
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Production of a recyclable nanobiocatalyst to synthesize quinazolinone derivatives†
Meenakshi Budhirajaa,
Bhupendra Chudasamabc,
Amjad Ali *ab and
Vikas Tyagi*ab
*ab and
Vikas Tyagi*ab
aSchool of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology (TIET), Patiala, Punjab, India. E-mail: vikas.tyagi@thapar.edu; amjadali@thapar.edu
bCenter of Excellence for Emerging Materials, Thapar Institute of Engineering and Technology, Patiala-147004, India
cSchool of Physics and Materials Science, Thapar Institute of Engineering and Technology, Patiala-147004, India
First published on 16th November 2022
Abstract
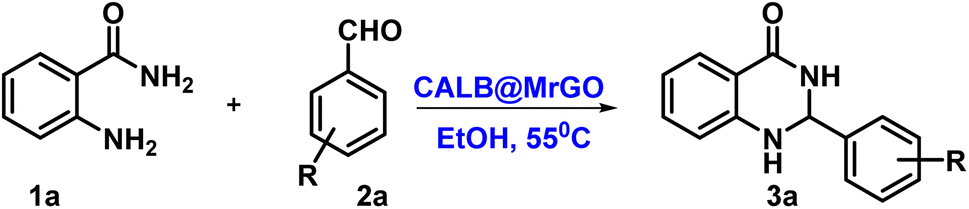
Nanobiocatalysts (NBCs) are an emerging innovation that paves the way toward sustainable and eco-friendly endeavors. In the quest for a robust and reusable nanobiocatalyst, herein, we report a nanobiocatalyst, namely CALB@MrGO, developed via immobilizing Candida antarctica lipase B onto the surface of Fe3O4-decorated reduced graphene oxide (MrGO). Next, the enormous potential of the NBC (CALB@MrGO) was checked by employing it to synthesize clinically important quinazolinone derivatives in good to excellent yield (70–95%) using differently substituted aryl aldehydes with 2-aminobenzamide. Further, the synthetic utility and generality of this protocol was proved by setting up a gram-scale reaction, which afforded the product in 87% yield. The green chemistry metrics calculated for the gram-scale reaction those prove the greenness of this protocol.
1 Introduction
Nanobiocatalysts (NBCs) are emerging as promising biomaterials in the development of sustainable strategies. They are a connecting bridge between nanotechnology and biotechnology, as they synergistically combine two different areas.1 Continuous efforts have been made to design new and environmentally benign methodologies, leading to the development of highly efficient and reusable nanobiocatalysts that work well in intense operational conditions.2 In this regard, enzymes have been used extensively with nanomaterials to develop nanobiocatalysts as they promote procedures under green conditions, have low chemical consumption and produce no or less toxic by-products.3 Moreover, the excellent activity, specificity, and selectivity of enzymes have made them promising biocatalysts with numerous fascinating applications, including the synthesis of moieties with pharmacological applications.4 In this context, hydrolase enzymes such as lipase and amylase have been widely used by chemists to synthesize clinically important molecules using several organic reactions, such as Michael addition, the Aldol reaction, the Henry reaction, and a range of multicomponent reactions.5 Nevertheless, numerous challenges are associated with enzymatic reactions, such as high operational costs, low thermal stability of the pure enzyme in the reaction conditions, low solubility, low tolerance in the presence of an organic solvent, and low/no reusability, which hinder large-scale applications.6 To overcome these shortcomings, the immobilization of the enzyme has gathered interest in recent years. It involves binding the enzyme to a solid support in order to make it a heterogenous catalyst, which aids in its easy separation from the reaction mixture. The greatest advantage of immobilization is to make the enzyme reusable for successive catalytic cycles, making it cost effective. Moreover, immobilization of the enzyme to a matrix enhances its structural stability, activity, specificity, and selectivity, which have been very well reported in the literature by several research groups.4a,7 Still, it has been observed in a few cases that enzyme immobilization may reduce the catalytic efficiency of an enzyme;7 however, this has not been observed in the case of lipase enzyme by us7g,h or others in previous studies.5a,b The various strategies for immobilizing enzymes onto nanostructured materials (such as hybrid nanoflowers, nanofibers (NFs), nanotubes, magnetic nanoparticles (NPs) and nanocomposites) made from polymers, silicas, carbons and metals include physical adsorption, entrapment, encapsulation, covalent binding, cross-linking, etc.3a,8 Furthermore, the use of a carbon-based material like graphene oxide (GO) and reduced graphene oxide (rGO) for the immobilization of enzymes has gained considerable attention from the chemical community.9 Graphene oxide (GO) is an allotrope of carbon, comprised of a 2-D honeycomb-like structure with tight packing of the sp2-hybridized carbon atoms along with random distribution of oxygen-containing functional groups.10 Interestingly, the conversion of graphene oxide (GO) to reduced graphene oxide (rGO) by removing the oxygen-containing functionalities improved many properties of the material, such as conductivity, elasticity, tensile strength, and a solid form for composites.11 The formation of nanocomposites by uniting rGO with metal oxide nanoparticles such as Fe3O4 offers several benefits, like improved conductivity, planar geometry with a large surface area and strong magnetism.12 In addition, the magnetic separation of a nanocomposite hybrid from the medium is more efficient, fast and economical in comparison to traditional separation methods. Hence, the use of rGO@Fe3O4 for the immobilization of the enzyme provides eco-friendly and magnetically-separable support.13The quinazolinone unit is part of many drug molecules and natural products (Fig. 1).14 In particular, 2,3-dihydroquinazolinone-4(1H)-one (DHQ) is the building block of many important therapeutic agents, such as anti-tumor, anti-convulsant, anti-microbial, anti-depressant, anti-viral, etc.15 In this context, a number of methods have been reported to synthesize 2,3-dihydroquinazolinone-4(1H)-one derivatives (Scheme 1). The condensation of 2-aminobenzamide with an aldehyde (aryl/alkyl) is a traditional method for synthesizing DHQ derivatives using a variety of catalysts such as Cp2TiCl2, Y(OTf)3, H[Gly2B], CAN, TiCl4–Zn, CNTs, H3PW12O40, etc.16 In 2014, Rangappa et al. reported an efficient one-pot method for the conversion of substituted 2-aminobenzamide and gem-dibromomethylarenes into the corresponding 2,3-dihydroquinazolin-4(1H)-ones in the presence of potassium tert-butoxide (t-BuOK) using pyridine-dimethyl formamide as the solvent mixture (Scheme 1a).17 Shankarling and group reported a simple protocol using choline hydroxide (ChOH) in an aqueous medium as a catalyst for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones derivatives via the cyclo-condensation reaction of 2-aminobenzonitrile and alkyl/aryl/hetero-aryl aldehydes (Scheme 1b).18 In 2018, Mosavian and coworkers documented an atom efficient one-pot multicomponent protocol utilizing isatoic anhydride and aromatic aldehydes with ammonium acetate or primary amines to synthesize mono- or disubstituted 2,3-dihydroquinazolin-4(1H)-ones in the presence of perchlorated zirconia (HClO4/ZrO2) nanoparticles (Scheme 1c).19 Badathala et al. reported one-pot cyclo-condensation of 2-aminobenzamide and aryl aldehydes by employing boronic acid supported over montmorillonites (H3BO3/montmorillonite K10) as the catalyst to synthesize 2,3-dihydroquinazolinone-4(1H)-one derivative (Scheme 1d).20 These methods are able to provide DHQ efficiently, but have certain drawbacks, such as required high reaction temperature, longer reaction time, tedious workup, generation of toxic chemical waste, and use of a non-reusable catalyst. In continuation of our efforts in the area of biocatalysis,21 herein, we have developed a reusable nanobiocatalyst via immobilization of lipase onto Fe3O4-decorated reduced graphene oxide to synthesize 2,3-dihydroquinazolinone-4(1H)-ones using 2-aminobenzamide and aryl aldehydes (Scheme 1e).
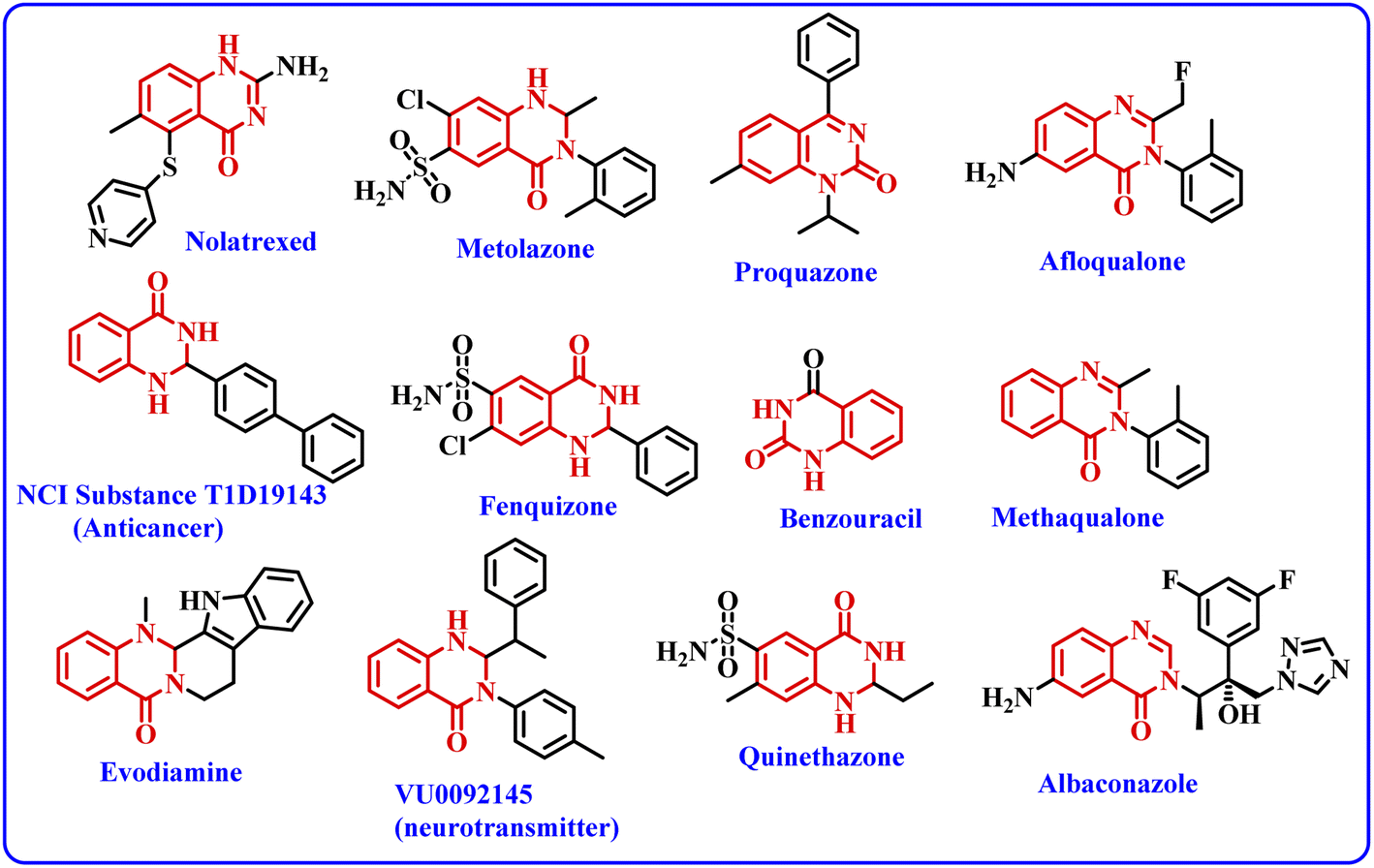 | ||
| Fig. 1 Examples of selective drugs and biologically active compounds containing the DHQ moiety. | ||
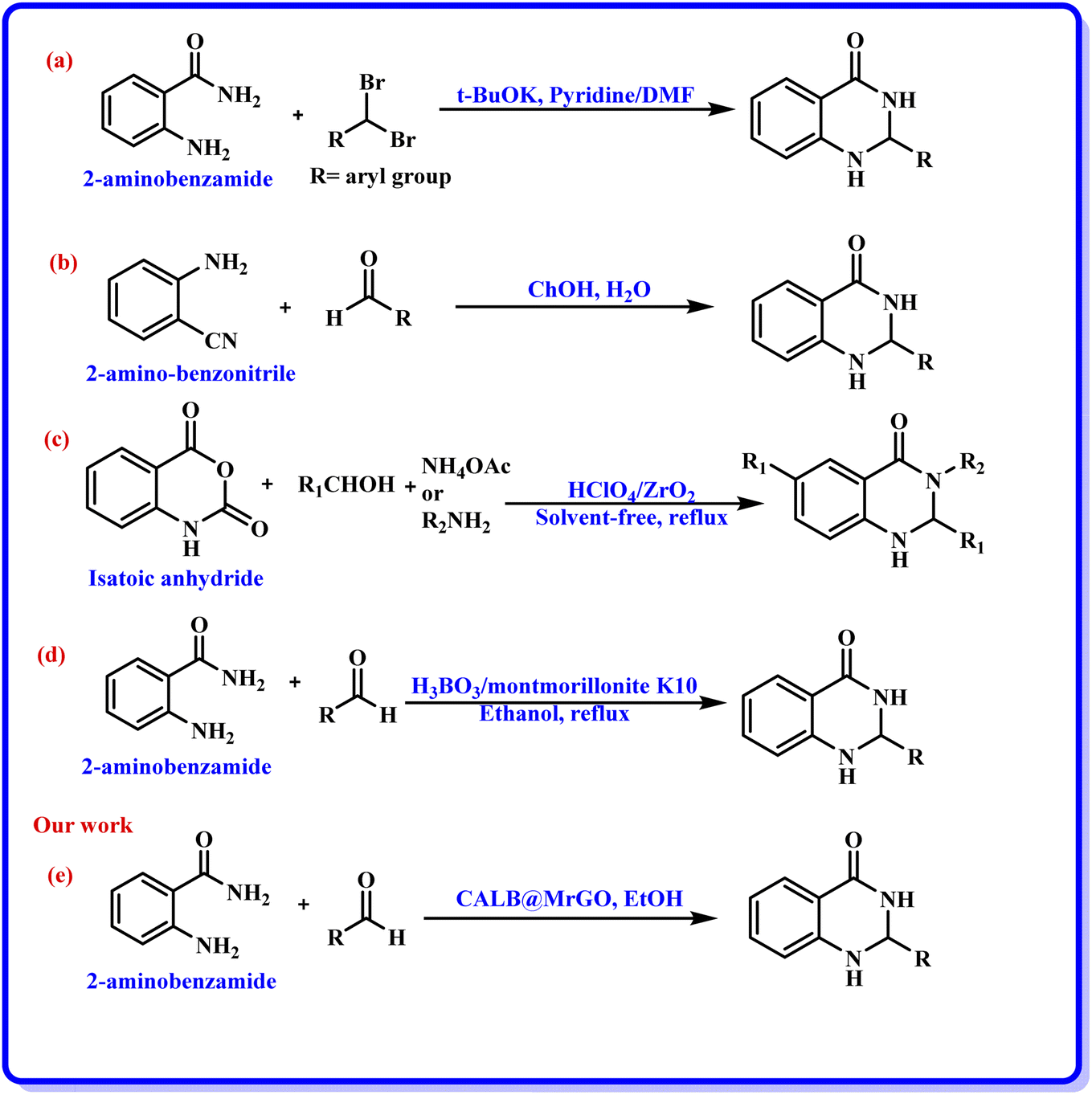 | ||
| Scheme 1 (a)–(d) Various synthetic pathways reported for the synthesis of DHQ, (e) present study. | ||
2 Results and discussion
We started by synthesizing the catalyst as depicted in Scheme 2. The first step was the synthesis of graphene oxide from powdered graphite using Tour's method with slight modifications.22 In the second step, the graphene oxide was chemically reduced by L-ascorbic acid, which works both as a reducing agent and a protecting agent and makes the procedure more economical, non-toxic, and environmentally friendly in comparison of hydrazine, hydroquinone, NaOH, or NaBH4, as hydrazine and others are toxic to the environment and cannot be used for large scale production of rGO due to their explosive nature.23 The GO reduction method proposed by Gao and group membes used NaBH4 and conc. H2SO4, which demands careful handling due to the evolution of flammable H2. The rGO reduced by L-ascorbic acid is used for large-scale production and has applications in the fields like sensors, flexible graphene fibres, and dye-sensitized solar cells.23 The Fe3O4-particles were grafted at the surface of reduced graphene oxide (rGO) to make it magnetic, which helps in the easy separation from the reaction mixture as depicted in step 3 (Scheme 2). Next, to improve the binding of magnetic reduced graphene oxide (MrGO) with the enzyme, the surface of MrGO was modified by adding the cross-linker 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) which activates the carboxyl groups of MrGO and produces an active O-acylisourea intermediate (step 4, Scheme 2). Afterward, in the fifth step, N-hydroxysuccinimide (NHS) was used, which rapidly reacts with the O-acylisourea intermediate to form reactive esters and considerably reduces the formation of side products.24 In the last step, the amine group present in the Candida antarctica lipase B (CALB) enzyme reacted with N-hydroxysuccinimide ester to make an amide linkage and immobilize CALB over the surface-modified magnetic reduced graphene oxide (MrGO) via covalent bonding to generate the final catalyst, i.e., CALB@MrGO (Scheme 2).24 The synthesized CALB@MrGO was characterized using techniques such as FTIR, XRD, XPS, SEM-EDS, HR-TEM and elemental mapping studies.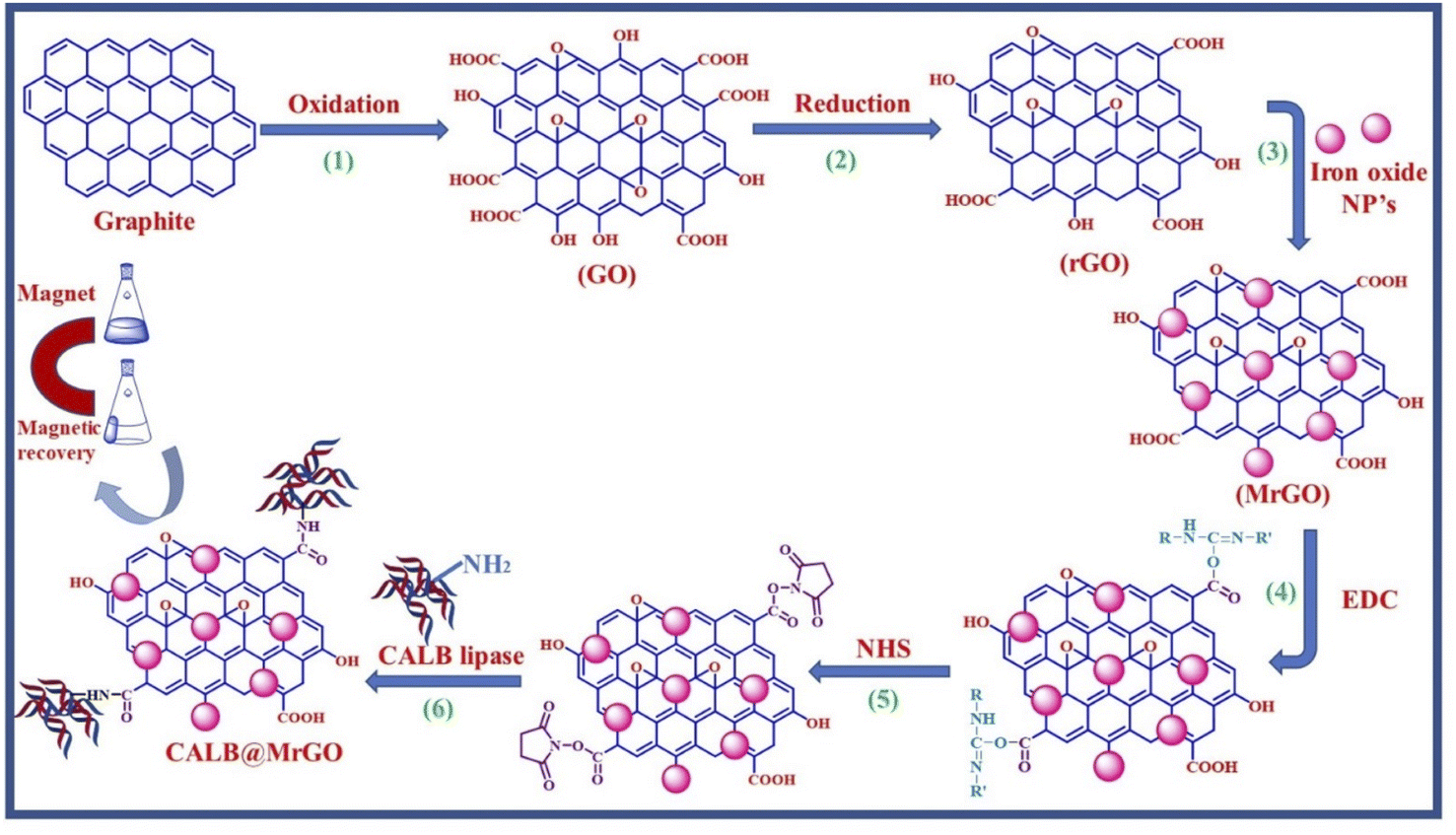 | ||
| Scheme 2 A diagrammatic representation of catalyst preparation. | ||
2.1 Characterization of catalyst
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonding in accordance with reported literature.26 In the GO spectrum, the characteristic bands positioned at ∼3432 cm−1 (OH stretching), 3060 cm−1 (C–H stretching), 1644 cm−1 (CC stretching), 1437 cm−1 (C–H bending), 1091 cm−1 (C–O stretching) and 599 cm−1 (OH out-of-plane bending) strongly indicate the oxidation of graphite powder into GO.27 The reduction of GO into rGO was supported by the bands at 1223 cm−1 (H–CC–H) and 1628 cm−1 (CC stretching) and by the fading of the broad band between 2700–3000 cm−1 (C–H stretching).28 Also, all the absorption bands related to oxidized groups disappear in the FT-IR spectrum of rGO, indicating the reduction of the groups containing oxygen by L-ascorbic acid (Fig. 2a). In the spectrum of pure Candida antarctica lipase B, the band at ∼1648 cm−1 was mainly due to the amide-I functionality present. Similar amide linkage bands at ∼1648 cm−1 and 1548 cm−1 were noticed in the IR spectrum of CALB@MrGO, indicating successful immobilization of the CALB enzyme over the MrGO surface (Fig. 2a).29
O bonding in accordance with reported literature.26 In the GO spectrum, the characteristic bands positioned at ∼3432 cm−1 (OH stretching), 3060 cm−1 (C–H stretching), 1644 cm−1 (CC stretching), 1437 cm−1 (C–H bending), 1091 cm−1 (C–O stretching) and 599 cm−1 (OH out-of-plane bending) strongly indicate the oxidation of graphite powder into GO.27 The reduction of GO into rGO was supported by the bands at 1223 cm−1 (H–CC–H) and 1628 cm−1 (CC stretching) and by the fading of the broad band between 2700–3000 cm−1 (C–H stretching).28 Also, all the absorption bands related to oxidized groups disappear in the FT-IR spectrum of rGO, indicating the reduction of the groups containing oxygen by L-ascorbic acid (Fig. 2a). In the spectrum of pure Candida antarctica lipase B, the band at ∼1648 cm−1 was mainly due to the amide-I functionality present. Similar amide linkage bands at ∼1648 cm−1 and 1548 cm−1 were noticed in the IR spectrum of CALB@MrGO, indicating successful immobilization of the CALB enzyme over the MrGO surface (Fig. 2a).29
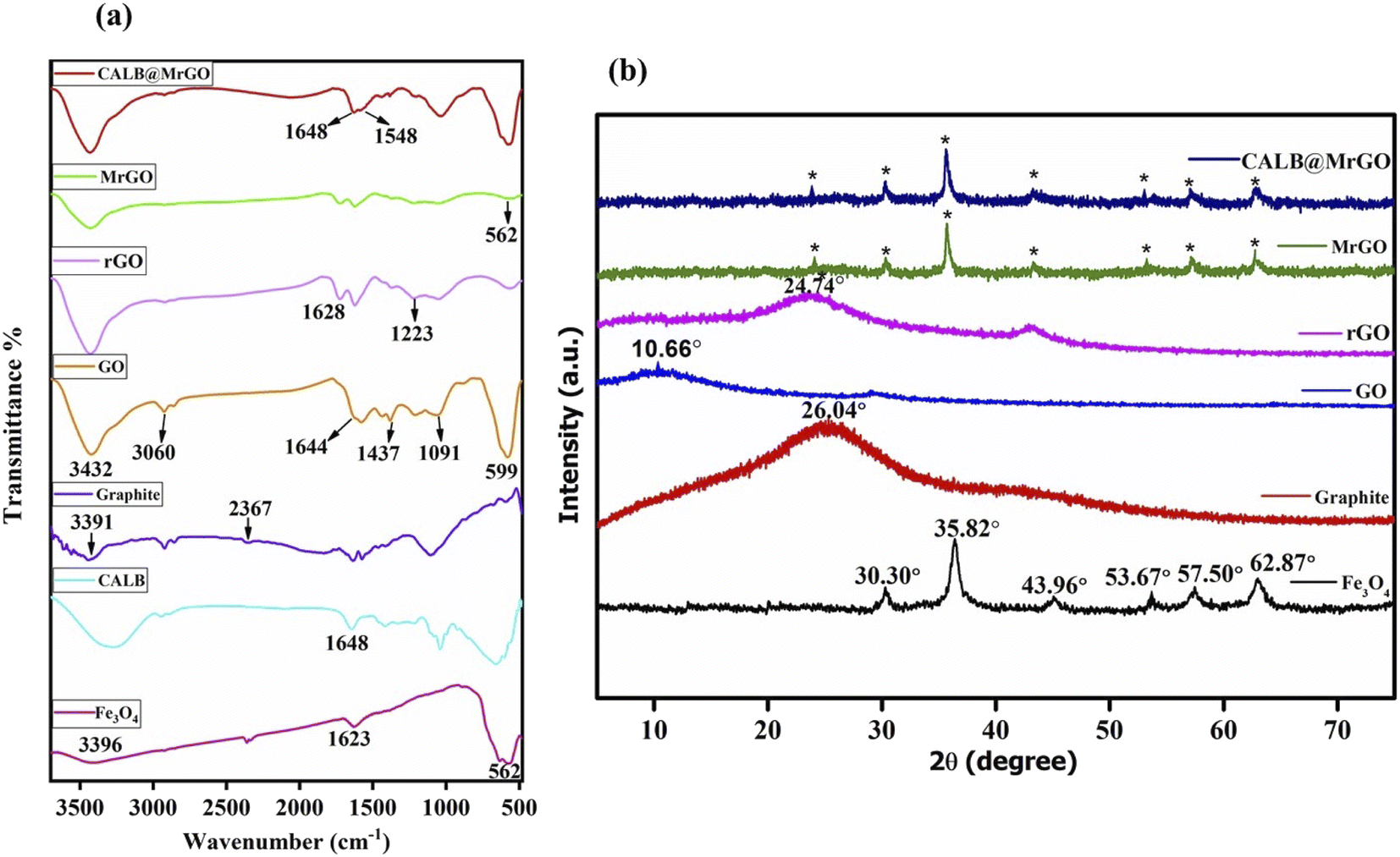 | ||
| Fig. 2 (a) FTIR spectra of Fe3O4 nanoparticles, pure CALB, graphite, GO, rGO, MrGO and CALB@MrGO. (b) Comparison of XRD results of Fe3O4 nanoparticles, graphite, GO, rGO, MrGO and CALB@MrGO. | ||
C), 285.1 eV (C–OH), 286.1 eV (C–O–C or C–OH) and 287.1 eV (CO) (Fig. 3b).33 As reported by Thomas Wagberg et al., the photoelectron peak at 286 eV could be due to C–N bonding,34 thus suggesting the binding of lipase over MrGO. Next, the deconvoluted N(1s) spectrum displays strong signals at 399.1 eV, 400 eV and 402 eV (Fig. 3c). The first peak at 399.1 eV could correspond to –CN bonding or to the amine groups present in the lipase enzyme, while the peak at 402 eV could be ascribed to either –N–CO or protonated (H–NH2) amine bonding. The peak at 400 eV could be assigned to the –CO–NH– bond and amine groups.7g,35 Thus, these peaks indicated that lipase is successfully immobilized on the nano-magnetic support. Further, the high-resolution spectrum of Fe(2p) can be resolved into two major peaks located at 724.1 eV and 726 eV, which are due to Fe 2p1/2 (Fig. 3d). The binding energy peaks between 710.3 eV and 712.1 eV are due to Fe 2p3/2, validating the existence of Fe3O4 nanoparticles in the biocatalyst.35b,36 The deconvoluted spectrum of O(1s) shows two peaks at 531 eV and 532 eV which are assigned to OC and O–C, respectively (Fig. 3e).37
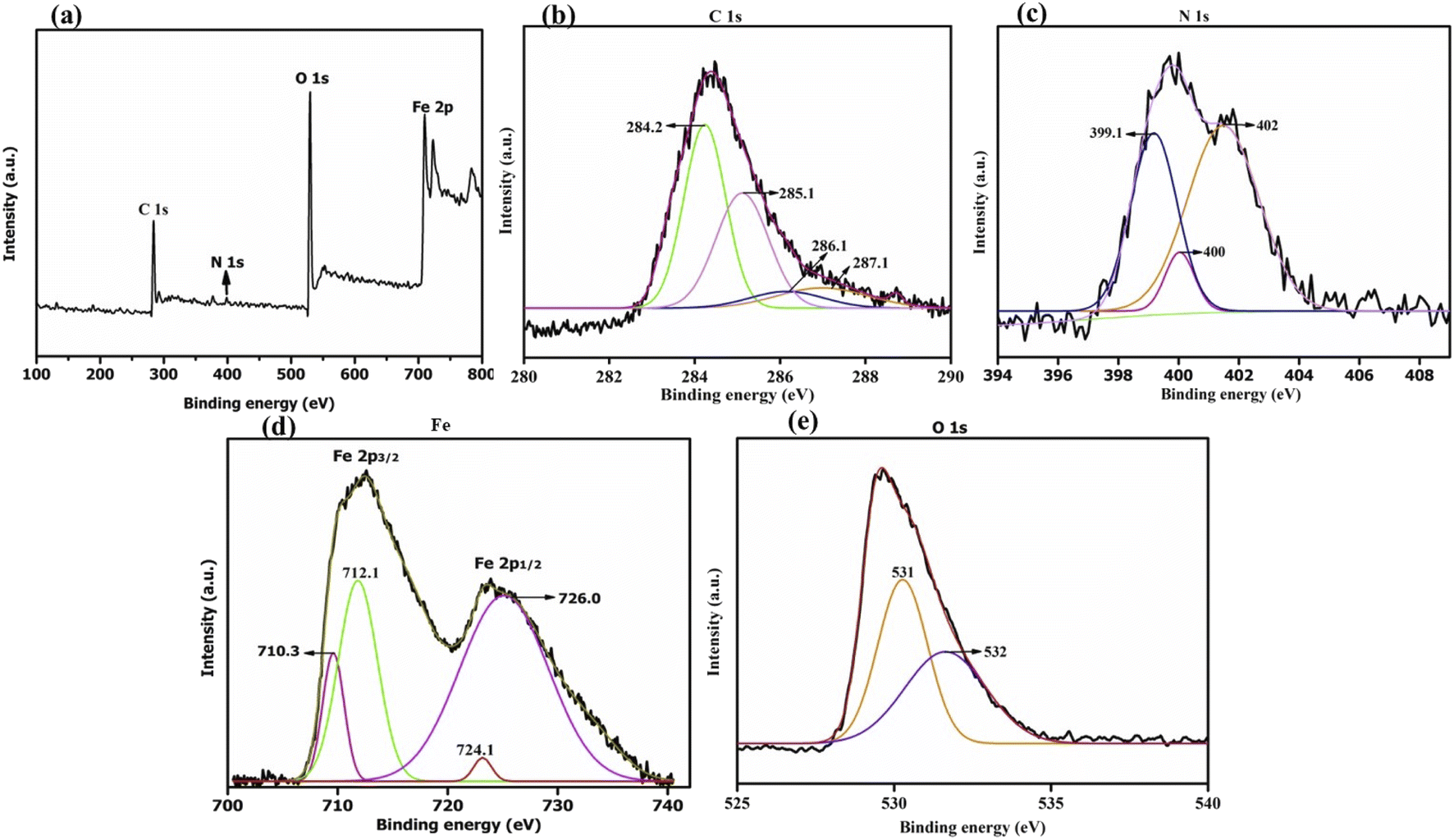 | ||
| Fig. 3 XPS spectra of catalyst CALB@MrGO showing (a) survey scan and (b–e) deconvoluted high-resolution scans of C, N, Fe and O, respectively. | ||
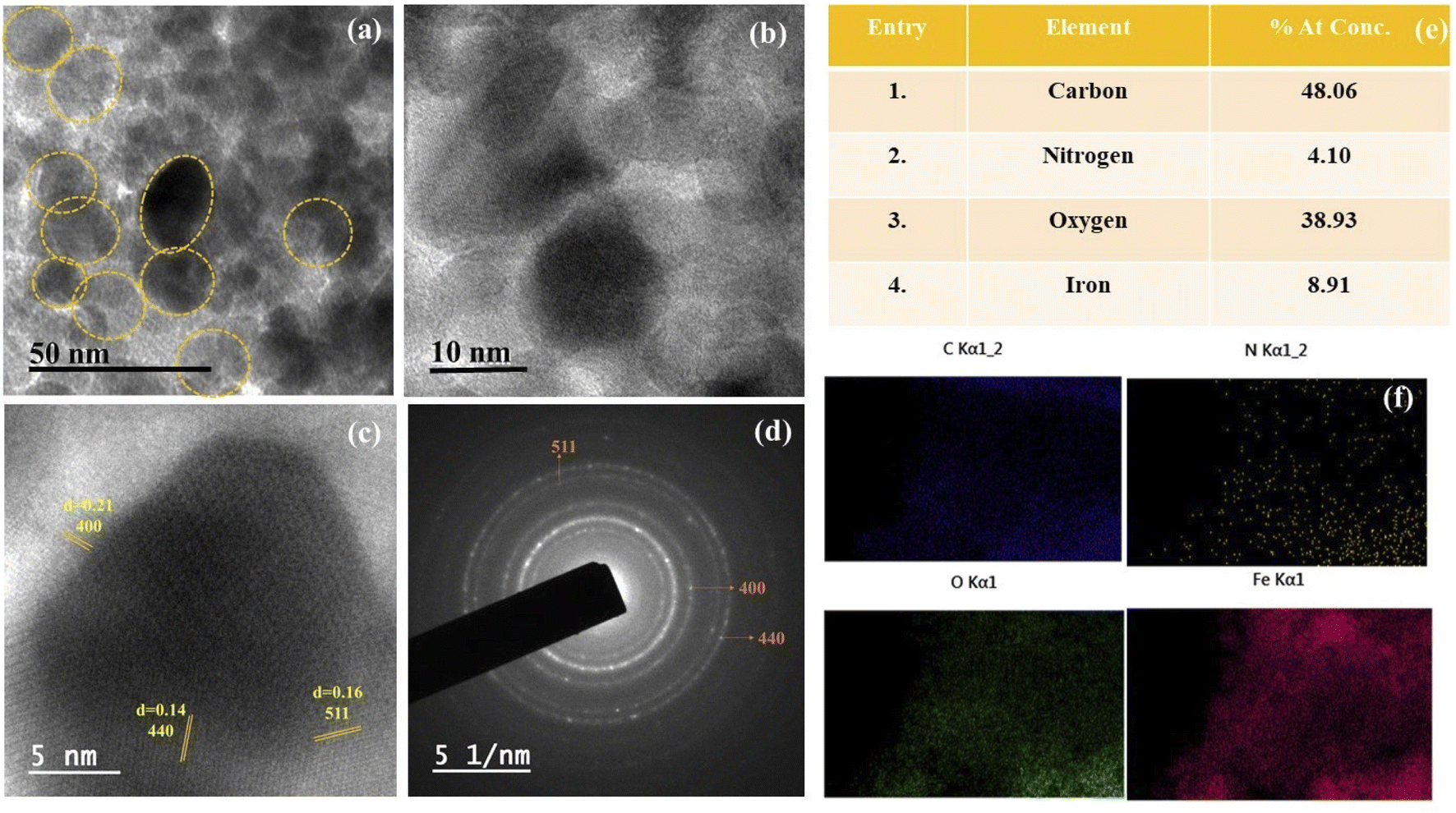 | ||
| Fig. 4 (a and b) HR-TEM micrographs of CALB@MrGO showing densely decorated iron-oxide nanoparticles over rGO surface. (c) Corresponding high-resolution TEM image of CALB@MrGO showing lattice fringe. (d) SAED image showing several crystallographic planes of Fe3O4. (e) EDS data of CALB@MrGO. (f) All image show the elemental mapping of the selected region of the catalyst. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe to −10000 Oe. The coercivity and remanence of the samples were found to be nearly zero, representing a typical superparamagnetic sample. The magnetic sample (MrGO) shows a saturation magnetization (Ms) of ∼19 emu g−1 before immobilization, while the catalyst (CALB@MrGO) shows a decrement in saturation magnetization up to ∼10 emu g−1 after lipase immobilization. The decrease in the magnetization value could be due to the increase in diamagnetic content within the magnetic biocatalyst. Despite the low Ms value, the nanobiocatalyst was easily recoverable by solid–liquid phase parting and effectively responded to an external magnet, as shown in Fig. 5.38
000 Oe to −10000 Oe. The coercivity and remanence of the samples were found to be nearly zero, representing a typical superparamagnetic sample. The magnetic sample (MrGO) shows a saturation magnetization (Ms) of ∼19 emu g−1 before immobilization, while the catalyst (CALB@MrGO) shows a decrement in saturation magnetization up to ∼10 emu g−1 after lipase immobilization. The decrease in the magnetization value could be due to the increase in diamagnetic content within the magnetic biocatalyst. Despite the low Ms value, the nanobiocatalyst was easily recoverable by solid–liquid phase parting and effectively responded to an external magnet, as shown in Fig. 5.38
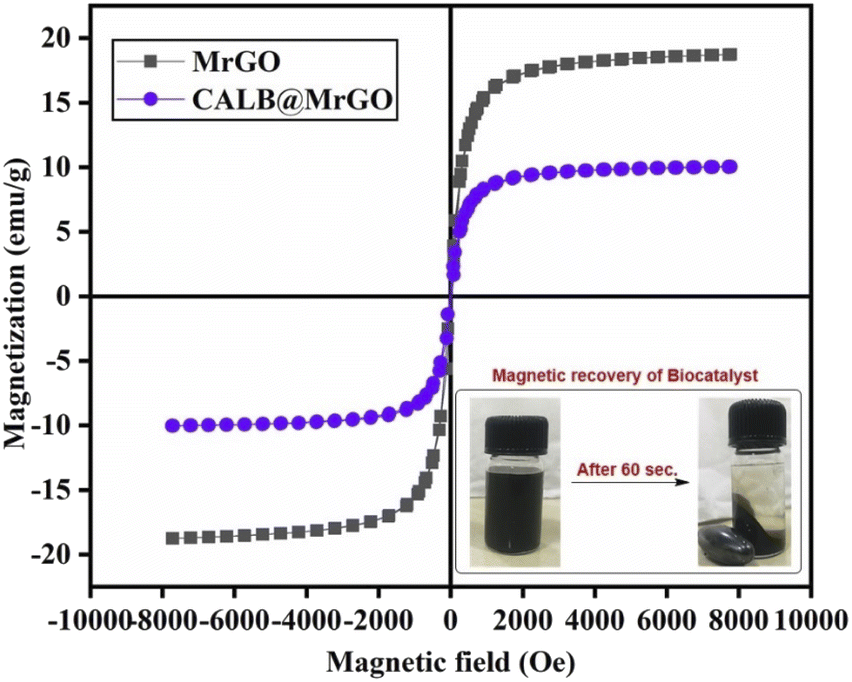 | ||
| Fig. 5 Magnetic hysteresis loops of pre-treated MrGO and CALB@MrGO demonstrating the magnetization potential. | ||
2.1.6.1 Effect of enzyme concentration. The immobilization of the CALB over MrGO (50 mg) was optimized by varying the concentration of CALB from 1–5 mg mL−1. As shown in Fig. 6a, saturation of lipase immobilization occurred at a 4 mg mL−1 concentration as calculated by Bradford assay.39 In this context, the amount of enzyme loaded over the solid support was found to be ∼356 mg per gram of MrGO. Further, the immobilization efficiency (IE) was calculated in respect to the varying concentration of enzyme using the formula reported by Al-Zuhair et al.40 and the results are summarized in Fig. 6b. The maximum efficiency of 36% was obtained when 4 mg mL−1 of enzyme was used, which was similar to the result obtained by Bradford assay.
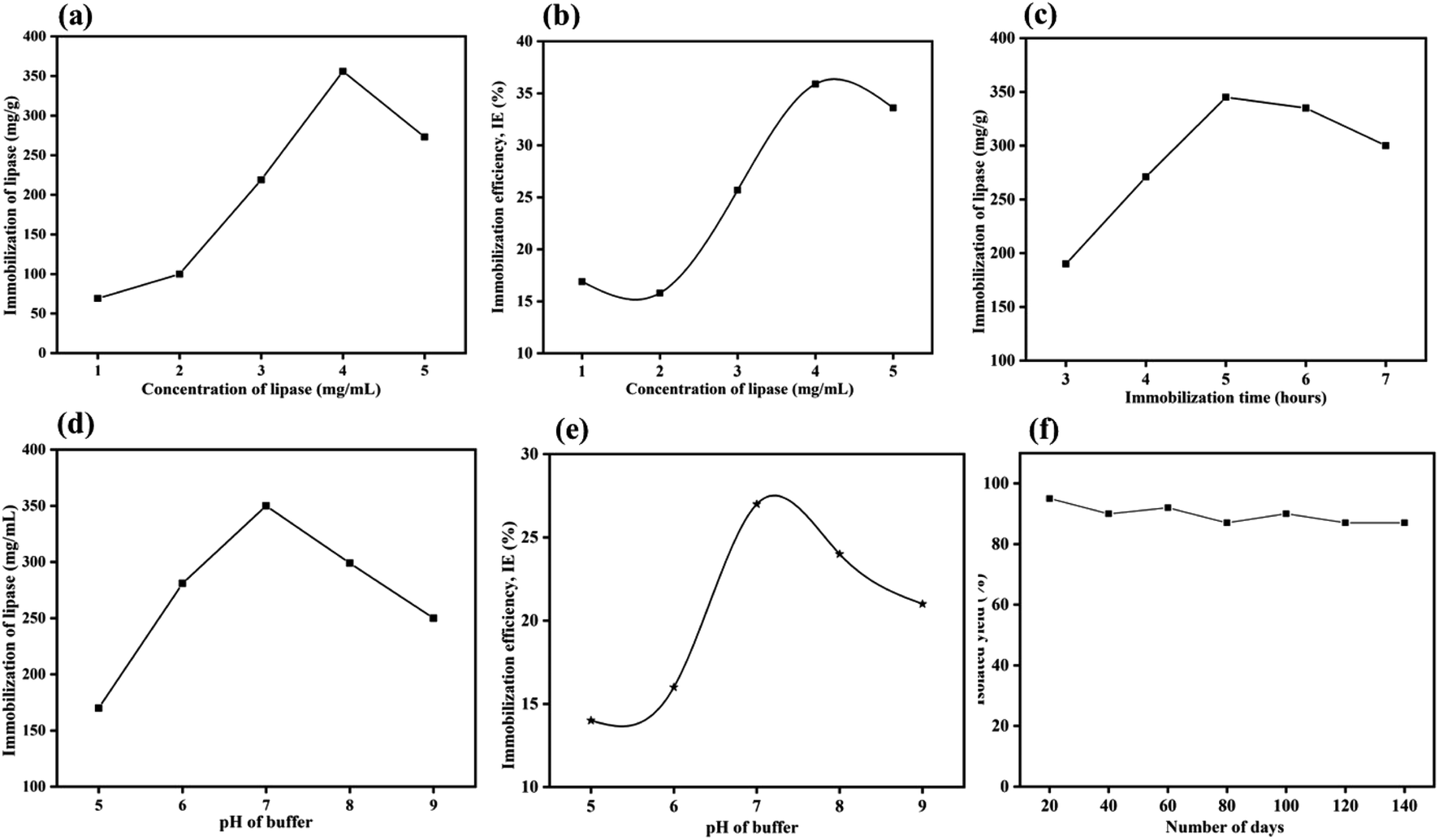 | ||
| Fig. 6 Effect of various parameters on lipase loading at diffenrent (a) concentration of CALB, (c) immobilization time and (d) pH of buffer. Effect of various parameters on immobilization efficacy at diffenrent (b) concentration of CALB (e) pH of buffer and (f) shelf life of catalyst. | ||
2.1.6.2 Effect of buffer pH. To study the influence of pH on the immobilization, solutions of CALB enzyme having a final concentration 4 mg mL−1 were prepared by mixing 40 mg CALB in 10 mL phosphate buffer solution (0.1 M) with pH = 5 to 9 at 25 °C. The CALB solutions thus obtained were stored in the refrigerator for future use. Then, 5 mL of the enzyme solutions at different pH values were added to 50 mg of solid support and mixed at 180 rpm and 25 °C for 5 hours. As shown in Fig. 6c, the maximum lipase loading occurs at pH ∼ 7, which is close to the value reported in the literature.41 Again, the immobilization efficiencies at the varying pH values were calculated (Fig. 6d) and the maximum efficiency was found at pH ∼ 7. A similar trend was noticed by Xie and group when immobilizing lipase over a magnetic support.41b
2.1.6.3 Reaction time optimization. The time of immobilization was optimized by varying the time of reaction between lipase and pre-treated MrGO from 3 to 7 hours. The maximum enzyme loading onto MrGO was found after 5 hours of reaction time (Fig. 6e).
2.1.6.4 Catalyst shelf life and activity. A study was carried out to check the shelf life and activity of the developed catalyst for this non-natural organic transformation by storing it at room temperature for 140 days (Fig. 6f). Gratifyingly, it was found that the catalyst was adequately stable and gave 87% yield of product (3a) even after 140 days of storage.
After optimizing the conditions for enzyme immobilization, we obtained the best enzyme loading, i.e., 356 mg of enzyme per gram of MrGO, when 4 mg mL−1 enzyme in phosphate buffer (pH ∼ 7, 0.1 M) was used for 5 h reaction time. We obtained 37% enzyme immobilization efficiency (IE) under the aforementioned conditions.
In the second phase, initial efforts were made to find the best reaction conditions for the condensation reaction of 2-aminobenzamide (1a) and an aromatic aldehyde (2a) to synthesize 2-phenyl-2,3-dihydroquinazolin-4(1H)-one (3a). In this regard, several parameters, such as temperature, solvent, molar ratio of reactants and catalyst loading, were screened. The model reaction gave only traces of product when performed in the absence of the catalyst at room temperature and at 55 °C (entries 1 and 2, Table 1), which proves the role of the catalyst in this transformation; similar observations were made by Badathala et al.42a After confirming the role of the catalyst in the model reaction (entry 3, Table 1), a range of temperatures, i.e. rt to 75 °C, was screened. We observed increments in the reaction yield as temperature increased from room temperature to 55 °C (entries 3–5, Table 1), because as temperature increases there is the possibility of more interaction between the enzyme and reactant molecules. However, the reaction gave product 3a in only 50% yield when the temperature was raised to 75 °C (entry 6, Table 1) which might be due to the denaturation of enzyme molecules. The same observations were made by Liu et al. while working with the lipase enzyme.42 Hence, temperature plays a key role in deciding the advancement of this reaction (entry 5, Table 1).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Catalyst amount (mg) | Temperature | Yield % (3a)b |
| a Reaction conditions: 2-aminobenzamide (1 mmol, 1a), benzaldehyde (1 mmol, 2a), CALB@MrGO catalyst in 5 mL of ethanol taken in a round bottom flask and stirred for 10 h.b Isolated yield. | ||||
| 1 | — | — | rt | Trace |
| 2 | — | — | 55 °C | Trace |
| 3 | CALB@MrGO | 40 | rt | 19% |
| 4 | CALB@MrGO | 40 | 45 °C | 44% |
| 5 | CALB@MrGO | 40 | 55 °C | 61% |
| 6 | CALB@MrGO | 40 | 75 °C | 50% |
It has been reported previously that the reaction medium plays a prime role in enzymatic reactions as the dispersibility of substrates in the solvent decides the fate of the reaction.42 Additionally, sometimes the solvents can alter the conformation of the enzyme in a reaction.42 The pH of the reaction medium might also affect the outcome of an enzymatic reaction to some extent.42 In this context, initially, the effect of solvent on the model reaction was studied by screening different solvents such as EtOH, MeOH, CH3CN, THF, hexane, CH2Cl2, and 0.1 M phosphate buffer (pH = 7). The reaction gave product 3a in 61% and 53% yield when EtOH and MeOH, respectively, were used as the solvent (entries 1 and 2, Table 2). However, trace or negligible reaction was observed with an array of solvents including CH3CN, THF, hexane, and CH2Cl2 (entries 3–6, Table 2), which might be due to the low solubility of the substrates during the reaction. Additional attempts were made to carry out the reaction in H2O and a mixture of H2O:EtOH (1:1 v/v), but very low yield of corresponding product 3a was observed (entries 7 and 8, Table 2). Next, we tried phosphate buffer (0.1 M) with pH = 7 as the solvent and observed product 3a in only 31% yield (entry 9, Table 2). The above-mentioned results indicate that EtOH remains the best choice to get maximum conversion of 3a (entry 1, Table 2), as was previously observed by Luo and co-workers.16a Next, the effect of the molar ratio of substrates was investigated in order to further optimize the reaction conditions. The performance of the reaction was improved when the molar ratio of 2-aminobenzamide (1a) and benzaldehyde (2a) was changed (entries 10–13, Table 2). The highest yield of product (80%) was obtained with a 1:1.25 molar ratio of 2-aminobenzamide (1a) and benzaldehyde (2a) (entry 12, Table 2).
|
|||
|---|---|---|---|
| Entry | Solvent | Molar ratio of 1a:2a |
Yield % (3a)b |
| a Reaction conditions: 2-aminobenzamide (1a), benzaldehyde (2a), CALB@MrGO catalyst (40 mg) in 5 mL of solvent taken in a round bottom flask and stirred at 55 °C for 10 h.b Isolated yield. | |||
| 1 | EtOH | 1:1 |
61% |
| 2 | MeOH | 1:1 |
53% |
| 3 | CH3CN | 1:1 |
Trace |
| 4 | THF | 1:1 |
Trace |
| 5 | Hexane | 1:1 |
Trace |
| 6 | CH2Cl2 | 1:1 |
Trace |
| 7 | H2O | 1:1 |
17% |
| 8 | EtOH:H2O |
1:1 |
11% |
| 9 | Phosphate buffer (0.1 M) pH = 7 | 1:1 |
31% |
| 10 | EtOH | 1:0.75 |
42% |
| 11 | EtOH | 1:1 |
61% |
| 12 | EtOH | 1:1.25 |
80% |
| 13 | EtOH | 1:1.5 |
75% |
Next, we investigated the model reaction using a varied amount of CALB@MrGO, from 20 to 100 mg (entries 1–5, Table 3). We found that 60 mg of CALB@MrGO was the best amount to carry out the reaction efficiently (entry 3, Table 3). However, there was decrement in the yield of the reaction when the enzyme concentration was increased beyond 60 mg (entries 4 and 5, Table 3). The decrement in the yield of the reaction at a higher concentration of the enzyme is due to the aggregation of enzyme molecules, which affects the interaction between substrate and enzyme molecule due to the blockage of the active sites of the enzyme.43 Fu et al. have also reported that a higher enzyme loading did not raise the reaction yield.43b A set of control reactions was conducted to confirm the role of the developed catalyst (entries 6–8, Table 3). To our delight, only trace or no product was obtained with only rGO, MrGO and surface functionalized MrGO (entries 6–8, Table 3).
|
|||
|---|---|---|---|
| Entry | Catalyst | Catalyst amount (mg) | Yield % (3a)b |
| a Reaction conditions: 2-aminobenzamide (1 mmol, 1a), benzaldehyde (1 mmol, 2a), catalyst in 5 mL of ethanol taken in a round bottom flask and stirred at 55 °C for 10 h.b Isolated yield. | |||
| 1 | CALB@MrGO | 20 | 42% |
| 2 | CALB@MrGO | 40 | 55% |
| 3 | CALB@MrGO | 60 | 97% |
| 4 | CALB@MrGO | 80 | 68% |
| 5 | CALB@MrGO | 100 | 61% |
| 6 | rGO | 60 | Trace |
| 7 | MrGO | 60 | Trace |
| 8 | Surface activated MrGO | 60 | Trace |
Having optimized the conditions in hand, we further investigated the substrate scope to prove the generality of this transformation with the developed catalyst. It is noteworthy to mention that the reaction proceeds efficiently to furnish the corresponding 2,3-dihydroquinazolin-4(1H)-ones in good to excellent yield with a range of electronically divergent aromatic aldehydes, as summarized in Table 4. The reaction of unsubstituted benzaldehyde with 2-aminobenzamide showed the highest product conversion with 95% isolated yield (entry 1, Table 4). Next, the effect of electron donating groups such as 4-OMe, 4-Me, 2-OH and 4-OH at the aryl aldehyde was tested, obtaining the corresponding products in 71–77% yield (entries 2–5, Table 4). Further, halide-substituted aryl aldehydes with 4-Br, 2-Cl, 3-Cl and 4-Cl were employed in the reaction and the products were obtained in isolated yields in the range of 84–89% (entries 6–9, Table 4). Then, the effect of the electron withdrawing group on the aryl aldehyde was tested. In this context, the aromatic aldehyde with substitutions such as 2-NO2 and 4-NO2 on the ring provided the isolated products at 75 and 70% yields, respectively (entries 10 and 11, Table 4). In addition, when 4-CN benzaldehyde reacted with 2-aminobenzamide, the product (3l) was obtained at 82% isolated yield (entry 12, Table 4).
|
||
|---|---|---|
| Entry | R | Product, yieldb |
| a Reaction conditions: 2-aminobenzamide (1 mmol, 1a), benzaldehyde (1.25 mmol, 2a) using CALB@MrGO catalyst (60 mg) in 5 mL of ethanol in a round bottom flask and stirred at 55 °C for 10 h.b Isolated yield. | ||
| 1 | H | 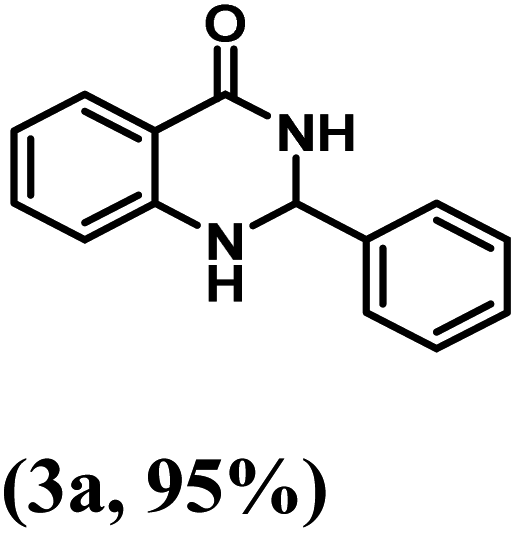 |
| 2 | 4-Me |  |
| 3 | 4-OMe |  |
| 4 | 4-OH |  |
| 5 | 2-OH |  |
| 6 | 2-Cl | 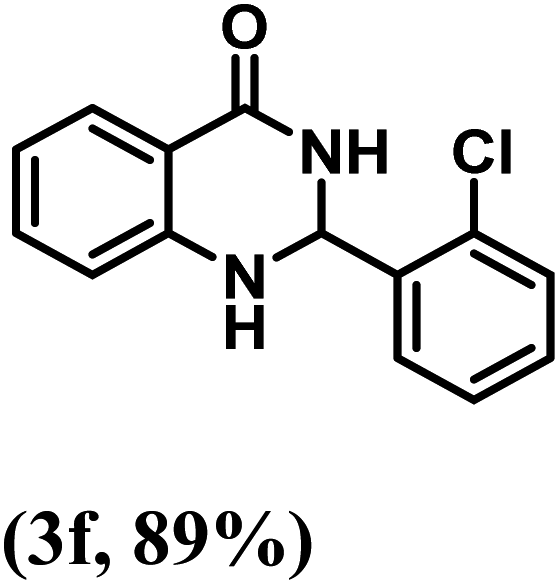 |
| 7 | 3-Cl | 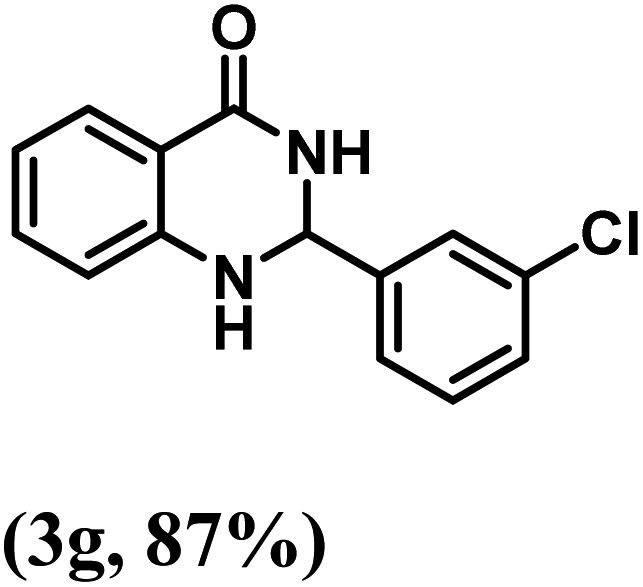 |
| 8 | 4-Cl | 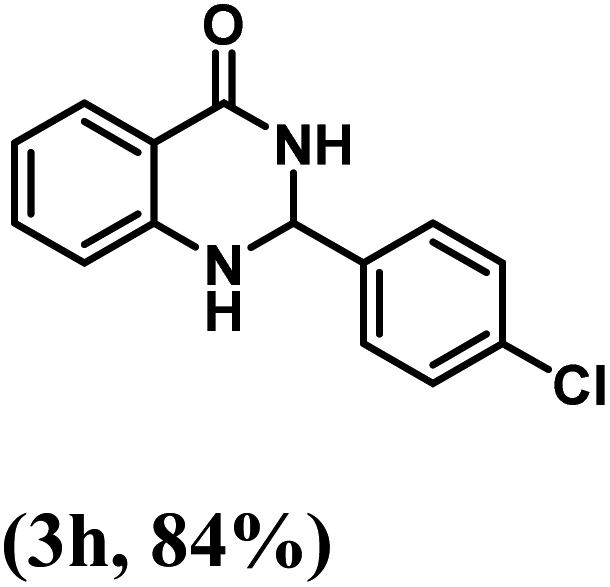 |
| 9 | 4-Br | 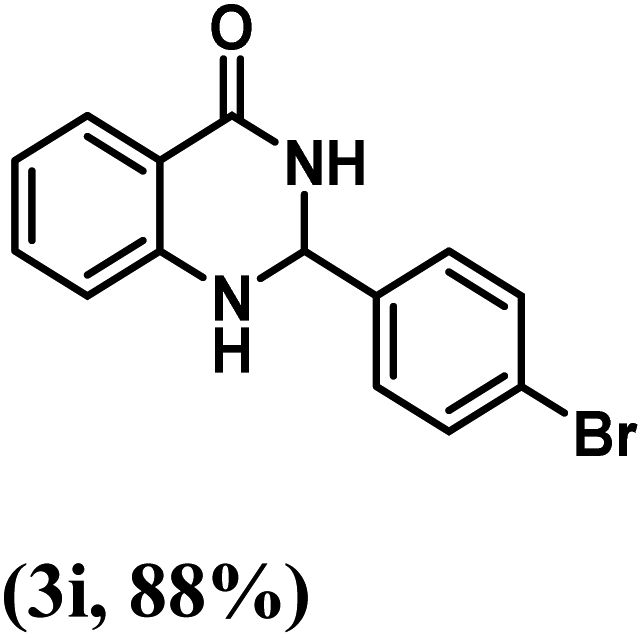 |
| 10 | 4-NO2 | 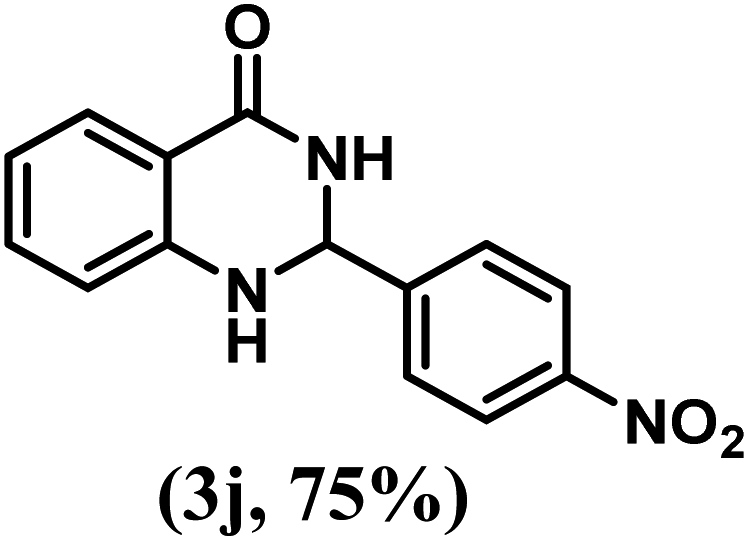 |
| 11 | 2-NO2 | 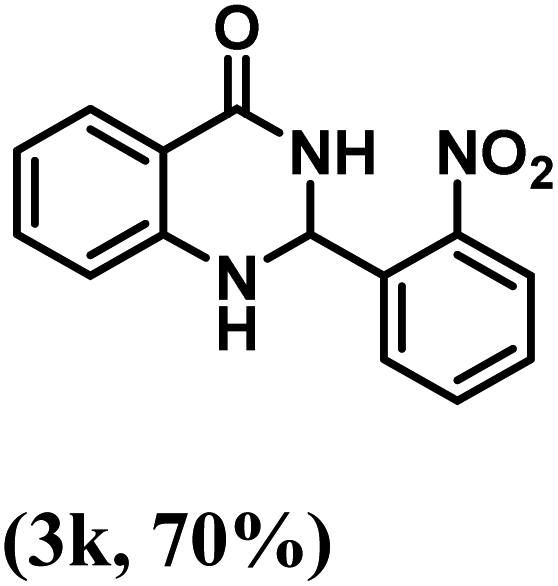 |
| 12 | 4-CN | 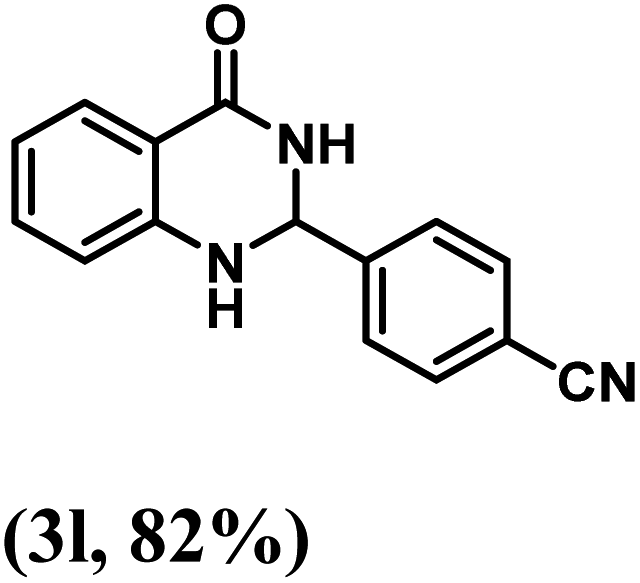 |
3 Scale-up and reusability of the catalyst
Next, we showed the synthetic utility of this transformation by setting up a gram-scale reaction of 2-aminobenzamide (1.0 g, 0.0073 mol, 1.0 equiv.) and benzaldehyde (0.973 g, 0.0087 mol, 1.25 equiv.) and obtained 2,3-dihydroquinazolin-4(1H)-one derivative (3a) in 87.1% isolated yield (1.43 g). After the completion of the reaction, the catalyst CALB@MrGO was easily separated from the reaction mixture using an external magnet followed by decantation. The separated catalyst was washed with EtOH, air-dried, and reused for up to 10 consecutive catalytic cycles to obtain the desired 2,3-dihydroquinazolin-4(1H)-one (Fig. 7). The results showed that the catalytic activity remained unchanged for up to five consecutive cycles and gradual decrement was observed after that. Furthermore, we calculated green chemistry metrics such as E-factor, atom economy, atom efficiency, process mass index and reaction mass efficiency to demonstrate the greenness of this protocol (entries 1–6, Table 5).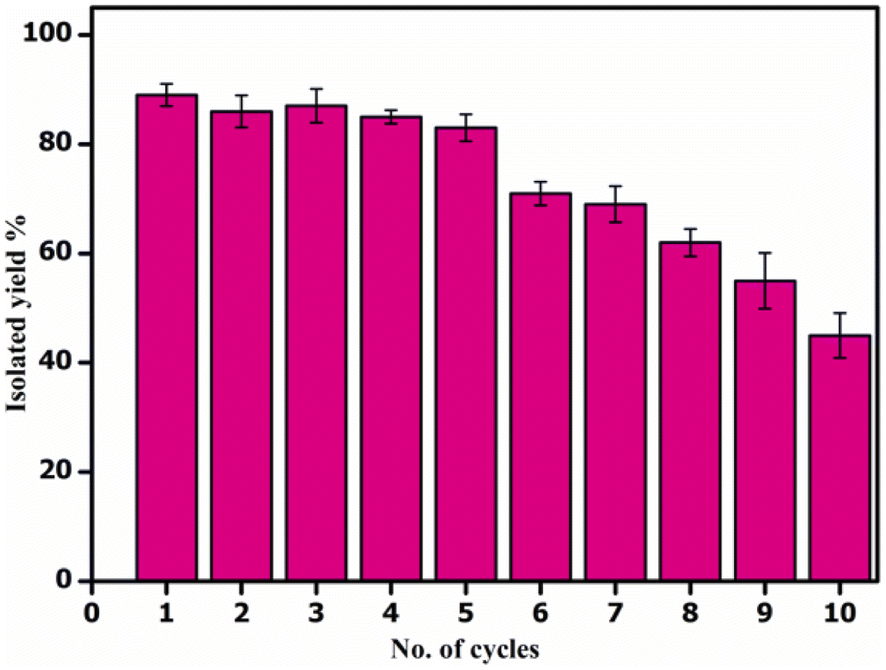 | ||
| Fig. 7 Reusability test of catalyst (CALB@MrGO) for the gram scale reaction. | ||
|
||
|---|---|---|
| Entry | Metrics | Results |
| 1 | Isolated yield | 87.1% |
| 2 | E-Factor | 0.37 |
| 3 | Atom economy (AE) | 92.5% |
| 4 | Atom efficiency | 80.6% |
| 5 | Process mass index (PMI) | 1.37 |
| 6 | Reaction mass efficiency (RME) | 92.5% |
3.1 Study of various kinetic parameters
To compare the catalytic efficiency of the immobilized enzyme CALB@MrGO vs. pure Candida antarctica lipase B enzyme, we designed an experiment to calculate various kinetic parameters, such as Vmax, KM, Kcat and catalytic efficiency, by varying the concentration of benzaldehyde from 0.25 mM to 1.50 mM while keeping the concentration of the enzyme in both pure form and immobilized form exactly the same, i.e. 4 mg mL−1 (entries 1–5, Table 6). Both pure CALB enzyme and CALB@MrGO followed the Michaelis–Menten kinetics model as depicted in Fig. 8a. Fascinatingly, immobilized lipase showed a catalytic efficiency (1082.5 mM s−1) approximately 1.5 times better than that of pure lipase (714.1 mM s−1) (entry 5, Table 6). These results clearly rule out the possibility of enzyme deactivation by the Fe3O4-decorated reduced graphene oxide.| Entry | Immobilized lipase | Pure lipase |
|---|---|---|
| 1 | Vmax = 2.0 mM min−1 | Vmax = 1.9 mM min−1 |
| 2 | KM = 0.40 mM | KM = 0.42 mM |
| 3 | R2 = 0.991 | R2 = 0.996 |
| 4 | Kcat = 433.1 s−1 | Kcat = 300.8 s−1 |
| 5 | Catalytic efficiency = 1082.5 mM s−1 | Catalytic efficiency = 714.1 mM s−1 |
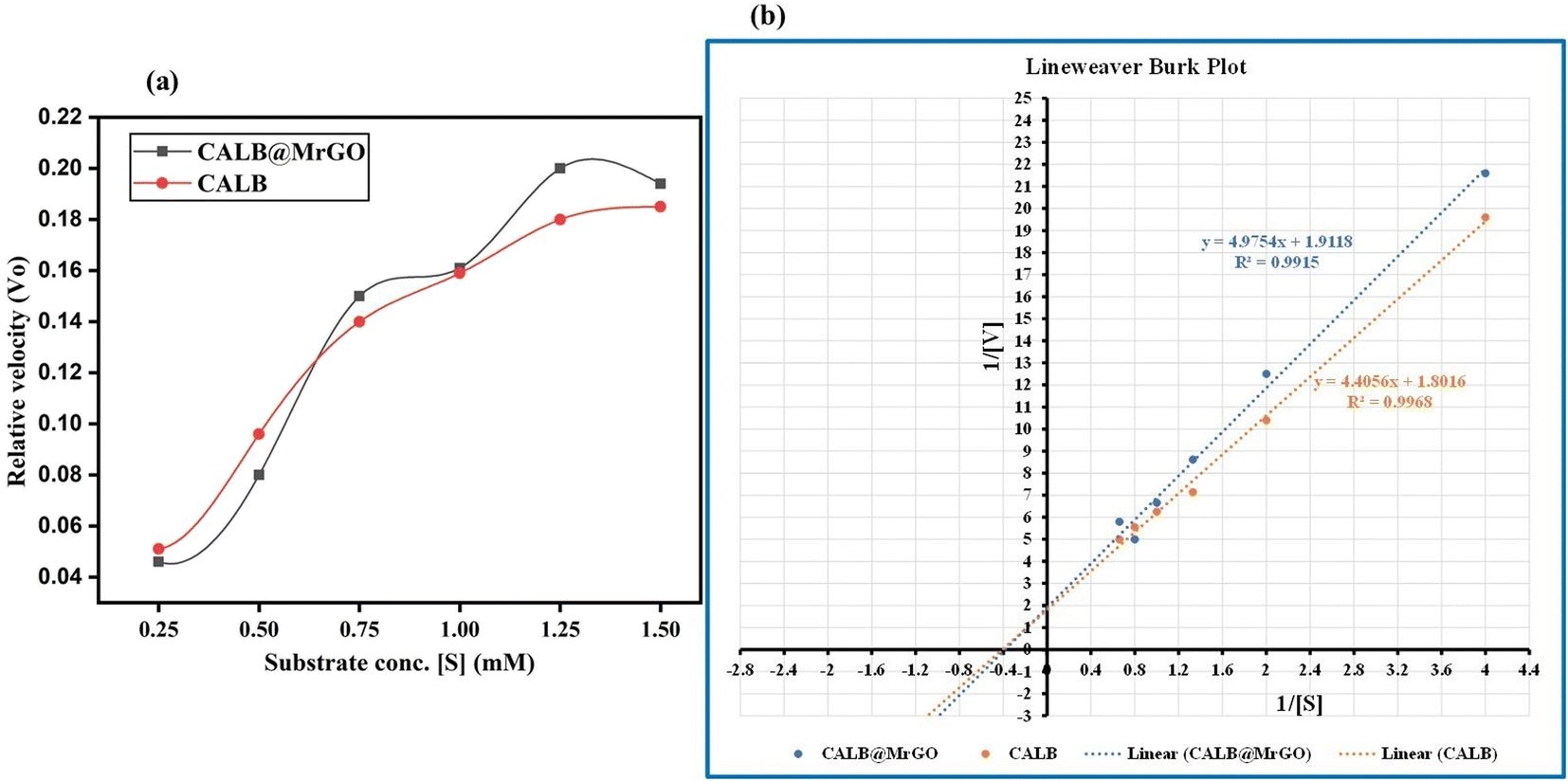 | ||
| Fig. 8 (a) Michaelis–Menten plot for CALB and CALB@MrGO atvarying concentrations of benzaldehyde. (b) Lineweaver Burk plot of CALB and CALB@MrGO. | ||
4 Conclusion
In summary, we report an efficient preparation of a highly reusable and eco-friendly nanobiocatalyst (CALB@MrGO). The magnetic character was imparted to NBC by anchoring Fe3O4 nanoparticles over reduced graphene oxide, onto which Candida antarctica lipase B was immobilized. The successful functionalization and post-immobilization changes in the nanobiocatalyst were characterized using the FTIR, XRD, XPS, HR-TEM, SEM-EDS and VSM techniques. The loading of the lipase enzyme over MrGO was studied using the Bradford assay, which exhibited 356 mg of enzyme per gram of solid support. Next, we employed the developed NBC (CALB@MrGO) in the one-pot synthesis of 2,3-dihydroquinazolin-4(1H)-one derivatives using 2-aminobenzamide with various substituted aromatic aldehydes and obtained the corresponding products in good to excellent yields. Moreover, the magnetically separable CALB@MrGO was found to be stable after the completion of the reaction and was reused in up to ten catalytic cycles. The catalytic activity of the synthesized NBC remained unchanged for five consecutive cycles, but afterward a slight decrement in the catalytic efficiency was noted. Further, the scalability of the transformation was proved by a gram-scale reaction. Additionally, the kinetics studies revealed that immobilized lipase CALB@MrGO was 1.5 times more active than pure Candida antarctica lipase B.5 Experimental
5.1 General information
All chemicals and solvents were purchased from commercial suppliers and used without any further purification. Lipase enzyme (EC 3.1.1.3) Candida antarctica lipase B (CALB) was purchased from commercial sources. The reaction progress was monitored using thin layer chromatography (TLC, thin silica layer coated on glass slide). The compounds were purified by column chromatography using silica (particle size 200–400) as the stationary phase and ethyl acetate in hexane as the mobile phase. NMR spectra were collected on a JEOL or Bruker NMR using deuterated DMSO solvent with TMS as an internal reference. The coupling constant (J) is expressed in hertz (Hz) and the chemical shift (δ) is expressed in parts per million (ppm). Multiplicities are abbreviated as s: singlet, d: doublet, dd: doublet of doublet, t: triplet, br s: broad singlet, and m: multiplet. The Fourier transform infrared (FT-IR) spectrum was collected using PerkinElmer Spectrum software, version 10.4.00. The X-ray analysis was done using an X-ray diffractometer (PanAlytical). The high resolution transmission electron microscopy (HR-TEM) was done using a JEOL JEM 2100 PLUS. The X-ray photon spectroscopy (XPS) was done using a Thermo Fisher Scientific ESCALAB Xi+ instrument.5.1.1.1 Synthesis of reduced graphene oxide (rGO). The rGO was synthesized using the Tour's method with slight modifications.20 The procedure was comprised three stages: (a) oxidation or intercalation, (b) exfoliation and (c) reduction.
5.1.1.1.1 Oxidation. In a typical preparation, 100 mL of concentrated sulfuric acid/ortho-phosphoric acid mixture (9
:1 v/v) was added slowly to 0.5 g of graphite powder under constant stirring in a 500 mL conical flask equipped with a magnetic stirrer and water bath. To this, 4.5 g of potassium permanganate was added and the resulting mixture was magnetically stirred for 12 h at 55 °C. After the stipulated time, a thick, dark green colored paste was obtained and allowed to cool to room temperature. To this, 250 mL of deionized water was added slowly, followed by the addition of 10 mL hydrogen peroxide, resulting in a bright yellow mixture that indicated the oxidation of the graphite powder. Finally, the mixture was centrifuged at 7000 rpm and the obtained solid was washed with a 5% hydrochloric acid aqueous solution 6–7 times and dried in an oven at 60 °C for 12 h.
5.1.1.1.2 Exfoliation. Exfoliation includes the formation of layered GO from the oxidized form of graphite synthesized in the previous step. Here, 0.4 g of oxidized graphite was added to 200 mL deionized water in a beaker and stirred at 60 °C in a water bath for 12 h. After the stipulated time, the obtained black colored paste was allowed to cool to room temperature, centrifuged at 7000 rpm, and then dried in an oven at 60 °C for 24 h.
5.1.1.1.3 Reduction. This is the final step, where the GO obtained from the above step was converted to rGO. In this procedure, 4 g of ascorbic acid was added to 0.4 g of GO powder in a beaker with 400 mL of deionized water and stirred for 45 minutes at 60 °C. After the specified time, the mixture was allowed to cool to room temperature and centrifuged at 7000 rpm. The obtained rGO was treated with excess 30 wt% hydrogen peroxide for 45 min at 60 °C to oxidize the remaining ascorbic acid. Next, the obtained mixture of rGO was allowed to cool to room temperature, centrifuged at 7000 rpm, washed with ethanol and deionized water (5 times) and dried in oven for 24 h at 120 °C.
5.1.1.2 Synthesis of magnetic reduced graphene oxide (MrGO). In a typical procedure, 500 mg of rGO was dispersed in 25 mL of deionized water and then ultrasonicated to get a stable rGO suspension. Meanwhile, 0.6 g FeSO4·7H2O and 1.16 g FeCl3·6H2O were individually dissolved in 10 mL deionized water each. Both solutions were transferred to the prepared rGO suspension along with 2.5 g sodium acetate and then stirred for 30 minutes. Afterwards, the mixture was transferred into a 50 mL Teflon lined stainless-steel autoclave, sealed and heated at 200 °C for 12 h. After that, the autoclave was allowed to cool to room temperature and the supernatant was decanted off. The magnetic reduced graphene oxide (MrGO) was obtained in the form of black particles. The MrGO particles were washed with ethanol multiple times and dried in the oven for 12 h at 60 °C.
5.1.1.3 Immobilization of CALB enzyme. To immobilize the lipase on MrGO, 100 mg of MrGO support was dispersed in 100 mL deionized water in a beaker and ultrasonicated for 1 h. Subsequently, 100 mg of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 60 mg of N-hydroxysuccinimide (NHS) were added to the above mixture with constant stirring for 2 h. After 2 h, the activated support was recovered using an external magnet, washed with deionized water three times and dried for 12 h at 60 °C. Next, 0.05 g of pre-treated MrGO was added to 10 mL CALB suspension (4 mg mL−1 in phosphate buffer, 0.1 M, pH = 7) and stirred gently (180 rpm) at 25 °C for 5 hours. Then, the CALB immobilized catalyst (CALB@MrGO) was recovered using an external magnet and the supernatant was also collected in a separate beaker for protein estimation by the Bradford method. Finally, the obtained magnetic catalyst was washed with phosphate buffer, dried at 40 °C and stored in an air-tight container for further use.
5.2 General procedure for the synthesis of quinazoline
In a round-bottom flask equipped with a stir bar, 2-aminobenzamide (1a, 1.0 equiv.) and aryl aldehyde (2a, 1.25 equiv.) were added. Then, 60 mg of CALB@MrGO in 5 mL of ethanol was added and the resulting mixture was gently stirred at 55 °C. The progress of the reaction was monitored by thin-layer chromatography (TLC) using ethyl acetate in hexane (30:70). Upon the completion of the reaction, as indicated by TLC, the catalyst was separated from the reaction mixture using an external magnet. The reaction mixture was cooled to room temperature and water (10 mL) was added to provide a solid precipitate. The solid precipitate was filtered and washed 3–4 times with 5 mL of EtOH to get the pure product. In some cases, the oily product was also obtained and was further purified using column chromatography with ethyl acetate and hexane as eluents.
Author contributions
MB, AA and VT designed and executed all the experiments and prepared the manuscript. BC contributed to the VSM studies.Conflicts of interest
The authors declare that they have no conflict of interest to report.Acknowledgements
The financial supports from Department of Science and Technology (DST/INSPIRE/04/2017/000095), India is extremely acknowledged.References
- (a) M. Misson, H. Zhang and B. Jin, J. R. Soc., Interface, 2014, 12, 20140891–20140911 CrossRef PubMed; (b) Q. Husain, S. A. Ansari, F. Alam and A. Azam, Int. J. Biol. Macromol., 2011, 49, 37–43 CrossRef CAS PubMed.
- (a) M. L. Verma, C. J. Barrow and M. Puri, Appl. Microbiol. Biotechnol., 2012, 97, 23–39 CrossRef PubMed; (b) A. Illanes, A. Cauerhff, L. Wilson and G. R. Castro, Bioresour. Technol., 2012, 115, 48–57 CrossRef CAS PubMed.
- (a) R. Reshmy, E. Philip, R. Sirohi, A. Tarafdar, K. B. Arun, A. Madhavan, P. Binod, M. Kumar Awasthi, S. Varjani, G. Szakacs and R. Sindhu, Bioresour. Technol., 2021, 337, 125491–125502 CrossRef CAS PubMed; (b) S. A. Ansari and Q. Husain, Biotechnol. Adv., 2012, 30, 512–523 CrossRef CAS PubMed; (c) F. Wang, C. Guo, L. R. Yang and C. Z. Liu, Bioresour. Technol., 2010, 101, 8931–8935 CrossRef CAS PubMed; (d) L. Wang and R. Jiang, Methods Mol. Biol., 2011, 743, 9–106 CrossRef PubMed; (e) J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS PubMed.
- (a) C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS; (b) G. Brahmachari, Lipase-Catalyzed Organic Transformations: A Recent Update, Elsevier Inc., 2017 Search PubMed; (c) M. Kapoor and M. N. Gupta, Process Biochem., 2012, 47, 555–569 CrossRef CAS; (d) R. Mu, Z. Wang, M. C. Wamsley, C. N. Duke, P. H. Lii, S. E. Epley, L. C. Todd and P. J. Roberts, Catalysts, 2020, 10, 832–857 CrossRef CAS.
- (a) N. D. Fessner, C. P. S. Badenhorst and U. T. Bornscheuer, ChemCatChem., 2022, 14, e202200156 CAS; (b) C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed; (c) H. Renata, Z. J. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351–3367 CrossRef CAS PubMed; (d) M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed; (e) B. P. Dwivedee, S. Soni, M. Sharma, J. Bhaumik, J. K. Laha and U. C. Banerjee, ChemistrySelect, 2018, 3, 2441–2466 CrossRef CAS; (f) M. Lõpez-Iglesias and V. Gotor-Fernández, Chem. Rec., 2015, 15, 743–759 CrossRef PubMed; (g) E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504–4523 RSC.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef CAS PubMed.
- (a) R. K. Singh, M. K. Tiwari, R. Singh and J. K. Lee, Int. J. Mol. Sci., 2013, 14, 1232–1277 CrossRef CAS PubMed; (b) U. Guzik, K. Hupert-Kocurek and D. Wojcieszynska, Molecules, 2014, 19, 8995–9018 CrossRef PubMed; (c) S. Datta, L. R. Christena and Y. R. S. Rajaram, Biotech, 2013, 3, 1–9 Search PubMed; (d) V. L. Sirisha, A. Jain and A. Jain, Adv. Food Nutr. Res., 2016, 79, 179–211 CAS; (e) T. de Andrade Silva, W. J. Keijok, M. C. C. Guimarães, S. T. A. Cassini and J. P. de Oliveira, Sci. Rep., 2022, 12, 1–11 CrossRef PubMed; (f) H. Dong, J. Li, Y. Li, L. Hu and D. Luo, Chem. Eng. J., 2012, 181–182, 590–596 CrossRef CAS; (g) M. Budhiraja, R. Kondabala, A. Ali and V. Tyagi, Tetrahedron, 2020, 76, 131643–131651 CrossRef CAS; (h) M. Budhiraja, A. Ali and V. Tyagi, New J. Chem., 2022, 46, 4837–4849 RSC.
- (a) R. A. Wahab, N. Elias, F. Abdullah and S. K. Ghoshal, React. Funct. Polym., 2020, 152, 104613–104639 CrossRef CAS; (b) B. Thangaraj and P. R. Solomon, ChemBioEng Rev., 2019, 6, 157–166 CrossRef CAS; (c) D. N. Tran and K. J. Balkus, ACS Catal., 2011, 1, 956–968 CrossRef CAS; (d) J. Liu, R. T. Ma and Y. P. Shi, Anal. Chim. Acta, 2020, 1101, 9–22 CrossRef CAS PubMed; (e) S. Aggarwal, A. Chakravarty and S. Ikram, Int. J. Biol. Macromol., 2021, 167, 962–986 CrossRef CAS PubMed.
- (a) K. T. Sriwong, R. Kamogawa, C. S. Castro Issasi, M. Sasaki and T. Matsuda, Biochem. Eng. J., 2022, 177, 108263–108272 CrossRef; (b) K. Chaudhary, K. Kumar, P. Venkatesu and D. T. Masram, Adv. Colloid Interface Sci., 2021, 289, 102367–102384 CrossRef CAS PubMed; (c) M. B. Vineh, A. A. Saboury, A. A. Poostchi and A. Ghasemi, Int. J. Biol. Macromol., 2020, 164, 4403–4414 CrossRef CAS PubMed; (d) S. Rassi and R. Baharfar, Polyhedron, 2019, 174, 114153–114162 CrossRef; (e) G. Coşkun, Z. Çıplak, N. Yıldız and Ü. Mehmetoğlu, Appl. Biochem. Biotechnol., 2021, 193, 430–445 CrossRef PubMed; (f) M. Adeel, M. Bilal, T. Rasheed, A. Sharma and H. M. N. Iqbal, Int. J. Biol. Macromol., 2018, 120, 1430–1440 CrossRef CAS PubMed.
- (a) X. Xiao, T. E. Beechem, M. T. Brumbach, T. N. Lambert, D. J. Davis, J. R. Michael, C. M. Washburn, J. Wang, S. M. Brozik, D. R. Wheeler, D. B. Burckel and R. Polsky, ACS Nano, 2012, 6, 3573–3579 CrossRef CAS PubMed; (b) K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed; (c) P. Chammingkwan, K. Matsushita, T. Taniike and M. Terano, Materials, 2016, 9, 240–254 CrossRef PubMed.
- (a) R. Tarcan, O. Todor-Boer, I. Petrovai, C. Leordean, S. Astilean and I. Botiz, J. Mater. Chem. C, 2020, 8, 1198–1224 RSC; (b) X. Yu, H. Cheng, M. Zhang, Y. Zhao, L. Qu and G. Shi, Nat. Rev. Mater., 2017, 2, 1–14 Search PubMed.
- (a) L. Liu, X. Zhu, Y. Zeng, H. Wang, Y. Lu, J. Zhang, Z. Yin, Z. Chen, Y. Yang and L. Li, Polymers, 2018, 10, 1329–1342 CrossRef PubMed; (b) Y. Sun, W. Zhang, H. Yu, C. Hou, D. Sen Li, Y. Zhang and Y. Liu, J. Alloys Compd., 2015, 638, 182–187 CrossRef CAS.
- S. K. S. Patel, S. H. Choi, Y. C. Kang and J. K. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 2213–2222 CrossRef CAS PubMed.
- M. Badolato, F. Aiello and N. Neamati, RSC Adv., 2018, 8, 20894–20921 RSC.
- (a) M. J. Hour, L. J. Huang, S. C. Kuo, Y. Xia, K. Bastow, Y. Nakanishi, E. Hamel and K. H. Lee, J. Med. Chem., 2000, 43, 4479–4487 CrossRef CAS PubMed; (b) D. C. White, T. D. Greenwood, A. L. Downey, J. R. Bloomquist and J. F. Wolfe, Bioorg. Med. Chem., 2004, 12, 5711–5717 CrossRef CAS PubMed; (c) R. J. Alaimo, J. Med. Chem., 1972, 15, 335–336 CrossRef CAS PubMed; (d) X. Cheng, S. Vellalath, R. Goddard and B. List, Synfacts, 2009, 2009, 0147 CrossRef; (e) K. Srivalli and K. Satish, Chem. Sci. Trans., 2012, 1, 624–631 CrossRef.
- (a) Y. Luo, Y. Wu, Y. Wang, H. Sun, Z. Xie, W. Zhang and Z. Gao, RSC Adv., 2016, 6, 66074–66077 RSC; (b) Y. H. Shang, L. Y. Fan, X. X. Li and M. X. Liu, Chin. Chem. Lett., 2015, 26, 1355–1358 CrossRef CAS; (c) H. R. Safaei, M. Shekouhy, V. Shafiee and M. Davoodi, J. Mol. Liq., 2013, 180, 139–144 CrossRef CAS; (d) M. Wang, J. J. Gao, Z. G. Song and L. Wang, Chem. Heterocycl. Compd., 2011, 47, 851–855 CrossRef CAS; (e) G. Cai, X. Xu, Z. Li, W. P. Weber and P. Lu, J. Heterocycl. Chem., 2002, 39, 1271–1272 CrossRef CAS; (f) J. Safari and S. Gandomi-Ravandi, J. Mol. Catal. A: Chem., 2014, 390, 1–6 CrossRef CAS; (g) Y. X. Zong, Y. Zhao, W. C. Luo, X. H. Yu, J. K. Wang and Y. Pan, Chin. Chem. Lett., 2010, 21, 778–781 CrossRef CAS.
- K. H. Narasimhamurthy, S. Chandrappa, K. S. Sharath Kumar, K. B. Harsha, H. Ananda and K. S. Rangappa, RSC Adv., 2014, 4, 34479–34486 RSC.
- P. N. Borase, P. B. Thale and G. S. Shankarling, RSC Adv., 2016, 6, 63078–63083 RSC.
- Z. Hamidi, M. Abdollahi-Alibeik and S. Y. Mosavian, Silicon, 2018, 10, 2491–2497 CrossRef CAS.
- M. Kancherla, M. R. Katlakanti, K. Seku and V. Badathala, Iran. J. Chem. Chem. Eng., 2019, 38, 37–49 CAS.
- (a) S. Dutt, V. Goel, N. Garg, D. Choudhury, D. Mallick and V. Tyagi, Adv. Synth. Catal., 2020, 362, 858–866 CrossRef CAS; (b) S. Dutt and V. Tyagi, Tetrahedron Lett., 2021, 87, 153527–153532 CrossRef CAS.
- A. T. Habte and D. W. Ayele, Adv. Mater. Sci. Eng., 2019, 2019 DOI:10.1155/2019/5058163.
- (a) R. Sadek, M. S. Sharawi, C. Dubois and H. Tantawy, RSC Adv., 2022, 12, 22608–22622 RSC; (b) K. K. H. De Silva, H. H. Huang and M. Yoshimura, Appl. Surf. Sci., 2018, 447, 338–346 CrossRef CAS; (c) M. R. Keshavarz and S. Hassanajili, Iran. J. Chem. Chem. Eng., 2021, 40, 731–742 Search PubMed.
- (a) S. Liu, M. Bilal, K. Rizwan, I. Gul, T. Rasheed and H. M. N. Iqbal, Int. J. Biol. Macromol., 2021, 190, 396–408 CrossRef CAS PubMed; (b) N. Z. Prlainović, D. I. Bezbradica, J. R. Rogan, P. S. Uskoković, D. Mijin and A. D. Marinković, C. R. Chim., 2016, 19, 363–370 CrossRef.
- L. Nalbandian, E. Patrikiadou, V. Zaspalis, A. Patrikidou, E. Hatzidaki and C. N. Papandreou, Curr. Nanosci., 2015, 12, 455–468 CrossRef.
- I. O. Faniyi, O. Fasakin, B. Olofinjana, A. S. Adekunle, T. V. Oluwasusi, M. A. Eleruja and E. O. B. Ajayi, SN Appl. Sci., 2019, 1, 1–7 Search PubMed.
- Y. Jiao, H. Zhang, T. Dong, P. Shen, Y. Cai, H. Zhang and S. Zhang, J. Mater. Sci., 2017, 52, 3233–3243 CrossRef CAS.
- T. R. B. Ramakrishna, D. P. Killeen, T. D. Nalder, S. N. Marshall, W. Yang and C. J. Barrow, Appl. Spectrosc., 2018, 72, 1764–1773 CrossRef CAS PubMed.
- (a) R. Arunkumar, C. J. Drummond and T. L. Greaves, Front. Chem., 2019, 7, 1–11 CrossRef PubMed; (b) W. Xie and M. Huang, Energy Convers. Manage., 2018, 159, 42–53 CrossRef CAS.
- T. Kamakshi, G. S. Sundari, H. Erothu and T. P. Rao, Rasayan J. Chem., 2018, 11, 1113–1119 CrossRef CAS.
- T. K. Ghosh, S. Gope, D. Rana, I. Roy, G. Sarkar, S. Sadhukhan, A. Bhattacharya, K. Pramanik, S. Chattopadhyay, M. Chakraborty and D. Chattopadhyay, Bull. Mater. Sci., 2016, 39, 543–550 CrossRef CAS.
- K. Sa, P. C. Mahakul, B. V. R. S. Subramanyam, J. Raiguru, S. Das, I. Alam and P. Mahanandia, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 338, 012055–012062 Search PubMed.
- (a) B. D. Ossonon and D. Bélanger, RSC Adv., 2017, 7, 27224–27234 RSC; (b) M. K. Rabchinskii, A. T. Dideikin, D. A. Kirilenko, M. V. Baidakova, V. V. Shnitov, F. Roth, S. V. Konyakhin, N. A. Besedina, S. I. Pavlov, R. A. Kuricyn, N. M. Lebedeva, P. N. Brunkov and A. Y. Vul', Sci. Rep., 2018, 8, 1–11 CrossRef CAS PubMed.
- E. Gracia-Espino, G. Hu, A. Shchukarev and T. Wagberg, J. Am. Chem. Soc., 2014, 136, 6626–6633 CrossRef CAS PubMed.
- (a) Y. Ren, J. G. Rivera, L. He, H. Kulkarni, D. K. Lee and P. B. Messersmith, BMC Biotechnol., 2011, 11, 63–71 CrossRef CAS PubMed; (b) W. Xie and M. Huang, Energy Convers. Manage., 2018, 159, 42–53 CrossRef CAS.
- R. Liang, L. Shen, F. Jing, N. Qin and L. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 9507–9515 CrossRef CAS PubMed.
- N. Díez, A. Śliwak, S. Gryglewicz, B. Grzyb and G. Gryglewicz, RSC Adv., 2015, 5, 81831–81837 RSC.
- B. Sahoo, S. Dutta and D. Dhara, J. Chem. Sci., 2016, 128, 1131–1140 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 76, 248–254 CrossRef.
- A. A. Qayoudi and S. Al-zuhair, Sustainability, 2022, 14, 8399–8416 CrossRef.
- (a) J. J. Jacob and K. Suthindhiran, Biotechnol. Rep., 2020, 25, e00422–e00429 CrossRef PubMed; (b) W. Xie and M. Huang, Catalysts, 2019, 9, 850–869 CrossRef CAS.
- (a) M. Kancherla, M. R. Katlakanti, K. Seku and B. Badathala, Iran. J. Chem. Chem. Eng., 2019, 38, 37–49 CAS; (b) J. Liu, F. Li, X. Zheng, J. Su, Y. Yu, L. Wang and H. Zhuang, Process Biochem., 2021, 101, 99–103 CrossRef CAS.
- (a) H. Aghaei, A. Yasinian and A. Taghizadeh, Int. J. Biol. Macromol., 2021, 178, 569–579 CrossRef CAS PubMed; (b) Y. Fu, Z. Lu, K. Fang, X. He, H. Xu and Y. Hu, RSC Adv., 2020, 10, 10848–10853 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra04405f |
| This journal is © The Royal Society of Chemistry 2022 |