
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel deep eutectic solvent-based liquid phase microextraction for the extraction of estrogenic compounds from environmental samples
Nazir Fattahi *a,
Mojtaba Shamsipurb,
Ziba Nematifara,
Nasrin Babajanib,
Masoud Moradia,
Shahin Soltania and
Shahram Akbaria
*a,
Mojtaba Shamsipurb,
Ziba Nematifara,
Nasrin Babajanib,
Masoud Moradia,
Shahin Soltania and
Shahram Akbaria
aResearch Center for Environmental Determinants of Health (RCEDH), Health Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran. E-mail: nazirfatahi@yahoo.com; Fax: +988338263048; Tel: +989183364311
bDepartment of Analytical Chemistry, Faculty of Chemistry, Razi University, Kermanshah, Iran
First published on 13th May 2022
Abstract
Steroid hormones, such as estrone (E1), 17β-estradiol (E2), 17β-ethinylestradiol (EE2) and estriol (E3) are a group of lipophilic active substances, synthesized biologically from cholesterol or chemically. A pH-switchable hydrophobic deep eutectic solvent-based liquid phase microextraction (DES-LPME) technique was established and combined with gas chromatography-mass spectroscopy for the determination of estrogenic compounds in environmental water and wastewater samples. A DES was synthesized using l-menthol as HBA and (1S)-(+)-camphor-10-sulfonic acid (CSA) as HBD, and used as a green extraction solvent. By adjusting the pH of the solution, the unique behavior of the DES in the phase transition and extraction of the desired analytes was investigated. The homogenization process of the mixture is done only by manual shaking in less than 30 seconds and the phase separation is done only by changing the pH and without centrifugation. Some effective parameters on the extraction and derivatization, such as molar ratio of DES components, DES volume, KOH concentration, HCl volume, salt addition, extraction and derivatization time and derivatization prior or after extraction were studied and optimized. Under the optimum conditions, relative standard deviation (RSD) values for intra-day and inter-day of the method based on 7 replicate measurements of 20 ng L−1 of estrogenic compounds and 10 ng L−1 for internal standard in different samples were in the range of 2.2–4.6% and 3.9–5.7%, respectively. The calibration graphs were linear in the range of 0.5–100 ng L−1 and the limits of detection (LODs) were in the range of 0.2–1.0 ng L−1. The relative recoveries of environmental water and wastewater samples which have been spiked with different levels of target compounds were 91.0–108.8%.
1 Introduction
Estrogen hormones are a class of endocrine disrupting compounds (EDCs) that, when released into the environment, contaminate water, disrupt ecosystem stability, and endanger human health.1 Cholesterol-derived steroid hormones have a four-ring molecular structure and these four rings include a phenolic ring, two hexamethylenediamine rings and a cyclopentane ring. Estrogens exist naturally and artificially and are a large group of environmental pollutants that are mostly known for their negative effects on aquatic ecosystems. Most of the estrogens released into the environment are the estrone (E1) and 17-β-estradiol (E2), which are excreted by all humans and animals.2 Other estrogens that are released into the environment to a lesser extent include estriol (E3), 17-α-ethinyl estradiol (EE2), and mestranol. Among the above compounds, EE2 and E2 are the most important estrogenic compounds, followed by E1 and E3.3,4E1 is one of the sex hormones in the estrogen group in women, which is secreted by the ovaries, has low blood levels, and is not one of the main estrogenic hormones.5 E1 is less important than other estrogens, but in women, it can cause breast cancer, breast pain and tenderness, nausea, headache and high blood pressure. The main role of this hormone is in reproductive tissues such as uterus and breast.6 E2 is one of the major sex hormones in women and is prescribed as a medicine for estrogen deficiency and other diseases. Although the main role of this hormone is in reproductive tissues such as the uterus, but it also affects other tissues such as bone, liver, blood vessels and brain. E2 is produced in very small amounts in men from testosterone metabolism.7 E3 is an important female sex hormone from the estrogen group, plays an important role in pregnancy, and is most produced by the placenta. E3 also has therapeutic applications and is produced from the metabolism of two estrogens, E1 and E2.8 Synthetic estrogen EE2, which is widely used as a contraceptive, can bind to estrogen receptors and interfere with the development of natural biological responses. It inhibits the growth of hormone-sensitive tissue in advanced and non-surgical prostate cancer in men and breast cancer in postmenopausal women. EE2 is converted to E2 by the addition of an ethinyl group, and the presence of this additional functional group makes the compound resistant to biodegradation.9 The concentration of these compounds varies in environmental matrices in different regions. A review article that compiled the results of previous research reported the concentrations of E1, E2, E3 and EE2 in the range of 2.4–670, 4–150, 23–660 and 0.3–14.4 ng L−1, respectively.10 The method of determining the amount of steroid hormones in a variety of samples, despite the development of several methods for measuring organic compounds in small amounts, is still being studied. Most research has been done to measure these pollutants in complex environmental matrices by methods such as high performance liquid chromatography (HPLC),11–13 gas chromatography (GC)14–16 and gas chromatography-mass spectrometry (GC-MS).17–19 The GC-MS method has been used more in recent years due to its better resolution and higher detection power.
Due to the very low concentration of estrogens in the environmental samples and the complexity of the wastewater matrices, an extraction/preconcentration method is required before analysis with GC-MS. Dispersive liquid–liquid microextraction based on solidification of floating organic droplet (DLLME–SFOD),20,21 dispersive liquid–liquid microextraction (DLLME),22–24 solid-phase microextraction (SPME),25,26 stir bar sorptive extraction (SBSE),27 liquid–liquid extraction (LLE)28 and solid-phase extraction (SPE)29 have been employed for the determination of estrogens from different samples. Advantages and disadvantages of these methods have already been discussed.30,31
DLLME is the latest version of liquid phase microextraction, first introduced by Dr Asadi et al. In 2006.32 This method, despite its many advantages over other microextraction methods, has disadvantages such as high consumption of disperser solvent and the use of toxic and harmful organic solvents as extraction solvent. So far, many developments have been made to DLLME to solve these problems. Vertex, ultrasonic, pH and temperature changes are methods that have replaced disperser solvents.33–35 Also, lighter-than-water organic solvents (less toxic and cheaper), ionic liquids, and deep eutectic solvents have replaced toxic organic solvents as extractants.36–39
In recent years deep eutectic solvents (DESs) have rapidly emerged as a new type of green and sustainable solvent.40,41 A DES consists of a hydrogen bond acceptor (HBA) and a hydrogen bond donor (HBD). Compared to ionic liquids, DESs are ecologically friendly, biodegradable, inexpensive, non-toxic and easy to produce. In addition, the physical and chemical properties of DESs can be adjusted by changing the types and ratios of used HBAs and HBDs. Generally, at first most DESs are hydrophilic, as they consist of hydrophilic HBDs and HBAs and their application to hydrophobic compounds, is restricted. One of the most important features of DESs is that by selecting different types of HBDs and HBAs, the hydrophilicity of these solvents can be adjusted and many studies have been reported on hydrophobic DESs.42–44 Recently, researchers have synthesized the types of DESs whose hydrophilicity can be changed by changing environmental conditions such as temperature and pH. Limited studies have been reported in this area.33,34
This research aims to develop a fast, simple, and environmentally friendly pretreatment method called pH-switchable DES based liquid phase microextraction (DES-LPME) for the extraction/preconcentration of strogenic compounds in environmental water and wastewater samples combined with GC-MS. A pH-switchable DES was synthesized using l-menthol as HBA and (1S)-(+)-camphor-10-sulfonic acid (CSA) as HBD, and used as a green extraction solvent. By adjusting the pH of the solution, the unique behaviour of the DES in the phase transition and extraction of the desired analytes was investigated. The parameters affecting the extraction efficiency of DES-LPME was thoroughly evaluated and optimized.
2 Experimental
2.1 Reagents and materials
Estrogenic compounds (E1, E2, E3 and EE2 with purity higher than 97%), strone 3-methyl ether (internal standard, I.S.), N,O-bis(trimethylsilyl) trifluoroacetamide (BSTFA) with trimethylchlorosilane (BSTFA + TMSC, 1%), l-menthol and (1S)-(+)-camphor-10-sulfonic acid (CSA) were obtained from Sigma-Aldrich Chemie (St. Louis, Missouri, USA). Anhydrous pyridine (99%), acetone, acetonitrile, methanol, sodium chloride, KOH and HCl (37%) were purchased from Merck (Darmstadt, Germany).2.2 Instrumentation
The analysis of target strogens were performed by an Agilent GC (Model 6890, Agilent Technologies, CA, USA) coupled to a 5973 mass-selective detector and equipped with a splitless/split injector operated at 280 °C in the splitless mode with splitless time of 0.50 min. Chromatographic separation was achieved on a DB-5 MS capillary column (30 m × 0.25 mm i.d., 0.25 μm film thickness) from SGE, Victoria, Australia. Helium (99.9999%, Air Products, West Sussex, UK) at a constant flow rate of 1.0 mL min−1 and passing through a molecular sieve trap and oxygen trap (Chromatography Research Supplies, Louisville, USA) was used as the carrier gas. The column oven temperature program is described as follows: initial temperature 100 °C (held for 1 min), ramped at 20 °C min−1 to 200 °C and then increasing at a rate of 5 °C min−1 to 280 °C (hold for 2 min) with a total run of 24 min and 10 min of solvent delay. The ion source temperature of mass spectrometry (MS) was 230 °C, and the electron ionization (EI) was 70 eV.Fourier Transform Infrared (FT-IR) spectrum of DES was obtained on a Bruker PS-15 spectrometer with wave numbers in the range of 400–4000 cm−1. Bruker SP-400 Avance spectrometer was used to the 1H and 13C NMR analysis of DES structure. In order to infer the DES's hydrophobicity, the water content of the synthesised DES was quantified using a Karl–Fischer titrator (720-KSS-Metrohm Herisau, Switzerland) for volumetric water content determination. Thermogravimetric analysis (TGA) was performed on the synthesised DES and its components using TGA (Mettler Toledo Instrument Model TGA/SDTA 851 e, Switzerland) purged with of nitrogen, at the temperature range of 50–700 °C and the heating rate of 10 °C min−1.
2.3 Sampling and sample preparation
Two samples of tap water in the north and south of Kermanshah, two samples of agricultural well water in Mian Darband region, two samples of river water taken from Qarasu and Sirvan rivers, and two samples of raw sewage (taken from the entrance of Kermanshah water treatment plant) were collected in dark glass bottles were maintained at 4 °C until analysis. Well, tap and river water samples analyzed without any pretreatment or filtration. Due to the presence of solid particles, the wastewater samples were passed through a 0.42 μm filter paper before analysis. The pH of all samples was close to neutral and none had strong acidic or alkaline properties.2.4 Preparation of DESs
l-Menthol and CSA were accurately weighed, as required to achieve the molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and mixed in a 100 mL conical flask. The mixture was stirred at a constant temperature of 40 °C for 30 min until a clear and homogeneous liquid was obtained. The synthesized DES was stored in a desiccator after cooling down to room temperature. The obtained DES was used as extractant in DES–LPME procedure without any further purification.
:1, and mixed in a 100 mL conical flask. The mixture was stirred at a constant temperature of 40 °C for 30 min until a clear and homogeneous liquid was obtained. The synthesized DES was stored in a desiccator after cooling down to room temperature. The obtained DES was used as extractant in DES–LPME procedure without any further purification.
2.5 DES-LPME procedure
For the presented procedure, an aliquot of 10.0 mL of the sample solution (spiked or not with target analytes) containing 10.0 ng L−1 strone 3-methyl ether as internal standard was placed in a 20 mL glass test tube, and 50.0 μL of DES was added as the extraction phase. Then 80 μL of potassium hydroxide solution (7 mol L−1) was added to the test tube and shaken manually for 30 seconds to dissolve the DES in the sample solution. After that, to break the homogeneity, 120 μL of HCl solution (7 mol L−1) was added and shaken manually for a few seconds. The DES containing the desired analytes was separated from the sample solution and collected on the surface of the solution without centrifugation. Subsequently, the test tube was there after put into a freezer for 2 min; at this time, the DES was solidified because of the low melting point and was transferred into a conical glass vial. After melting at room temperature, derivatization of target estrogens was done using 60 μL of BSTFA + 1%TMCS and 30 μL of pyridine at 70 °C for 25 min. The derivatives were cooled at room temperature and after evaporation of the mixture to dryness under a gentle flow of N2, the residue was dissolved in 50 μL of n-hexane and 1.00 μL of it was injected into the GC–MS.2.6 Enrichment factor, extraction recovery and relative recovery
The enrichment factor (EF) was calculated as the ratio between the concentration of analytes in the DES (CDES) and initial concentration of analytes within the aqueous phase (C0):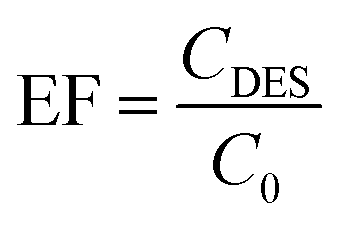
The extraction recovery (ER%) was calculated as the ratio between the amount of the analyte in the DES (nDES) and the initial amount of the analyte within the aqueous solution (n0):
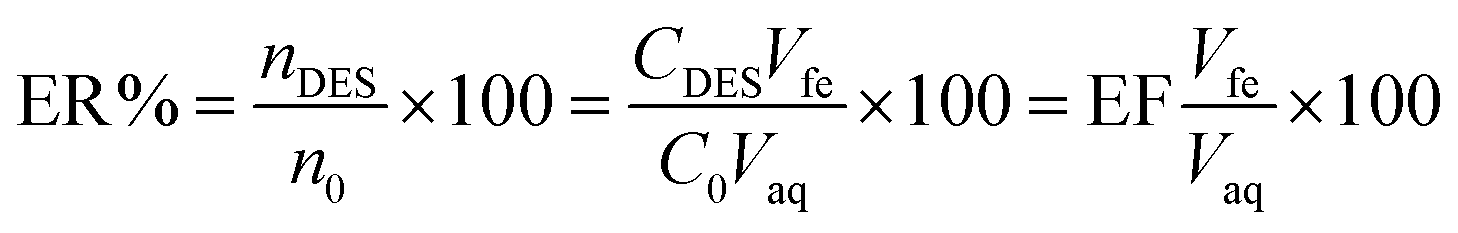
The relative recovery (RR%) of the spike samples was determined using the following formula:
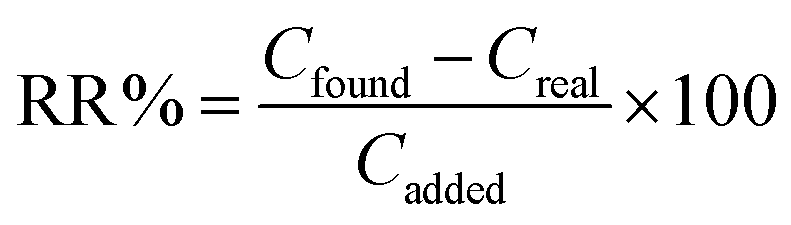
3 Results and discussion
3.1 Characterization of DES
:1. The functional groups of this synthesised DES and its components was confirmed with FT-IR analysis. Fig. 1(A) shows the FT-IR spectrum of l-menthol, CSA and DES before and after transition. The FT-IR spectra demonstrated the most significant peaks observed at 1368, 2929, 2959 and 3263 cm−1 for l-menthol as basic component that attributed to the isopropyl, methyl and hydroxyl groups, respectively. The broad band due to O–H stretching vibration of pure l-menthol was observed at around 3263 cm−1. In contrast, the O–H stretching vibration in the DES before and after the phase transition was observed at approximately 3346 cm−1. This can be attributed to CSA's ability to disrupt intermolecular hydrogen bonding in l-menthol molecules, which means that l-menthol and CSA act as HBA and HBD, respectively. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of CSA and DES were observed at arounds 1741 cm−1 and 1745 cm−1, respectively. The FT-IR spectrum quite similar for DES (before and after phase transition) shows that acid and base treatments had little effect on the structure of DES.
O stretching vibration of CSA and DES were observed at arounds 1741 cm−1 and 1745 cm−1, respectively. The FT-IR spectrum quite similar for DES (before and after phase transition) shows that acid and base treatments had little effect on the structure of DES.
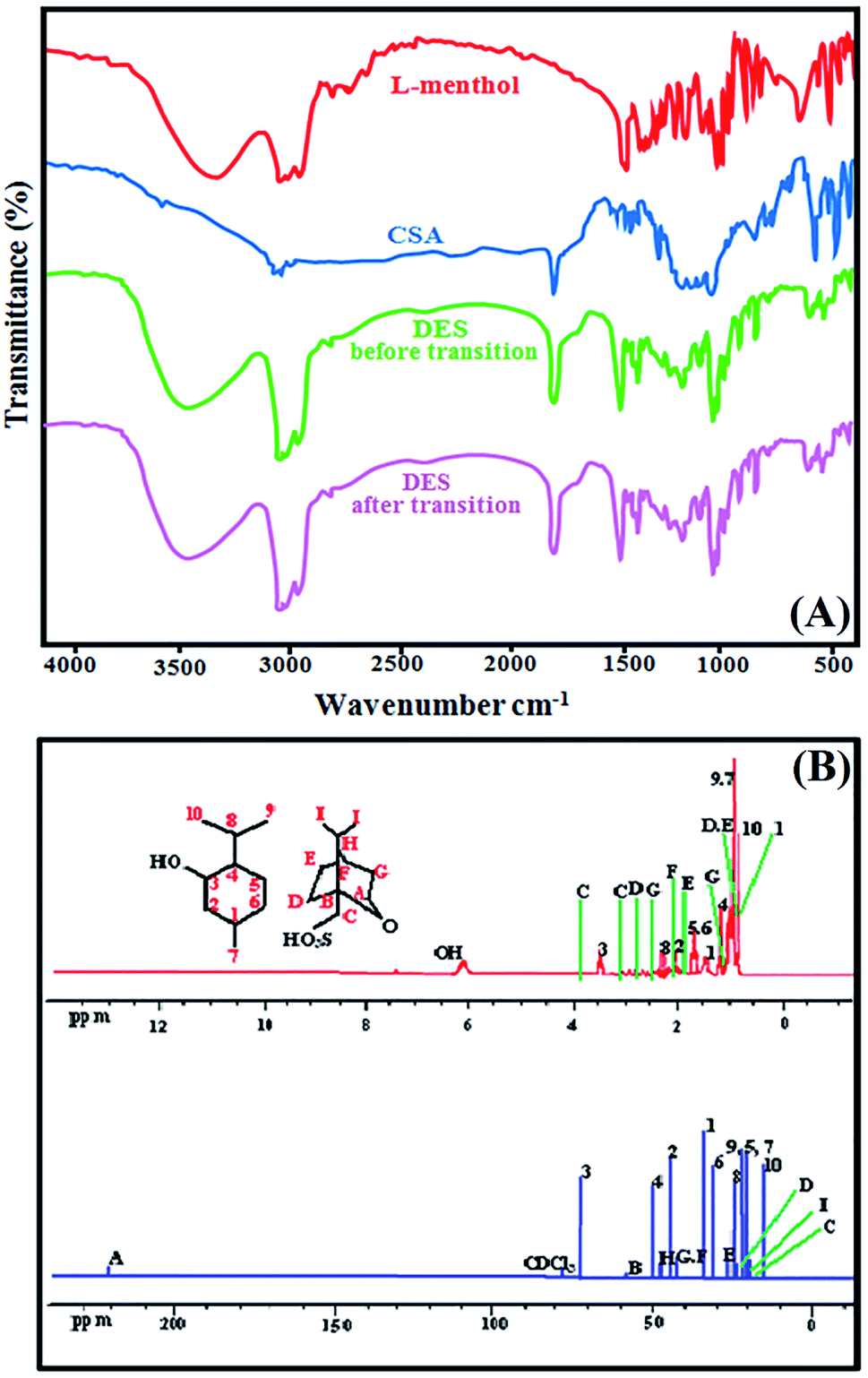 | ||
| Fig. 1 FT-IR spectra of the pure l-menthol, (1S)-(+)-camphor-10-sulfonic acid and the DES mixture of l-menthol and (1S)-(+)-camphor-10-sulfonic acid before and after transition (A), and NMR spectra of l-menthol:(1S)-(+)-camphor-10-sulfonic acid DES in CDCl3 (B). | ||
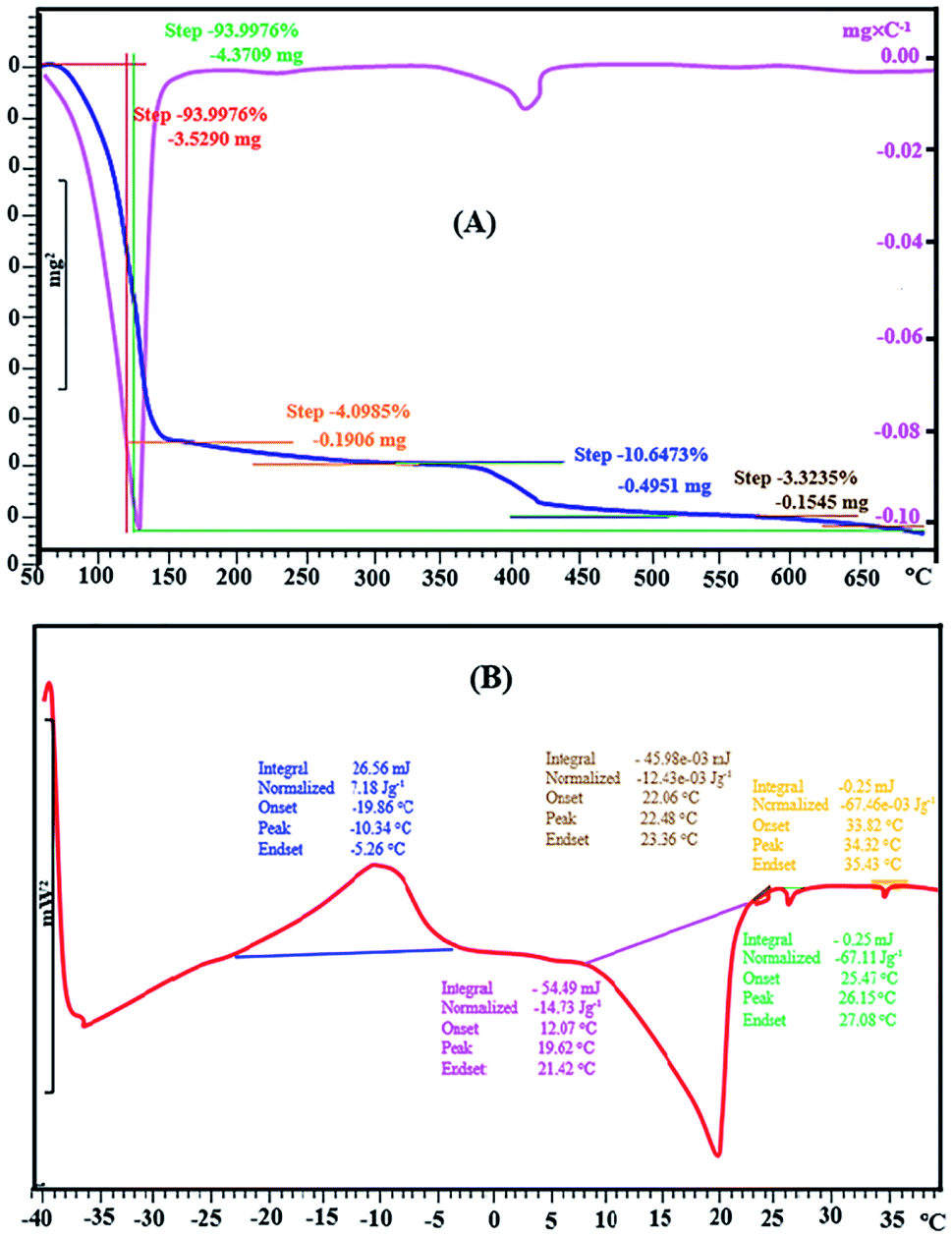 | ||
| Fig. 2 TGA/DTG thermogram of l-menthol:(1S)-(+)-camphor-10-sulfonic acid DES (A) and DSC thermograms of l-menthol:(1S)-(+)-camphor-10-sulfonic acid DES (B). | ||
3.2 Selection of HBA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :HBD molar ratio
:HBD molar ratio
The physical properties of the synthesized DES depend on the molar ratio of HBA and HBD and affect the extraction efficiency. Therefore, in order to synthesize a suitable DES, the molar ratios of l-menthol and CSA were changed in the range of 1:10 to 10:1. The results showed that only in the molar ratios of 3:1, 4:1, 5:1, 6:1 and 7:1 of HBA:HBD a suitable DES was formed and in other ratios the composition was in the form of gelatin or paste. As a result, the extraction efficiency of estrogen compounds was investigated by changing these molar ratios. As shown in Fig. 3(A), the extraction efficiency of the desired compounds increases with increasing the ratio of l-menthol:CSA to 5:1. By further increasing the menthol ratio, the extraction efficiency decreases, which is probably due to the weakened interaction between the DES and the analytes. Therefore, DES consisting of l-menthol and CSA at a 5:1 molar ratio was the most suitable extraction solvent for estrogenic compounds in the presented method.
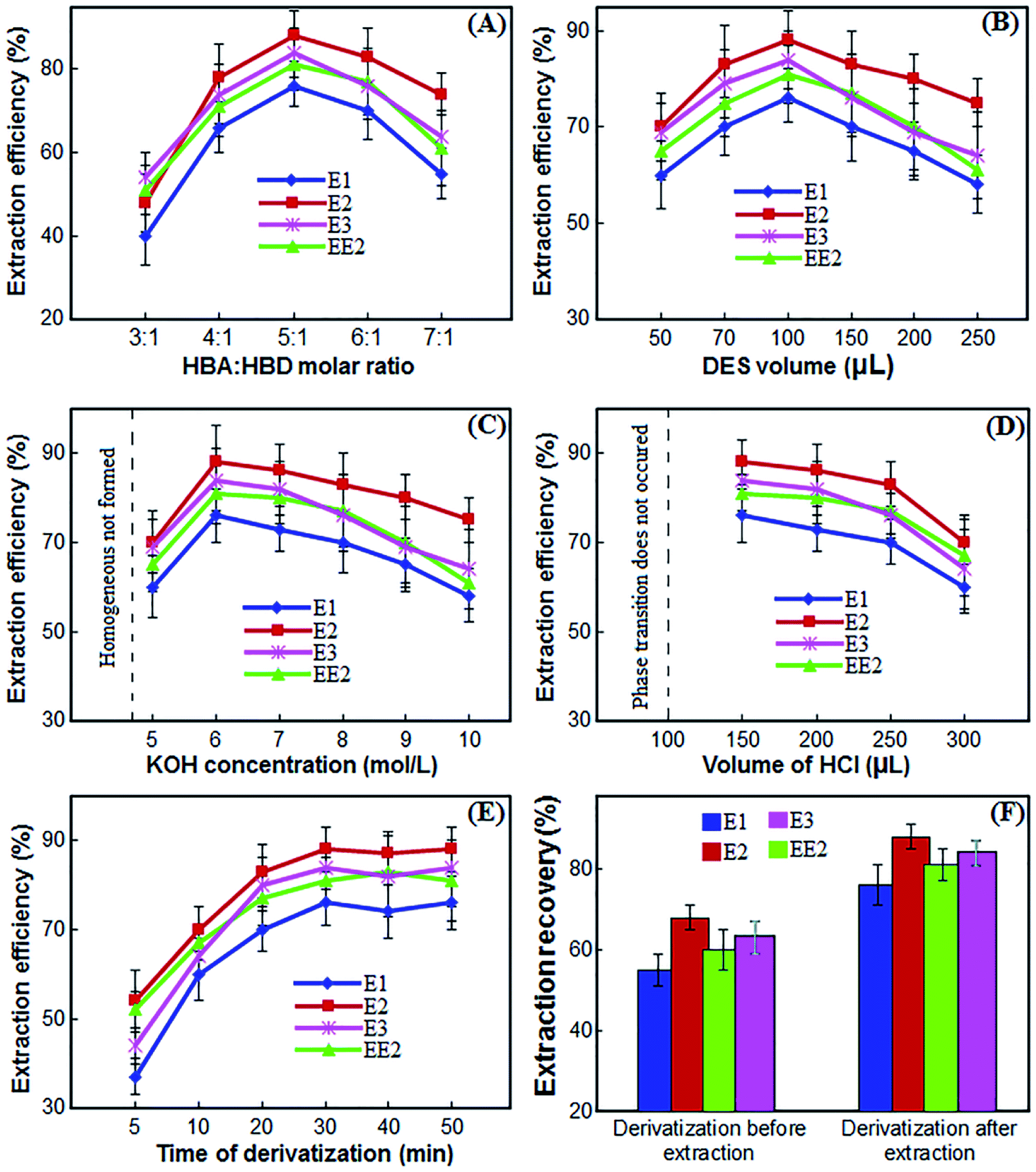 | ||
| Fig. 3 The effect of the molar ratio of HBA to HBD (A), volume of DES (B), KOH concentration (C), volume of HCl (D), time of derivatization (E) and derivatization before and after extraction (F) on the extraction efficiency of estrogenic compounds obtained from DES-LPME/GC-MS. | ||
3.3 Selection of DES volume
An appropriate volume of DES is a crucial parameter in the performance of liquid phase microextraction. To investigate the influence of the volume of DES on the extraction efficiency of the strogenic compounds, the volume was changed from 50 μL to 250 μL. According to the experimental results (Fig. 3(B)), the gradual rise in the volume of the DES from 50 μL to 100 μL led to an increase of extraction efficiency. However, when the volume of the DES was more than 100 μL, the extraction efficiency was reduced. Volumes of 50 μL and less were often associated with unstable extraction efficiencies and significant fluctuations due to their difficulty in collection of them. Thus, 100 μL of DES was chosen as the optimum volume for further experiments.3.4 Selection of KOH concentration
The synthesized DES used as the extractant in the proposed procedure was hydrophobic and immiscible with water. In the proposed method, the phase transition of DES into aqueous phase was performed by alkalizing the medium using KOH solution. By using KOH solution, the water-immiscible DES is dissolved in aqueous phase to form a homogeneous mixture with a high contact surface that directly affects the extraction efficiency. To investigate the effect of KOH concentration on the performance of proposed procedure, various experiments were performed by adding 100 μL of KOH with different concentrations ranged from 1–10 mol L−1 in sample solution. Other experimental conditions were kept constant. The results in Fig. 3(C) show that by increasing the KOH concentration from 1 to 4 mol L−1, a homogeneous mixture is not formed and acceptable efficiencies are not obtained. By increasing the KOH concentration to 6 mol L−1, the extraction efficiency increases because the phase transition is complete and the homogeneous mixture is well formed. As the KOH concentration increases further, the extraction efficiency decreases because breaking the homogeneous mixture and biphasing the system requires more acid. Therefore, a 6 mol L−1 concentration of KOH solution was selected for further experiments.3.5 Selection of HCl volume
In the previous section, a concentration of 6 mol L−1 was selected for KOH. Therefore, we used different volumes of HCl solution with the same concentration of 6 mol L−1 to make the water-miscible DES immiscible and separation of DES containing the target strogenic compounds from the homogeneous phase by neutralizing the alkaline environment. Accordingly, to select the optimum volume of HCl solution, several experiments were performed using different volumes of HCl solution, i.e. 100, 150, 200, 250 and 300 μL. As can be seen from Fig. 3(D), at volumes less than 100 μL, KOH solution is not neutralized and phase transition does not occur. At a volume of 100 μL, although the KOH solution is theoretically neutralized, breaking the homogenous mixture and phase transition is not complete and collecting the DES is very difficult. When 150 μL are used, the system becomes completely two-phase and the maximum extraction efficiency is achieved. With further increase in the volume of HCl solution, phase transition and collection of DES is well done but the extraction efficiency gradually decreases. Therefore, 150 μL of the HCl solution was chosen for further experiments.3.6 Effect of salt
The proposed method is based on changing the pH and adding acid and base and it is logical that when the KOH solution is neutralized by HCl solution, KCl solution is obtained. Thus, the ionic strength of the sample solution as a potential factor affects the extraction efficiency due to the effect of salting-out. To study the effect of salt addition on the extraction efficiency of the strogenic compounds, different concentrations of NaCl were added to the sample solution in the range of 0–5% (w/v). The results demonstrated that salt addition has no effect on extraction efficiency of the target analytes. Consequently, further extractions were performed in the absence of the salt.3.7 Selection of extraction and derivatization time
In DES-LPME procedure, extraction time is the time interval between adding the solutions of KOH and HCl. The effect of extraction time on extraction efficiency was investigated in the range of 0 to 10 min intervals. The results showed that after adding KOH, only 30 seconds of shaking the vessel is sufficient to phase transition and dissolve the DES in the aqueous phase, and with further increase in time, the extraction efficiency remains almost constant. In less than 30 seconds, the phase transition and homogenization of the system are not performed well. Therefore, 30 seconds was chosen as the optimal extraction time.Analysis of estrogenic compounds with GC requires derivatization due to the presence of hydroxyl groups in their structure, and this derivatization is usually performed slowly. As a result, derivation time must be evaluated and optimized. The effect of derivatization time on the extraction efficiency was investigated in the range of 5 to 50 min intervals. The results in Fig. 3(E) show that by increasing the derivatization time from 5 to 30 minutes, the extraction efficiency increases and by further increasing the derivatization time, the extraction efficiency remains almost constant. Therefore, 30 minutes was selected for further experiments.
3.8 Selection of derivatization prior or after extraction
The reagent of BSFTA + 1% TMCS in pyridine was used for derivatization of strogenic compounds. To investigate the derivatization effect before or after extraction, two series of experiments were arranged. First, 80 μL of BSFTA + 1% TMCS and 40 μL of pyridine were added to 10 mL of water, and placed at 70 °C for 30 minutes to give the derivatization reaction. After extraction of the target analytes according to presented procedure, the final extractant was evaporated to dryness under a gentle flow of N2 and the residue was redissolved in 50 μL of n-hexane. Finally, 1.00 μL of n-hexane was injected into the GC-MS. For the second time, after extraction of the analytes by DES-LPME procedure, derivatization of target analytes was done using 80 μL of BSTFA + 1%TMCS and 40 μL of pyridine at 70 °C for 30 min. The derivatives were cooled at room temperature and after evaporation of the mixture to dryness under a gentle flow of N2, the residue was dissolved in 50 μL of n-hexane and 1.00 μL of it was injected into the GC-MS. The results showed (Fig. 3(F)) that derivation after extraction gives better extraction efficiency.3.9 Quantitative analysis
The performance of the proposed DES-LPME method was evaluated under the optimal conditions determined above. Table 1 presents the repeatability and reproducibility, ranges of linearity, coefficients of determination (r2), limits of detection (LODs), enrichment factor (EF) and extraction efficiency (EE) thus derived. The repeatability and reproducibility is calculated based on 7 replicate measurements. The repeatability and reproducibility of the DES-LPME combined with GC–MS by for 20 ng L−1 of estrogens and 10 ng L−1 for internal standard were determined to be 2.2–4.6 and 3.9–5.7%, respectively. Linearity was observed in the range of 0.5–100 ng L−1 of target compounds with coefficients of determination better than 0.9991. The LODs, based on signal-to noise ratio (S/N) of 3 ranged from 0.2 to 1.0 ng L−1. The EF and the EE of strogens were from 152 to 176 and 76 to 88%, respectively.| Compounds | RSDa (intra-day, n = 7) | RSD (inter-day, n = 7) | r2 | LODb (ng L−1) | LRc (ng L−1) | EFd | EEe (%) |
|---|---|---|---|---|---|---|---|
| a Percent relative standard deviation for seven replicate measurements of the estrogens by using internal standard at concentration of 20 ng L−1 for estrogenic compounds and 10 ng L−1 for internal standard.b Limit of detection for S/N = 3.c Linear range.d Enrichment factor.e Extraction efficiency. | |||||||
| E1 | 2.2 | 4.2 | 0.9992 | 1.0 | 3.0–100 | 152 | 76 |
| E2 | 3.5 | 5.3 | 0.9991 | 0.5 | 1.5–100 | 176 | 88 |
| EE2 | 2.8 | 3.9 | 0.9995 | 0.2 | 0.5–50 | 162 | 81 |
| E3 | 4.6 | 5.7 | 0.9993 | 0.2 | 0.5–50 | 168 | 84 |
3.10 Real samples analysis
To test the practicability of the presented method, experiments were conducted to determine the concentration of strogenic compounds in tap water, agricultural well water, river water and wastewater samples under optimized conditions. The result showed that the tap, well and river water samples did not contain strogens, while both wastewater samples contained these compounds. In the sample of wastewater no. 1, E1 and E2 were detected at 53.5 and 88.6 ng L−1, respectively. In the sample of wastewater no. 2, E1, E2, EE2 and E3 were detected at 16.1, 28.3, 49.6 and 77.4 μg L−1, respectively.In order to examine the matrix effect in real samples, all samples were spiked with target analytes at different concentration levels. The analyses were performed in triplicate to ensure the accuracy. The results in Table 2 showed that the relative recoveries of strogens in real samples are in the range of 91.0–108.8%, with RSD < 10. The high recoveries indicated a negligible matrix effect on DES-LPME efficiency in different environmental samples. Fig. 4 shows the GC-MS chromatograms of well water (no. 2) and wastewater (no. 1) samples, and the corresponding spiked ones. Furthermore, four strogens-free water and wastewater samples from different sources were selected randomly to investigate the selectivity (matrix effect) of the presented method. Interferences were assessed by comparing chromatograms of the blank samples and the blank samples spiked with the target strogens. The acceptance criteria for the experiment were achieved. The retention times of E1, E2, EE2 and E3 were 15.82, 16.88, 19.79 and 21.52 min, respectively. No other peaks corresponding to these retention times were observed in the chromatograms of the blank samples. The method is selective as it was able to differentiate and quantify the strogens in the matrix. These results demonstrate that the environmental water and wastewater matrices, in our present context, have no significant effect on the DES-LPME followed by GC-MS for preconcentration and analysis of the estrogenic compounds.
| Type of samples | Analyte | Added (ng L−1) | Found, mean ± SDa (n = 3) (ng L−1) | Relative recovery (%) |
|---|---|---|---|---|
| a Standard deviation.b Not detected. | ||||
| Tap water (no. 1) | E1 | 0 | n.d.b | — |
| 10 | 9.6 ± 0.8 | 96.0 | ||
| E2 | 0 | n.d. | — | |
| 10 | 10.4 ± 1.03 | 104.0 | ||
| EE2 | 0 | n.d. | — | |
| 10 | 9.1 ± 0.7 | 91.0 | ||
| E3 | 0 | n.d. | — | |
| 10 | 10.6 ± 1.05 | 106.0 | ||
| Tap water (no. 2) | E1 | 0 | n.d. | — |
| 20 | 19.1 ± 1.5 | 95.5 | ||
| E2 | 0 | n.d. | — | |
| 20 | 20.65 ± 1.2 | 103.2 | ||
| EE2 | 0 | n.d. | — | |
| 20 | 18.76 ± 1.8 | 93.8 | ||
| E3 | 0 | n.d. | — | |
| 20 | 21.3 ± 2.1 | 106.5 | ||
| Well water (no. 1) | E1 | 0 | n.d. | — |
| 30 | 27.4 ± 2.6 | 91.3 | ||
| E2 | 0 | n.d. | — | |
| 30 | 29.2 ± 1.3 | 107.3 | ||
| EE2 | 0 | n.d. | — | |
| 30 | 31.4 ± 2.7 | 104.7 | ||
| E3 | 0 | n.d. | — | |
| 30 | 30.5 ± 2.2 | 101.7 | ||
| Well water (no. 2) | E1 | 0 | n.d. | — |
| 50 | 47.9 ± 3.3 | 95.8 | ||
| E2 | 0 | n.d. | — | |
| 50 | 52.5 ± 4.1 | 105.0 | ||
| EE2 | 0 | n.d. | — | |
| 50 | 53.7 ± 4.5 | 107.4 | ||
| E3 | 0 | n.d. | — | |
| 50 | 49.1 ± 3.8 | 98.2 | ||
| River water (from sirvan river) | E1 | 0 | n.d. | — |
| 60 | 62.6 ± 4.8 | 104.3 | ||
| E2 | 0 | n.d. | — | |
| 60 | 56.4 ± 5.3 | 94.0 | ||
| EE2 | 0 | n.d. | — | |
| 60 | 60.4 ± 5.1 | 100.7 | ||
| E3 | 0 | n.d. | — | |
| 60 | 58.6 ± 4.5 | 97.7 | ||
| River water (from Qarasu river) | E1 | 0 | n.d. | — |
| 80 | 76.1 ± 5.5 | 95.1 | ||
| E2 | 0 | n.d. | — | |
| 80 | 83.5 ± 6.2 | 104.4 | ||
| EE2 | 0 | n.d. | — | |
| 80 | 87.1 ± 5.8 | 108.8 | ||
| E3 | 0 | n.d. | — | |
| 80 | 77.3 ± 4.3 | 96.6 | ||
| Wastewater (no. 1) | E1 | 0 | 53.5 ± 3.9 | — |
| 20 | 75.1 ± 5.1 | 108.0 | ||
| E2 | 0 | 88.6 ± 6.3 | — | |
| 20 | 106.8 ± 8.3 | 91.0 | ||
| EE2 | 0 | n.d. | — | |
| 20 | 21.3 ± 2.2 | 106.5 | ||
| E3 | 0 | n.d. | — | |
| 20 | 19.5 ± 1.6 | 97.5 | ||
| Wastewater (no. 2) | E1 | 0 | 16.1 ± 0.8 | — |
| 40 | 57.4 ± 4.3 | 103.2 | ||
| E2 | 0 | 28.3 ± 1.5 | — | |
| 40 | 71.1 ± 5.2 | 107.0 | ||
| EE2 | 0 | 49.6 ± 3.7 | — | |
| 40 | 88.3 ± 6.5 | 96.8 | ||
| E3 | 0 | 77.4 ± 5.4 | — | |
| 40 | 119.1 ± 7.8 | 104.2 | ||
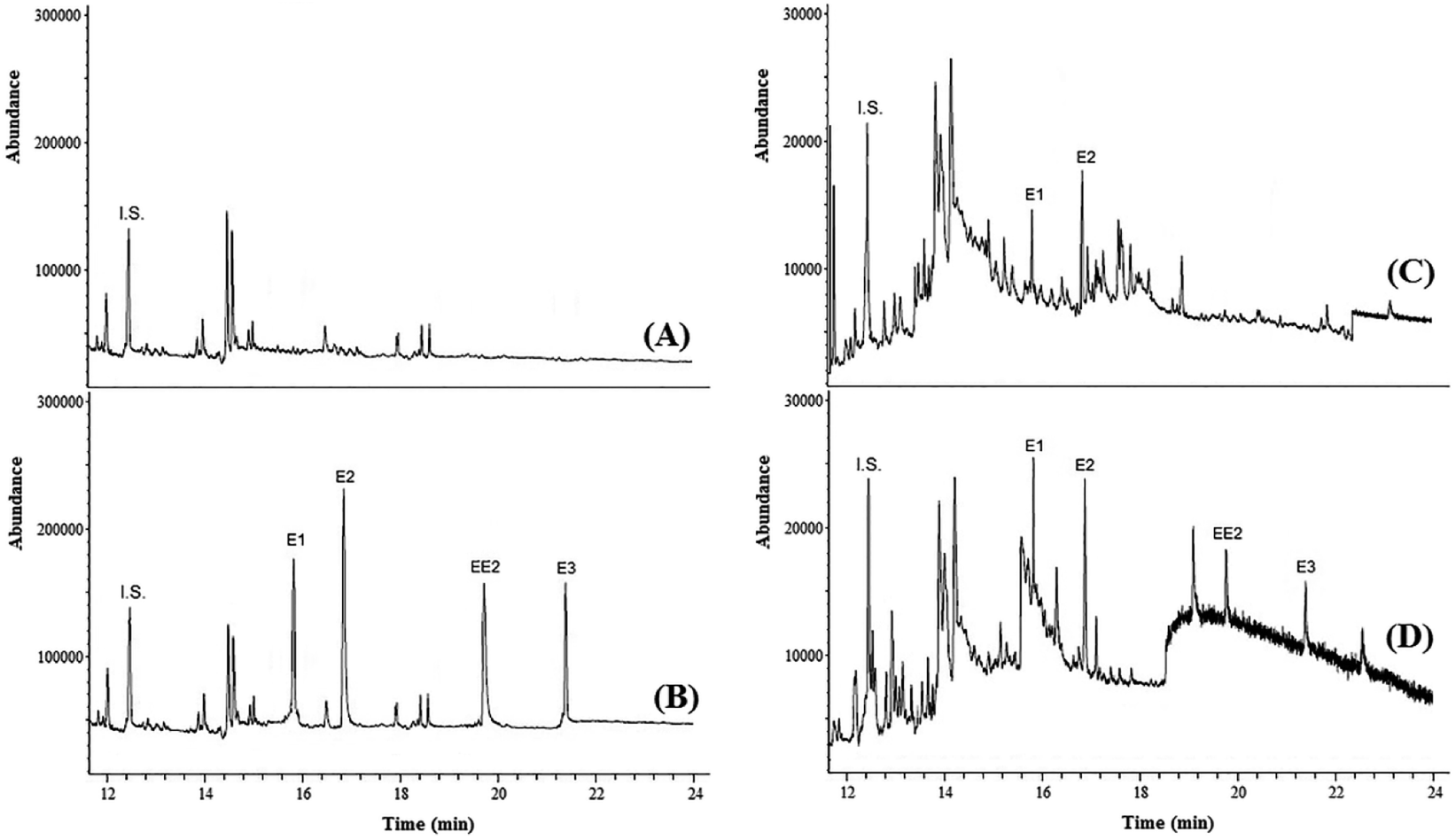 | ||
| Fig. 4 Chromatograms of well water (A), spiked well water at concentration level of 50 ng L−1 for strogens (B), wastewater sample (C) and spiked wastewater at concentration level of 20 ng L−1 for strogens (D) obtained by using DES-LPME combined GC-MS. | ||
3.11 Comparison with other methods
The performance of the proposed DES-LPME pretreatment method was compared with the performances obtained using the existing pretreatment methods for strogenic compounds in Table 3. As can be seen from Table 3, LOD is lower than all of the mentioned techniques, considering sample volume of 10 mL. The RSD and linear range of the proposed method are superior to those reported before. Consumption of toxic organic solvents has been drastically reduced and the use of disperser solvent, vortex, ultrasonic and centrifuge is eliminated. Therefore, DES-LPME is a fast, simple, and environmentally friendly method for extraction and preconcentration of strogenic compounds.| Method | LOD (ng L−1) | RSD (%) | LR (ng L−1) | EF | Sample type | Sample volume (mL) | Reference |
|---|---|---|---|---|---|---|---|
| DLLME-SFO-HPLC-UV | 800–2700 | 7–14 | 5000–1000000 |
121–329 | Water | 5 | 16 |
| MSPE-UPLC-HR | 0.2–3 | 2.3–8.1 | 0.5–1000 | 500 | Water | 3 | 29 |
| SPME-LC-MS/MS | 50–150 | 7.8–12.7 | 50–50000 |
20 | Environmental waters | 1 | 25 |
| UAE-E-MOF-5-SPME-HP | 170–560 | 3.5–6.1 | 500–200000 |
— | Milk | 2 | 26 |
| BHF-LPME-UHPLC-MS/MS | 0.251–0.440 | 7.25–8.13 | 5–1000 | 77–137 | Aqueous matrices | 10 | 45 |
| DLLME-HPLC-FLD | 2–6.5 | 5–10 | 10–500 | 145–178 | Water | 8 | 24 |
| DLLME-SFO-HPLC-UV | 30300–666700 |
0.4–1.8 | 100–50000 |
18–22 | Urine and water | 10 | 21 |
| SCSE-HPLC-DAD | 24–57 | 0.92–5.41 | 100–200000 |
— | Environmental waters | 100 | 46 |
| DES-LPME-GC-MS | 0.2–1.0 | 2.2–4.6 | 0.5–100 | 152–176 | Environmental water and wastewater samples | 10 | This work |
4 Conclusions
In this research, a new DES-LPME procedure combined with GC-MS was introduced for the extraction and preconcentration of strogenic compounds in environmental water and wastewater samples. A pH-switchable hydrophobic DES was synthesized using l-menthol as HBA and (1S)-(+)-camphor-10-sulfonic acid (CSA) as HBD, and used as a green extraction solvent. Solutions of potassium hydroxide and hydrochloric acid were used to induce a phase transition of DES. This method is simple, fast and low cost and the whole process is done in few minutes. The homogenization process of the mixture is done only by manual shaking in less than 30 seconds and the phase separation is done only by changing the pH and without centrifugation. The method has good linearity, sensitivity, accuracy, and precision. It is also environmentally friendly and was successfully used to determine the concentrations of strogens in water and wastewater samples. The method can therefore be potentially applied to various analytes in environmental samples. The development of the presented method in future studies can lead to a reduction in the consumption of toxic organic solvents and the complete elimination of those that act as disperser solvent.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Research Council of Kermanshah University of Medical Sciences (Grant Number: 4000185) for the financial. Also, the authors appreciate the Research Center for Environmental Determinants of Health (RCEDH) for their cooperation in the preparation and analysis of real samples.References
- R. F. Brocenschi, R. C. Rocha-Filho, B. Duran and G. M. Swain, Talanta, 2014, 126, 12–19 CrossRef CAS PubMed.
- A. L. Capriotti, C. Cavaliere, V. Colapicchioni, S. Piovesana, R. Samperi and A. Lagana, J. Chromatogr. A, 2013, 1313, 62–77 CrossRef CAS PubMed.
- A. Glineur, M. Beccaria and G. Purcaro, J. Chromatogr. A, 2021, 1652, 462359 CrossRef CAS PubMed.
- M. Pedrouzo, F. Borrull, E. Pocurull and R. M. Marcé, Talanta, 2009, 78, 1327–1331 CrossRef CAS PubMed.
- Z. Liu, Y. Kanjo and S. Mizutani, Sci. Total Environ., 2009, 407, 731–748 CrossRef CAS PubMed.
- H. Chang, Y. Wan and J. Hu, Environ. Sci. Technol., 2009, 43, 7691–7698 CrossRef CAS PubMed.
- L. Wang, G. G. Ying, J. L. Zhao, S. Liu, B. Yang, L. J. Zhou, R. Tao and H. C. Su, Environ. Pollut., 2011, 159, 148–156 CrossRef CAS PubMed.
- Y. Hu, Y. Wang, X. Chen, Y. Hu and G. Li, Talanta, 2010, 80, 2099–2105 CrossRef CAS PubMed.
- K. I. Ekpeghere, W.-J. Sim, H.-J. Lee and J.-E. Oh, Sci. Total Environ., 2018, 640–641, 1015–1023 CrossRef CAS PubMed.
- H. Hamid and C. Eskicioglu, Water Res., 2012, 46, 5813–5833 CrossRef CAS PubMed.
- B. Socas-Rodríguez, M. Asensio-Ramos, J. Hernández-Borges and M. A. Rodríguez-Delgado, Food Chem., 2014, 149, 319–325 CrossRef PubMed.
- C. Hu, M. He, B. Chen, C. Zhong and B. Hu, J. Chromatogr. A, 2013, 1310, 21–30 CrossRef CAS PubMed.
- M. B. Woudneh, J. P. Benskin, G. Wang, R. Grace, M. C. Hamilton and J. R. Cosgrove, J. Chromatogr. A, 2015, 1400, 149–155 CrossRef CAS PubMed.
- A. Gonzalez, J. Avivar and V. Cerda, J. Cromatogr. A, 2015, 1413, 1–8 CrossRef CAS PubMed.
- R. Jeannot, H. Sabik, E. Sauvard, T. Dagnac and K. Dohrendorf, J. Chromatogr. A, 2002, 974, 143–159 CrossRef CAS PubMed.
- K. Zhang and Y. Zuo, Anal. Chim. Acta, 2005, 554, 190–196 CrossRef CAS.
- R. Davoodi, R. N. Nodehi, N. Rastkari, A. A. Zinatizadeh, A. H. Mahvi and N. Fattahi, New J. Chem., 2020, 44, 9844–9851 RSC.
- M. T. García-Córcoles, M. Cipa, R. Rodríguez-Gómez, A. Rivas, F. Olea-Serrano, J. L. Vílchez and A. Zafra-Gómez, Talanta, 2018, 178, 441–448 CrossRef PubMed.
- A. González, S. Clavijo and V. Cerda, Talanta, 2019, 194, 852–858 CrossRef PubMed.
- R. Zhang, C. Wang, Q. Yue, T. Zhou, N. Li, H. Zhang and X. Hao, J. Sep. Sci., 2014, 37, 3133–3141 CrossRef CAS PubMed.
- Z. Niu, J. Zhang, D. Wu and Y. Wen, Int. J. Environ. Anal. Chem., 2019, 99, 444–453 CrossRef CAS.
- F. R. Mansour and M. A. Khairy, J. Chromatogr. B, 2017, 1061–1062, 382–391 CrossRef CAS PubMed.
- E. Kupcová and K. Reiffová, J. Sep. Sci., 2017, 40, 2620–2628 CrossRef PubMed.
- D. L. D. Lima, C. P. Silva, M. Otero and V. I. Esteves, Talanta, 2013, 115, 980–985 CrossRef CAS PubMed.
- C. Wang, L. Yang, N. Li, X. Zhang, Y. Guo and C. Li, J. Chromatogr. B, 2017, 1061–1062, 41–48 CrossRef CAS PubMed.
- H. Lan, D. Pan, Y. Sun, Y. Guo and Z. Wu, Anal. Chim. Acta, 2016, 937, 53–60 CrossRef CAS PubMed.
- H. Liu, L. Qiao, N. Gan, S. Lin, Y. Cao, F. Hu, J. Wang and Y. Chen, J. Chromatogr. B, 2016, 1027, 50–56 CrossRef CAS PubMed.
- X. Li, S. Li and G. Kellermann, Talanta, 2018, 178, 464–472 CrossRef CAS PubMed.
- Y. Zhao, X.-L. Bai, T. Song, G.-L. Zhang, Y.-C. Yuan, Y.-M. Liu and X. Liao, Microchem. J., 2019, 151, 104212 CrossRef CAS.
- M. Karimaei, K. Sharafi, M. Moradi, H. R. Ghaffari, H. Biglari, H. Arfaeinia and N. Fattahi, Anal. Methods, 2017, 9, 2865–2872 RSC.
- S. Taheri, F. Jalali, N. Fattahi, R. Jalili and G. Bahrami, J. Sep. Sci., 2015, 38, 3545–3551 CrossRef CAS PubMed.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- D. Xiong, Q. Zhang, W. Ma, Y. Wang, W. Wan, Y. Shi and J. Wang, Sep. Purif. Technol., 2021, 265, 118479 CrossRef CAS.
- W. Ma and K. H. Row, Microchem. J., 2021, 160, 105642 CrossRef CAS.
- R. Akramipour, N. Fattahi and M. R. Golpayegani, J. Chromatogr. B, 2021, 1171, 122628 CrossRef CAS PubMed.
- K. Sharafi, N. Fattahi, A. H. Mahvi, M. Pirsaheb, N. Azizzadeh and M. Noori, J. Sep. Sci., 2015, 38, 1010–1016 CrossRef CAS PubMed.
- M. Pirsaheb, N. Fattahi, R. Rahimi, K. Sharafi and H. R. Ghaffari, Food Chem., 2017, 231, 148–155 CrossRef CAS PubMed.
- P. Hashemi, S. Beyranvand, R. Siah Mansur and A. R. Ghiasvand, Anal. Chim. Acta, 2009, 655, 60–65 CrossRef CAS PubMed.
- D. Cao, X. Xu, S. Xue, X. Feng and L. Zhang, Talanta, 2019, 199, 212–219 CrossRef CAS PubMed.
- F. Shakirova, A. Shishov and A. Bulatov, Talanta, 2022, 237, 122968 CrossRef CAS PubMed.
- R. Akramipour, M. R. Golpayegani, S. Gheini and N. Fattahi, Talanta, 2018, 186, 17–23 CrossRef CAS PubMed.
- M. R. Golpayegani, R. Akramipour, S. Gheini, M. V. Amini, F. Fattahi, A. Mohebbi and N. Fattahi, RSC Adv., 2022, 12, 3611–3617 RSC.
- T. Ahmadi-Jouibari, Z. Shaahmadi, M. Moradi and N. Fattahi, Food Addit. Contam. Part A, 2022, 39, 105–115 CrossRef CAS PubMed.
- M. Nemati, M. A. Farajzadeh, M. R. Afshar Mogaddam, A. Mohebbi, A. R. Azimi, N. Fattahi and M. Tuzen, Microchem. J., 2022, 175, 107196 CrossRef CAS.
- S. X. L. Goh and H. K. Lee, J. Chromatogr. A, 2017, 1488, 26–36 CrossRef CAS PubMed.
- L. Chen, M. Mei, X. Huang and D. Yuan, Talanta, 2016, 152, 98–104 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |