. Sun and D. Su, ChemPhysChem, 2015, 17, 6691–6694 CAS.
- T. Kondo, S. Casolo, T. Suzuki, T. Shikano, M. Sakurai, Y. Harada, M. Saito, M. Oshima, M. I. Trioni, G. F. Tantardini and J. Nakamura, Phys. Rev. B, 2021, 86, 035436 CrossRef.
- J. Yu, Q. Liu, W. Qiao, D. Lv, Y. Li, C. Liu, Y. Yu, Y. Li, H. Niemantsverdriet, B. Zhang and R. Su, ACS Catal., 2021, 11, 6656–6661 CrossRef CAS.
- B. Lippert, Coord. Chem. Rev., 1999, 182, 263–295 CrossRef.
- W. Bao, H. Chen, H. Wang, R. Zhang, Y. Wei and L. Zheng, ACS Appl. Nano Mater., 2020, 3, 2614–2624 CrossRef CAS.
- B. B. Chen, C. Shi, M. Crocker, Y. Wang and A.-M. Zhu, Appl. Catal., B, 2013, 132–133, 245–255 CrossRef CAS.
- X. Sun, J. Lin, Y. Chen, Y. Wang, L. Li, S. Miao, X. Pan and X. Wang, Commun. Chem., 2019, 2, 27 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00538g |
| This journal is © The Royal Society of Chemistry 2022 |