
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide therapeutics in the management of metastatic cancers
Debopriya Bose†
,
Laboni Roy†
and
Subhrangsu Chatterjee
 *
*
Department of Biophysics Bose Institute, Unified Academic Campus EN 80, Sector V, Bidhan Nagar, Kolkata 700091, WB, India. E-mail: subhrangsu@gmail.com
First published on 2nd August 2022
Abstract
Cancer remains a leading health concern threatening lives of millions of patients worldwide. Peptide-based drugs provide a valuable alternative to chemotherapeutics as they are highly specific, cheap, less toxic and easier to synthesize compared to other drugs. In this review, we have discussed various modes in which peptides are being used to curb cancer. Our review highlights specially the various anti-metastatic peptide-based agents developed by targeting a plethora of cellular factors. Herein we have given a special focus on integrins as targets for peptide drugs, as these molecules play key roles in metastatic progression. The review also discusses use of peptides as anti-cancer vaccines and their efficiency as drug-delivery tools. We hope this work will give the reader a clear idea of the mechanisms of peptide-based anti-cancer therapeutics and encourage the development of superior drugs in the future.
 Debopriya Bose | Debopriya Bose is currently pursuing her PhD in the laboratory of Dr Subhrangsu Chatterjee, Associate Professor, Department of Biophysics, Bose Institute, Calcutta, India. She received her M.Sc. degree from Bose Institute, University of Calcutta, India (2018) with a specialization in Molecular and Cellular Biology. Her M.Sc. dissertation deals with cancer immunity. She is highly specialized in the field of tetraplex biology and her research focuses on non-canonical structures in DNA and the role of proteins in maintaining these structures. |
 Laboni Roy | Laboni Roy obtained her B.Sc. degree from University of North Bengal in 2015 and M.Sc. degree in 2017 from Vidyasagar University, Midnapur, India. She is currently pursuing her PhD from the Department of Biophysics at Bose Institute under the principle supervision of Dr Subhrangsu Chatterjee. Her focus of research is to design DNA-based aptamers that alter cellular biology through G-quadruplex regulation. |
 Subhrangsu Chatterjee | Dr Subhrangsu Chatterjee is an Associate Professor in the Department of Biophysics, Bose Institute, Calcutta, India. He has obtained his PhD degree in 2008 from Uppsala University, Department of Organic and Biochemistry, Uppsala, Sweden. After that he was a postdoctoral fellow at the Department of Biochemistry, University of Alberta, Alberta, Canada. Deing astructural and chemical biologist his current research interest focusses on the development of peptide and small-molecule based therapeutic molecules. He is also interested in uncovering the underlying disease mechanisms of various human disorders including Cancer and Diabetes. He has published many papers and review articles in high impact peer reviewed journals including JACS, Nucleic Acid Research, PNAS, JMC etc. covering high quality frontier research. He invented a tetrapeptide which locks therapeutic insulin and makes it thermostable and bioactive upto 65 °C. |
Introduction
Peptides have emerged as a fascinating tool in the field of therapeutics due to their enhanced specificity, extensive biological activity, high membrane permeability and low manufacturing costs. Intense research has contributed various short therapeutic peptides, many of which are undergoing clinical trials.1 These short peptides (20–30 residues) are significantly less immunogenic and more stable at room temperature compared to full length proteins and antibodies. They also show enhanced organ and tumor permeability. Therapeutic peptides can be classified as natural or synthetic based on their source.2Cancer remains a leading health concern threatening the lives of millions of patients worldwide. Although the last two decades have witnessed a phenomenal development in modes and tools of cancer therapeutics, the most frequent treatment modalities still include chemotherapy along with surgery and radiotherapy.1 Drug resistance, off-target effects and other adverse physiological effects often prohibit the use of many of these agents.1 Peptide drugs may provide a valuable alternative to existing solutions as tumor cells have been found to harbour receptors for peptides.1 Anti-cancer peptides (ACPs) have traditionally been grouped into three categories, namely: pro-apoptosis, inhibitory and necrosis inducing.1 Pro-apoptotic and necrosis inducing peptides bring about apoptosis or necrosis of cancer cells respectively. Inhibitor peptides function by binding to the integrin receptors on cancer cells. Unlike pro-apoptotic or necrotic peptides, these peptides are unable to penetrate within the cells. Instead they function as adhesion molecules having the Arg–Gly–Asp (RGD) sequence along with other functional groups derived from the conserved sequences of integrin binding ligands. Peptide binding to integrins compromises the motility of cancer cells, thereby inhibiting migration and blocking metastasis.3 Deaths in cancer patients are most often associated with complications due to metastasis. During malignant transformation, cells generate a host of mutations which allow them to obtain the properties of cancer. These features allow a potential tumorigenic cell to surmount machineries that limit unchecked growth and division of healthy cells.4
Developments in the last two decades has shed light on unique modes via which ACPs block metastasis. Peptides have been used as vaccines or fused with anti-metastatic agents. Additionally, peptides designed to target a specific pathway or protein involved in the metastatic cascade have been reported (Fig. 1). In this review, we will discuss how peptides have been used in various strategies to manage and inhibit metastatic tumors. In this regard, we will give special importance to ACPs that have already reached clinical trial. Additionally, we will try to shed light on the uses of peptides as drug-delivery agents.
Types of ACPs and their mode of action
ACPs have been grouped as pro-apoptotic, necrotic and inhibitory based on their mode of action. Recently peptides have been utilised in novel strategies to manage and eradicate metastatic cancers. In this section, we will discuss the functioning of these ACP classes along with their advantages and drawbacks.Pro-apoptotic peptides
Apoptosis allows the removal of irreparably damaged or stressed cells in a programmed manner. The extrinsic pathway of apoptosis is initiated when ligands bind to cellular death-causing receptors, thus generating the death-inducing signalling complex (DISC).5 The intrinsic apoptotic pathway is initiated when a cell undergoes irrecoverable stress, thus causing apoptotic proteins to be released from mitochondrial inter-membrane space.5 Different players of the apoptotic cascade serve as targets for anti-cancer therapy and peptides have been designed to target them. These cause restoration of a defective apoptotic pathway and lead to death of cancer cells. Pro-apoptotic peptides have been reviewed in detail elsewhere.5 Researchers have developed mimics of ligands that bind death receptors, which cause the formation of the DISC complex.5 Examples of such ACPs include the TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) mimicking M1 peptide. This peptide binds the TRAIL receptor 2 (TR2) thus generating pro-apoptotic downstream signalling processes. The peptide is effective in inducing apoptosis only in its multimeric form, as TRAIL works as a homotrimer.6 The Bcl-2 family composed of 25 pro and anti-apoptotic proteins, are major determinants of fate of a cell. A disruption in the balance of pro and anti-apoptotic members leads to either apoptosis or unregulated cell survival. Anti-apoptotic Bcl-2 family members have been targeted by BH3 mimetic peptides. Interaction between these peptides and anti-apoptotic proteins free the pro-apoptotic Bcl-2 members. Further, this leads to the formation of oligomeric Bax and Bak, thereby causing apoptosis.7 In addition to serving as ACPs, BH3 mimetic peptides have also been used to characterize the interaction between Bcl-2 family members. Pro-apoptotic peptides based on the BH3 domains of the Bim protein were used for this purpose. The peptides were found to intrinsically self-associate in aqueous environments and this interaction was alleviated in the presence of the anti-apoptotic Bcl-xL component.8The p53 protein is mutated in more than 50% of human cancers and various agents have been designed based on p53.9 Peptides that have similar biological functions as p53 or can restore p53 function are used.5 A few examples of such peptides include the peptide C1 which binds one of the two negative regulatory regions (NRR2) of p53, leading to its activation.10 Another example is given by the azurin based peptide P28. Azurin is a protein from Pseudomonas aeruginosa that binds and stabilizes p53. The P28 peptide is a short fragment from azurin which penetrates the cell and stabilizes p53.10 Inhibitor of apoptosis proteins (IAPs) contain a conserved baculovirus IAP repeat (BIR) region. The BIR region in these proteins bind pro-caspases and convert them to caspases, thus promoting apoptosis. The expression of IAP proteins is altered in various cancers.5 The mitochondrial SMAC (second mitochondria-derived activator of caspases) proteins could bind IAPs, thus freeing the active caspases. Thus, peptidomimetics mimicking SMAC proteins based on the AVPF sequence have been designed as ACPs.11 The effector proteins in the apoptotic cascade are the caspases. Caspases once activated cleave various target proteins bringing about cell death. RGD peptides, although mainly used as inhibitory peptides, may activate pro-caspase 3 and function as a pro-apoptotic peptide.12 The microtubule targeting agent (MTA) paclitaxel (PTX) causes both cell-cycle arrest in the G2/M stage and induces apoptosis by binding to Bcl-2. It was found that the binding site of PTX on Bcl-2 and β-tubulin, mimics that of Nur77 receptor. Two peptides (PEP1 and PEP2) mimicking Nur77 domains were designed as pro-apoptotic agents. Of these PEP2 showed higher biological activity and later cyclic peptides derived from it were found to induce high levels of apoptosis in both leukemic and solid tumor cell lines.13
Cells in the tumor micro-environment such as the tumor-associated macrophages (TAMs) are altered to aid cancer progression. TAMs consist of both M1 and M2 macrophages and lead to a poor prognosis. An M1 macrophage is a pro-inflammatory form that is reported to be anti-cancer while the M2 form is pro-cancer.14,15 M2-based TAMs have been targeted by the pro-apoptotic peptide MEL-dKLA. Melittin (MEL) binds to M2 while d(KLAKLAK)2 (dKLA) induces apoptosis. Death of these TAMs inhibit tumor growth and angiogenesis.16 The KLAK peptide has been fused with several peptide and non-peptide conjugates for targeted apoptosis of cancer cells. For example, it has recently been conjugated with an AP1-ELP domain. AP1 specifically targets the IL4 receptor overexpressed in many cancers and ELP (elastin-like polypeptide) forms a micelle containing these two peptides in active state.17 Antimicrobial peptides (AMPs) such as the peptide B1 can selectively bind tumor cells by electrostatic interaction and induce apoptosis by destabilizing the mitochondrial membrane. The sequence of this peptide was further modified either by replacing lysine residues with arginine or by insertion of hydrophobic residues to obtain highly specific pro-apoptotic cell penetrating peptides.18 Another human AMP that has shown anti-cancer properties is the human cathelicidin LL-37 that is secreted from monocytes, macrophages and various epithelial cells. The LL-37 peptide has been hypothesised to have dual effects on cancer cells via interaction with the cell-membrane and various receptors such as ERBb2 (or HER2), EGFR and FRP2.19 Various shorter peptides have also been designed from the LL37 scaffold, the shortest of which is named: 17BIPHE2. This peptide has lower toxicity compared to LL37 and induces apoptosis of cancer cells via modulation of multiple targets.20
Pro-apoptotic peptides have shown great potential as ACPs but there remain hurdles in their development and clinical application. Often these peptides show poor abilities to be internalized into tumor cells and thus they have been conjugated with various compounds like antibodies and nanoparticles to allow better uptake. Additionally, these compounds suffer from drawbacks such as lack of selectivity against cancer cells, poor pharmacokinetic profiles, generation of an immunogenic response, and sub-optimal accumulation within the subcellular components.5
Necrotic peptides
A huge number of necrotic ACPs have been reported over the years. Additionally, several peptides have been reported which cause both apoptotic and necrotic cell death.15 The MDM2 binding site of p53 has been used to design the p53(15)Ant peptide, wherein the p53 residues 12–26 was fused to Antennapedia (Drosophila carrier protein). The conjugated peptide assumes a highly hydrophobic, α-helical structure and exhibits increased membrane disruptive functions leading to necrosis. The necrotic ability of the peptide does not depend on the p53 status of the tumor cells.21 The p53 residues in the above peptide when conjugated with a membrane targeting sequence independently bind plasma membrane localized MDM2 leading to necrosis of cancer cells.22,23 This peptide was named PNC-27 and other p53 residues also showed lesser but significant anti-cancer activities.23 AMPs have been used widely to create both pro-apoptotic and necrotic peptides. These possess a net positive charge and interact with bacterial cell membranes to kill the pathogen. Cancer cells are also marked by a net negative charge on their membranes due to the enhancement of anionic membrane components. Membrane disruption due to AMPs is dependent on factors such as secondary structure, overall charge, size, amphipathicity and hydrophobicity.24 The effect of hydrophobicity is clearly demonstrated by the AMP chensinin-1B and its lipoanalog PA-C1b which has a higher hydrophobicity. It was observed in comparative studies that while the parent peptide induce cell membrane disruption, PA-C1b mainly induced apoptosis via upregulation of pro-apoptotic Bcl2 family members. PA-C1b also increased ROS levels and both peptides caused the loss of mitochondrial membrane potential.25 Recently a tool named A-CaMP has been developed by scientists which can identify AMPs and ACPs by analysis of medical data throughout the literature. This tool may be highly useful in characterization of novel AMPs useful in various cancers.26 Harmoniasin, an AMP obtained from Harmonia axyridis (ladybug), was used to generate synthetic analogues including the homodimer HaA4 which was found to exhibit high anti-cancer activity. HaA4 induced both caspase-dependent apoptosis and necrosis in leukemic cell lines. The prevalence of apoptosis or necrosis, in response to peptide treatment, depends on HaA4 concentration.27 A similar mechanism has also been observed for piscidin-1, an AMP from Morone saxatilis × M. chrysops (hybrid striped bass).28 A range of AMPs causing necrosis in tumor cells have been reported including D-K6-L9,29 Dermaseptin B2,29 MPI-1,30 etc. Lactoferrin (LF) is an AMP found in mammals which is cleaved into lactoferricin (LFC) upon digestion by pepsin. The LF11-322 peptide designed from sequences of human LFC shows necrosis of tumor cells.31 The peptide brevinin-1RL1 obtained from skin secretions of the frog Rana limnocharis, induces both apoptosis and necrosis of cancer cells due to its cationic nature and intermolecular disulphide bridge.32 The reader is directed to other reviews for knowledge on the action of AMPs against cancer.33 A novel necrosis inducing peptide was reported by Seo et al., involving the MTD (mitochondrial targeting domain) of the Noxa protein. This peptide induces necrosis by promoting calcium release from the mitochondria to the cytosol.34 Later Ji-Young et al., fused this peptide with a tumor homing sequence, binding to Neuropilin-1 (NRP-1) receptor. NRP-1 functions as a co-receptor for VEGF and is present in most tumor types and thus serves as a good target. The TU17:MTD peptide causes necrosis of tumor cells within 30 minutes of application and thus its serum-stability is not an issue.35 Nisin ZP, an AMP from the Gram positive bacterium Lactococcus lactis induces pore formation in the plasma membrane of cancer cells. This peptide has shown to be effective against various cancers and leads to a decrease in the mitochondrial membrane potential leading to apoptosis. Additionally, the peptide causes higher levels of intracellular ROS in cancer cells which may lead to necrosis or apoptosis.36 Another cathelicidin, named BMAP-27 from bovine also has necrotic properties against cancer cells. This peptide targets the cancer cell membrane leading to disruption.37 BMAP was recently conjugated with a second peptide melittin which increased its anti-cancer properties. This conjugated peptide showed both apoptosis and necrosis of cancer cells.38 The human AMP defensin beta-1 is also involved in suppressing cancer cells in vivo and may serve as good target for the development of therapeutic peptides. The peptide targets a range of factors including proteins of the MAPK pathway and also regulates other immune cells. The exact mode of action of the peptide is as yet unknown and should be studied further.39 The α-helical peptides with high hydrophobicity induces necrosis through disruption of the cancer cell membrane. The specificity of these peptides towards cancer cell membranes is due to a lower deposition of cholesterol and different distribution of phospholipids compared to normal cells.29A major obstacle in the development and use of AMPs in therapeutics is presented by their polyfunctional nature. Structural optimization of AMPs to overcome this problem proves difficult as well. Additionally, AMPs are often unstable in vivo and are cleared rapidly from the system. Scientists have addressed this issue by inserting D-amino acids in the AMP sequence. Additionally, cyclisation, PEGylation and liposomal delivery are used to protect AMPs from digestive enzymes and other proteases.40
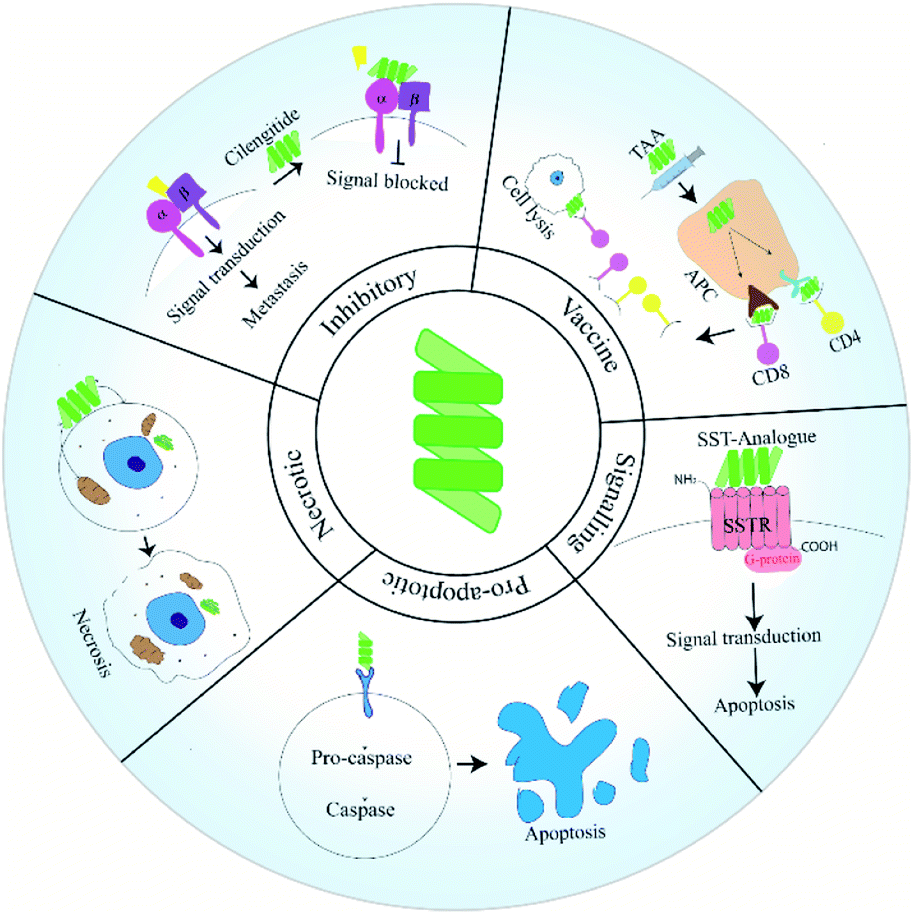 | ||
| Fig. 1 Types of peptides used in anti-cancer therapy. | ||
Integrins as targets for anti-cancer therapy
Cancer cells are often characterized by an increased expression of integrins and these mediate binding to extracellular matrix (ECM) and act as receptors for extracellular signalling molecules. Intracellular signals lead to integrin activation causing exaggerated interaction with ECM ligands. The bidirectional signalling via integrin receptors are categorised as outside-in and inside-out respectively.41 Inside-out signals activate proteins like talins and kindlins which alter the conformation of integrins to an active form. Activated integrins bind ligands which generate outside in signals and trigger oligomerization of integrins. These oligomeric complexes lead to formation of an adhesome which triggers pathways for cancer cell survival and epithelial to mesenchymal transition (EMT)41 (Fig. 2). Ligand binding to integrins also switches on several survival pathways.42 Proliferation of a cancerous mass necessitates the formation of new blood vessels to provide nutrients to growing cells. So angiogenesis is initiated and thereafter the cancer mass proliferates rapidly and metastatic cells are generated. Angiogenesis thus serves as a precursor to metastasis and involves various types of integrins. The induction of angiogenesis enables cell-cycle re-entry of vascular cells and these often overexpress specific integrins. Thus peptides inhibiting angiogenesis also block metastasis. ACPs specifically inhibiting angiogenesis have been discussed in detail elsewhere.43 All integrins contain α- and β-subunits bonded by non-covalent interactions. Metastatic cells have a predominance of β3 integrins.44 18 α- and 8 β-subunits interact in several stoichiometries to form 24 heterodimers which interact with various ECM ligands via Arg–Gly–Asp (RGD), Glu–Ile–Leu–Asp–Val (EILDV), or Arg–Glu–Asp–Val (REDV) sequences.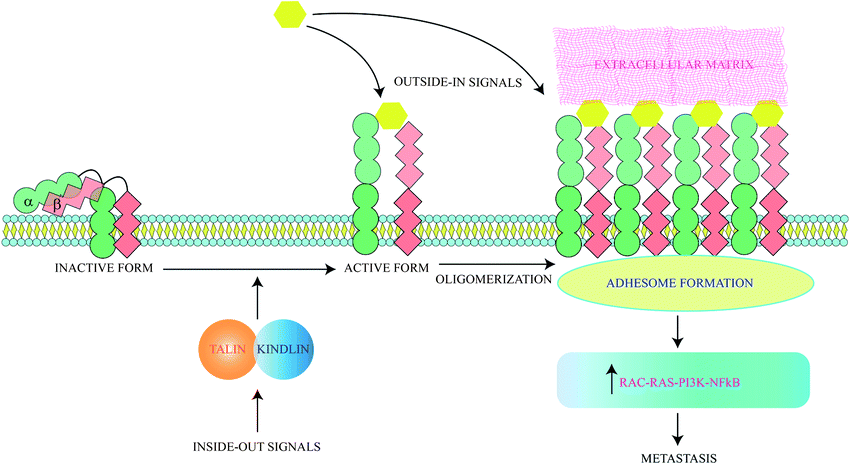 | ||
| Fig. 2 Integrins activate metastasis through both intracellular and extracellular signals. Both types of signals ultimately lead to oligomerization of integrins, leading to adhesome formation and metastasis. | ||
Anti-metastatic peptides targeting integrins
The integrins most commonly targeted by ACPs for anti-cancer therapy are the αvβ3 integrins, which are vital for EMT and cell survival.45 These integrins are also used for treatment of thrombosis46 and wound healing.47 RGD based peptides bind αvβ3 integrins mainly through electrostatic interactions supported by van der Waals forces and formation of water bridges.48,49 It has been observed that tryptophan residues although less abundant in protein sequences are widespread in protein–ligand interacting motifs. The binding affinity of integrins with RGD peptides can be improved via incorporation of tryptophan residues modified with halogen atoms. This modified tryptophan also enhances selectivity among various integrin subtypes.50 These peptides may be used alone or with nanoparticles.51,52 Along with acting as anti-metastatic therapeutic probes, the RGD peptide also acts an efficient targeting domain. For example, recently a RGD peptide was conjugated with an ACP obtained from bullfrog skin (modified temporin-La peptides) for better tumor targeting. Temporin-La is an AMP that kills cancer cells by membrane destabilization and the chimeric peptide showed higher efficacy by altering mitochondrial membrane potential.53,54 The specificity and efficacy of RGD based ACPs are controlled by the flanking amino acids and cyclised forms of these molecules are often more potent, specific and stable as cyclisation preserves specific RGD conformations. Incorporation of one D-amino acid into a cRGD (cyclic-RGD) induces kinks in the peptide backbone increasing αvβ3 and αvβ1 selectivity.55 Among the various d-cRGDs studied, cRGDfC showed highest integrin affinity followed by cRGDyK, cRGDfK and cRGDfV respectively.55 cRGD motifs like RGDC4 and RGD10 linked by di-sulphide bonds are also used as ACPs.56 RGD10 was used to generate liposomes for selective drug-delivery.57 N-Methylation enhances RGD specificity and combined with cyclisation was used to develop cilengitide (cyclo[Arg–Gly–Asp–D-Phe–N(Me)Val]).58 Cilengitide was developed from cRGDfV and binds αvβ3, αvβ1 and αvβ5 integrins.59 Cilengitide reached phase III clinical trials for treatment of glioblastoma but failed,60 although it is being investigated for other cancers. Cilengitide has also been found to increase the anti-cancer properties of other drugs such as erlotinib (TGF-β induced EMT inhibitor) when used as a combination therapy.61 Cilengitide was further modified to develop five new analogues which showed enhanced integrin binding.62 Multimeric cRGD peptides have higher integrin binding efficiency63 and previous studies have explained the higher efficacy of poly-RGD moieties.64 Lucie et al., generated a cyclo-decapeptide scaffold containing four cRGD molecules. The tetrameric cRGD showed improved αvβ3 binding and suppressed their lateral motion on the membrane. This causes clustering and aggregation followed by co-internalisation of the peptide and the integrins leading to simultaneous drug-delivery and reduction of αvβ3 mediated angiogenesis65 (Fig. 3). It is now known that the anti-cancer properties of multivalent ligands such as multimeric RGD moieties are strongly influenced by the nature of the linker used. Liu et al., have previously demonstrated that multivalent RGD peptides have the highest binding affinity to αvβ3 when linked by four units of ethylene glycol.66,67 Since activity of these ACPs are dependent on both association and dissociation kinetics between ligand and integrin receptors, Mizuno et al., have studied the effect of a semi-rigid (DPro–Gly)n linker on dissociation kinetics of hexavalent RGD peptides. They showed that this linker slowed the dissociation of hexavalent RGD via formation of additional bonds with αvβ3 integrins and resulted in better cellular uptake.68 RGD peptidomimetics like S247 also show antimetastatic and antiangiogenic activities in several cancers.58 RGD peptides are also used for cancer cell imaging69–71 and gene delivery.72–74 Internalizing RGD (iRGD) peptides are used to deliver therapeutic compounds to target cells. iRGD peptides contain an RGD sequence fused with the cell-penetrating C-end rule (CendR) motif.75 The CendR motif has a sequence RXXR/K and binds the NRP receptor initiating bulk import within cancer cells. This interaction inhibits metastasis independent to the RGD domain.75 The iRGD system allowed dual imaging and drug delivery when conjugated with the cisplatin pro-drug and a fluorescent ligand. The specific delivery of cisplatin reduced its associated side-effects.76 The iRGD peptide may also improve the efficiency of irradiation therapy by decreasing hypoxia at the tumor micro-environment. Hypoxia plays a key role in mediating irradiation resistance and iRGD allows the diffusion of oxygen molecules into tumor tissues alleviating hypoxia.77 The ProteoChip system was used to develop twelve αvβ3-antagonistic peptides from the hexapeptide library. Interestingly, the two hexapeptides with the highest antiangiogenic and antimetastatic activities contained the Ser–Asp–Val (SDV) and Gly–Asp–Val (GDV) sequences.78 Disintegrins are a family of non-enzymatic proteins found in snake venom that strongly inhibit integrin receptors. Disintegrins like atragin were used to generate synthetic peptides which inhibit cell migration by binding to integrins. These are enriched with cysteine disulphide bonds, resulting in enhanced proteolysis resistance.79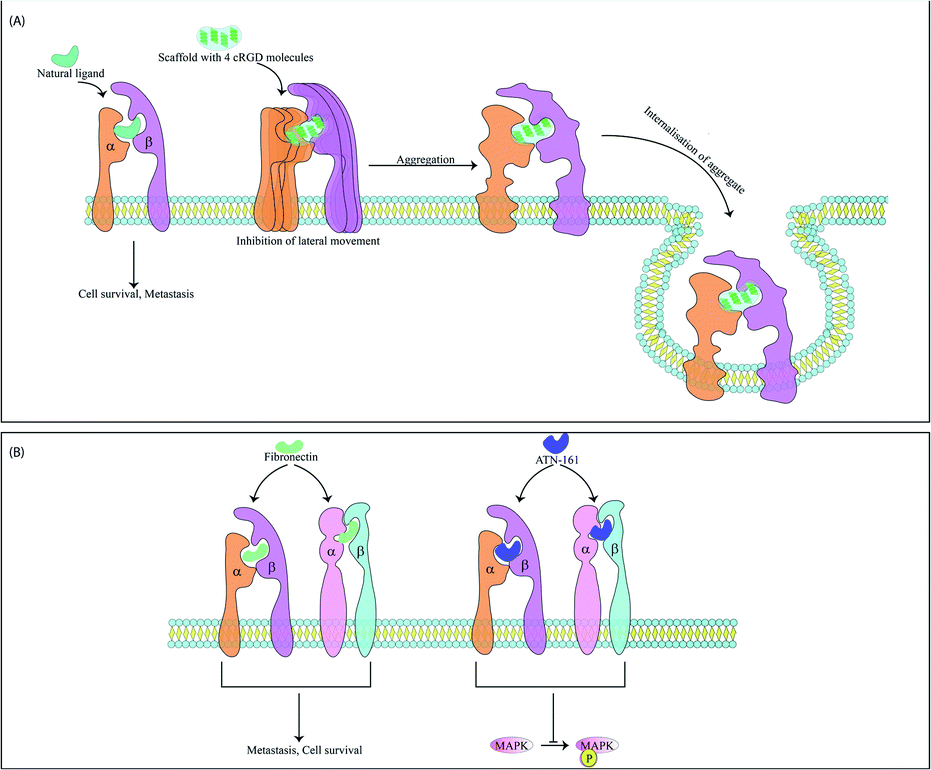 | ||
| Fig. 3 Mode of action of two peptide-based motifs targeting αvβ3 integrins and αvβ1 integrins. (A) A tetrameric cRGD cyclo-decapeptide scaffold binds multiple integrins and inhibits lateral movement of integrins. Arrested integrins are then aggregated and internalized within the cell. The scaffold is also internalized with the receptor. (B) ATN-161 has the binding motif of fibronectin and upon binding to fibronectin receptor downregulates MAPK phosphorylation and angiogenesis. | ||
αvβ1 serves as a receptor for fibronectin and is overexpressed in metastatic cells. The ACP ATN-161 has a non-RGD based sequence which matches the binding motif of fibronectin and upon integrin binding, significantly downregulated MAPK phosphorylation and several angiogenic cascades. The drug is interesting as it does not inhibit cell motility and migration in vitro but causes regression of tumor size in vivo and has reached phase II clinical trial80,81 (Fig. 3). Peptidomimetic ligands with specific functional groups endowing higher specificity to either αvβ3 or αvβ1 have also been developed.82–84 Another integrin overexpressed in several cancers and related to a poor outcome in patients is the αvβ6 integrin. This integrin has been targeted by Annette Altmann's group by two ACPs, namely, SFITGv6 and SFLAP3 which were developed by incorporating RGD containing octameric sequences into the sunflower trypsin inhibitor 1 (SFTI1) scaffold. The ACPs are highly stable and were used for cancer cell imaging in patients but need further improvement as they show some accumulation in kidneys.85 Another study utilised a RGD peptide sequence from the foot and mouth disease virus as a starting point to develop enzymatically stable cyclic peptide antagonists of αvβ6. The final candidate had a sub-nanomolar range affinity for αvβ6 without any significant cross-reactivity for other integrins.86 Another peptide has also been found in this virus that binds the αvβ6 integrins. This 20mer peptide named A20FMDV2 has previously proved useful as an imaging agent and αvβ6 antagonist but suffers due to its limited serum stability.87 To alleviate this problem, A20FMDV2 has been modified via cyclisation, retro-inversion and amino acid substitutions to yield more stable agents with higher therapeutic potential.88 Serum stability of this peptide has also been improved via conjugation with albumin binding components which enhances the half-life and tumor uptake of the peptide. These conjugates were further radiolabelled for aggravated and targeted tumor inhibition.89 The αvβ5 integrin heightens angiogenesis and metastasis and has recently been targeted by an ACP named RGDechi15D. RGDechi15D was developed by modification of the RGDechi peptide which contains a cRGD motif fused to echistatin C-terminal domain. RGDechi selectively binds αvβ3 without any cross-reactivity and causes tumor cell death while also reducing EMT.90 RGDechi15D was synthesised by mutating the 15th residue of echistatin C-terminal domain from homocitrulline to Asp. This homocitrulline residue is key to αvβ3 selectivity and aspartic acid shifts selectivity to αvβ5. RGDechi15D reduces cell adhesion, migration, invasion, angiogenesis and proliferation of cancer cells overexpressing αvβ5.91
Recently, a peptide mediated imaging technique for glioblastoma cells was reported. Transformed cells in glioblastoma show high intracellular levels of reactive oxygen species (ROS) and various imaging probes target these high ROS levels.92 The novel peptide RWrNR (Arg–Trp–(D-Arg)–Asn–Arg) was fused with a hydrocyanine moiety which serves as a ROS sensitive fluorophore. RWrNR shows higher affinity for αvβ3 compared to RGD-peptides and the fluorescent conjugate has high specificity for glioblastoma cells both in vitro and in vivo. Although highly efficient, the RWrNR peptide was limited due to its rapid clearance from the system. To circumvent this it was fused to 8-arm PEG to form PEGylated RWrNR which consisted of eight RWrNR peptides and thus has increased antiangiogenic and antimetastatic properties. PEGylation also reduces its renal clearance and increased its solubility and half-life.93 RWrNR when fused with the pro-apoptotic peptide KLA increased its specificity. Further, this conjugate was encapsulated in a biodegradable acid-sensitive nanogel, which allowed controlled peptide release within the acidic tumor cell microenvironment thus improving tumor specificity. Poly-L-arginine was a component of this nanogel and it upregulated the production of nitric oxide from TAMs. This increased the apoptosis of cancer cells expressing αvβ3 further.94
ACPs which alter tumor growth and block metastasis
The interaction of VEGF with its receptors switches on several proliferative and angiogenic pathways. VEGF binding to KDR/Flk-1 (kinase domain receptor) leads to cancer progression. The K237 peptide acts as a competitor for VEGF binding to KDR and reduces angiogenesis and metastasis in immunodeficient mice.95 The peptide CPQPRPLC derived from VEGF was analysed to identify the minimal motif for VEGF receptor binding. The RPL domain was thus identified and modified by retro-inversion to D(LPR). D(LPR) targets both VEGFR1 and NRP1 strongly inhibiting angiogenesis.43,96 The VEGF165 isoform binds VEGFR2 and the 6a-exon of VEGF has been used to develop competitors for this isoform43 (Fig. 4). Other growth factors such as FGF and TGFβ also play vital roles in cancer progression. ACPs inhibiting these factors bind either their soluble form or their receptors.43 The VEGF signalling axis is strongly inhibited by the tryptophan containing dipeptide WL leading to anti-angiogenic effects.97 The dipeptide drug IM862 (L-glutamine–L-tryptophan) also inhibits angiogenesis and reached phase III clinical trial for AIDS-Kaposi's sarcoma. The molecule failed but may be used for design of modified candidates98,99 (Fig. 4). NRP-1 acts as a co-receptor of VEGF and its extracellular domain has been targeted by several ACPs. Recently, its trans-membrane domain has also been targeted by the peptide MTP-NRP1 which blocks receptor dimerization thereby inhibiting proliferation and metastasis in breast cancer and glioblastoma100 (Fig. 4). Growth factor receptors such as EGFR also serve as cancer specific probes for targeting therapeutic molecules to tumor sites. For example, recently such a liposome probe has been designed which encapsulated doxorubicin and tariquidar. The probe contained an EGFR binding peptide named EMC along with a sequence cleaved by MMP2. The liposome specifically targeted cancer cells and showed high therapeutic potential.101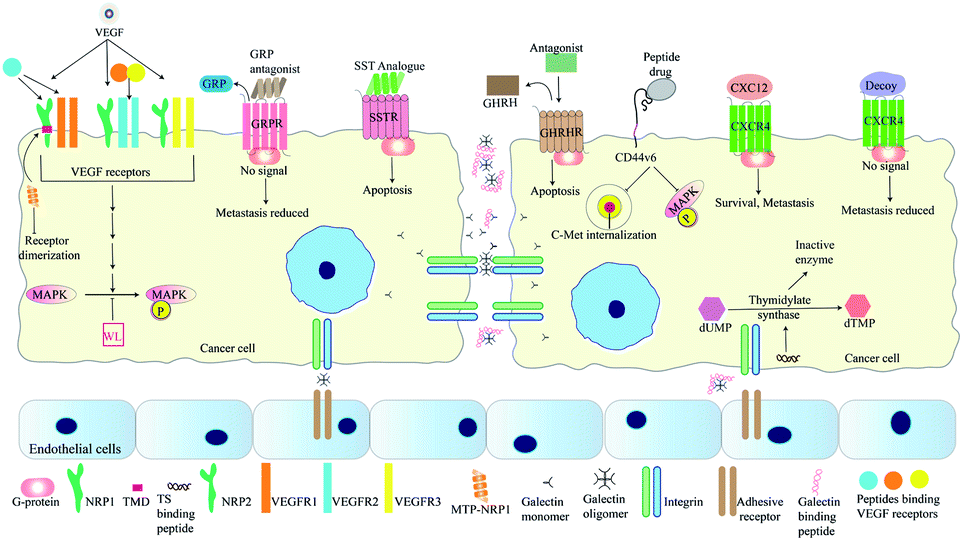 | ||
| Fig. 4 Peptides targeting cellular signalling pathways involved in metastasis. A range of peptides have been developed as antagonists for binding receptors such as VEGF receptors, GRPR, and CXCR4. MTP-NRP1 targets the TMD of NRP1, thereby inhibiting receptor dimerization. Analogues for SST with higher half-lives induces apoptosis of cancer cells and GHRH antagonists such as JMR-132 causes cell-cycle arrest followed by apoptosis. Peptide inhibitors of CD44v6 such as NLN and NEW inhibit C-Met internalization and MAPK phosphorylation. MAPK phosphorylation is also inhibited by the dipeptide WL. Inhibitors of thymidylate synthase reduce cancer proliferation as this enzyme is essential for DNA synthesis. Peptides blocking interaction between galectins and TFAg blocks cell-adhesion and reduces metastasis. | ||
The bombesin/gastrin-releasing peptides (BN/GRP) stimulate cancer progression through interaction with GPCRs. Peptide antagonists binding the GRP receptors inhibit cancer growth in various cancers98 (Fig. 4). Growth hormone releasing hormone (GHRH) interacts with its receptors in the pituitary gland and helps to control the levels of growth hormone (GH) secreted. Both GHRH and GH are involved in tumor growth, development and metastasis either directly or by controlling levels of other growth factors. Antagonists of GHRH like JMR-132 have been reported. This peptide arrests cancer cells in the S-phase and later induces apoptosis of cancer cells102 (Fig. 4). In 2011, Cardinale et al., designed multiple peptides from a small fragment of human thymidylate synthase (TS) which target the dimer interface of thymidylate synthase. These octapeptides trap this enzyme in an inactive confirmation. Inactivation of this enzyme reduces tumor growth as thymidylate synthase is vital for DNA synthesis98,103 (Fig. 4). XPNPEPZ a subtype of aminopeptidase-P serves as the receptor for the tumor metastasis targeting (TMT) peptide. The TMT decapeptide targets metastatic cancer cells specifically and has been used for targeted delivery of drugs. TMT conjugated liposomes were constructed and loaded with doxorubicin for specific inhibition of metastatic cancers.104 Lee et al., have developed a biomimetic peptide derived from collagen IV which is able to inhibit growth, angiogenesis and metastasis of triple negative breast cancer. This peptide targets β1 integrin and an unknown receptor to inhibit IGF1R and Met signalling pathways.105 The NGR (Asp–Gly–Arg) peptide motif has been used for drug-delivery through its receptor aminopeptidase-N (CD-13). CD-13 is widely expressed in metastatic cells. The NGR motif has been used for the targeted delivery of doxorubicin and c-myc siRNA.106,107 Besides the RGD and NGR based peptides, other peptides targeting tumor blood vessels include IF7, F3, cytotoxic T-lymphocyte (CTL), and CREKA. IF7 and F3 target annexin-1 and cell-surface nucleolins respectively, while CREKA and the cyclic decapeptides CLT1 and CLT2 target the proteins associated with blood clotting and induce more clotting thus amplifying the target for efficient drug delivery.108 The FAK–PAX interaction in breast cancer is associated with initiation of downstream signalling processes that lead to cell-adhesion, migration and survival. FAK (focal adhesion kinase) is activated by signalling cascades when extracellular ligands bind receptors such as integrins. This protein has a FAT domain that is then bound by the paxillin protein. This leads to the phosphorylation and activation of Pax which then serves as an adaptor for formation of multi-protein complexes leading to metastasis and cell-survival. Khan et al., have screened a range of reported anti-cancer peptides to identify those able to bind Pax protein and inhibit FAK–Pax interaction. By bioinformatics approaches they identified magainin as the best candidate along with two other peptides with high binding efficiency. Magainins were initially identified as ionophoric AMPs from Xenopus laevis which caused lysis of cancer cells.109 Magainin interacts with those Pax residues that are essential for FAT binding, thus inhibiting their interaction.110
CXC cytokines are involved in maintaining the migratory processes of both normal and cancer cells. The CXC12 cytokine is vital for metastasis and is over-expressed in both breast and prostate cancer cells. Additionally, its receptor CXCR4 is over-expressed in breast cancer while normal breast cells are devoid of both. The CXC12/CXCR4 signalling axis activates a range of pro-cancer responses in cancer associated fibroblasts (CAFs) of prostate carcinoma as well. The siRNA for CXC12 was specifically targeted to the CAFs by loading it onto a R9 cell penetrating peptide and coating the surface with antibodies against fibroblast activation protein-α, which is present on CAF surface.111 CXCR4 is a GPCR which activates several downstream cascades ultimately contributing to tumor growth and metastasis. Understandably, a wide variety of peptide decoys that bind CXCR4 has been reported. These function by binding CXCR4 and blocking its ability to activate downstream processes (Fig. 4). Other articles have described the types and functions of CXCR4 antagonists in vivid detail.112 Peptide density may play an important role in reduction of CXCR4 based signalling. CXCR4 molecules are present on lipid rafts and function as homodimers. Liposomes conjugated with a CXCR4 binding peptide named DV-1 shows maximum anti-migratory activity when the density of the peptide was 24k molecules per μm2. This effect is attributed to the similar distance of the binding pocket in the CXCR4 homodimer and the consecutive peptides.113 Peptide antagonists of CXCR4 like NT21MP and Nef-M1 reduce migratory properties of breast cancer and colon cancer cells. They also somewhat reverse the process of EMT and reduce drug-resistance properties.114–116 Another CXCR4 antagonist named peptide R inhibits cancer progression by altering the phenotype of TAMs from the M2 (pro-tumor) to the M1 (anti-tumor) form.117,118 The effectiveness of CXCR4 antagonists may further be increased by their dimerization. This has recently been achieved by Harms et al., who have shown that Y-shaped peptide conjugates linked by an albumin binding compound increased the efficacy and serum stability of the CXCR4 peptide antagonist EPI-X4 JM#21. This trend was also observed when other linkers such as PEG, PVA and PVP were used.119
In addition to receptors on the cell surface, intracellular signalling proteins have also been targeted via ACPs. For example, adaptor proteins such as Grb7 mediate important signalling cascades and serves as a therapeutic target. This protein contains a Src homology 2 (SH2) domain through which it interacts with cell-membrane based receptors such as HER2, HER3 and HER4. Interaction leads to phosphorylation and activation of Grb7 followed by mediation of cascades enhancing cell migration, invasion, growth and proliferation.120 The SH2 domain of this protein has been targeted by the 11 residue peptide G7-18NATE. This cyclic peptide although highly active was unable to appropriately penetrate the cell and thus was co-delivered by fusion with the CPP, penetratin.121,122 The G7-18NATE peptide has since been modified to increase its affinity and selectivity for Grb7 and the peptide G7-B7 was found to have the highest affinity but it was not delivered into the cell even in presence of penetratin.123 Recently, further modifications have been made to allow better permeability with increased affinity and specificity.120 It has now been recognised that many proteins promoting cell-survival and metastasis may contain cryptic segments that serve as growth inhibiting domains. These domains may be naturally exposed due to the presence of high concentrations of specific ligands in the microenvironment or due to other factors. These may be chemically synthesized to obtain the same effect. For example, alpha-fetoprotein (AFP) promotes cell growth but contains a growth inhibitory peptide (GIP) fragment which is exposed when AFP is in the denatured molten globular state. This GIP fragment has been chemically synthesized and has growth inhibiting and anti-metastatic properties.124
The somatostatin peptide (SST) binds its receptor (SSTR) thereby inhibiting proliferation and inducing apoptosis of cells.125 The SST peptide has a nominal half-life of only about 3 minutes in plasma. Agonists of the SST peptide with higher stabilities have been developed. The cyclic octapeptide, octreotide is a SST agonist with a half-life close to 113 minutes.117 Another SST analogue is RC-160, which has a half-life lower than 2 hours. The half-life of RC-160 was further increased by conjugating it with fatty-acids of various lengths. Addition of up to 16 carbons increases the stability of RC-160 by up to 10-fold, thereby reducing the need for repeated dosage117,126 (Fig. 4).
A cyclic pentapeptide called PSAP was designed from the sequence of the glycoprotein prosaposin. This glycoprotein induces the secretion of thrombospondin-1 which is a tumor suppressor. This peptide shows anti-neoplastic activities in case of ovarian cancer.114,127 Lymph nodes serve as reservoirs of metastatic cells and these cells then spread to other parts of the body. Lymph node metastasis can be targeted by a nanovehicle developed using the D-oligoarginine peptide r9, which is a cell-penetrating peptide (CPP). The nanovehicle contained cabazitaxel (CTX), an anticancer drug which causes microtubule disruption. The r9-CN nanovehicle showed efficient targeting to cancer cells followed by deep penetration allowing CTX to work more effectively. This 13 nm nanovehicle targets the primary tumor mass along with the metastatic lymph nodes and distant lung metastases.128 A 17 residue lysine–tryptophan rich cationic peptide, KT2 inhibits migration in both colon cancer and melanoma tissues. KT2 acts as a CPP due to its lysine rich nature and suppresses the activity of various proteins involved in cancer progression. The KT2 peptide displays higher toxicity for cancer cells and induces minimal morphological change.129
Another family of proteins involved in mediating cellular adhesion are the galectins. Galectin-3 participates in angiogenesis and metastasis of cancer cells by mediating adhesion among cancer cells or among a cancer cell and the endothelium. This is mediated via a galactose β1,3 N-acetylgalactosamine disaccharide known as the Thomsen–Friedenreich glycoantigen (TFAg). Metastasis can be inhibited by peptides blocking the interaction between galectin-3 and TFAg. Such peptides were designed by Zou et al. and those blocked cell-adhesion by 60–70% in MDA-MB-435 thereby inhibiting metastasis130 (Fig. 4). Like integrins, CD44 also maintains cellular contact with the ECM and controls cell differentiation, migration and maintenance of tissue integrity.131 The CD44 isoform CD44v6 (CD44 variant 6) is selectively expressed on highly metastatic cancer cells and cancer stem cells.131 This variant enables the interaction between hepatocyte growth factor and c-Met receptor tyrosine kinase. Recently two peptide inhibitors of CD44v6 were reported by Khan et al. with sequences CNLNTIDTC (NLN) and CNEWQLKSC (NEW). These have nanomolar range affinities for CD44v6 and inhibit c-Met internalization and phosphorylation of Erk and c-Met. This results in inhibition of tumor cell migration and invasion. Additionally, the efficacy of these peptides have been checked in combination with crizotinib, a c-Met inhibitor and by fusion with the pro-apoptotic peptide KLAKLAKKLAKLAK. Dual treatment with crizotinib led to more vigorous metastatic reduction while the NLN–KLA and NEW–KLA conjugates caused both apoptosis of tumor cells and inhibition of metastasis (Table 1).132
| ACP | Source | Target | Mechanism of action | Class | Reference |
|---|---|---|---|---|---|
| M1 peptide | Binding domains of TRAIL protein | TR2 | Mimics TRAIL and induces pro-apoptotic signals | Pro-apoptotic | 6 |
| BH3 mimetics | BH3 domains of pro-apoptotic Bcl-2 family members | Anti-apoptotic Bcl-2 proteins | Mimics the BH3 domains of pro-apoptotic Bcl-2 family proteins | 7 | |
| P28 | Azurin protein from Pseudomonas aeruginosa | p53-DNA binding domain | Stabilizes p53 | 10 | |
| Peptide C1 | C-terminus of p53 | p53-NRR2 | Restores p53 function by binding its NRR2 | 10 | |
| AVPF-based peptides | Binding domains of SMAC proteins | BIR region of IAP proteins | Peptides mimic SMAC, binding IAP proteins and freeing active caspases | 11 | |
| PEP2 based cyclic peptides | Binding domains of Nur77 receptor | Bcl-2 and β-tubulin | Mimics the Nur77 domains binding Bcl-2 and β-tubulin | 13 | |
| MEL-dKLA | Melittin from honey bee venom fused to dKLA (engineered) | M2 macrophage | Melittin binds M2-like TAMs and dKLA induces mitochondrial death | 16 | |
| AMPs modified from peptide B1 | Cathelicidin-BF 15 | Cancer cell membranes | Cell penetration followed by destabilization of mitochondrial membrane | 18 | |
| LL-37, 17BIPHE2 | LL-37 is a human cathelicidin and 17BIPHE2 is its shortest derivative | Cell membrane and various receptors | Apoptosis via membrane depolarization and DNA fragmentation | 19 and 20 | |
| Temporin-La | Bullfrog skin | Mitochondrial membrane | Increase in ROS levels and decrease in mitochondrial membrane potential. The ratio of pro and anti-apoptotic Bcl2 family members were also altered | 53 and 54 | |
| PA-C1b | Skin secretions of Rana chensinensis | Cell, mitochondrial membrane and Bcl2 family proteins | Pore formation in cell-membrane along with upregulation of pro-apoptotic Bax and downregulation of anti-apoptotic Bcl2. Loss of mitochondrial membrane potential and increase in ROS levels | 25 | |
| p53(15)Ant peptide | p53 residues 12–26 | Cancer cell membranes | Highly hydrophobic, α-helical structure leads to membrane disruptive functions | Necrotic peptide | 21 |
| PNC-27 | p53 residues 12–26 | Plasma membrane localized MDM2 | Membrane disruption | 23 | |
| D-K6-L9 | Artificially engineered | Negatively charged phosphatidylserines in cell membrane | Induces membranolysis | 29 | |
| MPI-1 | Venom of Polybia paulista | Possibly phosphatidylserines on cell membrane | Membrane destabilization | 30 | |
| Dermaseptin B2 | Skin secretions of Phyllomedusa bicolor | Sulphated glycosaminoglycans on cancer cell membranes | Membrane destabilization | 29 | |
| LF11-322 (PFR peptide) | Human lactoferricin | Anionic lipids on cancer cell membrane | Membrane destabilization | 31 | |
| MTD peptide | MTD of Noxa protein | Mitochondrial permeability transition pore protein | Induces calcium release from mitochondria to cytosol | 34 | |
| TU17:MTD | MTD of Noxa protein fused to a tumor homing sequence binding NRP-1 | Same as MTD peptide with the TU17 domain targeting NRP1 receptor | Same as above | 35 | |
| BMAP-27 | Bovine cathelicidin | Cell membrane | Membrane disruption | 37 | |
| Chensinin-1B | Skin secretions of Rana chensinensis | Cell and mitochondrial membrane | Disruption of cell membrane and loss of mitochondrial membrane potential | Pro-apoptotic and necrotic | 25 |
| HaA4 | Analog of the protein harmoniasin from Harmonia axyridis | Cancer cell membrane and caspase family proteins | Induces caspase-dependent apoptosis and necrosis of cancer cells | 27 | |
| Piscidin-1 | AMP from Morone saxatilis × M. chrysops (hybrid striped bass) | Cellular and mitochondrial membrane | Membrane destabilization | 28 | |
| Brevinin-IRL1 | Skin secretions of Rana limnocharis | Lipids or proteins on the plasma membrane | Induces caspase-dependent apoptosis and necrosis of cancer cells | 32 | |
| Nisin ZP | AMP from Lactococcus lactis | Mitochondrial membrane | Decreases the mitochondrial membrane potential and increases the levels of intracellular ROS | 36 | |
| MELITININ-BMAP27 | Bovine cathelicidin | Cell membrane and pro-apoptotic Bax | Membrane destabilization and apoptosis induction | 38 | |
| Defensin beta-1 | Human AMP | Multiple cancer associated proteins and immune cells | Unknown | Not clear | 39 |
| RGD peptides | Integrin binding ligands | αvβ3 integrins and caspase-3 | Inhibition of metastasis, angiogenesis via blockage of integrin–ligand interaction and apoptosis | Inhibitory and pro-apoptotic peptide | 12, 48 and 49 |
| d-cRGD peptides | Integrin binding ligands | αvβ3 integrins and αvβ1 integrins | Blockage of integrin–ligand interactions | Inhibitory peptides | 55 |
| RGD4, RGD10 | Integrin binding ligands | αvβ3 integrins and αvβ1 integrins | Blockage of integrin–ligand interactions | 56 | |
| Cilengitide | Integrin binding ligands | αvβ3, αvβ5 and αvβ1 integrins | Blockage of integrin–ligand interactions | 58 | |
| cRGD tetramer | Integrin binding ligands | αvβ3 integrins | Aggregation and internalisation of integrins | 65 | |
| iRGD peptide | Integrin binding ligands | αvβ3 integrins and NRP receptor | Inhibition of metastasis and initiation of bulk import within cancer cells | 75 | |
| SDV and GDV peptides | Identified from hexapeptide library by ProteoChip | αvβ3 integrins | Blockage of integrin–ligand interactions | 78 | |
| Atragin based peptides | Snake venom | Integrin family proteins | Inhibition of cell migration | 79 | |
| ATN-161 | Binding domains of fibronectin | αvβ1 integrins | Mimics fibronectin and downregulates MAPK phosphorylation upon integrin binding | 80 and 81 | |
| SFITGv6 and SFLAP3 | RGD containing octameric sequences incorporated into SFTI1 scaffold | αvβ6 integrins | Blockage of integrin–ligand interactions | 85 | |
| FRGDLA-Fp(NMe)K and FRGDLA-Fp(NMe)K(Ac) based peptides | Foot and mouth disease virus | αvβ6 integrins | Blockage of integrin–ligand interactions | 86 | |
| A20FMDV2 and derivatives | Foot and mouth disease virus | αvβ6 integrins | Blockage of integrin–ligand interactions | 87–89 | |
| RGDechi peptide | cRGD peptide fused to echistatin C-terminal domain | αvβ3 integrins | Blockage of integrin–ligand interactions | 90 | |
| RGDechi15D peptide | Same as above but 15th residue of echistatin mutated to aspartate | αvβ5 integrins | Blockage of integrin–ligand interactions | 91 | |
| RWrNR peptide | Identified by structure based drug design | αvβ3 integrins | Blockage of integrin–ligand interactions. PEGylation of this peptide increases anti-metastatic and anti-angiogenic activities. Conjugation to KLA peptide provides pro-apoptotic nature | 93 and 94 | |
| K237 peptide | Engineered VEGF mimic | Kinase domain receptor (KDR) | Acts as a competitor for VEGF | Signalling modulator | 95 |
| D(LPR) peptide | Minimal motif for VEGF binding | VEGFR1 and NRP1 | Acts as a competitor for VEGF | 43 and 96 | |
| 6a-exon based peptides | 6a-exon of VEGF | VEGFR2 | Acts as a competitor for the VEGF165 isoform | 43 | |
| PTX3 derived peptides | Long-pentraxin3, a soluble pattern recognition receptor | FGF2 | Inhibits binding of FGF to receptor | 43 | |
| P144 peptide | Membrane-proximal ligand-binding domain of b-glycan | Soluble TGFβ | Sequesters TGFβ disallowing receptor binding | 43 | |
| WL dipeptide | Sequence present in the primary structure of multiple proteins | VEGFR2 | Inhibition of VEGFR2 phosphorylation and downstream signalling | 97 | |
| IM862 | Isolated from thymus | Unknown | Inhibits production of VEGF and activation of natural killer (NK) cell function | 98 and 99 | |
| MTP-NRP1 | Transmembrane sequence of NRP1 | NRP1 | Blocks receptor dimerization of NRP1 inhibiting downstream signalling | 100 | |
| GRP antagonistic peptides | Engineered using structure of GRP | GRP receptor GPCRs | Inhibition of ligand–receptor interaction | 98 | |
| JMR-132 | Artificially engineered, structure-based design | Growth hormone releasing hormone receptor (GHRHR) | Inhibition of ligand–receptor interaction | 102 | |
| hC20 derived octapeptides | Engineered using sequences from the dimer interface of thymidylate synthase | Dimer interface of thymidylate synthase | Enzyme is trapped in an inactive conformation and DNA synthesis is restricted | 98 and 103 | |
| Collagen IV biomimetic peptide | Engineered from sequences of transmembrane protein 45A | β1 integrin and an unknown receptor | Inhibition of IGF1R and Met signalling pathways | 105 | |
| IF7 | Engineered peptide | Annexin-1 | Inhibition of hematogenous carbohydrate-dependent cancer cell colonization | 108 | |
| Magainins | Ionophoric AMP from Xenopus laevis | Cell membrane and binding domains of Pax protein | Formation of ion channels and inhibition of Fak–Pax signalling | 109 and 110 | |
| CXC12 decoys | Artificially engineered, structure-based design | CXCR4 | Inhibition of CXC12/CXCR4 signalling | 112 | |
| DV-1 | Engineered from chemokine peptides | CXCR4 | Inhibition of receptor–ligand interaction. Activity is maximum when peptide density is 24k molecules per μm2 | 113 | |
| NT21MP | Derived from the viral macrophage inflammatory protein II | CXCR4 | Inhibition of CXC12/CXCR4 signalling | 114 and 115 | |
| Nef-M1 | Residues 50–60 of Nef protein | CXCR4 | Inhibition of CXC12/CXCR4 signalling | 114 and 116 | |
| Peptide R | Engineered from the RRX motif of CXC12 | CXCR4 | Alters the phenotype of TAMs from M2 to M1 | 117 and 118 | |
| EPI-X4 JM#21 | Derived from a 16-mer fragment of human serum albumin | CXCR4 | Inhibition of CXC12/CXCR4 signalling | 119 | |
| G7-18NATE, G7-B7 | Identified by phage display and engineered for better action | SH2 domain of Grb7 | Inhibition of Grb7 interaction | 121–123 | |
| GIP fragment from AFP | GIP domain of AFP | Unknown | Inhibits the function of multiple cytosolic enzymes leading to alterations in MAPK cascades and regulation of apoptotic events | 124 | |
| Octreotide | Engineered synthetic SST analogue | SSTR | Acts as an SST agonist with a longer half-life | 117 | |
| RC-160 | Engineered synthetic SST analogue | SSTR | Acts as an SST agonist with a longer half-life | 117 and 126 | |
| PSAP based peptides | Sequences of the glycoprotein prosaposin | CD11b+/GR1+/Lys6Chi monocytes | Induces secretion of the tumor suppressor thrombospondin-1 by the monocytes which are recruited at metastatic lesions | 114 and 127 | |
| KT2 | Engineered from Crocodylus siamensis leucocyte peptide | Multiple cancer associated proteins | Acts as a CPP which downregulates the expression of several proteins leading to decreased cell migration and invasion | 129 | |
| Galectin binding peptides | Identified from phage display library | Galectins | Inhibits interaction between galectins and TFAg | 130 | |
| NLN and NEW | Identified from phage display library | CD44v6 | Inhibit c-Met internalisation and phosphorylation of MAPK and c-Met | 132 |
The ACPs discussed so far have been summarised in table for better understanding.
Anti-cancer peptide vaccines
Peptides are popular candidates for vaccine design as they are cheap, easy to synthesize, stable and free of bacterial pathogens or oncogenic properties. Antigens specifically expressed by cancer cells are known as tumor associated antigens (TAAs). These are presented by major histocompatibility complex (MHC) class I molecules and identified by cytotoxic T lymphocytes (CTLs)98,133 (Fig. 5). TAAs introduced into a hosts system as pre-immunization or post-operatively after primary tumor removal may protect the host from cancer development and metastasis. Since, TAAs alone do not confer significant immunogenicity in patients, they are often administered with adjuvants like toll-like receptors, cytokines and APCs to increase their efficiency.134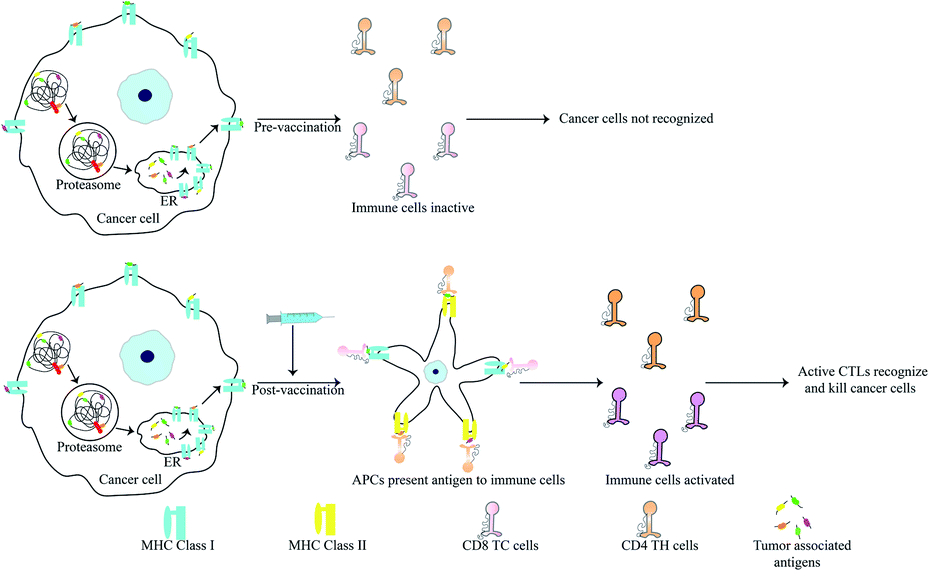 | ||
| Fig. 5 Mode of action of anti-cancer peptide vaccines. Peptide vaccines contain tumor-associated antigens. Upon vaccination, APCs present these antigens to cytotoxic and helper T-cells, thereby activating them. Active T-cells can then recognise the TAAs on cancer cells. Active CTLs kill cancer cells presenting TAAs. | ||
TAAs are generally 8–10 amino acids in length and recognise the MHC class I molecules through interaction of 2–3 primary anchor residues. They also interact with the CTLs via 2–3 other residues.98,133 TAAs are most usually altered forms of normal proteins. Since the selection of potential TAA candidates from a range of differently mutated proteins can prove challenging, researchers have developed bioinformatics tools for the same. These tools calculate the affinity of various TAAs for the patients HLA and also consider their potential in activating a T-cell response amon other parameters. Some bioinformatic tools for this purpose include: NeoPredPipe, MuPeXI, pVAC-Seq and CloudNeo.135–139 Examples of TAAs include the mutated forms of the ras p21 proto-oncogenes. Immunization of mice and human with synthetic mutant p21 leads to development of a T cell based immunity.140–142 13-mer synthetic ras peptides with mutations in codon 12 similar to the patient's own mutation generated an immune response in 3 out of 10 patients.142 Various TAAs have now been characterized and many vaccines based on these epitopes have reached clinical trials. It is known that oncoviruses like Human Papillomavirus (HPV) harbour proteins which induce oncogenic transformation. E7 of HPV is one such protein and presence of antibodies against this protein leads to worsening disease. TAAs derived from E7 were used for immunization of patients with highly invasive cervical and vulvar cancer. Only a fraction of the patients showed complete disease regression but the CTL titre against E7 was increased in all patients.143 Other vaccines showing similar results include: a 100-mer peptide vaccine derived from the glycoprotein MUC1 which is overexpressed in cancer cells in hypoglycosylated state144,145 and the E75 peptide vaccine derived from HER2/neu that is involved in prostate cancer.146,147 Adjuvants also play a critical role in increasing the efficiency of vaccination. Since peptides by themselves are prone to degradation and lack sufficient antigenic properties, the choice of adjuvants becomes critical for successful immunization. Adjuvants can be as simple as water-in-oil emulsions which allow slow release and epitope persistence to cytokines such as GM-CSF.148 Recently the agonist of STING (stimulator of interferon gene) protein has also found use as an adjuvant. Activation of STING induces a strong interferon response leading to activation of macrophages and dendritic cells.148,149 The STING protein along with acting as an adjuvant has also been for delivery of peptide antigen. The C-terminal domain of STING was fused with the fusion protein cGAMP. This forms a tetrameric complex that delivers the peptide vaccine and initiates STING signalling.150
Anti-tumor immunity in humans is mainly CTL based but T-helper cells activated by MHC class II associated peptides are necessary for long-life and better immune clearance of tumor cells by CTL. Knutson et al., vaccinated patients with peptides derived from HER2/neu where the peptides contained TH-cell epitopes. Activation of both T-cell subsets resulted in 86% of the patients generating increased CTLs post-vaccination.151 HER2/neu based vaccines have been used for breast and ovarian cancers. Higher immunization efficacy in vaccines containing MHC class II targets was also observed for GV1001 derived from hTERT.152 This vaccine also acts a CPP enabling the delivery of macromolecules such as DNA, siRNA and proteins through extracellular heat-shock protein complexes. This confers direct anti-cancer activities to this peptide.153 Scientists have identified a range of epitopes known as the universal cancer peptides (UCPs) derived from TERT. These activate CD4 T cell responses in cancer patients and vaccination of A2/DR1 mice model with two such UCPs caused enormous TH1 activity leading to tumor clearance via elevated activation of CD8 T cells.154
Peptide-based vaccines have been developed against melanoma using various melanoma specific TAAs. It was found that GM-CSF works as a more efficient adjuvant for these TAAs.134 Melanoma vaccines that have shown efficacy in preliminary studies but failed clinical trials are addressed elsewhere.155 The MKC1106-PP vaccine targets multiple epitopes of melanoma and induces immune response against preferentially expressed antigen in melanoma (PRAME) and prostate-specific membrane antigen (PSMA). The vaccine consists of a recombinant plasmid which synthesises parts of both the antigens and peptides derived from each of the proteins mentioned. More than 62% of the patients were able to develop an immune response against either PRAME or PSMA and the immunity lasted more than 6 months.156 Dendritic cells have been used in many cases to stimulate the efficiency of peptide vaccines as they can activate dormant T-cells. A range of vaccines have been developed using dendritic cells including sipuleucel-T which reduces death risk by up to 22%. The vaccine although not peptide-based warrants a mention as it was sanctioned to be used clinically in prostate cancer.157,158 A study was conducted to find the efficiency of vaccines containing dendritic cells loaded with antigenic peptides from stage III melanoma. Patients were vaccinated after lymph node dissection to prevent relapse. It was found that CD8-T cells were generated in 15 of 22 vaccinated patients. Additionally, the 3 year overall survival and disease free survival rate in vaccinated patients increased by 45.2% and 26.4% respectively.159 Recently a neoantigen based vaccine dendritic cell vaccine was tested in patients with advanced metastatic lung cancer. The neoantigens selected were personalized for each patient using data from whole exome sequencing, RNA sequencing and HLA typing. The vaccine was found be safe, tolerable and could efficiently lead to disease control.160
A cyclic peptide vaccine was developed using B-cell epitopes from the tetraspanin family protein TM4SF5 (transmembrane 4 superfamily member 5) which is involved in EMT and metastasis. This was tested against hepatocellular carcinoma and colon cancer in addition with a humanized antibody which targeted TM4SF5 and induced expression of E-cadherin and β-catenin. Mice treated with the vaccine showed production of antibodies along with reduction in tumor growth. Both the synthetic monoclonal antibody and the vaccine reduced metastasis and may be used for further studies.161 Wanzun et al., have devised an ingenious method to identify TAAs by irradiating H22 cells. This causes the overexpression of proteins involved in cell survival and resistance to therapy. From the overexpressed proteins CD151 was chosen for immunization as it is overexpressed in various cancers and its expression correlates with poor prognosis. Two TAAs from CD151 were used for peptide synthesis and immunization of in vivo mouse models. Significant CTL responses against these were developed three weeks post vaccination suggesting CD151 based peptide vaccines could be used just after tumor resection with radiotherapy to inhibit tumor growth, progression and metastasis.162 The Prss14/ST14 membrane serine protease plays a vital role in promoting disease progression and metastasis of triple negative breast cancer (TNBC). Antigenic peptides derived from the autocatalytic loop of this protein were used to vaccinate relevant mice models. Vaccination significantly reduced the number of metastatic secondary tumors in vaccinated mice.163
Personalized peptide vaccines (PPVs) offer a new approach for more effective immunization in cancer patients. The peptides chosen generate a faster CTL response as they are HLA matched to the patient. PPV immunization in patients with advanced colorectal carcinoma showed positive outcomes in phase II clinical study.164 For metastatic renal cell carcinoma (RCC) patients, the UroRCC vaccine containing 10 TAAs were used for immunization after HLA matching. The vaccine generated significant anti-tumor immunity and increased the overall survival time.165 PPVs were also tested on a patient with metastatic intrahepatic cholangiocarcinoma. This patient had relapses twice in the liver and a metastatic secondary tumor in the lungs after the surgical removal of the primary mass. Patient was immunized with a vaccine containing 7 epitopes identified by NGS after removal of relapsed and metastatic tumors. The vaccine was improved also by the addition of CD4 binding peptides. The vaccine caused strong immune cell movement to the resected metastatic lesion along with high levels of perforin which acts as a cytotoxic molecule. The patient remained tumor free for three years before the study was published while vaccination was continued.166 Other PPVs like NeoVax had as many as 20 epitopes identified from 6 melanoma patients by whole exome sequencing. The vaccine elicited response from both the CTL and T-helper subsets and was able to prevent relapse in four of the six patients. The other two patients showed complete tumor clearance after treatment with immune checkpoint modulators.167,168 PPVs have also been used against anal cancer, kidney cancer, leukemia, lung cancer, glioblastoma, lymphoma, and brain tumors. Details about these can be found elsewhere.167 PPVs although highly efficient are limited in use due to higher cost as the patient is unable to develop T-cells against many of the antigens used. To circumvent this, TAAs are loaded on various formulations that allow precise delivery to APCs. Lynn et al., have designed a peptide-based conjugate which folds into nanoparticles of almost 20 nm size irrespective of the TAAs used. This allows to develop a universal protocol for delivery of peptides and adjuvants to increase T-cell response.169
Peptides as drug-delivery agents
Peptides were classified into three main categories by Boohaker et al., namely: (i) cell-permeable peptides; (ii) antimicrobial peptides; and (iii) tumour-targeting peptides.170 In this section we will focus on CPPs as these aid in the transport of cargo containing small molecules and thus play an important role in drug delivery.171 Several factors, like temperature, concentration, and size of the drug, are vital in the internalisation of the cargo.172 The majority of CPPs have basic, positively charged residues and hydrophobic amino acids, which increases the efficiency of their internalisation.173 The TAT (transactivator of transcription) peptide, discovered in the late 1980s derived from human immunodeficiency virus and the Drosophila Antennapedia homeodomain, is well known for its ability to easily penetrate the cell membrane.174 The ability of CPPs to form alpha helices or beta sheets determines whether they are incorporated into the cell.175 Cellular uptake of CPPs occurs in a variety of ways, only few of which encourage direct translocation across the lipid bilayer. A more common mechanism involves energy-dependent endocytosis, such as macropinocytosis, clathrin-mediated endocytosis, or caveolae-mediated endocytosis. This intake mechanism is followed by peptides such as CADY, MPG, and Pep-1.176,177 A novel dipeptide named melflufen was designed by combination of melphalan and para-fluoro-L-phenylalanine. This compound acts as a highly lipophilic tumor targeting CPP that crosses the plasma membrane in a passive and fast manner. Melflufen contains a binding domain for aminopeptidases and esterases which cleave the molecule thus releasing the encapsulated drug.178 These CPPs carry various types of cargo across the membrane, such as liposomes, antisense oligonucleotides, nucleic acids, therapeutic agents, and small interfering RNAs.179–181 Other mechanisms for peptide translocation include membrane thinning, pore formation, and inverted micelle formation.182Peptide insertion into the cell depends on a multitude of factors.
The alpha helical peptide aggregates and evolves into a nanofibre structure as well. This arrangement exposes the side chains, allowing the peptide to be easily accessed by the solvent. When interacting with the cell membrane, many well-known peptides, including PEP1, TP10, MPG, and SAP, adopt a helical conformation.189 van der Waals forces, hydrophobic interactions, and π-interactions (π–π stacking, cation and anion–π interactions) all help to organise self-assembled structures. A peptide can be modified by hybridising an alkyl or aromatic group, which serves as a driving force for self-assembly via hydrophobic interactions190 (Fig. 6).
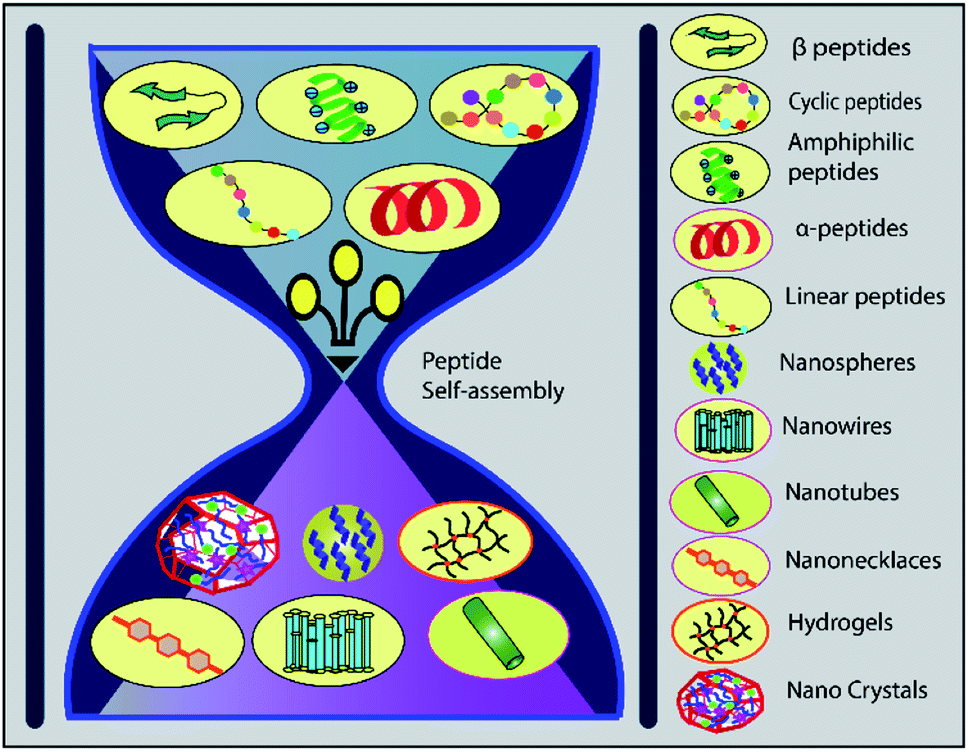 | ||
| Fig. 6 Different peptides can rearrange themselves into various nanostructures via self-assembly, offering a promising approach to drug internalisation. | ||
Pharmacokinetic profiling is critical in determining the biologically active conformation of peptides. To determine the structural elements of peptides responsible for biological activity, a systematic approach is required. After identifying the key amino acids, the next step is to determine the 3D relationship of the pharmacophore elements. Following this study, it may be possible to further modify the structure to overcome proteolytic degradation, or improve its penetrating ability through membrane.206
Internalization mechanisms of CPPs
Peptides are internalised in the cell by a variety of mechanisms.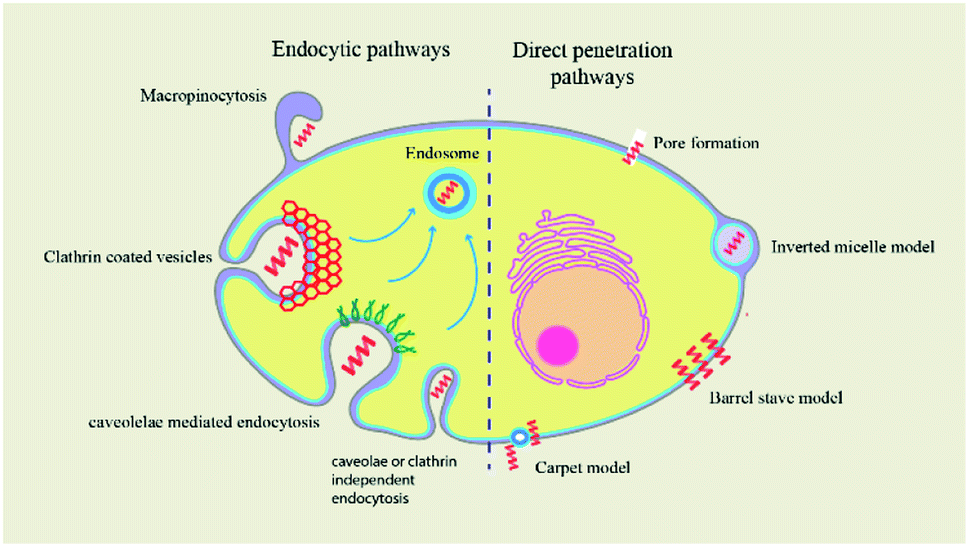 | ||
| Fig. 7 The mechanism of cellular uptake of CPP cargos occurs by two types of pathways: endocytosis and direct translocation. Direct translocation models include the barrel-stave model, the carpet-like model, and the inverted micelle model. Clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin- and caveolin-independent endocytosis are all endocytosis mechanisms. | ||
Conclusions
Peptides have been widely studied as anti-cancer agents targeting different cellular factors. They have also gained wide popularity in the development of vaccines and as drug-delivery vehicles. Although significant milestones have been crossed, there remains major hurdles in the development of peptide-based drugs. Small therapeutic peptides may sometimes harbour significant immunogenicity222 and overcoming the immunogenic nature of therapeutic peptides remains a major challenge in developing peptide therapeutics. Short peptides often have low enzyme resistance and blood–brain barrier penetration.223 This decreases its half-life while increasing its probable toxicity. Therapeutic peptides may also possess poly-functional properties. To overcome these problems, researchers have fused peptides with stabilizing groups or lipophilic radicals. Enzyme resistance of peptides have been increased by substituting L-amino acids with D-amino acids and by using unique drug delivery systems.223 Researchers are working tirelessly to develop highly efficient and specific peptides that have minimal side-effects. The field of peptide-based drugs has progressed enormously in the last two decades and we hope that this review gives a glimpse of all the ways in which peptides have been designed and modified to target cancer cells.Author contributions
DB and LR wrote the manuscript and made the figures. SC did the final editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Bose Institute and University Grants Commission for supporting our work with fellowships.Notes and references
- E. Fisher, K. Pavlenko, A. Vlasov and G. Ramenskaya, Pharm. Med., 2019, 33, 9–20 CrossRef CAS PubMed
.
- W. Chiangjong, S. Chutipongtanate and S. Hongeng, Int. J. Oncol., 2020, 57, 678–696 CrossRef CAS PubMed
- C. Hu, X. Chen and W. Zhao, Drug Des.: Open Access, 2016, 05, 1000138 Search PubMed
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed
- K. A. Min, P. Maharjan, S. Ham and M. C. Shin, Arch. Pharm. Res., 2018, 41, 594–616 CrossRef CAS PubMed
- G. Lamanna, C. R. Smulski, N. Chekkat, K. Estieu-Gionnet, G. Guichard, S. Fournel and A. Bianco, Chem.–Eur. J., 2013, 19, 1762–1768 CrossRef CAS PubMed
- L. Zhang, L. Ming and J. Yu, Drug Resist. Updat., 2007, 10, 207–217 CrossRef CAS PubMed
- T. E. Assafa, S. Nandi, D. Śmiłowicz, L. Galazzo, M. Teucher, C. Elsner, S. Pütz, S. Bleicken, A. Y. Robin, D. Westphal, I. Uson, R. Stoll, P. E. Czabotar, N. Metzler-Nolte and E. Bordignon, Structure, 2021, 29, 114–124.e3 CrossRef CAS
- M. J. Duffy, N. C. Synnott, P. M. McGowan, J. Crown, D. O'Connor and W. M. Gallagher, Cancer Treat. Rev., 2014, 40, 1153–1160 CrossRef CAS PubMed
- E. Teveroni, R. Lucà, M. Pellegrino, G. Ciolli, A. Pontecorvi and F. Moretti, Expert Opin. Ther. Pat., 2016, 26, 1417–1429 CrossRef CAS PubMed
- S. T. Le Quement, M. Ishoey, M. T. Petersen, J. Thastrup, G. Hagel and T. E. Nielsen, ACS Comb. Sci., 2011, 13, 667–675 CrossRef CAS PubMed
- C. D. Buckley, D. Pilling, N. V. Henriquez, G. Parsonage, K. Threlfall, D. Scheel-Toellner, D. L. Simmons, A. N. Akbar, J. M. Lord and M. Salmon, Nature, 1999, 397, 534–539 CrossRef CAS PubMed
- M. Brindisi, S. Maramai, S. Brogi, E. Fanigliulo, S. Butini, E. Guarino, A. Casagni, S. Lamponi, C. Bonechi, S. M. Nathwani, F. Finetti, F. Ragonese, P. Arcidiacono, P. Campiglia, S. Valenti, E. Novellino, R. Spaccapelo, L. Morbidelli, D. M. Zisterer, C. D. Williams, A. Donati, C. Baldari, G. Campiani, C. Ulivieri and S. Gemma, Eur. J. Med. Chem., 2016, 117, 301–320 CrossRef CAS PubMed
- A. Yuan, Y.-J. Hsiao, H.-Y. Chen, H.-W. Chen, C.-C. Ho, Y.-Y. Chen, Y.-C. Liu, T.-H. Hong, S.-L. Yu, J. J. W. Chen and P.-C. Yang, Sci. Rep., 2015, 5, 14273 CrossRef CAS
- E. W. C. Smith, J. Clin. Cell. Immunol., 2015, 06, 1–8 Search PubMed
- C. Lee, H. Jeong, Y. Bae, K. Shin, S. Kang, H. Kim, J. Oh and H. Bae, J. Immunother. Cancer, 2019, 7, 147 CrossRef
- V. Sarangthem, A. Yi, Y. Kim, A. Rehemtulla, B.-H. Lee, Y. H. Jeon, T. D. Singh and R.-W. Park, Int. J. Nanomed., 2021, 16, 5039–5052 CrossRef PubMed
- Y. Dai, X. Cai, W. Shi, X. Bi, X. Su, M. Pan, H. Li, H. Lin, W. Huang and H. Qian, Amino Acids, 2017, 49, 1601–1610 CrossRef CAS PubMed
- K. Kuroda, K. Okumura, H. Isogai and E. Isogai, Front. Oncol., 2015, 5 DOI:10.3389/fonc.2015.00144
- T. Yang, J. Li, Q. Jia, S. Zhan, Q. Zhang, Y. Wang and X. Wang, Oncol. Lett., 2021, 22, 501 CrossRef CAS PubMed
- T. N. Do, R. V. Rosal, L. Drew, A. J. Raffo, J. Michl, M. R. Pincus, F. K. Friedman, D. P. Petrylak, N. Cassai, J. Szmulewicz, G. Sidhu, R. L. Fine and P. W. Brandt-Rauf, Oncogene, 2003, 22, 1431–1444 CrossRef CAS PubMed
- A. Thadi, W. F. Morano, M. Khalili, B. D. Babcock, M. F. Shaikh, D. S. Foster, Y. Piazza, E. M. Gleeson, E. Goldstein, L. Steele, P. M. Campbell, B. Lin, M. R. Pincus and W. B. Bowne, Anticancer Res., 2021, 41, 27–42 CrossRef PubMed
- M. Kanovsky, A. Raffo, L. Drew, R. Rosal, T. Do, F. K. Friedman, P. Rubinstein, J. Visser, R. Robinson, P. W. Brandt-Rauf, J. Michl, R. L. Fine and M. R. Pincus, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12438–12443 CrossRef CAS PubMed
- C.-R. Chung, J.-H. Jhong, Z. Wang, S. Chen, Y. Wan, J.-T. Horng and T.-Y. Lee, Int. J. Mol. Sci., 2020, 21, 986 CrossRef CAS PubMed
- F. Guo, Y. Zhang, W. Dong, Y. Guan and D. Shang, Int. J. Biochem. Cell Biol., 2022, 143, 106156 CrossRef CAS PubMed
- A. C. Kaushik, A. Mehmood, S. Peng, Y.-J. Zhang, X. Dai and D.-Q. Wei, J. Biomol. Struct. Dyn., 2021, 39, 285–293 CrossRef CAS PubMed
- I.-W. Kim, J. H. Lee, Y.-N. Kwon, E.-Y. Yun, S.-H. Nam, M.-Y. Ahn, D.-C. Kang and J. S. Hwang, Int. J. Oncol., 2013, 43, 622–628 CrossRef CAS PubMed
- H.-J. Lin, T.-C. Huang, S. Muthusamy, J.-F. Lee, Y.-F. Duann and C.-H. Lin, Zool. Sci., 2012, 29, 327–332 CrossRef CAS
- A. L. Tornesello, A. Borrelli, L. Buonaguro, F. M. Buonaguro and M. L. Tornesello, Molecules, 2020, 25, 2850 CrossRef CAS PubMed
- W. Zhang, J. Li, L.-W. Liu, K. Wang, J. Song, J.-X. Yan, Z.-Y. Li, B.-Z. Zhang and R. Wang, Peptides, 2010, 31, 1832–1838 CrossRef CAS PubMed
- Y. Lu, T.-F. Zhang, Y. Shi, H.-W. Zhou, Q. Chen, B.-Y. Wei, X. Wang, T.-X. Yang, Y. E. Chinn, J. Kang and C.-Y. Fu, Sci. Rep., 2016, 6, 20823 CrossRef CAS PubMed
- X. Ju, D. Fan, L. Kong, Q. Yang, Y. Zhu, S. Zhang, G. Su and Y. Li, Molecules, 2021, 26, 2059 CrossRef CAS PubMed
- D. Gaspar, A. S. Veiga and M. A. R. B. Castanho, Front. Microbiol., 2013, 4, 294 Search PubMed
- Y.-W. Seo, H.-N. Woo, S. Piya, A. R. Moon, J.-W. Oh, C.-W. Yun, K.-K. Kim, J.-Y. Min, S.-Y. Jeong, S. Chung, P. I. Song, S.-Y. Jeong, E. K. Choi, D.-W. Seol and T.-H. Kim, Cancer Res., 2009, 69, 8356–8365 CrossRef CAS PubMed
- J.-Y. Kim, J.-H. Han, G. Park, Y.-W. Seo, C.-W. Yun, B.-C. Lee, J. Bae, A. R. Moon and T.-H. Kim, Oncotarget, 2016, 7, 32449–32461 CrossRef PubMed
- S. M. Patil and N. K. Kunda, Pharm. Res., 2022 DOI:10.1007/s11095-022-03220-2
- S. Yang, C. W. Lee, H. J. Kim, H.-H. Jung, J. I. Kim, S. Y. Shin and S.-H. Shin, Peptides, 2019, 118, 170106 CrossRef CAS PubMed
- R. O. Saleh, I. N. A. Essia and S. A. Jasim, J. Gastrointest. Cancer, 2022 DOI:10.1007/s12029-021-00799-4
- S. Li, H. Li, Y. Xu, W. Ning, S. Hu, S. Wei, H. Song, J. Sun, D. Ziebolz, G. Schmalz, X. Hu and M. Liu, Comput. Math. Methods Med., 2022, 2022, 2203615 Search PubMed
- B. Deslouches and Y. P. Di, Oncotarget, 2017, 8, 46635–46651 CrossRef PubMed
- H. Hamidi and J. Ivaska, Nat. Rev. Cancer, 2018, 18, 533–548 CrossRef CAS PubMed
- C.-C. Sun, X.-J. Qu and Z.-H. Gao, Am. J. Ther., 2016, 23, e198–207 CrossRef PubMed
- E. V. Rosca, J. E. Koskimaki, C. G. Rivera, N. B. Pandey, A. P. Tamiz and A. S. Popel, Curr. Pharm. Biotechnol., 2011, 12, 1101–1116 CAS
- K. Switala-Jelen, K. Dabrowska, A. Opolski, L. Lipinska, M. Nowaczyk and A. Gorski, Folia Biol., 2004, 50, 143–152 CAS
- J. D. Hood and D. A. Cheresh, Nat. Rev. Cancer, 2002, 2, 91–100 CrossRef
- B. Estevez, B. Shen and X. Du, Arterioscler. Thromb. Vasc. Biol., 2015, 35, 24–29 CrossRef CAS PubMed
- L. Koivisto, J. Heino, L. Häkkinen and H. Larjava, Adv. Wound Care, 2014, 3, 762–783 CrossRef PubMed
- Y.-P. Yu, Q. Wang, Y.-C. Liu and Y. Xie, Biomaterials, 2014, 35, 1667–1675 CrossRef CAS PubMed
- X. Dong, Y. Yu, Q. Wang, Y. Xi and Y. Liu, Mol. Inform., 2017, 36, 1600069 CrossRef PubMed
- I. Kemker, D. C. Schröder, R. C. Feiner, K. M. Müller, A. Marion and N. Sewald, J. Med. Chem., 2021, 64, 586–601 CrossRef CAS PubMed
- P. C. Brooks, A. M. P. Montgomery, M. Rosenfeld, R. A. Reisfeld, T. Hu, G. Klier and D. A. Cheresh, Cell, 1994, 79, 1157–1164 CrossRef CAS PubMed
- W. Liu, J. Su, Q. Shi, J. Wang, X. Chen, S. Zhang, M. Li, J. Cui, C. Fan, B. Sun and G. Wang, Front. Bioeng. Biotechnol., 2021, 9, 781608 CrossRef PubMed
- X. Jiang, X. Zhang, C. Fu, R. Zhao, T. Jin, M. Liu, C. Pan, L. A. Li, J. Ma, E. Yu, X. Yu and Y. Hu, Protein J., 2021, 40, 709–720 CrossRef CAS PubMed
- Y. Diao, W. Han, H. Zhao, S. Zhu, X. Liu, X. Feng, J. Gu, C. Yao, S. Liu, C. Sun and F. Pan, J. Pept. Sci., 2012, 18, 476–486 CrossRef CAS PubMed
- T. G. Kapp, F. Rechenmacher, S. Neubauer, O. V. Maltsev, E. A. Cavalcanti-Adam, R. Zarka, U. Reuning, J. Notni, H.-J. Wester, C. Mas-Moruno, J. Spatz, B. Geiger and H. Kessler, Sci. Rep., 2017, 7, 39805 CrossRef CAS PubMed
- A. L. Dunehoo, M. Anderson, S. Majumdar, N. Kobayashi, C. Berkland and T. J. Siahaan, J. Pharm. Sci., 2006, 95, 1856–1872 CrossRef CAS PubMed
- P. Hölig, M. Bach, T. Völkel, T. Nahde, S. Hoffmann, R. Müller and R. E. Kontermann, Protein Eng. Des. Sel., 2004, 17, 433–441 CrossRef PubMed
- F. Danhier, A. Le Breton and V. Préat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef CAS PubMed
- C. Mas-Moruno, F. Rechenmacher and H. Kessler, Anticancer Agents Med. Chem., 2010, 10, 753–768 CrossRef CAS PubMed
- W. P. Mason, Neuro. Oncol., 2015, 17, 634–635 CrossRef PubMed
- J. Jeong and J. Kim, Int. J. Mol. Sci., 2022, 23, 3423 CrossRef CAS PubMed
- D. Arosio, L. Manzoni, C. Corno and P. Perego, Recent Pat. Anticancer Drug Discov., 2017, 12, 148–168 CrossRef CAS PubMed
- C. Wängler, S. Maschauer, O. Prante, M. Schäfer, R. Schirrmacher, P. Bartenstein, M. Eisenhut and B. Wängler, ChemBioChem, 2010, 11, 2168–2181 CrossRef PubMed
- I. Saiki, J. Murata, K. Matsuno, R. Ogawa, N. Nishi, S. Tokura and I. Azuma, Jpn. J. Cancer Res., 1990, 81, 660–667 CrossRef CAS PubMed
- S. Lucie, G. Elisabeth, F. Stéphanie, S. Guy, H. Amandine, A.-R. Corinne, B. Didier, S. Catherine, G. Alexeï, D. Pascal and C. Jean-Luc, Mol. Ther., 2009, 17, 837–843 CrossRef PubMed
- S. Liu, Bioconjugate Chem., 2009, 20, 2199–2213 CrossRef CAS PubMed
- S. Liu, Bioconjugate Chem., 2015, 26, 1413–1438 CrossRef CAS PubMed
- Y. Mizuno, K. Kimura, S. Onoe, M. Shukuri, Y. Kuge and H. Akizawa, J. Med. Chem., 2021, 64, 16008–16019 CrossRef CAS PubMed
- J.-J. Liu, Z. Wang, L.-M. Nie, Y.-Y. Zhu, G. Li, L.-L. Lin, M. Chen and G.-J. Zhang, Eur. J. Nucl. Med. Mol. Imaging, 2022, 49, 847–860 CrossRef CAS PubMed
- L. Li, X. Chen, J. Yu and S. Yuan, Front. Oncol., 2022, 12, 837952 CrossRef PubMed
- A. Ouyang, D. Zhao, X. Wang, W. Zhang, T. Jiang, A. Li and W. Liu, J. Mater. Chem. B, 2022, 10, 306–320 RSC
- K. Temming, R. M. Schiffelers, G. Molema and R. J. Kok, Drug Resist. Updat., 2005, 8, 381–402 CrossRef CAS PubMed
- E. Khabazian, F. Vakhshiteh, P. Norouzi, Y. Fatahi, R. Dinarvand and F. Atyabi, J. Drug Target., 2022, 30(5), 522–533 CrossRef CAS PubMed
- S. Fu, X. Xu, Y. Ma, S. Zhang and S. Zhang, J. Drug Target., 2019, 27, 1–11 CrossRef CAS PubMed
- K. N. Sugahara, G. B. Braun, T. H. de Mendoza, V. R. Kotamraju, R. P. French, A. M. Lowy, T. Teesalu and E. Ruoslahti, Mol. Cancer Ther., 2015, 14, 120–128 CrossRef CAS PubMed
- H.-J. Cho, S.-J. Park, Y.-S. Lee and S. Kim, J. Controlled Release, 2019, 300, 73–80 CrossRef CAS PubMed
- F. Meng, J. Liu, J. Wei, J. Yang, C. Zhou, J. Yan and B. Liu, Cancer Sci., 2022, 113, 1417–1427 CrossRef CAS PubMed
- Y. Lee, D.-K. Kang, S.-I. Chang, M. H. Han and I.-C. Kang, J. Biomol. Screening, 2004, 9, 687–694 CrossRef CAS PubMed
- H. S. Selistre-de-Araujo, C. L. S. Pontes, C. F. Montenegro and A. C. B. M. Martin, Toxins, 2010, 2, 2606–2621 CrossRef CAS PubMed
- Attenuon, A Dose-Ranging, Phase II, Open Label Study of ATN 161 in Advanced Renal Cell Cancer, https://clinicaltrials.gov, 2007 Search PubMed
- P. Khalili, A. Arakelian, G. Chen, M. L. Plunkett, I. Beck, G. C. Parry, F. Doñate, D. E. Shaw, A. P. Mazar and S. A. Rabbani, Mol. Cancer Ther., 2006, 5, 2271–2280 CrossRef CAS PubMed
- D. Heckmann, A. Meyer, L. Marinelli, G. Zahn, R. Stragies and H. Kessler, Angew. Chem., Int. Ed., 2007, 46, 3571–3574 CrossRef PubMed
- S. Neubauer, F. Rechenmacher, R. Brimioulle, F. S. Di Leva, A. Bochen, T. R. Sobahi, M. Schottelius, E. Novellino, C. Mas-Moruno, L. Marinelli and H. Kessler, J. Med. Chem., 2014, 57, 3410–3417 CrossRef CAS PubMed
- R. Stragies, F. Osterkamp, G. Zischinsky, D. Vossmeyer, H. Kalkhof, U. Reimer and G. Zahn, J. Med. Chem., 2007, 50, 3786–3794 CrossRef CAS PubMed
- S. Roesch, T. Lindner, M. Sauter, A. Loktev, P. Flechsig, M. Müller, W. Mier, R. Warta, G. Dyckhoff, C. Herold-Mende, U. Haberkorn and A. Altmann, J. Nucl. Med., 2018, 59, 1679–1685 CrossRef CAS PubMed
- O. V. Maltsev, U. K. Marelli, T. G. Kapp, F. S. Di Leva, S. Di Maro, M. Nieberler, U. Reuning, M. Schwaiger, E. Novellino, L. Marinelli and H. Kessler, Angew. Chem., Int. Ed., 2016, 55, 1535–1539 CrossRef CAS PubMed
- S. H. Hausner, D. DiCara, J. Marik, J. F. Marshall and J. L. Sutcliffe, Cancer Res., 2007, 67, 7833–7840 CrossRef CAS PubMed
- I. I. Cardle, M. C. Jensen, S. H. Pun and D. L. Sellers, J. Biol. Chem., 2021, 296, 100657 CrossRef CAS PubMed
- T. T. Huynh, S. Sreekumar, C. Mpoy and B. E. Rogers, Pharmaceuticals, 2022, 15, 229 CrossRef CAS PubMed
- B. Farina, I. de Paola, L. Russo, D. Capasso, A. Liguoro, A. D. Gatto, M. Saviano, P. V. Pedone, S. Di Gaetano, G. Malgieri, L. Zaccaro and R. Fattorusso, Chem.–Eur. J., 2016, 22, 681–693 CrossRef CAS PubMed
- D. Capasso, A. Del Gatto, D. Comegna, L. Russo, R. Fattorusso, M. Saviano, S. Di Gaetano and L. Zaccaro, Molecules, 2020, 25, E4298 CrossRef PubMed
- L. Zhang, X. Meng, X. Shan, T. Gu, J. Zhang, S. Feng, X. An, J. Chen, Q. Jiang and X. Ning, Anal. Chem., 2019, 91, 12587–12595 CrossRef CAS PubMed
- X. Shan, L. Zhang, Y. Xu, L. Sun, L. Guo, S. Feng, H. Ge, T. Gu and X. Ning, Chem. Commun., 2020, 56, 2178–2181 RSC
- Y. Xu, L. Sun, S. Feng, J. Chen, Y. Gao, L. Guo, X. An, Y. Nie, Y. Zhang, X. Liu and X. Ning, ACS Appl. Mater. Interfaces, 2019, 11, 34663–34675 CrossRef CAS PubMed
- L. Hetian, A. Ping, S. Shumei, L. Xiaoying, H. Luowen, W. Jian, M. Lin, L. Meisheng, Y. Junshan and S. Chengchao, J. Biol. Chem., 2002, 277, 43137–43142 CrossRef PubMed
- R. J. Giordano, M. Cardó-Vila, A. Salameh, C. D. Anobom, B. D. Zeitlin, D. H. Hawke, A. P. Valente, F. C. L. Almeida, J. E. Nör, R. L. Sidman, R. Pasqualini and W. Arap, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5112–5117 CrossRef CAS PubMed
- S. Khedr, A. Klotzsche-von Ameln, M. Khedr, M. H. Elsayed, T. Sudha, S. A. Mousa, A. Deussen and M. Martin, Acta Physiol., 2021, 231, e13556 CAS
- J. Thundimadathil, J. Amino Acids, 2012, 2012, e967347 Search PubMed
- A. Noy, D. T. Scadden, J. Lee, B. J. Dezube, D. Aboulafia, A. Tulpule, S. Walmsley and P. Gill, J. Clin. Oncol., 2005, 23, 990–998 CrossRef CAS PubMed
- A. Arpel, C. Gamper, C. Spenlé, A. Fernandez, L. Jacob, N. Baumlin, P. Laquerriere, G. Orend, G. Crémel and D. Bagnard, Oncotarget, 2016, 7, 54723–54732 CrossRef PubMed
- C. Liu, Z. Zhao, R. Gao, X. Zhang, Y. Sun, J. Wu, J. Liu and C. Chen, ACS Biomater. Sci. Eng., 2022, 8(7), 2979–2994 CrossRef CAS PubMed
- F. Hohla, S. Buchholz, A. V. Schally, S. Seitz, F. G. Rick, L. Szalontay, J. L. Varga, M. Zarandi, G. Halmos, I. Vidaurre, A. Krishan, M. Kurtoglu, S. Chandna, E. Aigner and C. Datz, Cell Cycle, 2009, 8, 3149–3156 CrossRef CAS PubMed
- D. Cardinale, G. Guaitoli, D. Tondi, R. Luciani, S. Henrich, O. M. H. Salo-Ahen, S. Ferrari, G. Marverti, D. Guerrieri, A. Ligabue, C. Frassineti, C. Pozzi, S. Mangani, D. Fessas, R. Guerrini, G. Ponterini, R. C. Wade and M. P. Costi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E542–E549 CrossRef CAS PubMed
- Z. Wang, Y. Yu, W. Dai, J. Lu, J. Cui, H. Wu, L. Yuan, H. Zhang, X. Wang, J. Wang, X. Zhang and Q. Zhang, Biomaterials, 2012, 33, 8451–8460 CrossRef CAS PubMed
- E. Lee, S. J. Lee, J. E. Koskimaki, Z. Han, N. B. Pandey and A. S. Popel, Sci. Rep., 2014, 4, 7139 CrossRef CAS PubMed
- Y. Chen, J. J. Wu and L. Huang, Mol. Ther., 2010, 18, 828–834 CrossRef CAS PubMed
- S. Marqus, E. Pirogova and T. J. Piva, J. Biomed. Sci., 2017, 24, 21 CrossRef PubMed
- H. Qin, Y. Ding, A. Mujeeb, Y. Zhao and G. Nie, Mol. Pharmacol., 2017, 92, 219–231 CrossRef CAS PubMed
- R. A. Cruciani, J. L. Barker, M. Zasloff, H. C. Chen and O. Colamonici, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3792–3796 CrossRef CAS PubMed
- A. Khan, S. Shan, T. F. Toor, M. Suleman, Y. Wang, J. Zhou and D.-Q. Wei, Mol. Divers., 2022 DOI:10.1007/s11030-022-10438-0
- J. Lang, X. Zhao, Y. Qi, Y. Zhang, X. Han, Y. Ding, J. Guan, T. Ji, Y. Zhao and G. Nie, ACS Nano, 2019, 13, 12357–12371 CrossRef CAS PubMed
- G. J. Mizejewski, J. Neoplasms, 2018, 1, 1–9 Search PubMed
- D. Liu, P. Guo, C. McCarthy, B. Wang, Y. Tao and D. Auguste, Nat. Commun., 2018, 9, 2612 CrossRef PubMed
- V. Le Joncour and P. Laakkonen, Bioorg. Med. Chem., 2018, 26, 2797–2806 CrossRef CAS PubMed
- Q. Yang, F. Zhang, Y. Ding, J. Huang, S. Chen, Q. Wu, Z. Wang, Z. Wang and C. Chen, Br. J. Cancer, 2014, 110, 1288–1297 CrossRef CAS PubMed
- H. Bumpers, M.-B. Huang, V. Katkoori, U. Manne and V. Bond, J. Cancer Ther., 2013, 4, 898–906 CrossRef PubMed
- V. Le Joncour and P. Laakkonen, Bioorg. Med. Chem., 2018, 26, 2797–2806 CrossRef CAS PubMed
- L. Mercurio, M. A. Ajmone-Cat, S. Cecchetti, A. Ricci, G. Bozzuto, A. Molinari, I. Manni, B. Pollo, S. Scala, G. Carpinelli and L. Minghetti, J. Exp. Clin. Cancer Res., 2016, 35, 55 CrossRef PubMed
- M. Harms, R. F. Hansson, S. Carmali, Y. Almeida-Hernández, E. Sanchez-Garcia, J. Münch and A. N. Zelikin, Bioconjugate Chem., 2022, 33, 594–607 CrossRef CAS PubMed
- N. P. Sturre, R. N. Colson, N. Shah, G. M. Watson, X. Yang, M. C. J. Wilce, J. T. Price and J. A. Wilce, Biomedicines, 2022, 10, 1145 CrossRef CAS PubMed
- M. J. Gunzburg, N. D. Ambaye, M. P. Del Borgo, S. C. Pero, D. N. Krag, M. C. J. Wilce and J. A. Wilce, J. Mol. Recognit., 2012, 25, 57–67 CrossRef CAS PubMed
- S. C. Pero, L. Oligino, R. J. Daly, A. L. Soden, C. Liu, P. P. Roller, P. Li and D. N. Krag, J. Biol. Chem., 2002, 277, 11918–11926 CrossRef CAS PubMed
- G. M. Watson, K. Kulkarni, J. Sang, X. Ma, M. J. Gunzburg, P. Perlmutter, M. C. J. Wilce and J. A. Wilce, J. Med. Chem., 2017, 60, 9349–9359 CrossRef CAS PubMed
- G. J. Mizejewski, J. Cancer Metastasis Treat., 2018, 4, 27 CrossRef
- H. L. Watt, G. Kharmate and U. Kumar, Mol. Cell. Endocrinol., 2008, 286, 251–261 CrossRef CAS PubMed
- P. Dasgupta, A. Singh and R. Mukherjee, Biol. Pharm. Bull., 2002, 25, 8 CrossRef PubMed
- S. Wang, A. Blois, T. El Rayes, J. F. Liu, M. S. Hirsch, K. Gravdal, S. Palakurthi, D. R. Bielenberg, L. A. Akslen, R. Drapkin, V. Mittal and R. S. Watnick, Sci. Transl. Med., 2016, 8, 329ra34 Search PubMed
- H. Hu, J. Wang, H. Wang, T. Tan, J. Li, Z. Wang, K. Sun, Y. Li and Z. Zhang, Theranostics, 2018, 8, 3597–3610 CrossRef CAS PubMed
- P. Maraming, S. Klaynongsruang, P. Boonsiri, S.-F. Peng, S. Daduang, P. Rungsa, R. Tavichakorntrakool, J.-G. Chung and J. Daduang, In Vivo, 2021, 35, 215–227 CrossRef CAS PubMed
- J. Zou, V. V. Glinsky, L. A. Landon, L. Matthews and S. L. Deutscher, Carcinogenesis, 2005, 26, 309–318 CrossRef CAS PubMed
- M. Todaro, M. Gaggianesi, V. Catalano, A. Benfante, F. Iovino, M. Biffoni, T. Apuzzo, I. Sperduti, S. Volpe, G. Cocorullo, G. Gulotta, F. Dieli, R. De Maria and G. Stassi, Cell Stem Cell, 2014, 14, 342–356 CrossRef CAS PubMed
- F. Khan, S. Gurung, G. R. Gunassekaran, S. M. P. Vadevoo, L. Chi, U. Permpoon, M. E. Haque, Y.-K. Lee, S.-W. Lee, S. Kim and B. Lee, Theranostics, 2021, 11, 1326–1344 CrossRef CAS PubMed
- L. Eisenbach, O. Mandelboim, E. Bar-Haim, L. Carmon, H. Copcow, K. El-Shami, A. Paz, D. Popovic, E. Vadai, E. Tzehoval, M. Feldman and M. Fridkin, Lett. Pept. Sci., 1998, 5, 323–328 CAS
- H. L. Kaufman, Semin. Oncol., 2012, 39, 263–275 CrossRef CAS PubMed
- A. Nelde, H.-G. Rammensee and J. S. Walz, Mol. Cell Proteomics, 2021, 20, 100022 CrossRef CAS PubMed
- J. Hundal, B. M. Carreno, A. A. Petti, G. P. Linette, O. L. Griffith, E. R. Mardis and M. Griffith, Genome Med., 2016, 8(1), 11 CrossRef PubMed
- R. O. Schenck, E. Lakatos, C. Gatenbee, T. A. Graham and A. R. A. Anderson, BMC Bioinform., 2019, 20, 264 CrossRef PubMed
- A.-M. Bjerregaard, M. Nielsen, S. R. Hadrup, Z. Szallasi and A. C. Eklund, Cancer Immunol. Immunother., 2017, 66, 1123–1130 CrossRef CAS PubMed
- P. Bais, S. Namburi, D. M. Gatti, X. Zhang and J. H. Chuang, Bioinformatics, 2017, 33, 3110 CrossRef CAS PubMed
- S. I. Abrams, P. H. Hand, K. Y. Tsang and J. Schlom, Semin. Oncol., 1996, 23, 118–134 CAS
- M. k. Gjertsen and G. Gaudernack, Vox Sang., 1998, 74, 489–495 CrossRef CAS PubMed
- S. N. Khleif, S. I. Abrams, J. M. Hamilton, E. Bergmann-Leitner, A. Chen, A. Bastian, S. Bernstein, Y. Chung, C. J. Allegra and J. Schlom, J. Immunother., 1999, 22, 155–165 CrossRef CAS PubMed
- L. Muderspach, S. Wilczynski, L. Roman, L. Bade, J. Felix, L. A. Small, W. M. Kast, G. Fascio, V. Marty and J. Weber, Clin. Cancer Res., 2000, 6, 3406–3416 CAS
- R. K. Ramanathan, K. M. Lee, J. McKolanis, E. Hitbold, W. Schraut, A. J. Moser, E. Warnick, T. Whiteside, J. Osborne, H. Kim, R. Day, M. Troetschel and O. J. Finn, Cancer Immunol. Immunother., 2005, 54, 254–264 CrossRef CAS PubMed
- K. Yamamoto, T. Ueno, T. Kawaoka, S. Hazama, M. Fukui, Y. Suehiro, Y. Hamanaka, Y. Ikematsu, K. Imai, M. Oka and Y. Hinoda, Anticancer Res., 2005, 25, 3575–3579 CAS
- M. T. Hueman, Z. A. Dehqanzada, T. E. Novak, J. M. Gurney, M. M. Woll, G. B. Ryan, C. E. Storrer, C. Fisher, D. G. McLeod, C. G. Ioannides, S. Ponniah and G. E. Peoples, Clin. Cancer Res., 2005, 11, 7470–7479 CrossRef CAS PubMed
- K. Sole, Nat. Clin. Pract. Urol., 2006, 3, 6 Search PubMed
- N. Abd-Aziz and C. L. Poh, J. Oncol., 2022 DOI:10.1155/2022/9749363
- M. F. Gulen, U. Koch, S. M. Haag, F. Schuler, L. Apetoh, A. Villunger, F. Radtke and A. Ablasser, Nat. Commun., 2017, 8(1), 427 CrossRef PubMed
- Y. He, C. Hong, S. J. Fletcher, A. G. Berger, X. Sun, M. Yang, S. Huang, A. M. Belcher, D. J. Irvine, J. Li and P. T. Hammond, Adv. Healthc. Mater., 2200905 Search PubMed
- K. L. Knutson, K. Schiffman and M. L. Disis, J. Clin. Invest., 2001, 107, 477–484 CrossRef CAS PubMed
- P. Nava-Parada and L. A. Emens, Curr. Opin. Mol. Ther., 2007, 9, 490–497 CAS
- H. Kim, E.-H. Seo, S.-H. Lee and B.-J. Kim, Int. J. Mol. Sci., 2016, 17, 2054 CrossRef PubMed
- M. Dosset, Y. Godet, C. Vauchy, L. Beziaud, Y. C. Lone, C. Sedlik, C. Liard, E. Levionnois, B. Clerc, F. Sandoval, E. Daguindau, S. Wain-Hobson, E. Tartour, P. Langlade-Demoyen, C. Borg and O. Adotévi, Clin. Cancer Res., 2012, 18, 6284–6295 CrossRef CAS PubMed
- J. C. Yang, Cancer J., 2011, 17, 277–282 CrossRef CAS PubMed
- J. S. Weber, N. J. Vogelzang, M. S. Ernstoff, O. B. Goodman, L. D. Cranmer, J. L. Marshall, S. Miles, D. Rosario, D. C. Diamond, Z. Qiu, M. Obrocea and A. Bot, J. Immunother., 2011, 34, 556–567 CrossRef CAS PubMed
- P. W. Kantoff, C. S. Higano, N. D. Shore, E. R. Berger, E. J. Small, D. F. Penson, C. H. Redfern, A. C. Ferrari, R. Dreicer, R. B. Sims, Y. Xu, M. W. Frohlich and P. F. Schellhammer, N. Engl. J. Med., 2010, 363, 411–422 CrossRef CAS PubMed
- N. H. Akhtar, O. Pail, A. Saran, L. Tyrell and S. T. Tagawa, Adv. Urol., 2011, 2012, e973820 Search PubMed
- S. Markowicz, Z. I. Nowecki, P. Rutkowski, A. W. Lipkowski, M. Biernacka, A. Jakubowska-Mucka, T. Switaj, A. Misicka, H. Skurzak, H. Polowniak-Pracka and J. Walewski, Med. Oncol., 2012, 29, 2966–2977 CrossRef CAS PubMed
- Z. Ding, Q. Li, R. Zhang, L. Xie, Y. Shu, S. Gao, P. Wang, X. Su, Y. Qin, Y. Wang, J. Fang, Z. Zhu, X. Xia, G. Wei, H. Wang, H. Qian, X. Guo, Z. Gao, Y. Wang, Y. Wei, Q. Xu, H. Xu and L. Yang, Signal Transduct. Target. Ther., 2021, 6, 26 CrossRef CAS PubMed
- G. Wu, D. Kim, B. K. Park, S. Park, J.-H. Ha, T. H. Kim, A. Gautam, J. N. Kim, S. I. Lee, H.-B. Park, Y.-S. Kim, H.-J. Kwon and Y. Lee, Oncotarget, 2016, 7, 79170–79186 CrossRef PubMed
- W. Lin, J. Liu, J. Chen, J. Li, S. Qiu, J. Ma, X. Lin, L. Zhang and J. Wu, EBioMedicine, 2019, 49, 133–144 CrossRef PubMed
- K. Y. Kim, M. Yoon, Y. Cho, K.-H. Lee, S. Park, S. Lee, S.-Y. Choi, D. Lee, C. Yang, E. H. Cho, S. D. Jeon, S.-H. Kim, C. Kim and M. G. Kim, J. Exp. Clin. Cancer Res., 2019, 38, 363 CrossRef PubMed
- T. Sasada, S. Kibe, Y. Akagi and K. Itoh, Oncoimmunology, 2015, 4, e1005512 CrossRef PubMed
- S. Rausch, C. Gouttefangeas, J. Hennenlotter, K. Laske, K. Walter, S. Feyerabend, P. A. Chandran, S. Kruck, H. Singh-Jasuja, A. Frick, N. Kröger, S. Stevanović, A. Stenzl, H.-G. Rammensee and J. Bedke, Eur. Urol. Focus, 2019, 5, 604–607 CrossRef PubMed
- M. W. Löffler, P. A. Chandran, K. Laske, C. Schroeder, I. Bonzheim, M. Walzer, F. J. Hilke, N. Trautwein, D. J. Kowalewski, H. Schuster, M. Günder, V. A. Carcamo Yañez, C. Mohr, M. Sturm, H.-P. Nguyen, O. Riess, P. Bauer, S. Nahnsen, S. Nadalin, D. Zieker, J. Glatzle, K. Thiel, N. Schneiderhan-Marra, S. Clasen, H. Bösmüller, F. Fend, O. Kohlbacher, C. Gouttefangeas, S. Stevanović, A. Königsrainer and H.-G. Rammensee, J. Hepatol., 2016, 65, 849–855 CrossRef PubMed
- L. Bezu, O. Kepp, G. Cerrato, J. Pol, J. Fucikova, R. Spisek, L. Zitvogel, G. Kroemer and L. Galluzzi, Oncoimmunology, 2018, 7, e1511506 CrossRef PubMed
- P. A. Ott, Z. Hu, D. B. Keskin, S. A. Shukla, J. Sun, D. J. Bozym, W. Zhang, A. Luoma, A. Giobbie-Hurder, L. Peter, C. Chen, O. Olive, T. A. Carter, S. Li, D. J. Lieb, T. Eisenhaure, E. Gjini, J. Stevens, W. J. Lane, I. Javeri, K. Nellaiappan, A. M. Salazar, H. Daley, M. Seaman, E. I. Buchbinder, C. H. Yoon, M. Harden, N. Lennon, S. Gabriel, S. J. Rodig, D. H. Barouch, J. C. Aster, G. Getz, K. Wucherpfennig, D. Neuberg, J. Ritz, E. S. Lander, E. F. Fritsch, N. Hacohen and C. J. Wu, Nature, 2017, 547, 217–221 CrossRef CAS PubMed
- G. M. Lynn, C. Sedlik, F. Baharom, Y. Zhu, R. A. Ramirez-Valdez, V. L. Coble, K. Tobin, S. R. Nichols, Y. Itzkowitz, N. Zaidi, J. M. Gammon, N. J. Blobel, J. Denizeau, P. de la Rochere, B. J. Francica, B. Decker, M. Maciejewski, J. Cheung, H. Yamane, M. G. Smelkinson, J. R. Francica, R. Laga, J. D. Bernstock, L. W. Seymour, C. G. Drake, C. M. Jewell, O. Lantz, E. Piaggio, A. S. Ishizuka and R. A. Seder, Nat. Biotechnol., 2020, 38, 320–332 CrossRef CAS PubMed
- R. J. Boohaker, M. W. Lee, P. Vishnubhotla, J. M. Perez and A. R. Khaled, Curr. Med. Chem., 2012, 19, 3794–3804 CrossRef CAS PubMed
- W. B. Kauffman, T. Fuselier, J. He and W. C. Wimley, Trends Biochem. Sci., 2015, 40, 749–764 CrossRef CAS PubMed
- M. M. Fretz, N. A. Penning, S. Al-Taei, S. Futaki, T. Takeuchi, I. Nakase, G. Storm and A. T. Jones, Biochem. J., 2007, 403, 335–342 CrossRef CAS PubMed
- G. L. Bidwell and D. Raucher, Expert Opin. Drug Deliv., 2009, 6, 1033–1047 CrossRef CAS PubMed
- E. Vivès, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef PubMed
- D. Kalafatovic and E. Giralt, Molecules, 2017, 22, 1929 CrossRef PubMed
- F. Simeoni, M. C. Morris, F. Heitz and G. Divita, Nucleic Acids Res., 2003, 31, 2717–2724 CrossRef CAS PubMed
- A. Rydström, S. Deshayes, K. Konate, L. Crombez, K. Padari, H. Boukhaddaoui, G. Aldrian, M. Pooga and G. Divita, PLoS One, 2011, 6, e25924 CrossRef
- J. Lindberg, J. Nilvebrant, P.-Å. Nygren and F. Lehmann, Molecules, 2021, 26, 6042 CrossRef CAS PubMed
- I. Nakase, H. Akita, K. Kogure, A. Gräslund, Ü. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 CrossRef CAS PubMed
- M. Lindgren, K. Rosenthal-Aizman, K. Saar, E. Eiríksdóttir, Y. Jiang, M. Sassian, P. Ostlund, M. Hällbrink and U. Langel, Biochem. Pharmacol., 2006, 71, 416–425 CrossRef CAS PubMed
- V. P. Torchilin, R. Rammohan, V. Weissig and T. S. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8786–8791 CrossRef CAS PubMed
- B. R. Meade and S. F. Dowdy, Adv. Drug Deliv. Rev., 2008, 60, 530–536 CrossRef CAS PubMed
- D. Tesauro, A. Accardo, C. Diaferia, V. Milano, J. Guillon, L. Ronga and F. Rossi, Molecules, 2019, 24, 351 CrossRef PubMed
- A. Brack and L. E. Orgel, Nature, 1975, 256, 383–387 CrossRef CAS PubMed
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed
- M. Nune, P. Kumaraswamy, U. M. Krishnan and S. Sethuraman, Curr. Protein Pept. Sci., 2013, 14, 70–84 CrossRef CAS PubMed
- S. Lu, W. F. D. Bennett, Y. Ding, L. Zhang, H. Y. Fan, D. Zhao, T. Zheng, P.-K. Ouyang, J. Li, Y. Wu, W. Xu, D. Chu, Y. Yuan, H. Heerklotz, M. Karttunen and P. Chen, Adv. Healthc. Mater., 2015, 4, 2709–2718 CrossRef CAS PubMed
- P. Nygren, M. Lundqvist, B. Liedberg, B.-H. Jonsson and T. Ederth, Langmuir, 2010, 26, 6437–6448 CrossRef CAS PubMed
- S. Li, R. Xing, R. Chang, Q. Zou and X. Yan, Curr. Opin. Colloid Interface Sci., 2018, 35, 17–25 CrossRef CAS
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS PubMed
- M. C. Morris, P. Vidal, L. Chaloin, F. Heitz and G. Divita, Nucleic Acids Res., 1997, 25, 2730–2736 CrossRef CAS PubMed
- S. R. Jean, M. Ahmed, E. K. Lei, S. P. Wisnovsky and S. O. Kelley, Acc. Chem. Res., 2016, 49, 1893–1902 CrossRef CAS PubMed
- S. Kobayashi, I. Nakase, N. Kawabata, H.-H. Yu, S. Pujals, M. Imanishi, E. Giralt and S. Futaki, Bioconjugate Chem., 2009, 20, 953–959 CrossRef CAS PubMed
- D. Weerakkody, A. Moshnikova, M. S. Thakur, V. Moshnikova, J. Daniels, D. M. Engelman, O. A. Andreev and Y. K. Reshetnyak, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5834–5839 CrossRef CAS PubMed
- Y. K. Reshetnyak, O. A. Andreev, M. Segala, V. S. Markin and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15340–15345 CrossRef CAS PubMed
- N. S. Shu, M. S. Chung, L. Yao, M. An and W. Qiang, Nat. Commun., 2015, 6, 7787 CrossRef CAS PubMed
- O. A. Andreev, D. M. Engelman and Y. K. Reshetnyak, Mol. Membr. Biol., 2010, 27, 341–352 CrossRef CAS PubMed
- T. Narayanan, D. Weerakkody, A. G. Karabadzhak, M. Anderson, O. A. Andreev and Y. K. Reshetnyak, J. Phys. Chem. B, 2016, 120, 11484–11491 CrossRef CAS PubMed
- W. Li, F. Nicol and F. C. Szoka, Adv. Drug Deliv. Rev., 2004, 56, 967–985 CrossRef CAS PubMed
- F. Heitz, M. C. Morris and G. Divita, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS PubMed
- S. Pujals, J. Fernández-Carneado, C. López-Iglesias, M. J. Kogan and E. Giralt, Biochim. Biophys. Acta, 2006, 1758, 264–279 CrossRef CAS PubMed
- I. D. Alves, N. Goasdoué, I. Correia, S. Aubry, C. Galanth, S. Sagan, S. Lavielle and G. Chassaing, Biochim. Biophys. Acta, 2008, 1780, 948–959 CrossRef CAS PubMed
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed
- A. Ayo and P. Laakkonen, Pharmaceutics, 2021, 13, 481 CrossRef CAS PubMed
- V. J. Hruby, Nat. Rev. Drug Discovery, 2002, 1, 847–858 CrossRef CAS PubMed
- G. Guidotti, L. Brambilla and D. Rossi, Trends Pharmacol. Sci., 2017, 38, 406–424 CrossRef CAS PubMed
- J. Xie, Y. Bi, H. Zhang, S. Dong, L. Teng, R. J. Lee and Z. Yang, Front. Pharmacol., 2020, 11, 697 CrossRef CAS PubMed
- A. Borrelli, A. L. Tornesello, M. L. Tornesello and F. M. Buonaguro, Molecules, 2018, 23, 295 CrossRef PubMed
- S. Kumari, S. Mg and S. Mayor, Cell Res., 2010, 20, 256–275 CrossRef CAS PubMed
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed
- J. P. Richard, K. Melikov, H. Brooks, P. Prevot, B. Lebleu and L. V. Chernomordik, J. Biol. Chem., 2005, 280, 15300–15306 CrossRef CAS PubMed
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310–315 CrossRef CAS PubMed
- S. Pujals and E. Giralt, Adv. Drug Deliv. Rev., 2008, 60, 473–484 CrossRef CAS PubMed
- C. Zhang, D. Pan, K. Luo, W. She, C. Guo, Y. Yang and Z. Gu, Adv. Healthc. Mater., 2014, 3, 1299–1308 CrossRef CAS PubMed
- B. M. Cooper, J. Iegre, D. H. O. Donovan, M. Ö. Halvarsson and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1480–1494 RSC
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed
- R. M. Sawant, J. P. Hurley, S. Salmaso, A. Kale, E. Tolcheva, T. S. Levchenko and V. P. Torchilin, Bioconjugate Chem., 2006, 17, 943–949 CrossRef CAS PubMed
- Y.-S. Zhu, K. Tang and J. Lv, Trends Pharmacol. Sci., 2021, 42, 857–869 CrossRef CAS PubMed
- N. Yao, W. Xiao, X. Wang, J. Marik, S. H. Park, Y. Takada and K. S. Lam, J. Med. Chem., 2009, 52, 126–133 CrossRef CAS PubMed
- L. Fernandez, R. H. Bustos, C. Zapata, J. Garcia, E. Jauregui and G. M. Ashraf, Curr. Protein Pept. Sci., 2018, 19, 958–971 CrossRef CAS PubMed
- Y. A. Haggag, Biomed. J. Sci. Tech. Res., 2018, 8(4) DOI:10.26717/BJSTR.2018.08.001694
Footnote |
| † These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |