
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photodegradation of decabromodiphenyl ether on oxygen vacancy-enriched Bi2MoO6†
Qin Lib,
Xueqing Jina,
Meiying Yanga,
Qi Shen *ac and
Chunyan Sun*a
*ac and
Chunyan Sun*a
aSchool of Chemistry and Chemical Engineering, Shaoxing University, Shaoxing 312000, Zhejiang, China. E-mail: shenqi@usx.edu.cn; sunchunyan@usx.edu.cn
bBeijing GeoEnviron Engineering & Technology, Lnc, Beijing 100000, China
cInstitute of New Energy, School of Chemistry and Chemical Engineering, Shaoxing University, Shaoxing 312000, China
First published on 13th May 2022
Abstract
Debromination is a primary and critical procedure in the treatment of polybrominated diphenyl ethers (PBDEs) in the environment. Herein, oxygen vacancy-enriched Bi2MoO6 is firstly applied in the photoreduction debromination of PBDEs under visible light illumination. The introduction of oxygen vacancies not only promotes the red-shift of the light absorption band by Bi2MoO6, but also activates the C–Br bond through the formation of Br–O halogen bonds, thus realizing efficient visible light reduction of decabromodiphenyl ether (BDE209). The activation adsorption mode inferred by tracking analysis of the degradation process shows that the meta-position adsorption mode is the main adsorption configuration during the activation process, while the ortho-position adsorption mode is the most difficult. Thence, the oxygen vacancy-dominated photocatalytic BDE209 process is a position-selective multi-electron reduction process. The study shows that oxygen vacancy assisted C–Br activation is an excellent strategy for photocatalytic treatment of halogenated persistent organic pollutants.
1. Introduction
Persistent organic pollutants (POPs) are harmful to human beings and are a global environmental issue that has attracted much attention. As a class of POPs, polybrominated diphenyl ethers (PBDEs) have been widely distributed in the atmosphere, silt and river sediments around the world and are endangering human health and affecting sustainable development.1,2 However, due to the lack of effective alternatives, PBDEs are still being produced and used in large quantities as efficient flame-retardants,3 and the content of PBDEs in the environment is increasing. Therefore, the environmental pollution of PBDEs and their treatment are also the concern of current environment science.The toxicity of brominated pollutants is mainly caused by bromine atoms in PBDEs. Meanwhile, the electron-withdrawing properties of bromine determine the resistance of PBDEs to advanced oxidation technologies. Therefore, debromination of PBDEs is a key step in the treatment of PBDEs in the environment. At present, the degradation methods of PBDEs include photocatalytic method,4 zero-valent iron reduction,5 microbial method6 and hydrothermal method.7 Among them, photocatalysis, as a green, efficient and low-cost catalytic technology, has been widely studied. Common photocatalysts, such as TiO2,8 Fe3O4/C3N4 (ref. 9) and other composites10,11 have been successfully applied for the degradation of decabromodiphenyl ether (BDE209) under UV and visible light. Our previous studies showed that constructing a catalytic interface to form weak interaction force with BDE209 would help to promote C–Br bond cleavage. For example, oxalic acid and BDE209 can form a complex with visible light absorption under the interaction of halogen bond.12 The electrons generated by the excitation of the complex under visible light irradiation could be transferred from oxalic acid to BDE209, which promoted the cleavage and debromination of C–Br. The π–π interaction and halogen-binding interaction between the PBDEs and reduced graphene oxide (RGO) enabled the photoinduced electron transferred from the RGO to PBDEs and leaded to the efficient reductive debromination of PBDEs.13
As point defects at the catalyst interface, oxygen vacancies are capable of reducing the reaction energy barrier and promoting the molecules activation by interacting with the reaction molecules. Research work has shown that oxygen vacancies can greatly enhance the efficiency of semiconductor for organic pollutant degradation,14,15 photocatalytic CO2 reduction,16–18 and photocatalytic nitrogen fixation.19,20 Lee et al. studied the adsorption of CO2 molecules on the oxygen vacancies of the TiO2{110} plane by scanning tunneling microscopy.21 One of the oxygen atoms in CO2 molecule can fill in the oxygen vacancy by forming a bridge bond, thereby activating CO2 and reducing the reaction energy barrier for reduction. Xie's group studied that on the oxygen vacancy introduced Co3O4 single-atom layer, the activation energy barrier for the rate-determining step of CO2 reduction could be reduced from 0.51 eV to 0.40 eV by stabilizing the formate anion radical intermediate.22 Interestingly, previous work has shown that the in situ generated oxygen vacancies at the BiOCl interface during photocatalysis help to activate C–Br bond and promote the degradation of BDE209.23 Therefore, the introduction of a reasonable number of oxygen vacancies into the catalyst surface helps to facilitate the debromination of BDE209. Meanwhile, the oxygen vacancies will induce new energy levels and promote the red shift of the light absorption band edge, thereby increasing the light utilization and improving the catalytic efficiency.
As a typical Aurivillius oxide, bismuth molybdate (Bi2MoO6) is a common bismuth metal oxide with a layered structure.24–26 Compared with wide bandgap photocatalysts such as TiO2 and ZnO, Bi2MoO6 has excellent visible light catalytic activity with a narrow band gap width of 2.70–2.80 eV. Although Bi2MoO6 can utilize more sunlight, its conduction band position is not negative enough, resulting in low reducing ability of photoelectron. In addition, limited by its own weak built-in electric field, the electron–hole pairs of Bi2MoO6 excited by light are very easy to recombine. The introduction of oxygen vacancies will promote the separation of photogenerated electron–hole pairs to a certain extent. At the same time, oxygen vacancies act as active sites to activate reactive molecules. Therefore, it is an effective strategy to improve the photocatalytic ability. Via the modulation of tetramethylethylenediamine (TMEDA) in solvothermal process, oxygen vacancies can be tunably introduced into Bi2MoO6 crystal lattice, which has been successfully applied in the photocatalytic oxidative degradation of ciprofloxacin.27
In this work, oxygen vacancies enriched Bi2MoO6 is firstly applied in the photoreduction debromination of BDE209 under visible light illumination. The introduction of oxygen vacancies into Bi2MoO6 not only promotes the red-shift of light absorption band, but also activates the C–Br bond through the formation of Br–O halogen bonds, thus realizing efficient visible light reduction of BDE209. The activation adsorption mode inferred by tracking analysis of the degradation process shown that meta-position adsorption mode is the main adsorption configuration during the activation process, while the ortho-position adsorption mode is the most difficult. Thence, oxygen vacancy-dominated photocatalytic BDE209 process is a position-selective multi-electron reduction process. The study shows that oxygen vacancy assisted debromination is an excellent strategy for photocatalytic treatment of halogenated POPs.
2. Materials and methods
2.1 Chemicals
Bi(NO3)3·5H2O (AR, 99.0%), glycol (AR, 99.5%), and methanol (AR, 99.5%) were purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Na2MoO4·2H2O (AR, 99.0%), TEMDA (AR, 99.5%), ethyl alcohol (AR, 99.5%) and tetrahydrofuran (AR, 99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd., SCRC, China. BDE209 (Fig. S1†) was obtained from Aldrich Chemical Company (USA). All chemicals were used without further purification.2.2 Preparation of photocatalysts
In a typical solvothermal synthesis of Bi2MoO6 photocatalysts,27 firstly, 0.3638 g Bi(NO3)3·5H2O and 0.0908 g Na2MoO4·2H2O were dissolved into 7.5 mL glycol under stirring to form homogenous solution. Then 45 mL alcohol was added slowly into the homogenous solution under vigorous magnetic stirring for 30 min. The solution was transformed to a 100 mL teflon-lined stainless autoclave, followed by a heating process at 160 °C for 6 h, and then cooled to room temperature naturally. The as-prepared precipitates were rinsed with ethanol and deionized water three times, and dried at 60 °C in oven for overnight. For preparation of oxygen vacancy enriched Bi2MoO6, keeping the rest of the experimental steps unchanged, a certain amounts of TMEDA (0.3 mL, 0.5 mL, 0.7 mL and 0.9 mL) were dropped into the reaction solution before hydrothermal treatment. The samples collected were denoted as BMO-3, BMO-5, BWO-7, BWO-9, and the sample without TMEDA was denoted as BMO-0.For preparation the photoelectrodes BMO-x (x = 0, 3, 5, 7, 9), 5 mg BMO-x was first dissolved in 1 mL of n-propanol and sonicated for 20 minutes, then 50 μL of Nafion solution was added to continue sonication for 10 minutes. And then 30 μL of the mixture was spin-coated onto the FTO electrode with a fixed area of 0.5 × 0.5 cm2. The prepared photoelectrodes were dried overnight for later use. The photoelectrochemical properties were measured in 0.5 M sodium sulfate solution using a common three-electrode system, with the prepared photoelectrode as the working electrode, platinum plate as the counter electrode and saturated calomel electrode (SCE) as the reference electrode.
2.3 Photocatalytic degradation of BDE209
Photocatalytic degradation of BDE209 was conducted with 5 mg Bi2MoO6 samples under visible light with Xe lamp (CEL-HXUV300, with 420 nm cutoff filter). In order to reach the adsorption equilibrium, the suspension was stirred for 30 min in methanol solution with 1 × 10−5 mol L−1 BDE209 before irradiation. The Pyrex vessel was purged with high purity N2 (99.9%) for 15 min to remove O2 and protected under a N2 atmosphere during the irradiation. At given time intervals, 0.5 mL of suspension was sampled and filtered through a 0.22 μm membrane to remove the catalyst particles for analysis. The reaction system without removal of oxygen was used for comparison.Products analysis and quantification were used by high performance liquid chromatography (HPLC) and gas chromatography (GC-μECD). HPLC (SHIMADZU) was equipped with a DIKMA Platisil ODS C-18 column (250 × 4.6 mm, 5 mm film thickness) and an UV-visible detector (UV/vis SPD-20A detector) set at a wavelength of 240 nm. The mobile phase was chromatographically pure methanol with a flow rate of 1 mL min−1. GC-μECD (Agilent 7890A, Agilent Technologies Co. U.S.A.) was equipped with a programmable pressure on-column injection port and a DB-5 capillary column (30 m × 50 μm, i.d. × 0.1 μm film thickness). A splitless 10 μL injection was used manually at 300 °C. The constant flow rate was 1.0 mL min−1 with helium as carrier gas. The oven temperature was set up as follows: 100 °C for 2 min, increased to 230 °C with 15 °C min−1; then to 270 °C with 5 °C min−1, and finally to 320 °C with 10 °C min−1, kept for 10 min.
3. Results and discussion
3.1 Photocatalytic performance of Bi2MO6 photocatalysts
Fig. 1a shows the photocatalytic BDE209 performance of Bi2MoO6 in N2-blanketed methanol under visible light (λ > 420 nm). BDE209 is highly resistant to visible light in the absence of photocatalyst. With the addition of photocatalyst, it can be observed that BDE209 was significantly degraded. The degradation efficiency of BDE209 is in the order of BMO-5 > BMO-7 > BMO-3 > BMO-0 ≈ BMO-9. The ability of photodegradation of BDE209 over Bi2MO6 increased firstly with the addition of TMEDA. Only less than 40% of BDE209 was removed after a 50 min light irradiation on BMO-0. The degradation efficiency was then increased on BMO-3. And the maximum degradation efficiency was achieved on BMO-5, with nearly 80% of BDE209 being degraded under the identical experimental conditions. Subsequently, with the increase of TMDEA, the degradation efficiency gradually weakened, which was weak to the lowest on BMO-9.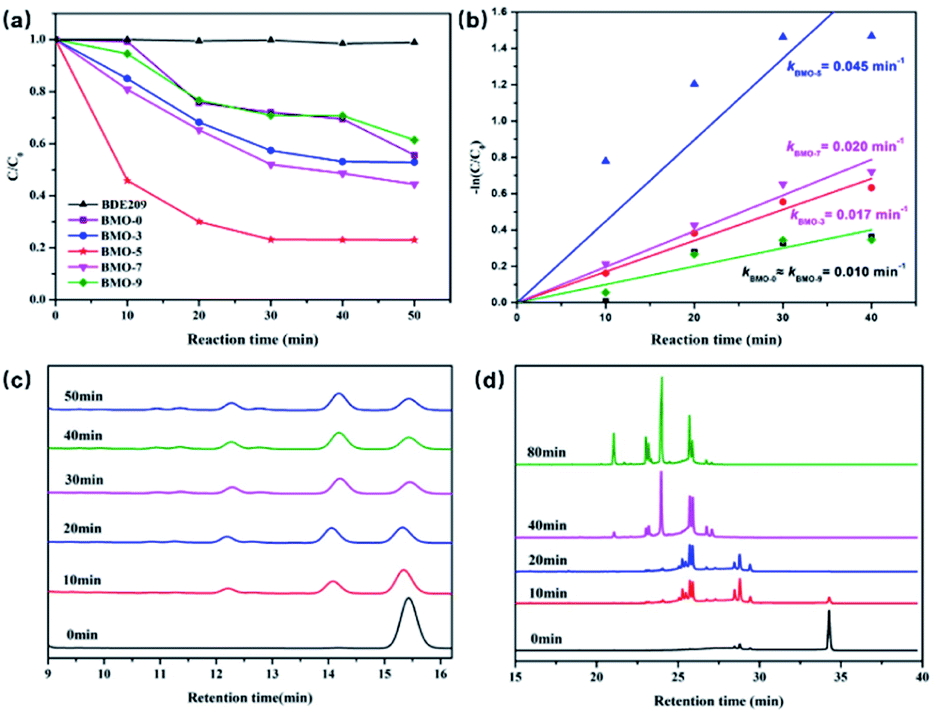 | ||
| Fig. 1 (a) Comparison of photocatalytic degradation of BDE209 over BMO-x (x = 0, 3, 5, 7, 9) in N2-saturated methanol system (λ > 420 nm); (b) dependence of −ln(C/C0) on irradiation time over BMO-x (x = 0, 3, 5, 7, 9) in methanol system; HPLC chromatograms (c) and GC-μECD (d) chromatograms of degradation products of BDE209 over BMO-5 in N2-saturated methanol system (λ > 420 nm). | ||
The degradation data fitted well to the pseudo-first-order kinetics model (Fig. 1b). The estimated reaction rate constant on BMO-5 (0.045 min−1) was 4.5 times greater than that on BMO-9 (0.010 min−1). The calculated reaction rate constants (k′) normalized by the SBET (Table S1†) were used to express activity per unit area of samples. The values of activity per unit area of BMO-5 was 1.28 × 10−3 g m−1 min−1, 7 times greater than that of BMO-9.
The debrominated products analysis during the degradation experiments was used by HPLC and GC-μECD on BMO-5. As shown in Fig. 1c, before the irradiation, only the BDE209 peak at retention time of 15.3 min was observed. Upon the visible light irradiation, the peaks of nona-BDEs at nearly 14 min began to increase as the peak of BDE209 decreased. More accurate product analysis is obtained from the GC-μECD (Fig. 1d). Similar to results of HPLC analysis, only a main peak of BDE209 at retention time of 34 min can be observed before light irradiation. And then the peak of BDE209 decreased rapidly and peaks of lower bromine congeners were observed with prolonged irradiation time, indicating a stepwise debromination of BDE209 on BMO-5. Herein, the peaks of nona-bromo products gradually disappeared, and peaks of other low-bromo products such as octa-, hepta-, and hexa-bromo products appeared with the further prolongation of the illumination time.8 Less brominated PBDEs such as 4-BDEs and 5-BDEs are expected to be further debrominated to the full-debrominated diphenyl ether in the sufficient time.
The photostability of catalysts is one of the important factors to be considered. Herein, repeated cycling experiments were carried out on the degradation of BDE209 over BMO-5, as shown in Fig. 2. After repeated use three times, the degradation rate of BDE209 by BMO-5 remained above 70%, and the reaction rate did not slow down significantly, indicating that the BMO-5 catalyst has good photostability. XRD characterization found that the crystal phase of the recycled catalyst material is consistent with the original crystal phase, which is consistent with the photocatalytic stability results (Fig. S2†).
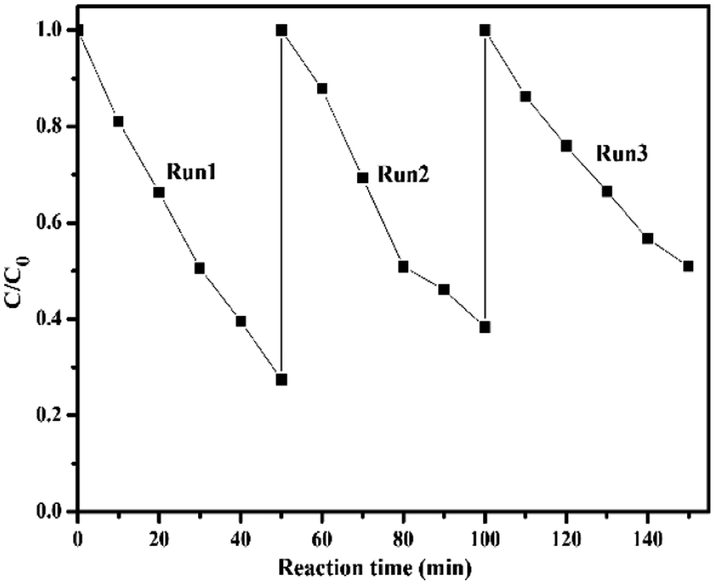 | ||
| Fig. 2 Cycling runs in the degradation of BDE209 over BMO-5 in N2-saturated methanol system (λ > 420 nm). | ||
3.2 Structural properties of Bi2WO6 photocatalysts
The catalytic performance of a photocatalyst is determined by its surface structure.28 As shown in Fig. 3a–e, BMO-x were consisted of large-scale nanosheets. As the amount of TMEDA increased, the size of the nanosheets did not change. However, the degree of aggregation had been changed. The increase in TMEDA helps to improve the dispersion of Bi2WO6. Fig. 1h shows the XRD patterns of as-prepared Bi2WO6. All XRD diffraction peaks were identified to the orthorhombic phase structure of γ-Bi2WO6 (JCPDS no. 21-0102).29 The addition of TMEDA did not change the crystal phase of Bi2MoO6, which is consistent with the HRTEM characterization results. From the HRTEM images, both BMO-0 and BMO-5 were observed to have clear orthorhombic lattice fringes with spacing of 0.270 nm and 0.274 nm, corresponding to the (060) and (200) crystal facets of the Bi2WO6, respectively. However, the difference is that compared with the lattice fringes of BMO-0, the lattice fringes displayed by BMO-5 had obvious lattice disorder (Fig. 3g), which is caused by the unsaturated coordination of metal atoms due to oxygen vacancies.30,31 | ||
| Fig. 3 TEM image of the BMO-0 (a), BMO-3 (b), BMO-5 (c), BMO-7 (d) and BMO-9 (e); HRTEM image of the BMO-0 (f) and BMO-5 (g); and (h) XRD patterns of BMO-x (x = 0, 3, 5, 7, 9). | ||
Electron paramagnetic resonance (EPR) spectroscopy was carried out at 77 K to confirm the formation of oxygen vacancies. Fig. 4a shows that a fingerprint signal with magnetic moment appeared at g value of 2.002 in all samples, which could be contributed to unpaired electrons at oxygen vacancies.32,33 In addition, the intensity of this signal increased with the addition of TMEDA, which is in the order of BMO-9 > BMO-7 > BMO-5 > BMO-3 > BMO-0. The X-ray photoelectron spectroscopy (XPS) was conducted to further identify the atomic valence states of BMO-0, BMO-5 and BMO-9 (Fig. S3†). In Mo 3d region, two strong peaks near at 235.7 and 232.5 eV can be assigned to Mo 3d3/2 and Mo 3d5/2, respectively.34,35 And peaks near at 164.5 and 159.2 eV appeared in Bi region can be assigned to Bi 4f5/2 and Bi 4f7/2 respectively. The high-resolution O 1s spectra can be divided into three peaks near at 529.8, 530.6 and 532.2 eV, attributing to the binding energy of oxygen in Bi–O, Mo–O and adsorbed oxygen,36,37 respectively (Fig. 4b). The peak area of the adsorbed oxygen peak is proportional to the number of oxygen vacancies at the catalyst interface, which is in the order of BMO-9 > BMO-5 > BMO-0 and is consistent with the results of EPR. Appropriate oxygen vacancy content is beneficial to promote the separation of photogenerated electron–hole pairs, while too high content is unfavorable for the separation of photoelectrons and holes because oxygen vacancies are also electron–hole recombination sites in photolysis.38 As a result, the reduction efficiency of BDE209 on BMO-9 is greatly reduced. The chronoamperometric i–t curves of BMO-x photoelectrodes were recorded at −0.4 V vs. SCE under chopped light irradiation to further verify the photocatalytic activity after inducing oxygen vacancies (Fig. S5†). The photocurrent density of the BMO-x photocatalysts increased first and then decreased with the increase of oxygen vacancies. The fastest separation of photogenerated electrons and holes occurred at the BMO-5 photoelectrode upon illumination. Such a result indicated that a suitable amount of oxygen vacancies can increase the photocatalytic efficiency, and excessive oxygen vacancies will lead to a decrease in the photocatalytic ability.
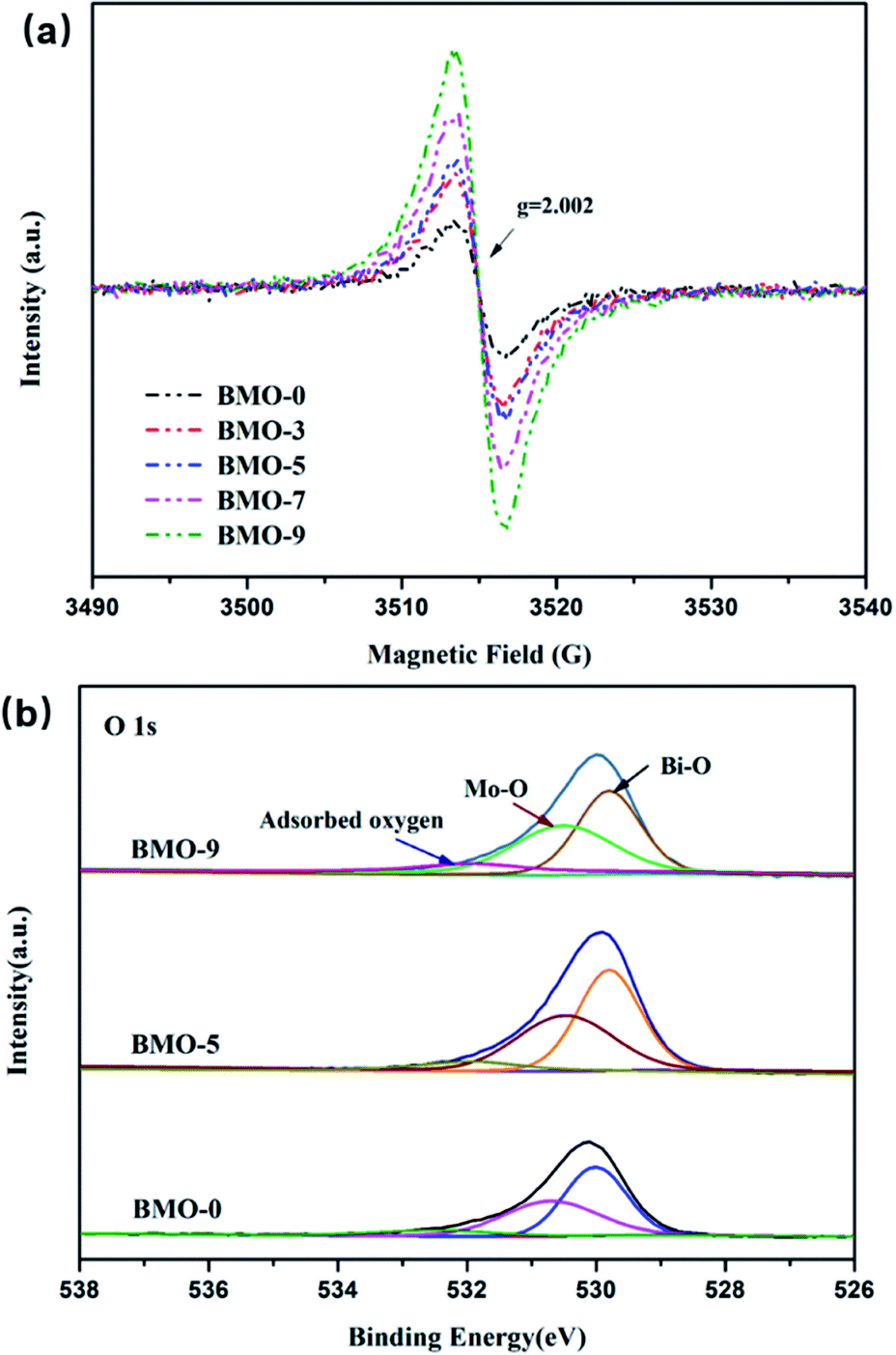 | ||
| Fig. 4 (a) EPR spectra of BMO-x (x = 0, 3, 5, 7, 9) collected at 77 K. (b) O 1s XPS spectra of BMO-0, BMO-5 and BMO-9. | ||
UV-vis diffuse reflectance spectroscopy (DRS) displays the variation of oxygen vacancy-induced light absorption (Fig. 5). It can be seen that all Bi2MoO6 samples exhibited fairly strong light absorption from the UV region to the visible region with wavelengths less than 490 nm. In addition, with the increase of oxygen vacancies content in the samples, the absorption band edge of the BMO samples exhibited an obvious red shift. The corresponding band-gap energy of BMO-0 was calculated to be 2.98 eV, which of BMO-3 was 2.91 eV, while the band gaps of BMO-5, BMO-7 and BMO-9 were red-shifted to 2.77 eV. This is due to defect states in the band gap by introducing oxygen vacancies.39 Apparently, the increase in the amount of oxygen vacancies broadens the light absorption of Bi2MoO6 to a certain extent, which is beneficial to the improvement of its photocatalytic performance. At the same time, the study also found that with the occurrence of the photoreduction reaction, the oxygen vacancies on the surface of the Bi2MoO6 would be further increased, resulting in the color of the Bi2MoO6 changing from the light yellow at the beginning of the reaction to gray-black,33 as shown in Fig. S4.†
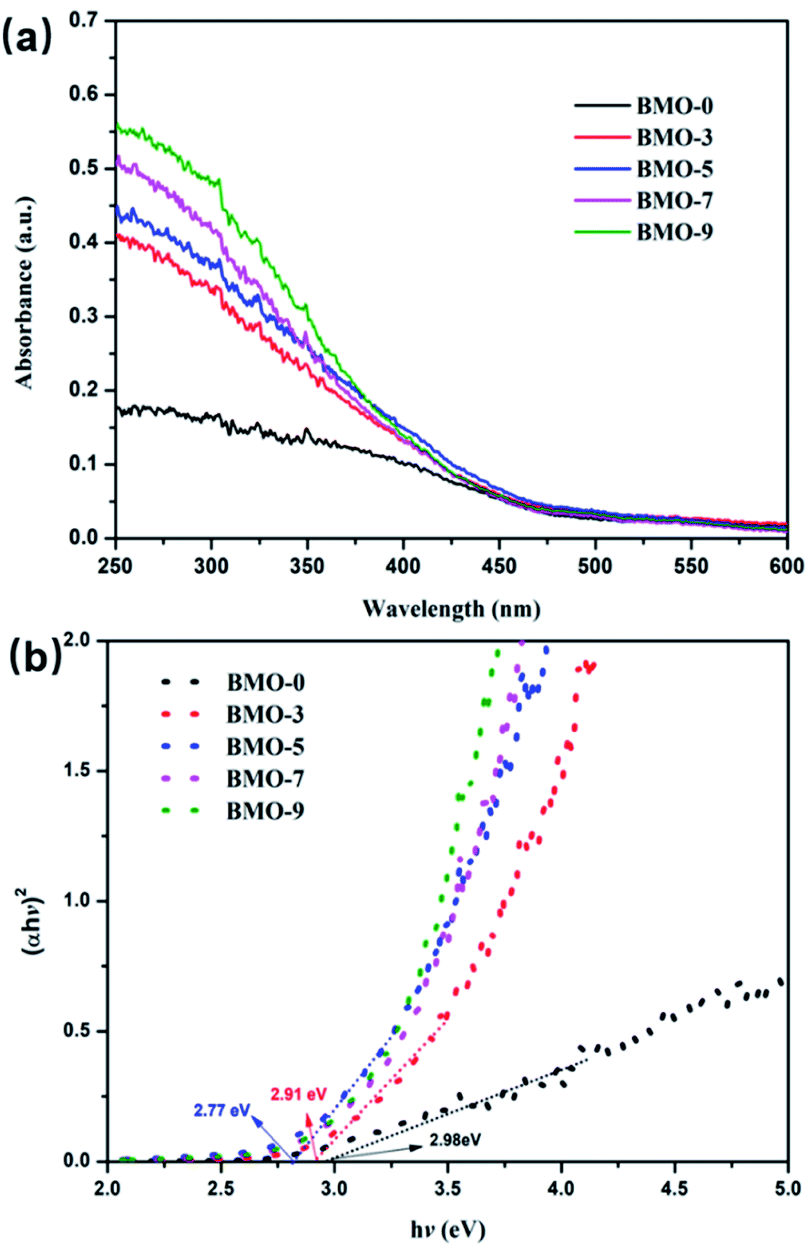 | ||
| Fig. 5 (a) UV-vis DRS spectrum of BMO-x (x = 0, 3, 5, 7, 9); (b) the plot of transformed KM function vs. photon energy of BMO-x (x = 0, 3, 5, 7, 9). | ||
3.3 Possible photocatalytic degradation mechanism
The photoelectron scavengers, such as O2, H2O2 and 1,4-benzoquinone, were used in the degradation of BDE209 as control experiments.40–42 As shown in Fig. S6,† only 10–20% of BDE209 were degraded in the presence of photoelectrons scavengers. The photocatalytic ability of BWO-5 to BDE209 was largely prohibited compared with that conducted in the sole N2 atmosphere. As we all know, O2˙− and ˙OH are usually derived from the reduction of O2 and H2O2 by photoelectrons in photo-oxidation systems.43,44 Therefore, it can be proved that photoelectrons are mainly involved in the degradation of BDE209 directly and play decisive role under an oxygen-free atmosphere, rather than O2˙− and ˙OH reactive oxide species.The relative intensities of the three kinds of nona-BDEs obtained by removing one bromine from BDE209 can be used to speculate on the debromination mechanism of BDE209.23,37 As shown in Fig. 6a, the para-, meta- and ortho-position debromination products of BDE209 are labeled as BDE208, BDE207 and BDE206. In general, C–Br bond in the ortho-position has the lowest dissociation energy,8 compared with that in the meta- and para-position, resulting in a preferred debromination in the ortho-position. Herein, the relative peak area was in the order of BDE207 > BDE208 > BDE206, indicating that the C–Br bond in meta-position is weakened during the photocatalytic process. A possible catalytic BDE209 debromination mechanism is proposed as follows. Oxygen vacancies at the bismuth tungstate interface can provide defects for the adsorption of BDE209 in a Br–O bond mode. Among the three adsorption modes shown in the Fig. 6b, the ortho-Br atom is difficult to access the catalyst surface due to the steric hindrance effect, resulting in the lowest yield of BDE206 among the three nona-BDEs products. And the meta-Br atom is easy to be absorbed by oxygen atoms around oxygen vacancies in the end-on mode, thereby realizing the activation of the C–Br bond. Upon visible light irradiation, the photogenerated electrons will transfer to the oxygen vacancies through the conduction band, and then through the Br–O halogen bond to break the weakened C–Br bond, thereby realizing the first step of debromination.
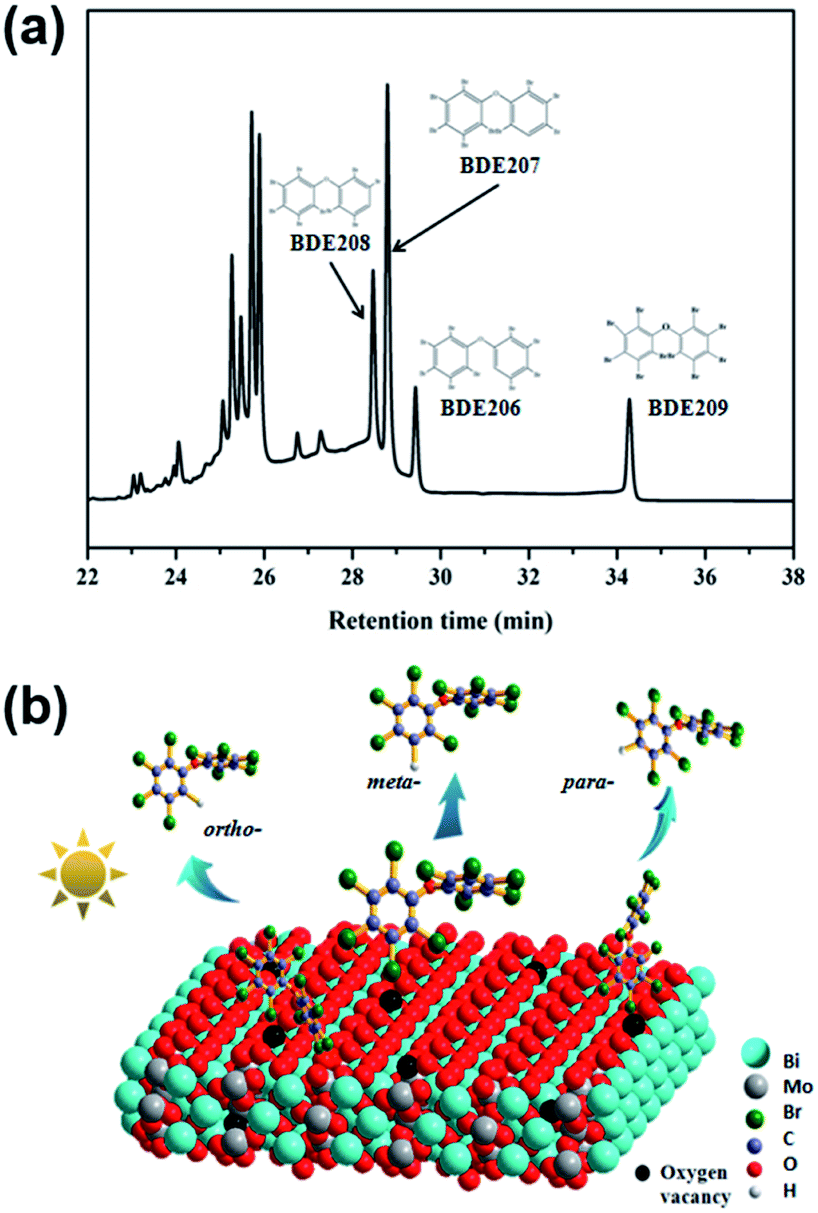 | ||
| Fig. 6 (a) GC-μECD chromatograms of BDE209 degradation products over BMO-5 at 20 min. (b) Possible adsorption and debromination model of BDE209 on oxygen vacancies enriched Bi2MoO6. | ||
4. Conclusions
In conclusion, oxygen vacancies enriched Bi2MoO6 is firstly applied in the photoreduction debromination of PBDEs under the visible light. The introduction of oxygen vacancies not only promotes the red-shift of light absorption band by Bi2MoO6, but also activates the C–Br bond through the formation of Br–O halogen bonds, thus realizing efficient visible light reduction of BDE209. The photocatalytic efficiency showed a trend of increasing first and then inhibiting with the increase of oxygen vacancies. Among the BMO-x (x = 0, 3, 5, 7, 9) materials, BMO-5 displayed the highest degradation rate for BDE209. In addition, BMO-5 exhibited a good photocatalytic activity and light stability after repeated three times. The activation adsorption mode inferred by tracking analysis of the degradation process showed that meta-position adsorption mode is the main adsorption configuration during the activation process, while the ortho-position adsorption mode is relatively difficult. Thence, oxygen vacancy-dominated photocatalytic BDE209 process is a position-selective multi-electron reduction process. The study provides a feasible strategy for the tunable introduction of oxygen vacancies on semiconductors for efficient photocatalytic treatment of halogenated POPs.Author contributions
Qin Li: investigation, visualization, writing-original draft. Xueqing Jin: investigation, writing-original draft. Meiying Yang: investigation, visualization, Qi Shen: supervision, resources, writing-review & editing, Chunyan Sun: conceptualization, supervision, resources.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The generous financial is supported by the National Natural Science Foundation of China (No. 22006100) and the Natural Science Foundation of Zhejiang Province (No. LQ19B070003). The authors gratefully acknowledge Prof. ChunCheng Chen from Institute of Chemistry, Chinese Academy of Sciences for his guidance on this research.Notes and references
- C. Ibeto, E. Aju and B. Imafidon, Environ. Sci. Pollut. Res., 2021, 28, 32374–32385 CrossRef CAS PubMed.
- S. M. Tongu, R. Sha'Ato, J. O. Okonkwo, O. I. Olukunle and I. S. Eneji, Environ. Monit. Assess., 2021, 193, 244 CrossRef CAS PubMed.
- K. S. Betts, Environ. Sci. Technol., 2000, 34, 222A–226A CrossRef PubMed.
- Y. K. Zhao, Y. Y. Wang, H. W. Ji, W. H. Ma, C. C. Chen and J. C. Zhao, Prog. Chem., 2017, 29, 911–918 CAS.
- Y. S. Keum and Q. X. Li, Environ. Sci. Technol., 2005, 39, 2280–2286 CrossRef CAS PubMed.
- A. C. Gerecke, P. C. Hartmann, N. V. Heeb, H. P. E. Kohler, W. Giger, P. Schmid, M. Zennegg and M. Kohler, Environ. Sci. Technol., 2005, 39, 1078–1083 CrossRef CAS PubMed.
- K. Nose, S. Hashimoto, S. Takahashi, Y. Noma and S. Sakai, Chemosphere, 2007, 68, 120–125 CrossRef CAS PubMed.
- C. Y. Sun, D. Zhao, C. C. Chen, W. H. Ma and J. C. Zhao, Environ. Sci. Technol., 2009, 43, 157–162 CrossRef CAS PubMed.
- Y. Y. Shao, W. D. Ye, C. Y. Sun, C. L. Liu, Q. Wang, C. C. Chen, J. Y. Gu and X. Q. Chen, RSC Adv., 2018, 8, 10914 RSC.
- M. Lei, N. Wang, S. Guo, L. H. Zhu, Y. B. Ding and H. Q. Tang, Chem. Eng. J., 2018, 345, 586–593 CrossRef CAS.
- E. B. Miller, E. M. Zahran, M. R. Knecht and L. G. Bachas, Appl. Catal., B, 2017, 213, 147–152 CrossRef CAS.
- C. Y. Sun, W. Chang, W. H. Ma, C. C. Chen and J. C. Zhao, Environ. Sci. Technol., 2013, 47, 2370–2377 CrossRef CAS PubMed.
- G. D. Sheng, Y. Y. Shao, W. D. Ye, C. Y. Sun, C. C. Chen, J. C. Crittenden and C. L. Liu, ACS Sustainable Chem. Eng., 2018, 6, 6711–6717 CrossRef CAS.
- J. S. Hu, J. Li, J. F. Cui, W. J. An, L. Liu, Y. H. Liang and W. Q. Cui, J. Hazard. Mater., 2020, 384, 121399 CrossRef CAS PubMed.
- F. Fu, H. D. Shen, X. Sun, W. W. Xue, A. Shoneye, J. N. Ma, L. Luo, D. J. Wang, J. G. Wang and J. W. Tang, Appl. Catal., B, 2019, 15, 150–162 CrossRef.
- L. Zhang, W. Z. Wang, D. Jiang, E. P. Gao and S. M. Sun, Nano Res., 2015, 8, 821–831 CrossRef CAS.
- X. W. Zhu, J. M. Yang, X. L. Zhu, J. J. Yuan, M. Zhou, X. J. She, Q. Yu, Y. H. Song, Y. B. She, Y. J. Hua, H. M. Li and H. Xu, Chem. Eng. J., 2021, 422, 129888 CrossRef CAS.
- J. Yu, F. Chen, X. W. Li, H. W. Huang, Q. Y. Zhang, S. Q. Su, K. Y. Wang, E. Y. Mao, B. Mei, G. Mul, T. Y. Ma and Y. H. Zhang, Nat. Commun., 2021, 12, 4594 CrossRef PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- X. Z. Chen, N. Li, Z. Z. Kong, W. J. Ong and X. J. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC.
- J. Lee, D. C. Sorescu and X. Y. Deng, J. Am. Chem. Soc., 2011, 133, 10066–10069 CrossRef CAS PubMed.
- S. Gao, Z. T. Sun, W. Liu, X. C. Jiao, X. L. Zu, Q. T. Hu, Y. F. Sun, T. Yao, W. H. Zhang, S. Q. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- T. T. Xu, M. Y. Yang, C. C. Chen, R. Duan, Q. Shen and C. Y. Sun, Appl. Surf. Sci., 2021, 548, 149243 CrossRef CAS.
- W. Z. Yin, W. Z. Wang and S. M. Sun, Catal. Commun., 2010, 11, 647–650 CrossRef CAS.
- L. J. Xie, J. F. Ma and G. J. Xu, Mater. Chem. Phys., 2008, 110, 197–200 CrossRef CAS.
- H. P. Li, J. Y. Liu, W. G. Hou, N. Du, R. J. Zhang and X. T. Tao, Appl. Catal., B, 2014, 160–161, 89–97 CrossRef CAS.
- X. Xu, X. Ding, X. L. Yang, P. Wang, S. Li, Z. X. Lu and H. Chen, J. Hazard. Mater., 2019, 364, 691–699 CrossRef CAS PubMed.
- L. X. Wu, C. Fu and W. X. Huang, Phys. Chem. Chem. Phys., 2020, 22, 9875–9909 RSC.
- M. G. Raj, E. Vijayakumar, B. Neppolian, S. K. Lakhera and A. J. Bosco, RSC Adv., 2021, 11, 25511 RSC.
- Q. D. Li, X. W. Zhu, J. M. Yang, Q. Yu, X. L. Zhu, J. Y. Chu, Y. S. Du, C. T. Wang, Y. J. Hua, H. M. Li and H. Xu, Inorg. Chem. Front., 2019, 7, 597–602 RSC.
- S. C. Chen, H. Wang, Z. X. Kang, S. Jin, X. D. Zhang, X. S. Zheng, Z. M. Qi, J. F. Zhu, B. C. Pan and Y. Xie, Nat. Commun., 2019, 10, 788 CrossRef CAS PubMed.
- M. Y. Yang, T. T. Xu, X. Q. Jin, Q. Shen and C. Y. Sun, Appl. Surf. Sci., 2022, 581, 152439 CrossRef CAS.
- A. Sarkar and G. G. Khan, Nanoscale, 2019, 11, 3414–3444 RSC.
- Q. He, Y. H. Ni and S. Y. Ye, RSC Adv., 2017, 7, 27089–27099 RSC.
- Y. C. Hao, X. L. Dong, S. R. Zhai, X. Y. Wang, H. C. Ma and X. F. Zhang, RSC Adv., 2016, 6, 35709–35718 RSC.
- X. Y. Kong, Y. Y. Choo, S. P. Chai, A. K. Soh and A. R. Mohamed, Chem. Commun., 2019, 52, 14242–14245 RSC.
- Y. H. Lv, W. Q. Yao, R. L. Zong and Y. F. Zhu, Sci. Rep., 2016, 6, 19347 CrossRef CAS PubMed.
- Y. H. Lv, Y. F. Liu, Y. Y. Zhu and Y. F. Zhu, J. Mater. Chem. A, 2014, 2, 1174 RSC.
- C. Zhou, N. F. Ye, X. H. Yan, J. J. Wang, J. M. Pan, D. F. Wang, Q. Wang, J. X. Zu and X. N. Cheng, J. Materiomics, 2018, 3, 238–246 CrossRef.
- C. H. Chiou, C. Y. Wu and R. S. Juang, Chem. Eng. J., 2008, 139, 322–329 CrossRef CAS.
- R. Scotti, M. D'Arienzo, F. Morazzoni and I. R. Bellobono, Appl. Catal., B, 2009, 88, 323–330 CrossRef CAS.
- H. Wu, X. Y. Xu, L. Shi, Y. Yin, L. C. Zhang, Z. T. Wu, X. G. Duan, S. B. Wang and H. Q. Sun, Water Res., 2019, 167, 115110 CrossRef CAS PubMed.
- L. Y. Ding, M. Li, Y. K. Zhao, H. N. Zhang, J. T. Shang, J. B. Zhong, H. Sheng, C. C. Chen and J. C. Zhao, Appl. Catal., B, 2020, 266, 118634 CrossRef CAS.
- Y. X. Wang, X. Li, S. N. Liu, Y. Liu, T. Kong, H. Y. Zhang, X. G. Duan, C. M. Chen and S. B. Wang, ACS Catal., 2022, 12, 2770–2780 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01762h |
| This journal is © The Royal Society of Chemistry 2022 |