
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, antitumor activity, 3D-QSAR and molecular docking studies of new iodinated 4-(3H)-quinazolinones 3N-substituted†
Marcia Pérez-Fehrmann *a,
Víctor Kesternicha,
Arturo Puellesa,
Víctor Quezadaa,
Fernanda Salazara,
Philippe Christenbc,
Jonathan Castillod,
Juan Guillermo Cárcamode,
Alejandro Castro-Alvarezfg and
Ronald Nelson*a
*a,
Víctor Kesternicha,
Arturo Puellesa,
Víctor Quezadaa,
Fernanda Salazara,
Philippe Christenbc,
Jonathan Castillod,
Juan Guillermo Cárcamode,
Alejandro Castro-Alvarezfg and
Ronald Nelson*a
aDepartamento de Química, Facultad de Ciencias, Universidad Católica del Norte, Av. Angamos 0610, Antofagasta 1270709, Chile. E-mail: rnelson@ucn.cl
bSchool of Pharmaceutical Sciences, University of Geneva, 1211 Geneva 4, Switzerland
cInstitute of Pharmaceutical Sciences of Western Switzerland, University of Geneva, 1211 Geneva 4, Switzerland
dInstituto de Bioquímica y Microbiología, Facultad de Ciencias, Universidad Austral de Chile, Campus Isla Teja, Valdivia, Chile
eCentro FONDAP, Interdisciplinary Center for Aquaculture Research (INCAR), Chile
fLaboratorio de Bioproductos Farmacéuticos y Cosméticos, Centro de Excelencia en Medicina Traslacional, Facultad de Medicina, Universidad de La Frontera, Av. Francisco Salazar 01145, Temuco, 4780000, Chile
gDepartamento de Química de los Materiales, Facultad de Química y Biología, Universidad de Santiago de Chile, Casilla 40, Correo 33, Santiago, Chile
First published on 2nd August 2022
Abstract
A novel series of 6-iodo-2-methylquinazolin-4-(3H)-one derivatives, 3a–n, were synthesized and evaluated for their in vitro cytotoxic activity. Compounds 3a, 3b, 3d, 3e, and 3h showed remarkable cytotoxic activity on specific human cancer cell lines when compared to the anti-cancer drug, paclitaxel. Compound 3a was found to be particularly effective on promyelocytic leukaemia HL60 and non-Hodgkin lymphoma U937, with IC50 values of 21 and 30 μM, respectively. Compound 3d showed significant activity against cervical cancer HeLa (IC50 = 10 μM). The compounds 3e and 3h were strongly active against glioblastoma multiform tumour T98G, with IC50 values of 12 and 22 μM, respectively. These five compounds showed an interesting cytotoxic activity on four human cancer cell types of high incidence. The molecular docking results reveal a good correlation between experimental activity and calculated binding affinity on dihydrofolate reductase (DHFR). Docking studies proved 3d as the most potent compound. In addition, the three-dimensional quantitative structure–activity relationship (3D-QSAR) analysis exhibited activities that may indicate the existence of electron-withdrawing and lipophilic groups at the para-position of the phenyl ring and hydrophobic interactions of the quinazolinic ring in the DHFR active site.
1 Introduction
4-(3H)-Quinazolinones are an important group of fused heterocycles found in interesting natural alkaloids and drugs (Fig. 1). These structures are of high interest due to their wide range of biological and pharmacological properties,1 including antibacterial,2 antifungal,3 antitubercular,4 antimalarial,5 antitoxoplasma,6 anti-inflammatory,7 anti-ulcer,8 and kinase inhibitor,9 and anticancer activities.10 Quinazolines as anticancer agents have received considerable attention since the development of the thymidylate synthetase inhibitors altitrexed and thymitaq.11 Since then, several quinazolines have been described with anticancer activity,10 among which the following can be highlighted: inhibitors of epidermal growth factor receptor (EGFR),12 inhibitors of angiogenesis by inhibiting the vascular endothelial growth factor receptor (VEGFR-2)13 and dihydrofolate reductase (DHFR) inhibitors that prevent the growth of cancer cells and depletes the cell from thymine causing cell death.14 Consequently, DHFR inhibition played an essential role in medicine clinical as antitumor agents and becomes a target for the development of new antitumor agents.15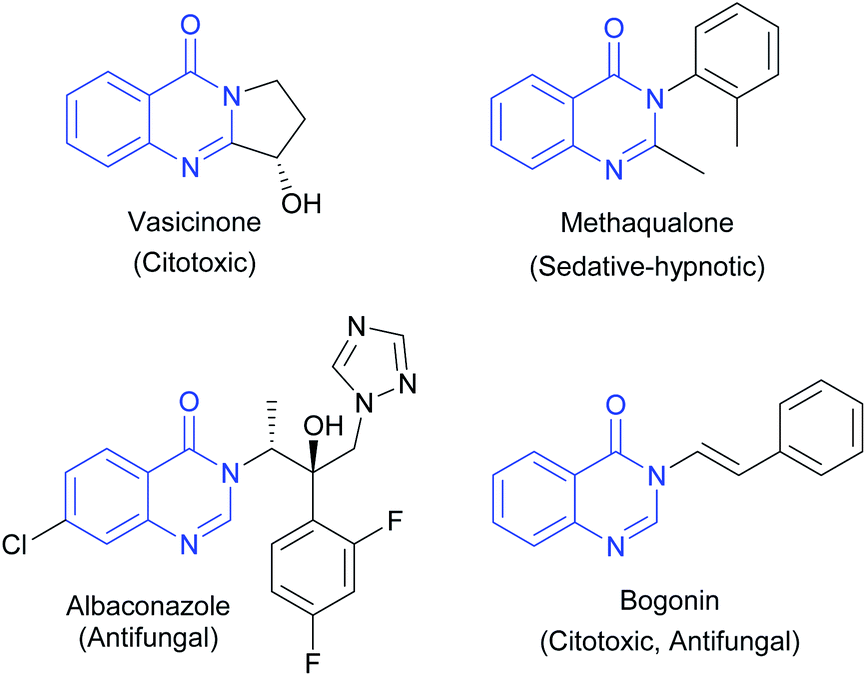 | ||
| Fig. 1 Selected examples of 4-(3H)-quinazolinones. | ||
Additionally, they are important intermediates in natural product preparation and are used as structural scaffolds in drug discovery.16 Due to the extensive biological properties associated with quinazolinone moieties in drug design, obtaining these compounds is of great relevance and has motivated the development of multiple synthetic strategies such as ring opening of isatoic anhydride by nitrogen nucleophiles followed by the oxidative cyclocondensation by electrophiles,17 aza-Wittig/cyclization reactions of iminophosporanes,18 oxidative olefin bond cleavage,19 transition metal dehydrogenative coupling,20 transition metal-free dehydrogenative coupling,21 transition metal-catalyzed cross-coupling,22 palladium-catalyzed carbonylative methods23 and miscellaneous transition metal-free methods.24 Likewise, interest in these compounds has also led to spectroscopic structural studies, and X-ray diffraction analysis of this class of heterocycles.25
Here we report the synthesis of novel 6-iodo-2-methylquinazolin-4-(3H)-one derivatives (3a–n), carrying different substituents in the 3N position. The cytotoxic activity of these new derivatives against various cancer cell lines was evaluated in vitro, and molecular docking and 3D-QSAR studies of the compounds were completed to correlate the structures with their cytotoxic activities. The IC50 values obtained for the derivatives were contrasted with those obtained with paclitaxel, a chemotherapy drug used to treat many different types of cancer. The objective of forming these compounds is to develop an active antitumor agent with potential activity and selectivity toward human cancer cells in vivo.
The synthesis of iodine quinazolinones was developed because iodinated derivatives are characterized as stable, non-toxic, and relatively easy to obtain.26,27 Previous work has shown that the atom in position six increases lipophilicity and molecular absorption.27,28 On the other hand, iodinated compounds offer an interesting starting point for the realization of various synthetized analogues.29
2 Results and discussion
2.1 Chemistry
For the synthesis of the quinazolinones 3N-substituted obtained in the present work, many reactions were tested.30 However, the method described by Grimmel et al.,31 allowed us to achieve better yields. Thus, the synthesis of 6-iodo-2-methylquinazolin-4-(3H)-one 3N-substituted were carried out using the 5-iodoanthranilic acid (1) as key intermediate. The iodination of anthranilic acid in presence of hydrogen peroxide in ethanol at reflux, gave the compound 1 with 88% yield. Then, the reaction of acid 1 with acetic anhydride and subsequent hydrolysis under reflux, it was possible to obtain 2-acetamido-5-iodobenzoic acid (2) in with 75% yield (Scheme 1). Finally, the reaction of intermediate 2 with different amine derivatives (R-NH2) in the presence of PCl3 as dehydrating agent, allowed us to obtain the quinazolinones 3a–n with variated yields (Scheme 2).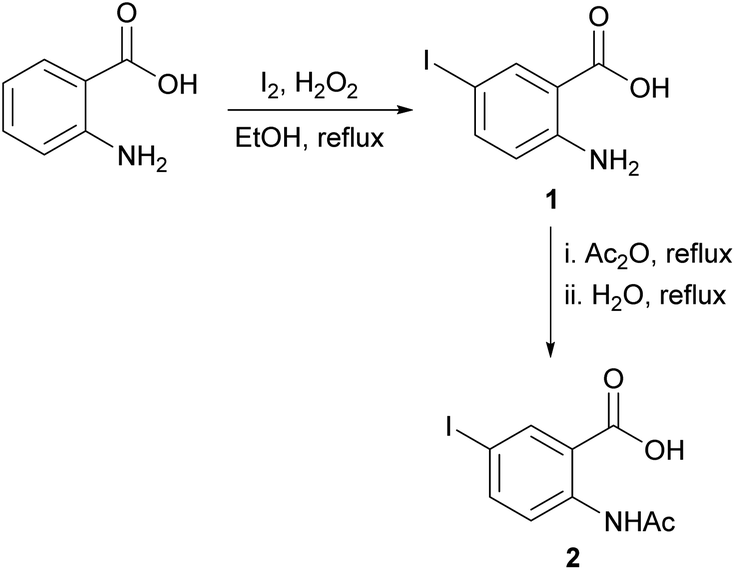 | ||
| Scheme 1 Synthesis of 2-acetamido-5-iodobenzoic acid (2). | ||
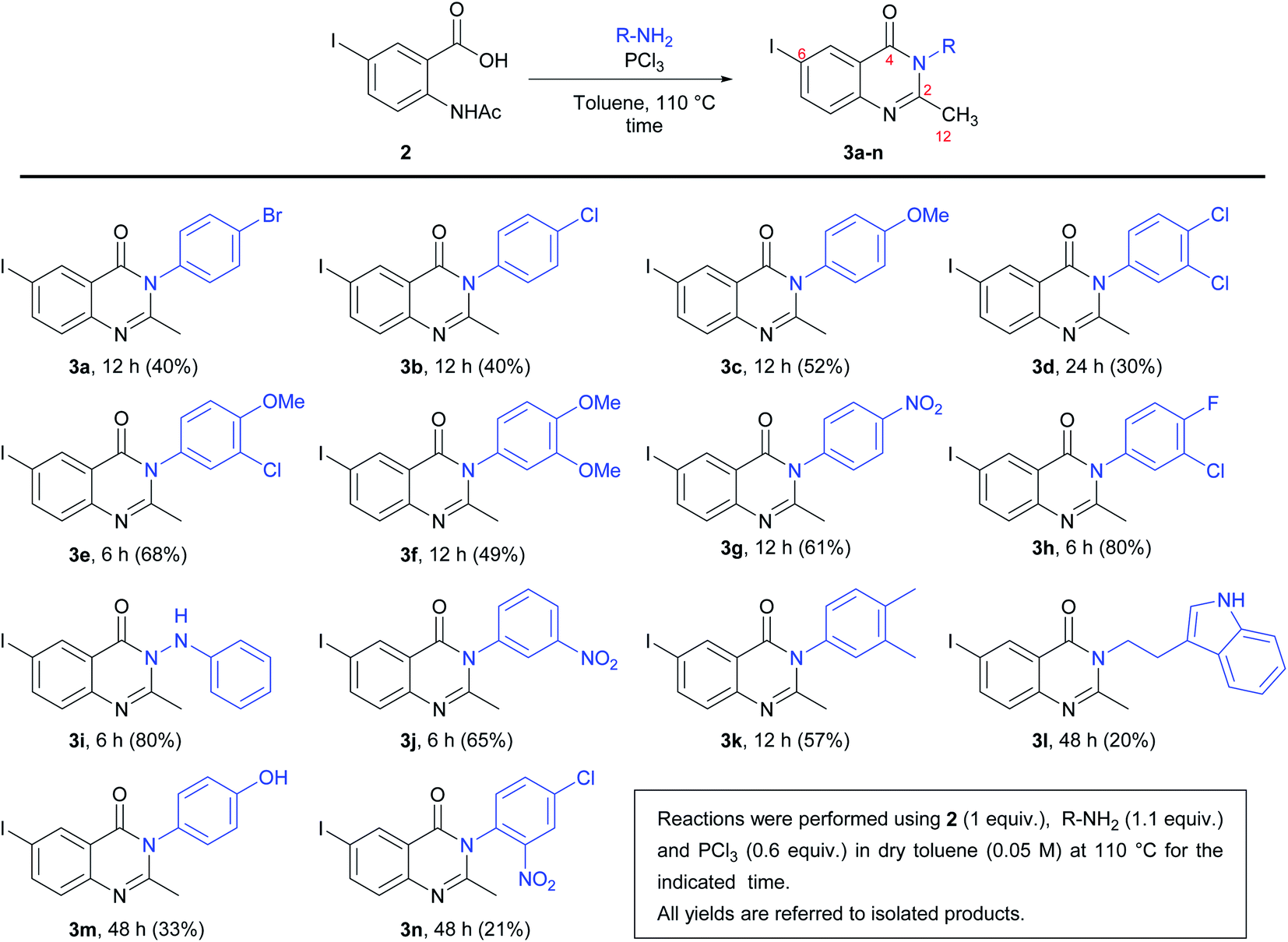 | ||
| Scheme 2 Synthesis of 6-iodo-2-methylquinazolin-4(3H)-one derivatives 3a–n. | ||
As can be seen from Scheme 2, the lowest yield occurred in the synthesis of compound 3n (21%). It seems that the presence of a nitro group in the ortho position of the phenyl ring has an unfavourable effect on the reactivity because this behaviour was not observed when obtaining compounds 3g (p-NO2) and 3j (m-NO2), with respective yields of 61 and 65%. On the other hand, the highest yields were obtained in the synthesis of 3h (m-Cl-p-F) and 3i (hydrazine derivative), both with 80% yields.
The structures of quinazolinones 3a–n was confirmed by IR, 1H NMR, 13C NMR-APT, and high-resolution electrospray-ionisation mass spectrometry (HRESIMS) methods. In general, the IR spectra of all compounds showed a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band in the 1652–1685 cm−1 range, as well as a CN stretching band of the quinazolinone ring in the 1598–1616 cm−1 range. In the 1H NMR spectra, the most characteristic signal was a singlet in the range of δH = 2.11–2.45, corresponding to the protons of the methyl group H12 at C2. In the 13C NMR-APT spectra, CO signals were seen at δC = 160.3–158.9. In addition, a chemical shift was seen at δC = 158.5–154.2 and 91.8–90.9, corresponding to C2 and C6, respectively. These spectroscopic data confirmed the formation of the quinazolinone ring (see the ESI† for details).
O stretching band in the 1652–1685 cm−1 range, as well as a CN stretching band of the quinazolinone ring in the 1598–1616 cm−1 range. In the 1H NMR spectra, the most characteristic signal was a singlet in the range of δH = 2.11–2.45, corresponding to the protons of the methyl group H12 at C2. In the 13C NMR-APT spectra, CO signals were seen at δC = 160.3–158.9. In addition, a chemical shift was seen at δC = 158.5–154.2 and 91.8–90.9, corresponding to C2 and C6, respectively. These spectroscopic data confirmed the formation of the quinazolinone ring (see the ESI† for details).
2.2 Cytotoxic activity
The cytotoxic activities of quinazolinones 3a–n were measured using the MTT colorimetric method against six cancer cell lines: G415, Gbd1, T98G, HeLa, HL60, and U937.32 The IC50 values are summarized in Table 1 and compared to paclitaxel as a positive control. The IC50 values fall over a wide range of concentrations, from 10 μM to over 200 μM, demonstrating an important variation in the cytotoxicity of the quinazolinones on different cancer cell lines. The cytotoxic effects were cell line dependent. The human cervical adenocarcinoma cell line, HeLa, was sensitive to 3c, 3d, 3e, 3f, 3g, 3h, 3i, 3k, and 3n, with IC50 values of 180, 10, 60, 86, 110, 148, 70, 193, and 175 μM, respectively. For all the other quinazolinones, the IC50 values were greater than 200 μM. The human glioblastoma cell line T98G was sensitive to 3e and 3h and, to a much lesser extent, 3c, with IC50 values of 12, 22, and 114 μM, respectively. The human promyelocytic leukemia cell line, HL60, showed a weak sensitivity to compounds 3a and 3b, with IC50 values of 21 and 50 μM, respectively. The human non-Hodgkin lymphoma cell line, U937, was sensitive to 3a and 3b, with IC50 values of 30 and 58 μM, respectively. All synthetic compounds were inactive against human gallbladder carcinoma cell lines, G415 and Gbd1 (IC50 > 200 μM). Furthermore, compounds 3j–n were inactive or very weakly active against all cancer cell lines.| Comp. | Adherent cells | Nonadherent cells | ||||
|---|---|---|---|---|---|---|
| G415 | Gbd1 | T98G | HeLa | HL60 | U937 | |
| a 50% inhibitory concentration values are an average of three individual experiments. | ||||||
| 3a | >200 | >200 | >200 | >200 | 21 ± 1.1 | 30 ± 1.5 |
| 3b | >200 | >200 | >200 | >200 | 50 ± 0.7 | 58 ± 5.6 |
| 3c | >200 | >200 | 114 ± 22 | 180 ± 65 | >200 | >200 |
| 3d | >200 | >200 | >200 | 10 ± 0.7 | >200 | >200 |
| 3e | >200 | >200 | 12 ± 3.4 | 60 ± 1.3 | >200 | >200 |
| 3f | >200 | >200 | >200 | 86 ± 12 | >200 | >200 |
| 3g | >200 | >200 | >200 | 110 ± 51 | >200 | >200 |
| 3h | >200 | >200 | 22 ± 1.7 | 148 ± 91 | >200 | >200 |
| 3i | >200 | >200 | >200 | 70 ± 5.4 | >200 | >200 |
| 3j | >200 | >200 | >200 | >200 | >200 | >200 |
| 3k | >200 | >200 | >200 | 193 ± 120 | >200 | >200 |
| 3l | >200 | >200 | >200 | >200 | >200 | >200 |
| 3m | >200 | >200 | >200 | >200 | >200 | >200 |
| 3n | >200 | >200 | >200 | 175 ± 98 | >200 | >200 |
| Paclitaxel | 10 ± 0.7 | 6 ± 0.6 | 21 ± 3.0 | 6.2 ± 1.9 | 3.1 ± 0.9 | 41 ± 7.0 |
From the results summarized in Table 1, it can be deduced that the bromine atom in the para-position could be an important factor for the cytotoxicity of compound 3a against HL60 (IC50 = 21 μM) and U937 (IC50 = 30 μM) cell lines (Fig. 2). Regarding the inhibitory effect on U937, 3a showed greater cytotoxic activity than paclitaxel (IC50 = 41 μM). However, when the bromine atom is replaced by a chlorine atom, such as in compound 3b, the cytotoxic effect over HL60 and U937 decreases with IC50 values changing to 50 and 58 μM, respectively. A methoxy group in the para-position (compound 3c) has no cytotoxic activity on these two cell lines. Compound 3d (m,p-di-Cl) was very effective on HeLa cells (IC50 = 10 μM). Compounds 3e and 3h showed good efficiency in T98G (IC50 = 12 and 22 μM, respectively), considering that paclitaxel showed an IC50 value of 21 μM. These three molecules (3d, 3e, and 3h) have the presence of a chlorine atom in the meta-position in common. Other substitutions and analogues studied did not show a promising level of activity in the evaluated cell lines.
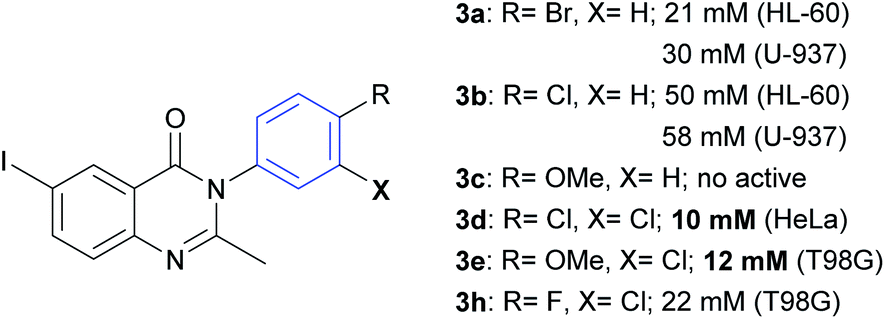 | ||
| Fig. 2 Structures and IC50 of-substituted phenyl rings. | ||
2.3 Molecular docking of compound 3d with DHFR
The calculations of molecular docking for model compounds were performed with the aim of elucidating the elements determining the biological activity. The protein selected for docking studies was dihydrofolate reductase (DHFR), which has an important role in the evolution of several human cancers.33 Moreover, quinazoline and quinazolinone derivatives have been previously described as human DHFR inhibitors.33 DHFR is an enzyme involved in the synthesis of pyrimidinic base timidin, a structural component of DNA, therefore a molecule that inhibits this enzyme inhibit the DNA synthesis and it can be potentially useful as a drug against several types of cancer.33 Moreover, as overexpression of DHFR occurs in breast, prostate, gastric/gastroesophageal, ovarian, endometrial, bladder, lung, colon, and head and neck cancers, it is a target for therapies pointing to inhibition of this protein to decrease tumour growth.34 Principally, the overexpression of this protein is associated in HeLa cell lines.35The molecular docking studies were performed in the active site of DHFR. The results of induced-fit molecular docking, considering flexible residues from 6 Å of the best docked position, describe good correlation among experimental data and calculated values (Fig. 3).
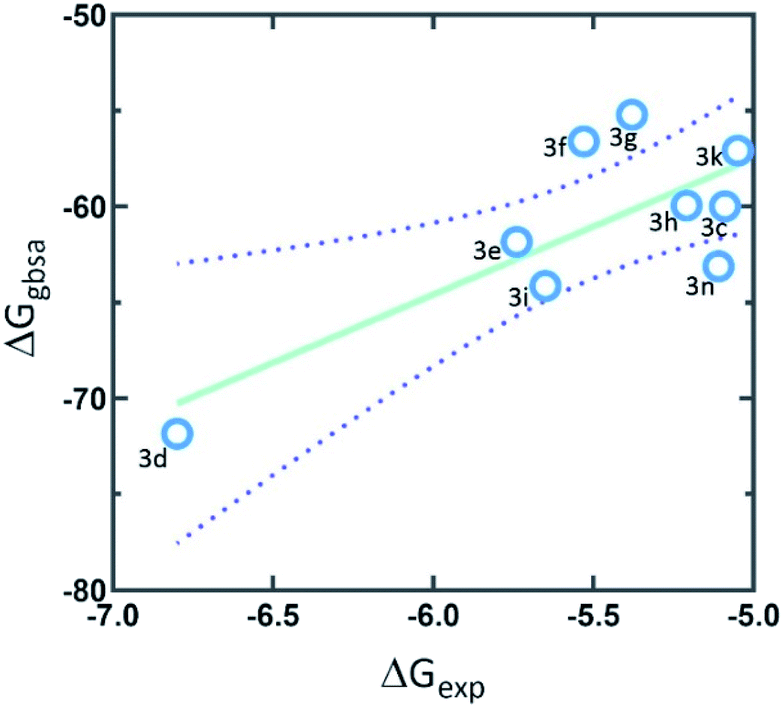 | ||
| Fig. 3 The correlation graph between experimental activity (ΔGexp) and predicted docking activity (ΔGgbsa). | ||
The interaction of synthetized 4-(3H)-quinazolinones was observed to get the view of ligand binding modes while docking since a co-crystal ligand was absent for DFHR. Prediction of the size and spatial orientation of the ligand binding sites of proteins was a major challenge due to the small size of the ligand. The active site of the DFHR crystal structure reported in literature was characterized.36 The important interactions are between residue R71 with an iodine atom, and van der Waals interactions are predominant with amino acids L23, L68, and T57, especially π-stacking with residues F32 and F35 with the quinazolinic aromatic ring (Fig. 4). Altogether, the molecular docking result is in accordance with the binding mechanism of natural ligands (DHF and NADPH), inhibiting catalytic activity of the protein.
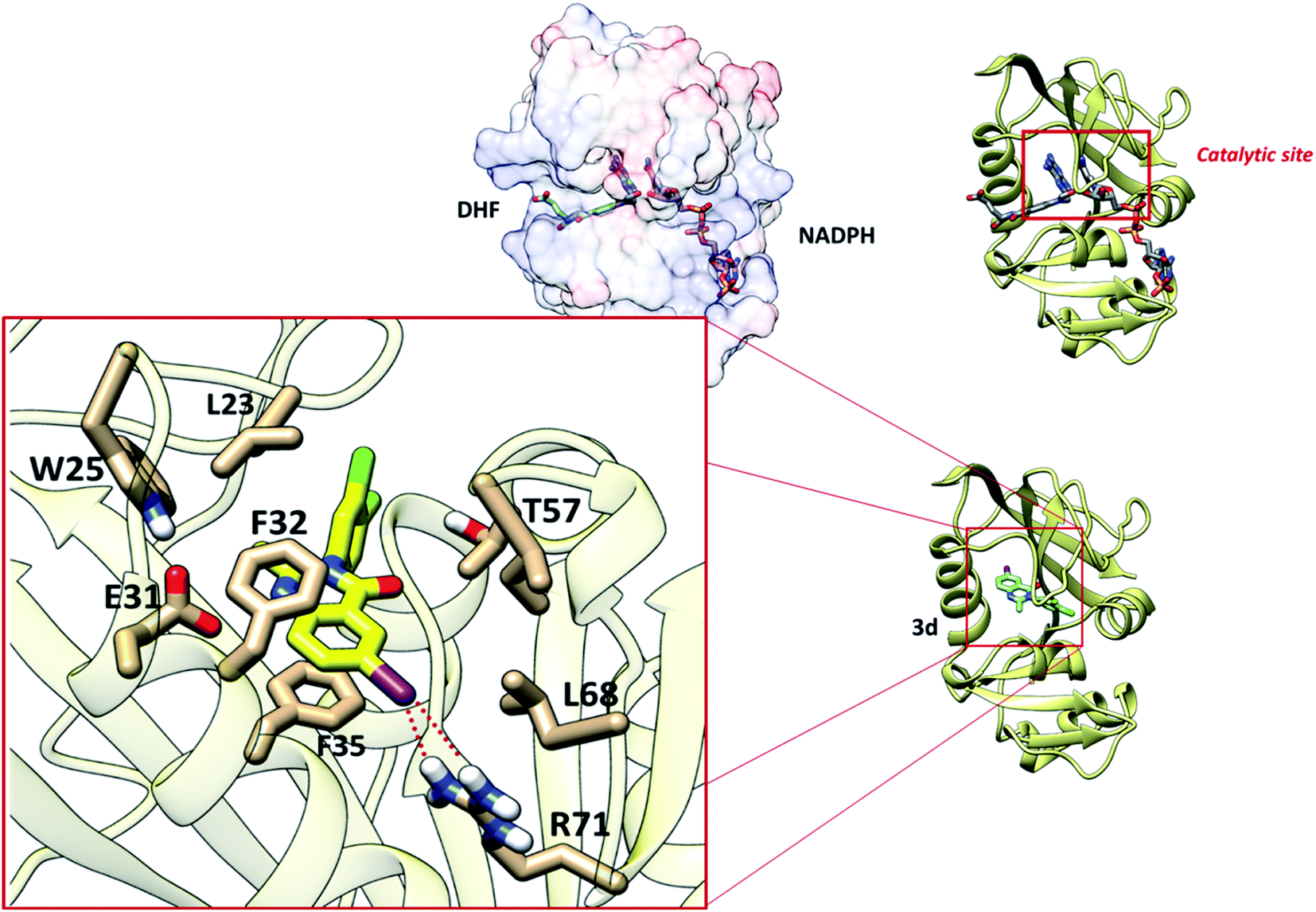 | ||
| Fig. 4 The binding mode of compound 3d in the catalytic site of DHFR. | ||
2.4 3D-QSAR study
Our first challenge was to understand the small structural differences of these synthetized compounds with their notorious biological activity in distinct cell lines, principally in the HeLa cell line. Therefore, we used a ligand set from Pathak's article.37 The Pathak's compounds have quinazoline fragments, similar to our synthetized compounds. Alignments of the 24 structures (9 synthetized compounds and 15 compounds of Pathak, see Fig. S1 in ESI†) were carried out from the more active compounds. Compound Pathak_11 has two halogenated-aromatic fragments, so our compounds were aligned, considering this feature (Fig. 5).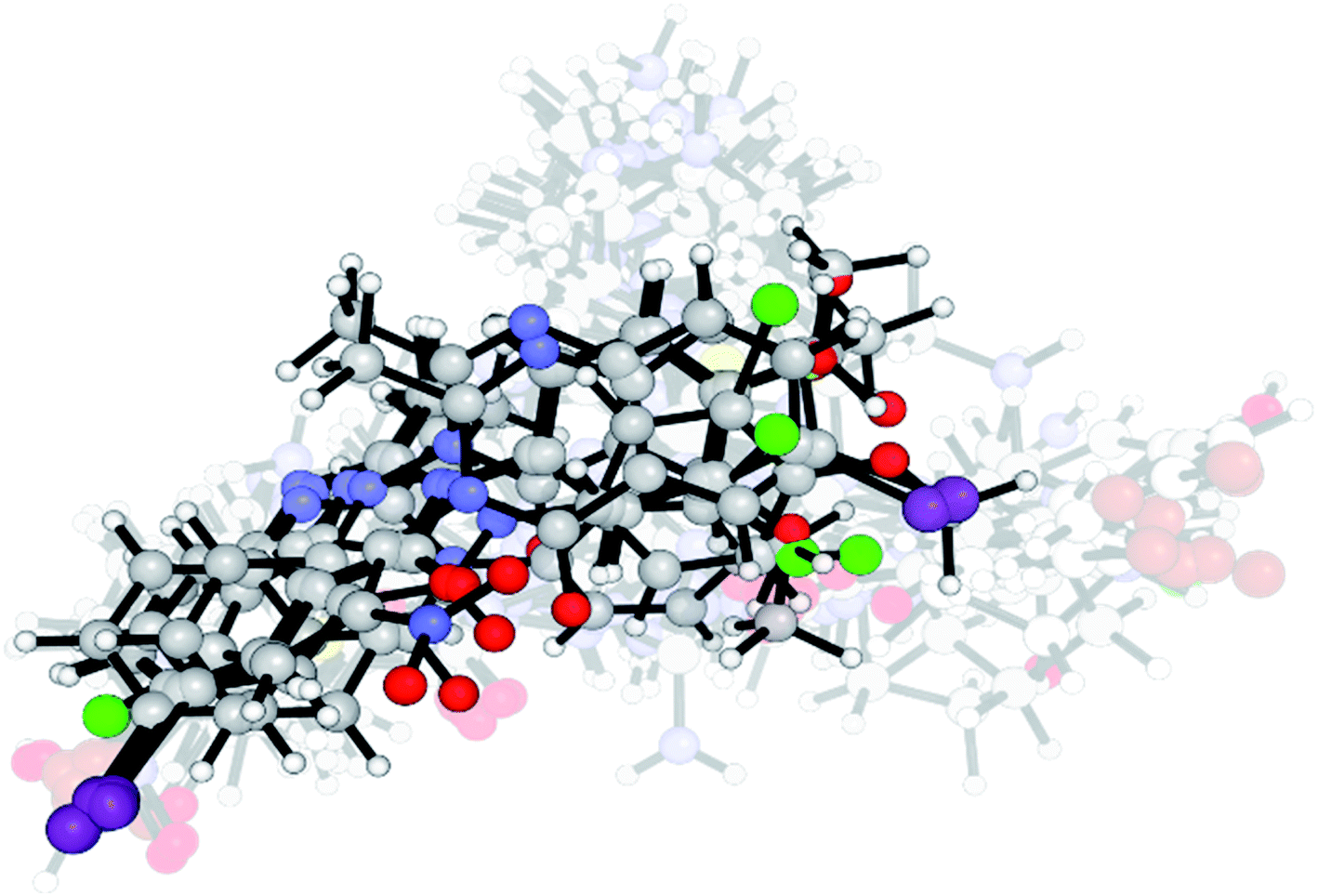 | ||
| Fig. 5 Aligned structures of all compounds for the 3D-QSAR study. Highlighted structures are synthetized compounds in this work, and transparent structures are from Pathak's dataset. | ||
The Comparative Molecular Field Analysis (COMFA)38 is based on 3D-structured features of molecules, such as electrostatic and hydrophobic properties. Indeed, it becomes necessary to develop a QSAR model to predict biological activity before the synthesis of new cytotoxic quinazolinones. The success of 3D-QSAR and molecular docking studies help to understand relationships between the physicochemical properties and biological activity.
These contour maps give us some general insight into the nature of the receptor-ligand binding region. The training set of 18 compounds, COMFA model with five Partial Least Square (PLS) components, was built, and then, the external test set including six compounds was used to evaluate the reliability and applicability of the built model. Statistical quality parameters associated with COMFA models were based on Fractional Factorial Design (FFD) procedures for noise reduction. The COMFA model gave a good cross-validated correlation coefficient (Q2) for Leave-One-Out (LOO), Leave-Two-Out (LTO), and Leave-Many-Out (LMO) as 0.854, 0.826, and 0.841 respectively, indicating an excellent internal predictive power of the established model. The PLS analysis with the five components resulted in a conventional R2 of 0.996, F-test of 666.323, and SDEC of 0.032. Thus, the COMFA model was found to be reasonable (Table 2).
| R2 | SDEC | F-test | Q2 | SDEP | ||
|---|---|---|---|---|---|---|
| a R2 = non-cross validated for determination; Q2 = coefficient of determination for internal validation; SDEC = standard deviation error in calculation; SDEP = standard deviation error in prediction. | ||||||
| Training set | PLS | 0.996 | 0.032 | 666.323 | ||
| LOO | — | — | — | 0.854 | 0.205 | |
| LTO | — | — | — | 0.841 | 0.2.13 | |
| LMO | — | — | — | 0.826 | 0.146 | |
| Test set | 0.852 | 0.210 | ||||
The values of experimental and predicted activities, along with the residual values of the training set and test set molecules, are summarized in Table 3. The scatter plot of the observed versus predicted values of pIC50 for both the training and test set of COMFA models is shown in Fig. 6. This data shows that the experimental and predicted activities of inhibitors are very close to each other. This graphical representation, again, confirms the good predictive power of the established model and also indicates that the developed COMFA model is reliable.
| Compounds | pIC50 exp | pIC50 calc | Set |
|---|---|---|---|
| 3c | 3.745 | 3.733 | Training |
| 3d | 5 | 4.9849 | Training |
| 3e | 4.222 | 4.2706 | Training |
| 3f | 4.066 | 3.9808 | Training |
| 3g | 3.959 | 3.7844 | Test |
| 3h | 3.83 | 3.884 | Training |
| 3i | 4.155 | 4.1573 | Training |
| 3k | 3.714 | 3.8836 | Test |
| 3n | 3.757 | 3.7665 | Training |
| Pathak_1 | 5.017 | 4.8399 | Test |
| Pathak_2 | 5.126 | 5.1072 | Training |
| Pathak_3 | 4.95 | 4.969 | Training |
| Pathak_4 | 5.15 | 5.1361 | Training |
| Pathak_5 | 4.903 | 5.1317 | Test |
| Pathak_6 | 5.013 | 5.1289 | Test |
| Pathak_7 | 5.02 | 5.0128 | Training |
| Pathak_8 | 4.888 | 5.2175 | Test |
| Pathak_9 | 5.022 | 5.0288 | Training |
| Pathak_10 | 5.177 | 5.2045 | Training |
| Pathak_11 | 5.147 | 5.1017 | Training |
| Pathak_12 | 5.124 | 5.133 | Training |
| Pathak_13 | 5.142 | 5.1462 | Training |
| Pathak_14 | 5.075 | 5.111 | Training |
| Pathak_15 | 4.782 | 4.7626 | Training |
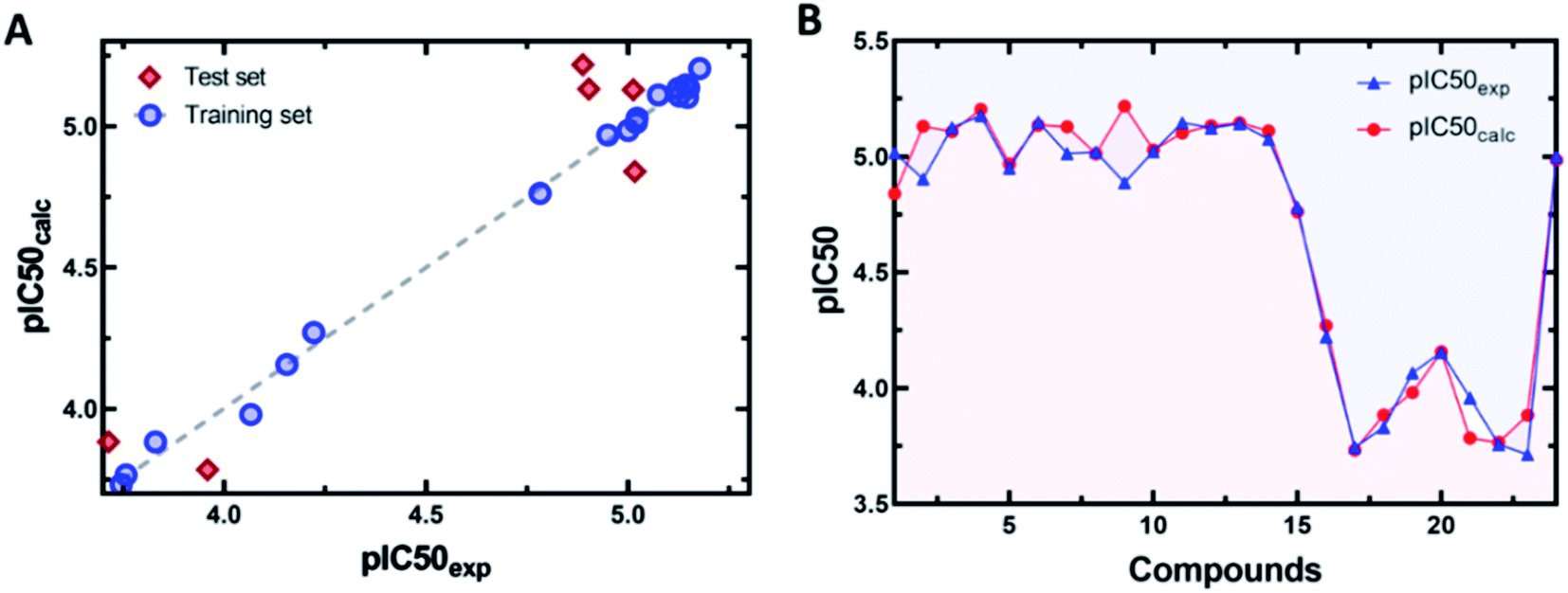 | ||
| Fig. 6 (A) Scatter plots of predicted versus experimental activity. (B) Residual plots between predicted and experimental values. | ||
Contributions by steric effects are observed mainly in the vicinities of the halogen atom (region (a) in Fig. 7) and C-2 methyl group (region (b) in Fig. 7B), where steric interaction tends to increase activity in these regions. On the other hand, the phenyl ring in position 2 tends to increase activity with a second halogen atom in the meta substitution and decrease activity with methoxy/nitro group in ortho substitution by a sterically unfavourable interaction, and this, obviously, reduces the activity.
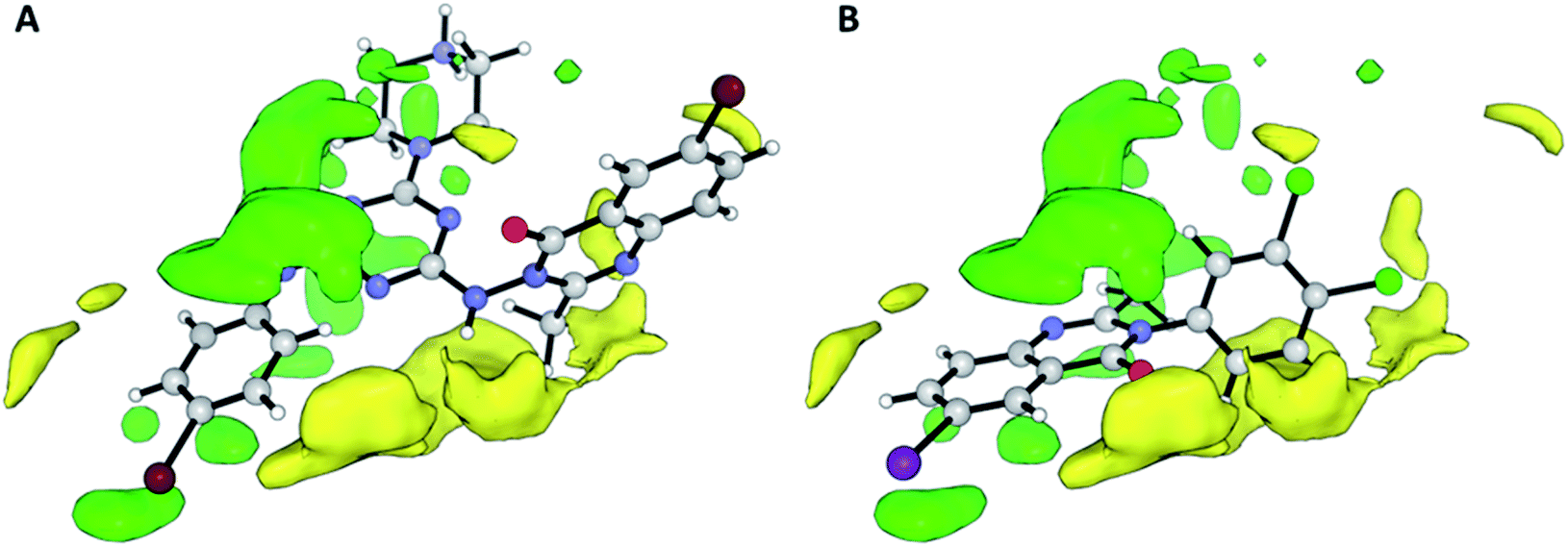 | ||
| Fig. 7 Steric contour maps representing the COMFA model for cytotoxic activity. The compounds shown are the strongest (A; Pakhar_3) and weakest (B; 3d). Green and yellow regions indicate areas where steric interactions increase and decrease activity, respectively. | ||
Nevertheless, the electrostatic effects observed are unfavourable interactions around the quinazolinic ring (Fig. 8, red area in region (a)). The electrostatic interaction, with a positive charge on the putative receptor, is favourable for cytotoxic activity, mainly in the vicinity of the aromatic group at position 2.
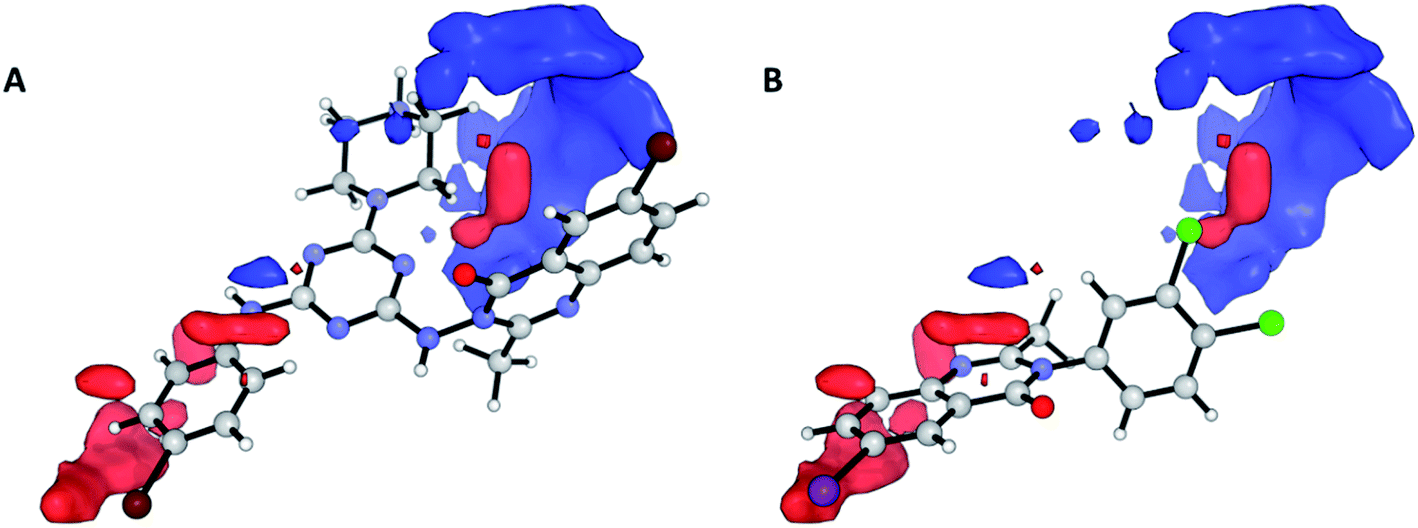 | ||
| Fig. 8 Electrostatic contour maps representing the COMFA model for cytotoxic activity. Compounds shown are the strongest (A; 3) and weakest (B; 17). Blue and red regions denote enhancing and detrimental electrostatic effects with the positively charged probe, respectively. | ||
2.5 Solubility predictions
Physicochemical and pharmacokinetic properties were predicted using the SwissADME Web Service (accessed July 5, 2022) (Table 4). As expected, the synthesized compounds have good liposolubility due to the halogen atoms, essentially the iodine and chlorine atoms, with compound 3d being the most lipophilic compound with characteristics responsive to the dichlorophenyl group. In contrast, the least lipophilic compounds were compounds 3g and 3j (2.55 and 2.57 of consensus log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, respectively), which have nitrophenyl structures.39–41
P, respectively), which have nitrophenyl structures.39–41
| Comp. | iLOGP39 | XLOGP3 (ref. 40) | MLOGP41 | Consensus logP |
|---|---|---|---|---|
| 3a | 3.28 | 3.48 | 4.26 | 3.95 |
| 3b | 3.17 | 3.41 | 4.15 | 3.86 |
| 3c | 3.20 | 2.76 | 3.30 | 3.31 |
| 3d | 3.30 | 4.34 | 4.65 | 4.43 |
| 3e | 3.42 | 3.68 | 3.80 | 3.90 |
| 3f | 3.36 | 3.03 | 2.97 | 3.34 |
| 3g | 2.53 | 2.61 | 2.63 | 2.55 |
| 3h | 3.16 | 3.81 | 4.53 | 4.21 |
| 3i | 2.60 | 3.40 | 3.87 | 3.25 |
| 3j | 2.61 | 2.61 | 2.63 | 2.57 |
| 3k | 3.36 | 3.81 | 4.38 | 4.09 |
| 3l | 3.02 | 3.61 | 3.83 | 3.95 |
| 3m | 2.52 | 2.43 | 3.06 | 2.90 |
| 3n | 2.86 | 3.54 | 3.13 | 3.16 |
The prediction methods of J. Delaney42 and J. Ali43 were used to predict water solubility (Table 5), and their results indicate that the synthesized structures have moderate solubility and are soluble in water. The prediction of J. Delaney states that all structures are moderately soluble, while J. Ali suggests that some structures are soluble in an aqueous solvent. Both methods suggest that compound 3d is moderately soluble in water, as are compounds 3e, 3h, 3i, 3k, 3l, and 3n.
| Comp. | ESOL42 | Ali43 | ||
|---|---|---|---|---|
| logS |
Class | logS |
Class | |
| a Mod. soluble = Moderately soluble. | ||||
| 3a | −5.29 | Mod. soluble | −3.90 | Soluble |
| 3b | −4.97 | Mod. soluble | −3.82 | Soluble |
| 3c | −4.44 | Mod. soluble | −3.34 | Soluble |
| 3d | −5.74 | Mod. soluble | −4.79 | Mod. soluble |
| 3e | −5.21 | Mod. soluble | −4.30 | Mod. soluble |
| 3f | −4.68 | Mod. soluble | −3.82 | Soluble |
| 3g | −4.41 | Mod. soluble | −3.95 | Soluble |
| 3h | −5.31 | Mod. soluble | −4.24 | Mod. soluble |
| 3i | −4.78 | Mod. soluble | −4.06 | Mod. soluble |
| 3j | −4.41 | Mod. soluble | −3.95 | Soluble |
| 3k | −5.16 | Mod. soluble | −4.24 | Mod. soluble |
| 3l | −5.16 | Mod. soluble | −4.36 | Mod. soluble |
| 3m | −4.24 | Mod. soluble | −3.23 | Soluble |
| 3n | −5.19 | Mod. soluble | −4.92 | Mod. soluble |
Finally, the drug-likeness of the synthesized compounds was evaluated based on the Lipinski, Ghose, Veber, Egan, and Muegge rules (Table 6), of which the most active compound (3d) satisfying all rules except the Lipinski rule; however, compound 3e (second slightly active compound) does comply with all five drug-likeness rules.
| Comp. | Lipinski | Ghose | Veber | Egan | Muegge |
|---|---|---|---|---|---|
| 3a | No | Yes | Yes | Yes | Yes |
| 3b | Yes | Yes | Yes | Yes | Yes |
| 3c | Yes | Yes | Yes | Yes | Yes |
| 3d | No | Yes | Yes | Yes | Yes |
| 3e | Yes | Yes | Yes | Yes | Yes |
| 3f | Yes | Yes | Yes | Yes | Yes |
| 3g | Yes | Yes | Yes | Yes | Yes |
| 3h | No | Yes | Yes | Yes | Yes |
| 3i | Yes | Yes | Yes | Yes | Yes |
| 3j | Yes | Yes | Yes | Yes | Yes |
| 3k | No | Yes | Yes | Yes | Yes |
| 3l | Yes | Yes | Yes | Yes | Yes |
| 3m | Yes | Yes | Yes | Yes | Yes |
| 3n | Yes | Yes | Yes | Yes | Yes |
3 Experimental
3.1 Synthesis
Melting points were determined on a Kofler-type apparatus and were uncorrected. The IR spectra were taken on a PerkinElmer 200 spectrophotometer with KBr. NMR spectra were collected in DMSO-d6 or CD3OD with a Varian Unity Inova 500 MHz spectrometer. Chemical shifts were reported in parts per million (δ) using the residual solvent signals (DMSO-d6: δH 2.50, δC 39.5 or CD3OD; δH H 3.31, δC C 49.0) as the internal standards for the 1H and 13C NMR-APT spectra and coupling constants (J) in Hz. HRMS spectra were recorded on a Micromass-LCT Premier Time-of-Flight ESI spectrometer with an acquity ultra-high-performance liquid chromatography interface system. TLC was performed on Si gel Merck 60 F254 (Al plates) and the TLC plates were visualized by spraying with phosphomolybdic acid reagent and heating. The starting materials and reagents were purchased from Sigma–Aldrich or Merck.:32:7.6:0.4). mp. 228–230 °C. IR (cm−1) ν: 3501, 3388 (N–H); 2919, 2623 (O–H); 1677 (CO); 1613 (CAr–NH2); 1579 (CAr–CAr); 1228 (Csp2–OH). 1H NMR (500 MHz, CD3OD) δ (ppm): 8.05 (s, 1H), 7.43 (d, J = 8.73 Hz, 1H), 6.56 (d, J = 8.76 Hz, 1H). 13C NMR-APT (126 MHz, CD3OD) δ (ppm): 169.9 (CO), 147.9 (C), 141.6 (CH), 139.2 (CH), 120.7 (CH), 112.2 (C), 79.5 (C). HRMS-ESI calculated for C7H7INO2 [M + H]+: 263.99215, found 263.95447.:32:7.6:0.4). mp. 239–241 °C. IR (cm−1) ν: 3235 (N–H); 3120 (CAr–H); 2923, 2866, 2763 (O–H); 1687, 1650 (CO); 1592 (CAr–NH2); 1572 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.94 (s, 1H), 8.25 (d, J = 8.84, 1H), 8.18 (s, 1H), 7.84 (d, J = 8.83 Hz, 1H), 2.12 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 168.5 (CO), 168.1 (CO), 142.1 (CH), 140.4 (C), 138.9 (CH), 122.1 (CH), 118.6 (C), 85.5 (C), 25.0 (CH3). HRMS-ESI Calculated for C9H9INO3 [M + H]+: 305.96271, found 305.96454.3.1.3.1 6-Iodo-2-methyl-3-(4-bromophenyl)-4-(3H)-quinazolinone (3a). Rf: 0.83 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3066 (CAr–H); 2925 (Csp3–H); 1685 (CO); 1598 (CN); 1575 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.34 (s, 1H), 8.11 (d, J = 8.54 Hz, 1H), 7.77 (d, J = 8.58, 2H), 7.45 (d, J = 8.59 Hz, 2H), 7.43 (d, J = 8.65 Hz, 1H), 2.12 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 159.9 (CO), 154.9 (C), 146.5 (C), 142.9 (CH), 136.9 (C), 134.4 (CH), 132.6 (2 CH), 130.7 (2 CH), 128.9 (C), 122.3 (CH), 122.2 (C), 91.1 (C–I), 24.1 (CH3). HRMS-ESI calculated for C15H11BrIN2O [M + H]+: 440.90994, found 440.91055.
3.1.3.2 6-Iodo-2-methyl-3-(4-chlorophenyl)-4-(3H)-quinazolinone (3b). Rf: 0.80 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3086, 3058, 3034 and 3007 (CAr–H); 2930 (Csp3–H); 1676 (CO); 1604 (CN); 1587 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.33 (s, 1H), 8.10 (d, J = 8.54 Hz, 1H), 7.64 (d, J = 8.74 Hz, 2H), 7.51 (d, J = 8.77 Hz, 2H), 7.44 (d, J = 8.55 Hz, 1H), 2.12 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 159.9 (CO), 154.9 (C), 146.5 (C), 142.9 (CH), 136.4 (CH), 134.4 (C), 133.7 (C), 130.3 (2 C), 129.6 (2 C), 128.8 (CH), 122.2 (C), 91.1 (C–I), 24.1 (CH3). HRMS-ESI calculated for C15H11ClIN2O [M + H]+: 396.96046, found 396.96112.
3.1.3.3 6-Iodo-2-methyl-3-(4-methoxiphenyl)-4-(3H)-quinazolinone (3c). Rf: 0.83 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3081, 3052 and 3003 (CAr–H); 2960 and 2934 (Csp3–H); 1672 (CO); 1610 (CN); 1598 (CAr–CAr); 1246, 1027 (C–O). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.36 (s, 1H), 8.11 (d, J = 8.54 Hz, 1H), 7.45 (d, J = 8.55 Hz, 1H), 7.34 (d, J = 8.84 Hz, 2H), 7.09 (d, J = 8.88 Hz, 2H), 3.83 (s, 3H), 2.12 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 160.2 (CO), 159.3 (C), 155.9 (C), 146.6 (C), 142.8 (CH), 134.5 (CH), 130.1 (C), 129.4 (2 C), 128.9 (CH), 122.4 (C), 114.7 (2 C), 90.9 (CI), 55.4 (CH3), 24.1 (CH3). HRMS-ESI calculated for C16H14IN2O2 [M + H]+: 393.00999, found 393.01059.
3.1.3.4 6-Iodo-2-methyl-3-(3,4-dichlorophenyl)-4-(3H)-quinazolinone (3d). Rf: 0.85 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3061 (CAr–H); 2928 (Csp3–H); 1671 (CO); 1603 (CN); 1587 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.35 (d, J = 2.0 Hz, 1H), 8.13 (dd, J = 8.5, 2.0 Hz, 1H), 7.91 (d, J = 2.3 Hz, 1H), 7.86 (d, J = 8.5 Hz, 1H), 7.54 (dd, J = 8.5, 2.3 Hz, 1H), 7.46 (d, J = 8.5 Hz, 1H),2.15 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 159.9 (CO), 154.7 (C), 146.5 (C), 143.0 (CH), 137.4 (C), 134.4 (CH), 132.1 (CH), 131.9 (C), 131.5 (C), 130.8 (CH), 129.1 (CH), 128.9 (CH), 122.2 (C), 91.2 (C–I), 24.0 (CH3). HRMS-ESI calculated for C15H10Cl2IN2O [M + H]+: 430.92148, found 430.91843.
3.1.3.5 6-Iodo-2-methyl-3-(3-chloro-4-methoxiphenyl)-4-(3H)-quinazolinone (3e). Rf: 0.78 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3062, 3006 (CAr–H); 2965, 2942, 2927 (Csp3–H); 1675 (CO); 1609 (CN); 1592 (CAr–CAr); 1264, 1058 (C–O). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.35 (d, J = 2.0 Hz, 1H), 8.12 (dd, J = 8.5, 2.1 Hz, 1H), 7.63 (d, J = 2.5 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H), 7.42 (dd, J = 8.7, 2.5 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 3.94 (s, 3H), 2.14 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 160.2 (CO), 155.5 (C), 154.9 (C), 146.6 (C), 142.9 (CH), 134.5 (CH), 130.4 (C), 129.7 (CH), 128.9 (CH), 128.4 (CH), 122.3 (C), 121.3 (C), 113.2 (CH), 91.0 (C–I), 56.4 (CH3), 24.1 (CH3). HRMS-ESI calculated for C16H13ClIN2O2 [M + H]+: 426.97102, found 426.97101.
3.1.3.6 6-Iodo-2-methyl-3-(3,4-dimethoxiphenyl)-4-(3H)-quinazolinone (3f). Rf: 0.80 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3062 (CAr–H); 2997, 2962, 2962, 2934 (Csp3–H); 1673 (CO); 1608 (CN); 1590 (CAr–CAr); 1249, 1132 (C–O). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.36 (d, J = 2.0 Hz, 1H), 8.11 (dd, J = 8.5, 2.1 Hz, 1H), 7.44 (d, J = 8.6 Hz, 1H), 7.11–7.07 (m, 2H), 6.95 (dd, J = 8.4, 2.4 Hz, 1H),3.83 (s, 3H), 3.74 (s, 3H), 2.16 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 160.1 (CO), 155.9 (C), 149.3 (C), 148.9 (C), 146.6 (C), 142.7 (CH), 134.5 (CH), 130.2 (CH), 128.8 (CH), 122.4 (CH), 120.2 (C), 111.9 (CH), 111.8 (CH), 90.9 (C–I), 55.7 (CH3), 55.6 (CH3), 23.9 (CH3). HRMS-ESI calculated for C17H16IN2O3 [M + H]+: 423.02056, found 423.01840.
3.1.3.7 6-Iodo-2-methyl-3-(4-nitrophenyl)-4-(3H)-quinazolinone (3g). Rf: 0.85 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3112, 3071 (CAr–H); 2976, 2930 (Csp3–H); 1679 (CO); 1602 (CN); 1579 (CAr–CAr); 1520 (C–NO2); 1354 (C–NO2). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.42 (d, J = 9.0 Hz, 2H), 8.37 (d, J = 2.0 Hz, 1H), 8.15 (dd, J = 8.5, 2.1 Hz, 1H), 7.82 (d, J = 9.0 Hz, 2H), 7.48 (d, J = 8.6 Hz, 1H),2.13 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 159.9 (CO), 154.3 (C), 147.7 (C), 146.6 (C), 143.3 (CH), 143.1 (C), 134.4 (CH), 130.3 (2 CH), 128.9 (CH), 124.8 (2 CH), 122.2 (C), 91.3 (C–I), 24.0 (CH3). HRMS-ESI calculated for C15H11IN3O3 [M + H]+: 407.98450, found 407.98343.
3.1.3.8 6-Iodo-2-methyl-3-(3-chloro-4-fluorophenyl)-4-(3H)-quinazolinone (3h). Rf: 0.82 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3063 (CAr–H); 2926 (Csp3–H); 1671 (CO); 1603 (CN); 1558 (CAr–CAr).1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.36 (d, J = 2.0 Hz, 1H), 8.13 (dd, J = 8.5, 2.1 Hz, 1H), 7.87 (dd, J = 6.7, 2.5 Hz, 1H), 7.64 (t, J = 9.0 Hz, 1H), 7.58–7.52 (m, 1H), 7.46 (d, J = 8.5 Hz, 1H), 2.14 (s, 3H). 13C NMR-APT (DMSO-d6, 126 MHz) δ (ppm): 160.0 (CO), 157.3 (d, J = 248.9 Hz, C), 154.9 (C), 146.5 (C), 143.0 (CH), 134.5 (CH), 134.4 (C), 130.9 (CH), 129.6 (d, J = 8.0 Hz, CH), 128.9 (CH), 122.2 (C), 120.2 (d, J = 18.8 Hz, C), 117.8 (d, J = 22.1 Hz, CH), 91.1 (C–I), 24.1 (CH3). HRMS-ESI calculated for C15H10ClFIN2O [M + H]+: 414.95103, found 414.95044.
3.1.3.9 6-Iodo-2-methyl-3-(1-phenylamino)-4-(3H)-quinazolinone (3i). Rf: 0.80 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3239 (N–H); 3066, 3021 (CAr–H); 2964, 2929 (Csp3–H); 1672 (CO); 1600 (CN); 1588 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.12 (s, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.12 (dd, J = 8.6, 2.1 Hz, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.19 (dd, J = 8.4, 7.5 Hz, 2H), 6.85 (t, J = 7.3 Hz, 1H), 6.66 (d, J = 7.7 Hz, 2H), 2.49 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 158.9 (CO), 158.5 (C), 146.5 (C), 146.0 (C), 143.0 (CH), 134.3 (CH), 129.2 (2 CH), 129.1 (CH), 122.9 (CH), 120.4 (C), 112.4 (2 CH), 91.3 (C–I), 21.5 (CH3). HRMS-ESI calculated for C15H13IN3O [M + H]+: 378.01032, found 378.01184.
3.1.3.10 6-Iodo-2-methyl-3-(3-nitrophenyl)-4-(3H)-quinazolinone (3j). Rf: 0.85 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3073 (CAr–H); 2932 (Csp3–H); 1673 (CO); 1616 (CN); 1599 (CAr–CAr); 1531, 1349 (NO2). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.49 (t, J = 2.0 Hz, 1H), 8.41–8.37 (m, 1H), 8.36 (d, J = 1.5 Hz, 1H), 8.14 (dt, J = 8.5, 1.7 Hz, 1H), 8.01–7.95 (m, 1H), 7.88 (t, J = 8.1 Hz, 1H), 7.47 (dd, J = 8.5, 0.8 Hz, 1H), 2.13 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 160.1 (CO), 154.6 (C), 148.5 (C), 146.6 (C), 143.0 (CH), 138.6 (C), 135.4 (CH), 134.4 (CH), 130.9 (CH), 128.9 (CH), 124.1 (2 CH), 122.3 (C), 91.2 (C–I), 24.1 (CH3). HRMS-ESI calculated for C15H11IN3O3 [M + H]+: 407.98450, found 407.98532.
3.1.3.11 6-Iodo-2-methyl-3-(3,4-dimethylphenyl)-4-(3H)-quinazolinone (3k). Rf: 0.83 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3048, 3019 (CAr–H); 2972, 2915 (Csp3–H); 1667 (CO); 1596 (CN); 1576 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.34 (d, J = 2.0 Hz, 1H), 8.10 (dd, J = 8.5, 2.1 Hz, 1H), 7.44 (d, J = 8.6 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.12 (dd, J = 7.9, 2.0 Hz, 1H), 2.30 (s, 3H), 2.27 (s, 3H), 2.12 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 160.0 (CO), 155.5 (C), 146.6 (C), 142.8 (CH), 137.7 (C), 137.3 (CH), 135.1 (C), 134.4 (C), 130.4 (CH), 128.8 (CH), 128.8 (CH), 125.3 (CH), 122.3 (C), 90.9 (C–I), 24.1 (CH3), 19.3 (CH3), 19.0 (CH3). HRMS-ESI calculated for C17H16IN2O [M + H]+: 391.03073, found 391.03262.
3.1.3.12 6-Iodo-2-methyl-3-[-2-(3-indolyl)-ethyl]-4-(3H)-quinazolinone (3l). Purified by column chromatography. Rf: 0.78 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3329 (N–H); 3062, 3010 (CAr–H); 2979, 2917 (Csp3–H); 1656 (CO); 1616 (CN); 1589 (CAr–CAr). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.90 (s, 1H), 8.43 (d, J = 2.1 Hz, 1H), 8.07 (dd, J = 8.5, 2.1 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.36 (dd, J = 8.3, 5.8 Hz, 2H), 7.18 (d, J = 2.3 Hz, 1H), 7.08 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 6.98 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 4.30–4.23 (m, 2H), 3.12–3.05 (m, 2H), 2.45 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) (ppm): 159.88 (CO), 155.86 (C), 146.36 (C), 142.58 (CH), 136.22 (CH), 134.44 (C), 128.79 (C), 127.07 (CH), 123.34 (CH), 121.91 (CH), 121.14 (C), 118.50 (CH), 118.15 (CH), 111.52 (C), 110.47 (CH), 90.98 (C–I), 45.33 (CH2), 23.53 (CH2), 22.81 (CH3). HRMS-ESI calculated for C19H17IN3O [M + H]+: 430.04162, found 430.03720.
3.1.3.13 6-Iodo-2-methyl-3-(4-hydroxyphenyl)-4-(3H)-quinazolinone (3m). Purified by column chromatography. Rf: 0.85 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3202 (O–H); 3081, 3033 (CAr–H); 2935 (Csp3–H); 1652 (CO); 1613 (CN); 1598 (CAr–CAr); 1270 (C–O). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.85 (s, 1H), 8.33 (d, J = 2.1 Hz, 1H), 8.09 (dd, J = 8.5, 2.1 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.19 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 2.11 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 160.3 (CO), 157.7 (C), 156.1 (C), 146.6 (C), 142.8 (CH), 134.5 (CH), 129.2 (C), 128.9 (2 CH), 128.6 (CH), 122.4 (C), 116.0 (2 CH), 91.0 (C–I), 24.2 (CH3). HRMS-ESI calculated for C15H12IN2O2 [M + H]+: 378.99434, found 378.99564.
3.1.3.14 6-Iodo-2-methyl-3-(4-chloro2-nitrophenyl)-4-(3H)-quinazolinone (3n). Purified by column chromatography. Rf: 0.88 (benzene/acetone/methanol/acetic acid 60
:32:7.6:0.4). IR (cm−1) ν: 3212, 3082, 3018 (CAr–H); 2967, 2923 (Csp3–H); 1681 (CO); 1603 (CN); 1592 (CAr–CAr); 1527, 1348 (C–NO2). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.44 (d, J = 2.4 Hz, 1H), 8.34 (d, J = 2.0 Hz, 1H), 8.18 (dd, J = 8.5, 2.1 Hz, 1H), 8.12 (m, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 2.23 (s, 3H). 13C NMR-APT (126 MHz, DMSO-d6) δ (ppm): 159.6 (CO), 154.3 (C), 146.4 (C), 146.0 (CH), 143.6 (C), 135.4 (C), 135.2 (CH), 134.4 (CH), 133.0 (CH), 129.2 (C), 129.0 (CH), 126.0 (CH), 121.4 (C), 91.8 (C–I), 23.7 (CH3). HRMS-ESI calculated for C15H10ClIN3O3 [M + H]+: 441.94553, found 441.94547.
3.2 Cytotoxicity assay
Human gallbladder adenocarcinoma (G415), human gallbladder adenocarcinoma (Gbd1), human promyelocytic leukemia (HL60), human histiocytic lymphoma (U937), human cervix adenocarcinoma (HeLa) and human brain glioblastoma multiforma (T98G) cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). All cell lines were grown at 37 °C in a humidified atmosphere of 5% CO2 environment and the adherent cells were removed from culture plates by trypsinization (0.5 mM EDTA, 0.05% trypsin). G-415 and Gbd1 were maintained in RPMI 1640 supplemented with 2 mM glutamine, 10% FBS, penicillin/streptomycin/amphotericin-B (100 units per mL; 100 μg mL−1; 0.25 μg mL−1). Confluent cultures of these two adherent cell lines were split 1:3 to 1:6 by trypsinization and seeded at 2–4 × 104 cells per cm2. HL60 and U937 cells were grown in the same medium and seeded at 1–5 × 105 and 2–9 × 105 cells per mL, respectively. Three times per week, the culture cells were diluted under the same conditions to maintain density and were harvested in the exponential phase of growth. HeLa cells were cultured in MEM, containing 2 mM glutamine, 10% FBS, 1% non-essential amino acids, penicillin/streptomycin/amphotericin-B (100 units per mL; 100 μg mL−1; 0.25 μg mL−1). T98G cells were grown in DMEM-F12 supplemented with 2 mM glutamine, 10% FBS, 1% non-essential amino acids, 1% sodium pyruvate, penicillin/streptomycin/amphotericin-B (100 units per mL; 100 μg mL−1; 0.25 μg mL−1). Confluent cultures of these last two adherent cell lines were split 1:3 to 1:6 by trypsinization and seeded at 2–4 × 104 cells per cm2. For cytotoxicity studies, cells were seeded in a 96-well microtiter plate at a density of 5 × 105 mL−1 and allowed to adhere for 24 h in a CO2 incubator. One day after seeding, cells were treated with fresh medium containing the compounds, dissolved in DMSO (1% final concentration in the well) plus culture medium, incubating by 24 h at 37 °C. The compound concentrations ranged from 0 μM up to 200 μM. After incubation, 10 μL aliquots of MTT solution (5 mg mL−1 in PBS) were added to each well and re-incubated for 4 h at 37 °C, followed by low centrifugation at 800 rpm for 5 minutes. Cell viability was determined by means of MTT reduction and the cells incubated in culture medium alone and 1% DMSO served as control for cell viability (untreated cells). 200 μL of supernatant was carefully aspirated and 200 μL aliquots of 100% DMSO were added to each well to dissolve the formazan crystals, followed by incubation of 10 minutes at 37 °C to dissolve air bubbles. The culture plate was placed on an Emax model micro-plate reader (Molecular Devices) and the absorbance was measured spectrophotometrically at 650 nm. The amount of color produced is directly proportional to the number of viable cells. Untreated cells and the controls containing 1% DMSO were used as 100% viability controls (negative controls, see ESI†). Paclitaxel (T7191, Sigma-Aldrich) was used as reference compound (positive controls, see ESI†). All assays were performed twice with three replicates and processed independently. Mean ± SD was used to estimate the cell viability. Cell viability rate was calculated as the percentage of MTT absorption as follows:| % survival = (mean experimental absorbance/mean control absorbance) × 100 |
The compound concentration was plotted against the corresponding percentage (%) of cell viability obtained with MTT assays, and the 50% inhibitory concentration (IC50) was calculated by non-linear regression. The curve fittings were performed using GraphPad Prism®6 from Systat Software, Inc. Compounds with IC50 > 200 μM were considered as inactive.
3.3 Computational method
The preparations of iodinated 4-(3H)-quinazolinone 3D structures were obtained with OpenBabel44 from SMILES annotations for each ligand. Protonation states were adjusted to pH 7.2 using FixpKa and AM1BCC charges implemented in the QUACPAC package,45 followed by conformer generation using OMEGA.46 Docking was performed using FRED47 and the coordinates of the co-crystal structure of DFHR (PDB code 4M6J),36 keeping 20 poses for each docked molecule. Optimization of the docked poses was carried out using a two-step protocol and SZYBKI.48 First, optimization of the ligand's Cartesian coordinates was performed using a constraint of 1 kcal mol−1, followed by optimization of the complex using MMFF94 s as a force field, a Poisson–Boltzmann model, and AM1BCC charges for the ligands. Flexibility of residue side chains were kept within 6 Å of the ligand. The resulting poses were ranked according to the predicted ligand-protein energy, and the pose with the lowest score was selected as the best pose for each molecule.3D-QSAR models were obtained with the Open3DQSAR package,49 which performs partial least squares (PLS) regression models from molecular interaction fields (MIF). Unless otherwise noted, default parameters were employed for Open3DQSAR. The input to Open3DQSAR is a set of aligned conformers of the dataset with associated bioactivities. A grid was constructed around the aligned molecules in such a way that its box exceeded 5 Å in the largest molecule, and grid spacing was set to 0.5 Å. Steric and electrostatic molecular mechanics of MIFs were computed using the Merck force field (MMFF94). The pictures were obtained with Chimera UCSF and PyMol software.
The prediction of drug likeness of synthesized molecules is estimated using parameters based on Lipinski, Ghose, Veber, Egan, and Muegge rules and, their lipid and water solubility by applying the SwissADME web tool (https://www.swissadme.ch, accessed on 5 July 2022).50 The SwissADME synthetic accessibility score is mainly based on the assumption of the molecular.
4 Conclusions
A novel series of iodinated 4-(3H)-quinazolinones 3N-substituted were synthesized and screened for their in vitro cytotoxic activity against six cancer cell lines (G415, Gbd1, T98G, HeLa, HL60, and U937). Some of these compounds, 3e and 3h, showed remarkable cytotoxic activity against the T98G cell line with IC50 values similar or slightly lower than the control, paclitaxel (IC50 = 21 μM). Similarly, marked inhibitory activity was also observed for compounds 3a and 3b on cell line U937, with one of them showing greater efficiency than paclitaxel (IC50 = 41 μM). Additionally, compound 3a showed important activity on HL60 cells (IC50 = 21 μM). Moreover, 3d showed significant cytotoxic activity on HeLa cells (IC50 = 10 μM), on same order of magnitude as paclitaxel (IC50 = 6.2 μM). Therefore, the experimental study of these compounds should move towards in vivo studies, using animal models of glioblastoma (3e and 3h), lymphoma (3a and 3b), promyelocytic leukemia (3a), and cervical adenocarcinoma (3d). The cytotoxic activities of the new iodinated 4-(3H)-quinazolinones 3N-substituted were screened, and the molecular docking between compound 3d and the active site of DHFR was performed through the FRED software. The results revealed that the molecular coupling of compound 3d to the active site is similar to that of natural DHFR ligands, suggesting the inhibitory mechanism of compound 3d on the catalytic activity of this enzyme. Moreover, based on the results of the 3D-QSAR calculations for the synthesized compounds, the structure activity relationship (SAR) can be summarized by the change of substituents at positions of the quinozoline rings. This change has an effect on cytotoxic activity. Docking results showed that higher lipophilic character and the presence of electron-withdrawing groups at the para position of phenyl ring has constructive impacts toward the development of new iodinated-quinazolinic compounds. It is known that the inhibition of DHFR by quinazolinones derivatives leads to cell death, for this reason the compounds of this chemical nature and that also show cytotoxic activity against various lineages of cancer cells would have possibilities not only as antineoplastic drugs, but also as biocides to various protozoan, fungal and microbial pathogens, in addition to their use in arthritis treatment. Therefore, studies of the effect of these quinazolinones on the catalytic activity of DHFR, as well as their ability as antiprotozoa, antifungal and antimicrobial agents are required, as well as testing the main active compounds in an in vivo study in animal models for the malignant neoplasms evaluated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VK thanks the European Union Project ChemBioFight (grant 269301). The biological studies were supported by grants awarded by the “Fondo Nacional de Desarrollo Científico y Tecnológico de Chile” (Fondecyt 1150934) and “Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias” (FONDAP 15110027). A. C.-A. thanks the OpenEye company for academic licenses. Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02).Notes and references
- (a) M. F. Grundon, Nat. Prod. Rep., 1984, 1, 196–200 Search PubMed; (b) D. He, M. Wang, S. Zhao, Y. Shu, H. Zeng, C. Xiao, C. Lu and Y. Liu, Fitoterapia, 2017, 119, 136–149 CrossRef CAS PubMed; (c) J. Bartroli, E. Turmo, M. Algueró, E. Boncompte, M. L. Vericat, L. Conte, J. Ramis, M. Merlos, J. García-Rafanell and J. Forn, J. Med. Chem., 1998, 41, 1869–1882 CrossRef CAS PubMed; (d) I. Khan, S. Zaib, S. Batool, N. Abbas, Z. Ashraf, J. Iqbal and A. Saeed, Bioorg. Med. Chem., 2016, 24, 2361–2381 CrossRef CAS PubMed; (e) I. Khan, A. Ibrar, N. Abbas and A. Saeed, Eur. J. Med. Chem., 2014, 76, 193–244 CrossRef CAS PubMed; (f) V. Alagarsamy, K. Chitra, G. Saravanan, V. R. Solomon, M. T. Sulthana and B. Narendhar, Eur. J. Med. Chem., 2018, 151, 628–685 CrossRef CAS PubMed.
- (a) M. Hrast, K. Rožman, M. Jukič, D. Patin, S. Gobec and M. Sova, Bioorg. Med. Chem. Lett., 2017, 27, 3529–3533 CrossRef CAS PubMed; (b) A. K. Nanda, S. Ganguli and R. Chakraborty, Molecules, 2007, 12, 2413–2426 CrossRef CAS PubMed; (c) R. Bouley, D. Ding, Z. Peng, M. Bastian, E. Lastochkin, W. Song, M. A. Suckow, V. A. Schroeder, W. R. Wolter, S. Mobashery and M. Chang, J. Med. Chem., 2016, 59, 5011–5021 CrossRef CAS PubMed; (d) M. S. Mohameda, M. M. Kamel, E. M. Kassem, N. Abotaleb, S. I. AbdEl-Moez and M. F. Ahmed, Eur. J. Med. Chem., 2010, 45, 3311–3319 CrossRef PubMed; (e) S. Gatadi, J. Gour, M. Shukla, G. Kaul, S. Das, A. Dasgupta, S. Malasala, R. S. Borra, Y. V. Madhavi, S. Chopra and S. Nanduri, Eur. J. Med. Chem., 2018, 157, 1056–1067 CrossRef CAS PubMed.
- (a) B. S. Kuarm, Y. T. Reddy, J. V. Madhav, P. A. Crooks and B. Rajitha, Bioorg. Med. Chem. Lett., 2011, 21, 524–527 CrossRef CAS PubMed; (b) M. M. Ghorab, S. M. Abdel-Gawad and M. S. A. El-Gaby, Farmaco, 2000, 55, 249–255 CrossRef CAS PubMed; (c) S. N. Pandeya, D. Sriram, G. Nath and E. De Clercq, Arzneim. Forsch., 2000, 50, 55–59 CAS.
- (a) S. Gatadi, J. Gour, M. Shukla, G. Kaul, A. Dasgupta, Y. V. Madhavi, S. Chopra and S. Nanduri, Eur. J. Med. Chem., 2019, 175, 287–308 CrossRef CAS PubMed; (b) C. Couturier, C. Lair, A. Pellet, A. Upton, T. Kaneko, C. Perron, E. Cogo, J. Menegotto, A. Bauer, B. Scheiper, S. Lagrange and E. Bacqué, Bioorg. Med. Chem. Lett., 2016, 26, 5290–5299 CrossRef CAS PubMed.
- (a) S. Zhu, J. Wang, G. Chandrashekar, E. Smith, X. Liu and Y. Zhang, Eur. J. Med. Chem., 2010, 45, 3864–3869 CrossRef CAS PubMed; (b) H. Kikuchi, K. Yamamoto, S. Horoiwa, S. Hirai, R. Kasahara, N. Hariguchi, M. Matsumoto and Y. Oshima, J. Med. Chem., 2006, 49, 4698–4706 CrossRef CAS PubMed.
- Y. Deng, H. Mu, H. B. Li, L. Z. Fu, D. Tang, T. Wu, S. H. Huang and C. H. Li, Chem. Biodiversity, 2021, 18, e202100687 CrossRef PubMed.
- (a) B. De Filippis, A. Ammazzalorso, M. Fantacuzzi, L. Giampietro, C. Maccallini and R. Amoroso, ChemMedChem, 2017, 12, 558–570 CrossRef CAS PubMed; (b) K. I. Ozaki, Y. Yamada, T. Oine, T. Ishizuka and Y. Iwasawa, J. Med. Chem., 1985, 28, 568–576 CrossRef CAS PubMed; (c) A. A. M. Abdel-Aziz, L. A. Abou-Zeid, K. E. H. ElTahir, M. A. Mohamed, M. A. Abu El-Enin and A. S. El-Azab, Bioorg. Med. Chem., 2016, 24, 3818–3828 CrossRef CAS PubMed.
- (a) S. E. Abbas, F. M. Awadallah, N. A. Ibrahin, E. G. Said and G. M. Kamel, Eur. J. Med. Chem., 2012, 53, 141–149 CrossRef CAS PubMed; (b) K. P. Rakesh, C. S. Shantharam and H. M. Manukumar, Bioorg. Chem., 2016, 68, 1–8 CrossRef CAS PubMed.
- (a) G. Marzaro, I. Castagliuolo, G. Schirato, G. Palu, M. Dalla Via, A. Chilin and P. Brun, Eur. J. Med. Chem., 2016, 115, 416–425 CrossRef CAS PubMed; (b) X. Fang, Y. T. Chen, E. H. Sessions, S. Chowdhury, T. Vojkovsky, Y. Yin, J. R. Pocas, W. Grant, T. Schröter, L. Lin, C. Ruiz, M. D. Cameron, P. Lograsso, T. D. Bannister and Y. Feng, Bioorg. Med. Chem. Lett., 2011, 21, 1844–1848 CrossRef CAS PubMed; (c) T. O. Mirgany, A. N. Abdalla, M. Arifuzzaman, A. F. M. Motiur Rahman and H. S. Al-Salem, J. Enzyme Inhib. Med. Chem., 2021, 36, 2055–2067 CrossRef CAS PubMed.
- (a) P. M. Chandrika, T. Yakaiah, A. R. R. Rao, B. Narsaiah, N. C. Reddy, V. Sridhar and J. V. Rao, Eur. J. Med. Chem., 2008, 43, 846–852 CrossRef CAS PubMed; (b) L. Mosca, D. Rotili, I. Tempera, A. Masci, M. Fontana, R. Chiaraluce, P. Mastromarino, M. d'Erme and A. Mai, ChemMedChem, 2011, 6, 606–611 CrossRef CAS PubMed; (c) B. Marvania, P. C. Lee, R. Chaniyara, H. Dong, S. Suman, R. Kakadiya, T. C. Chou, T. C. Lee, A. Shah and T. L. Su, Bioorg. Med. Chem., 2011, 19, 1987–1998 CrossRef CAS PubMed; (d) M. A. Mohamed, R. R. Ayyad, T. Z. Shawer, A. A. M. Abdel-Aziz and A. S. El-Azab, Eur. J. Med. Chem., 2016, 112, 106–113 CrossRef CAS PubMed; (e) N. M. Abdel Gawad, H. H. Georgey, R. M. Youssef and N. A. El-Sayed, Eur. J. Med. Chem., 2010, 45, 6058–6067 CrossRef CAS PubMed; (f) N. Manhas, P. Singh, C. Mocktar, M. Singh and N. Koorbanally, Chem. Biodiversity, 2021, 18, e2100096 CrossRef CAS PubMed; (g) C. J. Wang, X. Guo, R. Q. Zhai, C. Sun, G. Xiao, J. Chen, M. Y. Wei, C. L. Shao and Y. Gu, Eur. J. Med. Chem., 2021, 224, 113671 CrossRef CAS PubMed; (h) E. R. Mohammed and G. F. Elmasry, J. Enzyme Inhib. Med. Chem., 2022, 37, 686–700 CrossRef CAS PubMed.
- (a) V. Bavetsias, J. H. Marriott, C. Melin, R. Kimbell, Z. S. Matusiak, F. T. Boyle and A. L. Jackman, J. Med. Chem., 2000, 43, 1910–1926 CrossRef CAS PubMed; (b) J. B. Smaill, G. W. Rewcastle, J. A. Loo, K. D. Greis, O. H. Chan, E. L. Reyner, E. Lipka, H. D. H. Showalter, P. W. Vincent, W. L. Elliott and W. A. Denny, J. Med. Chem., 2000, 43, 1380–1397 CrossRef CAS PubMed; (c) A. Wissner, D. M. Berger, D. H. Boschelli, M. Brawner Floyd, L. M. Greenberger, B. C. Gruber, B. D. Johnson, N. Mamuya, R. Nilakantan, M. F. Reich, R. Shen, H. R. Tsou, E. Upeslacis, Y. F. Wang, B. Wu, F. Ye and N. Zhang, J. Med. Chem., 2000, 43, 3244–3256 CrossRef CAS PubMed.
- (a) H. S. A. ElZahabi, M. S. Nafie, D. Osman, N. H. Elghazawy, D. H. Soliman, A. A. H. EL-Helby and R. K. Arafa, Eur. J. Med. Chem., 2021, 222, 113609 CrossRef CAS PubMed; (b) A. M. Soliman, A. Khalil, E. Ramadan and M. M. Ghorab, Bioorg. Med. Chem. Lett., 2021, 49, 128308 CrossRef CAS PubMed; (c) M. W. Aziz, A. M. Kamal, K. O. Mohamed and A. A. Elgendy, Bioorg. Med. Chem. Lett., 2021, 41, 127987 CrossRef CAS PubMed.
- (a) K. El-Adl, A. G. A. El-Helby, R. R. Ayyad, H. A. Mahdy, M. M. Khalifa, H. A. Elnagar, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly and I. H. Eissa, Bioorg. Med. Chem., 2021, 29, 115872 CrossRef CAS PubMed; (b) H. A. Mahdy, M. K. Ibrahim, A. M. Metwaly, A. Belal, A. B. M. Mehany, K. M. A. El-Gamal, A. El-Sharkawy, M. A. Elhendawy, M. M. Radwan, M. A. Elsohly and I. H. Eissa, Bioorg. Chem., 2020, 94, 103422 CrossRef CAS PubMed; (c) I. H. Eissa, A. G. A. El-Helby, H. A. Mahdy, M. M. Khalifa, H. A. Elnagar, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly and K. El-Adl, Bioorg. Chem., 2020, 105, 104380 CrossRef CAS PubMed.
- (a) M. A. Sabry, H. A. Ewida, G. S. Hassan, M. A. Ghaly and H. I. El-Subbagh, Bioorg. Chem., 2019, 88, 102923 CrossRef CAS PubMed; (b) S. M. El-Messery, G. S. Hassan, M. N. Nagi, E. S. E. Habib, S. T. Al-Rashood and H. I. El-Subbagh, Bioorg. Med. Chem. Lett., 2016, 26, 4815–4823 CrossRef CAS PubMed; (c) S. T. Al-Rashood, G. S. Hassan, S. M. El-Messery, M. N. Nagi, E. S. E. Habib, F. A. M. Al-Omary and H. I. El-Subbagh, Bioorganic Med. Chem. Lett., 2014, 24, 4557–4567 CrossRef CAS PubMed.
- A. Gangjee, J. Yu, J. J. McGuire, V. Cody, N. Galitsky, R. L. Kisliuk and S. F. Queener, J. Med. Chem., 2000, 43, 3837–3851 CrossRef CAS PubMed.
- J.-F. Liu, Curr. Org. Synth., 2007, 4, 223–237 CrossRef CAS.
- (a) H. T. B. Bui, K. M. Do, H. T. D. Nguyen, H. Van Mai, T. L. D. Danh, D. Q. Tran and H. Morita, Tetrahedron, 2021, 98, 132426 CrossRef CAS; (b) S. Y. Abbas, K. A. M. El-Bayouki and W. M. Basyouni, Synth. Commun., 2016, 46, 993–1035 CrossRef CAS; (c) Z. Tashrifi, M. Mohammadi-Khanaposhtani, M. Biglar, B. Larijani and M. Mahdavi, Curr. Org. Chem., 2019, 23, 1090–1130 CrossRef CAS.
- (a) J. A. Bleda, P. M. Fresneda, R. Orenes and P. Molina, Eur. J. Org. Chem., 2009, 4, 2490–2504 CrossRef; (b) X. Yang, M. Wu, S. Sun, M. Ding and J. Xie, J. Heterocycl. Chem., 2008, 45, 1365–1369 CrossRef CAS; (c) C. Xie, H. X. Li, M. G. Liu and M. W. Ding, Chin. Chem. Lett., 2008, 19, 505–508 CrossRef CAS; (d) M. M. Vögtle and A. L. Marzinzik, QSAR Comb. Sci., 2004, 23, 440–459 CrossRef.
- (a) M. Sharif, Appl. Sci., 2020, 10, 2815 CrossRef CAS; (b) M. Abdullaha, S. Mohammed, M. Ali, A. Kumar, R. A. Vishwakarma and S. B. Bharate, J. Org. Chem., 2019, 84, 5129–5140 CrossRef CAS PubMed; (c) Q. H. Teng, Y. Sun, Y. Yao, H. T. Tang, J. R. Li and Y. M. Pan, ChemElectroChem, 2019, 6, 3120–3124 CrossRef CAS; (d) J. An, Y. Wang, Z. Zhang, Z. Zhao, J. Zhang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 12308–12312 CrossRef CAS PubMed; (e) J. Clayden, N. Greeves and S. Warren, Org. Chem. Front., 2012, 58, 1261 Search PubMed.
- (a) A. Philips, D. Raja, A. Arumugam and W. Lin, Asian J. Org. Chem., 2021, 10, 1795–1800 CrossRef CAS; (b) F. Li, L. Lu and P. Liu, Org. Lett., 2016, 18, 2580–2583 CrossRef CAS PubMed; (c) P. S. Kerdphon, T. S. Sanghong, J. Chatwichien, V. Choommongkol, P. Rithchumpon and P. Meepowpan, Eur. J. Org. Chem., 2020, 2730–2734 CrossRef; (d) S. Balaji, G. Balamurugan, R. Ramesh and D. Semeril, Organometallics, 2021, 40, 725–734 CrossRef CAS; (e) S. R. Vemula, D. Kumar and G. R. Cook, Tetrahedron Lett., 2018, 59, 3801–3805 CrossRef CAS PubMed.
- (a) H. Hou, X. Ma, Y. Lin, J. Lin, W. Sun, L. Wang, X. Xu and F. Ke, RSC Adv., 2021, 11, 17721–17726 RSC; (b) V. Nomula and S. N. Rao, Synth. Commun., 2021, 51, 2602–2612 CrossRef CAS.
- (a) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348–351 CrossRef CAS PubMed; (b) D. Yang, H. Fu, L. Hu, Y. Jiang and Y. Zhao, J. Comb. Chem., 2009, 11, 653–657 CrossRef CAS PubMed; (c) T. Liu, C. Zhu, H. Yang and H. Fu, Adv. Synth. Catal., 2012, 354, 1579–1584 CrossRef CAS; (d) L. Xu, Y. Jiang and D. Ma, Org. Lett., 2012, 14, 1150–1153 CrossRef CAS PubMed; (e) B. Li, L. Samp, J. Sagal, C. M. Hayward, C. Yang and Z. Zhang, J. Org. Chem., 2013, 78, 1273–1277 CrossRef CAS PubMed.
- (a) B. Ma, Y. Wang, J. Peng and Q. Zhu, J. Org. Chem., 2011, 76, 6362–6366 CrossRef CAS PubMed; (b) F. Zeng and H. Alper, Org. Lett., 2010, 12, 3642–3644 CrossRef CAS PubMed; (c) F. Zeng and H. Alper, Org. Lett., 2010, 2, 1188–1191 CrossRef PubMed; (d) Z. Zheng and H. Alper, Org. Lett., 2008, 10, 829–832 CrossRef CAS PubMed; (e) L. He, H. Li, H. Neumann, M. Beller and X. Wu, Angew. Chem., Int. Ed., 2014, 53, 1420–1424 CrossRef CAS PubMed; (f) X. F. Wu, L. He, H. Neumann and M. Beller, Chem.–Eur. J., 2013, 19, 12635–12638 CrossRef CAS PubMed; (g) S. W. Tao, R. Q. Liu, J. Y. Zhou and Y. M. Zhu, ChemistrySelect, 2020, 5, 7332–7337 CrossRef CAS.
- (a) T. Ghosh, I. Mandal, S. J. Basak and J. Dash, J. Org. Chem., 2021, 86, 14695–14704 CrossRef CAS PubMed; (b) H. L. Liu, X. T. Li, H. Z. Tian and X. W. Sun, Org. Lett., 2021, 23, 4579–4583 CrossRef CAS PubMed; (c) A. S. Hussen, S. Bagchi and A. Sharma, ChemistrySelect, 2019, 4, 10169–10173 CrossRef CAS; (d) D. Cheng, X. Yan, Y. Pu, J. Shen, X. Xu and J. Yan, Eur. J. Org. Chem., 2021, 2021, 944–950 CrossRef CAS.
- (a) V. Kesternich, M. Pérez-Fehrmann, A. Puelles, I. Brito, A. Cárdenas, M. Bolte and M. López-Rodríguez, Z. Krystallog., 2013, 228, 383–384 CAS; (b) M. Pérez-Ferhmann, V. Kesternich, R. Fernández, F. Verdugo, I. Brito, A. Cárdenas and M. Bolte, Z. Krystallog., 2014, 229, 401–402 Search PubMed; (c) M. Pérez-Ferhmann, V. Kesternich, R. Fernández, F. Verdugo, I. Brito, A. Cárdenas and M. Bolte, Z. Krystallog., 2014, 229, 419–420 Search PubMed.
- M. F. Zayed, H. E. A. Ahmed, A. S. M. Omar, A. S. Abdelrahim and K. El-Adl, Med. Chem. Res., 2013, 22, 5823–5831 CrossRef CAS.
- Y. Wang, C. A. Mathis, G. F. Huang, M. L. Debnath, D. P. Holt, L. Shao and W. E. Klunk, J. Mol. Neurosci., 2003, 20, 255–260 CrossRef PubMed.
- (a) V. G. Ugale, H. M. Patel, S. G. Wadodkar, S. B. Bari, A. A. Shirkhedkar and S. J. Surana, Eur. J. Med. Chem., 2012, 53, 107–113 CrossRef CAS PubMed; (b) C. Parkanyi and D. S. Schmidt, J. Heterocycl. Chem., 2000, 37, 725–729 CrossRef CAS.
- (a) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC; (b) R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed; (c) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed; (d) S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC; (e) J. P. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31–44 RSC; (f) F. Christoffel and T. R. Ward, Catal. Lett., 2018, 148, 489–511 CrossRef CAS.
- (a) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348–351 CrossRef CAS PubMed; (b) C. Bingi, K. Y. Kola, A. Kale, J. B. Nanubolu and K. Atmakur, Tetrahedron Lett., 2017, 58, 1071–1074 CrossRef CAS; (c) J. F. Liu, J. Lee, A. M. Dalton, G. Bi, L. Yu, C. M. Baldino, E. McElory and M. Brown, Tetrahedron Lett., 2005, 46, 1241–1244 CrossRef CAS; (d) M. Sharif, J. Opalach, P. Langer, M. Beller and X. F. Wu, RSC Adv., 2014, 4, 8–17 RSC.
- (a) H. W. Grimmel, A. Guenther and J. F. Morgan, J. Am. Chem. Soc., 1946, 68, 542–543 CrossRef CAS PubMed; (b) S. Xue, J. Mckenna, W. Shieh and O. Repic, J. Org. Chem., 2004, 69, 6474–6477 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- M. V. Raimondi, O. Randazzo, M. La Franca, G. Barone, E. Vignoni, D. Rossi and S. Collina, Molecules, 2019, 24, 1–19 CrossRef PubMed.
- J. R. Bertino, Best Pract. Res., Clin. Haematol., 2009, 22, 577–582 CrossRef PubMed.
- E. Ercikan-Abali, H. Celikkaya, Y.-C. Hsieh, D. Banerjee and J. R. Bertino, Curr. Enzyme Inhib., 2012, 8, 107–117 CrossRef.
- G. Bhabha, D. C. Ekiert, M. Jennewein, C. M. Zmasek, L. M. Tuttle, G. Kroon, H. J. Dyson, A. Godzik, I. A. Wilson and P. E. Wright, Nat. Struct. Mol. Biol., 2013, 20, 1243–1249 CrossRef CAS PubMed.
- P. Pathak, P. K. Shukla, V. Kumar, A. Kumar and A. Verma, Inflammopharmacology, 2018, 26, 1441–1453 CrossRef CAS PubMed.
- (a) R. D. Cramer, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS PubMed; (b) M. Clark, R. D. Cramer, D. M. Jones, D. E. Patterson and P. E. Simeroth, Tetrahedron Comput. Methodol., 1990, 3, 47–59 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, J. Chem. Inf. Model., 2014, 54, 3284–3301 CrossRef CAS PubMed.
- T. Cheng, Y. Zhao, X. Li, F. Lin, Y. Xu, X. Zhang, Y. Li, R. Wang and L. Lai, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS PubMed.
- Y. M. Ikuo Moriguchi, Shuichi Hirono, Qian Liu and Izumi Nakagome, Chem. Pharm. Bull., 1992, 127–130 CrossRef.
- J. S. Delaney, J. Chem. Inf. Comput. Sci., 2004, 44, 1000–1005 CrossRef CAS PubMed.
- J. Ali, P. Camilleri, M. B. Brown, A. J. Hutt and S. B. Kirton, J. Chem. Inf. Model., 2012, 52, 2950–2957 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminform., 2011, 3, 1–14 CrossRef PubMed.
- QUACPAC 2.0.1.2: OpenEye Scientific Software, Santa Fe, NM, https://www.eyesopen.com Search PubMed.
- P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS PubMed.
- M. McGann, J. Chem. Inf. Model., 2011, 51, 578–596 CrossRef CAS PubMed.
- SZYBKI 1.10.1.2: OpenEye Scientific Software, Santa Fe, NM, https://www.eyesopen.com Search PubMed.
- P. Tosco and T. Balle, J. Mol. Model., 2011, 17, 201–208 CrossRef PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03684c |
| This journal is © The Royal Society of Chemistry 2022 |