
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic valorisation of biomass levulinic acid into gamma valerolactone using formic acid as a H2 donor: a critical review†
Ayman Hijazi
 *,
Nidal Khalaf
,
Witold Kwapinski
and
J. J. Leahy
*
*,
Nidal Khalaf
,
Witold Kwapinski
and
J. J. Leahy
*
Chemical and Environmental Science Department, University of Limerick, Limerick, V94 T9PX, Ireland. E-mail: j.j.leahy@ul.ie; Ayman.Hijazi@ul.ie; Tel: +353-83-3783841
First published on 6th May 2022
Abstract
This review sheds light on the catalytic valorisation of agroforestry biomass through levulinic acid and formic acid towards γ-valerolactone and other higher-value chemicals. γ-Valerolactone is produced by the hydrogenation of levulinic acid, which can be achieved through an internal hydrogen transfer reaction with formic acid in the presence of catalyst. By reviewing corresponding catalysts, the paper underlines the most efficient steps constituting an integrated sustainable process that eliminates the need for external H2 sources while producing biofuels as an alternative energy source. Furthermore, the review emphasizes the role of catalysts in the hydrogenation of levulinic acid, with special focus on heterogeneous catalysts. The authors highlighted the dual role of different catalysts by comparing their activity, morphology, electronic structure, synergetic relation between support and doped species, as well as their deactivation and recyclability. Acknowledging the need for green and sustainable H2 production, the review extends to cover the role of photo catalysis in dissociating H2-donor solvents for reducing levulinic acid into γ-valerolactone under mild temperatures. To wrap up, the critical discussion presented enables readers to hone their knowledge about different schools and emphasizes research gaps emerging from experimental work. The review concludes with a comprehensive table summarizing the recent catalysts reported between the years 2017–2021.
 Ayman Hijazi | Ayman Hijazi is currently pursuing his postgraduate studies in the Bernal Institute, University of Limerick, Republic of Ireland. He is accommodated in Prof. James J. Leahy's lab, where his work focuses on developing novel heterogeneous catalysts for biomass hydrolysis processes along with developing robust analytical methods (HPLC, LC-MS, GC-MS, and GC-FID) for analysing organic acids and furans products. Hijazi earned his BSc degree in chemical engineering from the Middle East Technical University, Turkey, in 2011. Afterwards, he held a process engineer job at a waste treatment plant, which receives municipal solid waste biomass feedstock and valorises it into biogas using anaerobic digestion technology. At the same time, he attended master's program at the American University of Beirut, where he received his Msc. in chemical engineering with a focus on developing waste to energy catalytic processes. The agenda of research was based on synthesizing heterogeneous catalysts capable of optimizing pyrolytic gas from the pyrolysis of scrap tires. In 2022, Hijazi joined Dairy Processing technology Centre (DPTC) in Ireland-Limerick where he conducts applied research on the valorisation of oil and grease from dairy industry into biofuels. |
 Nidal Khalaf | Nidal Khalaf is a PhD researcher in Chemical Engineering at the University of Limerick, Ireland. Nidal is a researcher in the REFLOW group, which is an interdisciplinary cross-sectoral European Training Network initiated by Marie Skłodowska-Curie Actions (MSCA) H2020. Nidal's main research interest revolves around the valorisation of dairy processing waste to produce valuable end products. Nidal has earned his bachelor in Chemical Engineering from the American University of Beirut, followed by a job experience at ITCAN solutions working as a Chemical Engineering with the responsibility of designing and digitizing process flow diagrams for regional oil companies. |
 Witold Kwapinski | Dr. Witold Kwapinski works as an Associate Professor at the University of Limerick in the Chemical Sciences Department. He is also Course Director of the IChemE-accredited Chemical & Biochemical Engineering (HonsBEng) programme. Witold is co-leader of the ‘Carbolea’ research group on biomass/biowaste thermochemical conversions at UL. Carbolea is a leading research group on advanced biowaste strategies and technologies in Ireland and Europe. Work in his laboratory concentrates on pyrolysis, gasification, hydrothermal processing of bio-waste, product upgrading and catalytic conversion. Witold has successfully supervised 14 PhD students. Currently (2021) he supervises 2 PhD students as main supervisor and 5 as co-supervisor. In Ireland he has participated in four EU projects and five national, publicly-funded projects. He is author of over ninety papers in ISI journals (h-index = 31), and more than 100 communications in international conferences. Witold graduated from Lodz Technical University and in his career worked in Germany and the UK. He also acted as a Visiting Lecturer at: Cambridge University (UK); Aalto University (Finland); American University of Beirut (Lebanon); Belarusian State Technological University; National Technical University of Ukraine. He is an Editorial Board member and occasional guest editor of three ISI journals. |
 J. J. Leahy | J. J. Leahy is professor of process chemistry at the University of Limerick in Ireland. He has worked for 30 years on the valorisation of agri-waste as biofuels, fertilizers and intermediates for the production of chemicals. He currently coordinates two EU H2020 projects, (1) BIOWILL which aims to be a flagship for rural economies through a zero-waste biorefinery utilising all fractions of willow trees for the production of high value pharmaceutically active ingredients and bio-based packaging materials, renewable energy in the form of bio-methane and natural fertilisers with potential for carbon offset; (2) REFLOW which is a collaboration between academic and industrial organisations in seven countries focused on the recycling of phosphorous from the wastewater from dairy factories and using it in CE approved fertilizers. Future common agricultural policy (CAP) recognises that it is essential to build stronger agricultural knowledge and innovation systems so that farmers and rural communities can benefit directly. EU analysis has shown that key to the success of links between the CAP Reform and the Green Deal are concrete examples such as BIOWILL and REFLOW. Nationally, JJ has forged links with major and small stakeholders with the agro-food and biofuels industry and is a principal researcher with the industry led Dairy Processing technology Centre (DPTC) based in Limerick as well as BIORBIC the Bio-economy research center Biorbic, where he leads several projects on biomass valorisation. |
1 Introduction
The reliance of the world on petroleum as a source of energy has been a subject of controversy for a while. It was previously reported that the oil peak expectations fall in 2020; however, an updated model expects that the fall of the oil age is not before 2040.1 Moreover, the high dependence on non-renewable fossil fuels raises concerns about their fast depletion rate.2,3 Additionally, a continued concern is marked due to the extensive burning of fossil fuels for energy supply. This came out as a devastating blow, which is characterized by a drastic increase of CO2 levels from 280 ppm before preindustrial to 409.5 ppm in 2017.4 Needless to say, finding a sustainable alternative to fossil fuels is an urgent need. Consequently, lignocellulosic biomass is pronounced as a prime candidate to be used directly as an energy source or to be valorised into added value chemicals as well as gasoline blends that are compatible with current infrastructure.5 In that case, upgrading biomass generally requires a combination of O2 removal reactions and molecular weight modifications via C–C coupling reactions.6 A wide variety of platform molecules are thereby produced, having distinctive properties and applications. To illustrate, levulinic acid (LA), functionalized by a ketone and a carbonyl group is involved in chemical industries such as resin precursors and polymers;7 formic acid (FA), known as methanoic acid, is an alternative direct source of energy in fuel cells;8 γ-valerolactone (GVL) is a famous fuel additive and an important monomer for polyester and bioplastic industries;7,9,10 methyltetrahydrofuran (MTHF) is also used as a gasoline blend.7The derived biomass molecules are symbolized by their functional groups, which facilitate their use in producing chemicals compared to saturated alkanes.11 LA is known for facilitating the production of added-value molecules. This can be achieved through its catalytic hydrogenation using secondary alcohols or FA as a H2 donor.12,13 Therefore, FA is a promising biomass-derived compound that presents a potential pool of energy source of H2.
Here in this review, we mainly focus on the valorisation of cellulosic food waste into LA and FA, as part of establishing sustainable production of higher-value chemicals and liquid fuels. The valorisation routes starting from cellulose and hemicellulose are defined in Section 2. Our approach is economic-oriented, and we seek to shed light on one-pot processes as presented in Section 3.2.1, wherein the in situ produced H2 via FA decomposition provokes the hydrogenation of LA into further useful compounds, eliminating the need for an external source of H2. The decomposition of FA into H2 is highlighted in Section 3.1 (Table 1), while an overview of different H2 sources is presented in Section 4, with an insight into the photocatalytic hydrogenation of LA into GVL using alcohol solvents presented in Section 4.1. To add, using H2 for hydrogenation reactions remains a challenging step as it possesses low mass transfer in many organic solvents.14 Heterogeneous catalysts featuring a dual role of dehydrogenation/hydrogenation for driving the above process are also emphasized in Sections 3.1 and 3.2 Table 2. Moreover, insight into the nature of catalysts that have been used to date is gained by relating activity, deactivation, and recyclability to their morphological structure, as shown in Section 3.2.1.
| Catalyst | TOF (H2) | Solvent | Temp. (°C) | Ref. |
|---|---|---|---|---|
| a TOF: Turn over frequency (s−1). | ||||
| Pd–P–SiO2 | 719 h−1 | Formate salts HCOONa | 85 | 36 |
| Pd–S–SiO2 | 959 h−1 | K2SO4 | 85 | 36 |
| Pd shell@Ag core | 252 h−1 | H2O | 50 | 40 |
| Pd–Au/C | 27 h−1 | Formate salts HCOONa | 90 | 45 |
| Pd–Au/C–CeO | 832 h−1 | Formate salts HCOONa | 102 | 38 |
| Pd/γ-Al2O3 | 600 h−1 | NaOH, 0.1 vol% O2 | 20 | 52 |
| 1 wt% Pd/C | 255 h−1 | Gaseous phase | 100 | 46 |
| Au/TiO2 | 201 h−1 | Gaseous phase | 100 | 50 |
| Au/SiO2 | 7023 h−1 | Gaseous phase | 100–300 | 51 |
Finally, in Section 5, we highlight recent studies that suggest promising catalysts, enabling simple down streaming and upgrading of GVL into significant compounds such as pentanoic acids (PA). Ultimately, bio-solvents are the candidate final product to which further analysis could be done to substitute petroleum-derived solvents involved in pharmaceutical industries. Nevertheless, this review provides attention on gaps and challenges to shift individual catalytic reaction steps into a process that extends from upstream treatment of lignocellulosic biomass to higher value end products.
2 Valorisation of lignocellulosic biomass into levulinic acid and formic acid
The valorisation of lignocellulosic biomass feedstock offers a sustainable approach that combines the low-cost production of LA–FA mixture with energy recovery, where the end products are a variety of liquid compounds including biofuels and bio solvents. It is reported in literature that LA and FA are obtained in equimolar quantities by the fermentation of cellulosic biomass.15–17 The aqueous solution of LA and FA is achieved by acidic hydrolysis of the cellulose.3 Moreover, LA and FA can also be formed from the hemicellulose fraction of biomass. In either case, liberating carbohydrates polymers from the rigid structure of lignin is a crucial preliminary step before any subsequent reaction.182.1 Route from cellulose
As mentioned previously, FA and LA are obtained in equimolar ratios by acid hydrolysis of cellulose, which constitutes 40–50% of the whole biomass. This route comprises the dehydration of hexoses (C6 sugars) to obtain 5-hydroxymethylfurfural (HMF). Further hydration of HMF results in the acid mixture (LA& FA).3 The reaction mechanism of the hydrolysis of C6 sugars, in which FA is formed along with LA, takes place in the presence of a strong acid catalyst, e.g., H2SO4, HCl, and HNO3.19–21 Heeres et al. made a breakthrough by using triflouroacetic acid (TFA) as an acid catalyst to hydrolyse D-glucose and D-fructose sugars. The results signified TFA with high potential in attaining competitive yields of acids compared to using conventional H2SO4.22 The authors had previously examined the role of TFA as an acid catalyst for hydrolysis of D-glucose. At 180 °C, the aforementioned reaction yielded 57 mol% of LA. Furthermore, the hydrolysis of D-fructose in the presence of TFA acid catalyst under the same conditions resulted in a higher yield (70 mol%) of LA than D-glucose substrate.20,23 These results came in agreement with the literature24 and proved that LA is produced in higher yields from D-fructose-rich feeds than from D-glucose ones. Before proceeding, it is worth noting that humins, the insoluble by-products, are formed by acid hydrolysis side reaction of the intermediate HMF. However, Heeres et al.22 experienced minor formation of humins due to fast conversion of D-fructose using TFA into LA along with FA.2.2 Route from hemicellulose
LA is also produced from the hemicellulose constituent, which composes 25–35% of biomass. This route is typically performed in aqueous acidic medium. The reaction includes the formation of furfural by the dehydration of pentoses (xylose C5 sugars), the building rocks of hemicellulose.3,20 Therefore, furfural is formed as an intermediate, representing an excellent platform molecule which can be converted into valuable chemicals or fuel additives blended with currently used fuels.25,26 Furthermore, the produced furfural is hydrogenated into furfuryl alcohol, which is readily hydrolysed under acidic medium into LA.3 However, hemicellulose might experience acid hydrolysis to obtain xylose oligomers that undergo acid hydrolysis side reaction to obtain FA and acetic acid. That being said, both carboxylic acids are obtained from xylose formulated and acetylated oligomers, respectively.27 The latter reaction chemistry was proposed to take place via side acidic reaction inside a continuous biphasic reactor, where feedstocks are mainly xylose oligomers derived from hardwood chips biomass.63 Further research can be devoted to investigating the separation of the co-produced FA to be utilized as a H2 donor. This suggests an integrated process in which LA streamline, produced from furfural is mixed with a streamline of the separated FA for hydrogenation into further biomass platform compounds.3 Formic acid decomposition
It is essential to understand the mechanism of FA decomposition to design a robust catalytic process for hydrogenating LA in the presence of FA as H2 donor. FA decomposition follows two competitive routes. In detail, FA is disintegrated either by dehydrogenation (eqn (1)) or dehydration (eqn (2)).| HCOOH → CO2 + H2 | (1) |
| HCOOH → CO + H2O | (2) |
It was reported that both pathways are thermodynamically favoured. Indeed, both reactions are easily shifted forward due to negative Gibbs free energy of −32.9 kJ mol−1 and −20.7 kJ mol−1, respectively.28 The slight difference in Gibbs free energy as well as enthalpy results in competitive production of CO as a by-product, when H2 is targeted.29 Selective production of CO-free H2 is the ultimate goal to obtain an enhanced overall catalytic performance of LA hydrogenation via internal H2 transfer using FA. Therefore, designing a heterogeneous catalyst that favours dehydrogenation of HCOOH (eqn (1)) on the expense of the dehydration step (eqn (2)) is a key approach for LA/FA system to avoid unfavourable CO by-product, which leads to the poisoning of heterogeneous catalyst.30,31
3.1 Formic acid decomposition into free hydrogen using heterogeneous catalysis
In this section, we herein report a variety of heterogeneous catalysis that are engaged in FA decomposition, starting with heterogeneous catalysts for aqueous FA decomposition. Afterwards, we will emphasize on heterogeneous catalysts that are involved in gas phase FA decomposition.FA decomposition in the presence of metals, alloys, and oxides heterogeneous catalysis dates back as far as the 1950s and 1960s.32 Heterogeneous catalytic systems were developed to seek appreciable reaction kinetics of FA decomposition while saving energy demand for the reaction. Generally speaking, noble metal heterogeneous catalysts favour dehydrogenation decomposition of FA (eqn (1)), whilst oxides and base metals catalysts are engaged in the dehydration route (eqn (2)).33–35 Zhao Y. et al. synthesized a group of immobilized catalysts by anchoring Ru and Pd metals using functionalized groups onto silica supports. Amongst, Pd–P–SiO2 attained the highest activity of TOF of 719 h−1 at 85 °C followed by Ru–S–SiO2 with TOF of 344 h−1. Decreasing temperature resulted in lower activity, which reached TOF of 16 h−1 for Pd–S–SiO2 at 45 °C. It was found that the inclusion of sulfates by using Na2SO4, MgSO4, and K2SO4, boosted the catalytic activity to exceed 940 h−1 with sulphur functionalized catalysts.36 These results bear Pd(II) as an active species, which is stabilized by mercaptopropyl groups.36 Nevertheless, performing FA decomposition in the presence of heterogeneous catalysts suffers from drawbacks owing to the presence of CO contamination, water vapour formation, and therefore catalyst deactivation.37,38 To illustrate, elevated temperature favoured decarbonylation reaction, generating CO gas despite using active Pd–Au/C core–shell catalyst.38,39 For this reason, Tedsree K et al. optimized a core–shell catalyst by the inclusion of core metal Ag into Pd shell metal to be active for FA decomposition at low temperatures. The attractive activity of TOF = 252 h−1 at 50 °C was observed for Ag@Pd (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) core–shell catalyst. This trait is attributed to altering Pd active sites via direct charge transfer from Ag core metal, which thereby stimulates the bridging adsorption of FA to promote hydrogen production.40 Further on bimetallic catalysts, the morphology is influenced by the followed preparation method. Bimetallic Pd–Au can be constructed as a mixture, clusters to clusters, random alloy, and core–shell.41,42 These options can be achieved through calcination. For instance, Pd is detected in the outer framework of the particle surface, resulting in a rich Pd shell on top of an Au rich core. This structure could be flipped by further calcination that leads to Pd rich core and Au rich shell morphology.43 Additionally, the application of subsequent calcination processes yields further structural changes signified by three shell layers of core–shell catalyst: Pd–Au alloy as the inner core, Au as intermediate layer, and a rich Pd outer shell.44 Haung Y. et al. prepared a core–shell Pd/Au@Au/C catalyst, which showed a high anti-poisoning activity versus CO and an attractive activity represented by 90% conversion of FA and only 30 ppm CO after 6 h at a temperature of 92 °C.39 Moreover, they proposed the crystallization process for making Pd/Au@Au/C system (Fig. 1).
:1) core–shell catalyst. This trait is attributed to altering Pd active sites via direct charge transfer from Ag core metal, which thereby stimulates the bridging adsorption of FA to promote hydrogen production.40 Further on bimetallic catalysts, the morphology is influenced by the followed preparation method. Bimetallic Pd–Au can be constructed as a mixture, clusters to clusters, random alloy, and core–shell.41,42 These options can be achieved through calcination. For instance, Pd is detected in the outer framework of the particle surface, resulting in a rich Pd shell on top of an Au rich core. This structure could be flipped by further calcination that leads to Pd rich core and Au rich shell morphology.43 Additionally, the application of subsequent calcination processes yields further structural changes signified by three shell layers of core–shell catalyst: Pd–Au alloy as the inner core, Au as intermediate layer, and a rich Pd outer shell.44 Haung Y. et al. prepared a core–shell Pd/Au@Au/C catalyst, which showed a high anti-poisoning activity versus CO and an attractive activity represented by 90% conversion of FA and only 30 ppm CO after 6 h at a temperature of 92 °C.39 Moreover, they proposed the crystallization process for making Pd/Au@Au/C system (Fig. 1).
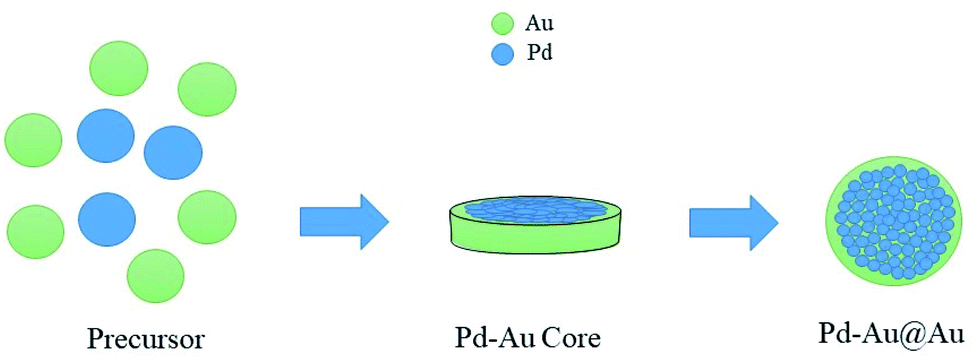 | ||
| Fig. 1 Crystallization process of Pd/Au@Au/C system.39 | ||
Another bimetallic catalytic system was studied by Zhou X. et al. in which they discovered that the highest FA dehydrogenation was attributed to Pd–Au alloy system as well as Pd–Ag alloy on a carbon support. Considering the loading ratio, reaction temperature and HCOOH/HCOONa ratio, Pd–Au/C showed the highest TOF of 27 h−1 followed by Pd–Ag/C with a value of 17 h−1 at 365 K. An attractive catalyst stability for 240 h−1 with only 80 ppm of CO formation is attributed to the basicity of HCOONa solution. The resulting reforming gas qualifies for PEMFC standard as it doesn't exceed 100 ppm CO.45 The inclusion of CeO into both catalysts improved their activity further due to the formation of more oxidized Pd that can oxidize adsorbed CO. Indeed, the TON attained was 227 h−1 and 76 h−1 for Pd–Au/C–CeO and Pd–Ag/C–CeO, respectively.38 The improved catalytic activity of TON of 832 h−1 was observed at a higher temperature of 375 K using Pd–Au/C–CeO alloy catalyst.
Bulushev et al. compared the activity of Pd and Au active sites over carbon and TiO2 supports in gaseous phase FA decomposition at 373 K. The highest TOF of 255 h−1 was attributed to 1 wt% Pd/C followed by 10 wt% Pd/C with a TOF of 180 h−1. The higher decomposition rate of FA was due to the higher dispersion of Pd nanoparticles. On the other hand, lower activities were observed for 1 wt% Au/TiO2 and 0.8 wt% Au/C at TOFs of 57 h−1 and 21 h−1, respectively. Despite the higher activity of Au/TiO2 catalyst in decomposing FA, a lower selectivity of H2 was noticed,46 which comes in agreement with TiO2 support engaged in favouring FA decomposition through decarbonylation route (eqn (1)).47,48 It was reported elsewhere in literature that Au comprises low areal rates in comparison with other noble metals, which is represented by first-order kinetic of FA decomposition,49 whereas well-dispersed Au is capable of decomposing FA.50 It is worthy to note that Pd particles were inhibited by CO which exhibits strong adsorption. Whereas, Au showed weaker CO adsorption and improved WGS reaction, producing additional H2.46 Further interesting work was done on investigating the role of Au species in FA decomposition. The thermal treatment of Au/Al2O3 caused Au cluster size to increase (sintering effect) accompanied by a drastic decrease in HCOOH decomposition rate. Consequently, FA dissociation is favoured by well spread Au species only. Despite similar cluster size of Au/TiO2 and Au/Al2O3 of 3–4 nm, the TOF for HCOOH decomposition were 201 mol h−1 g−1 and 7 mol h−1 g−1, respectively. In this context, Au is less dispersive over TiO2 than on Al2O3 support. It is worth noting that none of the used catalysts; Au/TiO2, Au/Al2O3, and Pt/Al2O3 yielded CO and H2O, indicating the absence of dehydration of HCOOH and reverse WGS reaction.50 On another end, the authors proposed that the irreversible production of H2 is achieved by the adsorbed H produced by the preceding step as shown below.50
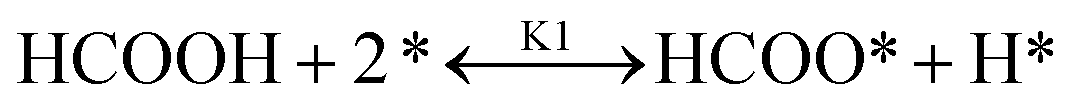 | (3) |
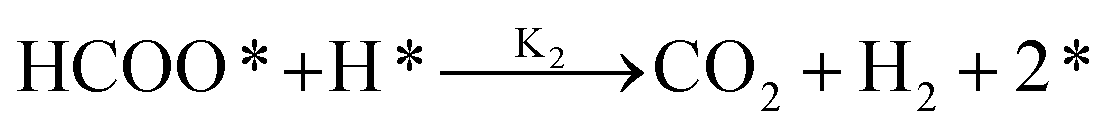 | (4) |
Eqn (3) and (4) further studies investigated the catalytic performance of Au species over different oxide supports. Gazsi A. et al. found that Au/SiO2, Au/CeO2, and Au/Norit favoured FA decomposition through dehydrogenation route (eqn (1)). Whilst dehydration of FA (eqn (2)) was dominated in the presence of Au/Al2O3, Au/ZSM-5, and Au/TiO2. The most active catalyst regarding FA conversion was Au/SiO2 with a TOF 7023 h−1 of 100% H2 selectivity between 373 K and 523 K. They also reported that the FA rate of decomposition is not dependent on Au particle size.51 However, their results came in contrary to conclusions about the Au cluster size effect investigated by Manuel Ojeda and Enrique Iglesia as mentioned previously.50
Regarding reaction kinetics, FA decomposition follows a zero-order pattern over Au/SiO2, which depends on formate intermediates instead of HCOOH substrate initial quantities.51 This comes in harmony with the suggested kinetics by Ojeda M. et al. who also recognized that changing HCOOH partial pressure did not affect FA decomposition.50 In both studies, the proposed mechanisms involve the cleavage of C–H bond of formate, which falls in the rate-determining step.50,51 Another contradiction arises regarding H2 selectivity. A very low yield of H2 (TOFH2 = 18 h−1) was observed for Au/Al2O3 at 473 K,51 whereas much higher H2 was produced with only 10 ppm of CO traces detected in the presence of Au/Al2O3 at ambient temperatures.50 In both cases, Au inhibited catalyst deactivation due to weak CO–Aux complex formation.
A very recent breakthrough was achieved by Xu P. et al. on removal of adsorbed CO over Pd/γ-Al2O3 by adding traces of oxygen. The effect of the concentration of O2 on the conversion and selectivity of the FA decomposition was also investigated. The optimal concentration of O2 of 0.1 vol% yielded an activity of TOFH2 = 600 h−1 beyond which the selectivity of H2 drops down, while conversion was preserved at 60%. The yield of H2 dropped to 500 h−1 at O2 concentration of 2 vol% due to the consequent oxidation of H2 into H2O on the expense of CO oxidation.52 Sneka-Płatek O. et al. used another approach to inhibit CO poisoning in which tuning the alloy ratio of Ag:Pd at 4% Ag–1% Pd resulted in isolated Pd atoms and thereby in the highest conversion of HCOOH at 34%.53
3.2 Formic acid: a hydrogen donor for hydrogenation of levulinic acid into higher value platform compounds
Here in this section, we emphasize heterogeneous catalytic routes that govern the transformation of levulinic acid into higher-value compounds in the presence of FA as a hydrogen donor. As mentioned earlier in this paper, FA comprising 4.4 wt% H2 and a volumetric capacity of 53 g H2 L−1, is produced in equimolar stoichiometry with LA.54 Hydrogenation is a key factor that is involved in upgrading biomass into higher-value chemicals and fuel additives. For instance, LA (4-oxypentanoic acid) represents an attractive platform molecule, which is produced easily and inexpensively from biomass, undergoes sequential deoxygenating steps to yield higher value liquid alkanes.3,55 To add, hydrogenation of LA yields several compounds, having distinctive properties and applications. To list, γ-valerolactone (GVL) is a valuable polyester monomer, 1,4-pentanediol is also used in polyester production, methyltetrahydrofuran (MTHF) is a valuable liquid hydrocarbon that can be used as a gasoline blend, and diphenolic acid which is involved in polycarbonate production,7 and pentanoic acid (PA).56–58Owing to the difficulty of handling conventional storage systems as an external source of hydrogen, shifting towards the in situ production of hydrogen from FA for hydrogenating reactions has become crucial.59,60
Biomass compounds processing has been well discussed in literature from different perspectives. A good review was published elsewhere about upgrading biomass-derived compounds from different cellulosic biomass fractions, e.g. cellulose, hemicellulose, and lignin through C–C coupling reactions (Fig. 2).3
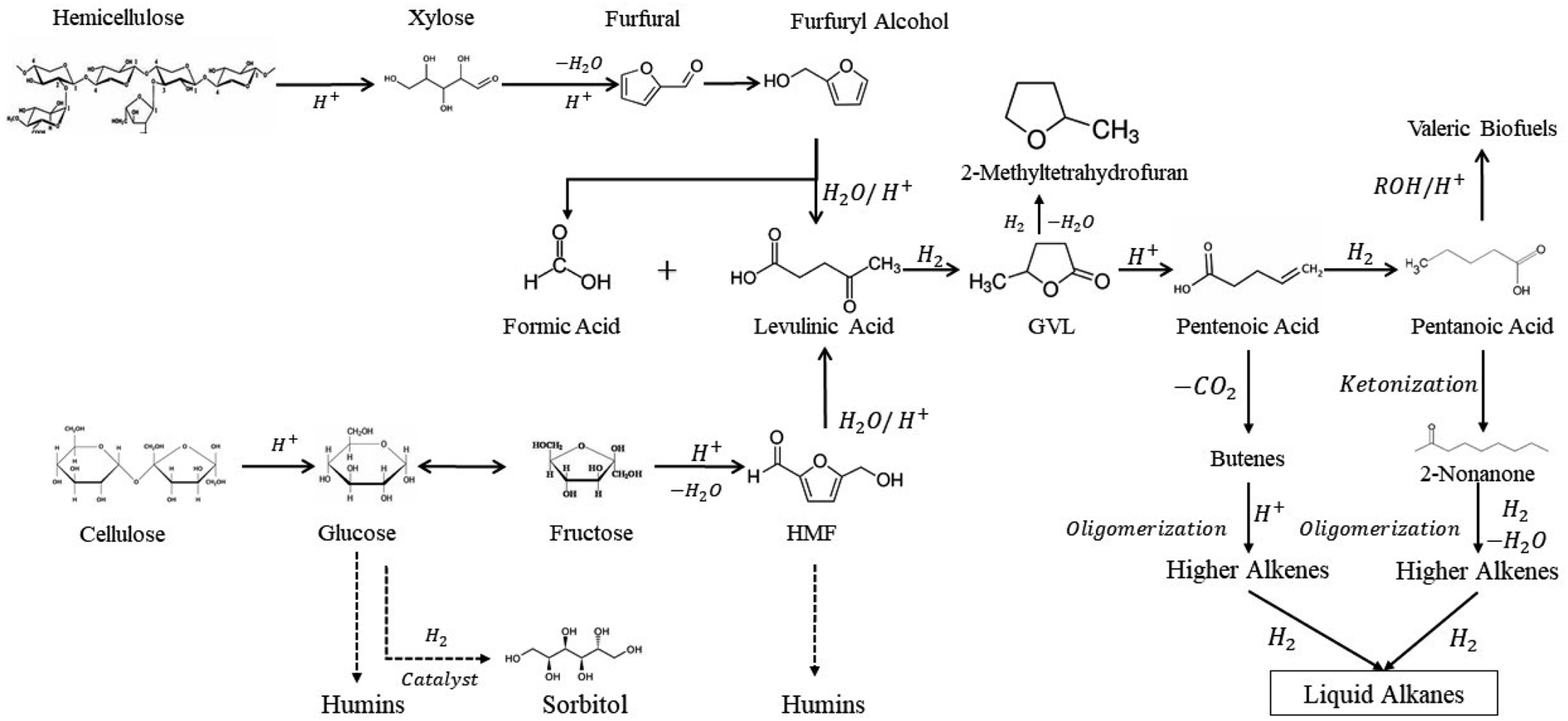 | ||
| Fig. 2 Biomass platform molecules upgrade into valuable products and fuel additives.3 | ||
Another review focuses on the conversion of carbohydrates using heterogeneous catalysts. For instance, hexoses and pentoses, the building rocks of carbohydrates, undergo dehydrogenation and dehydration catalytic reactions to yield HMF and furfural (Fig. 3), respectively, which are subsequently upgraded into fine chemicals.61
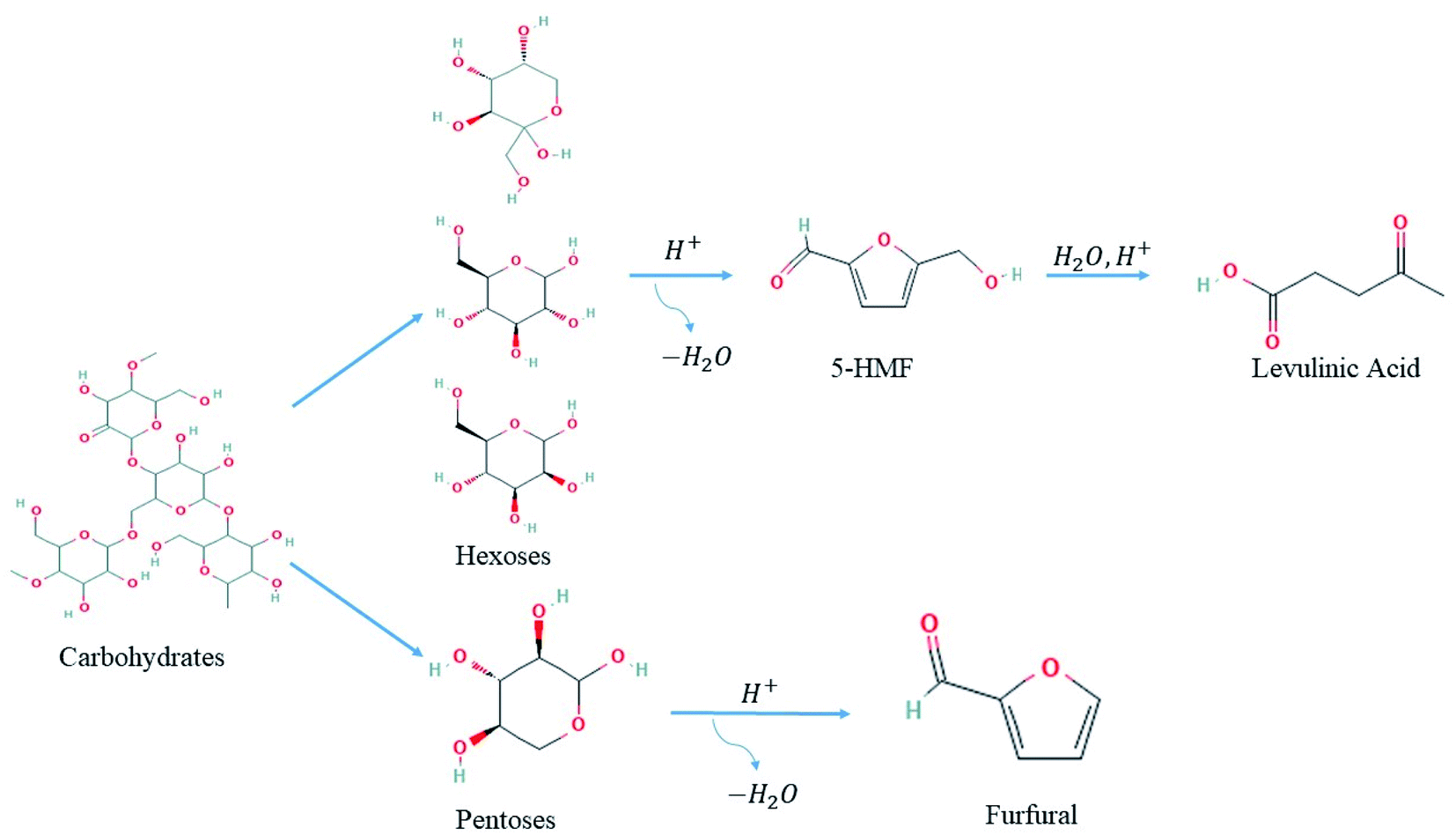 | ||
| Fig. 3 Possible routes for dehydration of carbohydrates into levulinic acid and furfural platform compounds.61 | ||
Huber W. G. et al. wrapped up all known routes to produce bio-oils from lignocellulosic biomass. They reported a wide range of liquid fuels and gasoline additives that are formed through molecular weight adjustments.62
Levulinic acid has been widely reported as an attractive platform chemical from which higher-value chemicals are produced, including levulinate esters, GVL, HMF, and MTHF. Noteworthy, designing a process with the least number of deoxygenating steps as one-pot transformation is shifting to a more economic route. As an added advantage for a cost-effective biomass process, replacing the external source of hydrogen by in situ molecular H2 produced from FA is applied in addition to designing a catalyst capable of performing dehydrogenation and hydrogenation.
LA platform chemical along with FA is produced commercially from sugars that are produced subsequently from cellulose (Fig. 4). The production cost is estimated to be 0.09–0.11 $ per kg for a throughput of 1000–2000 dry tons per day.63
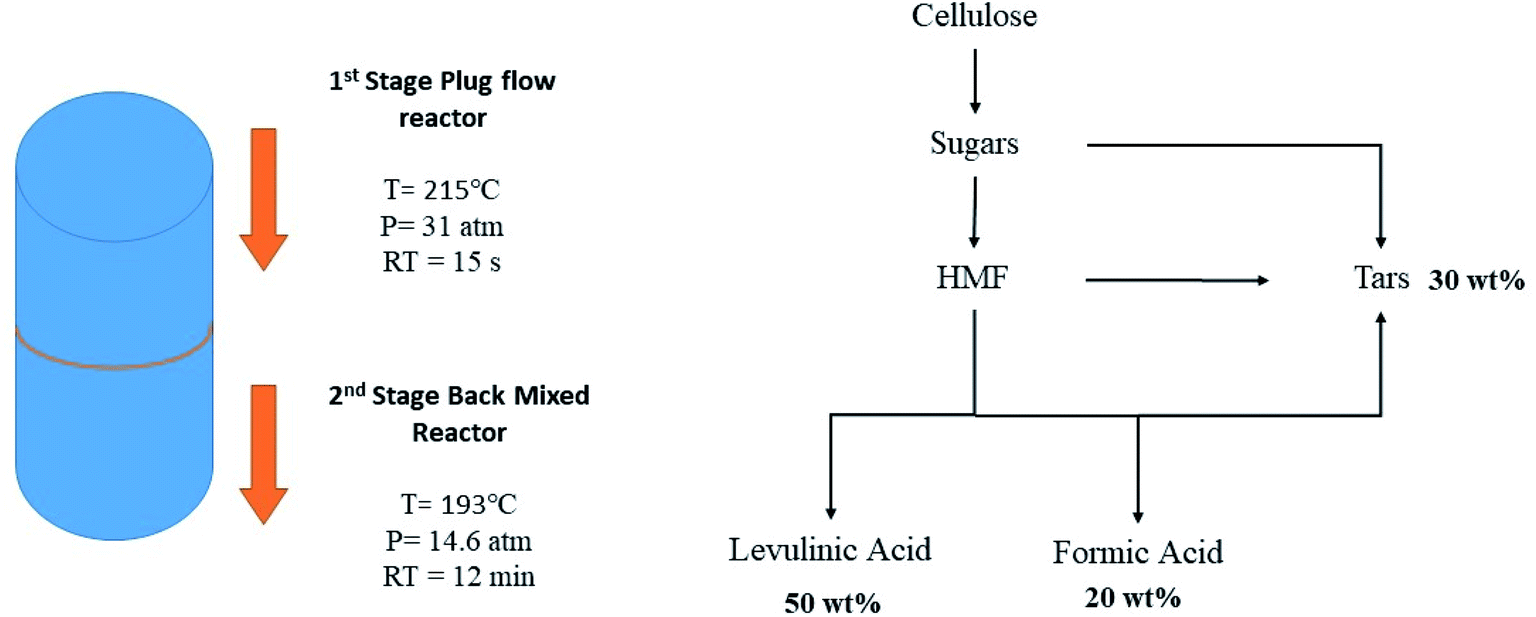 | ||
| Fig. 4 Two-stage production of levulinic acid and formic acid from cellulose.63 | ||
LA comprises distinctive properties due to the presence of carbonyl and ketone functional groups. Consequently, LA can undergo different reactions, mainly at its carbonyl group to yield cyclic compounds. Owing to its distinct products including but not limited to GVL, levulinate esters, 2-methyltetrahydrofuran, valeric biofuels, and pentanoic acid.64
Deng Li et al. developed a catalytic route that comprises the production of GVL from biomass carbohydrates with a yield of 93 mol% by using RuCl3.3H2O/PPh3 with pyridine base at 423 K. The novelty is pronounced by eliminating the need to separate levulinic acid from FA in the intermediate mixture. Noteworthy, it was found that introducing CO2 to the reaction medium boosted the catalytic activity of Ru catalyst to reach a yield of 100 mol% GVL, whilst H2O did not have any effect.65
Sequential production of deoxygenated compounds is achieved by Ru/C catalyst followed by a dual catalytic bed containing Pd/Nb2O5 & Ce–Zr. The former catalyst completely converted LA and FA into GVL at 423 K. In the second reaction stage, Pd/Nb2O5 catalyst resulted in 100% conversion of GVL with 78% selectivity of pentanoic acid. Further on, they observed that the addition of zirconia led to further improvements. Indeed, the dual catalyst system Pd/Nb2O5 & Ce–Zr produced 89% selectivity of nonanone (C9) from GVL.66 Even though sulphuric acid was reported as a major inhibitor for catalysts, Ru/C catalyst showed good activity in the presence of H2SO4 medium.67,68 The authors explained this by emphasizing the importance of using optimum H2SO4 proportions for cellulose pretreatment during the upstream process.66
In another study done by Deng Li et al., their previous work including one-step process using RuCl3/PPh3 catalyst is substituted by two-step processes using immobilized Ru–P/SiO2 catalyst for FA dehydrogenation and Ru/TiO2 for intramolecular hydrogenation of LA into GVL. The immobilized Ru–P/SiO2 catalyst showed highest TOF of LA of 447 h−1 at 423 K (GVL yield 96%).69 However, the team encountered contradictory results with Ruiz et al.66 regarding the activity of Ru/C catalyst. According to Deng Li et al.,69 the Ru/C catalyst did not show promising results in FA decomposition and in situ hydrogenation of LA into GVL (TOF 69 h−1, 7% GVL yield). Instead, Ru/TiO2 catalyst exhibited 63% GVL yield from LA at 4 MPa H2 at 423 K.69 An important aspect of the effect of CO2 on Ru-catalysed hydrogenation of LA arose from this research; reinjecting the CO2 produced from FA did not show any improvement of Ru/C in catalysing LA hydrogenation.69 Dongmei H et al. (2017) also evaluated the activity of immobilized Shvo's catalyst over SiO2 surface as well as covalently grafted catalyst in reducing LA into GVL using FA while avoiding the external use of H2. The highest activity of 504 TON, 21 TOF(h−1) and 88% GVL yield was attributed to immobilized Shvo's catalyst over SiO2 surface. The reaction was held in the presence of 1,4-dioxane solvent under nitrogen gas for 24 h at 90 °C.70
Feng J. et al.71 proposed a new reaction pathway for the production of GVL from LA using FA solely as H2 source. A blend of Ru/C catalyst and trimethylamine accelerated LA hydrogenation and resulted in considerable activity at 87.26% as LA conversion and 80.75% as GVL yield. The XRD pattern showed an overlap between the characteristic peaks of Ru and graphite at 2q = 44.6°, implying the high dispersion of Ru over carbon support. The proposed reaction route was exhibited using experiment probe of HPLC, 1H NMR & 13C NMR by which LA, triethylamine, GVL, ethanol, 4-hydroxyvaleric acid, and ethyl levulinate are the compounds present at the end of the reaction. This features two important observations, the first of which is that FA was completely consumed, and that the esterification reaction is possible due to some produced ethanol that reacts with LA. The postulated mechanism is a good tool that helps researchers in investigating for the possible production of CO on the expense of CO2 from FA decomposition (Fig. 5).
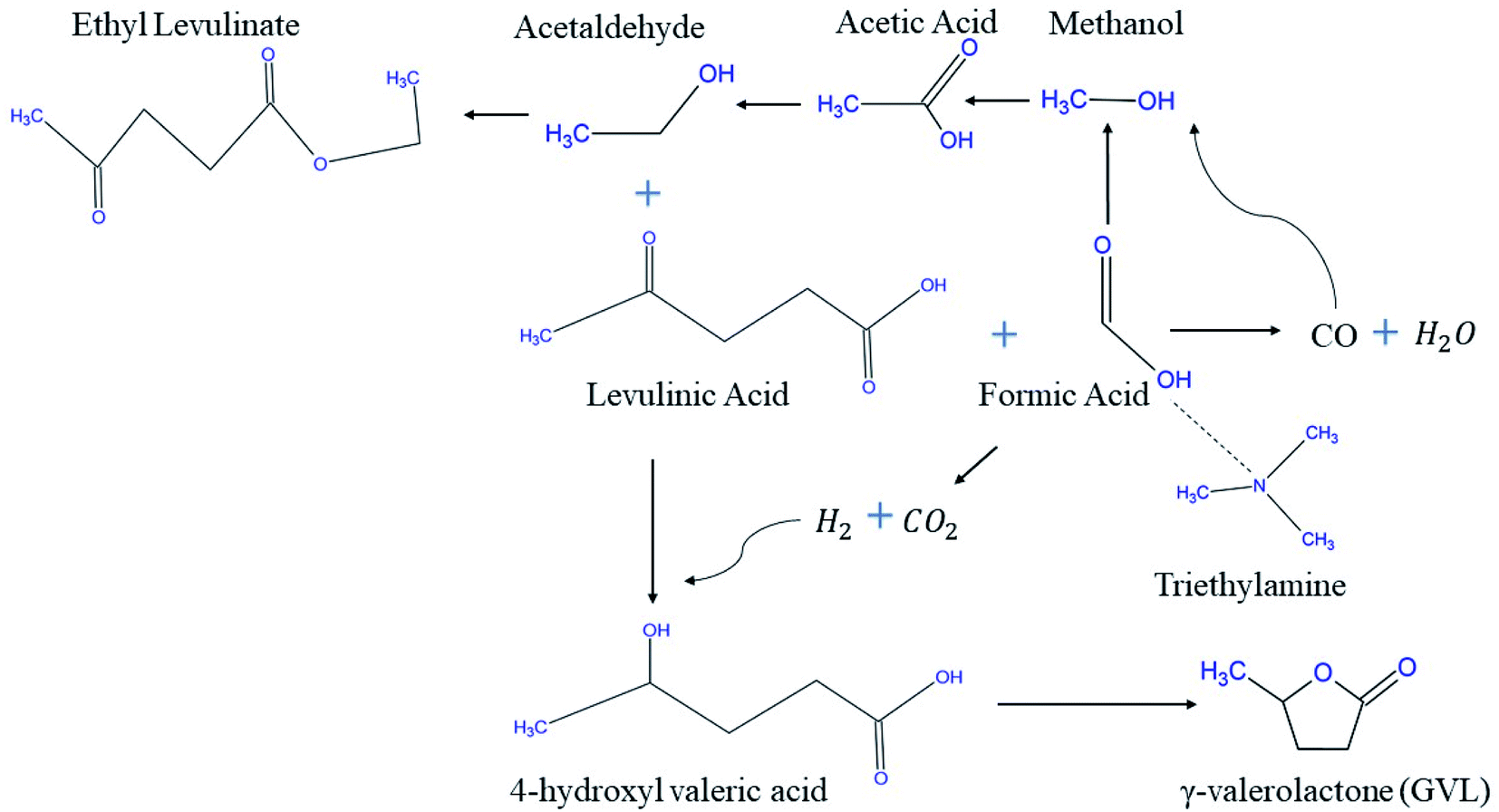 | ||
| Fig. 5 Proposed reaction pathway of LA into GVL and ethyl levulinate using FA.71 | ||
Due to the continued concern over the inhibiting effect of H2SO4 on heterogeneous catalysts,68,72 Heeres H. et al.22 proposed the use of TFA instead. It was reported that TFA has a pKa value of 0.5.73 Indeed, TFA resulted by a yield of 57 mol% of GVL22 slightly higher than the yield of GVL of 55 mol% in the presence of H2SO4.20 The team studied extensively the conditions of conducting a one-pot process starting from C6-sugars to yield GVL. It was found that Ru/C catalyst resulted in 11 mol% LA yield and 52 mol% yield of GVL.22 On another note, the authors also shed light on further improvement of GVL yield% while minimizing hydrogenation side reactions, which can be achieved by tuning reactor parameters; temperature and mixing.22 An alternative strategy to avoid catalyst inhibition by the acidic medium is using reactive extraction in which butene is added to LA, FA, and H2SO4 aqueous solution to yield formate esters and levulinate esters. The aforesaid aqueous solution is separated easily into an organic phase (hydrophobic esters) and pure H2SO4 upon its contact with water. The group proposed a dual catalytic system containing Pd/C followed by Ru/C catalysts. H2 is generated in situ from FA and formate esters over Pd/C hydrogenate LA and its ester over Ru/C catalyst.74 This comes in harmony with sulphur tolerant Pd support material used for catalysts.75,76
Interestingly, SnCl4 catalyst was used as a substitute for H2SO4 in breaking down furfural biomass carbohydrates into LA and FA, which subsequently undergo catalytic transfer hydrogenation over Au–Ni/ZrO2.77
Kopetzki D et al. suggested the use of Na2SO4 salt in reducing LA into GVL using H2 produced from FA. The salt catalyst activity relies on its dissociation constant in which SO 2−4 ions switch into a weak base at elevated temperatures (548 K) and eventually deprotonate FA. Hence, the use of a conventional base was avoided.78 It was reported elsewhere that salt additives induce autocatalysis in aqueous solutions.79 Accordingly, this approach should be given more attention towards either improving its activity further or blending it with other heterogeneous catalysts to boost up the GVL yield.
The high sensitivity of Ru/C catalyst amid acidic conditions inspired Demesic et al. to synthesize 15 wt% RuRe (3:4)/C catalyst for the hydrogenation of LA by FA in the presence of H2SO4, with no observed deactivation. Indeed, the utilized catalyst resulted in a GVL yield of more than 95% at 423 K and higher stability, which is demonstrated by the increased TOF from 50 h−1 to 150 h−1 for Ru/C and RuRe/C catalysts, respectively.6 Likewise, the concept of Ru/C catalyst deactivation due to presence of acids drove Cao et al. to study the catalytic hydrogenation of LA by consuming FA as a reducing agent using Au nanoparticles on different catalyst supports. Their results presented ZrO2 doped with Au as an attractive catalyst in comparison with Pd, Pt, Ru, and Pd as well as Au/C, Au/SiO2, and immobilized Au/TiO2.80 The activity of Au nanoparticles was linked to its particle size (1.2–2.5 nm). Both studies come in agreement regarding the particle size effect of Au particles; larger particle size has limited the activity of Au catalyst.50,80 Therefore, the good catalytic activity of Au nanoparticles over ZrO2 could be attributed to its value-added activity in HCOOH decomposition step.81 The high dispersion of Au nanoparticles of size 2.4 nm in their metallic state (Au0) was found to be improved by the inclusion of Ce into Au doped ZrO2 catalyst. These improvements play a key role in enhancing the decomposition of FA and thereby the hydrogenation of LA. The addition of Ce facilitated the formation of tetragonal phase of Zr along with the increase of weak and medium acidic sites, thus the formation of GVL was considerable at yield 83.5% due to upgraded dehydration reaction of the intermediate derived from LA hydrogenation reaction at conversion 90.8%. Despite the use of a high reaction temperature of 240 °C, the catalyst showed robustness with a limited decrease in its activity after 5 consecutive runs.82 Another study performed by Zhou C. et al.77 confirmed the improvement of Au/ZrO2 activity in hydrogenating LA by means of FA in the presence of highly dispersed Au0 due to the incorporation of Ni metal. The role of Ni comes in agreement with the above-mentioned Ce106 in reducing Au particles to provide highly dispersed doped Au0 with less aggregation.
A strong relation between the method of preparation of nano-alloy Ni-based Au doped catalyst and its activity in hydrogenating LA into GVL in the presence of FA was demonstrated by M. Ruppert A. et al. It was found that highest LA conversion and highest GVL yield at 89% and 86%, respectively, were attributed to co-impregnation method of both metal precursors which are then thermally reduced under H2. The group utilized DFT calculations and thorough characterization for the nano-alloy surface and concluded that selective FA decomposition is due to high energetic span of FA adsorption was favoured by Ni atoms incorporated within Au nanoparticles.83
The effect of Au nanoparticles on different oxidic supports was studied thoroughly on LA decomposition into GVL using hydrogen transfer reaction in the presence of FA. Different Au@oxides catalysts were synthesized using co-precipitation and conventional impregnation techniques. Various oxide supports including ZrO2, Al2O3, SiO2, TiO2, MgO were used for fabricating Au doped catalysts by which Au/ZrO2 achieved highest GVL yield of 85% and LA conversion of 93% at 210 °C after 5 h.84 Unlike Au–Ce/ZrO2 catalyst reported above,82 the presence of Au3+ species improved the activity of the catalyst. XPS analysis elucidated the decrease in Au3+ species from 49.2% in fresh catalyst to 10% in spent one due to reducing agent FA. The Au/ZrO2 catalyst synthesized by co-precipitation experienced a slight drop in GVL yield to 69% after 3 reaction runs.84
The activity of Ru/C showed the highest conversion values of LA into GVL, and the highest selectivity compared with Pd/C, RANEY® Ni, and Urushibara Ni. Nevertheless, Ru/C exhibited low stability which necessitates adding a portion of fresh catalyst for further runs. LA conversion and GVL selectivity dropped from 92% and 99% on its first run to 61% and 70% after the second run, respectively. Above all, the authors emphasized the hydrogenation mechanisms over Ru/C catalyst (Fig. 6).
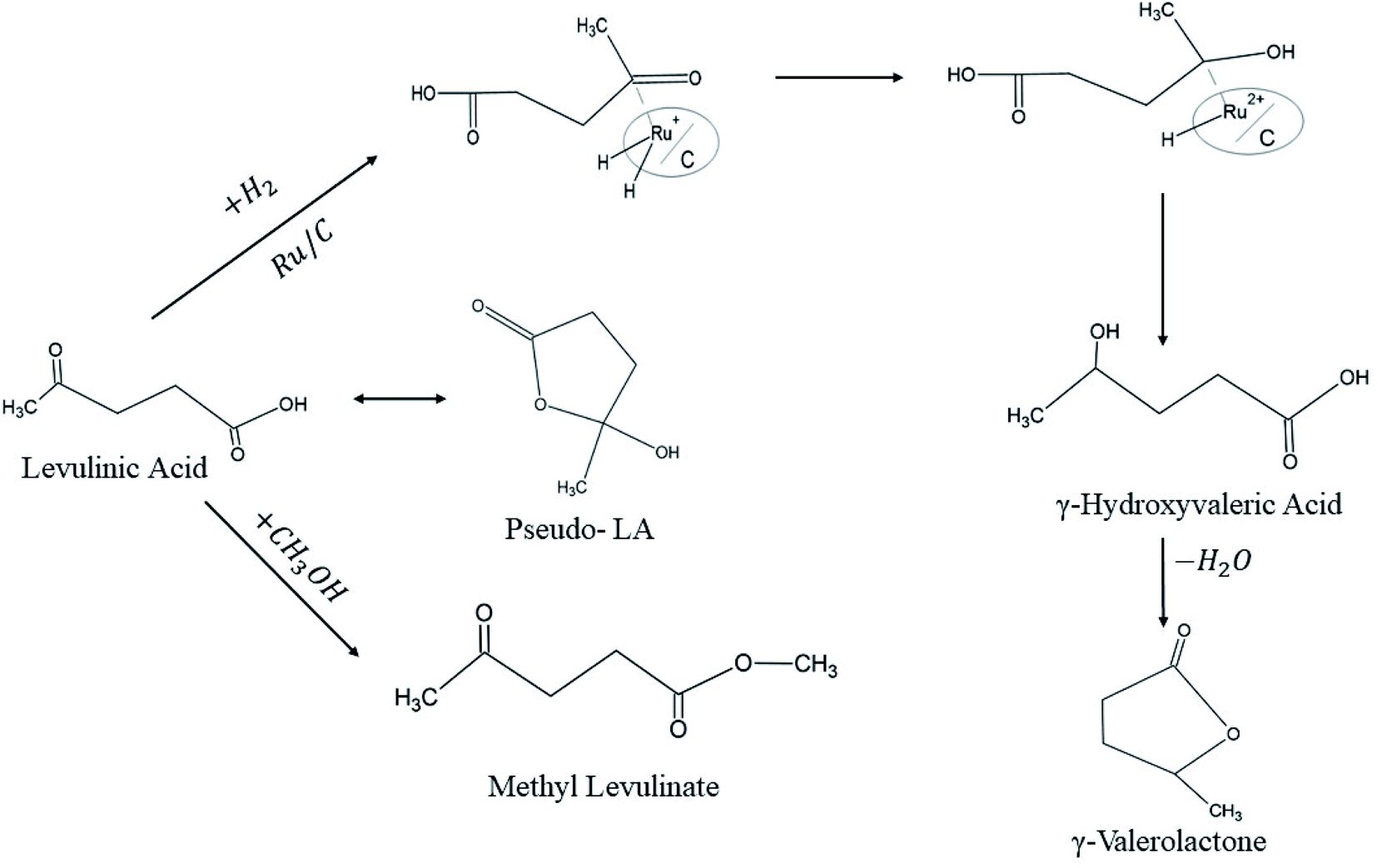 | ||
| Fig. 6 Levulinic acid hydrogenation mechanism over Ru/C catalyst.11 | ||
Both LA and H2 are chemisorbed on Ru/C before H–H bond cleavage step takes place, where one H atom will be transferred onto the carboxyl oxygen in LA and consequently an intermediate is produced. Thereafter, a second H atom transfers to the carbonyl carbon produces γ-hydroxyvaleric acid which undergoes subsequent dehydration into γ-valerolactone.11
Recently, Al Naji M. et al. made a breakthrough in the field of LA hydrogenation in the presence of FA as a reducing agent in aqueous solution. The novelty of their work was pronounced by synthesizing a robust catalyst, comprising high thermal stability and can withstand low pH conditions (pH = 2). The most active catalyst, 1.6 wt% Pt/MP-ZrO2 achieved a LA conversion of 84% and a GVL yield of 82% at 493 K after 24 h reaction time. Noteworthy, pentanoic acid of 22% wt was discovered at a higher temperature (533 K).85 On a different note, the authors confirmed that FA acid decomposition into H2 and CO2 is a rate-limiting step which precedes LA hydrogenation over the same Pt active sites (Fig. 7).
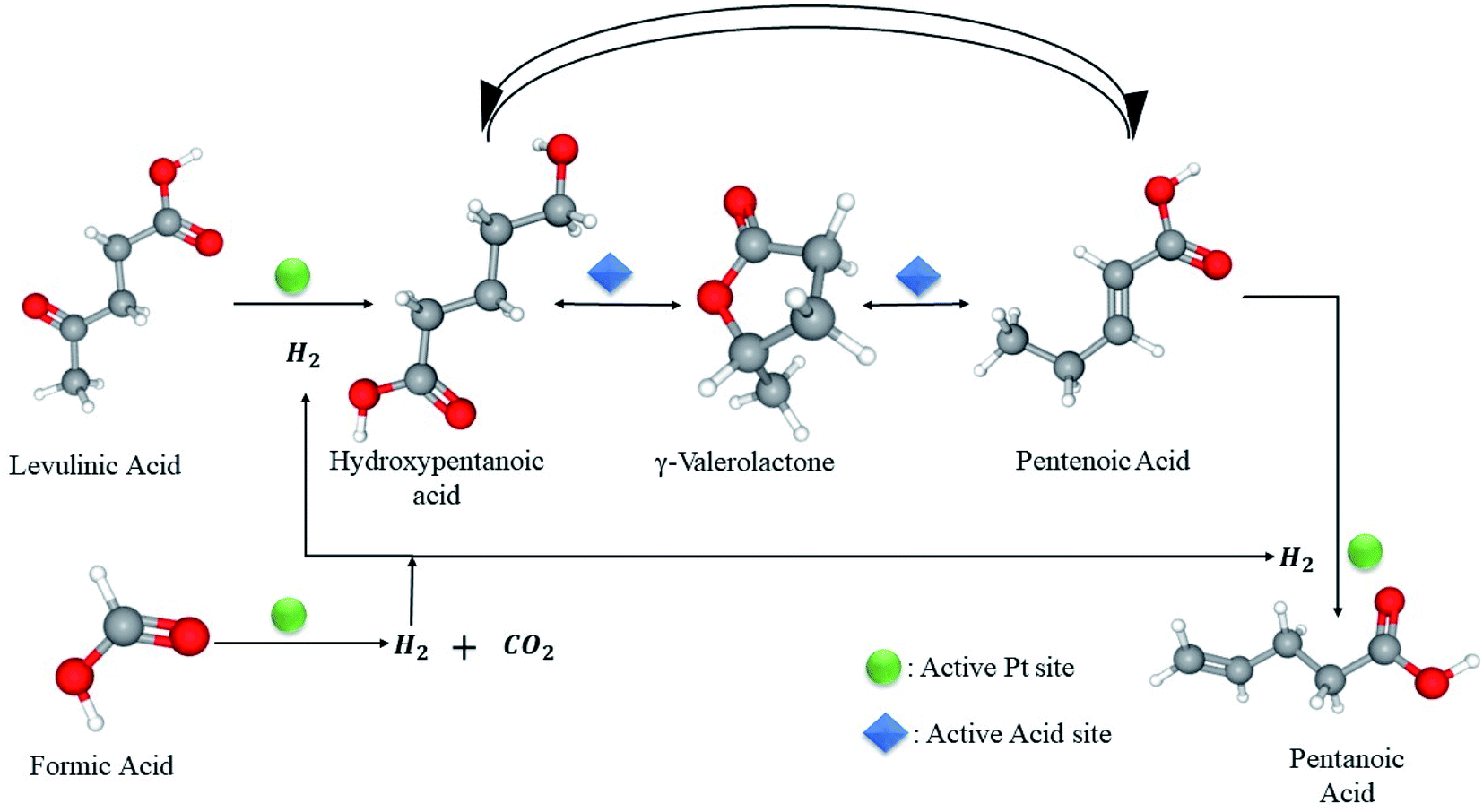 | ||
| Fig. 7 Competitive FA dehydrogenation and LA hydrogenation over Pt active sites to GVL and PA.85 | ||
Their findings come in line with Gazsi et al.51 and Ojeda et al.50 as it proved that C–H bond cleavage contained by formate intermediates controls the overall reaction. To add, the reaction follows zero-order kinetics, same as mentioned in the literature, which can be attributed to the desorption of CO2 & H2 to the liberation of Pt active sites for LA hydrogenation (Fig. 7). This suggests a competitive adsorption between LA and FA on the active sites that provoke hydrogenation,31 featuring FA decomposition the rate limiting step of the overall reaction.
It was previously emphasized that TiO2 and ZrO2 supports resulted in good activity regarding LA conversion to GVL using external molecular H2 at 473 K.86 Whereas, ZrO2 exhibited higher activity and stability in comparison with TiO2 support when doped with Ru active sites.11,87 In this context, Al Naji et al. also synthesized a bifunctional catalyst by way of doping noble metal Pt on zeolite support. The discovered catalyst was characterized by its acidic property, which thereupon permitted its ability to convert GVL into pentanoic acid. Briefly, this conversion was accomplished via a moderate number of acidic sites of zeolite, on which GVL ring-opening into pentanoic esters is favoured. At the same time, PEAs are hydrogenated using H2 produced from FA decomposition, where both reactions (PEAs hydrogenation and FA decomposition) occurred over dual Pt active sites. The enhanced activity of hydrogenation/dehydrogenation reactions using dual active noble Pd metal supported on acidic zeolite catalyst were also demonstrated in pyrolysis of waste rubber, cracking down complex hydrocarbons into simpler compounds.88 The conversion of LA achieved via the dehydration of 4-hydroxypentanoic acid intermediate, which is considered as a rate determining step, was reportedly enhanced by tuning Zr/Al ratio of Ru/(AlO)(ZrO)n catalyst. The improved activity was characterized by enhanced strength and distribution of Lewis acidic sites of (AlO)(ZrO)0.1 and calcination temperature of 400 °C.89
Designing an economic LA hydrogenation system is shifting from using sustainable FA as a hydrogen donor alone to the use of inexpensive metal catalysts. For instance, non-noble metals were tested in LA hydrogenation in the presence of catalytic transfer hydrogenation using nickel, zirconium, and iron.90–93 Amongst, Zr comprises the required acidity that stimulates LA hydrogenation into GVL.94,95 This was characterized by the high LA conversion due to high acidic density sites (136 μmol g−1) when Zr was used as a support in 1.6 Pt/MP-ZrO2.85
Non-noble metals heterogeneous catalysts have always been an attractive topic for researchers as they seek cost-effective biomass hydrogenation processes. A recent study investigated the relation between the catalyst performance of MnCo oxide catalyst and its crystal structure. Series experiments were made using several MnCo oxide catalysts using different Mn:Co ratios. The highest activity corresponded to Mn2Co0.1Ox of 83.4% LA conversion and 70% GVL selectivity achieved at 230 °C. The improved activity was attributed to the structural distortion induced at MnCoO3 and MnOx interface, which facilitated the adsorption of FA by H–H formation and π–π interaction with ketone group of LA simultaneously. Previous attempts for applying non-noble metals in hydrogenating LA into GVL using FA in vapour phase96–101 and in aqueous phase102–108 are published elsewhere. Amongst, deactivation was studied by Hussain Sk et al.101 who found that using pure MgO catalyst exhibited good activity at the start-up before it declines due to deactivation. However, using calcined Mg–Al hydrotalcite catalysts yielded complete conversion of LA for 10 h and appreciable selectivity of GVL of 98%. The catalyst showed a good extent of stability conveyed by the reusability under same reaction conditions. The conversion of LA dropped to 86% while GVL selectivity was preserved near 98% after 4th cycle of the reaction.109
A recent study also aimed at designing cost-efficient catalyst that is effective in hydrogenating LA into GVL at mild reaction conditions. The group used hard-soft template-assisted electro-deposition method to engineer Ni78Pt22 nanowires based on polycarbonate membranes that accommodate Ni–Pt mesoporous wires. Experiments showed that a TOF of 74 h−1 (100% conversion) and GVL selectivity near 99% were achieved at 140 °C during 120 min110. Ni and Cu cheap metals showed an excellent dispersion over SiO2 support, featuring good catalytic activity in terms of hydrogenation of LA into GVL at a yield of 96% along with 4% Angelina lactone at 285 °C. The bimetallic Ni–Cu/SiO2 catalyst characterized good stability for prolonged runs of 200 h. This robustness is attributed to nanocomposite morphology nature of Cu–Si, which prevents metallic sintering. Indeed, the highest H2-uptake chemisorption of 0.039 mmol g−1 corresponds to the optimal catalyst loading of 20% Ni & 60% Cu–SiO2, which proved a homogeneous dispersion of metallic nanoparticles.99 In another study, doping Ni metal on RANEY® catalyst resulted in a complete conversion of LA (100%) and a yield of GVL at 68.5% using a ratio FA/LA of 4:1 under 200 °C for 48 h. The RANEY® Ni catalyst was able to catalyse cellulose under same conditions into GVL at yield 23.3%.111
Considering the synergetic relation between Cu & SiO2 and the improved catalytic activity obtained, Lomate S. et al. (2017)96 characterized the SiO2 supports and Cu-doped SiO2 catalysts, thereby studying the relation between the morphology and its activity. The physio-chemical properties of the catalyst were investigated and found to affect its activity. For instance, the highest TOF of 0.0707 moles LA h−1 g−1 corresponds to Cu–SiO2 catalyst with larger CuO clusters with stronger interaction with SiO2 surface. The strong interaction was also confirmed by the existence of the partial oxidation of Cu species represented as Cuδ+ with a binding energy 933.25 ± 0.35 eV located between Cu+ and Cu2+. Indeed, the higher activity of Cu–SiO2 catalyst was attributed to its morphology, having narrow pore diameter distribution that plays a role in confinement of smaller Cu clusters inside pore walls, thus easily accessible for LA hydrogenation into GVL. Indeed, this work has thoroughly investigated the complex role of acid nature of the catalyst surface and its redox properties have on its activity and product selectivity. The use of characterization tools provided solid details about the catalyst nature including its electronic structure, which facilitated drawing a conclusion that Lewis acid sites of medium acid strength played a key role in the conversion of LA into GVL. Whilst Brønsted acid sites provoked hydrogenolysis of GVL into further hydrocarbons over metal sites and thus, increasing the yield of pentanoic acid and angelica lactone (AL) on the expense of GVL. The presence of mononuclear Cu2+ over oligomeric (Cu–O–Cu)n+ led to higher activity of Cu/SiO2 catalyst at 66% LA conversion and 81% GVL yield (Fig. 8).96
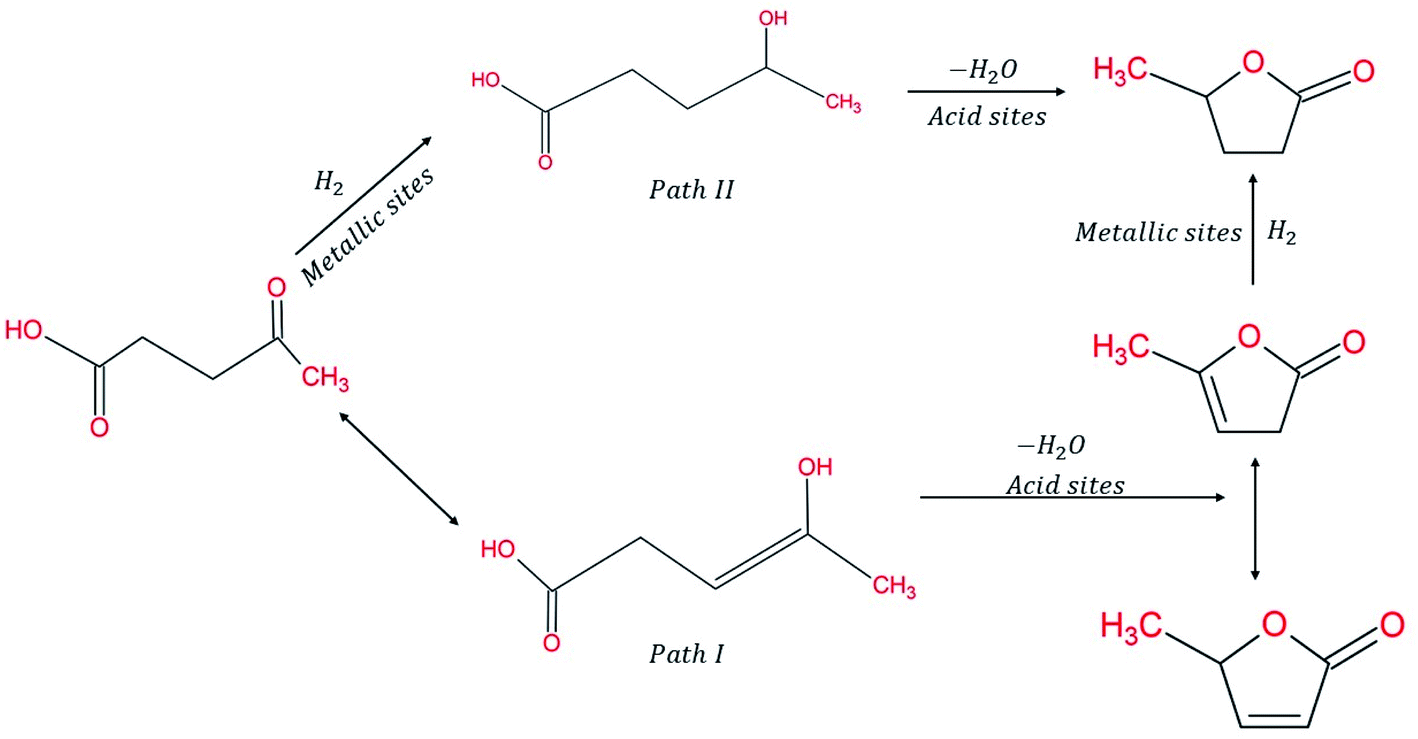 | ||
| Fig. 8 Conversion routes of LA into GVL.96 | ||
Designing a cost-effective route for hydrogenating LA into GVL using FA put it forward for considering the use of non-noble metal supported on oxides surfaces. For instance, ZnAl oxides were tested using different temperatures and LA/FA ratios, where it exhibited considerable GVL yield of 90% and LA conversion near 85% after 6 h reaction time at 140 °C and 5:1 FA/LA ratio.112 The work also featured kinetic modelling for proposed FA decomposition, LA hydrogenation as well as reversible formation of angelica lactone (AL) intermediates. The inhibitory effect of side reactions lies in the formation of lactones oligomers strongly adsorbed over basic sites of catalysts. Moreover, dehydrogenation of FA and hydrogenation of AL are key factors that establish the bounds within which LA dehydration reversible reaction is promoted in the forward direction without allowing the accumulation of intermediates (AL) (Fig. 8).112
From all the aforementioned information, LA can be transformed into its building blocks, which serve as valuable chemicals and biofuels additives. Together with GVL, LA can also be converted into other platform compounds such as pentanoic acid (valeric acid)57,113,114 or pentenoic acid115–118 (Fig. 3). In detail, GVL yielded from LA experiences ring opening to produce PA and pentanoic esters that undergo subsequent catalytic deoxygenating reactions to produce higher alkanes. As an illustration, Bond et al. established a dual catalytic system that comprises SiO2/Al2O3 followed by HZSM-5/Amberlyst acid catalysts, producing higher alkenes from butene and pentenoic acid intermediates.13,119 Likewise, pentanoic acid is also produced from GVL via hydrogenation/ring-opening before it experiences a consequent transformation into liquid alkanes, having distinctive properties which qualify it as a good diesel fuel blend.66 Lately, Zixiao Yi et al. introduced a comparative study of doping acidic catalyst by different metals; Ni and Ru. The two metals showed different reaction routes by which 3 wt% Ni/HZSM-5 catalyst resulted by limited hydrogenation of LA into GVL, whilst 3 wt% Ru/HZSM-5 catalyst facilitated further hydrogenation reaction, resulting in PA and pentanoic esters.120 Identically, the increase in acidic density due to Ru inclusion as well as 1.6 Pt MP−1-ZrO2 gave rise to PA production via GVL ring-opening mechanism.85 This comes in agreement with the findings of P. Sun et al. who reported that excessive acidity leads to decreasing GVL yields due to further ring opening reactions.117 Similarly, the dispersion of metal and the strong interaction between the metal and the doped acidic support MCM-49 can be adjusted by which a mesoporous Ru/MCM-49 can achieve LA conversion into GVL. This high conversion expressed by a TOF value of 3000 h−1 was attributed to the high amounts of lewis acid sites on which lactonization of 4-hydroxypentanoic acid into GVL is achieved.121 Metal active sites incorporated in heterogeneous catalysts experience some inhibitory factors that retard its activity, one of which is metal leaching due to the presence of the organic levulinic acid substrate. Interestingly, Bai X. et al. have developed a robust catalyst based on Ag–ZrO2–graphene oxide nanocomposite.122 In this perspective, Li J. et al. developed an attractive ternary catalyst Cu/ZrO2–Al2O3 for converting LA into GVL while avoiding the leaching of Cu. This was attributed to the existence of oxygen vacancies in the extra framework of the catalyst on which LA is adsorbed before it subsequently transforms into GVL.123 In the same context, it was reported that sol–gel entrapment of Ru in ZrO2 minimizes leaching of Ru/ZrO2 catalyst when used under acidic conditions (pH = 3.4). A conversion near 73% of LA and a complete decomposition of FA/HCOO− (100% conversion) were attained using only 2.5% wt Ru over ZrO2. Additionally, the authors tested the catalyst stability by incorporating 0.1 wt% SiO2, which eventually hindered any change of the tetragonal crystalline phase into monoclinic one.124
Generally speaking, designing non-noble metal-based catalysts has been an attractive material for researchers owing to its low cost and abundance that adds to the sustainability of the process. To demonstrate, Ni was one of the most used active metal species.91,125–127 With this in mind, Ni/NiO showed a quantitative LA conversion and GVL yield of 100% and 99.9%, respectively, at a relatively low temperature of 120 °C and 20 bar H2.126 Other research groups considered the acidity of catalyst support as a key factor in altering the conversion and yield of LA and GVL, respectively. In this context, acid supports, SiO2, Al2O3, Y–Al2O3 doped by Ni showed promising conversions of LA in the presence of external molecular H2.128–134 A complete conversion of LA and a GVL yield of 99.2% as well as a selectivity near 99.2% were exhibited using (40 wt%) Ni/Y–Al2O3 catalyst under dioxane solvent.132 In particular (15 wt%) Ni/Al2O3 showed high conversion without using a solvent.131 It is worth mentioning that active Ni species exist in the metallic state (Ni0), which comes in agreement with XPS results governing Ni0 (2p3/2) peaks,120 proving reduced catalyst improved activity.
Equally important, Cu non-noble metal is also doped on alumina and silica supports58,97,128,135 while other groups used zirconium oxide (ZrO2) supports.58,102,136 Amongst, Cu/SiO2 catalyzed intramolecular hydrogenation of LA by utilizing FA as an H2 donor with 28:22 wt% (LA:FA) ratio despite its low LA conversion of 48% and acceptable level of GVL selectivity of 80%.97 Nevertheless, Cu/ZrO2 catalyst outperformed Cu/SiO2 and achieved 60% LA conversion and 100% GVL selectivity by consuming FA. Furthermore, the former attained complete decomposition of LA at elevated temperatures of 200 °C for 5 h.102 Zirconium active species exist in both oxide states and Zr4+ on which LA hydrogenation takes place by using alcohol as solvents as well as H2 donor (2-propanol, 2-butanol, and 2-pentanol).94,95,137–141 The synergetic effect between Ni and Cu non-noble metals over SiO2 support was investigated in the vapour phase selective hydrogenation of LA into GVL using FA. The addition of Ni into Cu/SiO2 improved the hydrogenation of LA further compared to Cu/SiO2. The highest LA conversion of 99% and GVL selectivity of 96% was attributed to the optimal loading of 1:3 Ni to Cu at 285 °C. The temperature effect was demonstrated as both LA conversion and GVL selectivity increased with the increase of temperature. The good stability of the catalyst under high temperatures near Tamman temperature of Cu and for prolonged reactions up to 200 h was due to the presence of Ni. Interestingly, the excellent catalytic activity of nanocomposite was attributed to the excellent dispersion of the metallic nanoparticles and the absence of solid solution between Ni and Cu proved by the XRD and XPS.99
Based on the information mentioned above, two mechanisms characterize the hydrogenation of LA by means of in situ H2 donated by FA. The difference between the proposed mechanisms lies in different mode of adsorption of LA over the heterogeneous catalyst, resulting in a different intermediate compound. The first approach is outlined by forming 4-hydroxyvaleric acid intermediate as a result of LA hydrogenation over the metal active sites doped onto support. Thereafter, the 4-hydroxyvaleric acid undergoes dehydration step (lactonization) over acidic sites, for instance MP-ZrO2, to form GVL.85 Whereas the alternative route comprises the inverse order, where LA experiences dehydration step first over acidic sites to produce angelina lactone, which is readily hydrogenated into GVL over metal sites later.12,142,143 A conclusive table that sums the recent work in heterogeneous catalysis reported between 2017–2021 (Table 2).
| Year | Substrate | H2 Donor | Catalyst | T (°C) | t (h) | Xa/Yb/TOFc | Recyclability | S% GVLd/ALe/VAf | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a X = conversion (%); Y = yield (%); TOF = turnover frequency (s−1) S = selectivity (%); LA = levulinic acid; GVL = γ-valerolactone; AL = angelica lactone; VA = valeric acid. | |||||||||
| 2017 | LA | FA | Cu–SiO2 | 230 | 1 | 53%a | 82%d | 96 | |
| Cu–SiO2 | 270 | 1 | 66%a | 81%d | |||||
| Cu–SiO2 | 290 | 1 | 68%a | 75%d | |||||
| 2017 | LA | FA | Triethoxy-silylpropoxy-Shvo's catalys | 90 | 12 | 90.93%a | 99.90%d | 70 | |
| 28c | |||||||||
| 2018 | LA | FA | Au–Ni/γ-Al2O3 | 190 | 2 | 89%a, 86%b | 83 | ||
| 2018 | LA | FA | 2 wt% Au/Ce0.4Zr0.6O2 | 240 | 2 | 90.8%a | • Recovery of catalyst by centrifugation, then washing with pure water and ethanol and dry at room temperature | 100%d | 82 |
| 83.5%b | |||||||||
| Au/ZrO2 | 240 | 2 | 40.7%a | 89.5%d | |||||
| 36.4%b | |||||||||
| 0.6 wt% Au/Ce0.4Zr0.6O2 | 240 | 2 | 63.5%a | 77.3%d | |||||
| 49.7%b | |||||||||
| 240 | 2 | 13.1%a | • Good stability due to tetragonal phase attributing to strong metal–support interaction | 70.2%d | |||||
| Au/CeO2 | 9.2%b | • Limited decrease in activity after 5 runs | |||||||
| 2018 | LA | FA | Calcined hydrotalcite Mg/Al (3:1) |
270 | 5 | 100%a | • Cycle I TOS = 30 h | 98%d | 101 |
| 1%e | |||||||||
| Cycle II TOS = 22 h | |||||||||
| Cycle III TOS = 14 h | |||||||||
| Cycle IV TOS = 9 h | |||||||||
| • Regeneration of active sites under flowing air after each cycle | |||||||||
| • After cycle IV, LA conversion slightly decreased to 86% | |||||||||
| • After cycle IV, GVL selectivity was constant at 98% | |||||||||
| 2018 | LA | FA (1:1) |
RANEY® Ni catalyst | 200 | 24 | 52.6%a 20.4%b | 111 | ||
| FA (1:1) |
RANEY® Ni catalyst | 200 | 48 | 62.6%a | |||||
| 24.4%b | |||||||||
| FA (1:2) |
RANEY® Ni catalyst | 200 | 48 | 90.9%a | |||||
| 60.5%b | |||||||||
| FA (1:4) |
RANEY® Ni catalyst | 200 | 48 | 100%a | |||||
| 68.5%b | |||||||||
| 2018 | LA | FA | 0.2 g Ru/C + 3 mL triethyl-amine | 160 | 3 | 87.26%a | 71 | ||
| 80.75%b | |||||||||
| 2018 | LA | FA | 2.5% Ru/ZrO2 | 150 | 12 | 73%a | • Catalyst washed with water | >99%d | 124 |
| 2.5% Ru/ZrO2 (WI) | 150 | 12 | 63%a | • Activity significantly from 73% to 54% in the second run | >99%d | ||||
| • XRD showed transformation from tetragonal to the thermodynamically stable monoclinic structure | |||||||||
| 2018 | LA | FA | Cu–SiO2 | 250 | 1 | 48%a | • Deactivation of Cu–SiO2 catalyst studied at 250 °C with LA:FA ratio = 1:2 |
80%d | 97 |
| 17%e | |||||||||
| Cu–TiO2 | 250 | 1 | 8%a | • Consistent conversion of LA (48%) and selectivity of GVL | 25%d | ||||
| 55%e | |||||||||
| Cu-ZSM-5 | 250 | 1 | 38%a | • Strong interaction between Cu with SiO2 support | 4%d | ||||
| 82%e | |||||||||
| Cu–Al2O3 | 250 | 1 | 24%a | • Insignificant coke formation of Cu–SiO2 and Cu–TiO2 catalysts | 77%d | ||||
| 15%e | |||||||||
| 2019 | LA | FA | 6 wt% Au/ZrO2 | 210 | 5 | 93.3%a | • Catalyst washed by acetone 5 times and dried at 100 °C for 2 h | 91.1%d | |
| 85%b | • Stable after 3 cycles with 69% yield of GVL | ||||||||
| Selectivity of catalyst was not affected | |||||||||
| 4 wt% Au/ZrO2 | 210 | 5 | 80.8%a | 87.1%d | 84 | ||||
| 70.4%b | |||||||||
| 1 wt% Au/ZrO2 | 210 | 5 | 13.6%a | 98.5%d | |||||
| 13.4%b | |||||||||
| 2019 | LA | FA | 1.6 Pt/MP-ZrO2 | 220 | 24 | 82%a | 85 | ||
| 84%b | |||||||||
| 1.5 Pt/MP-ZrO2 | 220 | 24 | 53%a | ||||||
| 52%b | |||||||||
| 2020 | LA | FA | CoOx | 230 | 20 | 37.3%a | • Mn2CoOx catalyst showed durable performance after 5 cycles | 85.2%d | |
| 10%f | |||||||||
| 2020 | LA | FA | MnOx | 230 | 20 | 76.7%a | • Conversion of LA fluctuated slightly around 60% from the 1st to the 5th cycle | 74.7%d | 109 |
| 12%f | |||||||||
| Mn2Co0.1Ox | 230 | 20 | 78.9%a | • GVL yield fluctuated between 84% and 87% from the 1st to the 5th cycle | 76.7%d | ||||
| 11.8%f | |||||||||
| Mn2Co0.1Ox+ 0.1 mol L−1 Na2SO4 | 230 | 20 | 83.4%a | • Selectivity of VA did not change dramatically from the 1st to the 5th cycle | 70%d | ||||
| 14.1%f | |||||||||
| 2020 | LA | FA | 1Au/ZrO2 | 240 | 1 | 66.7%a | • Recyclability of 1Au–2Ni/ZrO2 catalyst | 90.9%d | 77 |
| 73.4 %b | |||||||||
| 1Au–2Ni/ZrO2 | 240 | 1 | 87.9%a | • Spent catalyst was recovered by simple filtration | 98.7%d | ||||
| 89.1%b | |||||||||
| 0.5Au–2Ni/ZrO2 | 240 | 1 | 62.2%a | • Calcined at 300 °C for 4 h. & reduced in H2 flow at 300 °C for 1h | 98.4%d | ||||
| 63%b | |||||||||
| 2Ni/ZrO2 | 240 | 1 | 1.2%b | • GVL yield showed a slight decrease after 3 cycles | |||||
| • Good recyclability of Au–Ni/ZrO2 | |||||||||
| 2021 | LA | FA | ZnAl oxide | 140 | <6 | 87%a | • Continuous but slight decrease in LA conversion during first 4 cycles | 90%d | 112 |
| 90%b | • LA is being directly or indirectly deposited on catalytic surface | ||||||||
| • After 4th cycle, used catalyst was calcined at 600 °C, and TPO results were similar to the fresh catalyst | |||||||||
| 2021 | Glucose | FA | Ru/TiO2 | 190 | 2 | 38.98%a | 144 | ||
The challenging task of valorisation of renewable biomass into its constituents, for example biofuels, places emphasis on synthesizing heterogeneous catalysis. Ultimately, researchers aim at developing a heterogeneous catalytic system where the conversion of hemicellulose part of biomass into biofuels such as GVL in a one-pot process is achieved, while avoiding the use of liquid acid. It was reported elsewhere that a catalyst blend of ZSM-5 and Au/ZrO2 achieved a complete conversion of xylose and hemicellulose into furfural, followed by a subsequent hydrogenation into GVL at a yield of 80% in the presence of 2-propanol and without the need of external H2. The reaction was performed at 120 °C for 30 h which therefore resulted in a maintained a GVL yield not less than 65% after 4 reaction cycles of 24 h each.147
4 Catalytic hydrogenation of LA using different H2 sources
Up until this point we have put a special emphasis on FA as a sole source of H2 for hydrogenating LA over heterogeneous catalyst. A concise overview on solvents, published in literature other than FA, is presented in this section. For instance, various alcohols such as methanol,148 isopropyl alcohol,10,149 2-butanol,94,150 and ethanol150 are used as H2 sources for LA hydrogenation. The use of isopropyl alcohol marked appreciated levels of LA conversion near 99% and an attractive selectivity of GVL at 99% in the presence of 50-Ni-MMt catalyst.91 In a different study, 2-butanol and 2-isoproyl alcohol were examined using F–ZrF and UiO-66-SO3H catalysts, respectively.151 For F–ZrF, high LA conversion and GVL selectivity were obtained by 2-isopropyl alcohol in the presence of DMF, which comes in agreement with Hengne A. M. et al.91 Despite achieving a complete conversion of LA and high GVL selectivity, the addition of solvents like DMF proposes additional environmental concerns. However, exploiting the presence of FA offers a greener process by avoiding the use of hazardous solvents. In addition, the complication of using conventional high pressure H2 puts a risk when shifting into large-scale applications, making FA a safer and a cost-saving substitute.4.1 Photocatalytic hydrogenation of LA into γ-valerolactone using alcohol solvents H2 donor
On another end, the use of photo catalysis is emerging as researchers are seeking to supplement catalytic processes with sustainable green energy. That being said, exploiting light energy has shown, for example, a promising role in catalytic pyrolysis of waste rubber tires.152 Photo catalysis are of vital importance to the degradation of contaminants and antibiotics traces in waste water.153 Interestingly, higher value solvents derived from biomass such as GVL can be successfully produced from LA using niobic acid photo catalyst using UV-assisted accumulated electron transfer technique.154 Moreover, TiO2 doped with Pt is suggested as a promising catalyst that features a dual role in the production of GVL from LA and levulinate esters using alcohol as H2 source and under mild reaction conditions. In brief, the catalyst showed a high GVL yield of 93% for thermocatalytic route using 2-propanol solvent but lower yields for photocatalytic one. Nevertheless, the addition of NaOH base promoted GVL yield up to 95% using ethanol as a green solvent.155 Noble metals Pt, Pd, & Au-loaded TiO2 photo catalyst were screened by Zhang H. et al. for hydrogenation of LA into GVL in presence of isopropanol. The corresponding order of LA conversion was Pt/TiO2 < Pd/TiO2 < Au/TiO2 of 69% (69.5% GVL yield), 71% (76.4% GVL yield), and 79% (85.3% GVL yield), respectively.156 The carboxyl functionalized Zn-porphyrin on polymer support yielded 72% GVL after 16 hours under visible light.157 The list of photo catalysts used for LA hydrogenation are tabulated in Table S1 (ESI document†).5 Discussion and further research recommendations
As mentioned earlier in this paper, FA is a major key factor in many applications as it houses valuable gases; H2 and CO. Therefore, HCOOH is treated as a hydrogen or CO storage system to be used for further purposes. Two methods define explicitly how both gases are charged into FA, particularly, CO2/CO and bicarbonate approaches.158,159 Additionally, heterogeneous and homogeneous catalysts are employed to drive the reaction towards the production of HCOOH.160–162 Charged FA readily liberates either H2 or CO. In fact, HCOOH decomposition is catalytically favoured by a variety of heterogeneous catalysts. Whenever FA is accounted as H2 donor, designing a catalyst capable of maximizing dehydrogenation reaction on the expense of dehydration is preferred. Even though homogeneous catalysts favour the decomposition of FA at relatively mild conditions, its separation, and the use of solvents pronounce major setbacks. It is of high significance that possible traces of CO, as a result of FA dehydration, react with catalyst precursor, producing further active species.163 Further research could be done on this concept, which would permit the presence of CO traces in the product stream. This provides the opportunity to operate at more cost-effective conditions with no need for producing pure H2. The inclusion of benzene into the catalyst led remarkably to further improvements due to its light dissociation under light irradiation. This suggests the use of photoactive catalysts along with benzene or other dissociative compounds that would assist in FA dehydrogenation. Photoactive catalysts using Pd & Pt@TiO2 support have shown a promising activity under harsh reactions represented by waste rubber cracking.152 A comprehensive literature review about approaches for designing photoactive catalysts based on TiO2, CdS, and C3N4 for HCOOH decomposition into H2 was published elsewhere.164 Photo catalysts reported in literature were evaluated using isopropanol, ethanol, and 2-propanol as H2 donor.155–157,165 These studies should direct toward testing FA decomposition for in situ H2 production in the presence of photo catalyst.On another end, the use of heterogeneous catalysts facilitates its separation from the yielded product, and thereby the reuse of the catalyst is possible. Besides, heterogeneous catalysts suffer deactivation due to CO poisoning yielded by dehydration reaction37,38 due to elevated temperatures. For that reason, some research groups tended to manipulate the crystalline frameworks by calcination processes, which allow the synthesis of alloy, core–shell, and bimetallic structures. As shown above, an increase in Au cluster size due to thermal treatment decreased catalyst activity thereupon. Unfortunately, few studies were reported regarding the sintering effect on the active sites of heterogeneous catalysts as a result of elevated temperatures. Morphology and dispersion analysis of the active sites must be further performed using in situ SEM & TEM at different temperatures to inspect sintering and contamination deposition.
Coupled with its ability to accommodate massive energy content (5.3 MJ kg−1) as well as its low cost of production, making it an ideal fuel substitute, FA is a key compound for catalytic hydrogenation reactions. Specifically, FA is a key factor in LA hydrogenation into GVL, which undergoes successive deoxygenating reactions to yield higher alkanes (Section 3). In real applications, a pretreatment cracking step using strong acid (H2SO4, HCl) is carried out for lignocellulosic biomass and therefore heterogeneous catalysts are prone to deactivation. Different catalyst structures experience different effects of metal leaching on catalyst activity. For Cu catalysts, the leaching of metals detected through ICP-OES of the liquid products can lead to a significant reduction in catalyst activity down to 70%. Copper metal leaching was confirmed using FT-IR for investigating soluble copper complex.58 In other studies, ICP analysis showed that metals such as Fe, Ni, Cr, Co and Mo leach into the liquid product upon the hydrogenation of LA in the aqueous phase.6
Owing to the setback of metal leaching, designing a robust support is essential for improved metal–support interaction. Catalysts with carbon supports have been proved overcome the issue of leaching on the expense of catalyst regeneration.166 On the other hand, stable ZrO2 support of tetragonal phase demonstrated a stronger binding to the active Cu species (<2 ppm) than Al2O3 (174 ppm), avoiding metal leaching into the solvent.58
Further research should be employed on using either alternative acids20 or using conventional acids in appropriate quantities to avoid catalytic inhibition.66 Hence, it would be essential to perform LA catalytic hydrogenation by using FA as an H2 donor but under acidic conditions. Separating acids from the reaction mixture is a costly process. Alternatively, more attention should be given to solvents such as butene that yield immiscible levulinate esters.74 Findings achieved by Deng Li et al. suggested a promising effect of the addition of CO2 on the catalytic activity which should be investigated further as it exists as a major by-product as a result of FA dehydrogenation reaction.65 Likewise, studying the possibility of optimizing WGS reaction by injecting CO paves the road for utilizing the traces of CO existing in the product.167
Generally speaking, executing an optimized catalytic hydrogenation of LA through intramolecular H2 necessitates designing a catalytic reactor that accounts for engineering perspectives. Particularly, the assessment of heterogeneous catalysts must take into consideration heat and mass transfer as well as mass transfer limitations between the inner pore surface and the bulk biomass. That being said, designing different mixing systems is useful to obtain benchmark values of potential conversions range instead of just one conversion value for the studied catalyst. Furthermore, the produced gas could be continuously bubbled throughout the reactor in which better mixing and higher catalytic activity could be achieved. Equally important, further studies that integrate experimental results and simulations should be implemented, aiming to optimize reactor design and operating conditions. Continuous processes from engineering perspective are hardly found in literature and more efforts should be devoted to bringing pilot scale applications on table such as the work performed by Zhou et al.77 Scaling-up the process of production of valuable biomass-derived chemicals and biofuels from lignocellulosic derived LA requires considering its economic feasibility. A comprehensive techno-economic analysis performed by Patel A. et al. (2010) addresses the feasibility of producing 5-nonanone from LA and also evaluates the relative purification costs, both of which are based on discounted cash flow analysis.168 The model uses molecular H2 raw material input at 1736.07 kg h−1 of a market value 0.116 $ per kg. Indeed, shifting towards using stored H2 in FA disregards near 201 $ per h of the running cost of hydrogenation processes.168
All things considered, the most active heterogeneous catalysts comprise noble metals, which are expensive and less abundant in nature. However, some findings exhibited promising LA + FA conversion into GVL by using cheaper metals doped over acidic catalysts.120 Establishing less number of deoxygenating steps that yields valuable chemicals and/or higher liquid alkanes render them as a cost-effective catalytic route. In this regard, some developed catalysts85,120 produced higher alkanes bio-solvents beyond GVL despite the fact that they contain noble metals, putting a whole new set of development options on the table.
6 Conclusion
The interest in GVL production has been increasing not only for its distinctive properties, but also for its potential as a feedstock to produce higher-value compounds. Heterogeneous catalysts lie at the heart of biomass deoxygenating mechanisms allowing efficient LA conversion, promoted GVL yield, and competent recyclability. It has been noticed that oxides were extensively used as supports for noble metals to hydrogenate LA into GVL through FA decomposition as a sole H2 source. Nevertheless, other authors shifted their focus to non-noble metals, for example, Ni and Cu to establish more economic processes. We have seen in the last 2 years that photo catalysis has been overarching the need to exploit light energy for fulfilling the production of GVL at mild conditions, which obviously is worth further investigations.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful to express their gratitude to the Science Foundation Ireland (SFI) for funding this project under grant number 16/RC/3889.Notes and references
- N. Norouzi, M. Fani and Z. K. Ziarani, The fall of oil Age: A scenario planning approach over the last peak oil of human history by 2040, J. Petrol. Sci. Eng., 2020, 188, 106827 CrossRef CAS.
- L. Yan, Q. Yao and Y. Fu, Conversion of levulinic acid and alkyl levulinates into biofuels and high-value chemicals, Green Chem., 2017, 19(23), 5527–5547 RSC.
- M. J. Climent, A. Corma and S. Iborra, Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels, Green Chem., 2014, 16(2), 516–547 RSC.
- M. L. Observatory Trends in Atmospheric Carbon Dioxide. 2016 [cited 2016 10 April, 2016]; Available from: https://www.esrl.noaa.gov/gmd/ccgg/trends/graph.html Search PubMed.
- K. Yan, et al., Catalytic reactions of gamma-valerolactone: A platform to fuels and value-added chemicals, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS.
- D. J. Braden, et al., Production of liquid hydrocarbon fuels by catalytic conversion of biomass-derived levulinic acid, Green Chem., 2011, 13(7), 1755–1765 RSC.
- J. J. Bozell, et al., Production of levulinic acid and use as a platform chemical for derived products, Resour. Conserv. Recycl., 2000, 28(3–4), 227–239 CrossRef.
- X. Yu and P. G. Pickup, Recent advances in direct formic acid fuel cells (DFAFC), J. Power Sources, 2008, 182(1), 124–132 CrossRef CAS.
- S. N. Derle and P. A. Parikh, Hydrogenation of levulinic acid and γ-valerolactone: steps towards biofuels, Biomass Convers. Biorefin., 2014, 4(4), 293–299 CrossRef CAS.
- M. G. Al-Shaal, et al., Microwave-assisted reduction of levulinic acid with alcohols producing γ-valerolactone in the presence of a Ru/C catalyst, Catal. Commun., 2016, 75, 65–68 CrossRef CAS.
- Z.-p. Yan, L. Lin and S. Liu, Synthesis of γ-valerolactone by hydrogenation of biomass-derived levulinic acid over Ru/C catalyst, Energy Fuels, 2009, 23(8), 3853–3858 CrossRef CAS.
- W. R. Wright and R. Palkovits, Development of heterogeneous catalysts for the conversion of levulinic acid to γ-valerolactone, ChemSusChem, 2012, 5(9), 1657–1667 CrossRef CAS PubMed.
- J. Q. Bond, et al., Integrated catalytic conversion of γ-valerolactone to liquid alkenes for transportation fuels, Science, 2010, 327(5969), 1110–1114 CrossRef CAS PubMed.
- Z. Yu, et al., Heterogeneous Catalytic Hydrogenation of Levulinic Acid to γ-Valerolactone with Formic Acid as Internal Hydrogen Source, ChemSusChem, 2020, 13(11), 2916–2930 CrossRef CAS PubMed.
- F. Valentini, et al., Formic acid, a biomass-derived source of energy and hydrogen for biomass upgrading, Energy Environ. Sci., 2019, 12(9), 2646–2664 RSC.
- J. Horvat, et al., Mechanism of levulinic acid formation, Tetrahedron Lett., 1985, 26(17), 2111–2114 CrossRef CAS.
- T. Flannelly, et al., Non-stoichiometric formation of formic and levulinic acids from the hydrolysis of biomass derived hexose carbohydrates, RSC Adv., 2016, 6(7), 5797–5804 RSC.
- J. P. Lange, Lignocellulose conversion: an introduction to chemistry, process and economics, Biofuels, Bioprod. Biorefin., 2007, 1(1), 39–48 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chemical routes for the transformation of biomass into chemicals, Chem. Rev., 2007, 107(6), 2411–2502 CrossRef CAS PubMed.
- B. Girisuta, L. Janssen and H. Heeres, Green chemicals: A kinetic study on the conversion of glucose to levulinic acid, Chem. Eng. Res. Des., 2006, 84(5), 339–349 CrossRef CAS.
- B. F. Kuster and H. S. van der Baan, The influence of the initial and catalyst concentrations on the dehydration of D-fructose, Carbohydr. Res., 1977, 54(2), 165–176 CrossRef CAS.
- H. Heeres, et al., Combined dehydration/(transfer)-hydrogenation of C6-sugars (D-glucose and D-fructose) to γ-valerolactone using ruthenium catalysts, Green Chem., 2009, 11(8), 1247–1255 RSC.
- B. Girisuta, L. Janssen and H. Heeres, A kinetic study on the decomposition of 5-hydroxymethylfurfural into levulinic acid, Green Chem., 2006, 8(8), 701–709 RSC.
- B. Girisuta, L. Janssen and H. Heeres, Kinetic study on the acid-catalyzed hydrolysis of cellulose to levulinic acid, Ind. Eng. Chem. Res., 2007, 46(6), 1696–1708 CrossRef CAS.
- O. Casanova, S. Iborra and A. Corma, Chemicals from biomass: Etherification of 5-hydroxymethyl-2-furfural (HMF) into 5, 5′(oxy-bis (methylene)) bis-2-furfural (OBMF) with solid catalysts, J. Catal., 2010, 275(2), 236–242 CrossRef CAS.
- K. Arias, et al., From Biomass to Chemicals: Synthesis of Precursors of Biodegradable Surfactants from 5-Hydroxymethylfurfural, ChemSusChem, 2013, 6(1), 123–131 CrossRef CAS PubMed.
- R. Xing, W. Qi and G. W. Huber, Production of furfural and carboxylic acids from waste aqueous hemicellulose solutions from the pulp and paper and cellulosic ethanol industries, Energy Environ. Sci., 2011, 4(6), 2193–2205 RSC.
- M. Grasemann and G. Laurenczy, Formic acid as a hydrogen source–recent developments and future trends, Energy Environ. Sci., 2012, 5(8), 8171–8181 RSC.
- N. Akiya and P. E. Savage, Role of water in formic acid decomposition, AIChE J., 1998, 44(2), 405–415 CrossRef CAS.
- X. L. Du, et al., Hydrogen-Independent reductive transformation of carbohydrate biomass into γ-valerolactone and pyrrolidone derivatives with supported gold catalysts, Angew. Chem., Int. Ed., 2011, 50(34), 7815–7819 CrossRef CAS PubMed.
- A. M. Ruppert, et al., Ru catalysts for levulinic acid hydrogenation with formic acid as a hydrogen source, Green Chem., 2016, 18(7), 2014–2028 RSC.
- F. Solymosi, Importance of the electric properties of supports in the carrier effect, Catal. Rev., 1968, 1(1), 233–255 CrossRef.
- J. Trillo, G. Munuera and J. Criado, Catalytic decomposition of formic acid on metal oxides, Catal. Rev., 1972, 7(1), 51–86 CrossRef CAS.
- P. Mars, J. Scholten, and P. Zwietering, The catalytic decomposition of formic acid, in Advances in Catalysis. 1963, Elsevier. pp. 35–113 Search PubMed.
- K. Kim and M. Barteau, Pathways for carboxylic acid decomposition on titania, Langmuir, 1988, 4(4), 945–953 CrossRef CAS.
- Y. Zhao, et al., Selective decomposition of formic acid over immobilized catalysts, Energy Fuels, 2011, 25(8), 3693–3697 CrossRef CAS.
- D. Ruthven and R. Upadhye, The catalytic decomposition of aqueous formic acid over suspended palladium catalysts, J. Catal., 1971, 21(1), 39–47 CrossRef CAS.
- X. Zhou, et al., High-quality hydrogen from the catalyzed decomposition of formic acid by Pd–Au/C and Pd–Ag/C, Chem. Commun., 2008,(30), 3540–3542 RSC.
- Y. Huang, et al., Novel PdAu@ Au/C core− shell catalyst: superior activity and selectivity in formic acid decomposition for hydrogen generation, Chem. Mater., 2010, 22(18), 5122–5128 CrossRef CAS.
- K. Tedsree, et al., Hydrogen production from formic acid decomposition at room temperature using a Ag–Pd core–shell nanocatalyst, Nat. Nanotechnol., 2011, 6(5), 302 CrossRef CAS PubMed.
- N. Toshima, et al., Catalytic activity and structural analysis of polymer-protected gold-palladium bimetallic clusters prepared by the simultaneous reduction of hydrogen tetrachloroaurate and palladium dichloride, J. Phys. Chem., 1992, 96(24), 9927–9933 CrossRef CAS.
- Y. Shiraishi, D. Ikenaga and N. Toshima, Preparation and catalysis of inverted core/shell structured Pd/Au bimetallic nanoparticles, Aust. J. Chem., 2003, 56(10), 1025–1029 CrossRef CAS.
- A. Herzing, et al., Microstructural development and catalytic performance of Au− Pd nanoparticles on Al2O3 supports: the effect of heat treatment temperature and atmosphere, Chem. Mater., 2008, 20(4), 1492–1501 CrossRef CAS.
- D. Ferrer, et al., Three-layer core/shell structure in Au−Pd bimetallic nanoparticles, Nano Lett., 2007, 7(6), 1701–1705 CrossRef CAS PubMed.
- T. Schmidt, et al., On the CO tolerance of novel colloidal PdAu/carbon electrocatalysts, J. Electroanal. Chem., 2001, 501(1–2), 132–140 CrossRef CAS.
- D. A. Bulushev, S. Beloshapkin and J. R. Ross, Hydrogen from formic acid decomposition over Pd and Au catalysts, Catal. Today, 2010, 154(1–2), 7–12 CrossRef CAS.
- F. Solymosi and A. Erdöhelyi, Decomposition of formic acid on supported Rh catalysts, J. Catal., 1985, 91(2), 327–337 CrossRef CAS.
- M. A. Henderson, Complexity in the decomposition of formic acid on the TiO2 (110) surface, J. Phys. Chem. B, 1997, 101(2), 221–229 CrossRef CAS.
- W. Sachtler and J. Fahrenfort. The catalytic decomposition of formic acid vapour on metals. in Proceedings of the 2nd International Congress on Catalysis, Paris, France. 1960 Search PubMed.
- M. Ojeda and E. Iglesia, Formic acid dehydrogenation on Au-based catalysts at near-ambient temperatures, Angew. Chem., 2009, 121(26), 4894–4897 CrossRef.
- A. Gazsi, T. Bánsági and F. Solymosi, Decomposition and reforming of formic acid on supported Au catalysts: Production of CO-free H2, J. Phys. Chem. C, 2011, 115(31), 15459–15466 CrossRef CAS.
- P. Xu, F. D. Bernal-Juan and L. Lefferts, Effect of oxygen on formic acid decomposition over Pd catalyst, J. Catal., 2020, 342–352 Search PubMed.
- O. Sneka-Płatek, et al., Understanding the influence of the composition of the AgPd catalysts on the selective formic acid decomposition and subsequent levulinic acid hydrogenation, Int. J. Hydrogen Energy, 2020, 45(35), 17339–17353 CrossRef.
- J. r. Eppinger and K.-W. Huang, Formic acid as a hydrogen energy carrier, ACS Energy Lett., 2017, 2(1), 188–195 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Technology development for the production of biobased products from biorefinery carbohydrates—the US Department of Energy's “Top 10” revisited, Green Chem., 2010, 12(4), 539–554 RSC.
- A. Villa, et al., Acid-functionalized mesoporous carbon: An efficient support for ruthenium-catalyzed γ-valerolactone production, ChemSusChem, 2015, 8(15), 2520–2528 CrossRef CAS PubMed.
- W. Luo, P. C. Bruijnincx and B. M. Weckhuysen, Selective, one-pot catalytic conversion of levulinic acid to pentanoic acid over Ru/H-ZSM5, J. Catal., 2014, 320, 33–41 CrossRef CAS.
- A. M. Hengne and C. V. Rode, Cu–ZrO 2 nanocomposite catalyst for selective hydrogenation of levulinic acid and its ester to γ-valerolactone, Green Chem., 2012, 14(4), 1064–1072 RSC.
- M. Felderhoff, et al., Hydrogen storage: the remaining scientific and technological challenges, Phys. Chem. Chem. Phys., 2007, 9(21), 2643–2653 RSC.
- J. Alazemi and J. Andrews, Automotive hydrogen fuelling stations: An international review, Renew. Sustain. Energy Rev., 2015, 48, 483–499 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Converting carbohydrates to bulk chemicals and fine chemicals over heterogeneous catalysts, Green Chem., 2011, 13(3), 520–540 RSC.
- G. W. Huber, S. Iborra and A. Corma, Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering, Chem. Rev., 2006, 106(9), 4044–4098 CrossRef CAS PubMed.
- L. E. Manzer, Biomass derivatives: a sustainable source of chemicals, ACS Publications, 2006 Search PubMed.
- D. J. Hayes, et al., The biofine process–production of levulinic acid, furfural, and formic acid from lignocellulosic feedstocks, Biorefineries: Ind. Processes Prod., 2006, 1, 139–164 CAS.
- L. Deng, et al., Catalytic conversion of biomass-derived carbohydrates into γ-valerolactone without using an external H2 supply, Angew. Chem., Int. Ed., 2009, 48(35), 6529–6532 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, et al., Conversion of cellulose to hydrocarbon fuels by progressive removal of oxygen, Appl. Catal., B, 2010, 100(1), 184–189 CrossRef CAS.
- M. Osada, et al., Effect of sulfur on catalytic gasification of lignin in supercritical water, Energy Fuel., 2007, 21(3), 1400–1405 CrossRef CAS.
- T. Miyazawa, et al., Glycerol hydrogenolysis to 1, 2-propanediol catalyzed by a heat-resistant ion-exchange resin combined with Ru/C, Appl. Catal., A, 2007, 329, 30–35 CrossRef CAS.
- L. Deng, et al., Conversion of levulinic acid and formic acid into γ-valerolactone over heterogeneous catalysts, ChemSusChem, 2010, 3(10), 1172–1175 CrossRef CAS PubMed.
- D. He and I. T. Horváth, Application of silica-supported Shvo's catalysts for transfer hydrogenation of levulinic acid with formic acid, J. Organomet. Chem., 2017, 847, 263–269 CrossRef CAS.
- J. Feng, et al., Production of γ-valerolactone from levulinic acid over a Ru/C catalyst using formic acid as the sole hydrogen source, Sci. Total Environ., 2018, 633, 426–432 CrossRef CAS PubMed.
- M. Osada, et al., Subcritical water regeneration of supported ruthenium catalyst poisoned by sulfur, Energy Fuel., 2008, 22(2), 845–849 CrossRef CAS.
- T. Marzialetti, V. Olarte, C. W. Jones, C. Sievers, T. J. C. Hoskins, P. K. Agrawal and C. W. Jones, Ind. Eng. Chem. Res., 2008, 47(19), 7131–7140 CrossRef CAS.
- E. I. Gürbüz, et al., Reactive Extraction of Levulinate Esters and Conversion to γ-Valerolactone for Production of Liquid Fuels, ChemSusChem, 2011, 4(3), 357–361 CrossRef PubMed.
- H. Jiang, et al., Effect of palladium on sulfur resistance in Pt–Pd bimetallic catalysts, Catal. Today, 2007, 125(3–4), 282–290 CrossRef CAS.
- K. Okamoto, et al., Formation of nanoarchitectures including subnanometer palladium clusters and their use as highly active catalysts, J. Am. Chem. Soc., 2005, 127(7), 2125–2135 CrossRef CAS PubMed.
- C. Zhou, et al., γ-Valerolactone Production from Furfural Residue with Formic Acid as the Sole Hydrogen Resource via an Integrated Strategy on Au-Ni/ZrO2, Ind. Eng. Chem. Res., 2020, 59(39), 17228–17238 CrossRef CAS.
- D. Kopetzki and M. Antonietti, Transfer hydrogenation of levulinic acid under hydrothermal conditions catalyzed by sulfate as a temperature-switchable base, Green Chem., 2010, 12(4), 656–660 RSC.
- M. Siskin and A. R. Katritzky, Reactivity of organic compounds in superheated water: General background, Chem. Rev., 2001, 101(4), 825–836 CrossRef CAS PubMed.
- H. Li, et al., One-pot transformation of polysaccharides via multi-catalytic processes, Catal. Sci. Technol., 2014, 4(12), 4138–4168 RSC.
- M. Boudart and G. Djéga-Mariadassou, Kinetics of heterogeneous catalytic reactions, Princeton University Press. 2014 Search PubMed.
- X. Li, et al., The Promoting Effect of Ce on the Performance of Au/CexZr1−xO2 for γ-Valerolactone Production from Biomass-Based Levulinic Acid and Formic Acid, Catalysts, 2018, 8(6), 241 CrossRef.
- A. M. Ruppert, et al., Supported gold–nickel nano-alloy as a highly efficient catalyst in levulinic acid hydrogenation with formic acid as an internal hydrogen source, Catal. Sci. Technol., 2018, 8(17), 4318–4331 RSC.
- P. A. Son, D. H. Hoang and K. T. Canh, The role of gold nanoparticles on different supports for the in-air conversion of levulinic acid into γ-valerolactone with formic acid as an alternative hydrogen source, Russ. J. Appl. Chem., 2019, 92(9), 1316–1323 CrossRef CAS.
- M. Al-Naji, et al., Aqueous-phase hydrogenation of levulinic acid using formic acid as a sustainable reducing agent over Pt catalysts supported on mesoporous zirconia, ACS Sustainable Chem. Eng., 2019, 8(1), 393–402 CrossRef.
- W. Luo, et al., Ruthenium-catalyzed hydrogenation of levulinic acid: Influence of the support and solvent on catalyst selectivity and stability, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- J. Ftouni, et al., ZrO2 is preferred over TiO2 as support for the Ru-catalyzed hydrogenation of levulinic acid to γ-valerolactone, ACS Catal., 2016, 6(8), 5462–5472 CrossRef CAS.
- A. Hijazi, et al., Pyrolysis of waste rubber tires with palladium doped zeolite, J. Environ. Chem. Eng., 2019, 7(6), 103451 CrossRef CAS.
- Y. Lu, et al., Hydrogenation of levulinic acid to γ-valerolactone over bifunctional Ru/(AlO)(ZrO) n catalyst: Effective control of Lewis acidity and surface synergy, Mol. Catal., 2020, 493, 111097 CrossRef CAS.
- Z. Yang, et al., RANEY® Ni catalyzed transfer hydrogenation of levulinate esters to γ-valerolactone at room temperature, Chem. Commun., 2013, 49(46), 5328–5330 RSC.
- A. Hengne, et al., Transfer hydrogenation of biomass-derived levulinic acid to γ-valerolactone over supported Ni catalysts, RSC Adv., 2016, 6(64), 59753–59761 RSC.
- X. Tang, et al., Conversion of biomass to γ-valerolactone by catalytic transfer hydrogenation of ethyl levulinate over metal hydroxides, Appl. Catal., B, 2014, 147, 827–834 CrossRef CAS.
- J. Song, et al., A new porous Zr-containing catalyst with a phenate group: an efficient catalyst for the catalytic transfer hydrogenation of ethyl levulinate to γ-valerolactone, Green Chem., 2015, 17(3), 1626–1632 RSC.
- Y. Kuwahara, H. Kango and H. Yamashita, Catalytic transfer hydrogenation of biomass-derived levulinic acid and its esters to γ-valerolactone over sulfonic acid-functionalized UiO-66, ACS Sustainable Chem. Eng., 2017, 5(1), 1141–1152 CrossRef CAS.
- M. Chia and J. A. Dumesic, Liquid-phase catalytic transfer hydrogenation and cyclization of levulinic acid and its esters to γ-valerolactone over metal oxide catalysts, Chem. Commun., 2011, 47(44), 12233–12235 RSC.
- S. Lomate, A. Sultana and T. Fujitani, Effect of SiO 2 support properties on the performance of Cu–SiO 2 catalysts for the hydrogenation of levulinic acid to gamma valerolactone using formic acid as a hydrogen source, Catal. Sci. Technol., 2017, 7(14), 3073–3083 RSC.
- S. Lomate, A. Sultana and T. Fujitani, Vapor phase catalytic transfer hydrogenation (CTH) of levulinic acid to γ-valerolactone over copper supported catalysts using formic acid as hydrogen source, Catal. Lett., 2018, 148(1), 348–358 CrossRef CAS.
- M. Ashokraju, et al., Formic acid assisted hydrogenation of levulinic acid to $$\upgamma $$ γ-valerolactone over ordered mesoporous $$\hbox {Cu/Fe} _ {2}\hbox {O} _ {3} $$ Cu/Fe 2 O 3 catalyst prepared by hard template method, J. Chem. Sci., 2018, 130(2), 1–8 CrossRef CAS.
- P. P. Upare, et al., Nickel-promoted copper–silica nanocomposite catalysts for hydrogenation of levulinic acid to lactones using formic acid as a hydrogen feeder, Appl. Catal., A, 2015, 491, 127–135 CrossRef CAS.
- M. Varkolu, et al., Gas phase hydrogenation of levulinic acid to γ-valerolactone over supported Ni catalysts with formic acid as hydrogen source, New J. Chem., 2016, 40(4), 3261–3267 RSC.
- S. Hussain, et al., Synthesis of γ-Valerolactone from Levulinic Acid and Formic Acid over Mg-Al Hydrotalcite Like Compound, ChemistrySelect, 2018, 3(22), 6186–6194 CrossRef CAS.
- J. Yuan, et al., Copper-based catalysts for the efficient conversion of carbohydrate biomass into γ-valerolactone in the absence of externally added hydrogen, Energy Environ. Sci., 2013, 6(11), 3308–3313 RSC.
- H. Guo, et al., Hydrogen gas-free processes for single-step preparation of transition-metal bifunctional catalysts and one-pot γ-valerolactone synthesis in supercritical CO2-ionic liquid systems, J. Supercrit. Fluids, 2019, 147, 263–270 CrossRef CAS.
- S. Gundekari and K. Srinivasan, Screening of Solvents, Hydrogen Source, and Investigation of Reaction Mechanism for the Hydrocyclisation of Levulinic Acid to γ-Valerolactone Using Ni/SiO 2–Al 2 O 3 Catalyst, Catal. Lett., 2019, 149(1), 215–227 CrossRef CAS.
- M. Varkolu, et al., Hydrogenation of levulinic acid using formic acid as a hydrogen source over Ni/SiO2 catalysts, Chem. Eng. Technol., 2017, 40(4), 719–726 CrossRef CAS.
- G. Metzker and A. C. Burtoloso, Conversion of levulinic acid into γ-valerolactone using Fe 3 (CO) 12: mimicking a biorefinery setting by exploiting crude liquors from biomass acid hydrolysis, Chem. Commun., 2015, 51(75), 14199–14202 RSC.
- R. R. Gowda and E. Y. X. Chen, Recyclable Earth-Abundant Metal Nanoparticle Catalysts for Selective Transfer Hydrogenation of Levulinic Acid to Produce γ-Valerolactone, ChemSusChem, 2016, 9(2), 181–185 CrossRef CAS PubMed.
- A. Yepez, et al., Microwave-assisted conversion of levulinic acid to γ-valerolactone using low-loaded supported iron oxide nanoparticles on porous silicates, Appl. Sci., 2015, 5(3), 532–543 CrossRef CAS.
- J. Wang, et al., Lattice distorted MnCo oxide materials as efficient catalysts for transfer hydrogenation of levulinic acid using formic acid as H-donor, Chem. Eng. Sci., 2020, 222, 115721 CrossRef CAS.
- A. Serrà, et al., Electrodeposited Ni-Rich Ni–Pt Mesoporous Nanowires for Selective and Efficient Formic Acid-Assisted Hydrogenation of Levulinic Acid to γ-Valerolactone, Langmuir, 2021, 37(15), 4666–4677 CrossRef PubMed.
- A. S. Amarasekara, et al., Conversion of levulinic acid and cellulose to γ-valerolactone over Raney-Ni catalyst using formic acid as a hydrogen donor, Biofuels, 2021, 12, 423–427 CrossRef CAS.
- M. N. Belguendouz, et al., Selective synthesis of γ-valerolactone from levulinic and formic acid over ZnAl mixed oxide, Chem. Eng. J., 2021, 414, 128902 CrossRef CAS.
- V. K. Velisoju, et al., Hydrodeoxygenation activity of W modified Ni/H-ZSM-5 catalyst for single step conversion of levulinic acid to pentanoic acid: An insight on the reaction mechanism and structure activity relationship, Appl. Catal., A, 2018, 550, 142–150 CrossRef CAS.
- A. Wang, et al., Selective Production of γ-Valerolactone and Valeric Acid in One-Pot Bifunctional Metal Catalysts, ChemistrySelect, 2018, 3(4), 1097–1101 CrossRef CAS.
- W. Li, et al., Role of surface cooperative effect in copper catalysts toward highly selective synthesis of valeric biofuels, ACS Sustainable Chem. Eng., 2017, 5(3), 2282–2291 CrossRef CAS.
- P. Sun, et al., Stabilization of cobalt catalysts by embedment for efficient production of valeric biofuel, ACS Catal., 2014, 4(11), 4136–4142 CrossRef CAS.
- P. Sun, et al., Acidity-regulation for enhancing the stability of Ni/HZSM-5 catalyst for valeric biofuel production, Appl. Catal., B, 2016, 189, 19–25 CrossRef CAS.
- J. P. Lange, et al., Valeric biofuels: a platform of cellulosic transportation fuels, Angew. Chem., Int. Ed., 2010, 49(26), 4479–4483 CrossRef CAS PubMed.
- J. Q. Bond, et al., Interconversion between γ-valerolactone and pentenoic acid combined with decarboxylation to form butene over silica/alumina, J. Catal., 2011, 281(2), 290–299 CrossRef CAS.
- Z. Yi, et al., Metal regulating the highly selective synthesis of gamma-valerolactone and valeric biofuels from biomass-derived levulinic acid, Fuel, 2020, 259, 116208 CrossRef CAS.
- W. Li, et al., Efficient and Sustainable Hydrogenation of Levulinic Acid to γ-Valerolactone in Aqueous Phase over Ru/MCM-49 Catalysts, Ind. Eng. Chem. Res., 2020, 59(39), 17338–17347 CrossRef CAS.
- X. Bai, et al., A Ag–ZrO 2–graphene oxide nanocomposite as a metal-leaching-resistant catalyst for the aqueous-phase hydrogenation of levulinic acid into gamma-valerolactone, New J. Chem., 2020, 44(38), 16526–16536 RSC.
- J. Li, et al., Construction of mesoporous Cu/ZrO2-Al2O3 as a ternary catalyst for efficient synthesis of γ-valerolactone from levulinic acid at low temperature, J. Catal., 2020, 381, 163–174 CrossRef CAS.
- Y. Gao, et al., Ru/ZrO2 catalysts for transfer hydrogenation of levulinic acid with formic acid/formate mixtures: importance of support stability, ChemistrySelect, 2018, 3(5), 1343–1351 CrossRef CAS.
- D. Liu, et al., One-step fabrication of Ni-embedded hierarchically-porous carbon microspheres for levulinic acid hydrogenation, Chem. Eng. J., 2019, 369, 386–393 CrossRef CAS.
- S. Song, et al., Heterostructured Ni/NiO composite as a robust catalyst for the hydrogenation of levulinic acid to γ-valerolactone, Appl. Catal., B, 2017, 217, 115–124 CrossRef CAS.
- V. V. Kumar, et al., Role of Brønsted and Lewis acid sites on Ni/TiO2 catalyst for vapour phase hydrogenation of levulinic acid: Kinetic and mechanistic study, Appl. Catal., A, 2015, 505, 217–223 CrossRef CAS.
- I. Obregón, et al., Levulinic acid hydrogenolysis on Al2O3-based Ni-Cu bimetallic catalysts, Chin. J. Catal., 2014, 35(5), 656–662 CrossRef.
- K. Jiang, et al., Hydrogenation of levulinic acid to γ-valerolactone in dioxane over mixed MgO–Al2O3 supported Ni catalyst, Catal. Today, 2016, 274, 55–59 CrossRef CAS.
- J. Lv, et al., Highly efficient conversion of biomass-derived levulinic acid into γ-valerolactone over Ni/MgO catalyst, RSC Adv., 2015, 5(88), 72037–72045 RSC.
- K. Hengst, et al., Synthesis of γ-valerolactone by hydrogenation of levulinic acid over supported nickel catalysts, Appl. Catal., A, 2015, 502, 18–26 CrossRef CAS.
- J. Fu, D. Sheng and X. Lu, Hydrogenation of levulinic acid over nickel catalysts supported on aluminum oxide to prepare γ-valerolactone, Catalysts, 2016, 6(1), 6 CrossRef.
- V. Mohan, et al., Vapour phase hydrocyclisation of levulinic acid to γ-valerolactone over supported Ni catalysts, Catal. Sci. Technol., 2014, 4(5), 1253–1259 RSC.
- V. V. Kumar, et al., An investigation on the influence of support type for Ni catalysed vapour phase hydrogenation of aqueous levulinic acid to γ-valerolactone, RSC Adv., 2016, 6(12), 9872–9879 RSC.
- L. Zhang, et al., Hydrogenation of levulinic acid into gamma-valerolactone over in situ reduced CuAg bimetallic catalyst: Strategy and mechanism of preventing Cu leaching, Appl. Catal., B, 2018, 232, 1–10 CrossRef CAS.
- Q. Xu, et al., Supported copper catalysts for highly efficient hydrogenation of biomass-derived levulinic acid and γ-valerolactone, Green Chem., 2016, 18(5), 1287–1294 RSC.
- L. Bui, et al., Domino reaction catalyzed by zeolites with Brønsted and Lewis acid sites for the production of γ-valerolactone from furfural, Angew. Chem., 2013, 125(31), 8180–8183 CrossRef.
- J. Wang, S. Jaenicke and G.-K. Chuah, Zirconium–Beta zeolite as a robust catalyst for the transformation of levulinic acid to γ-valerolactone via Meerwein–Ponndorf–Verley reduction, RSC Adv., 2014, 4(26), 13481–13489 RSC.
- S. S. Enumula, et al., ZrO 2/SBA-15 as an efficient catalyst for the production of γ-valerolactone from biomass-derived levulinic acid in the vapour phase at atmospheric pressure, RSC Adv., 2016, 6(24), 20230–20239 RSC.
- J. Song, et al., Porous zirconium–phytic acid hybrid: a highly efficient catalyst for Meerwein–Ponndorf–Verley reductions, Angew. Chem., Int. Ed., 2015, 54(32), 9399–9403 CrossRef CAS PubMed.
- X. Tang, et al., In situ generated catalyst system to convert biomass-derived levulinic acid to γ-valerolactone, ChemCatChem, 2015, 7(8), 1372–1379 CrossRef CAS.
- K. Yan, et al., Production and catalytic transformation of levulinic acid: A platform for speciality chemicals and fuels, Renew. Sustain. Energy Rev., 2015, 51, 986–997 CrossRef CAS.
- Z. Yu, et al., Transformation of levulinic acid to valeric biofuels: A review on heterogeneous bifunctional catalytic systems, ChemSusChem, 2019, 12(17), 3915–3930 CrossRef CAS PubMed.
- Y. Liu, et al., Conversion of glucose to levulinic acid and upgradation to γ-valerolactone on Ru/TiO 2 catalysts, New J. Chem., 2021, 45(32), 14406–14413 RSC.
- J. Cui, et al., Direct conversion of carbohydrates to γ-valerolactone facilitated by a solvent effect, Green Chem., 2015, 17(5), 3084–3089 RSC.
- J. Delgado, et al., Kinetic model assessment for the synthesis of γ-valerolactone from n-butyl levulinate and levulinic acid hydrogenation over the synergy effect of dual catalysts Ru/C and Amberlite IR-120, Chem. Eng. J., 2022, 430, 133053 CrossRef CAS.
- S. Zhu, et al., Integrated conversion of hemicellulose and furfural into γ-valerolactone over Au/ZrO2 catalyst combined with ZSM-5, ACS Catal., 2016, 6(3), 2035–2042 CrossRef CAS.
- R. Zhang, et al., Exploring to direct the reaction pathway for hydrogenation of levulinic acid into γ-valerolactone for future Clean-Energy Vehicles over a magnetic Cu-Ni catalyst, Int. J. Hydrogen Energy, 2017, 42(40), 25185–25194 CrossRef CAS.
- Z. Xue, et al., Zirconium–cyanuric acid coordination polymer: highly efficient catalyst for conversion of levulinic acid to γ-valerolactone, Catal. Sci. Technol., 2016, 6(14), 5374–5379 RSC.
- A. S. Amarasekara and M. A. Hasan, Pd/C catalyzed conversion of levulinic acid to γ-valerolactone using alcohol as a hydrogen donor under microwave conditions, Catal. Commun., 2015, 60, 5–7 CrossRef CAS.
- W.-C. Yun, M.-T. Yang and K.-Y. A. Lin, Water-born zirconium-based metal organic frameworks as green and effective catalysts for catalytic transfer hydrogenation of levulinic acid to γ-valerolactone: critical roles of modulators, J. Colloid Interface Sci., 2019, 543, 52–63 CrossRef CAS PubMed.
- A. Hijazi, et al., Solar pyrolysis of waste rubber tires using photoactive catalysts, Waste Manag., 2018, 77, 10–21 CrossRef CAS PubMed.
- M. Berkani, et al., Photocatalytic degradation of Penicillin G in aqueous solutions: Kinetic, degradation pathway, and microbioassays assessment, J. Hazard. Mater., 2022, 421, 126719 CrossRef CAS PubMed.
- B. José Filho, et al., A promising approach to transform levulinic acid into γ-valerolactone using niobic acid photocatalyst and the accumulated electron transfer technique, Appl. Catal., B, 2020, 119814 Search PubMed.
- A. Bunrit, et al., Photo–Thermo-Dual Catalysis of Levulinic Acid and Levulinate Ester to γ-Valerolactone, ACS Catal., 2022, 12, 1677–1685 CrossRef CAS.
- H. Zhang, et al., Hydrogenative cyclization of levulinic acid into γ-valerolactone by photocatalytic intermolecular hydrogen transfer, Green Chem., 2016, 18(8), 2296–2301 RSC.
- V. B. Khajone, et al., Recyclable polymer-supported carboxyl functionalized Zn–porphyrin photocatalyst for transfer hydrogenation of levulinic acid to γ-valerolactone, Biomass Convers. Biorefin., 2021, 1–11 Search PubMed.
- B. N. Pattanaik, The Advances In Processes And Catalysts For The Production Of Methyl Formate By Methanol Carbonylation–A Review, Petrochem. Technol., 2013, 3(2), 55–70 Search PubMed.
- K. Müller, K. Brooks and T. Autrey, Hydrogen storage in formic acid: a comparison of process options, Energy Fuels, 2017, 31(11), 12603–12611 CrossRef.
- H. Wiener, et al., The heterogeneous catalytic hydrogenation of bicarbonate to formate in aqueous solutions, J. Catal., 1988, 110(1), 184–190 CrossRef CAS.
- K. Koh, et al., Novel nanoporous N-doped carbon-supported ultrasmall Pd nanoparticles: Efficient catalysts for hydrogen storage and release, Appl. Catal., B, 2017, 203, 820–828 CrossRef CAS.
- Q. Y. Bi, et al., An Aqueous Rechargeable Formate-Based Hydrogen Battery Driven by Heterogeneous Pd Catalysis, Angew. Chem., Int. Ed., 2014, 53(49), 13583–13587 CrossRef CAS PubMed.
- M. Czaun, et al., Hydrogen generation from formic acid decomposition by ruthenium carbonyl complexes. Tetraruthenium dodecacarbonyl tetrahydride as an active intermediate, ChemSusChem, 2011, 4(9), 1241–1248 CrossRef CAS PubMed.
- M. Navlani-García, et al., Photocatalytic Approaches for Hydrogen Production via Formic Acid Decomposition, Heterog. Photocatal., 2020, 193–223 Search PubMed.
- B. Jose Filho, et al., A promising approach to transform levulinic acid into γ-valerolactone using niobic acid photocatalyst and the accumulated electron transfer technique, Appl. Catal., B, 2021, 285, 119814 CrossRef.
- J. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., 2010, 122, 4581–4585 CrossRef.
- L. Deng, et al., Catalytic conversion of biomass-derived carbohydrates into γ-valerolactone without using an external H2 supply, Angew. Chem., 2009, 121(35), 6651–6654 CrossRef.
- A. D. Patel, et al., Techno-economic analysis of 5-nonanone production from levulinic acid, Chem. Eng. J., 2010, 160(1), 311–321 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01379g |
| This journal is © The Royal Society of Chemistry 2022 |