
Carbonic anhydrase/formate dehydrogenase bienzymatic system for CO2 capture, utilization and storage
Ryohei
Sato
a and
Yutaka
Amao
 *ab
*ab
aGraduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
bResearch Centre for Artificial Photosynthesis (ReCAP), Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@osaka-cu.ac.jp
First published on 9th October 2021
Abstract
In order to establish carbon capture, utilization, and storage (CCUS) technology, we focused on a system consisting of two different biocatalysts (formate dehydrogenase from Candida boidinii; CbFDH and carbonic anhydrase from bovine erythrocytes; CA). CA catalyses the interconversion between CO2/water and dissociated bicarbonate ions/protons. CbFDH is a NAD+-dependent dehydrogenase that catalyzes CO2 reduction to formate by using the NAD+/NADH redox couple. The construction of a bienzymatic system consisting of CA and CbFDH (CA/CbFDH system) for a CCUS system was attempted. At 150 or 200 μM CA in the sample solution and a controlled pH of 6.3–6.5 by CO2 bubbling, due to the promotion of the conversion of CO2 to bicarbonate, the reaction rate for CbFDH-catalyzed CO2 reduction to formate decreased to about 50% as compared with that in the absence of CA. In the higher pH region (>9.5), despite the low CO2 concentration in this region, in contrast, it was found that the addition of CA promoted the reduction of CO2 catalyzed by CbFDH to formate to about 7 times higher than that under the conditions without CA. This shows that a CCUS system was constructed in which the conversion of bicarbonate to CO2 using CA and the reduction of CO2 to formate using CbFDH were coordinated.
Introduction
The carbon capture and storage (CCS) technique is the process of capturing CO2 before it enters the atmosphere, transporting it, and storing it for a long period.1–5 For the carbon storage technique, various approaches have been suggested for permanent storage including gaseous storage in deep geological formations.6–10 It is possible to physically eliminate CO2 on a large scale by using gaseous storage in deep geological formations. On the other hand, no productivity can be found with this method as it merely physically isolates CO2. Therefore, in recent years, the carbon capture, utilization, and storage (CCUS) technique, which is a combination of CCS technology and carbon capture and utilization (CCU) technology, has attracted a great deal of attention.11 The CCU technique is the process of capturing CO2 to be recycled for further usage. Various CCU techniques offer a response to the global challenge of significantly eliminating greenhouse gas emissions from major stationary emitters. The CCU techniques aim to convert captured CO2 into more valuable substances or products while maintaining the carbon neutrality of the production processes.12–20 Captured CO2 is converted to hydrocarbons such as methanol to use as biofuels and alternative and renewable sources of energy. Since the CCU technique immediately converts captured CO2 into other molecules, a method related to CO2 storage is required.21 As one candidate for the CO2 storage technique, there is a method of storing CO2 by conversion into bicarbonate or carbonate in an aqueous medium. Chemical absorption methods for CO2 capture and storage refer to the use of liquid alkaline solutions.22–30 CO2 can be efficiently captured by using a liquid alkaline solution, but energy is required to release the stored CO2 in the solution. For these reasons, a system that can easily convert CO2 to bicarbonate or carbonate is attractive for application to the CCUS technique. Among CCUS technologies, a CO2 conversion system using various enzymes is one of the promising candidates.31 Carbonic anhydrases (CAs) catalyse the interconversion between CO2/water and dissociated bicarbonate ions/protons as shown in Fig. 1.32–39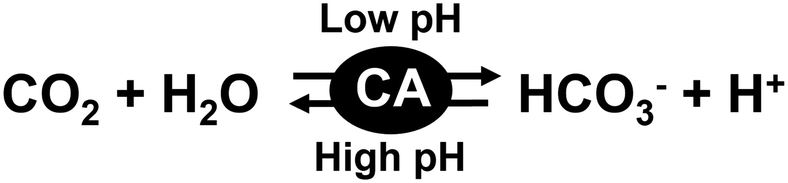 | ||
| Fig. 1 CA catalyzes the interconversion between CO2/water and bicarbonate/protons. | ||
By using CA, it is possible to capture CO2 and store it in an aqueous medium as bicarbonate. It is also possible to convert bicarbonate into CO2 with CA by increasing the pH of the aqueous medium. In other words, CA can be used to cover the capture, storage and utilization of CO2. Some CAs can withstand temperatures up to 107 °C and alkalinity of more than pH 10 (4.2 M N-methyldiethanolamine).40 It was reported that a pilot run with a more stable CA on a flue stream that consisted of 12–13% mol composition CO2 had a capture of 63.6% over a 60 h period with no noticeable effects on biocatalytic performance.40 Among various CAs, bovine erythrocyte-derived CA is commercially available and is easy to handle with various CCUS technologies. The bovine erythrocyte-derived CA has good catalytic activity between pH 4.5 and 9.5, with an optimal pH of 7.5.41 Therefore, it is expected that CA will be used for CCUS technology using an aqueous medium in a wide pH range.
On the other hand, candidates for biocatalysts involved in CO2 utilization include carbon monoxide dehydrogenase (CODH),42–45 formate dehydrogenase (FDH),46–52 and malic enzyme (ME).53–59 In particular, FDH from Candida boidinii (CbFDH) is a commercially available biocatalyst and is easy to handle with CO2 utilization technology. CbFDH is a NAD+-dependent dehydrogenase that catalyzes the interconversion between formate and CO2via the NAD+/NADH redox couple as shown in Fig. 2.
 | ||
| Fig. 2 CbFDH catalyzes the interconversion between CO2 and formate using the NAD+/NADH redox couple. | ||
The effect of pH on the stability of CbFDH has also been investigated and it was found that CbFDH has good catalytic activity between pH 5.5 and 11.0 for formate oxidation to CO2 in the presence of NAD+.60 In other words, CbFDH can be used as a catalyst in a wide range of pH values. The optimum pH for CbFDH-catalyzed reduction of CO2 to formate has been reported to be 6.0.61 CbFDH is also widely used as a catalyst for the reduction of CO2 to formate using a visible-light driven redox system consisting of an electron donor, a photosensitizer and an electron mediator.61–77 Most of the reported systems of CO2 reduction to formate catalyzed by CbFDH are carried out in neutral buffer (pH 7.0). There are several reports claiming that bicarbonate is directly reduced to formate by the catalytic function of CbFDH. It was not clear whether CO2, bicarbonate, or carbonate was reduced to formate with CbFDH. CO2, bicarbonate and carbonate are mixed in the reaction solution at a wide pH range. We have previously investigated the formate production by CbFDH in solution with different ratios of CO2, bicarbonate and carbonate. As a result, we found that CbFDH catalytically reduces only CO2 to formate among the three different carbonate species.78 In addition, it also has been reported that Mo/W-dependent FDH from Desulfovibrio vulgaris also directly reduces CO2 to formate rather than bicarbonate by an electrochemical method.79 For example, in a buffer solution of pH 7.0, the abundance ratios of CO2, bicarbonate and carbonate are estimated to be approximately 0.113, 0.885 and 0.002 by using the equation of Plummer and Busenberg.80–82 In order to increase the concentration of CO2, thus, it is necessary to lower the pH of the reaction solution. However, although the concentration of CO2 increases in the buffer solution in the low pH region, it immediately moves to the gas phase as CO2 gas. In other words, a system for CO2 reduction to formate by CbFDH can be constructed in the neutral pH region by capturing and storing CO2 as bicarbonate in an aqueous medium using CA and then controlling the pH as shown in Fig. 3.
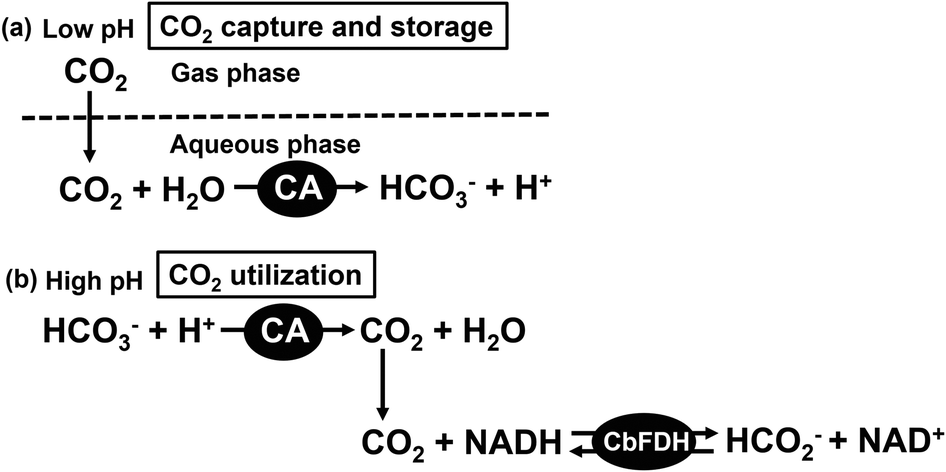 | ||
| Fig. 3 CO2 capture, storage and utilization system using CA and CbFDH (CA/CbFDH system). | ||
In this article, a bienzymatic system consisting of CA and CbFDH for a CO2 capture, storage and utilization system was developed. In this system, CO2 captured and stored in an aqueous medium by CA is reduced to formate by CbFDH over a wide pH region. Particularly, the rate of formate production catalyzed by CbFDH could be promoted in the high pH region at a lower CO2 concentration distribution by using this system.
Experimental
Materials
FDH from Candida boidinii (CbFDH; molecular weight: 74 kDa)83 and CA from bovine erythrocytes (molecular weight: 30 kDa)84 were supplied by Merck Ltd. NADH was supplied by Oriental Yeast Co. Ltd. 3,3-Dimethylglutaric acid, tris(hydroxymethyl)aminomethane, 2-amino-2-methyl-1,3-propanediol and 2-(N-morpholino)ethanesulfonic acid (MES) were obtained from Wako Pure Chemical Co. Ltd. The other chemicals were of analytical grade or the highest grade available and purchased from Wako Pure Chemical Co. Ltd.The 200 mM GTA buffer solution consisted of 3,3-dimethylglutaric acid (200 mM), tris(hydroxymethyl)aminomethane (200 mM) and 2-amino-2-methyl-1,3-propanediol (200 mM) dissolved in distilled water. The pH of the GTA buffer was controlled with an aqueous solution of HCl or NaOH.
Determination of the molar absorption coefficient of NADH using UV-visible absorption spectroscopy
The molar absorption coefficient at 386 nm of NADH was determined by the following method. A solution containing NADH in 200 mM GTA buffer at pH 10.0 was monitored using UV-vis absorption spectra from UV-visible absorption spectroscopy (HITACHI U-2910). The molar absorption coefficient at 386 nm of NADH was obtained from the gradient in the plot of the concentration of NADH (0–2.0 mM) and the absorbance maximum in the absorption spectrum using the Beer–Lambert law.Effect of adding CA on the CbFDH-catalyzed formate production in CO2 saturated buffer solution
The apparent formate concentration produced from CO2 with CbFDH was calculated from the absorbance change at 340 nm with the molar absorption coefficient of NADH (ε340 = 6300 M−1 cm−1)85 using UV-visible absorption spectroscopy (HITACHI U-2910) as equivalent to the consumed concentration of NADH during the reaction. The sample solution consisted of NADH (0.2 mM), CbFDH (40.0 μM) and CA in CO2 saturated 100 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer. The pH of the sample solution was adjusted to be 6.3–6.5. The concentration of CA was varied between 0 and 200 μM. The total volume and temperature of the reaction were set to 2.5 mL and room temperature, respectively.CbFDH-catalyzed CO2 reduction to formate in the presence of CA under various pH conditions
The CO2 reduction to formate with NADH and CbFDH in the presence of CA under the higher pH conditions was carried out as follows. The sample solution consisted of NADH (2.0 mM), sodium bicarbonate (50 mM) and CbFDH (44 ± 2 μM) in 200 mM GTA buffer. The pH of the GTA buffer solution was varied from 6.5 to 10.3. The initial CO2 concentration in the sample solution was calculated using the pH value of the GTA buffer and the equation of Plummer and Busenberg. The total volume and temperature of the reaction were set to 2.5 mL and room temperature, respectively. The apparent formate concentration was calculated using the absorbance change at 368 nm using UV-visible absorption spectroscopy.Results and discussion
Determination of the molar absorption coefficient of NADH using UV-visible absorption spectroscopy
In order to promote the reduction of CO2 to formate catalyzed by CbFDH using a high concentration of NADH (>1.0 mM) as a substrate, it is necessary to follow the accurate change in the concentration of NADH. As shown in Fig. 4, at a NADH concentration above 0.5 mM, the absorption band at 340 nm in the UV-visible absorption spectrum, which is commonly used for quantification, is very noisy. It is generally known that there is no quantification by UV-vis spectroscopy in the region where the absorbance exceeds 1.0.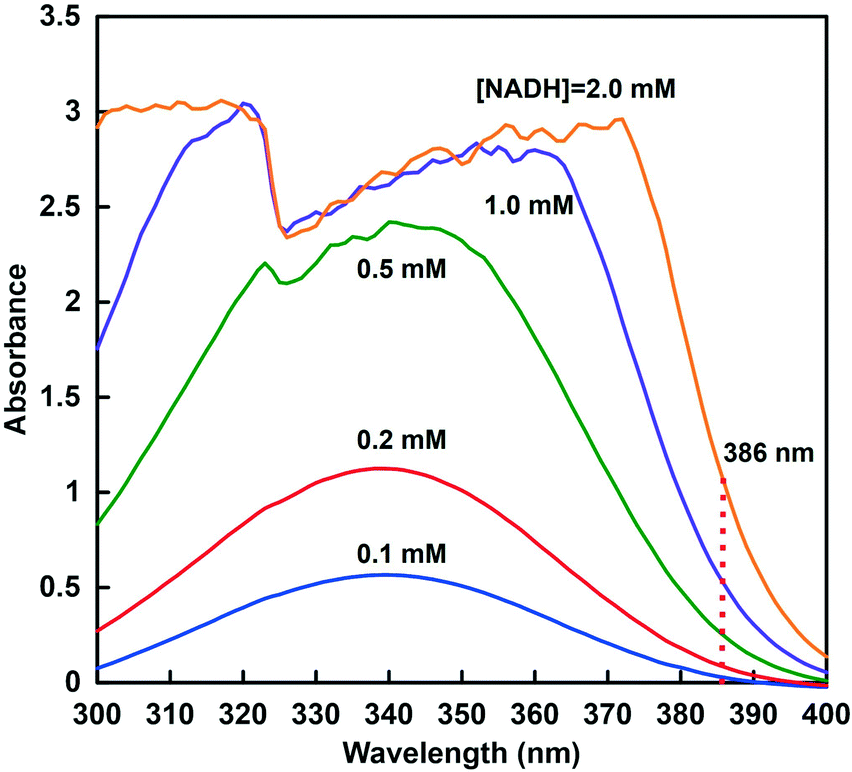 | ||
| Fig. 4 UV-vis absorption spectra changes in 200 mM GTA buffer at pH 10.0 containing various NADH concentrations (0.1–2.0 mM). | ||
Therefore, in order to ensure the quantification of NADH in this concentration region, the relationship between the NADH concentration and the change in absorbance at 386 nm was investigated. The molar absorption coefficient of NADH was determined from the absorbance at 386 nm in the absorption spectra using the Beer–Lambert law. Fig. 5 shows the relationship between NADH concentration and absorbance at 386 nm. This relationship showed good linearity (coefficient of determination r2 = 0.999). From the results of absorbance maxima in the spectra, the molar absorption coefficient of NADH at 386 nm was determined to be 518 M−1 cm−1. Therefore, it was suggested that CbFDH-catalyzed CO2 reduction using high-concentration NADH as a substrate can be quantified from the change in absorbance at a wavelength of 386 nm.
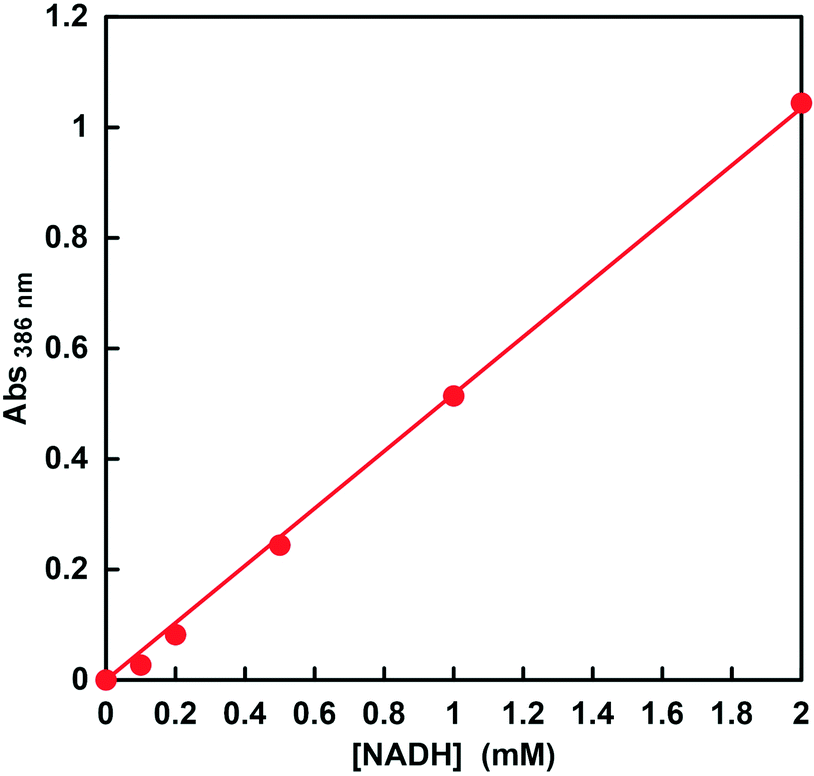 | ||
| Fig. 5 The relationship between NADH concentration and absorbance at 386 nm. | ||
Effect of adding CA on the CbFDH-catalyzed formate production in CO2 saturated buffer solution
The effect of adding CA on the CbFDH-catalyzed reduction of CO2 to formate with NADH is discussed. Fig. 6 shows the time dependence of the absorbance change at 340 nm with NADH and CbFDH at various concentrations of CA. The pH of the sample solution was adjusted to be 6.3–6.5.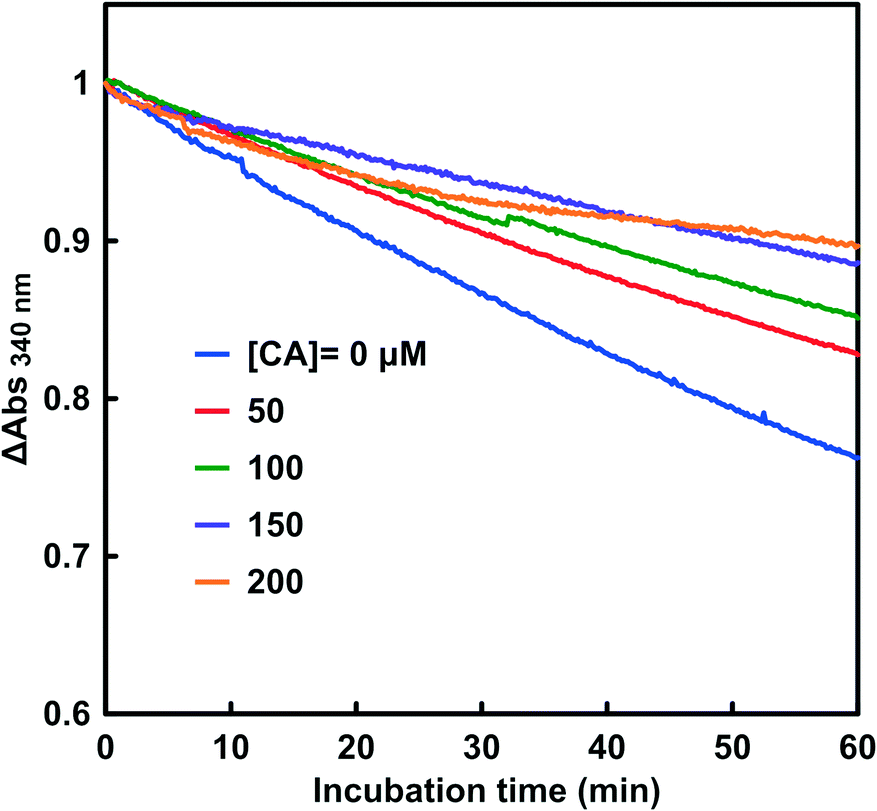 | ||
| Fig. 6 Time dependence of the absorbance change at 340 nm in the system of NADH and CbFDH in CO2 saturated MES buffer at various concentrations of CA (0–200 μM). | ||
As shown in Fig. 6, the absorbance at 340 nm based on NADH decreased with incubation time with and without CA. It was observed that increasing the amount of CA added to the reaction system tended to suppress the consumption of NADH due to the CbFDH-catalyzed CO2 reduction to formate.
Fig. 7 shows the time dependence of NAD+ production due to the CbFDH-catalyzed CO2 reduction to formate in the system of NADH, CbFDH and CA in the CO2 saturated MES buffer solution. Steady NAD+ production was observed with incubation time at all CA concentrations. While the NAD+ production concentration was the highest in the system of NADH and CbFDH, the NAD+ production concentration tended to decrease as the CA concentration increased in the system of NADH, CbFDH and CA. At 150 or 200 μM CA in the sample solution, the concentration of NAD+ produced in the system of NADH and CbFDH decreased to about 50% as compared with that in the absence of CA. By bubbling CO2 into the MES buffer, the pH of the sample solution is adjusted to around 6.3–6.5. It is predicted that the NAD+ production due to the CbFDH-catalyzed CO2 reduction was suppressed due to the decrease in CO2 in the sample because CA hydrolyzes CO2 to bicarbonate in the buffer solution adjusted to pH 6.3–6.5.
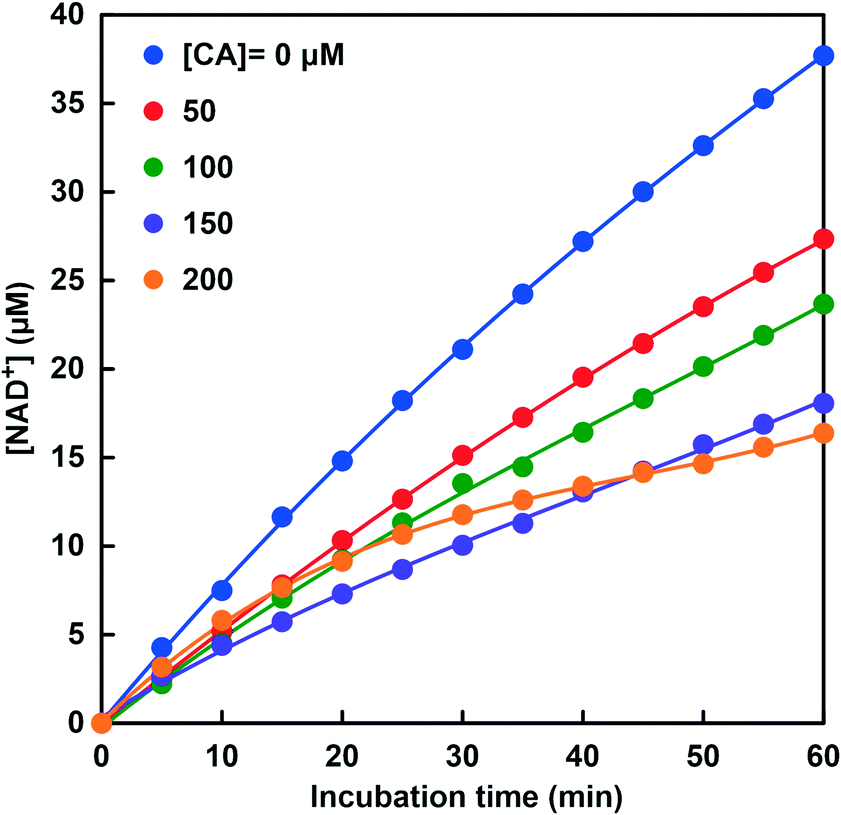 | ||
| Fig. 7 Time dependence of the concentration of produced NAD+ in the system of NADH and CbFDH in CO2 saturated MES buffer at various concentrations of CA (0–200 μM). | ||
Fig. 8 shows the relationship between CA concentration and the apparent formate production rate (Vapp) with CbFDH and NADH. The rate for the apparent formate production (Vapp) was calculated from the gradient of the NADH consumption up to 60 min incubation. It was found that the Vapp decreased as the CA concentration of the sample solution increased. The concentration distribution ratios of CO2, bicarbonate and carbonate in MES buffer at pH 6.5 are estimated to be approximately 0.29, 0.71 and 0 (almost), respectively. We previously reported that CbFDH catalytically reduces only CO2 to formate among the three different carbonate species. On the other hand, in the low pH region, CA catalyzes the rapid hydrolysis of CO2 to bicarbonate. From these results, it is predicted that the rapid hydrolysis of CO2 to bicarbonate by the catalytic function of CA suppressed the CbFDH-catalyzed CO2 reduction to formate.
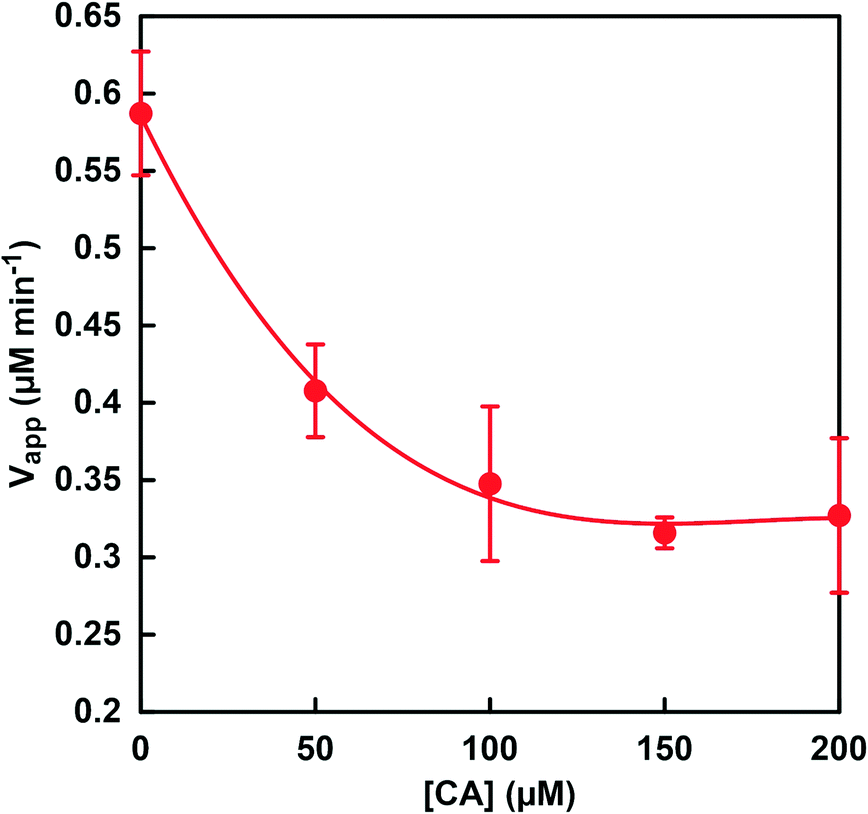 | ||
| Fig. 8 Relationship between CA concentration and the apparent formate production rate (Vapp). | ||
In this system, CO2 was bubbled into the reaction solution for a certain period of time, so it is difficult to precisely control the concentration of CO2, bicarbonate and carbonate in the solution. Next, the total carbonate concentration in the GTA buffer solution was adjusted to 50 mM and the pH was changed to specify the CO2 concentration for the CbFDH-catalyzed CO2 reduction to formate in the presence or absence of CA.
CbFDH-catalyzed CO2 reduction to formate in the presence of CA under lower pH conditions
First, the CbFDH-catalyzed CO2 reduction to formate in the presence or absence of CA at a lower pH of around 6.5 was discussed. Fig. 9 shows the time dependence of the absorbance change at 386 nm in the GTA buffer solution containing NADH, CbFDH and 50 mM sodium bicarbonate to control the pH at 6.5 in the presence or absence of CA.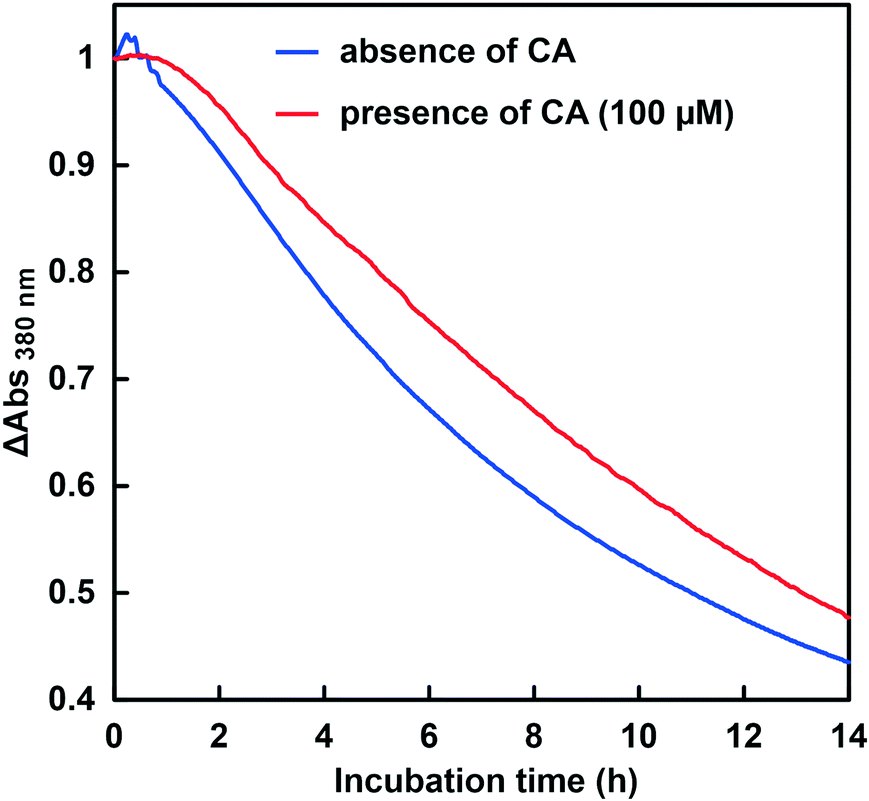 | ||
| Fig. 9 Time dependence of the absorbance change at 386 nm in the system of NADH and CbFDH in the GTA buffer solution containing NADH, CbFDH and 50 mM sodium bicarbonate to control the pH at 6.5 in the presence (red line) or absence (blue) of CA. | ||
As shown in Fig. 9, the absorbance at 386 nm attributed to NADH decreased with increasing incubation time in the absence or presence of CA. This indicates that NADH was consumed in the CbFDH-catalyzed reduction of CO2 to formate in the presence or absence of CA. There was no significant difference in NADH consumption during the CbFDH-catalyzed reduction of CO2 to formate in the presence or absence of CA. Fig. 10 shows the time dependence of the apparent concentration of produced formate in the system of NADH and CbFDH in the GTA buffer solution containing 50 mM sodium bicarbonate to control the pH at 6.5 in the presence or absence of CA.
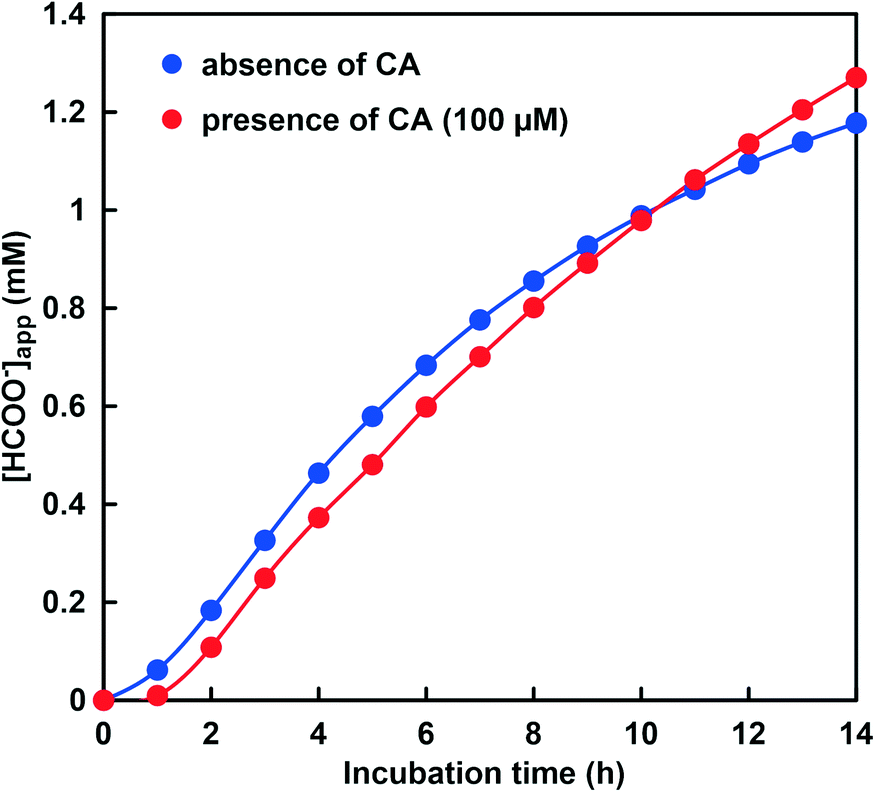 | ||
Fig. 10 Time dependence of the apparent concentration of produced formate in the system of NADH and CbFDH in the GTA buffer solution containing 50 mM sodium bicarbonate to control the pH at 6.5.  : absence of CA; : absence of CA;  : presence of 100 μM CA. : presence of 100 μM CA. | ||
The initial CO2 concentration in the sample solution at pH 6.5 was estimated to be 14.5 mM out of the 50 mM total carbonate species. As shown in Fig. 10, there was no significant difference in the apparent formate production due to the CbFDH-catalyzed CO2 reduction in the presence or absence of CA. CA catalyzes the hydrolysis of CO2 to bicarbonate in a low pH buffer solution, but due to the high initial concentration of CO2, CA has no effect on the CbFDH-catalyzed CO2 reduction.
CbFDH-catalyzed CO2 reduction to formate in the presence of CA under higher pH conditions
The CbFDH-catalyzed CO2 reduction to formate in the presence or absence of CA at a higher pH of around 9.5–10.5 was discussed. Fig. 11 shows the time dependence of the absorbance change at 386 nm in the GTA buffer solution containing NADH, CbFDH and 50 mM sodium bicarbonate to control the pH at 9.5 in the presence or absence of CA.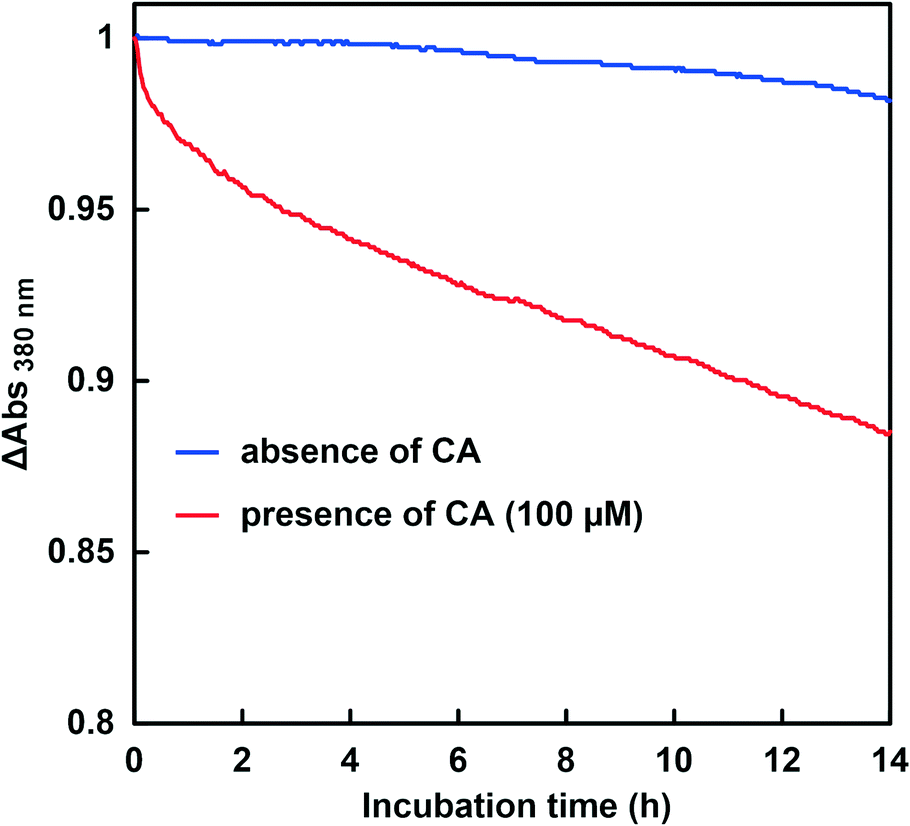 | ||
| Fig. 11 Time dependence of the absorbance change at 386 nm in the system of NADH and CbFDH in the GTA buffer solution containing NADH, CbFDH and 50 mM sodium bicarbonate to control the pH at 9.5 in the presence (red line) or absence (blue) of CA. | ||
As shown in Fig. 11, the absorbance at 386 nm attributed to NADH decreased with increasing incubation time in the absence or presence of CA. This indicates that NADH also was consumed in the CbFDH-catalyzed reduction of CO2 to formate under higher pH conditions with lower CO2 concentration in the presence or absence of CA. It was also found that the addition of CA increased NADH consumption during the CbFDH-catalyzed reduction of CO2 to formate compared with that in the system without CA.
Fig. 12 shows the time dependence of the apparent concentration of produced formate in the system of NADH and CbFDH in the GTA buffer solution containing 50 mM sodium bicarbonate to control the pH at 9.6–9.9 in the presence or absence of CA. The initial CO2 concentration in the sample solution at pH 9.6 and 9.9 were estimated to be 23.7 and 10.3 μM out of the 50 mM total carbonate species, respectively.
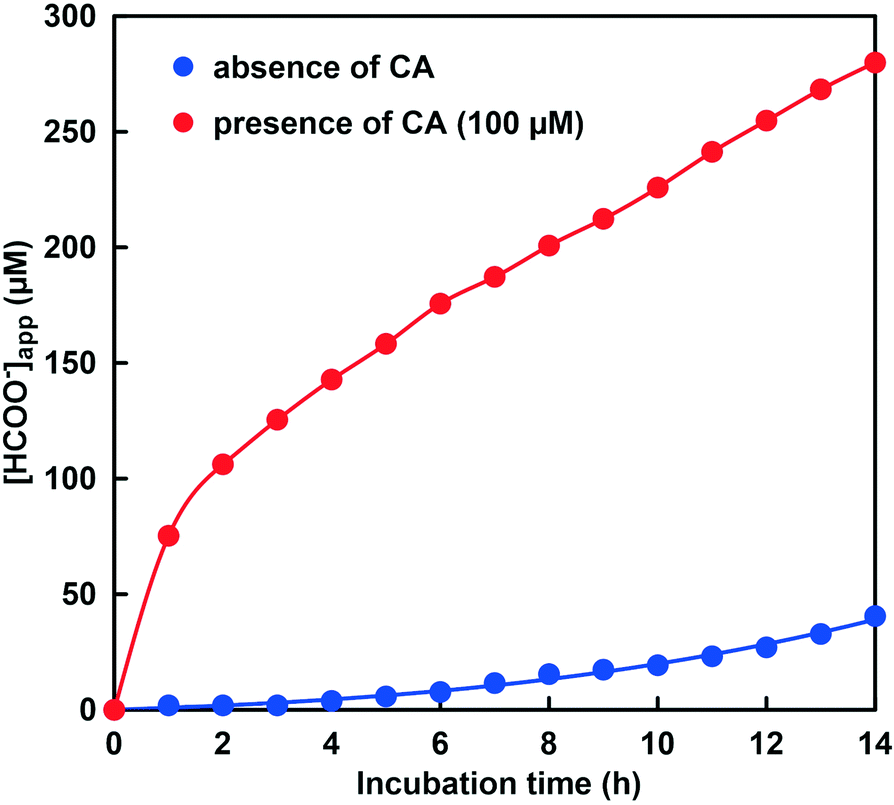 | ||
Fig. 12 Time dependence of the apparent concentration of produced formate in the system of NADH and CbFDH in the GTA buffer solution containing 50 mM sodium bicarbonate to control the pH at 9.6–9.9.  : absence of CA; : absence of CA;  : presence of 100 μM of CA. : presence of 100 μM of CA. | ||
As shown in Fig. 12, 40.5 μM formate was produced in the system of NADH and CbFDH in the absence of CA after 14 h incubation. In other words, formate production exceeding the initial CO2 concentration (10.3 μM) in the sample solution was observed. This result indicates that the CO2 concentration equivalent to the CbFDH-catalyzed formate production originates from bicarbonate or carbonate by the pH control function of the GTA buffer solution in the absence of CA. In contrast, 280 μM formate was produced in the system of NADH and CbFDH in the presence of CA (100 μM) after 14 h incubation. Formate production more than 10 times the initial CO2 concentration (23.7 μM) in the sample solution was observed in the presence of CA. Let's compare the pH of the sample solution with or without CA before and after the CbFDH-catalyzed formate production. In the absence of CA, the pH before the CbFDH-catalyzed formate production was adjusted to 9.9, but the pH value decreased to 9.7 after incubation for 14 h. In the presence of CA, in contrast, the pH before and after the CbFDH-catalyzed formate production was maintained at 9.6. Next, we investigated the reduction of CO2 to formate catalyzed by CbFDH in GTA buffer adjusted to pH 9.6 and 10.3 in the presence of CA. The initial CO2 concentration in the sample solution at pH 9.6 and 10.3 was estimated to be 23.7 and 0.85 μM out of the 50 mM total carbonate species, respectively.
Fig. 13 shows the time dependence of the absorbance change at 386 nm in the GTA buffer solution containing NADH, CbFDH and 50 mM sodium bicarbonate to control the pH at 9.6 and 10.3 in the presence of 100 μM CA.
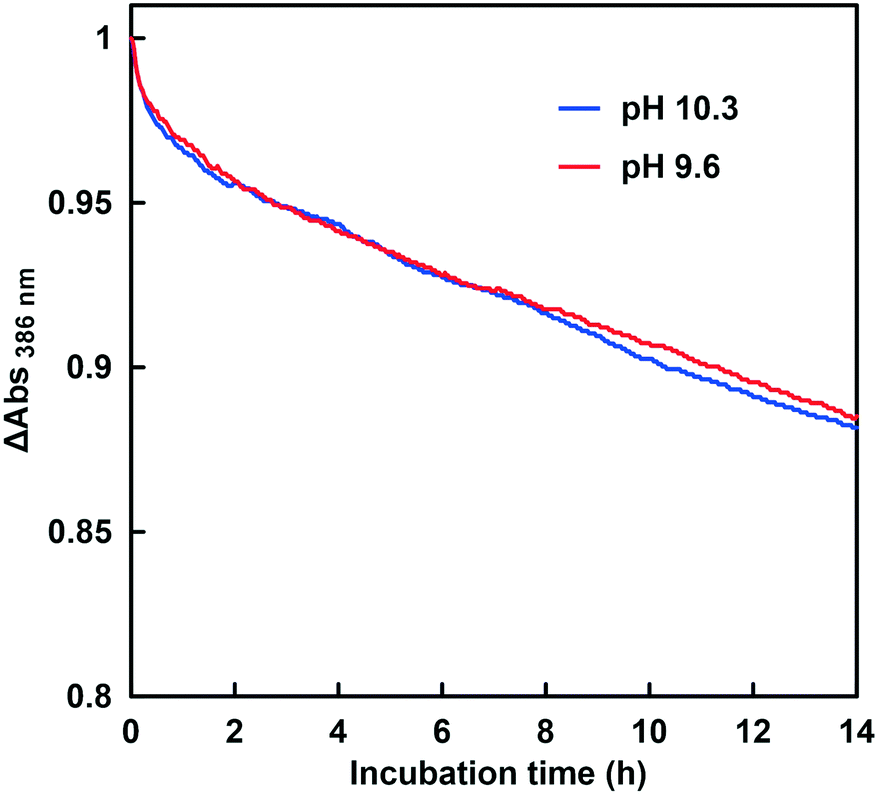 | ||
| Fig. 13 Time dependence of the absorbance change at 386 nm in the system of NADH and CbFDH in the GTA buffer solution containing NADH, CbFDH and 50 mM sodium bicarbonate to control the pH at 9.6 (red) and 10.3 (blue) in the presence of CA. | ||
As shown in Fig. 13, the absorbance at 386 nm attributed to NADH decreased with increasing incubation time in the presence of CA at pH 9.6 and 10.3. This indicates that NADH was consumed in the CbFDH-catalyzed reduction of CO2 to formate at pH 9.6 and 10.3 with CA. There was no significant difference in NADH consumption during the CbFDH-catalyzed reduction of CO2 to formate at pH 9.6 and 10.3 with CA. In particular, NADH consumption due to the CbFDH-catalyzed reduction of CO2 to formate proceeded under the initial concentration conditions of dilute CO2 (less than 1.0 μM) in the presence of CA. Moreover, it has been reported that CbFDH exhibits catalytic activity for the formate oxidation to CO2 between pH 5.5 and 11; however, the CbFDH-catalyzed reduction properties of CO2 to formate under high pH conditions (pH > 9.0) have not yet been clarified. From the present results, it has been found that CbFDH also exhibits CO2 reduction catalytic activity under conditions of pH higher than 10. Thus, it was found that CbFDH was not deactivated even under conditions of pH 10 or higher. From these results, it was shown that CbFDH functions stably as a catalyst even under conditions of pH higher than 10. The time dependence of the apparent concentration of produced formate in the system of NADH and CbFDH in the GTA buffer solution containing 50 mM sodium bicarbonate to control the pH at 9.6 or 10.3 in the presence of CA is shown in Fig. 14. Although the initial CO2 concentration at pH 10.3 was estimated to be 0.036 times that at pH 9.6, there was no significant difference in the CbFDH-catalyzed formate production in the presence of CA after 14 h incubation. At pH 10.3, thus, it is speculated that the catalytic function of CA promoted the conversion of bicarbonate to CO2 and the reduction of CO2 catalyzed by CbFDH to formate in a concerted manner. These results suggest that the coexistence of CA and CbFDH can reduce CO2 to formate in a basic solution storing CO2 as bicarbonate.
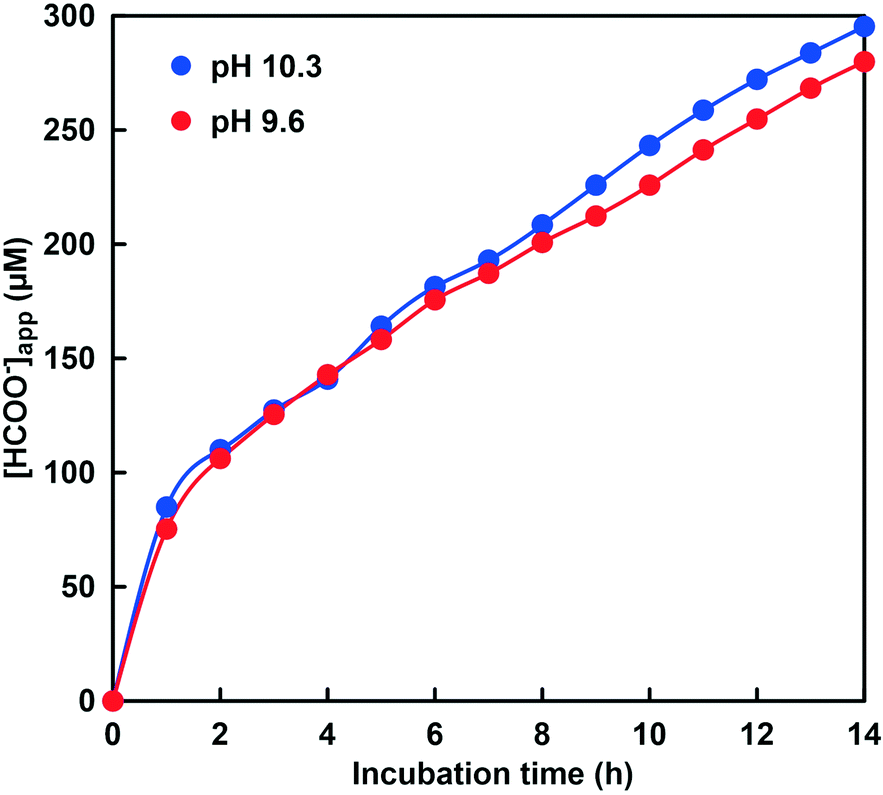 | ||
Fig. 14 Time dependence of the apparent concentration of produced formate in the system of NADH, CbFDH and CA in the GTA buffer solution containing 50 mM sodium bicarbonate to control the pH at 9.6 ( ) and 10.3 ( ) and 10.3 ( ). ). | ||
In the present system, no significant decrease in the enzyme activity of CbFDH or CA was observed within an incubation time of 14 h. CbFDH and CA are multimeric enzymes consisting of several subunits. In long-term experiments for practical use of the present system, it is necessary to suppress inactivation due to dissociation of multimeric enzymes. A method for enclosing an enzyme using various polymers has been reported as a method for stabilizing a multimeric enzyme by suppressing its dissociation.86 For example, it has been applied on the stabilization of multimeric enzymes by coating with ionic polymers and further immobilization of the coating-stabilized composites on a catalytic support. In addition, the stabilization of multimeric enzymes by the use of crosslinked enzyme crystals or aggregates or the preparation of copolymers also has been investigated. Even in our system, stabilization of CbFDH and CA with various polymers can be a useful method for further improving reaction efficiency.
Next, the effects of pH changes on CbFDH-catalyzed formate production in the absence of CA and in the presence of CA were discussed. Fig. 15 shows the pH dependence of the apparent concentration of formate produced due to the CbFDH-catalyzed CO2 reduction in the presence or absence of CA. Fig. 15 also plots the initial CO2 concentration in the sample solution at each pH value. In both cases, it was found that the pH of the sample solution increased and the apparent formate production due to the CbFDH-catalyzed CO2 reduction decreased. Since the initial concentration of CO2 decreases as the pH of the sample solution increases in the absence of CA, it can be easily predicted that the CbFDH-catalyzed CO2 reduction to formate will decrease. In contrast, in the presence of CA, formate production decreased with increasing pH, but CbFDH-catalyzed CO2 reduction was also observed at a pH higher than 9.5. An equivalent to the amount of formate produced by CbFDH-catalyzed CO2 reduction in the sample solution with pH of 7.4 in the absence of CA was also observed in the sample solution containing CA having more than pH 9.5. At pH 7.4 in the sample solution, the initial concentration of CO2 was estimated to be 2.4 mM out of the 50 mM total carbonate species. On the other hand, the initial CO2 concentration is estimated to be less than 23 μM out of the 50 mM total carbonate species in the sample solutions with a pH higher than 9.5. Nevertheless, the presence of CA promoted the reduction of CO2 to formate catalyzed by CbFDH. These results suggest that the ability of CA to convert bicarbonate to CO2 is far superior to the the pH control function of the GTA buffer solution to convert bicarbonate or carbonate to CO2.
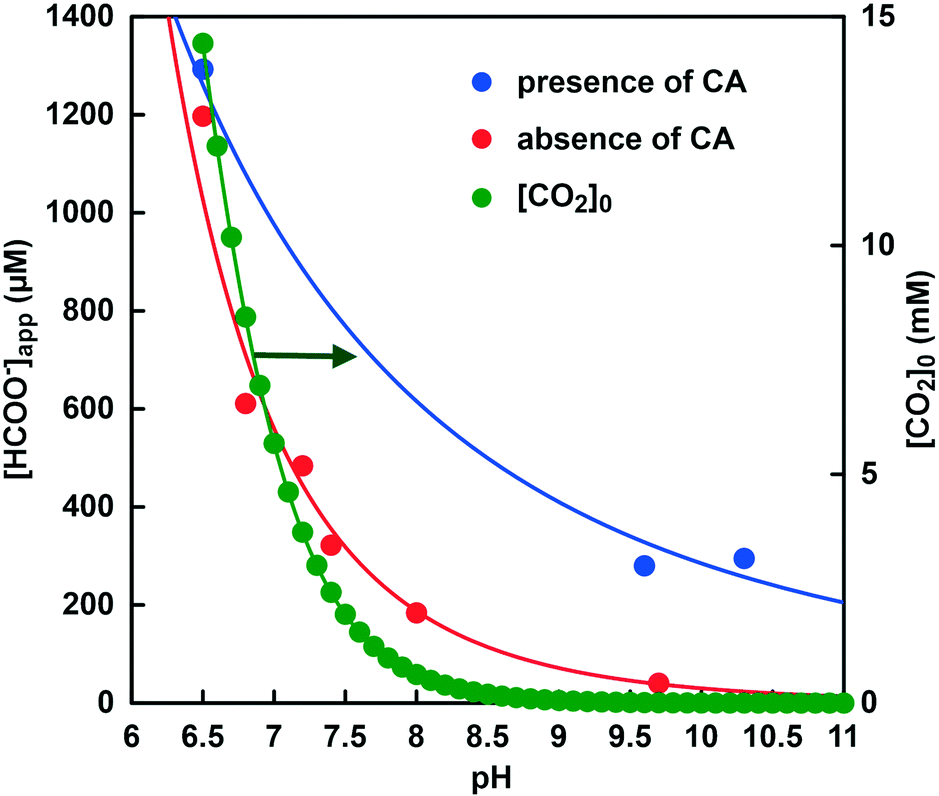 | ||
Fig. 15 Relationship between pH and the apparent concentration of formate produced due to the CbFDH-catalyzed CO2 reduction in the presence ( ) or absence ( ) or absence ( ) of CA. The initial CO2 concentration in the sample solution at each pH value ( ) of CA. The initial CO2 concentration in the sample solution at each pH value ( ). ). | ||
Challenges for practical application of the CCUS technology using CA and CbFDH
Here, the issues for the practical use of the CCUS technology using CA and CbFDH are mentioned. In general, in a reaction system using multiple different enzymes, it is necessary to determine the optimum conditions for the whole reaction. This is because the optimum conditions (pH, temperature, etc.) for each enzyme are different in the reaction. In the system using CA and CbFDH, it is possible to skillfully control the optimum conditions for each enzyme. As described above, under low pH conditions (pH 6.3–6.5), the addition of CA suppresses the reduction of CO2 to formate by CbFDH. Since CbFDH only catalyzes the reduction of CO2 to formate, this suggests that the conversion of CO2 to bicarbonate is promoted in solution by CA. Bicarbonate is more stable in aqueous solution than CO2. Thus, CO2 can be stored in solution as bicarbonate by demonstrating the function of CA at low pH. Next, let's focus on CCU with CbFDH. Since CbFDH reduces only CO2 in solution to formate, the amount of CO2 present in solution is less than that of bicarbonate under high pH conditions (pH = 9.3–10.6), and therefore efficient formate production cannot be expected. On the other hand, under high pH conditions, CA promotes the conversion of bicarbonate to CO2. By using this function of CA, CO2 can be generated in the solution even under high pH conditions, and it can be immediately reduced to formate by CbFDH. In other words, CO2 storage and utilization can be alternated with each other only by adjusting the pH without optimizing the whole reaction using the CA/CbFDH system.It has already been reported that CA is used to capture and store about 64% of a flue stream consisting of 12–13% mol CO2.40 CbFDH-catalyzed formate production also requires mM-order CO2 in solution. For these reasons, it is desirable to use CO2 from industrial flue gas rather than about 0.004% atmospheric CO2 in the CA/CbFDH system. Since it has not been reported that the catalytic activity of CA and CbFDH decreases due to other gases (NOx, SOx, etc.), it is expected that these enzymes can selectively use only CO2 in the reaction without pre-treatment.
Next, let us focus on the final application of the present system for CCUS technology. Enzyme stabilization and reusability/recyclability are important factors in the final application. As a solution to these problems, there is a technique for immobilizing an enzyme on a catalytic support.87,88 By using enzyme immobilization, enzymes are recovered and reused for many cycles, improving the economic balance of biocatalytic processes. Moreover, immobilization has been shown to be a potent tool to improve many enzyme features, like stability, selectivity, specificity, or activity.89–91 Also, enzyme immobilization decreases enzyme inhibition92 or widens the operating conditions.93 In addition, enzyme immobilization can be coupled to purification.94 Enzyme immobilization techniques are far more than a tool to recover and reuse enzymes and contribute a critical step in biocatalyst design.87 Here, some enzyme immobilization methods for FDH and CA and the future prospects of our system are discussed. The use of metal–organic frameworks (MOFs) such as zeolitic imidazolate framework-8 (ZIF-8),95,96 amine-functionalized Zr-MOF (UiO-66-NH2) membrane,97 and chromium(III) terephthalate MIL-101 MOF98 and cross-linking of CA and FDH based on amino acid specific recognition99 have been reported as enzyme immobilization methods. Immobilization methods focusing on the properties of enzymes (generation of favourable enzyme environments, prevention of enzyme subunit dissociation in multimeric enzymes, generation of more stable enzyme conformations, or enzyme rigidification via multipoint covalent attachment) are also being studied. The features of an ideal immobilization protocol to maximize the intensity of enzyme–support interactions also are proposed. By immobilizing an enzyme on a catalytic support, its thermal stability is improved, and the enzyme can be used repeatedly. There are many methods used for enzyme immobilization, but industry prefers simple and cost-effective methods. The most used methods are based on physical immobilization based on adsorption or physical entrapment and chemical immobilization due to covalent binding and cross linking. The advantages of enzyme immobilization for practical usage are easy separation of enzymes, reduced costs of downstream processing, multiple uses or recycling of enzymes, better stability, especially towards organic solvents and higher temperatures, the use of fixed bed or batch reactors without the need for membranes to isolate enzymes from the product and co-immobilization with other enzymes. In contrast, the disadvantages of enzyme immobilization are lower enzyme activity compared to that of native enzymes, additional costs for carriers and immobilization, lower reaction rates compared to that of native enzymes, being subject to fouling and disposal of exhausted immobilized enzymes.100 Therefore, we are planning the use of natural porous supports to prevent a decrease in enzyme activity. In the future, we also are planning research on co-immobilization of CA and CbFDH using porous protein crystal lysozymes similar to MOFs.101
Finally, in the present system, NADH is added as a co-enzyme for the reduction of CO2 to formate by CbFDH. However, since it is not preferable to use NADH as a sacrificial reducing agent, it is necessary to integrate a regeneration system for NADH. Several studies have proposed the integration of another NAD+-dependent enzyme as a regeneration system for NADH.102 Also, nanoscale multi-enzyme (CA and FDH) reactor consisting of ZIF-8 with cofactor, NADH regeneration with glutamate dehydrogenase for CO2 conversion has been reported.103 In these systems, cofactors are immobilized on a catalytic support together with multiple enzymes. These systems retain 50% of their original productivity after at least 8 cycles, demonstrating excellent reusability.103 On the other hand, a reduction system for NAD+ to NADH using a visible-light driven redox system composed of an electron donor (ED), a photosensitizer (PS) and colloidal rhodium as shown in Fig. 16 has been reported.104
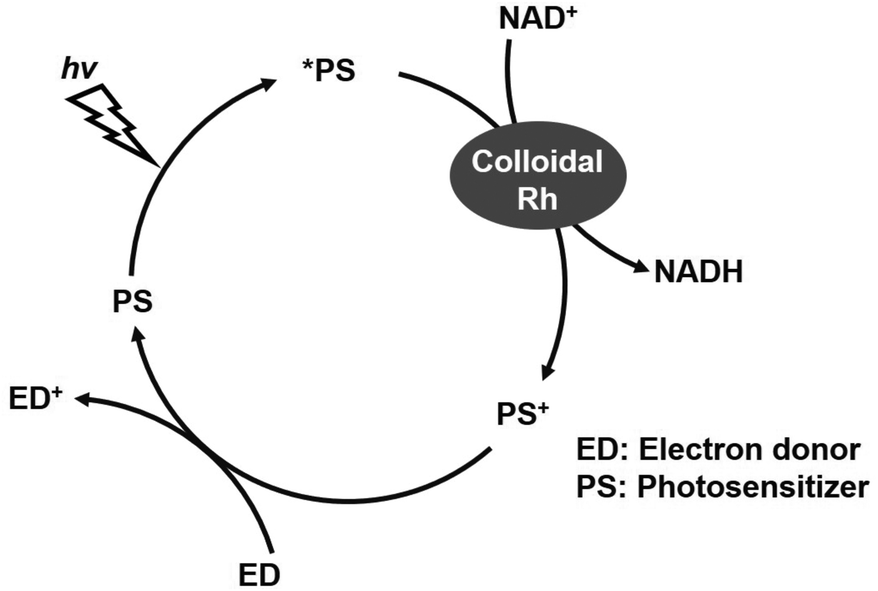 | ||
| Fig. 16 Visible-light driven redox system composed of an electron donor (ED), a photosensitizer (PS) and colloidal rhodium for NADH regeneration. | ||
We also are planning research on the combination of visible-light driven redox for NADH regeneration and a CA/CbFDH system for development of CCUS technology incorporating light energy utilization in the near future. Immobilization of the system and NAD+ as shown in Fig. 16 on a single support is also in view for the future.
Conclusion
In conclusion, development of a bienzymatic system consisting of CA and CbFDH for a CO2 capture, storage and utilization system was attempted. It was found that the reaction rate of CbFDH-catalyzed CO2 reduction to formate decreased as the CA concentration of the sample solution increased. At 150 or 200 μM CA in the sample solution and a controlled pH of 6.3–6.5 by CO2 bubbling, the reaction rate of CbFDH-catalyzed CO2 reduction to formate decreased to about 50% as compared with that in the absence of CA. It is predicted that the CbFDH-catalyzed CO2 reduction was suppressed due to the decrease in CO2 in the sample because CA hydrolyzes CO2 to bicarbonate in the buffer solution adjusted to pH 6.3–6.5. In particular, in the higher pH region (>9.5), it was found that the addition of CA promoted the reduction of CO2 to formate catalyzed by CbFDH. There was no significant difference in the CbFDH-catalyzed formate production at pH 9.6 and 10.3 in the presence of CA after 14 h incubation. At pH 10.3 (initial CO2 concentration: 0.85 μM out of the 50 mM total carbonate species), thus, it is speculated that the catalytic function of CA promoted the conversion of bicarbonate to CO2 and the CbFDH-catalyzed CO2 reduction to formate in a concerted manner. In particular, the rate of formate production catalyzed by CbFDH was promoted in the high pH region at a lower CO2 concentration distribution by using this system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Grant-in-Aid for Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (4906)” and the Fund for the Promotion of Joint International Research (Fostering Joint International Research (B)) (19KK0144).Notes and references
- A. Abdulla, R. Hanna, K. R. Schell, O. Babacanand and D. G. Victor, Environ. Res. Lett., 2020, 16, 014036 CrossRef.
- N. Mac Dowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. M. Dowell, Energy Environ. Sci., 2018, 11, 1062 RSC.
- GCCSI, Large-scale CCS projects, Global CCS Institute, http://www.globalccsinstitute.com/projects/large-scale-ccs-projects, accessed July, 2017.
- BEIS, UK carbon capture and storage: Government funding and support, Department for Business, Energy & Industrial Strategy (BEIS), London, UK, https://www.gov.uk/guidance/uk-carbon-capture-and-storage-government-funding-and-support, accessed June, 2017 Search PubMed.
- T. Ajayi, J. S. Gomes and A. Ber, Pet. Sci., 2019, 16, 1028 CrossRef CAS.
- W. A. Ambrose, S. Lakshminarasimhan, M. H. Holtz, V. Núñez-López, S. D. Hovorka and I. Duncan, Environ. Geol., 2017, 54, 1619 CrossRef.
- M. D. Aminu, S. A. Nabavi, C. A. Rochelle and V. A. Manovic, Appl. Energy, 2017, 208, 1389 CrossRef CAS.
- G. Aydin, I. Karakurt and K. Aydiner, Energy Policy, 2010, 38, 5072 CrossRef.
- S. Bachu and J. J. Adams, Energy Convers. Manage., 2003, 44, 3151 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- A. Dibenedetto, A. Angelini and P. Stufano, J. Chem. Technol. Biotechnol., 2014, 89, 334 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87 CrossRef CAS.
- Y. Xu, L. Isom and M. A. Hanna, Bioresour. Technol., 2010, 101, 3311 CrossRef CAS.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560 CrossRef CAS.
- A. Erdogan and Y. O. Ozge, Petroleum, 2017, 3, 109 CrossRef.
- F. Robert, S. Benjamin and R. Michael, Chem. Rev., 2018, 118, 4631 CrossRef.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer, M. Chi, C. Ma, B. G. Sumpter and A. J. Rondinone, ChemistrySelect, 2016, 1, 6055 CrossRef CAS.
- Z. Zhang, S. Y. Pan, H. Li, J. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Renewable Sustainable Energy Rev., 2020, 125, 109799 CrossRef CAS.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718 CrossRef.
- G. Garcia-Garcia, M. C. Fernandez, K. Armstrong, S. Woolass and P. Styring, ChemSusChem, 2021, 14, 995 CrossRef CAS PubMed.
- H. L. Zhang and M. G. Chen, Meitan Xuebao, 2007, 32, 748 Search PubMed.
- S. Masoumi, P. Keshavarz and Z. Rastgoo, J. Nat. Gas Sci. Eng., 2014, 18, 23 CrossRef CAS.
- J. Kuntz and A. Aroonwilas, Energy Procedia, 2009, 1, 205 CrossRef CAS.
- H. Bai and A. C. Yeh, Ind. Eng. Chem. Res., 1997, 36, 2490 CrossRef CAS.
- X. N. Li, E. Hagaman and C. Tsouris, Energy Fuels, 2003, 17, 69 CrossRef CAS.
- Y. F. Diao, X. Y. Zheng and B. S. He, Energy Convers. Manage., 2004, 45, 2283 CrossRef CAS.
- J. W. Lee and E. Hagaman, Energy Fuels, 2003, 17, 69 CrossRef.
- B. R. W. Pinsent, L. Pearson and F. J. W. Roughton, Trans. Faraday Soc., 1956, 52, 1594 RSC.
- Y. Peng, B. Zhao and L. Li, Energy Procedia, 2012, 14, 1515 CrossRef.
- A. Ünlü, Z. E. Duman-Özdamar, B. Çaloğlu and B. Binay, Protein J., 2021, 40, 489 CrossRef.
- M. R. Badger and G. D. Price, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1994, 45, 369 CrossRef CAS.
- C. T. Supuran, Carbonic Anhydrases: Catalytic and Inhibition Mechanisms, Distribution and Physiological Roles, Taylor & Francis, 2004 Search PubMed.
- T. H. Maren, Physiol. Rev., 1967, 47, 595 CrossRef CAS PubMed.
- M. R. Sawaya, G. C. Cannon, S. Heinhorst, S. Tanaka, E. B. Williams, T. O. Yeates and C. A. Kerfeld, J. Biol. Chem., 2006, 281, 7546 CrossRef CAS.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946 CrossRef CAS.
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed.
- S. Zhang, H. Lu and Y. Lu, Environ. Sci. Technol., 2013, 47, 13882 CrossRef CAS.
- A. S. Covarrubias, T. Bergfors, T. A. Jones and M. Högbom, J. Biol. Chem., 2006, 281, 4993 CrossRef PubMed.
- O. Alvizo, L. J. Nguyen, C. K. Savile, J. A. Bresson, S. L. Lakhapatri, E. O. P. Solis, R. J. Fox, J. M. Broering, M. R. Benoit, S. A. Zimmerman, S. J. Novick, J. Liang and J. J. Lalonde, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16436 CrossRef CAS PubMed.
- Y. Demir, N. Demir, H. Nadaroglu and E. Bakan, Prep. Biochem. Biotechnol., 2000, 30, 49 CrossRef CAS PubMed.
- W. Buckel and R. K. Thauer, Front. Microbiol., 2018, 9, 401 CrossRef PubMed.
- F. Kracke, B. Virdis, P. V. Bernhardt, K. Rabaey and J. O. Krömer, Biotechnol. Biofuels, 2016, 9, 249 CrossRef.
- W. A. van den Berg, W. R. Hagen and W. M. van Dongen, Eur. J. Biochem., 2000, 267, 666 CrossRef CAS PubMed.
- J. Hadj-Saïd, M. E. Pandelia, C. Léger, V. Fourmond and S. Dementin, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 1574 CrossRef PubMed.
- V. I. Tishkov and V. O. Popov, Biochemistry, 2004, 69, 1252 CAS.
- R. Wichmann, C. Wandrey, A. F. Buckmann and M. R. Kula, Biotechnol. Bioeng., 1981, 23, 2789 CrossRef CAS.
- W. Hummel and M. R. Kula, Eur. J. Biochem., 1989, 184, 1 CrossRef CAS.
- H. Zhao and W. A. van der Donk, Curr. Opin. Biotechnol., 2003, 14, 583 CrossRef CAS PubMed.
- J. G. Ferry, FEMS Microbiol. Rev., 1990, 7, 377 CrossRef CAS PubMed.
- Y. Amao, J. CO2 Util., 2018, 26, 623 CrossRef CAS.
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963 CrossRef CAS PubMed.
- S. Ochoa, A. H. Mehler and A. Kornberg, J. Biol. Chem., 1948, 174, 979 CrossRef CAS.
- I. Harary, S. R. Korey and S. Ochoa, J. Biol. Chem., 1953, 203, 595 CrossRef CAS.
- B. W. Geer, D. Krochko, M. J. Oliver, V. K. Walker and J. H. Williamson, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1980, 65, 25 CrossRef.
- W. J. Rutter and H. A. Lardy, J. Biol. Chem., 1958, 233, 374 CrossRef CAS.
- R. G. Strickland, Biochem. J., 1959, 73, 646 CrossRef PubMed.
- R. G. Strickland, Biochem. J., 1959, 73, 654 CrossRef PubMed.
- D. A. Walker, Biochem. J., 1960, 74, 216 CrossRef CAS PubMed.
- K. Schirwitz, A. Schmidt and V. S. Lamzin, Protein Sci., 2007, 16, 1146 CrossRef CAS PubMed.
- H. Choe, J. C. Joo, D. H. Cho, M. H. Kim, S. H. Lee, K. D. Jung and Y. H. Kim, PLoS One, 2014, 9, e103111 CrossRef PubMed.
- D. Mandler and I. Willner, J. Chem. Soc., Perkin Trans. 2, 1988, 997 RSC.
- M. Kodaka and Y. Kubota, J. Chem. Soc., Perkin Trans. 2, 1999, 891 RSC.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Mol. Catal. B: Enzym., 2004, 27, 121 CrossRef CAS.
- I. Tsujisho, M. Toyoda and Y. Amao, Catal. Commun., 2006, 7, 173 CrossRef CAS.
- Y. Amao, R. Abe and S. Shiotani, J. Photochem. Photobiol., A, 2015, 313, 149 CrossRef CAS.
- Y. Amao, Chem. Lett., 2017, 46, 780 CrossRef CAS.
- Y. Amao, Sustainable Energy Fuels, 2018, 2, 1928 RSC.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-J. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728 CrossRef CAS PubMed.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef CAS PubMed.
- W. S. Choi, S. H. Lee, J. W. Ko and C. B. Park, ChemSusChem, 2016, 9, 1559 CrossRef CAS PubMed.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J. K. Lee and C. B. Park, Angew. Chem., 2017, 129, 3885 CrossRef.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, New J. Chem., 2013, 37, 3721 RSC.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Nano, 2015, 9, 4600 CrossRef CAS PubMed.
- J. H. Kim, D. H. Nam and C. B. Park, Curr. Opin. Biotechnol., 2014, 28, 1 CrossRef CAS PubMed.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J. K. Lee and C. B. Park, Angew. Chem., Int. Ed., 2017, 56, 3827 CrossRef CAS PubMed.
- R. Sato and Y. Amao, New J. Chem., 2020, 44, 11922 RSC.
- M. Meneghello, A. R. Oliveira, A. Jacq-Bailly, I. A. C. Pereira, C. Leger and V. Fourmond, Angew. Chem., Int. Ed., 2021, 60, 9964 CrossRef CAS PubMed.
- K. Teramura, K. Hori, Y. Terao, Z. Huang, S. Iguchi, Z. Wang, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2017, 121, 8711 CrossRef CAS.
- E. Wilhelm, R. Battino and R. J. Wilcock, Chem. Rev., 1977, 77, 219 CrossRef CAS.
- L. N. Plummer and E. Busenberg, Geochim. Cosmochim. Acta, 1982, 46, 1011 CrossRef CAS.
- H. Slusarczyk, S. Felber, M.-R. Kula and M. Pohl, Eur. J. Biochem., 2000, 267, 1280 CrossRef CAS PubMed.
- J. da Costa Ores, L. Sala, G. P. Cerveira and S. J. Kalill, Chemosphere, 2012, 88, 255 CrossRef CAS PubMed.
- R. B. McComb, L. W. Bond, R. W. Burnett, R. C. Keech and G. N. Bowers Jr., Clin. Chem., 1976, 22, 141 CrossRef CAS.
- R. C. Rodrigues, A. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 52, 107821 CrossRef PubMed.
- R. Morellon-Sterling, D. Carballares, S. Arana-Peña, E.-H. Siar, S. A. Braham and R. Fernandez-Lafuente, ACS Sustainable Chem. Eng., 2021, 9, 7508 CrossRef CAS.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223 RSC.
- R. A. Wahab, N. Elias, F. Abdullah and S. K. Ghoshal, React. Funct. Polym., 2020, 152, 104613 CrossRef CAS.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451 CrossRef CAS.
- C. Mateo, R. Monti, B. C. C. Pessela, M. Fuentes, R. Torres, J. M. Guisán and R. Fernández-Lafuente, Biotechnol. Prog., 2004, 20, 1259 CrossRef CAS PubMed.
- L. Dal Magro, J. F. Kornecki, M. P. Klein, R. C. Rodrigues and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2020, 132, 109397 CrossRef CAS PubMed.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33, 435 CrossRef CAS PubMed.
- M. Chai, S. Razavi Bazaz, R. Daiyan, A. Razmjou, M. Ebrahimi Warkiani, R. Amal and V. Chen, Chem. Eng. J., 2021, 426, 130856 CrossRef CAS.
- S. Yu, P. Lv, P. Xue, K. Wang, Q. Yang, J. Zhou, M. Wang, L. Wang, B. Chen and T. Tan, Chem. Eng. J., 2021, 420, 127649 CrossRef CAS.
- M. Chai, A. Razmjou and V. Chen, J. Membr. Sci., 2021, 623, 118986 CrossRef CAS.
- Y. Li, L. Wen, T. Tan and Y. Lv, Front. Bioeng. Biotechnol., 2019, 7, 394 CrossRef PubMed.
- X. Zhang, W. Shao, B. Chen and M. Wang, Enzyme Microb. Technol., 2021, 146, 109763 CrossRef CAS PubMed.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- H. Tabe, H. Takahashi, T. Shiomi, S. Abe, T. Ueno and Y. Yamada, Appl. Catal., B, 2018, 237, 1124 CrossRef CAS.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771 CrossRef CAS PubMed.
- S. Ren, Z. Wang, M. Bilal, Y. Feng, Y. Jiang, S. Jia and J. Cui, Int. J. Biol. Macromol., 2020, 155, 110 CrossRef CAS PubMed.
- T. Katagiri and Y. Amao, New J. Chem., 2021, 45, 15748 RSC.
| This journal is © The Royal Society of Chemistry 2022 |