
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFighting metallodrug resistance through alteration of drug metabolism and blockage of autophagic flux by mitochondria-targeting AIEgens†
Yan
Su‡
 ,
Hai
Lin‡
,
Ying
Tu‡
,
Meng-Meng
Wang
,
Guan-Dong
Zhang
,
Jin
Yang
,
Hong-Ke
Liu
* and
Zhi
Su
*
,
Hai
Lin‡
,
Ying
Tu‡
,
Meng-Meng
Wang
,
Guan-Dong
Zhang
,
Jin
Yang
,
Hong-Ke
Liu
* and
Zhi
Su
*
Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, College of Chemistry and Materials Science, Nanjing Normal University, Nanjing, 210023, China. E-mail: zhisu@njnu.edu.cn; liuhongke@njnu.edu.cn
First published on 18th January 2022
Abstract
Metallodrug resistance has attracted a great deal of attention in cancer treatment. According to the cisplatin (cis-Pt) anticancer mechanism, a new strategy to overcome cis-Pt resistance through mitochondrial dysfunction is proposed. Two mitochondria-targeted aggregation-induced emission fluorogens (AIEgens) were first synthesized, named DP-PPh3 and TPE-PPh3, which showed superior capacities to overcome the cis-Pt resistance of lung cancer cells (A549R) by the alteration of drug metabolism (up-regulation of influx CTR1 and down-regulation of efflux MRP2) and blockage of autophagic flux (failure of the degradation of autophagosomes). This study is the first time that AIEgens are utilized in the treatment of cis-Pt resistant cancer cells. Moreover, the underlying molecular mechanism was fully revealed. Triphenylphosphonium (PPh3)-decorated AIEgens DP-PPh3 and TPE-PPh3 not only successfully realized aggregation and the imaging of mitochondria in A549R cells, but also activated cytotoxicity towards A549R cells. DP-PPh3 and TPE-PPh3 could induce ROS production, disrupt the mitochondrial structure, and impair mitochondrial and glycolytic metabolism. Furthermore, the anticancer efficacy of these drugs was demonstrated in 3D multicellular tumor spheroids (MCTSs) of A549R cells in vitro and in tumor-bearing nude mice in vivo. This AIE-PPh3 strategy not only promoted cytotoxicity towards cancer cells but also provided a new pathway for the treatment of metallodrug resistance.
Introduction
Metallodrugs have been utilized in cancer treatment for over 50 years since cis-Pt was first developed.1,2 Moreover, the second generation platinum drug carboplatin and the third generation oxaliplatin were also approved for use in clinical treatment.3,4 However, cancer cells generate metallodrug resistance after several rounds of treatment, which would deactivate the metallodrug and dramatically limit the treatment efficacy. For example, cis-Pt resistance involves several modes of action: reduced intracellular accumulation, enhanced efflux, increased inactivation by metallothionein (MT) and glutathione (GSH), and strengthened DNA repair capacity.3,5–7 In this case, alteration of cis-Pt metabolism by increasing the intracellular uptake and decreasing the efflux pump would be effective to overcome cis-Pt resistance. The copper transporter 1 (CTR1) and multidrug resistance-associated protein 2 (MRP2) were the two key transporters involved in cis-Pt influx and efflux.8–11 Thus, activation of CTR1 and inhibition of MRP2 would resolve the cis-Pt resistance during treatment.Aggregation-induced emission fluorogens (AIEgens) developed by Tang and co-workers have been applied in the imaging and therapy of cancer cells for over two decades, due to their high sensitivity and stability.12 Compared to conventional aggregation-caused quenching (ACQ), AIEgens would be lit up in the aggregation state due to the restriction of intramolecular motion.13 To generate the AIE effect inside the cancer cells, AIEgens have to target and aggregate in one specific organelle.14 Mitochondria, as the energy sources in cells, play important roles in cell signaling, biosynthesis, and cell differentiation.15,16 Due to the special structure of mitochondria, cation fragments help to realize the mitochondrial accumulation, as triphenylphosphonium (PPh3), pyridinium and so on.17 Most reported mitochondrial targeting AIEgens exhibit anticancer activity by inducing cell apoptosis, which was the same anticancer mechanism used by cis-Pt.18 Cancer cells can develop an antiapoptotic pathway to escape such treatments, which was also one of the reasons for the generation of cis-Pt resistance.19,20 In contrast to apoptosis, autophagy is a cytoprotective mechanism in response to cellular stress, where the cells capture cytoplasmic cargos in autophagosomes and then fused with lysosomes for degradation.21–23 However, autophagy is also a double-edged sword, either rescuing cells from damage or leading to autophagic cell death.24 Therefore, new AIEgens with novel autophagic pathways are needed to resolve the cis-Pt resistance.
In this research, two triphenylphosphonium (PPh3)-decorated mitochondrial-targeted AIEgens, namely, DP-PPh3 ((4-(3-(9,9-dimethylacridin-10(9H)-yl)-5-(pyridin-4-yl)phenoxy)butyl)triphenylphosphonium bromine) and TPE-PPh3 ((E)-(4-(4-(2-(4′-carboxy-[1,1′-biphenyl]-4-yl)-1,2-diphenylvinyl)phenoxy)butyl)triphenylphosphonium bromine), were designed and synthesized (Fig. 1a). Both DP-PPh3 and TPE-PPh3 showed 20-fold cytotoxicity towards cancer cells, when comparing to that of the precursors, DP-OH and TPE-Br. Mitochondrial accumulation and dysfunction after the treatments with DP-PPh3 and TPE-PPh3 were observed, including mitochondrial morphology alteration, mitochondrial membrane potential (MMP) reduction, and mitochondrial and glycolytic metabolism inhibition. With the illumination of the RNA-seq analysis, the superior capacity for DP-PPh3 and TPE-PPh3 to overcome cis-Pt resistance was due to the alteration of the drug metabolism (by up-regulation of influx CTR1 and down-regulation of efflux pump MRP2) and the blockage of the autophagic flux (by failure to degrade the autophagosomes). The 3D multicellular tumor spheroids (MCTSs) of A549R cells in vitro and the growth of tumor-bearing mice in vivo were also inhibited after treatment with DP-PPh3 or TPE-PPh3. This investigation not only demonstrated the efficacy of PPh3-decoration to achieve mitochondrial accumulation but also provided a new strategy for the treatment of metallodrug resistance.
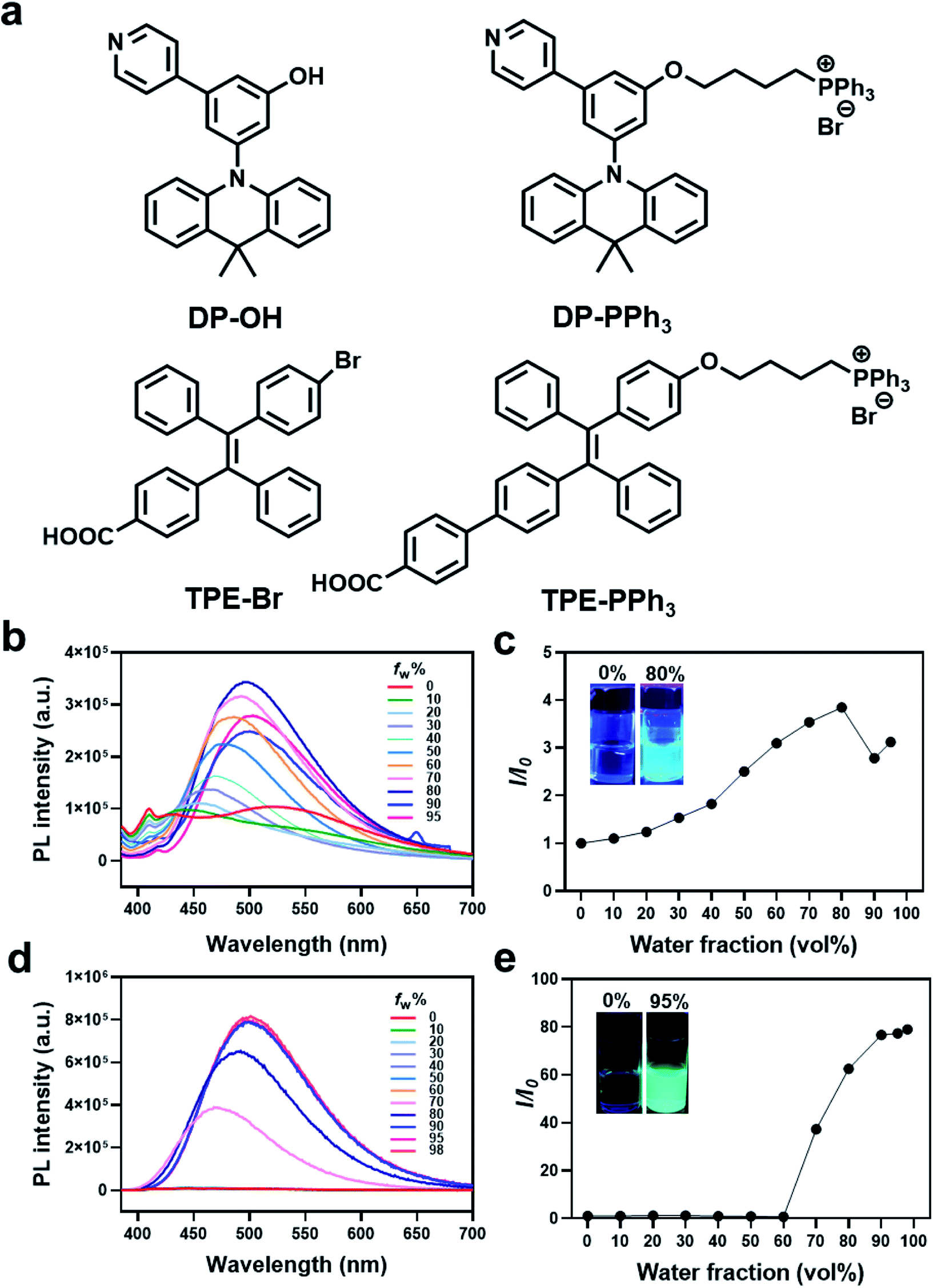 | ||
| Fig. 1 (a) Chemical structures of AIEgens including DP-OH, DP-PPh3, TPE-Br, and TPE-PPh3. Photoluminescence (PL) spectra and relative maximum intensity ratio I/I0 of DP-PPh3 ((b and c), 50 μM) and TPE-PPh3 ((d and e), 50 μM) in a DMSO/water mixture with different water volume fractions (fw). The I0 and I are the PL intensity in pure DMSO or in different compositions of DMSO/water. Ex = 405 nm. | ||
Results
Design, synthesis, and characterization
Two AIE fluorophores based on typical triphenylamine and tetraphenylethene groups were first prepared according to previously reported procedures (Schemes S1 and S2 (ESI†), Fig. 1). These AIE fluorophores were 3-(9,9-dimethylacridin-10(9H)-yl)-5-(pyridin-4-yl)phenol (DP-OH) and (E)-4-(2-(4-bromophenyl)-1,2-diphenylvinyl)benzoic acid (TPE-Br), respectively.25,26 Due to the extremely negative membrane potential (ca. 180 mV) of the mitochondria, a positively charged triphenylphosphonium (PPh3) group was introduced and conjugated to DP-OH and TPE-Br to achieve the aggregation in mitochondria and generate the AIE effect.27,28 Thus, DP-PPh3 and TPE-PPh3 were synthesized (Schemes S1 and S3, ESI†) and then fully characterized by 1H-NMR, 13C-NMR, and ESI-MS (Fig. S1–S6, ESI†).The photophysical properties of DP-OH, DP-PPh3, TPE-Br, TPE-PPh3 were studied. As shown in the UV-vis spectra (Fig. S7, ESI†), DP-OH, DP-PPh3, TPE-Br, and TPE-PPh3 exhibited main absorption peaks at 290, 275, 320, 340 nm, respectively, in DMSO/water (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99) at room temperature (Fig. S7, ESI†), which were due to an intramolecular charge transfer (ICT) transition.29
:99) at room temperature (Fig. S7, ESI†), which were due to an intramolecular charge transfer (ICT) transition.29
An obvious AIE phenomenon was observed for all the tested complexes in the DMSO/water mixed solvent with increasing water fractions (fw), due to the restriction of intramolecular motion. A gradually increased and red-shifted emission was detected for both the DP-PPh3 and TPE-PPh3 complexes, where the maximum emission for DP-PPh3 and TPE-PPh3 was defined at 500 nm with fw values of 80% and 98%, respectively (Fig. 1b–e). Complexes DP-OH and TPE-Br also exhibited typical AIE properties in the DMSO/water solvent, where the fw values for the maximum emission were 60% and 90%, respectively (Fig. S8, ESI†).
Imaging and cellular localization
Due to their negative charge, both DP-PPh3 and TPE-PPh3 would pass thorough the phospholipid bilayers of the mitochondria, and bind to the inner mitochondrial membrane. To further verify the mitochondria-targeting capacities of the PPh3-decorated complexes, DP-PPh3 and TPE-PPh3, a commercial mitochondrial dye, MitoTracker red was used to co-stain the DP-PPh3 or TPE-PPh3 complexes in A549R cells (cis-Pt resistant human non-small-cell lung cancer cells). The green fluorescence from DP-PPh3 or TPE-PPh3 greatly overlapped with the red fluorescence from MitoTracker with Pearson's correlation coefficients (PCC) of 0.84 and 0.88, respectively, (Fig. 2a). In contrast, the PCC of DP-PPh3 and TPE-PPh3 with LysoTracker red were only 0.11 and 0.04 in A549R cells, respectively, which further suggested that DP-PPh3 and TPE-PPh3 have an excellent mitochondria-targeting capacity (Fig. S9, ESI†). However, the fluorescence of complexes DP-OH and TPE-Br was not observed in A549R cells from the confocal images, which may be due to the low cellular uptake and even distribution of DP-OH and TPE-Br in A549R cells to minimize the AIE effect.27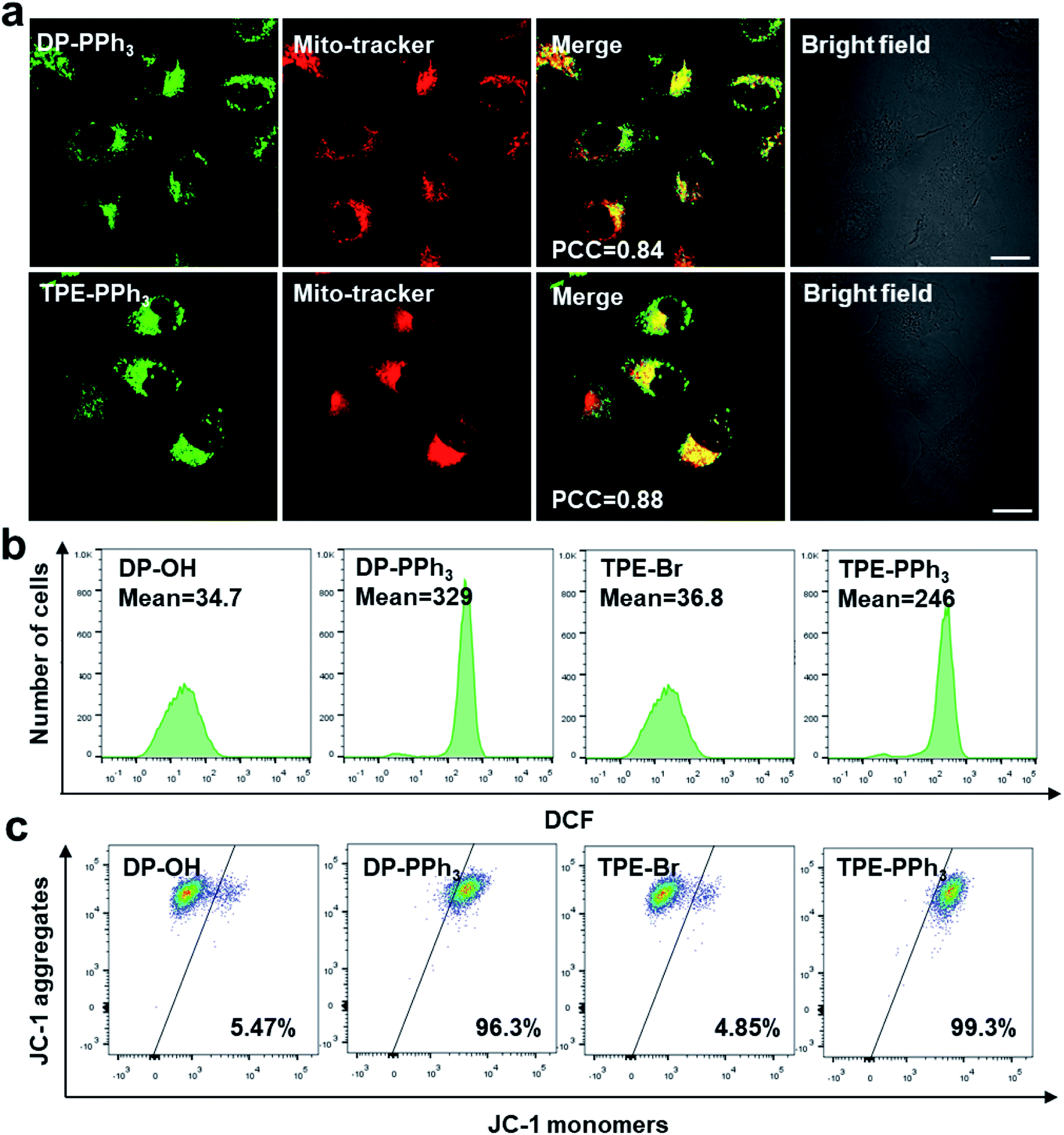 | ||
| Fig. 2 (a) The confocal microscopy images of A549R cells treated with DP-PPh3 (10 μM) and TPE-PPh3 (20 μM) after a 1 h incubation. The cells were co-stained with MitoTracker-Red (200 nM). Scale bar: 20 μm. The flow cytometry quantification of ROS generation in A549R cells (b) and JC-1-labeled A549R cells (c), with DP-OH (2 μM), DP-PPh3 (2 μM), TPE-Br (4 μM) and TPE-PPh3 (4 μM) for 24 h. | ||
In vitro cytotoxicity evaluation
Because both DP-PPh3 and TPE-PPh3 could localize in mitochondria, the cytotoxicities of DP-PPh3 and TPE-PPh3 together with those of DP-OH, TPE-Br were determined (by using the MTT assay) against different human cancer cell lines, including A549 (human non-small-cell lung cancer), A549R (cis-Pt resistant A549), A2780 (human ovarian cancer), MCF-7 (human breast adenocarcinoma), and Hep G2 (human liver cancer), as well as the normal cell line BEAS-2B (human lung epithelial cell), and HLF (human embryo-lung fibroblasts), with cis-Pt as a reference (Tables 1 and S1, ESI†). The results demonstrated that the cytotoxicity of DP-PPh3 or TPE-PPh3 was increased by at least 20-fold when compared to the corresponding results for DP-OH and TPE-Br. For example, the IC50 values of DP-OH and TPE-Br for A549 cells were 38.74 μM and 115.49 μM, whereas the IC50 values of DP-PPh3 or TPE-PPh3 for A549 cells were 1.19 μM and 3.39 μM, respectively. These results further illustrated the PPh3 moiety could facilitate the accumulation of the complex in the mitochondria and this was decisive for the activation of the cytotoxicity of DP-PPh3 or TPE-PPh3.The most striking feature for the DP-PPh3 or TPE-PPh3 complexes was their capacity to overcome the cis-Pt resistance of the A549R cells. The IC50 values of DP-PPh3 or TPE-PPh3 for A549R cells were 1.25 μM and 3.60 μM, respectively. The resistance factor (RF, the ratio of the IC50 values for A549R to that for A549) of DP-PPh3 and TPE-PPh3 were 1.05 and 1.06, respectively, which were significantly decreased compared to the RF value of cis-Pt (3.65).
ROS generation and mitochondrial membrane potential
Because both of the DP-PPh3 and TPE-PPh3 complexes accumulated in mitochondria, their impacts on mitochondria were first studied to understand the high cytotoxicity of the DP-PPh3 and TPE-PPh3 complexes. Mitochondria are the main organelle for reactive oxygen species (ROS) production and MMP is the basic characteristic which reflects mitochondrial integrity.30,31 Firstly, the ROS generation in the cis-Pt resistant A549R cells was determined using a green fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFDA), which could be oxidized by ROS to yield a strongly green fluorescent dichlorofluorescein (DCF).32 No obvious green fluorescence was observed in A549R cells in the presence of DP-OH or TPE-Br (Fig. S10, ESI†). However, a strong green fluorescence was observed when the A549R cells were treated with DP-PPh3 or TPE-PPh3 under the same conditions. Flow cytometry further confirmed that about 10-fold or 6-fold increase of ROS was generated in A549R cells after DP-PPh3 or TPE-PPh3 treatment when compared to the treatment with DP-OH or TPE-Br, respectively (Fig. 2b).The MMP was investigated using the mitochondria-selective aggregate dye JC-1, which emitted a strong green fluorescence once the MMP was dissipated.33 The DP-PPh3 or TPE-PPh3 treated A549R cells emitted a strong green fluorescence, which indicated the dissipation of MMP (Fig. S11, ESI†). Flow cytometric analysis was also performed to quantify the loss of MMP. In Fig. 2c, a significant loss of MMP for A549R cells in the presence of DP-PPh3 or TPE-PPh3 after 24 h incubation was observed, decreasing from 5% to 96% and 99%, respectively. These results suggested that both DP-PPh3 and TPE-PPh3 could induce ROS generation and severe depolarization of MMP.
Mitochondrial morphology and bioenergetics
The effects of DP-PPh3 and TPE-PPh3 on mitochondrial morphology in the A549R cells were then examined by transmission electron microscopy (TEM). The mitochondria in the untreated A549R cells showed a clear-outlined mitochondrial double-layer membrane and the cristae structure (Fig. 3a). In contrast, vacuolation was observed in the mitochondria after treatment with DP-PPh3 or TPE-PPh3, and most of the mitochondrial cristae structures were destroyed or had disappeared, indicating that the mitochondrial morphology was severely damaged.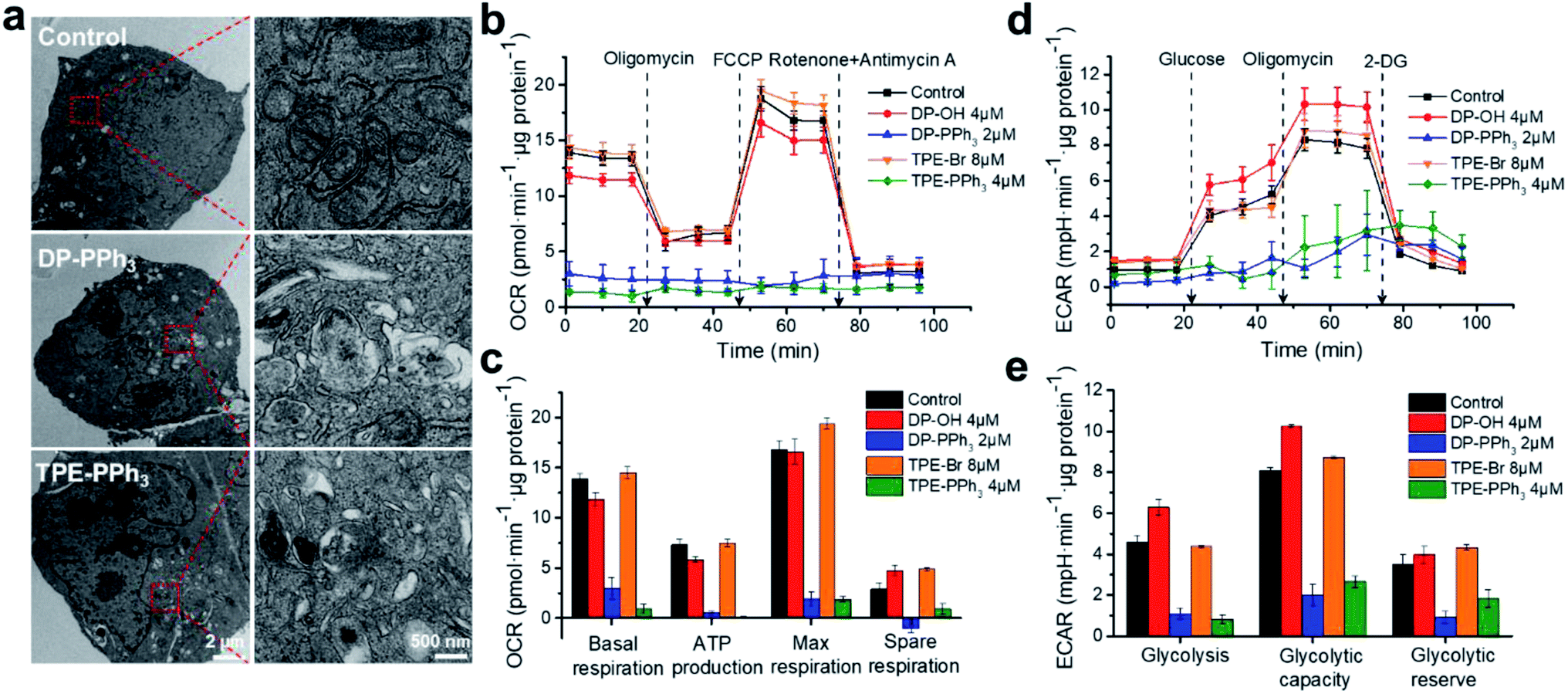 | ||
| Fig. 3 (a) Typical TEM images indicating the alterations in mitochondrial morphology in the A549R cells induced by DP-PPh3 (2 μM) and TPE-PPh3 (4 μM) for 24 h at 37 °C. (b) Kinetic profiles of OCR in A549R cells after treatment with DP-OH, DP-PPh3, TPE-Br, or TPE-PPh3 for 24 h. (c) Quantification of basal respiration, ATP production, maximum respiration, and spare respiratory capacity from the kinetic profiles of OCR. (d) Kinetic profiles of ECAR in A549R cells after treatment with DP-OH, DP-PPh3, TPE-Br, or TPE-PPh3 for 24 h. (e) Quantification of glycolysis, glycolytic capacity, and glycolytic reserve from the kinetic profiles of ECAR. Data are shown as mean ± SD (n = 3). | ||
To verify whether both of the DP-PPh3 or TPE-PPh3 complexes could be translocated to other organelles after the mitochondria were destroyed, a time dependent co-localization assay was performed for both complexes after 12 h and 24 h treatments (Fig. S12 and S13, ESI†). Excellent superimposition patterns between DP-PPh3/TPE-PPh3 and the MitoTracker channels were observed, whereas the overlap patterns of DP-PPh3/TPE-PPh3 with the LysoTracker channels were much more inferior (Fig. S12 and S13, ESI†). These results demonstrated that both the DP-PPh3 and TPE-PPh3 complexes remained in the mitochondria, and even the mitochondrial integration was destroyed.
As is already known, the inner membrane of mitochondria involves all the respiratory enzyme complexes, which are critical for oxidative phosphorylation (OXPHOS) and ATP production.34 The impact of DP-PPh3 and TPE-PPh3 on mitochondrial respiration of the A549R cells was then determined by the quantitative analysis of the oxygen consumption rate (OCR) using a Seahorse XFe24 Extracellular Flux Analyzer (Agilent). As shown in Fig. 3b and c, no obvious influence was observed for the DP-OH or TPE-Br treated cells, whereas mitochondrial respiration in the DP-PPh3 or TPE-PPh3 treated cells were dramatically suppressed. The basal respiration was decreased over 80% after the DP-PPh3 or TPE-PPh3 treatment, and the ATP product was even completely inhibited after the TPE-PPh3 treatment. Furthermore, it significantly ruined the reverse capacity of the maximal oxygen consumption of the cells, as there was no response to the carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP), an uncoupler of the proton gradient across the inner mitochondrial membrane. The mitochondrial respiration was shut down after the addition of rotenone and antimycin A (inhibitors for the mitochondrial complexes I and III, respectively).
Although mitochondrial respiration is strongly suppressed by DP-PPh3 and TPE-PPh3, the metabolism of the cancer cells was flexible. For example, the Warburg effect was well recognized, as the aerobic glycolysis would be activated instead of OXPHOS to supply the ATP production.35 Therefore, the extracellular acidification rate (ECAR) in DP-PPh3 and TPE-PPh3 treated A549R cells was also measured, which reflected the glycolytic capacity of the cells. As shown in Fig. 3d and e, the ECAR response to the glucose was considered as the basal condition. Once the oligomycin (the inhibitor for mitochondrial ATP production) was added, the energy product turned to glycolysis and this was considered as the maximum glycolytic capacity of the cells. The glycolytic reserve was calculated from the variation of ECAR before and after the treatment with oligomycin. According to the results indicated in Fig. 3e, the ECAR was dramatically decreased in A549R cells after the treatment with DP-PPh3 or TPE-PPh3. Thus, DP-PPh3 and TPE-PPh3 could not only efficiently destroy the mitochondrial structure but also lead to mitochondrial dysfunction including ATP production and aerobic glycolysis.
RNA-seq transcriptome analysis
To investigate the underlying molecular mechanisms of DP-PPh3 and TPE-PPh3 antitumor activity, RNA sequencing (RNA-seq) transcriptome analysis was used to compare the different gene expression profiles in DP-PPh3- or TPE-PPh3-treated A549R cells with that of untreated A549R cells.36 The PCCs between every two individual replicates from the same group were all above 0.99 (Fig. S14a, ESI†), which confirmed the reliability of the results. The cluster analysis further verified that the experiment was reproducible (Fig. S14b, ESI†). In the volcano diagram of all the compared data, it was found that the expression levels of 1161 genes were changed significantly after DP-PPh3 treatment, including 600 up-regulated genes, and 561 down-regulated genes (Fig. S15a, ESI†). The heat-map of the RNA-seq also showed similar gene expression patterns across replicates in each group, among which the blue color represented the lower transcription of genes, and the red color indicated high gene expression (Fig. 4a). Similarly, in the TPE-PPh3-treated A549R cells, the expression levels of 600 and 594 genes were found to be up-regulated and down-regulated, respectively, as shown in the volcano diagram (Fig. S15b, ESI†) and the heat-map diagram (Fig. S16a, ESI†).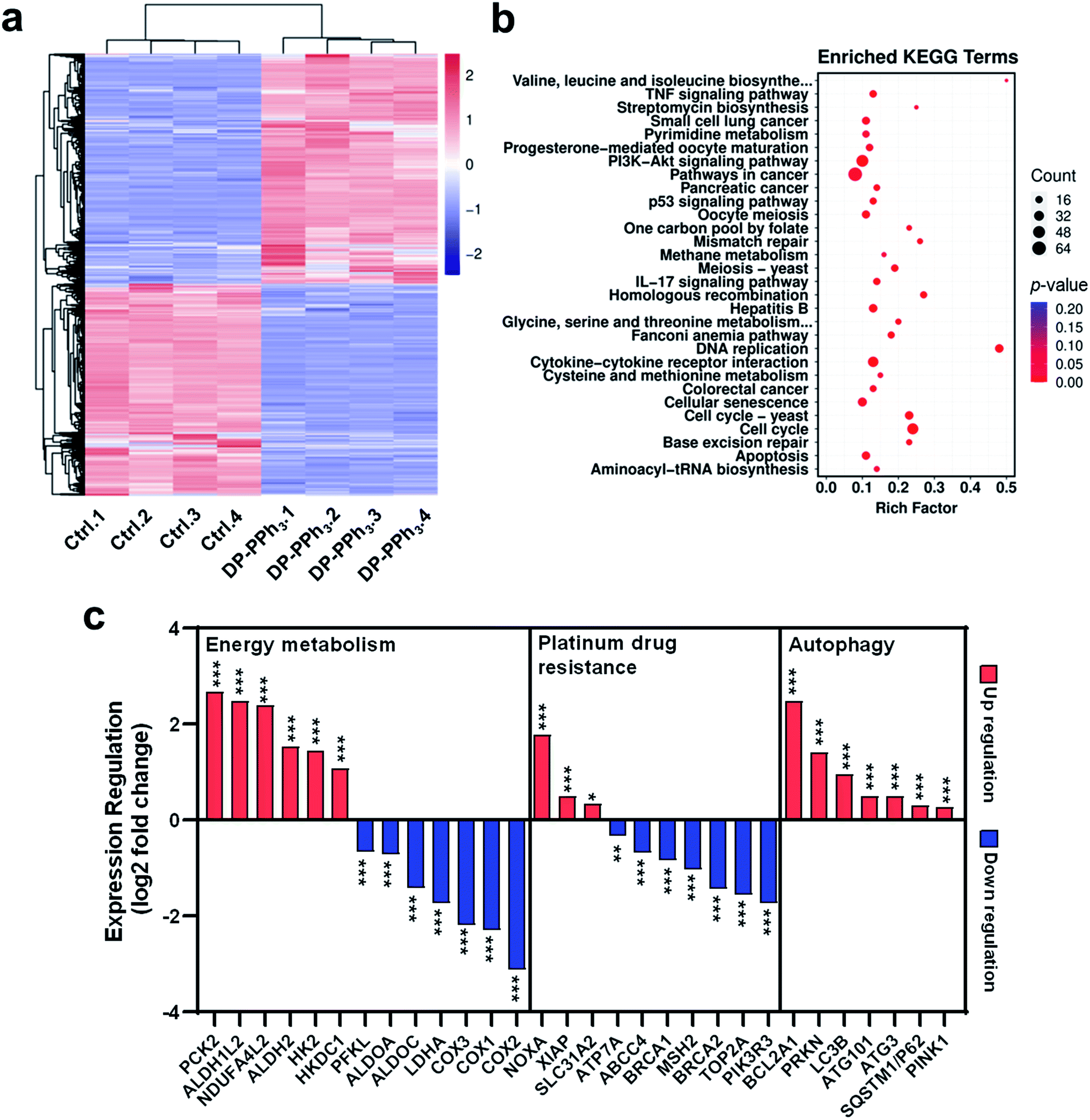 | ||
| Fig. 4 The RNA sequence analysis of differential gene expression (a), the KEGG enrichment analysis to identify signaling pathways (b), and expression regulation (c) in A549R cells after DP-PPh3 treatment (2 μM). *p < 0.05, ***p < 0.001. | ||
Gene ontology (GO) analysis was applied to perform enrichment analysis on the gene sets, where the DP-PPh3 and TPE-PPh3 treatment caused significant overall changes of A549R cells in gene categories, such as biological process, cellular component, and molecular function (Fig. S17, ESI†). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotation is a knowledge base for systematic analysis of gene functions, linking the genomic information with higher-order functional information.37 The KEGG analysis indicated that various pathways for cancer progression, such as the TNF signaling pathway, the PI3K-Akt signaling pathway, the IL-17 signaling pathway, and the p53 signaling pathway were regulated after the DP-PPh3 and TPE-PPh3 treatments (Fig. 4b and S16b, ESI†).
It is worth mentioning that the expression of oxidative phosphorylation-related genes (COX1, COX2, COX3, NDUFA4L2) and aerobic glycolysis-related genes (HK2, HKDC1, ALDH1L2, ALDHA, PCK2, PFKL, ALDOA, ALDOC) of the A549R cells were all significantly altered (either up-regulated or down-regulated) after the DP-PPh3 treatment (Fig. 4c).38–40 These results indicated that the DP-PPh3 treatment has been an effective influence on the energy metabolism of the A549R cells, which was consistent with the previous observations of the mitochondrial dysfunction (Fig. 3).
Moreover, several platinum resistance-related pathways of the A549R cells also showed severe alteration, either blocked or activated, after the DP-PPh3 treatment. For example, the expressions of the copper transporter family SLC31A2 were up-regulated significantly, and the expressions of the ATP-binding cassette (ABC) transporter family ABCC4 and ATPase copper transporter ATP7B were down-regulated, which could activate drug influx and block drug efflux. Furthermore, the mismatch repair systems including MSH2 and MSH6, and nucleotide excision repair systems including ERCC6, TOP2A, BRCA1, and BRCA2 were also deactivated.6,7 Similar results were also observed for the TPE-PPh3 treated A549R cells (Fig. S16, ESI†). These results are an obvious clue to explaining the mechanism for both DP-PPh3 and TPE-PPh3 complexes to overcome the cis-Pt resistance.
The autophagic pathway relative gene expressions (PRKN, PINK1, LC3, and SQSTM1) were also found to be enriched in the DP-PPh3- and TPE-PPh3-treated groups (Fig. 4c and S14, ESI†). In conclusion, the RNA-seq analysis suggested that the DP-PPh3 and TPE-PPh3 complexes could prevent cancer progression by the energy metabolism inhibition and autophagy, and overcome cis-Pt drug resistance by activation of drug influx and the blockade of the efflux.
Autophagic flux blockage
Mitochondrial damage could induce mitophagy or autophagy.41 Mitophagy is a unique form of selective macroautophagic/autophagy, where the mitochondrion is captured in an autophagosome and delivered to a lysosome for further degradation.42 In response to mitochondrial damage, PINK1 (PTEN induced kinase 1) accumulates on the outer mitochondrial membrane, where both ubiquitin and Parkin/PRKN (parkin RBR E3 ubiquitin protein ligase) were phosphorylated.43,44 Then, the autophagic adaptor protein sequestosome-1 (SQSTM1/p62) was recruited to the clustered mitochondria and interacted with LC3-II (microtubule-associated protein 1 light chain 3 II), and subsequently initiated the recognition of autophagosomes by the damaged mitochondria. Eventually, the damaged mitochondria was delivered to the lysosomes mediated by autophagosomes to form the autolysosome, which were degraded together with SQSTM1/p62.45,46The previous RNA-seq results showed that the relative gene expressions for autophagy were dramatically up-regulated after the DP-PPh3 and TPE-PPh3 treatment. To further demonstrate the observed alteration in the autophagic pathways, a western blotting assay was performed to quantify the relative protein expression (Fig. 5). The immunoblots showed that DP-PPh3 and TPE-PPh3 significantly increased the expression levels of PINK1 and Parkin/PRKN of the A549R cells in a concentration-dependent manner (Fig. 5a), when comparing to that in the DP-OH- and TPE-Br-treated A549R cells. The conversion from LC3-I to its lipidation form LC3-II (a specific marker of autophagy) was then also detected in A549R cells after treatment with DP-PPh3 and TPE-PPh3, respectively.22,47 The increasing LC3-II/LC3-I ratio in the DP-PPh3- and TPE-PPh3-treated A549R cells was also shown to occur in a highly dose-dependent manner (Fig. 5a).
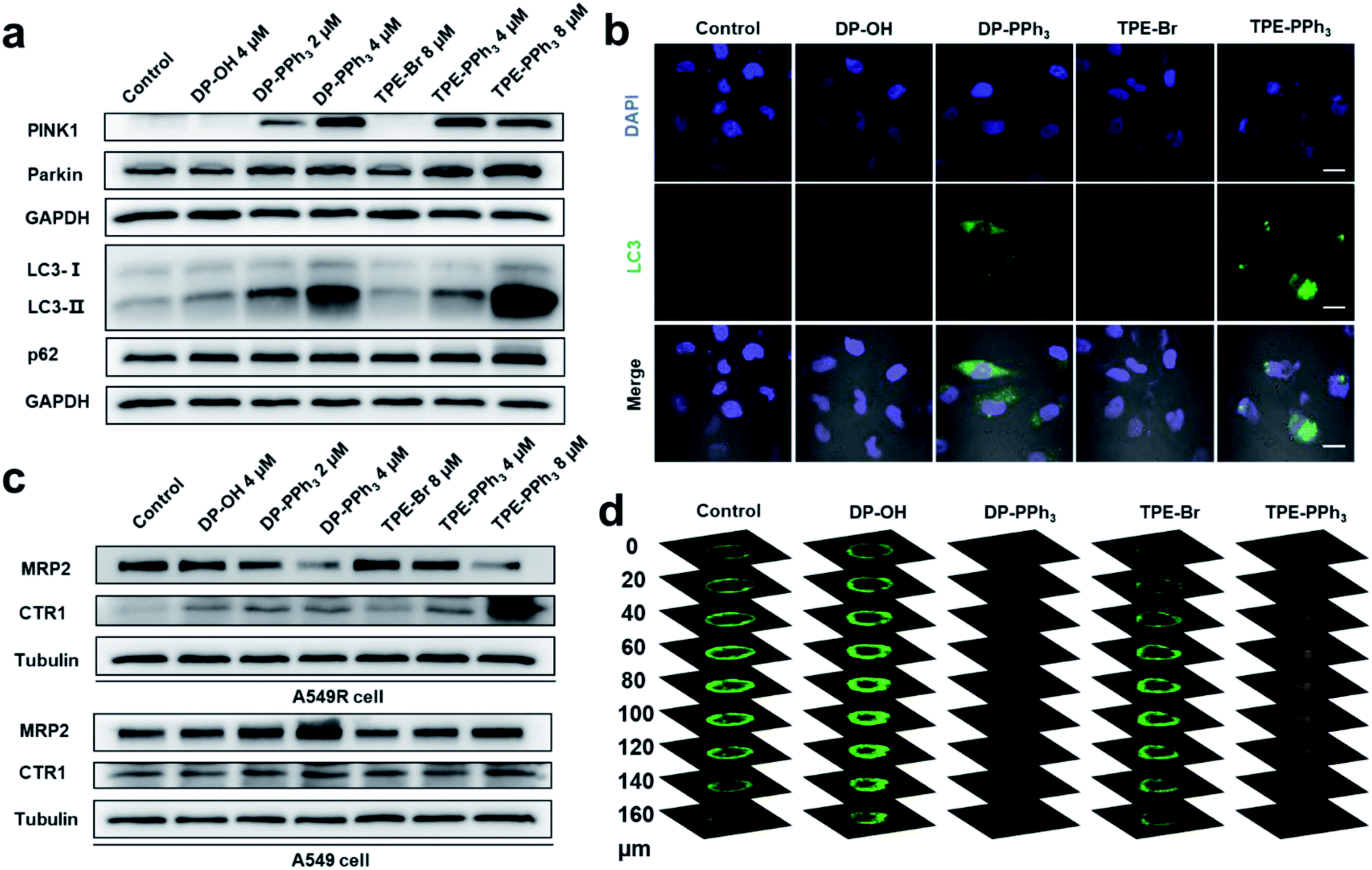 | ||
| Fig. 5 The results of the immunoblotting of PINK1, Parkin, LC3B, and p62 (a), and the expression levels of MRP2 and CTR1 (c) in A549R cells treated with DP-OH, DP-PPh3, TPE-Br, or TPE-PPh3 for 48 h. (b) Confocal microscopy images of A549R cells stained with LC3B after treatment with DP-OH (2 μM), DP-PPh3 (2 μM), TPE-Br (4 μM) or TPE-PPh3 (4 μM) for 48 h. Scale bar: 20 μm. (d) Depth of view of the confocal images of the 3D A549R tumor spheroids after treatment with DP-OH (10 μM), DP-PPh3 (10 μM), TPE-Br (20 μM) or TPE-PPh3 (20 μM) after 7 d. The live/dead cells were stained with calcein AM/PI. | ||
The onset of autophagy in the DP-PPh3- and TPE-PPh3-treated A549R cells was further demonstrated by the immunostaining of MAPLC3B (microtubule-associated proteins 1A/1B light chain 3B, a marker for autophagosomes).22,48 The punctate structures of LC3B were visualized in the confocal images of DP-PPh3- and TPE-PPh3-treated A549R cells (Fig. 5b), suggesting that autophagosomes had been generated.22,48 This was in good agreement with the TEM observations (Fig. 3a), where the typical vacuole-like morphology of autophagosomes was found in A549R cells after treatment with DP-PPh3 and TPE-PPh3.
According to previous reports, the p62 protein would degrade with the autophagosomes to complete the autophagic flux, which then lead to a decrease in the expression level of p62.22,46,48 However, no obvious alteration of the expression level of p62 was observed between the DP-PPh3- and TPE-PPh3-treated and the DP-OH- and TPE-Br-treated A549R cells (Fig. 5a). This suggested that the p62 degradation had been prevented and the autolysosome formation had been blocked by the treatment with DP-PPh3 and TPE-PPh3.22,48 The failure of the autophagic flux would result in the accumulation of damaged organelles, which is fatal to the cells.49 Thus, the high cytotoxicity of DP-PPh3 and TPE-PPh3 towards the A549R cells resulted from the autophagic flux blockage.
Drug metabolism pathway evaluation
Encouraged by the alternated expression levels of the copper transporter family and the ABC transporter family in RNA-seq results, next the expression of CTR1 and MRP2 were examined using a western blot assay (Fig. 5c). Proteins CTR1 and MRP2 were the two key transporters involved in the resistance to cis-Pt-based anticancer agents.8–11 In a previous study, Liu and co-workers reported obvious down-streamed expression of CTR1 in the A549R cells, where the cis-Pt resistance was overcome by using a conjugate of a cis-Pt prodrug and PEGylated gold nanorods to facilitate the delivery of a platinum-based prodrug into the cells.9 The MRP2 is an ABC transporter that contributes to multidrug resistance in various cancers by mediating the efflux of chemotherapy drugs.10As shown in Fig. 5c, the expression level of CTR1 in the untreated A549R cells was inhibited compared to that of the A549 cells, which was consistent with previous reports.9 The most noteworthy feature to mention here is that the drug metabolism in the A549R cells was completely reversed after the DP-PPh3 and TPE-PPh3 treatment, where the influx CTR1 was highly expressed and the efflux MRP2 was suppressed in a concentration-dependent manner (Fig. 5c). In contrast, in the DP-PPh3- and TPE-PPh3-treated A549 cells, the expression of CTR1 maintained nearly no change, whereas the MRP2 was highly expressed when compared to the untreated A549 cells (Fig. 5c). This suggested that the underlying mechanism for DP-PPh3 and TPE-PPh3 to overcome the cis-Pt resistance was due to the reverse of the metallodrug metabolism in the A549R cells.
The expression of P-glycoprotein (P-gp) was also examined, which was also known as multidrug resistance protein 1 (MDR1). As a member of the ABC superfamily, P-gp could efficiently expel a wide spectrum of chemotherapeutic agents out of cells, thereby decreasing their intracellular accumulation and therapeutic potency.50–52 Western blot results showed that there was no obvious suppression of P-gp after the DP-PPh3 or TPE-PPh3 treatment (Fig. S18, ESI†), indicating that P-gp was not affected and had little contribution to the capacity of either of the complexes to overcome resistance.
Biological evaluation of 3D A549R multicellular tumor spheroids
To evaluate the anti-proliferation activity of DP-PPh3 and TPE-PPh3 on the tumor spheroids, 3D A549R MCTSs with an average diameter of 500 nm were used to mimic the microenvironment of the solid tumor (Fig. 5d and S19, ESI†).53 After 7 d of incubation, the control spheroids and the TPE-Br-treated ones (20 μM, IC50 = 69.34 μM) grew to a diameter of about 650 nm, which demonstrated great survival conditions for the A549R spheroids (Fig. S19, ESI†). The DP-OH-treated ones (10 μM, IC50 = 29.96 μM) maintained a similar size due to their moderate cytotoxicity towards the A549R cells (Fig. S16, ESI†). Even though there was no obvious shrinkage of the size of the DP-PPh3- (10 μM, IC50 = 1.25 μM) or TPE-PPh3-treated spheroids (20 μM, IC50 = 3.60 μM), a double staining assay with calcein-AM (live cells, green fluorescence) and PI (dead cells, red fluorescence) demonstrated that most of the A549R cells were dead after the 7 d of treatment with DP-PPh3 or TPE-PPh3 (Fig. 5d). A red fluorescence was observed at a great depth of 160 nm of the DP-PPh3- or TPE-PPh3-treated A549R spheroids. In contrast, the control and the DP-OH- and TPE-Br-treated A549R cells were shown to be surviving well by the light green fluorescence (Fig. 5d). Thus, the DP-PPh3 and TPE-PPh3 complexes are proved to be promising anticancer agents to treat cis-Pt-resistant lung cancers.In vivo antitumor study
Encouraged by the results of the in vitro studies, the in vivo antitumor effect of DP-PPh3 and TPE-PPh3 in A549 tumor-bearing mice models were further evaluated. The tumor-bearing mice were randomly divided into four groups (n = 6) and injected intratumorally with DP-PPh3 or TPE-PPh3 or CDDP (cis-Pt) (5 mg kg−1 mice weight) or saline once every other day for 14 d. As shown in Fig. 6a–c, the growth of the tumors was significantly inhibited after the mice were treated with DP-PPh3 or TPE-PPh3. The average tumor volumes for the DP-PPh3 and TPE-PPh3 group after 14 d treatment were 528.19 mm3 and 667.97 mm3, respectively. The control group was characterized with rapid growth of the volume of the tumor to 1565.49 mm3. Moreover, the efficacy of the DP-PPh3 and TPE-PPh3 group were also superior to that of the positive CDDP-treated group (757.97 mm3). In addition, the body weight of the mice treated with DP-PPh3 and TPE-PPh3 showed no obvious change during the cancer treatment, unlike the control and CDDP-treated groups (Fig. 6d), which indicated that DP-PPh3 and TPE-PPh3 displayed potent antitumor activity in vivo and low toxicity to the mice.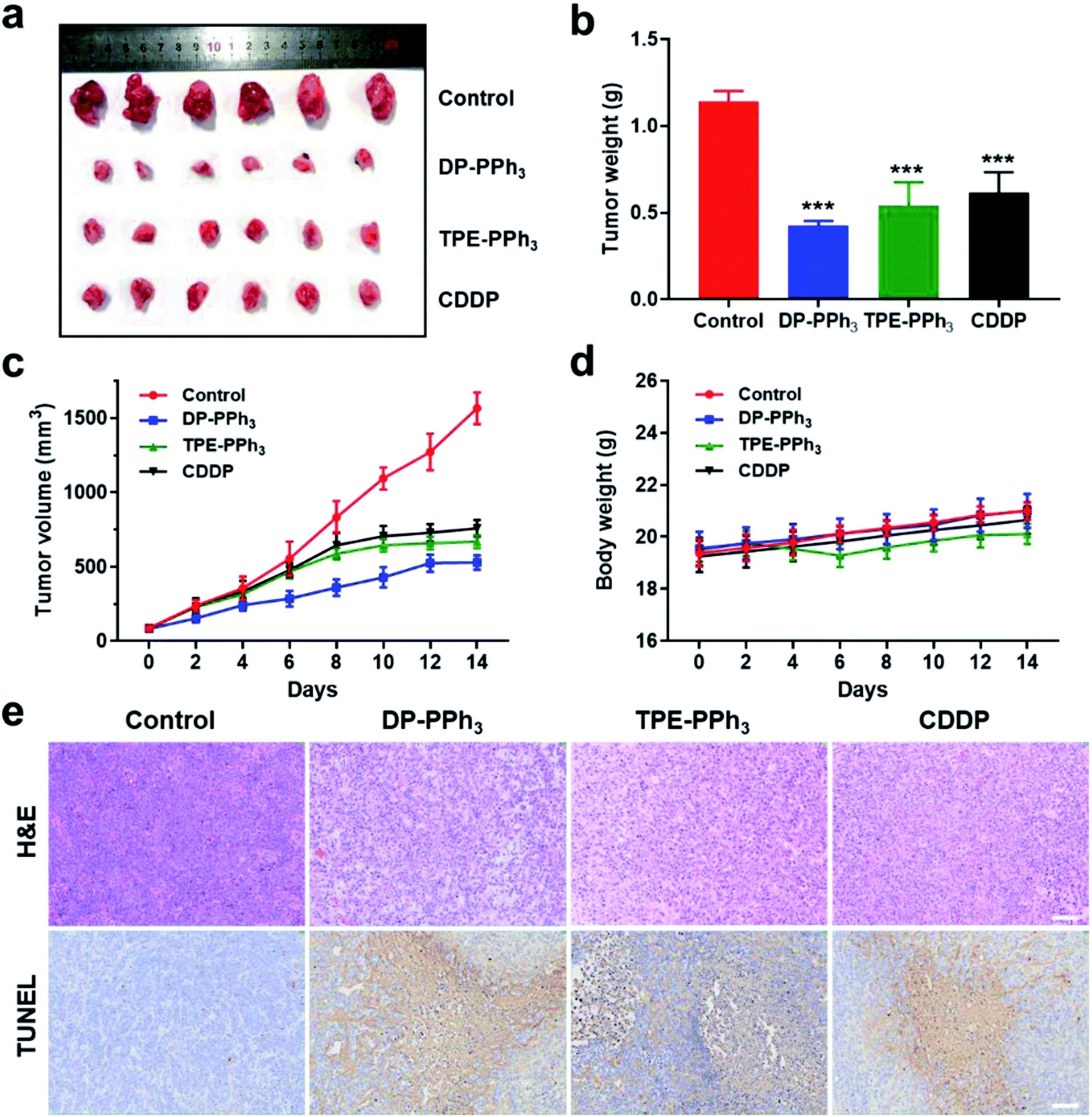 | ||
| Fig. 6 (a) Representative tumor samples collected from the mice after the injection of saline or DP-PPh3 or TPE-PPh3 or CDDP, for 14 d (n = 6). (b) Tumor weight of mice after the last determination, ***p < 0.001. (c) Time-dependent tumor growth curves after treatment. (d) Body weight variations during the treatment. (e) The H&E and TUNEL analyses of tumor tissues after treatment with different drugs. Scale bar: 100 μm. The dosage of DP-PPh3, TPE-PPh3, and CDDP was 5 mg per kg every second day. Saline was used as a control. | ||
Hematoxylin and eosin (H&E) staining and terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) staining assays were performed to evaluate the therapeutic efficacy of the different treatments. As can be seen in Fig. 6e, the images of the H&E and TUNEL-stained tumor tissue show that the DP-PPh3 and TPE-PPh3 induced a high level of cell death in the mice. However, the control group did not show obvious tumor necrosis. Notably, no obvious change in weight and no apparent lesions of the major organs were observed in mice treated with DP-PPh3 or TPE-PPh3 (Fig. S20, ESI†), indicating the minimal adverse effects and the excellent biocompatibility of DP-PPh3 or TPE-PPh3. All these results demonstrated the success of the strategy using mitochondrial targeting AIEgens for cancer therapy.
Discussion
In this work, the results of the experiments demonstrated that two mitochondria-targeted AIEgens, DP-PPh3 and TPE-PPh3 could accumulate in the mitochondria and gave a superior anticancer performance. Even though mitochondrial targeting AIEgens have been continuously reported over the last 20 years, cis-Pt -resistant cancer cells have still not been investigated.12,54 The work carried out here showed the underlying molecular mechanism, and for the first time revealed the remarkable capacity of AIEgens to overcome cis-Pt resistance. For example, in 2014, Liu and co-workers developed an AIE-mito-PPh3 that could selectively kill HeLa cancer cells by the destruction of MMP and elevation of the intracellular ROS levels.27 Lately, Zhang and co-workers reported several TPE-based pyridinium salts which induced ROS elevation and damaged the oxidative phosphorylation, leading to mitophagy in Hep G2 cells.49 The failure of autophagosome-lysosome fusion in the downstream autophagic flux resulted in cancer cell death. In addition, Tang and co-workers reported three pyridinium-functionalized TPE derivatives that could inhibit the expression of cell cycle-promoting genes to prevent cancer progression.29 Compared to the other reported mitochondria-targeted AIEgens, DP-PPh3 and TPE-PPh3 showed high cytotoxicity to cis-Pt-resistant A549R cancer cells with IC50 values of 1.19 μM and 3.39 μM, respectively. As far as is known, this is the first example of AIE-based probes used for cis-Pt-resistant cancer therapy.Incorporation of PPh3 into Pt-anticancer agents has been reported previously. Dhar and co-workers55 constructed mitochondria-targeted Pt(IV)-prodrug nanoparticles (Platin-M, with two lipophilic PPh3 cations in the axial positions), which could be used to precisely deliver the drug into the mitochondria. In the microenvironment of the tumor cells, the Pt(IV)-prodrug could be reduced, and then it binds to the mitochondrial DNA, destroys its structure, and then exhibits cellular activity against the cis-Pt resistant tumor cells. Wang and co-workers56 developed three mono-functional mitochondria-targeted Pt(II) complexes to suppress lung cancer by involving energy metabolism. Most PPh3 modified Pt-anticancer agents were designed to target mitochondrial DNA and metabolism. Once the PPh3 and the AIEgens were conjugated, the self-emitting fluorescence of the aggregates in the mitochondria could be observed, which then was able to achieve image-guided chemotherapy and the capacity to overcome cis-Pt resistance. This is the biggest difference to the PPh3 modified Pt-anticancer agents.
However, drug metabolism plays an important role in metallodrug resistance, which mainly involves the reduced drug uptake and enhanced efflux.3,5 The CTR1 and MRP2 were the two key transporters involved in cis-Pt influx and efflux to decrease the cis-Pt accumulation in cancer cells.8–10 In this work, the expression of CTR1 and the downstream expression of MRP2 in A549R cells it was successfully demonstrated. Furthermore, the mismatch repair systems including MSH2 and MSH6, and nucleotide excision repair systems including ERCC6, TOP2A, BRCA1, BRCA2 were all also deactivated in response to the treatment with DP-PPh3 and TPE-PPh3, which could also be responsible for their capacity to overcome the cis-Pt and this will be investigated in the future.6,7
Autophagy was a cytoprotective mechanism in response to cellular stress, however, unregulated autophagy can also be lethal.24,49 In a common scenario, the autophagosome would be swallowed by the lysosome and then be digested.23 Here, it was found that mitophagy was initiated, and the latter-stage autolysosome formation was inhibited after DP-PPh3 and TPE-PPh3 treatment, resulting in the accumulation of autophagic substrates. These observations suggested that the high cytotoxicity of these two AIEgens partially resulted from the autophagic flux blockage. It is worth noting that DP-PPh3 and TPE-PPh3 showed relatively low IC50 value in the normal BEAS-2B and HLF cells, which was even more toxic than cis-Pt. More research on the improving the selectivity to tumor cells with the tumor-targeting groups are needed in the future.
Conclusions
In summary, two PPh3-functionalized AIE-derivatives DP-PPh3 and TPE-PPh3 were developed and their anticancer activity against cis-Pt-resistant lung A549R cancer cells, is reported. The cytotoxicity experiments demonstrated that PPh3-decoration would be a feasible strategy to enhance AIEgens' cytotoxicity towards cancer cells due to their accumulation in the mitochondria. Specifically, DP-PPh3 and TPE-PPh3 could induce ROS production, decrease the mitochondrial membrane potential, disrupt the mitochondrial structure, and impair mitochondrial and glycolytic metabolism. More importantly, DP-PPh3 and TPE-PPh3 have shown the capacity to overcome the cis-Pt resistance of A549R cells through the alteration of the drug metabolism pathway (up-regulation of influx transporter CTR1 and down-regulation of efflux pump MRP2) and blockage of the autophagic flux (failure to degrade the autophagosomes). Moreover, DP-PPh3 and TPE-PPh3 displayed a good anticancer effect and undetectable systemic toxicity in vivo. This study not only provides two potent anticancer AIEgens but also demonstrates an effective strategy for the treatment of metallodrug resistant cancer cells through mitochondrial dysfunction. In this work, the linker may play some role, which would affect the lipophilic, cellular uptake, and photophysical properties of the molecules, which will be studied further in the future.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Investigation: Y. S., H. L., Y. T., M. M. W., G. D. Z. and J. Y. Writing: Y. S. for the original draft, Z. S. for the revised draft. Supervision and funding acquisition: Z. S. and H. K. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the National Natural Science Foundation of China (NSFC) (Grant No. 21771109, 21778033, 21977052) and the program of Jiangsu Specially-Appointed Professor for Prof. Z. Su.References
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- T. W. Hambley, Science, 2007, 318, 1392 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- A. H. Calvert, D. R. Newell, L. A. Gumbrell, S. O'Reilly, M. Burnell, F. E. Boxall, Z. H. Siddik, I. R. Judson, M. E. Gore and E. Wiltshaw, J. Clin. Oncol., 1989, 7, 1748–1756 CrossRef CAS PubMed.
- Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2536–2544 RSC.
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298 CrossRef CAS PubMed.
- Y. Min, C.-Q. Mao, S. Chen, G. Ma, J. Wang and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6742–6747 CrossRef CAS PubMed.
- A. D. Guminski, R. L. Balleine, Y.-E. Chiew, L. R. Webster, M. Tapner, G. C. Farrell, P. R. Harnett and A. deFazio, Gynecol. Oncol., 2006, 100, 239–246 CrossRef CAS PubMed.
- K. Katano, A. Kondo, R. Safaei, A. Holzer, G. Samimi, M. Mishima, Y. M. Kuo, M. Rochdi and S. B. Howell, Cancer Res., 2002, 62, 6559–6565 CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/b105159h.
- Y. Wang, J. Nie, W. Fang, L. Yang, Q. Hu, Z. Wang, J. Z. Sun and B. Z. Tang, Chem. Rev., 2020, 120, 4534–4577 CrossRef CAS PubMed.
- N. Song, P. Xiao, K. Ma, M. Kang, W. Zhu, J. Huang, D. Wang and B. Z. Tang, Chem. Res. Chin. Univ., 2021, 37, 52–65 CrossRef CAS.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- S. Vyas, E. Zaganjor and M. C. Haigis, Cell, 2016, 166, 555–566 CrossRef CAS PubMed.
- Y. Huang, G. Zhang, R. Zhao and D. Zhang, ChemMedChem, 2020, 15, 2220–2227 CrossRef CAS PubMed.
- K. Yu, J. Pan, E. Husamelden, H. Zhang, Q. He, Y. Wei and M. Tian, Chem.–Asian J., 2020, 15, 3942–3960 CrossRef CAS PubMed.
- N. Busschaert, S.-H. Park, K.-H. Baek, Y. P. Choi, J. Park, E. N. W. Howe, J. R. Hiscock, L. E. Karagiannidis, I. Marques, V. Félix, W. Namkung, J. L. Sessler, P. A. Gale and I. Shin, Nat. Chem., 2017, 9, 667–675 CrossRef CAS PubMed.
- K. Peynshaert, B. B. Manshian, F. Joris, K. Braeckmans, S. C. De Smedt, J. Demeester and S. J. Soenen, Chem. Rev., 2014, 114, 7581–7609 CrossRef CAS PubMed.
- I. Dikic and Z. Elazar, Nat. Rev. Mol. Cell Biol., 2018, 19, 349–364 CrossRef CAS PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef CAS PubMed.
- Z. Yang and D. J. Klionsky, Nat. Cell Biol., 2010, 12, 814–822 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069–1075 CrossRef CAS PubMed.
- X. Zhou, H. Yang, Z. Chen, S. Gong, Z.-H. Lu and C. Yang, J. Mater. Chem. C, 2019, 7, 6607–6615 RSC.
- W. Zhang, Y. Huang, Y. Chen, E. Zhao, Y. Hong, S. Chen, J. W. Y. Lam, Y. Chen, J. Hou and B. Z. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 10567–10577 CrossRef CAS PubMed.
- Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225–14229 CrossRef CAS PubMed.
- C.-J. Zhang, Q. Hu, G. Feng, R. Zhang, Y. Yuan, X. Lu and B. Liu, Chem. Sci., 2015, 6, 4580–4586 RSC.
- H.-T. Feng, S. Zou, M. Chen, F. Xiong, M.-H. Lee, L. Fang and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 11442–11450 CrossRef CAS PubMed.
- H. P. Indo, M. Davidson, H.-C. Yen, S. Suenaga, K. Tomita, T. Nishii, M. Higuchi, Y. Koga, T. Ozawa and H. J. Majima, Mitochondrion, 2007, 7, 106–118 CrossRef CAS PubMed.
- R. C. Scaduto and L. W. Grotyohann, Biophys. J., 1999, 76, 469–477 CrossRef CAS PubMed.
- D. Wu and P. Yotnda, J. Visualized Exp., 2011, 3357, DOI:10.3791/3357.
- S. T. Smiley, M. Reers, C. Mottola-Hartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3671 CrossRef CAS PubMed.
- S. Jin, Y. Hao, Z. Zhu, N. Muhammad, Z. Zhang, K. Wang, Y. Guo, Z. Guo and X. Wang, Inorg. Chem., 2018, 57, 11135–11145 CrossRef CAS PubMed.
- P. E. Porporato, N. Filigheddu, J. M. B.-S. Pedro, G. Kroemer and L. Galluzzi, Cell Res., 2018, 28, 265–280 CrossRef CAS PubMed.
- Y. Zheng, Y. Ling, D.-Y. Zhang, C.-P. Tan, H. Zhang, G.-G. Yang, H. Wang, L.-N. Ji and Z.-W. Mao, Small, 2021, 17, 2005086 CrossRef CAS PubMed.
- M. Kanehisa, Y. Sato, M. Kawashima, M. Furumichi and M. Tanabe, Nucleic Acids Res., 2016, 44, D457–D462 CrossRef CAS PubMed.
- Y.-D. Hu, H.-Z. Pang, D.-S. Li, S.-S. Ling, D. Lan, Y. Wang, Y. Zhu, D.-Y. Li, R.-P. Wei, H.-M. Zhang and C.-D. Wang, Gene, 2016, 592, 303–307 CrossRef CAS PubMed.
- T. R. Schmidt, D. E. Wildman, M. Uddin, J. C. Opazo, M. Goodman and L. I. Grossman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6379 CrossRef CAS PubMed.
- Y. Xu, Y. Guo, L. Chen, D. Ni, P. Hu and J. Shi, Chem. Sci., 2021, 12, 7763–7769 RSC.
- J. W. Harper, A. Ordureau and J.-M. Heo, Nat. Rev. Mol. Cell Biol., 2018, 19, 93–108 CrossRef CAS PubMed.
- W.-X. Ding and X.-M. Yin, Biol. Chem., 2012, 393, 547–564 CAS.
- A. Eiyama and K. Okamoto, Curr. Opin. Cell Biol., 2015, 33, 95–101 CrossRef CAS PubMed.
- L. A. Kane, M. Lazarou, A. I. Fogel, Y. Li, K. Yamano, S. A. Sarraf, S. Banerjee and R. J. Youle, J. Cell Biol., 2014, 205, 143–153 CrossRef CAS PubMed.
- H. Kong, C.-Y. Jiang, L. Hu, P. Teng, Y. Zhang, X.-X. Pan, X.-D. Sun and W.-T. Liu, J. Mol. Cell Biol., 2019, 11, 1056–1068 CrossRef CAS PubMed.
- G. Bjørkøy, T. Lamark, A. Brech, H. Outzen, M. Perander, A. Øvervatn, H. Stenmark and T. Johansen, J. Cell Biol., 2005, 171, 603–614 CrossRef PubMed.
- L. Laraia, K. Ohsawa, G. Konstantinidis, L. Robke, Y.-W. Wu, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2145–2150 CrossRef CAS PubMed.
- K.-H. Baek, J. Park and I. Shin, Chem. Soc. Rev., 2012, 41, 3245–3263 RSC.
- Y. Huang, X. You, L. Wang, G. Zhang, S. Gui, Y. Jin, R. Zhao and D. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10042–10051 CrossRef CAS PubMed.
- P. Zahedi, R. De Souza, L. Huynh, M. Piquette-Miller and C. Allen, Mol. Pharm., 2011, 8, 260–269 CrossRef CAS PubMed.
- J. I. Fletcher, M. Haber, M. J. Henderson and M. D. Norris, Nat. Rev. Cancer, 2010, 10, 147–156 CrossRef CAS PubMed.
- F. Bai, C. Wang, Q. Lu, M. Zhao, F.-Q. Ban, D.-H. Yu, Y.-Y. Guan, X. Luan, Y.-R. Liu, H.-Z. Chen and C. Fang, Biomaterials, 2013, 34, 6163–6174 CrossRef CAS PubMed.
- L. Sun, Y. Chen, S. Kuang, G. Li, R. Guan, J. Liu, L. Ji and H. Chao, Chem.–Eur. J., 2016, 22, 8955–8965 CrossRef CAS PubMed.
- S. Liu, G. Feng, B. Z. Tang and B. Liu, Chem. Sci., 2021, 12, 6488–6506 RSC.
- S. Marrache, R. K. Pathak and S. Dhar, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10444 CrossRef CAS PubMed.
- Z. Zhu, Z. Wang, C. Zhang, Y. Wang, H. Zhang, Z. Gan, Z. Guo and X. Wang, Chem. Sci., 2019, 10, 3089–3095 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06722b |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |