
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManufacturing silica aerogel and cryogel through ambient pressure and freeze drying †
Massimigliano Di Luigia,
Zipeng Guo b,
Lu Ana,
Jason N. Armstronga,
Chi Zhou*b and
Shenqiang Ren*acd
b,
Lu Ana,
Jason N. Armstronga,
Chi Zhou*b and
Shenqiang Ren*acd
aDepartment of Mechanical and Aerospace Engineering, University at Buffalo, The State University of New York, Buffalo 14260, New York, USA. E-mail: shenren@buffalo.edu
bDepartment of Industrial and Systems Engineering, University at Buffalo, The State University of New York, Buffalo 14260, New York, USA. E-mail: chizhou@buffalo.edu
cDepartment of Chemistry, University at Buffalo, The State University of New York, Buffalo 14260, New York, USA
dResearch and Education in Energy, Environment & Water (RENEW), University at Buffalo, The State University of New York, Buffalo 14260, New York, USA
First published on 1st August 2022
Abstract
Achieving a mesoporous structure in superinsulation materials is pivotal for guaranteeing a harmonious relationship between low thermal conductivity, high porosity, and low density. Herein, we report silica-based cryogel and aerogel materials by implementing freeze-drying and ambient-pressure-drying processes respectively. The obtained freeze-dried cryogels yield thermal conductivity of 23 mW m−1 K−1, with specific surface area of 369.4 m2 g−1, and porosity of 96.7%, whereas ambient-pressure-dried aerogels exhibit thermal conductivity of 23.6 mW m−1 K−1, specific surface area of 473.8 m2 g−1, and porosity of 97.4%. In addition, the fiber-reinforced nanocomposites obtained via freeze-drying feature a low thermal conductivity (28.0 mW m−1 K−1) and high mechanical properties (∼620 kPa maximum compressive stress and Young's modulus of 715 kPa), coupled with advanced flame-retardant capabilities, while the composite materials from the ambient pressure drying process have thermal conductivity of 28.8 mW m−1 K−1, ∼200 kPa maximum compressive stress and Young's modulus of 612 kPa respectively. The aforementioned results highlight the capabilities of both drying processes for the development of thermal insulation materials for energy-efficient applications.
1 Introduction
Lightweight aerogels are often referred to as super-insulation materials due to an exceptional combination of extremely low thermal conductivity and remarkably high porosity (typically above 95%1,2). Mesoporosity constitutes a crucial feature in these materials to ensure a congruent relationship between thermal conductivity, surface area, and density. A mesoporous material can be defined as that class of nanoporous materials with pores sizes between 2 and 50 nm,3 being these below the mean free path of air particles (at approximately 68 nm) at room temperature and atmospheric pressure, hence effectively avoiding the movement of air particles through the material. Furthermore, high values of specific surface area (>500 m2 g−1)4 constitute another significant characteristic of aerogels, which, in conjunction with the previously mentioned features, position this material as a preferred choice for applications related to energy-saving purposes.5–8Amongst all the materials that have been successfully used for the manufacturing of aerogels, silica has emerged as one of the most frequently used alternatives largely due to its distinctive simplicity of processing.9 Sodium silicate solution (also commonly referred to as waterglass), on the other hand, has gained extensive popularity as a result of its low cost, as well as its high versatility and excellent stability in different types of solutions, hence making it the ideal option for scalable processes.10 A critical aspect that has a determinant structural effect on porous materials is the type of drying methodology employed. Capillary forces occurring during the drying stage, depending on the nature and geometry of the pore in the gel, may reach values up to 200 MPa,4 which, when occurring inside pores of small size, could lead to severe shrinkage and culminate in the cracking or rupture of the gel network. The freeze-drying (FD) method could potentially avoid damage to the 3D solid network that is formed during condensation.11 The fundamental reason for this is the absence of the phase boundary between liquid and gas phase that typically occurs when ambient pressure drying (APD) is used, hence resulting in no capillary forces being present within the pores. Cryogels are produced using the FD methodology, during which the liquid within the pores of the wet gel is initially frozen and subsequently removed by sublimation under vacuum conditions. The occurrence of this freezing-sublimation phase eradicates the detrimental effect that the capillary pressure exerts on the nanopores of the gel network,12 being the latter a typical occurrence during the ambient pressure drying (APD) procedure.13 In order to avoid collapse of the gel when the solvent within the pores is extracted during APD,14 both solvent exchange and surface modification procedures – occurring either simultaneously15–20 or non-simultaneously,21–23 an even in multiple treatments during synthesis24 – are typically required to minimize the incidence of the capillary pressure. Hence, it is indispensable to achieve scalable manufacturing of insulation materials by mitigating the surface exchange and surface modification of the wet-gel precursor.25
Here, we report that the silica cryogels obtained from the FD process show a thermal conductivity as low as 23 mW m−1 K−1, with a specific surface area of 369.4 m2 g−1, and a porosity of 96.7%. Ambient-pressure-dried aerogels (referred as silica aerogels), on the other hand, have a thermal conductivity of 23.6 mW m−1 K−1, a specific surface area of 473.8 m2 g−1, and a porosity of 97.4% respectively. The developed fiber/silica cryogel composite material from the FD process possesses an extraordinary combination of significantly low thermal conductivity (28.0 mW m−1 K−1) and improved mechanical properties (∼620 kPa maximum compressive stress and Young's modulus of 715 kPa in compression at 40% strain), while the fiber/silica aerogel composite materials from APD show values of 28.8 mW m−1 K−1, ∼200 kPa maximum compressive stress and Young's modulus of 612 kPa in compression at 40% strain, which, in any case, results in an advantageous thermal/mechanical performance relationship.
2 Materials and methods
2.1 Preparation of diluted ion-exchanged sodium silicate solution (waterglass)
Sodium silicate solution – a.k.a. waterglass (Technical Grade 40°–42° Bé | Fisher Scientific) is diluted to 10% v/v using deionized water (DI H2O) in a beaker, which is followed by stirring of the resulting solution for at least 15 min. Then, the diluted ion-exchanged waterglass is passed through an ion-exchange column that has been previously loaded with cation-exchange resin (Amberlite® IRC120H – hydrogen form | Sigma Aldrich). The diluted-sodium-silicate/Amberlite® IRC120H – hydrogen form resin mix was performed to an approximate 0.75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. The required cation-exchange resin is washed 4–5 times for 10 minutes with deionized water before use.
:1 volume ratio. The required cation-exchange resin is washed 4–5 times for 10 minutes with deionized water before use.
2.2 Synthesis of ion-exchange-sodium-silicate-based aerogel precursor
Initially, urea [CH4N2O; 98% | Beantown Chemical (BTC)] is mixed with deionized water [DI H2O] in a beaker to a 6.875 mol L−1 molar ratio and stirred until complete dissolution takes place (typically for at least 60 min). Following this, Sodium Dodecyl Sulfate - SDS – [C12H25NaSO4; ≥ 90% | Sigma Aldrich] and Cetrimonium Bromide - CTAB – [C19H42BrN; High purity | VWR], to a 1:2 composition ratio (0.04 and 0.08 mol L−1 respectively), are added to the previously obtained D-I Water/Urea solution until a homogeneous, white-color solution is achieved. This will be referred to as Solution A, which will be typically stirred for at least 6 hours or overnight to ensure a thorough dissolution of the surfactants. Further to this, the previously obtained diluted ion-exchanged sodium silicate is added to Solution A at a proportion of 25 wt% and stirring is continued until completely mixed to form Solution B. Hydrochloric Acid [HCl(aq); 36.5–38.0% | Capitol Scientific] – to a proportion of 25% v/v with respect to the quantity of sodium silicate used – is mixed with deionized water [DI H2O] in a beaker to a 50% v/v HCl/DI H2O ratio. The diluted HCl is then added to Solution B and stirring is continued until fully blended (approx. 1–2 min) to form the pre-aerogel precursor (target pH of solution ≈ 1.6–1.8). The precursor is then transferred to a sealed plastic container and placed into a pre-heated oven to 80 °C for a period of 24 hours.
After the condensation stage has been completed, an aerogel precursor of white color and fine texture has formed, which is then transferred into a beaker and fully immersed in D-I water for aging purposes. The aging precursor is placed on a hot plate at approximately 50–55 °C, and D-I water is thoroughly replaced 4–5 times during a period of at least 24 hours as part of the washing process. As a result, the D-I water covering the aerogel precursor becomes almost fully transparent, suggesting that all ammonia, and possible unreacted surfactant, have been removed during the process. The excess of water is then removed from the beaker, and the precursor is now ready for post processing. For this research work, post-processing includes (a) drying of the precursor using the ambient pressure drying (APD) method in a pre-heated oven at 60 °C for approximately 48 hours until all the remaining solvent has been removed, to be followed by the respective sintering of the aerogels to 600 °C, (b) drying of the precursor using the freeze-drying (FD) method (as explained in the next sub-section), followed by the respective sintering of the cryogels to 600 °C as well as (c) the preparation of fiber/silica aerogel and fiber/silica cryogel composite materials including several types of fiber as detailed in the following sections. It is worth mentioning that 600 °C has been selected as the optimal sintering temperature following results from thermal conductivity tests of aerogel specimens (sintered to 300 °C, 400 °C, 500 °C, and 600 °C respectively) performed in previous work.25 Moreover, BET/BJH tests also confirm that better results are obtained for samples that have been sintered to 600 °C, as shown in Table S1 and Fig. S11† respectively. Temperatures above 600 °C are generally not used, since (a) the organic template is expected to have been extensively removed from the material at this point (as confirmed by the weight % reduction in Fig. S3†), and (b) degradation of the silica particles and damage to the silica network would begin.
2.3 Preparation of sodium-silicate-based composite samples
In regards to composite samples, different amounts of cellulose-based fiber and glass (acid resistant borosilicate) microfiber have been used in order to study the effect of the fiber/silica aerogel ratio on the physical, mechanical, and thermal properties of the composite samples. The overall quantity of fiber used ranges from 4 g to 12 g, which is blended with approximately 1.25 L of DI water – to ensure full immersion of the fiber in the liquid – for about 20 seconds using a blender. An overhead liquid mixer is used to thoroughly mix the blended fiber with the required volume of aerogel precursor in a larger container, with additional D-I water gradually incorporated to complete a volume of approximately 3 L to ensure complete dispersion of the fiber. The volume of aerogel precursor used ranges from 35 mL to 300 mL, which, alongside the different amounts of fiber previously cited, provides the means to systematically obtain composite specimens featuring an ample spectrum of aerogel weight percentage (wt%). The obtained fiber/aerogel precursor mixture is processed by means of a centrifugal-pump-operated composite sample making unit to extract the water through a filtration-like technique, and the wet composite specimens are subsequently dried in a pre-heated oven at 60 °C for a period of 24–48 hours. In order to increase the water removal rate, couch sheets are used to both sides of the wet specimens during the drying process, which also helps preserve the integrity of the samples.2.4 Post-processing using freeze-drying (FD) methodology
A freeze drier (Labconco FreeZone Triad 7400) is used to fabricate the cryogels and the fiber-cryogel composites. The as-prepared water-glass precursor and the fiber-precursor mixture are pre-frozen to −240 °C in a freezer. Then, the samples are transferred into the freeze drier chamber. The pressure in the chamber is decreased by the vacuum pumps and maintained at 0.014–0.018 mBar during the entire drying process. The temperature profile during the freeze-drying process can be found in Fig. S9.† After freeze-drying, the samples are sintered in ambient pressure to remove organic residues.2.5 Physical, Structural, thermal, and mechanical properties characterization
For calculation of the bulk or tapped density of aerogel samples, the relationship between the mass of the sample and the volume containing such amount of sample is used, which can be represented by eqn (1) shown below:
 | (1) |
Skeletal density (ρs), on the other hand, is measured by using a pycnometry system (Micromeritics Accu-Pyc II 1340 Gas Pycnometer). The system determines density by means of the gas displacement method, which employs highly pressurized helium gas (as the measuring medium) to measure the volume of the solid matter contained inside the measuring chamber. Further to this, values of porosity are calculated from eqn (2):
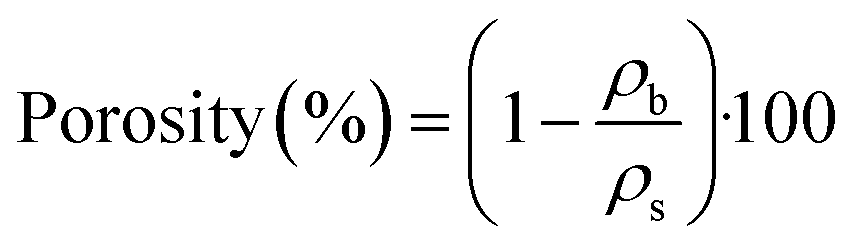 | (2) |
Through analysis of nitrogen sorption isotherms (adsorption/desorption), obtained from a Surface Area and Porosity Analyzer (Micromeritics TriStar II), specific surface area (SSA), pore size, and cumulative pore volume of aerogel/cryogel specimens are determined. Pre-treatment of previously sintered samples (to 600 °C) is performed by heating these to 250 °C during approximately 3 hours of degassing of a flowing gas used to remove any form of moisture, impurities, and contaminants. As next step, the degassed samples are cooled to cryogenic temperatures (−195 °C) under vacuum conditions, during which data in relation to the quantity of the absorbent gas adhering to the solid adsorbate for different values of relative pressure (P/Po) is collected. Calculation of the specific surface area (SSA) of the adsorbate is then performed from the data given by the adsorption isotherm plot at relative pressure (P/Po) from 0.003 to 0.3, which is based on the Brunauer–Emmett–Teller (BET) theory. The Barrett, Joyner, and Halenda (BJH) method, based on the Kelvin model of pore filling, is used for calculation of the pore size and pore volume of the samples, by analyzing the data from the desorption branch of the isotherm curve. The afore-mentioned test is also employed to establish nanoparticle size of the aerogel/cryogel samples.
An X-ray Diffraction System (XRD - Rigaku Ultima IV) is employed to investigate the structure of the silica aerogel/cryogel specimens. Copper is used as target material for single-crystal diffraction, with the X-ray detector rotating at an angle of 2θ from 5° to 80°, which are typical parameters used for powder patterns. In order to analyze the chemical bonding state of the aerogel/cryogel specimens, Fourier-transform infrared spectroscopy (FTIR – Agilent Cary 630 FTIR spectrometer) is used. This method is also utilized to investigate the interfacial bonding of aerogel-based composite samples, which could be key to discern relevant aspects in regards to their mechanical performance. The microstructure of aerogels, cryogels, and composite materials deriving from these are examined by Focused Ion Beam Scanning Electron Microscope (FIB-SEM, Carl Zeiss AURIGA CrossBeam).
Thermal conductivity of all specimens (aerogels, cryogels and composite materials) is determined through instruments using the steady-state methodology. For aerogel/cryogel samples, an in-house setup including a PHFS-01e Heat Flux Sensor from FluxTeq and complying with the ASTM C518 standard (related to Standard Test Methods for Steady-State Thermal Transmission Properties by means of a heat flow meter apparatus) is employed. After pertinent calibration of the system using commercial aerogels as reference, thermal conductivity of the specimens can be determined from the readings for temperature that are obtained from top and bottom plates, and once a steady heat flux through the sample has been achieved. The Heat Flow Meter – 100 series (HFM-100) Thermal Conductivity Measurement System from Thermtest is used for composite specimens. This system, which also complies with the ASTM C518 standard, requires rigorous calibration that can be accomplished by means of commercial products of the appropriate thickness and with a specified value of thermal conductivity. During testing of the composite materials between upper and lower plates within the testing chamber, the thermal conductivity of the specimen is calculated by the system once the heat flux between the plates, and through the sample, has converged to a constant value over time.
Thermogravimetric analysis and differential scanning calorimetry (TGA/DSC) tests are performed – using the TA Instrument DSC SDT Q600 – to evaluate the thermal stability of aerogel/cryogel specimens. Weight change (TGA) and true differential heat flow (DSC) of the samples, which are heat-treated in a nitrogen atmosphere (purge rate of 100 mL min−1) from room temperature (RT) to 800 °C at a rate of 20 °C min−1, are provided by the instrument.
Uniaxial compression tests of composite materials (both aerogel- and cryogel-based), with bulk dimensions of 15 mm × 15 mm and thickness in the region of 4–6 mm, are performed to characterize their mechanical properties. The equipment used is a universal test system (Model SSTM-20KN from United Testing Systems – Nominal capacity up to 20 kN or 4500 lbf) that is equipped with a sensitive load cell with a maximum capacity of 10 kgf (∼22 lbf), which is typically utilized for materials with significantly low yield strength.
3 Results and discussion
The schematic drawing shown in Fig. 1 includes a flowchart of the synthesis of both aerogel and fiber/aerogel composite materials that are obtained by means of the ambient-pressure-drying (referred as silica aerogel and fiber/silica aerogel composites going forward) and freeze-drying methods (referred as silica cryogel and fiber/silica cryogel composites going forward). It also highlights the difference between the surfactant molecules, in particular due to the dissimilar ionic head groups, while confirming the self-association and formation of spherical micelles - mixed micellization - once the critical micelle concentration (CMC) has been reached. Following the addition of ion-exchange waterglass, part of the silica source comes together in the inner core of the micelles due to its hydrophobicity associated to a reasonable lack of defined polarity, while the rest remains as suspended silica particles in the solution. Incorporation of an acid catalyst (HCl(aq); 36.5–38.0%) is expected to promote the rapid transformation of the precursor components into different types of silanol groups during hydrolysis, while eventually triggering the aggregation of silica particles into clusters to initiate the silica network structure that occurs during the solvent condensation stage.26 Such clustering of particles, alongside the attachment of the micelles to the formed chains, results in the development of a reinforced network structure. Drying of this pre-aerogel solution (also referred as wet gel) is performed through both FD and APD methods to obtain the highly porous specimens as shown in Fig. 1. Composite materials involve the rigorous mix of the wet gel with fiber to different composition ratios, with the representative resulting specimens from both FD and APD processes shown in Fig. 1.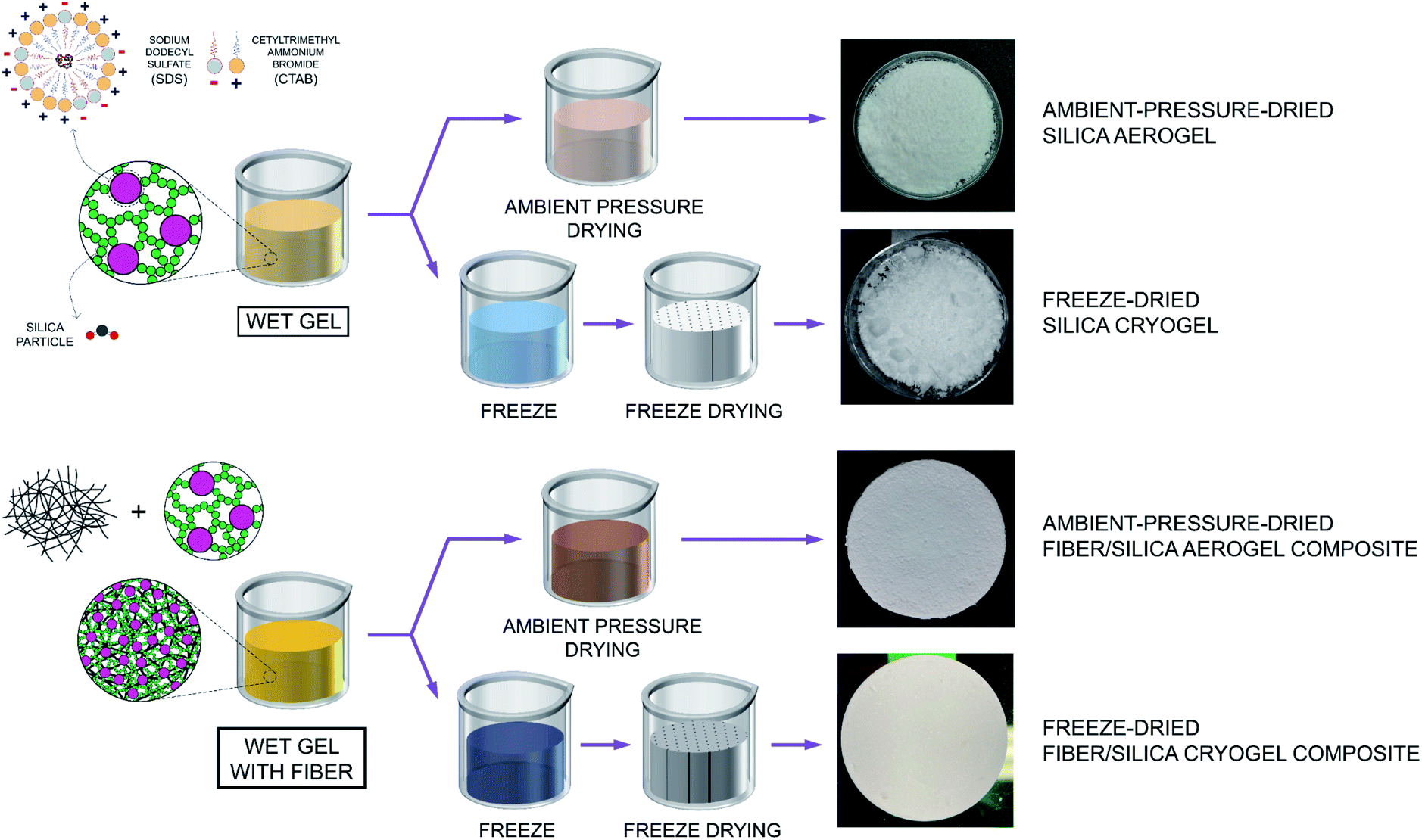 | ||
| Fig. 1 Schematic illustration of the processes used for the fabrication of both ambient-pressure-drying aerogels and freeze-drying cryogels, as well as composite materials. | ||
Aerogels and cryogels obtained by means of an ion-exchanged waterglass silica source would typically feature a uniform, dense microstructure in the nanoscale range. The extent of the resulting three-dimensional network structure that can be observed from the scanning electron microscopy (SEM) images included in Fig. 2a and b could vary, with the ambient-pressure-dried material comprising a highly ordered, intricate microstructure when compared to a layered-type configuration that is more noticeable from the freeze-dried specimen – a microstructural arrangement that can be further confirmed from Fig. S1a and b† respectively. This could be a direct consequence of the crystallization phenomena that takes place during the rather accelerated freezing rate of the solvent present in the wet gel precursor that is subject to the FD methodology. It is worth noticing that, even though the FD process effectively avoids the capillary pressure that builds up within the pore that is typical of the APD method, it would nevertheless tend to damage the three-dimensional network that was formed during condensation of the aerogel precursor. Hence, the implementation of the FD process would result in the transformation of the network structure to a certain extent once the sublimation process has completed.
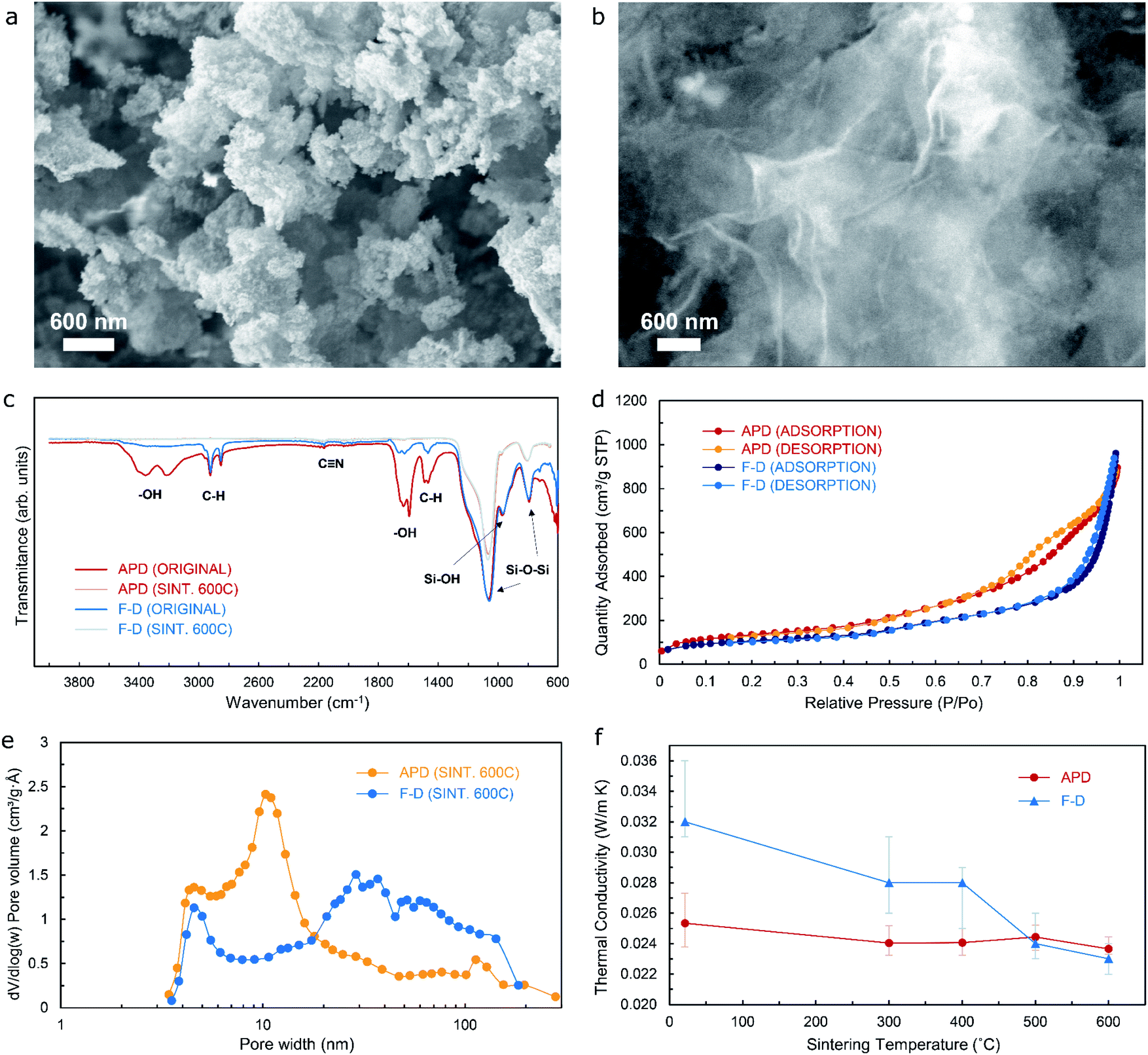 | ||
| Fig. 2 (a) and (b) SEM images showing typical microstructure of silica aerogels (from APD) and silica cryogels (from FD) following the sintering process; (c) Fourier transform infrared (FTIR) spectroscopy for silica aerogels and silica cryogels (before and after the sintering treatment); (d) isotherm curves from BET/BJH tests for APD and FD specimens after sintering to 600 °C; (e) pore volume plot from BET/BJH tests for APD and FD specimens after sintering to 600 °C, and (f) thermal conductivity vs. sintering temperature for both silica aerogels and silica cryogels. | ||
Fourier Transform Infrared (FTIR) spectra performed on the two materials being analyzed are shown in Fig. 2c (for both non-sintered and sintered-to-600 °C specimens). The rationale behind selecting 600 °C as the optimal temperature for sintering of both aerogels and cryogels has been thoroughly explained in the Materials and Methods section. The peaks given by the functional groups present in the materials would provide valuable information in order to determine the structural changes. Sharp peaks at 1066 cm−1 and 793 cm−1 – for both non-sintered and sintered specimens – are the result of antisymmetric and symmetric stretching vibrations of Si–O–Si bonds typical of siliceous materials, hence indicating the presence of SiO2. An adjacent peak at approximately 970 cm−1 can be associated to rocking bending vibrations of Si–OH groups, that usually present as an adsorption, single broad band that is typical of unmodified waterglass-based organosilicon compounds,17 which is less noticeable after the sintering heat treatment. A double-peak configuration at 3355/3220 cm−1 and 1630/1591 cm−1 corresponds to the pronounced hydrogen bonds between urea and water on stretching and bending vibration of –OH functional groups respectively, which can be attributed to the deformation vibrations of the adsorbed urea water solution molecules. A higher percentage of transmittance by the freeze-dried specimen in the non-sintered condition when compared to the ambient-pressure-dried material in the non-sintered condition suggests that a much higher proportion of water molecules are effectively removed through the FD methodology. Sintered samples show virtually no peaks due to stretching and bending vibration of –OH functional groups, which means that all absorbed water molecules have effectively been removed following the heat treatment. Some additional peaks can be seen in the spectra for samples before sintering, namely 2920/2855 cm−1 that are due to the C–H stretching vibration, 1460 cm−1 that corresponds to the C–H bending vibration, and a weak but clear peak at 2160 cm−1 that could possibly be attributed to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds (both C and N as ions resulting from the breakdown of carbon dioxide and ammonia that formed as by-products from the reaction of urea with water, sodium silicate, and hydrochloric acid during hydrolysis), although it is much weaker compared with the other stretching modes. The above-mentioned results suggest that the organic template can be removed by means of the sintering treatment, whilst the mesoporous silica skeleton can be successfully maintained to a high extent. The presence of organic residues and the various by-products (namely salt by-products, surfactant ions, carbon dioxide, and ammonia) within the pores of the aerogel could be detrimental to its overall performance as superinsulation material, hence timely washing of the precursor and subsequent sintering of the aerogel become critical steps prior to its use in any post-processing procedure.
N bonds (both C and N as ions resulting from the breakdown of carbon dioxide and ammonia that formed as by-products from the reaction of urea with water, sodium silicate, and hydrochloric acid during hydrolysis), although it is much weaker compared with the other stretching modes. The above-mentioned results suggest that the organic template can be removed by means of the sintering treatment, whilst the mesoporous silica skeleton can be successfully maintained to a high extent. The presence of organic residues and the various by-products (namely salt by-products, surfactant ions, carbon dioxide, and ammonia) within the pores of the aerogel could be detrimental to its overall performance as superinsulation material, hence timely washing of the precursor and subsequent sintering of the aerogel become critical steps prior to its use in any post-processing procedure.
From the analysis of Fig. 2e, two shallow peaks can be seen in the pore size distribution curve for the freeze-dried material (at around 5 and 30 nm), while a much prominent peak is evident in the pore size distribution curve for the ambient-pressure-dried material at approximately 11 nm. Hence, the freeze-dried specimen would have pores of different sizes within the range of 3.5–185 nm, which is confirmed by the results shown in Table 1 with an average pore width obtained from the BET/BJH test of 16.3 nm, whilst the resulting average pore width for the ambient-pressure-dried material is 10.1 nm respectively – in good agreement with the pore size distribution curve presented above. These results indicate that the materials are of the mesoporous type – which is also confirmed by the seemingly type IV isotherms with H3 type hysteresis loop from Fig. 2d. Additionally, the more uniformly distributed pore size across the mesoporous range for the freeze-dried sample would yield lower values of thermal conductivity as the sintering temperature increases and the organic matter is eventually removed due to the effects of the heat treatment, as suggested by the relationship between thermal conductivity versus sintering temperature for both silica aerogels and silica cryogels shown in Fig. 2f. It can be noticed that, although the trend of a decrease of the thermal conductivity as the sintering temperature increases occurs for both materials, the silica cryogel yields a larger improvement (from 32 mW m−1 K−1 for the non-sintered material to 23 mW m−1 K−1 for the specimen that has been sintered to 600 °C) when compared to the silica aerogel (from 25.3 mW m−1 K−1 for the non-sintered material to 23.6 mW m−1 K−1 for the specimen that has been sintered to 600 °C), which can be attributed to the slightly different pore size distribution for these as a consequence of the dissimilar procedure taking place for the removal of solvent from the porous network. Fig. S2† shows optical images of the freeze-dried material as the sintering temperature increases, showcasing a noticeable change in colour of the specimen as the organic matter – that has remained after the synthesis process – is removed, with the same phenomena occurring for the ambient-pressure-dried material.
| Drying method | Freeze-drying | Ambient pressure drying |
|---|---|---|
| Drying temperature | −20 °C | 60 °C |
| Thermal conductivity (W m−1 K−1) | 0.0230 | 0.0236 |
| BET surface area (m2 g−1) | 369.4 | 473.8 |
| Average pore width (nm) | 16.3 | 10.1 |
| Average particle size (nm) | 16.2 | 12.7 |
| Porosity (%) | 96.7 | 97.4 |
| Bulk density (g cm−3) | 0.102 | 0.063 |
| Skeletal density (g cm−3) | 3.092 | 2.468 |
Thermal stability of both silica cryogels and silica aerogels were investigated by means of TGA/DSC analysis performed from room temperature (RT) to 800 °C. In all cases, a consistent pattern could be observed, with weight % versus temperature curves shown in Fig. S3† indicating that initial weight loss occurs at around 70 °C due to removal of water molecules that are still present in the specimens. This is followed by a sharp weight loss that begins at around 160 °C and that continues until approximately 550 °C for both samples, which could be attributed to the thermal degradation of organic matter existing in the specimens. Thermal degradation of both specimens beyond 550 °C is almost negligible, with the resulting weight loss totalling approximately 35% and 55% of their initial mass (for the silica cryogel and the silica aerogel respectively) once the temperature has reached 800 °C. This discrepancy in weight loss could be associated to the dissimilar rate of removal of organic matter, which is present as by-products resulting from the reaction of the surfactants and urea with other compounds in the precursor during hydrolysis and solvent condensation stages, that takes place during the application of the different drying processes. From the X-ray diffraction (XRD) spectra obtained – and shown in Fig. S4,† only a rather broad peak at 2θ approximately equal to 22° is observed in the materials, which is typical of their amorphous structure and directly related to the bonding angle of the Si–O–Si siloxane functional groups27 that form the backbone of the silica aerogels. These results emphasize that the drying method used for the manufacturing of the silica-based materials has no determinant effect on their inherent structure.
Incorporating fibers with the wet-gel precursor represents one of the promising strategies to produce composite materials that overcome the brittleness and fragility of aerogel specimens. This technique primarily introduces some degree of elasticity to the resulting composites, while it also offers the possibility of improving the thermal performance of fiber-based products as a direct result of the aerogel precursor being used during the production of the composite material.25 In this work, both cellulose nanofiber and glass (acid resistant borosilicate) microfiber (referred as ceramic-fiber for simplicity) will be used. Through the incorporation of optimal quantities of aerogel precursor, an advanced cross-linking scheme between fiber and aerogel can be achieved as illustrated in the SEM images included in Fig. 3a and b, which is evidenced by an abundant presence of silica aerogel/cryogel fully adhered to cellulose fiber threads for specimens from both APD and FD processes. Fig. S12,† on the other hand, shows SEM images of a cellulose fiber specimen where no silica precursor has been used during its synthesis, hence highlighting the difference between the fiber specimen and the composite materials shown in Fig. 3a and b.
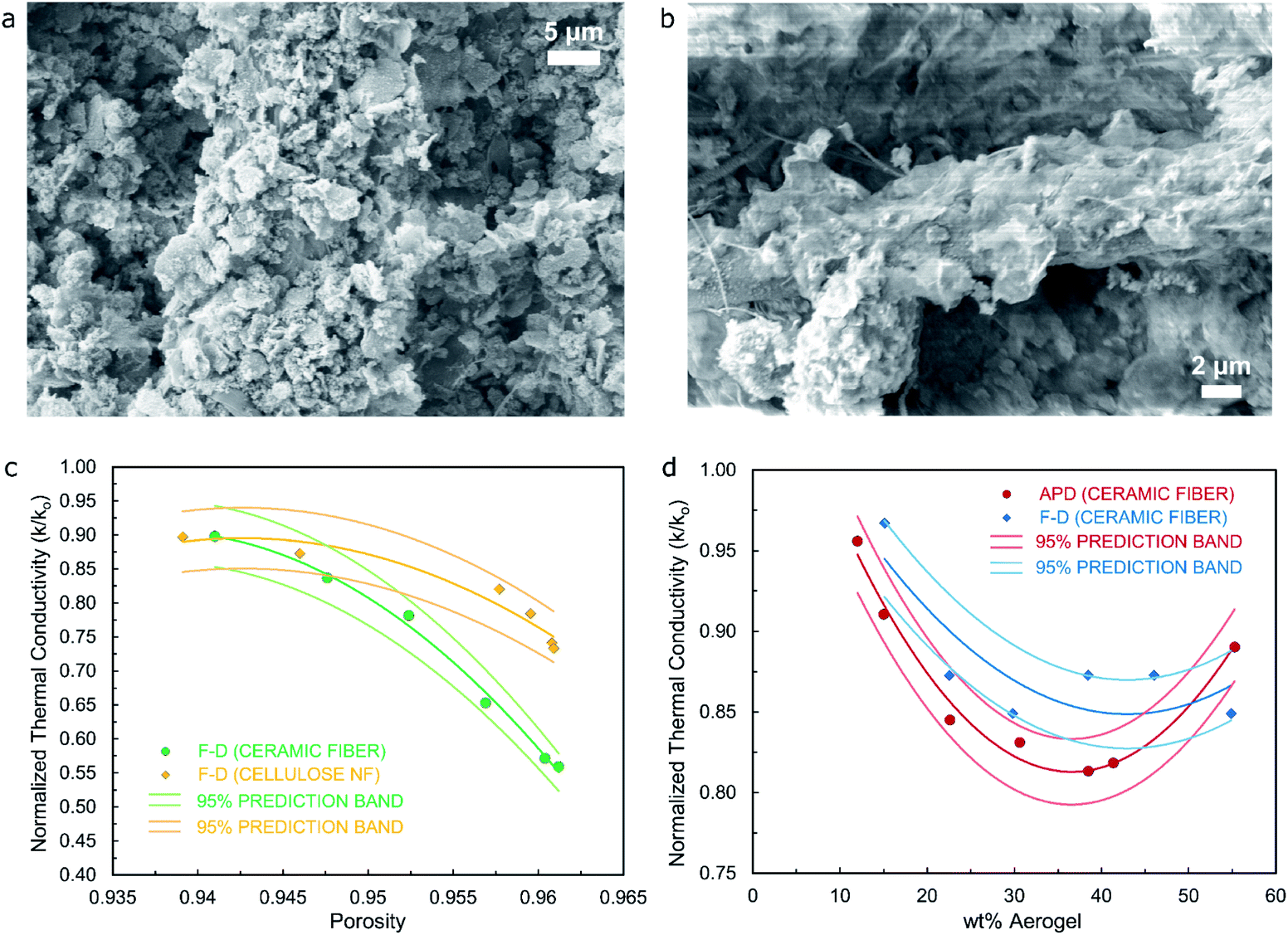 | ||
| Fig. 3 (a) and (b) SEM images showing typical microstructure of cellulose-fiber/silica aerogel and cellulose-fiber/silica cryogel specimens from APD and FD processes respectively; (c) normalized thermal conductivity vs. porosity for fiber/silica cryogel materials using both ceramic-fiber and cellulose nanofiber; and (d) normalized thermal conductivity vs. wt% or aerogel for both fiber/silica aerogel and fiber/silica cryogel specimens using ceramic-fiber. | ||
Fig. 3c correlates the value of normalized thermal conductivity (k/ko) with the value of porosity for composite materials (fiber/silica cryogel) from the freeze-drying process, where k corresponds to the value of thermal conductivity of the composite material and ko is the value of thermal conductivity of the control sample produced using only fiber. Values of porosity have been obtained by means of pycnometry tests, with bulk and skeletal densities determined for each of the specimens that are then used in eqn (2) (as defined in the Materials and Methods section) accordingly. Results show that as the porosity in the composite material increases, the value of normalized thermal conductivity decreases for both types of specimens (ceramic-fiber- and cellulose-nanofiber-based), being this a reasonable trend since the higher the porosity of the material, the more significant the gas state of the thermal conductivity would be expected to become with respect to the solid state as the secondary main component when breaking down the constituents defining this critical property. It should be noted that all values of normalized thermal conductivity are below the value of 1.0, hence showing that in all instances the silica cryogel introduces an improvement in the thermal performance of the material, indistinctively of the fiber used. Additionally, Fig. S5† confirms that as the wt% of aerogel used for the synthesis of the ceramic-fiber-based composite materials obtained from the freeze-dried process increases, their porosity would also increase while the value of thermal conductivity would decrease accordingly, with the majority of the pore sizes in the specimen seemingly being less than the mean free path of air molecules to accomplish this trend.
Fig. 3d, on the other hand, correlates the value of normalized thermal conductivity (k/ko) with the value of wt% of aerogel used for composite materials (fiber/silica cryogel and fiber/silica aerogel) from FD and APD processes, which includes the use of ceramic-fiber in order to assess its effect on the thermal performance of the specimens. Results show that as the wt% of the aerogel in the composite material increases, the value of normalized thermal conductivity decreases for both types of specimens (freeze-dried and ambient-pressure-dried) up to a minimum value to then increase slightly as the wt% of aerogel reaches 55% approximately. The lowest value of normalized thermal conductivity is in the order of 0.80 for the APD material, whilst the FD material shows a minimum value close to 0.85, results that are further confirmed by the plot shown in Fig. S6† with values of thermal conductivity for the specimens obtained from the APD process being regularly lower (lowest value of thermal conductivity of 31.8 mW m−1 K−1 at approximately 39 wt% of aerogel) than those for the materials obtained from the FD method (36 mW m−1 K−1 at approximately 30 wt% of aerogel). This trend is in full agreement with the average pore width reported in Table 1 for silica aerogel/cryogel specimens from both drying processes (10.1 nm for the APD material versus 16.3 nm for the FD material), hence confirming the crucial effect of the silica aerogel/cryogel on the thermal performance of the resulting composite materials. Moreover, Fig. S10† highlights the flame-retardant capabilities of the ceramic-fiber/silica cryogel composite materials.
Fig. 4a and b include SEM images of the typical microstructure of composite (ceramic-fiber/silica aerogel) specimens from the ambient-pressure-drying (APD) process, while Fig. 4d and e show the microstructure representative of composite (ceramic-fiber/silica cryogel) specimens from the freeze-drying (FD) methodology. In all cases, the main image refers to the top surface of the sample, with inserts showcasing the cross-section area of the materials. Although specimens shown in Fig. 4a and b feature a different weight% of aerogel (approximately 14% and 55% respectively), there exists perceptible similarities between the microstructure from their surface, which reveal a moderate extent of fiber–aerogel interaction during cross-linking of the components throughout the drying stage. The microstructure that can be seen from the cross-section images, nevertheless, clearly demonstrates that there is a difference in the contents of aerogel precursor during production of the materials, leading to an evident optical difference in the density observed for each of them. In regards to the materials produced from the FD process, a more consistent pattern can be seen from the main and insert images in Fig. 4d and e (including SEM images from specimens with approximately 10 and 50 wt% of cryogel respectively), with the microstructures accurately representing the difference in the composition ratio of the samples. For the material with the highest contents of cryogel, on the other hand, a noticeable extent of fiber–silica cryogel interaction can be observed from the SEM image of the specimen surface, whilst its cross-section shows occasional voids and air pockets. This trend, that was also present in the materials produced from APD, could be attributed to a layer-upon-layer fiber/aerogel arrangement that is typically achieved during the production of the specimens and that develops throughout the drying stage.
 | ||
| Fig. 4 (a) and (b) SEM images showing typical microstructure of composite ceramic-fiber/silica aerogel specimens from the ambient-pressure-drying (APD) process with different aerogel contents (14 and 55 wt% respectively); (c) stress–strain curve from uniaxial compression tests on specimens from ambient-pressure-drying (APD) and freeze-dried (FD) processes including an approximate 20 wt% aerogel/cryogel; (d) and (e) SEM images showing typical microstructure of composite ceramic-fiber/silica cryogel specimens from the freeze-drying (FD) process with different cryogel contents (10 and 50 wt% respectively); and (f) plot relating wt% aerogel with Young's modulus for both APD and FD composite materials featuring different aerogel contents. | ||
Uniaxial compression tests were performed to characterize mechanical performance, with stress–strain curves for composite samples (ceramic-fiber/silica aerogel and ceramic-fiber/silica cryogel) obtained from both APD and FD processes shown in Fig. S7 and S8† respectively. In order to more effectively acquire meaningful results from the data obtained after the tests, any readings related to both slack and alignment/seating of the sample (which is not a true response of the sample, but an artifact of its geometry) has been disregarded by including a “toe compensation” portion of the curve that would help us obtain more accurate values of strain. The calculation of the elastic or Young's modulus has been performed as the slope of the best linear fit over the region of the stress–strain curve up to 10% strain, hence ensuring that the sample is still in the elastic regime. Fig. 4c presents stress–strain curves from uniaxial compression tests for composite materials (ceramic-fiber-based) obtained from both APD and FD processes – both including approximately 20 wt% aerogel, which, irrespective of a rather different performance at low strain, clearly highlight marked similarities in the overall mechanical properties under compression loads (analogous elastic modulus – 353.3 and 367.9 kPa respectively, and maximum compressive stress at 40% strain that are in close agreement at around 175 kPa).
Furthermore, Fig. 4f provides the relationship between wt% of aerogel and Young's modulus for both APD and FD composite materials featuring different aerogel/cryogel contents. The plot shows a clear and well-defined diverse trend between the materials being analyzed, with the Young's modulus for the APD specimens increasing as the wt% of aerogel increases, while the tendency for the Young's modulus of the FD specimen would tend to decrease as the wt% of cryogel increases. This phenomenon can be attributed to a different architecture of the fiber-silica aerogel/fiber-silica cryogel network that results from the implementation of either APD or FD processes during the drying stage of the composite specimens. As the weight% of aerogel in the composite material obtained from the FD procedure increases, and given that the flake-shape particles that arise from the freeze-drying of the wet gel precursor (as evidenced by Fig. S1b†) would actually have weak bonding (possibly comparable to random loose packing for non-spherical grains) to both other particles and the fiber, the increase in the compression load would translate into a considerable collapse of both the silica and silica-fiber network structure due to a high incidence of particle detachment taking place, hence resulting in lower values of elastic modulus. Since a more typical spherical-shape particles are present in ambient-pressure-dried materials, as the wt% of aerogel increases – and the compression load imposed on the APD composite material also increases, the elastic modulus and the overall mechanical strength will improve as the silica network gets compressed and the packing of particles develop into a random-close-type arrangement. Notwithstanding, each process has its own advantages and critical features, which unquestionably open up a vast range of thermal insulation applications during the production of silica-aerogel-based products.
4 Conclusions
Freeze-drying is an effective manufacturing approach during the production of aerogels and composite materials that derive from these. Through the FD method, the detrimental effect that the capillary pressure exerts on the nanopores of the gel network is avoided, fundamentally because of the absence of the phase boundary between the liquid and the gas phase that typically occurs during the APD method. The resulting silica cryogels (waterglass-based) obtained from the FD approach typically feature a higher density (0.102 g cm−3 as bulk density and 3.092 g cm−3 as skeletal density), a lower specific surface area (369.4 m2 g−1), but a much lower shrinkage rate than the silica aerogels produced by means of the ambient-pressure-drying method. These materials produced via the FD process, in addition, present a more uniform 3D solid network structure due to freezing of the existing solvent within the pores and subsequent removal by sublimation that takes place. Composite materials obtained by means of this drying method, on the other hand, show a valuable combination of considerably low thermal conductivity and improved mechanical properties (28.0 mW m−1 K−1, ∼620 kPa maximum compressive stress and Young's modulus of 715 kPa in compression at 40% strain respectively) that results in an advantageous thermal/mechanical performance relationship, as well as favourable flame-retardant capabilities.Utilization of the APD method, nonetheless, can produce composite materials with rather similar properties (28.8 mW m−1 K−1, ∼200 kPa maximum compressive stress and Young's modulus of 612 kPa in compression at 40% strain), and aerogels with even slightly better thermal performance and physicochemical properties (0.063 g cm−3 as bulk density and 2.468 g cm−3 as skeletal density, as well as specific surface area of 473.8 m2 g−1) than those obtained from the FD process, with the additional advantage of being significantly cost-effective. Given all these benefits, both the FD and the APD methods can thus be considered as plausible, genuine drying alternatives in the development of thermal-performance-enhanced products, providing a promising pathway towards energy-efficiency technologies for applications in thermal management.
Author contributions
All authors discussed the results and commented on the manuscript. M. D., Z. G., and S. R. designed all experiments. M. D. and Z. G. prepared the materials and conducted experiments regarding mechanical and thermal properties, as well as structural and physicochemical characterization. J. A. supervised the mechanical properties study. S. R. conceived the project and directed the research. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The aerogel insulation work at University at Buffalo (S.R.) is supported by the U.S. Department of Energy’s Office of Energy Efficiency and Renewable Energy (EERE) under the Building Technology Office (BTO) Award Number DE-EE0008675. The additive manufacturing related work at University at Buffalo (C. Z) is supported by the NSF with the award no. CMMI-1846863.References
- B. M. Gauthier, S. D. Bakrania, A. M. Anderson and M. K. Carroll, A fast supercritical extraction technique for aerogel fabrication, J. Non-Cryst. Solids, 2004, 350, 238–243 CrossRef CAS.
- J.-H. Kim, J.-H. Ahn, J.-D. Kim, D.-H. Lee, S.-K. Kim and J.-M. Lee, Influence of Silica-Aerogel on Mechanical Characteristics of Polyurethane-Based Composites: Thermal Conductivity and Strength, Materials, 2021, 14(7), 1790 CrossRef CAS PubMed.
- J. Rouquerol, D. Avnir, C. Fairbridge, D. Everett, J. Haynes, N. Pernicone, J. Ramsay, K. Sing and K. Unger, Recommendations for the characterization of porous solids (Technical Report), Pure Appl. Chem., 1994, 66(8), 1739–1758 CrossRef CAS.
- A. S. Dorcheh and M. Abbasi, Silica aerogel; synthesis, properties and characterization, J. Mater. Process. Technol., 2008, 199(1–3), 10–26 CrossRef.
- M. de Fátima Júlio, A. Soares, L. M. Ilharco, I. Flores-Colen and J. de Brito, Silica-based aerogels as aggregates for cement-based thermal renders, Cem. Concr. Compos., 2016, 72, 309–318 CrossRef.
- P. C. Thapliyal and K. Singh, Aerogels as promising thermal insulating materials: An overview, J. Mater, 2014, 2014(1), 1–10 Search PubMed.
- C. Garnier, T. Muneer and L. McCauley, Super insulated aerogel windows: Impact on daylighting and thermal performance, Building and Environment, 2015, 94, 231–238 CrossRef.
- U. Berardi, The development of a monolithic aerogel glazed window for an energy retrofitting project, Appl. Energy, 2015, 154, 603–615 CrossRef.
- R. Gellert, Inorganic mineral materials for insulation in buildings, in Materials for Energy Efficiency and Thermal Comfort in Buildings, Elsevier, 2010, pp. 193–228 Search PubMed.
- N. Minju, B. N. Nair and S. Savithri, Sodium silicate-derived aerogels: effect of processing parameters on their applications, RSC Adv., 2021, 11(25), 15301–15322 RSC.
- L. Keshavarz, M. R. Ghaani and N. J. English, The Importance of Precursors and Modification Groups of Aerogels in CO2 Capture, Molecules, 2021, 26(16), 5023 CrossRef CAS PubMed.
- J. L. Gurav, I.-K. Jung, H.-H. Park, E. S. Kang and D. Y. Nadargi, Silica aerogel: synthesis and applications, J. Nanomater., 2010, 2010(1), 1–11 CrossRef.
- Y. Pan, S. He, L. Gong, X. Cheng, C. Li, Z. Li, Z. Liu and H. Zhang, Low thermal-conductivity and high thermal stable silica aerogel based on MTMS/Water-glass co-precursor prepared by freeze drying, Mater. Des., 2017, 113, 246–253 CrossRef CAS.
- L. E. Nita, A. Ghilan, A. G. Rusu, I. Neamtu and A. P. Chiriac, New trends in bio-based aerogels, Pharmaceutics, 2020, 12(5), 449 CrossRef CAS PubMed.
- P. B. Sarawade, G. N. Shao, D. V. Quang and H. T. Kim, Effect of various structure directing agents on the physicochemical properties of the silica aerogels prepared at an ambient pressure, Appl. Surf. Sci., 2013, 287, 84–90 CrossRef CAS.
- S. Iswar, S. Galmarini, L. Bonanomi, J. Wernery, E. Roumeli, S. Nimalshantha, A. M. B. Ishai, M. Lattuada, M. M. Koebel and W. J. Malfait, Dense and strong, but superinsulating silica aerogel, Acta Mater., 2021, 213, 116959 CrossRef CAS.
- Z. Shao, F. Luo, X. Cheng and Y. Zhang, Superhydrophobic sodium silicate based silica aerogel prepared by ambient pressure drying, Mater. Chem. Phys., 2013, 141(1), 570–575 CrossRef CAS.
- U. K. Bangi, I.-K. Jung, C.-S. Park, S. Baek and H.-H. Park, Optically transparent silica aerogels based on sodium silicate by a two step sol–gel process and ambient pressure drying, Solid State Sci., 2013, 18, 50–57 CrossRef CAS.
- S. He, D. Huang, H. Bi, Z. Li, H. Yang and X. Cheng, Synthesis and characterization of silica aerogels dried under ambient pressure bed on water glass, J. Non-Cryst. Solids, 2015, 410, 58–64 CrossRef CAS.
- S. De Pooter, S. Latré, F. Desplentere and D. Seveno, Optimized synthesis of ambient pressure dried thermal insulating silica aerogel powder from non-ion exchanged water glass, J. Non-Cryst. Solids, 2018, 499, 217–226 CrossRef CAS.
- K. Kanamori, M. Aizawa, K. Nakanishi and T. Hanada, New transparent methylsilsesquioxane aerogels and xerogels with improved mechanical properties, Adv. Mater., 2007, 19(12), 1589–1593 CrossRef CAS.
- M. V. Khedkar, S. B. Somvanshi, A. V. Humbe and K. Jadhav, Surface modified sodium silicate based superhydrophobic silica aerogels prepared via ambient pressure drying process, J. Non-Cryst. Solids, 2019, 511, 140–146 CrossRef CAS.
- Y. Huang, S. He, M. Feng, H. Dai, Y. Pan and X. Cheng, Organic solvent-saving preparation of water glass based aerogel granules under ambient pressure drying, J. Non-Cryst. Solids, 2019, 521, 119507 CrossRef CAS.
- T. Y. Wei, T. F. Chang, S. Y. Lu and Y. C. Chang, Preparation of monolithic silica aerogel of low thermal conductivity by ambient pressure drying, J. Am. Ceram. Soc., 2007, 90(7), 2003–2007 CrossRef CAS.
- M. Di Luigi, L. An, J. Armstrong and S. Ren, Scalable and robust silica aerogel materials from ambient pressure drying, Mater. Adv., 2022, 3(6), 2726–2736 RSC.
- S. Dervin and S. C. Pillai, An introduction to sol-gel processing for aerogels. in Sol-Gel Materials for Energy, Environment and Electronic Applications, Springer, 2017; pp. 1–22 Search PubMed.
- L. Wa, L. Fengyun, Z. Fanlu, C. Mengjing, C. Qiang, H. Jue, Z. Weijun and M. Mingwei, Preparation of silica aerogels using CTAB/SDS as template and their efficient adsorption, Appl. Surf. Sci., 2015, 353, 1031–1036 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of freeze-drying method, microstructure, thermal conductivity, porosity, surface area, pore width, average particle size, and structural characterization of aerogel and cryogel specimens; in addition to mechanical properties of aerogel- and cryogel-based composite specimens. See https://doi.org/10.1039/d2ra03325a |
| This journal is © The Royal Society of Chemistry 2022 |