
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibitory effect of phenolic extract from squirting cucumber (Ecballium elaterium (L.) A. Rich) seed oil on integrin-mediated cell adhesion, migration and angiogenesis†
Imen Touihri-Barakatia,
Olfa Kallech-Ziri‡
a,
Maram Morjen‡b,
Naziha Marrakchib,
José Luisc and
Karim Hosni *a
*a
aLaboratoire des Substances Naturelles (LR10INRAP02), Institut National de Recherche et d'Analyse Physico-chimique, Sidi Thabet, 2020 Ariana, Tunisia. E-mail: karim.hosni@inrap.rnrt.tn
bLaboratory of Biomolecules, Venoms and Theranostic Applications, LR20IPT01, Pasteur Institute of Tunis, University of Tunis El Manar, Tunis 1002, Tunisia
cCNRS-UMR 7051, Institut de Neuro Physiopathologie (INP), Université Aix-Marseille, 27 Bd Jean Moulin, 13385 Marseille, France
First published on 7th November 2022
Abstract
Integrin targeted therapies by natural bioactive compounds have attracted attention in the field of oncology and cancer treatment. This study evaluates the potential of phenolic extract from the medicinal herb Ecballium elaterium L. seed oil (PEO) to inhibit the adhesion and migration of the highly invasive human fibrosarcoma cell line HT1080. At safe concentrations (up to 40 μg mL−1), results show that PEO dose-dependently inhibits adhesion and migration of HT1080 to fibronectin (IC50 = 18 μg mL−1) and fibrinogen (IC50 = 12.86 μg mL−1). These observations were associated with the reduction of cell motility and migration velocity as revealed in the Boyden chamber and random motility using two-dimensional assays, respectively. Additional experiments using integrin blocking antibodies showed that PEO at the highest safe concentration (40 μg mL−1) competitively inhibited the attachment of HT1080 cell to anti-αvβ3 (>98%), anti-α5β1 (>86%), and to a lesser extent anti-α2 (>50%) immobilized antibodies, suggesting that αvβ3 and α5β1 integrins were selectively targeted by PEO. Moreover, PEO specifically targeted these integrins in human microvascular endothelial cells (HMEC-1) and dose-dependently blocked the in vitro tubulogenesis. In the CAM model, PEO inhibited the VEGF-induced neoangiogenesis confirming its anti-angiogenic effect. Collectively, these results indicate that PEO holds promise for the development of natural integrin-targeted therapies against fibrosarcoma.
1. Introduction
Cancer metastasis and proliferation relies on complex interactions between cancer cells and the extracellular matrix (ECM) that represents the structural support for integrin-mediated cell adhesion.1 The second key event for cancer metastasis involves proteolysis of the basement membrane (BM) and ECM by metalloproteinases (MMPs) forming the migration path.2 The disturbance of BM and ECM leads to the creation of a microenvironment that enables angiogenesis, invasion and metastasis that represent the main cancer hallmarks.1 Therefore, the formation of a metastatic niche depends primarily on the binding ability of integrin receptors with BM and ECM ligands including laminins, fibronectins, vitronectins, tenscins, nephronectins, osteopontins, thrombospondins, fibrinogens and collagens.3,4 Upon encountering a ligand, integrins undergo conformational changes thereby improving the binding affinity to the actin cytoskeleton forming a complex termed focal adhesion (FA). In addition to their signalling function, FAs are responsible for the control of tumor cell adhesion, proliferation, migration, invasion and angiogenesis.5Given its central role in the tumorigenesis process, considerable efforts have been made to develop integrins inhibitors as anticancer drugs. As a consequence, integrins like αvβ3, αvβ5, α5β1, and αvβ6 have been selectively targeted and several selective inhibitors have been developed and tested on different cancers.6 In this framework, it has been found that the B6-3 diabody engineered with a C-terminal hexahistidine tag (His tag) and expressed in Pichia pastoris inhibited adhesion and migration of the αvβ6-transfected melanoma cell line and the pancreatic adenocarcinoma cell line in vitro and in vivo.7 The 264RAD, a human monoclonal antibody targeting αvβ6 and its functionally related αvβ8, dose-dependently inhibited Ki67 and phospho-ERK (pERK) as well as fibronectin expression in Detroit 562 tumor cell line inhibiting thereby the growth and matastasis of lung tumor.8 The efficacy of a chimeric antigen receptor (CAR) derived from T cells as a putative αvβ6 inhibitor has been proven in mice with established ovarian, breast, and pancreatic tumor xenografts.9 In light of these successes, some integrin inhibitors including etaracizumab, vitaxin, intetumumab, volociximab, cilengitide and ATN-161 have reached the phase II clinical trials.10 Although their good tolerance, integrin inhibitors failed unfortunately to ensure significant therapeutic effect in patients, and none of them have been registered as effective anti-cancer drug to date.11 Nevertheless, integrins are still interesting therapeutic targets.12,13
Natural integrin antagonists have recently been emerged as promising therapeutic opportunities. At this point, many studies have uncovered that disintegrins from snake's and arthropod's venoms,14 alkaloids,15 coumarins,16 flavonoids,17 stilbenes, secoiridoids, lignans,18 and terpenoids,19 among others, could effectively modulate a wide range of integrins, inhibiting thereby the key cancer hallmarks (adhesion, migration, proliferation, invasion, angiogenesis and metastasis). The latter chemical class has received particular attention because of its high antitumor and anti-metastasis activity.20 In this context, Zhou et al.21 demonstrated that the diterpenequinone salvicine activated extracellular signal-regulated kinase (ERK) and p38 mitogen activated protein kinase (MAPK) by triggering ROS generation, inhibiting thereby the β1 integrins and consequently the adhesion and metastatsis of human breast cancer MDA-MB-435 cells. The antiproliferative and anticancer activity of terpenoid-monoterpenes (i.e. limonene, cantharidin), terpenoid-sesquiterpenes (i.e. shizukaol, α-cubebene), terpenoid-diterpenes (i.e. tanshinone, oridonine, casearinols, casearinone, andrographolide) and terpenoid-triterpenes (i.e. euphol, celastrol, alisol, pachymic acid) have also been reported.19,20
Ecballium elaterium (L.) A. Rich, commonly known as squirting cucumber, is a member of the Cucurbitaceae family, and is traditionally used for the treatment of fever, sinusitis, chronic jaundice, liver cirrhosis, hypertension, rheumatic diseases, nocturia, lumbago and otalgia.22,23 The triterpenoid-rich fruit juice of E. elaterium has been shown to possess anti-ulcer activity.24
In our previous works, we have successfully showed that the seed oil (with linoleic and punicic acids as main components) from E. elaterium demonstrated potential inhibitory anti-adhesive, antiproliferative, anti-angiogenic, and antimetastasic effects by inhibiting αvβ3 and α5β1 integrins of gliomatumor cell line U87.25 Using the same cellular model, we have later demonstrated that the tetracyclic triterpene cucurbutacin B purified from E. elaterium, served as α5β1 integrin-antagonist, which in turn, could be responsible for its anti-adhesive and anti-proliferative effect.26 These inspiring results suggest that fatty acids and triterpenes from E. elaterium seed oil hold potential as natural therapeutic agent against glioblastoma. However, the anticancer effect of the phenolic fraction of E. elaterium seed oil (PEO) against fibrosarcoma has not been reported yet. Therefore, the present study was performed to evaluate the effectiveness of the (PEO) against highly tumorigenic HT100 human fibrosarcoma cells, and to elucidate if the effect relies on integrin modulatory mechanism.
2. Experimental
2.1. Chemicals, reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM) and RPMI 1640 medium were purchased from GIBCO (Cergy-Pontoise, France). Fetal calf serum (FCS) was from BioWhittaker (Fontenay-sous-Bois, France). Penicillin, streptomycin, human fibrinogen, human laminin and poly-L-lysine were from Sigma (St. Quentin Fallavier, France). Rat type I collagen was from Upstate (Lake Placid, NY, USA) and human fibronectin from Chemicon (Temecula, CA, USA). Human vitronectin was purified according to Yatogho et al.27 Rat monoclonal antibody (mAb) 69.6.5 against αv integrin was produced as previously described by Lehmann et al.28 Mouse mAbs LM609 (anti-αvβ3) and P1F6 (αvβ5) were purchased from Chemicon. Mouse mAbs Gi9 (anti-α2β1), SAM1 (anti-α5β1) and C3VLA3 (anti-α3) were from Immunotech (Marseille, France). Rabbit anti-rat was purchased from Sigma. MatrigelTM was from BD Biosciences (Pont de Claix, France). Hexane, acetonitrile and methanol of analytical and HPLC grade were purchased from Fluka Chemical Co (Buchs, Swizerland).2.2. Plant material, crude oil extraction and preparation of the phenolic extract
Mature fruits of E. elaterium were collected from spontaneous populations growing wild in the region of Sidi Thabet (Northern Tunisia, latitude 36°54′45.25′′ (N), longitude 10°06′02.10′′ (E), altitude 30 m) in March 2021. The plant material was authenticated by Pr. Boulila Abdennacer of the Laboratory of Natural Substances, INRAP, Technopark sidi Thabet, Tunisia, where a voucher specimen (BPn 0321) was deposited. After the removal of peels, seeds were oven-dried at 40 °C until constant weight and ground to a fine powder.The crude oil was extracted from the ground dried seeds (50 g) with hexane using a Soxhlet apparatus. After removal of hexane in a rotary evaporator at 40 °C, a crude oil was obtained with a yield of 16% (w/w) which was in turn extracted using 80% methanol to give the phenolic fraction of the oil (PEO).29 The PEO was recovered after solvent evaporation in vacuo (40 °C) followed by lyophilisation and the final crude extract (480 mg) was stored at −20 °C until used.
2.3. HPLC-DAD-ESI-MS analysis
The qualitative analysis of the PEO by LC-DAD-ESI-MS was performed using an Agilent 1100 series HPLC systems equipped with a vacuum degasser, diode array detector and a triple quadrupole mass spectrometer type MicromassAutospecUltimaPt (Kelso, UK) operating in negative mode. Separation was performed on a reverse-phase Uptisphere C18 (Interchim, Montfluçon, France) (2 mm × 100 mm, 5 μm particle size) using a mobile phase consisting of A (0.1% acetic acid in water) and B (acetonitrile) at a flow rate of 0.25 mL min−1. The gradient system was 0 min (5% B), 65 min (100% B), 67 min (100% B), and 70 min (5% B). The flow rate of the mobile phase was fixed at 0.25 mL min−1. The column temperature was 40 °C and the injection volume was 20 μL. UV spectra were recorded in the range 190–800 nm. The MS conditions were as follow: mass spectra were collected in negative ion mode from 135 and 700 m/z, capillary voltage, 3.2 kV; cone voltage, 40 V; probe temperature, 300 °C; and ion source temperature, 130 °C.2.4. Anticancer activities
The HT1080 and HT29-D4 were routinely cultured in DMEM containing 10% FCS. The K562 cells were cultured in RPMI 1640 medium containing 10% FCS. HMEC-1 cells were routinely maintained in MCDB-131 medium (Lonza, Levallois-Perret, France) containing 10% heat-inactivated fetal bovine serum, 2 mmol L−1 glutamine, 1% penicillin and streptomycin (all from Life Technologies, Paisley, UK), 1 mg mL−1 hydrocortisone (Pharmacia & Upjohn, St-Quentin Yvelines, France) and 10 ng mL−1 epithelial growth factor (R&D Systems, Minneapolis, MN). HMEC-1 cells were grown on 0.1% gelatin-coated flasks and were used between passages 3 and 12. Cell lines were maintained under humidified 5% CO2 and 95% air at 37 °C.
Cells were treated for 30 min with vehicle (0.1% DMSO) or with different concentrations of extract dissolved in 0.1% DMSO. The cells treated with DMSO served as a vehicle control group.
For adhesion assay on antibodies, 96-well plates were coated with 50 μL of rabbit anti-Rat IgG (50 μg mL−1), overnight at 4 °C. Wells were washed once with PBS and 50 μL of blocking anti-integrin antibodies (10 μg mL−1) were added for 5 h at 37 °C. Then, wells were blocked with 0.5% PBS/BSA and adhesion assay was continued as described above.
For cell motility, human fibrosarcoma cells were trypsinized and seeded on fibrinogen pre-coated 24-well plates at low confluence (104 cells per cm2), and then allowed to adhere at 37 °C for 2 h. Time-lapse video-microscopy was used to track cell motility for 3 h at 5 min intervals, using an automated Nikon inverted microscope equipped with a heated stage and 5% CO2 supply, and a Coolsnap HQ camera (Photometrics, Tucson, AZ) controlled with NIS-elements AR 2.30 software (Nikon) and a Metamorph® image analysis software (Roper Scientific, Evry, France) for manual single-cell tracking. Migration tracks were used to calculate total migration distance, distance to origin determined as the net translocation between the initial and the final position, velocity calculated as the total migration distance divided by 180 min, and directional persistence of cell migration calculated as the ratio of the distance to origin to the total distance migration.32
Tube formation assay. The in vitro anti-angiogenic activity of PEO was determined using the HMEC-1 cells capillary-like tube formation assay. Briefly, a 96-well plate pre-coated with 100 μL Matrigel per well was prepared and solidified for 1 h at 37 °C. HMEC-1 cells were plated on Matrigel-coated wells at a density of (5 × 103 cells per 100 mL) and incubated for 6 h at 37 °C with 5% CO2. The formation of capillary-like tubular networks was observed with a DM-IRBE microscope (Leica, Rueil-Malmaison, France) coupled with a digital camera (CCD camera coolsnap FX, Princeton Instruments, Trenton, NJ). The percentage of tubule area was quantified using Metamorph1 image analysis software (Roper Scientific, Evry, France) as previously described by Pasquier et al.33
Ex ovo assay. The cholioallantoic membrane (CAM) of chiken embryo was used as an ex ovo experimental model to evaluate the antiangiogenic effect of PEO. Briefly, fertilized chick embryos from 3 days-old eggs were disinfected with 70% ethanol, opened and incubated at 37 °C and 80% humidity for embryogenesis. On the 5th day of incubation, filter paper disks (diameter 6 mm) soaked in buffer (0.9% NaCl) or PEO (100 μg mL−1) were applied onto the developing CAM. To check its effect on growth factor-induced angiogenesis or vascular endothelial growth factor (VEGF), PEO (200 ng per embryo) was topically applied on the CAM of 8 days-old embryos and incubated for 48 h. The imaging of the spontaneous and induced vascularization were performed with a digital camera at 10× magnification.31
2.5. Statistical analysis
All data were expressed as mean ± standard deviation (SD) of three experiments repeated at least three times. One-way analysis of variance (ANOVA) followed by Tukey's HSD post-hoc test was applied to compare means. A P-value of <0.05 was considered statistically significant. All analyses were performed using RStudio V. 0.97 (Boston, MA. USA).3. Results
3.1. LDH-based cytotoxicity of PEO
The LDH-based cytotoxicity assay was used to evaluate the chemosensitivity of fibrosarcoma HT1080 cell line to PEO (Fig. 1). The LDH-release curve showed that exposure to PEO (10–40 μg mL−1) for 48 h did not exert any cytotoxic effect on the HT1080 cell line. Up to 50 μg mL−1, the PEO was relatively safe as the cell viability was maintained at 86%. In contrast, the cell viability was significantly decreased at concentrations >50 μg mL−1, reflecting probably, a loss of membrane integrity and cell metabolic disturbance.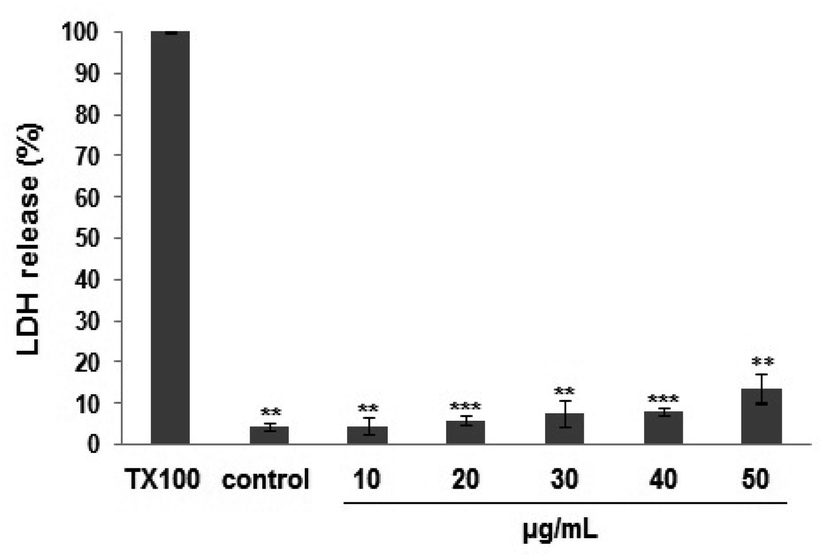 | ||
| Fig. 1 Dose-dependent cytotoxic effect of PEO in human HT1080 fibrosarcoma cells. The % LDH release is expressed as means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at *P < 0.05; **P < 0.01; ***P < 0.001. | ||
Based on the obtained results, all ensuing experiments were performed at 40 μg mL−1.
3.2. PEO inhibits HT1080 cell adhesion to fibronectin and fibrinogen
Data from Fig. 2a, showed that PEO significantly (p < 0.001) blocked the adhesion of HT1080 cells to fibrinogen and fibronectin, and to a lesser extent (p < 0.01) to type I collagen. In contrast, adhesion to on laminin-1 and vitronectin was not affected. In the presence of poly-L-lysine, a ligand-independent integrins, HT1080 cell adhesion was not affected. These results suggest that the inhibitory effect of PEO was mediated through the integrin family of adhesion receptors.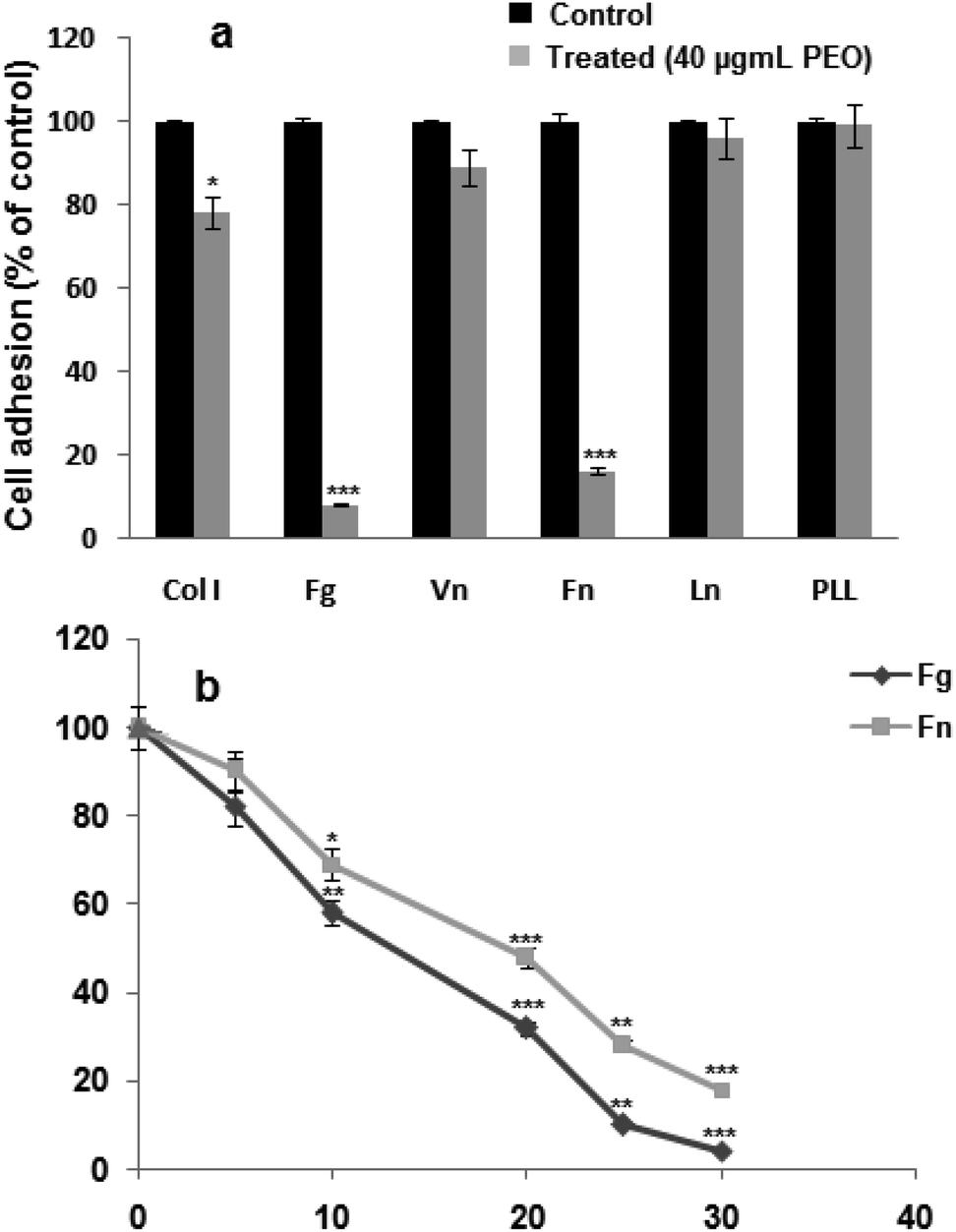 | ||
| Fig. 2 Inhibitory effect of PEO on HT1080 cell adhesion: (a) HT1080 cells were pre-incubated in the absence (black bars) or in the presence (grey bars) of 40 μg mL−1 of PEO for 30 min at room temperature. Cells were then added to 96-well microtiter plates coated with 10 μg mL−1 fibronectin (Fn) or collagen I (Coll I) or with 50 μg fibrinogen (Fg) or poly-L-lysine (PL) 1 h at 37 °C. After washing, adherent cells were stained with crystal violet, solubilized by SDS and absorbance was measured at 600 nm. (b) Dose-effect of oil on HT1080 cancer cell adhesion to fibrinogen and fibronectin. Data shown are means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at *P < 0.05; **P < 0.01; ***P < 0.001. | ||
Considering the modulatory effect of PEO on HT1080 adhesion on fibrinogen and fibronectin, the dose–response curve showed IC50 values of 12.04 and 20.28 μg mL−1 for fibrinogen and fibronectin, respectively (Fig. 2b).
3.3. PEO inhibits HT1080 cell migration by targeting αvβ3 and α5β1
To examine the effect of PEO on HT1080 migration ability, the modified Boyden chamber using haptotaxis assays towards fibrinogen, fibronectin, type I collagen and vitronectin was used. As shown in Fig. 3a, PEO dose-dependently inhibited the motility of HT1080 to fibrinogen (IC50 = 12.86 μg mL−1), fibronectin (IC50 = 18 μg mL−1) and type I collagen (IC50 = 38 μg mL−1). The migration of HT1080 cells to fibrinogen was completely impaired in the presence of PEO at 40 μg mL−1 (Fig. 3a and b).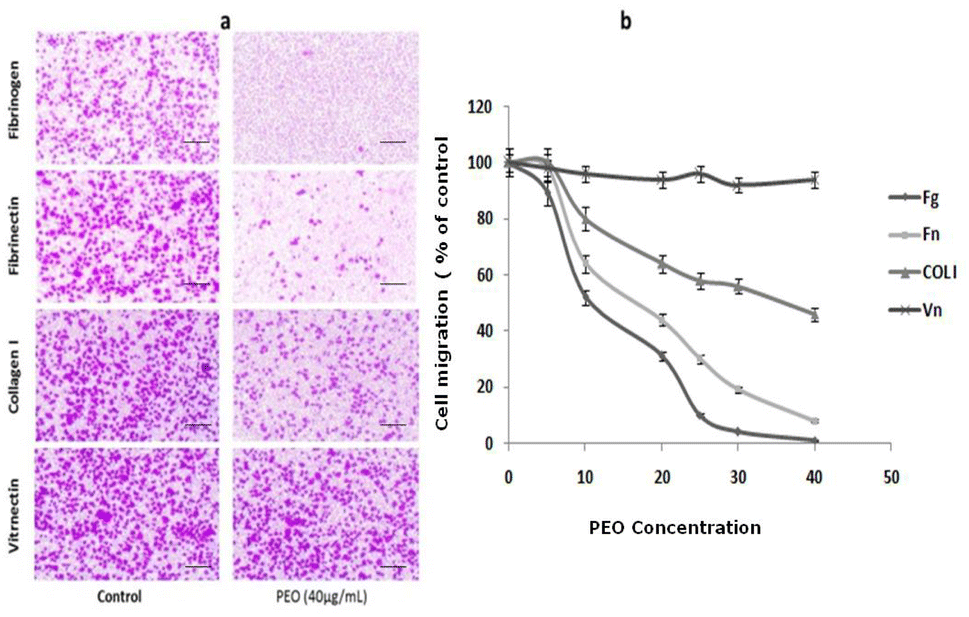 | ||
| Fig. 3 Inhibitory effect of PEO on HT1080 cell migration. (a) Cell motility was determined in a modified Boyden chamber. After treatment without or with 40 μg mL−1 PEO for 30 min at room temperature, HT1080 cells were seeded into the upper reservoir and allowed to migrate. Metamorph imaging software was used to capture images of cell that migrate through the filter (scale bar: 100 μm). (b) Dose-effect of PEO on cell migration to matrix. Data shown are means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at *P < 0.05; **P < 0.01; ***P < 0.001. | ||
The results of random motility on a two-dimensional fibronectin substrate (Fig. 4a) further confirmed the inhibitory effect of PEO on HT1080 cell motility on fibronectin. In fact, time-lapse recording for 3 h of single cells showed that untreated control cells and those treated with 5 μg mL−1 PEO exhibited a high migration velocity (0.99 ± 0.11 and 0.98 ± 0.18 μm min−1 for control and treated cells with 5 μg mL−1 PEO, respectively) (ESI, videos 1 and 2†). In contrast, treatment with 40 μg mL−1 PEO resulted in 75.75% reduction in the migration velocity as previewed in ESI, video 3.† At this point, it can be inferred that PEO inhibits the extension of protrusion reducing thereby the migration ability of HT1080 cells. Supporting evidences are given by data from Fig. 4b showing that PEO treatment greatly affected the distance from the point of origin (11 ± 0.8 μm) compared to the untreated control (49 ± 1 μm).
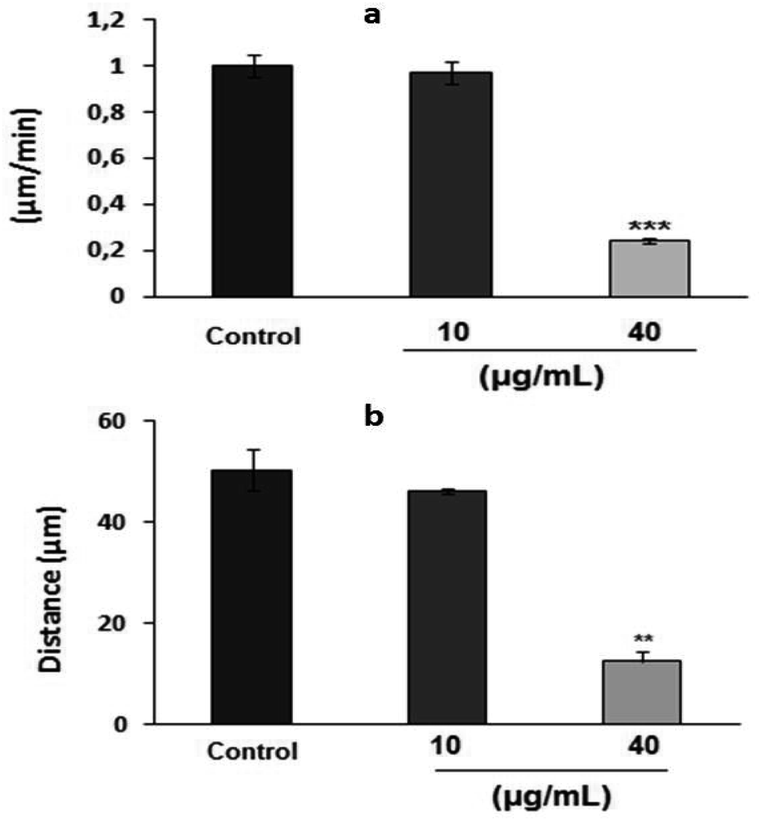 | ||
| Fig. 4 Effects of PEO on HT1080 cell migration as demonstrated on: (a) random motility on two-dimensional fibronectin substrate, and (b) the distance from the point of origin. Data shown are means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at **P < 0.01; ***P < 0.001. | ||
To decipher the mechanistic aspect of the inhibitory effect of PEO on HT1080 migration, several integrin-mediated cell adhesion were targeted. These include α1β1 (PC12/type IV collagen), α2β1 (HT1080/type I collagen), α5β1 (K562/fibronectin), α6β4 (HT29-D4/laminin-1), αvβ3 (HT1080/fibrinogen), αvβ5 (HT29-D4/vitronectin) and αvβ6 (HT29-D4/fibronectin). As illustrated in Fig. 5a, PEO did not affect cell adhesion through αvβ5, αvβ6, α1β1 and α6β4 integrins, but significantly reduced the adhesive function of α5β1, and αvβ3 integrins. Confirmatory assay using integrin blocking antibodies showed that PEO at 40 μg mL−1 competitively inhibited the attachment of HT1080 cell to anti-αvβ3 (>98%), anti-α5β1 (>86%), and to a lesser extend anti-α2 (>50%) immobilized antibodies (Fig. 5b). The attachment of HT1080 to the remaining anti-α3, anti-α6 and anti-αvβ5 antibodies was virtually not affected. These observations confirm that αvβ3 and α5β1 integrins were selectively targeted by PEO with IC50 values of 11.41 and 19.27 μg mL−1 for αvβ3 and α5β1, respectively (Fig. 5c).
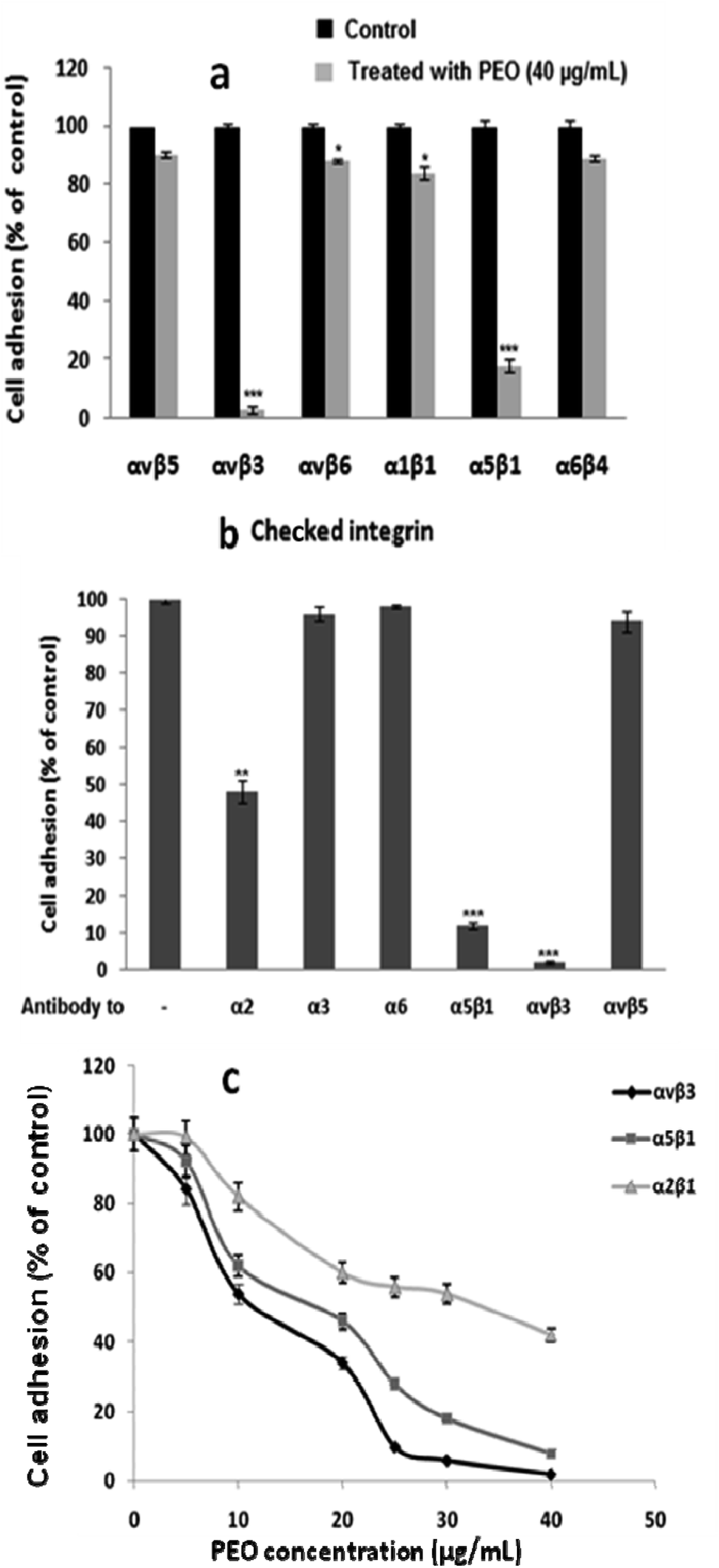 | ||
| Fig. 5 Selective inhibitory effect of PEO on HT1080 cell adhesion: HT1080 cells were preincubated without (black bars) or with 40 μg mL−1 PEO (grey bars). (a) Adhesion assays were performed with various cell/ECM protein pairs involving unique integrins: α1β1 (PC12/type IV collagen), α2β1 (HT1080/type I collagen), α5β1 (K562/fibronectin), αvβ3 (HT1080/fibrinogen), αvβ5 (HT29/vitronectin), α6β4 (HT29/laminin) and αvβ6 (HT29/fibronectin). (b) Adhesion was assessed using antibodies. (c) Dose–response curve of PEO induced inhibition of αvβ3, α5β1 and α2β1. Data shown are means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at *P < 0.05; **P < 0.01; ***P < 0.001. | ||
3.4. PEO exhibits in vitro and in vivo anti-angiogenic activity
To test whether PEO inhibited angiogenesis process, the in vitro tubulogenesis model of human microvascular endothelial cell (HMEC-1) cells capillary-like tube formation was used. As shown in Fig. 6a, PEO dose-dependently blocked the in vitro tubulogenesis with an IC50 value of 20 μg mL−1. By using function-blocking antibodies against various integrins, it has been demonstrated that PEO modulatory effect on the formation of capillary tube was mediated through the inhibition of αvβ3, α5β1 and αv integrins (Fig. 6b). These results strongly point to the anti-angiogenic effect of PEO.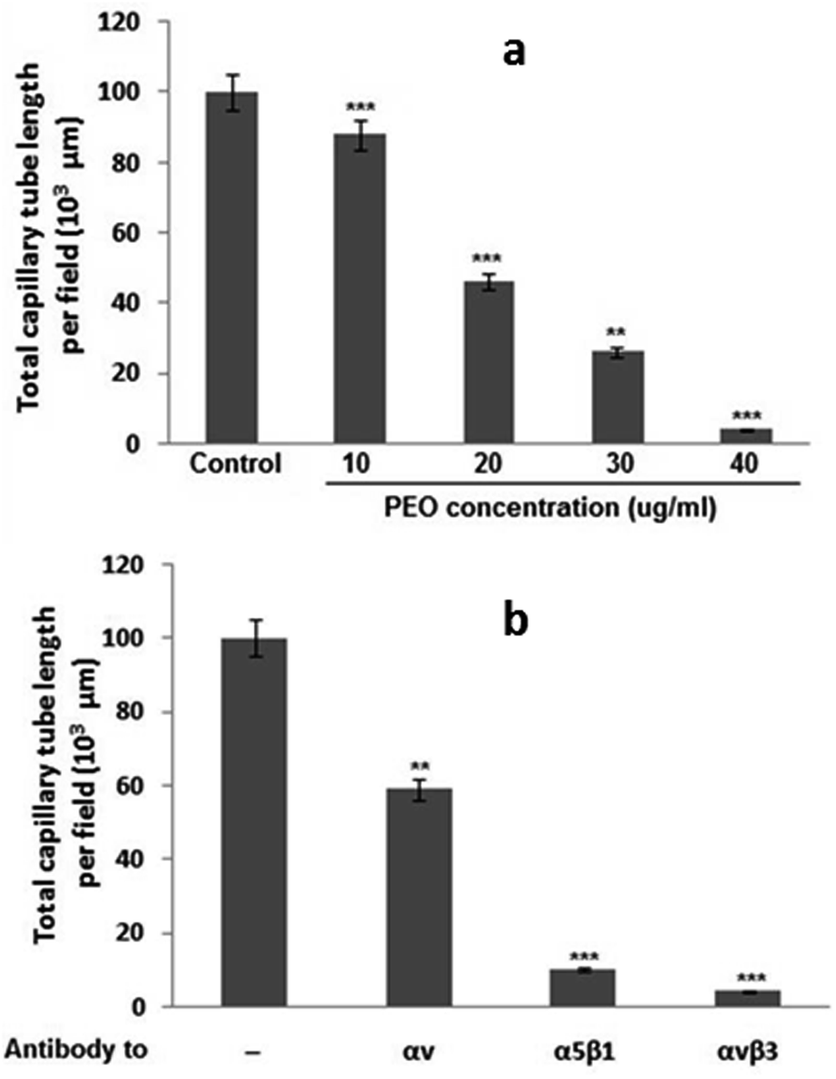 | ||
| Fig. 6 Dose–response curve (a) of the PEO inhibitory effect on (b) αv-, α5β1-, and αvβ3-induced tubulogenesis on human microvascular endothelial cell (HMEC-1). Data shown are means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at **P < 0.01; ***P < 0.001. | ||
To confirm whether this property was maintained in vivo, the CAM assay was used. Representative images (Fig. 7a) showed that PEO reduced the vascular plexus without affecting pre-existing vessels with the effect being more pronounced at concentration of 40 μg mL−1 PEO (Fig. 7b). Similar results were also observed in VEGF-induced angiogenesis assay. Importantly, it has been observed that PEO inhibited the formation of new blood vessels. These results suggest that PEO was a potent inhibitor of tubulogenesis and neoangiogenesis.
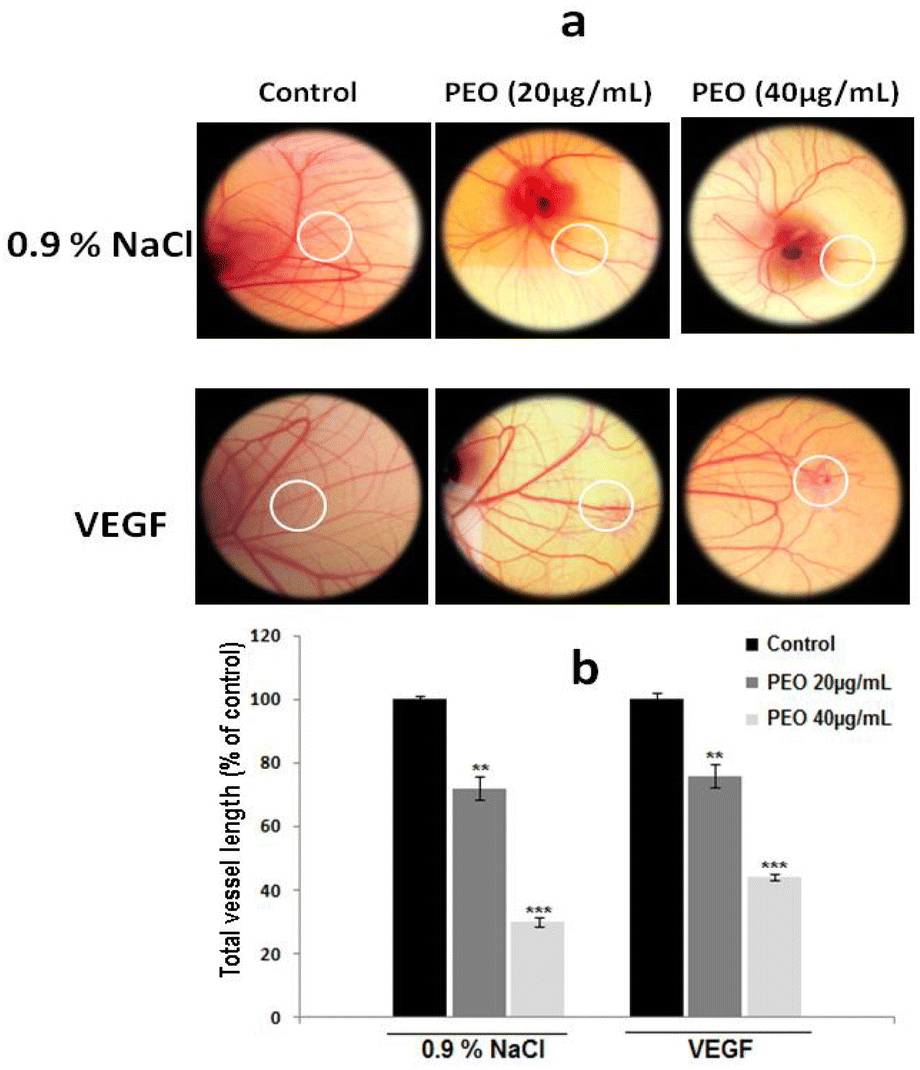 | ||
| Fig. 7 PEO inhibits in vivo angiogenesis. (a) The CAM models were prepared using 8 days-old chick embryos. White circles represent location of applied disks. After incubation for 48 h with or without PEO extract, CAMs were photographed with a digital camera. Each group contained 3 CAMs and the experiment was repeated three times. (b) The quantitative measurement of total vessel length was performed on 50% of the total CAM surface treated in the absence or in the presence of PEO extract. For total vessel length, results are expressed as means ± SD of three experiments performed in triplicate and difference between treated and untreated (control) cells was considered statistically significant at **P < 0.01; ***P < 0.001. | ||
To identify the putative inhibitors of the adhesion, migration and angiogenesis, the PEO was analysed by HPLC-ESI-MS/MS.
3.5. PEO analysis
The chromatographic analysis of the PEO by HPLC-DAD-ESI-MS/MS using the analytical procedure described in Section 2.3. A total of 22 compounds were tentatively identified based on their UV-vis absorption maxima, mass spectra recorded in negative mode and literature data (Fig. 8 and Table 1).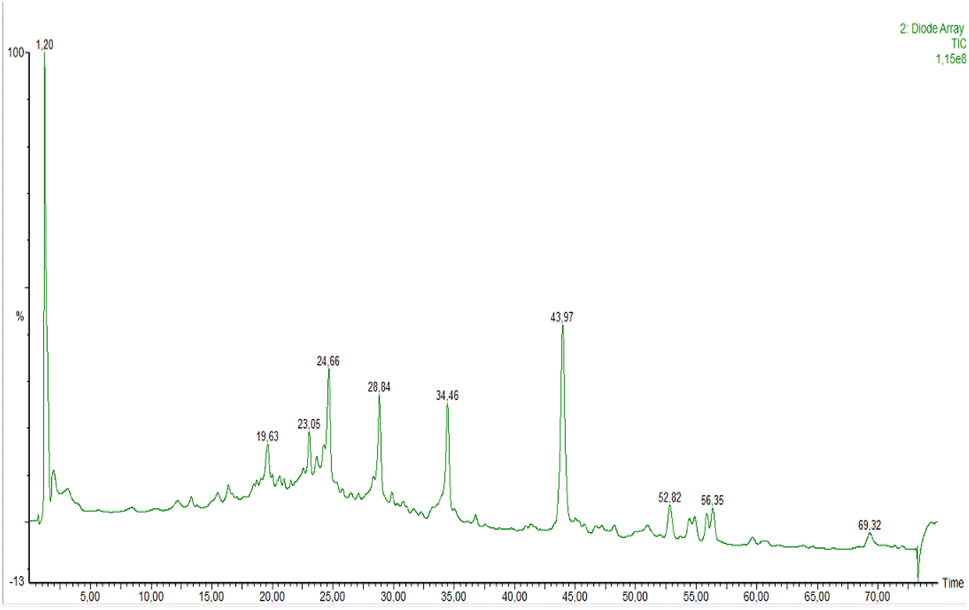 | ||
| Fig. 8 Representative LCMS total ion chromatogram (TIC) profile of bioactive ingredients of PEO. Peak assignment are given in Table 1. | ||
| Peak no. | RT (min) | UV | [M–H]− | Main fragments | Tentative identification |
|---|---|---|---|---|---|
| 1 | 13.35 | 279 | 289 | 245 | Catechin |
| 2 | 16.39 | 279 | 577 | 289 | Procyanidin dimer |
| 3 | 16.65 | 269 | 287 | 269 | 3,7,3′,4′′-Tetrahydroxyflavanone |
| 4 | 17.11 | 270 | 579 | 271, 151 | Trihydroxyflavanone-o-deoxyhexosyl-O-hexoside |
| 5 | 18.68 | 279 | 577 | 289 | Procyanidin dimer |
| 6 | 20.64 | 279 | 597 | 289 | Catechin-3-O-rutinoside |
| 7 | 21.52 | 321 | 367 | 173 | 4-Feruloylquinic acid |
| 8 | 23.05 | 269 | 271 | 135 | 7,3′,4′-Trihydroxyflavanone |
| 9 | 23.71 | 320 | 335 | 173 | Shikimic acid hexoside isomer I |
| 10 | 24.21 | 321 | 335 | 173 | Shikimic acid hexoside isomer II |
| 11 | 24.67 | 321 | 371 | — | Caffeoylglucaricacid |
| 12 | 28.85 | 229 | 327 | — | Trihydroxy-octadecadienoicacid |
| 13 | 29.90 | 229 | 329 | — | Trihydroxy-octadecenoicacid |
| 14 | 34.44 | 237 | 573 | — | Cucurbitacin A |
| 15 | 43.96 | 229 | 295 | — | 9-/or 13-Hydroxy-9Z,11E-octadecadienoic acid |
| 16 | 47.53 | 237 | 515 | — | Cucurbitacin D |
| 17 | 48.16 | 239 | 555 | 357 | Cucurbitacin E |
| 18 | 52.82 | 229 | 277 | — | Linolenic acid |
| 19 | 56.28 | 239 | 559 | — | Dihydro-cucurbitacin B |
| 20 | 59.55 | 237 | 807 | — | Colocynthoside B |
| 21 | 60.84 | 230 | 281 | — | Oleic acid |
| 22 | 63.94 | 239 | 557 | — | Cucurbitacin B |
The compounds identified included 4 flavan-3-ols (catechin, catechin-3-O-rutinoside, proanthocyanidin dimer I and proanthocyanidin II), 3 flavanones (calodendroside, trihydroxyflavanone-O-deoxyhexosyl-O-hexoside and 7,3′,4′-trihydroxyflavanone),34,35 4 phenolic acids (4-ferulyolquinic acid, shikimic acid hexoside I, shikimic acid hexoside II, and caffeoylglucaric acid),36–38 5 fatty acids and fatty acid derviatives (trihydroxy-octadecadienoic acid, trihydroxy-octadecenoic acid, 9-/13-hydroxy-9Z,11E-octadecadienoic acid and linolenic acid)39 and 6 cucurbitacin-type triterpenes (cucurbitacin A, B, D, E, dihydro-cucurbitacin B and colocynthoside B).39,40
4. Discussion
The malignant HT1080 human fibrosarcoma known for their high tumorigenic, migratory and invasion properties presents several challenges in relation to cancer cell proliferation, anti-angiogenesis and anti-metastatic therapies.41 In the present study, HT1080 cells were used as target cancer cell line to evaluate the potential anticancer activity of PEO in term of anti-adhesive, anti-migratory and anti-angiogenic effects.In the presence of PEO, HT1080 revealed significantly decreased adhesiveness to fibronectin and fibrinogen (Fig. 2). These observations were concomitantly associated with a remarkable reduction of HT1080 cell motility to both ECM ligands as observed in the modified Boyden chamber assay (Fig. 3). At this point, it can be inferred that the modulatory effect of PEO on HT1080 cell adhesion and motility was mediated through the integrin family of adhesion receptors. Additional experiments using two-dimensional fibronectin substrate (Fig. 4a) support these findings and showed that PEO at 40 μg mL−1 greatly reduced the migration velocity (ESI, videos 1 and 2†) and effected the distance from the point of origin (Fig. 4b). So, it is plausible to conclude that PEO inhibits the extension of protrusion reducing thereby the migration ability of HT1080 cells as previously described in fibrosarcoma (HS-913T; HT-1080) and leiomyosarcoma (SKUT-1)-derived cell lines.42 In the corresponding study, it has been observed that the migration and motility of the aforementioned cell lines were tightly associated with the expression of stathmin (a microtubule-destabilizing protein) and the cyclin-dependent kinase inhibitor p27kip1. Thus, the overexpression of stathmin and the low cytoplasmic expression of p27kip1 in these cell lines increased their fibronectin-directed motility, whereas, stathmin downregulation coupled with high cytoplasmic expression of p27kip1 strongly inhibited their migration.42 At this point, it is possible that PEO-induced inhibition of HT1080 migration and motility could imply molecular mechanisms that interfere with microtubules mass and dynamic, and destabilize proteins related to cytoskeleton.
The ability of PEO to modulate integrin-related signaling pathways (i.e. Src family kinase; focal adhesion kinase (FAK)), extracellular signal-regulated kinase 1/1 (ERK) and mitogen-activated protein kinase (PAPK), Wnt/β-catenin, nuclear factor-kappa B (NF-κB), yes-associated protein (YAP) and the expression of matrix metalloproteinase (MMP),43 is suggested too. However, additional in depth studies are warranted to explore these hypothesis.
The effect of some drugs, bioactive components and/or extracts in inhibiting integrin-mediated adhesion and migration of several tumor cell lines has been the focus of numerous studies and reviews.6,10,15,20,26,44
To determine the functional integrin-mediated inhibitory effect on H1080 cells adhesion and migration, PEO was screened across a range of integrin blocking antibodies. Results indicated that PEO specifically competed with anti-αvβ3, anti-α5β1 and to a lesser extent anti-α2 (Fig. 5), resulting in inhibition of HT1080 cell migration. The data support the conclusion that PEO is a potent inhibitor of αvβ3 and α5β1, with some activity against α2β1 integrin.
Given their crucial role in tumor blood vasculature and progression, αvβ3, α5β1 and α2β1 integrins were selectively targeted by PEO. In the in vitro HMEC-1 cellular model, PEO show the potential to inhibit the αvβ3-and α5β1-induced capillary-like tube formation in a dose-dependent manner (Fig. 6). It also inhibits VEGF-induced angiogenesis in the in vivo CAM assay (Fig. 7). From these data, it can be concluded that PEO could acts as a putative antagonist targeting both αvβ3 and α5β1 integrins, which in turn could have greater efficacy in inhibiting the formation and progression of tumor vasculature. The PEO-mediated inhibition of tubulogenesis and neoangiogenesis could be related to its high affinity binding to VEGF receptors resulting in inhibition of endothelial cell proliferation, migration and stromal degradation, as well as reduction of vascular permeability that hamper the formation of extravascular fibrin substrate for the growth of cancer cells.45 From practical standpoint, the observed results could provide baseline information for the possible consideration of PEO as potential source for the development of natural VEGF-targeted therapy.
Targeting VEGF and their corresponding receptors VEGFR as well as their associated signaling pathways to inhibit angiogenesis has been in the core of several pharmacological and clinical researches.42,46
Among the identified components in PEO, some of them have received particular attention because of their anticancer activity. In this context, the flavan-3-ols proanthocyanidins were found to inhibit tumor necrosis factor (TNF-α)-induced VEGF expression and its subsequent microvasculature.46 The study by Guruvayoorappan and Kuttan47 showed that catechin inhibited migration and tube formation in HUVECs (human umbilical vein endothelial cells), and reduced the number of tumor directed microvessels in C75BL/6 mice injected with B16F-melanoma cells. They attributed the anti-angiogenic activity of catechin to its ability to inhibit the production of pro-angiogenic factors like pro-nflammatory cytokines (IL-1β, IL-6, TNF-α, and GM-CSF), nitric oxide (NO), IL-2, tissue inhibitors of metalloproteinase-1 and VEGF in a dose-dependent manner.47 In other study using human neuroblastoma xenograph model, it was observed that the conjunction catechin–dextran prominently inhibited the formation of new blood vessels inhibiting thereby the angiogenesis and tumor progression in endothelial cells.48 The molecular mechanism behind such inhibition was found to be associated with the disruption of copper homeostasis by depleting its CTR-1 transporter and ATOX-1 trafficking protein in endothelial cells.48
Cucurbitacins-type triterpenes are the well-known anticancer agents with proven activity in multiple cancers types like breast, pancreatic, colon, ovarian, liver, lung, osteosarcoma, squamous skin carcinoma, and glioblastoma cancer cell lines.26,49 In addition to their integrin antagonistic, anti-neoangiogenic, pro-apoptotic, arresting cell cycle, and actin destroying effects, the anti-cancerous activity of cucurbitacins (namely cucurbitacin B, E, I, D, and 23,24-dihydrocucurbitacin B) was mediated through the inhibition of numerous signaling pathways including the JAK/STAT (Janus kinase/Signal Transducers and Activator of Transcription), MAPK, Ras/Raf/MEK/ERK mitogen activated protein kinase cascade.49–51
Regarding hydroxyflavanones, experimental studies showed that 2-hydroxyflavanone caused a dose-dependent suppression of VEGF, resulting in inhibition of neovascularization and ultimately the suppression of MDA-MB-231 xenograph.52
5. Conclusion
In light of these results, it can be concluded that the PEO could be considered as a consolidated source of anticancer agents. It efficiently inhibited adhesion, migration and proliferation of human fibrosarcoma HT1080 cell line in a dose-dependent manner through targeting αvβ3 and α5β1 integrins. PEO also inhibited the formation of HMEC-1 cell capillary-like tube in vitro, while in the CAM model in vivo, it strongly impaired VEGF-induced neoangiogenesis. Looking ahead, the phenolic fraction and/or some of its bioactive components of squirting cucumber may be used in the development of natural anti-migratory and anti-angiogenic drugs. However, an in depth exploration of the mechanistic aspects of its anticancer activity with a concomitant investigation of signalling pathway and the key targeted molecules will be performed.Author contributions
Touihri-Barakati: data curation, investigation, writing original draft; Kallech-Ziri and Morjen: data curation, methodology and visualization; Marrakchi and Luis: conceptualization, supervision, and data curation; Hosni: data curation, visualization, writing-original draft, writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Ministry of High Education and Scientific Research of Tunisia (MERST) (Grant number LR15INRAP02) and the Institut National de la Santé et de la RechercheMédicale (INSERM) and by grants from INCa (Institut National du Cancer) and ARCUS (Action en Région de CoopérationUniversitaire et Scientifique).References
- Y. Hashim, I. R. Rowland, R. Ian, H. Mcglynn, M. Servili, R. Selvaggini, A. Taticchi and C. Gill, Int. J. Cancer, 2008, 122, 495–500 CrossRef CAS PubMed.
- M. Janiszewska, M. C. Primi and T. Izard, J. Biol. Chem., 2020, 295, 2495–2505 CrossRef CAS PubMed.
- E. Askari, S. M. Naghib, A. Zahedi, A. Seyfoori, Y. Zare and K. Y. Rhee, J. Mater. Res. Technol., 2021, 12, 412–422 CrossRef CAS.
- M. Paolillo and S. Schinelli, Int. J. Mol. Sci., 2019, 20, 4947 CrossRef CAS PubMed.
- G. Lorusso, C. Rüegg and F. Kuonen, Front. Oncol., 2020, 1231 CrossRef PubMed.
- T. J. Slack, W. Li, D. Shi, J. B. McArthur, G. Zhao, Y. Li and X. Chen, Bioorg. Med. Chem., 2018, 26, 5751–5757 CrossRef CAS PubMed.
- H. Kogelberg, E. Miranda, J. Burnet, D. Ellison, B. Tolner, J. Foster and K. Chester, PLoS One, 2013, 8, e73260 CrossRef CAS PubMed.
- C. Eberlein, J. Kendrew, K. McDaid, A. Alfred, J. S. Kang, V. N. Jacobs and S. T. Barry, Oncogene, 2013, 32, 4406–4416 CrossRef CAS.
- M. Akce, M. Y. Zaidi, E. K. Waller, B. F. El-Rayes and G. B. Lesinski, Front. Immunol., 2018, 2166 CrossRef PubMed.
- M. Li, Y. Wang, M. Li, X. Wu, S. Setrerrahmane and H. Xu, Acta Pharm. Sin. B, 2021, 11, 2726–2737 CrossRef CAS.
- B. Alday-Parejo, R. Stupp and C. Rüegg, Cancers, 2019, 11, 978 CrossRef CAS PubMed.
- N. Aksorn and P. Chanvorachote, Anticancer Res., 2019, 39, 541–548 CrossRef CAS PubMed.
- C. Y. Su, J. Q. Li, L. L. Zhang, H. Wang, F. H. Wang, Y. W. Tao and J. Y. Zhang, Front. Pharmacol., 2020, 1435 Search PubMed.
- A. A. Vasconcelos, B. B. Succar, L. B. di Piero, E. Kurtenbach, R. B. Zingali and F. C. Almeida, Biomol. NMR Assignments, 2021, 1–4 Search PubMed.
- X. Wang, C. C. Decker, L. Zechner, S. Krstin and M. Wink, BMC Pharmacol. Toxicol., 2019, 20, 1–12 CrossRef PubMed.
- R. S. A. de Araújo, J. D. O. D. S. Carmo, S. L. de Omena Silva, C. R. A. Costa da Silva, T. P. M. Souza, N. B. D. Mélo and F. J. B. Mendonça-Junior, Pharmaceuticals, 2022, 15, 104 CrossRef PubMed.
- Y. Kang, W. He, C. Ren, J. Qiao, Q. Guo, J. Hu and L. Wang, Signal Transduction Targeted Ther., 2020, 1, 1–20 Search PubMed.
- B. Péter, I. Boldizsár, G. M. Kovács, A. Erdei, Z. Bajtay, A. Vörös and R. Horvath, Biomedicines, 2021, 9, 1781 CrossRef PubMed.
- M. Huang, J. J. Lu, M. Q. Huang, J. L. Bao, X. P. Chen and Y. T. Wang, Expert Opin. Invest. Drugs, 2012, 21, 1801–1818 CrossRef CAS PubMed.
- S. Takamatsu, J. Nat. Med., 2018, 72, 817–835 CrossRef CAS PubMed.
- J. Zhou, Y. Chen, J. Y. Lang, J. J. Lu and J. Ding, Mol. Cancer Res., 2008, 6, 194–204 CrossRef CAS PubMed.
- S. Felhi, A. Daoud, H. Hajlaoui, K. Mnafgui, N. Gharsallah and A. Kadri, Food Sci. Technol., 2017, 37, 483–492 CrossRef.
- I. Ibiloglu, U. Alabalik, A. N. Keles, G. Aydogdu, E. Basuguy and H. Buyukbayram, Biotechnol. Biotechnol. Equip., 2021, 35, 696–703 CrossRef CAS.
- S. Felhi, H. Hajlaoui, M. Ncir, S. Bakari, N. Ktari, M. Saoudi, N. Gharsallah and A. kadri, Food Sci. Technol., 2016, 36, 646–655 CrossRef.
- I. Touihri-Barakati, O. Kallech-Ziri, A. Boulila, K. Khwaldia, N. Marrakchi, B. Hanchi and J. Luis, Biomed. Pharmacother., 2016, 84, 1223–1232 CrossRef CAS PubMed.
- T. Touihri-Barakati, O. Kallech-Ziri, W. Ayadi, H. Kovacic, B. Hanchi, K. Hosni and J. Luis, Eur. J. Pharmacol., 2017, 797, 153–161 CrossRef PubMed.
- T. Yatogho, M. Izumi, H. Kashiwagi and M. Hayashi, Cell Struct. Funct., 1988, 13, 281–292 CrossRef PubMed.
- M. Lehmann, C. Rabenandrasana, R. Tamura, J. C. Lissitzky, V. Quaranta, J. Pichon and J. Marvaldi, Cancer Res., 1994, 54, 2102–2107 CAS.
- R. W. Owen, A. Giacosa, W. E. Hull, R. Haubner, B. Spiegelhalder and H. Bartsch, Eur. J. Cancer, 2000, 36, 1235–1247 CrossRef CAS.
- E. Delamarre, S. Taboubi, S. Mathieu, C. Bérenguer, V. Rigot, J. C. Lissitzky and J. Luis, Am. J. Pathol., 2009, 175, 844–855 CrossRef CAS PubMed.
- K. Z. Olfa, L. José, D. Salma, B. Amine, S. A. Najet, A. Nicolas and M. Naziha, Lab. Invest., 2005, 85(12), 1507–1516 CrossRef CAS PubMed.
- M. Morjen, O. Kallech-Ziri, A. Bazaa, H. Othman, K. Mabrouk, R. Zouari-Kessentini and N. Marrakchi, Matrix Biol., 2013, 32, 52–62 CrossRef CAS PubMed.
- E. Pasquier, M. Carré, B. Pourroy, L. Camoin, O. Rebaï, C. Briand and D. Braguer, Mol. Cancer Ther., 2004, 3, 1301–1310 CrossRef CAS PubMed.
- R. S. Darwish, O. A. Abdulmunem, A. Khairy, D. A. Ghareeb, A. M. Yassin, S. A. Abdulmalek and E. Shawky, RSC Adv., 2021, 11, 37049–37062 RSC.
- Y. Ye, Y. Liu, G. Zou and H. A. Aisa, Chem. Nat. Compd., 2012, 48, 681–682 CrossRef CAS.
- M. Clifford, K. L. Johnston, S. Knight and N. Kuhnert, J. Agric. Food Chem., 2003, 51, 2900–2911 CrossRef CAS PubMed.
- A. Ruiz, C. Mardones, C. Vergara, D. von Baer, S. Gómez-Alonso, M. V. Gómez and I. Hermosin-Gutierrez, J. Agric. Food Chem., 2014, 62, 6918–6925 CrossRef CAS PubMed.
- F. M. Mashitoa, T. Shoko, J. L. Shai, R. M. Slabbert and D. Sivakumar, Front. Nutr., 2021, 8, 86 Search PubMed.
- B. S. Levison, R. Zhang, Z. Wang, X. Fu, J. A. DiDonato and S. L. Hazen, Free Radical Biol. Med., 2013, 59, 2–13 CrossRef CAS PubMed.
- F. UlHaq, A. Ali, M. N. Khan, S. M. Z. Shah, R. C. Kandel, N. Aziz and S. G. Musharraf, Sci. Rep., 2019, 9, 1–11 CrossRef CAS PubMed.
- J. Zhang, Y. Song, Y. Liang, H. Zou, P. Zuo, M. Yan and Z. Wei, Food Chem. Toxicol., 2019, 132, 110654 CrossRef CAS PubMed.
- G. Baldassarre, B. Belletti, M. S. Nicoloso, M. Schiappacassi, A. Vecchione, P. Spessotto and A. Colombatti, Cancer Cell, 2005, 7, 51–63 CrossRef CAS PubMed.
- J. Hou, Y. L. Du Yan, P. Huang and H. Cui, OncoTargets Ther., 2020, 13, 13329 CrossRef CAS PubMed.
- M. C. Juan-River and M. Martínez-Ferrer, Cancers, 2018, 10, 44 CrossRef PubMed.
- A. C. Tufan and N. L. Satiroglu-Tufan, Curr. Cancer Drug Targets, 2005, 5, 249–266 CrossRef CAS PubMed.
- S. M. Sager, D. Yance and R. H. Wong, Curr. Oncol. Rep., 2006, 13, 14–26 CrossRef.
- C. Guruvayoorappan and G. Kuttan, Innate Immun., 2008, 14, 160–174 CrossRef CAS PubMed.
- E. M. Yee, M. B. Brandl, E. Pasquier, G. Cirillo, K. Kimpton, M. Kavallari and O. Vittorio, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- S. Jing, H. Zou, Z. Wu, L. Ren, T. Zhang, J. Zhang and Z. Wei, J. Funct. Foods, 2020, 72, 104042 CrossRef CAS.
- M. Zhou, C. Guo, X. Li, Y. Huang, M. Li, T. Zhang and N. Yang, J. Autoimmun., 2020, 109, 102424 CrossRef CAS PubMed.
- Y. Liu, H. Yang, Q. Guo, T. Liu, Y. Jiang, M. Zhao and P. Tu, Molecules, 2020, 25, 560 CrossRef CAS PubMed.
- J. Singhal, L. Nagaprashantha, S. Chikara, S. Awasth, D. Horne and S. S. Singhal, Oncotarget, 2017, 8, 75025 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra02593k |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |