
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the reactivity of a cationic iminoborane: avenues to unusual boron cations†
Rui
Guo
a,
Xin
Zhang
b,
Tong
Li
a,
Qianli
Li
 c,
David A.
Ruiz
b,
Liu Leo
Liu
b,
Chen-Ho
Tung
a and
Lingbing
Kong
*ad
c,
David A.
Ruiz
b,
Liu Leo
Liu
b,
Chen-Ho
Tung
a and
Lingbing
Kong
*ad
aSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: konglb@sdu.edu.cn
bDepartment of Chemistry and Shenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen 518055, P. R. China
cSchool of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252059, P. R. China
dState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 26th January 2022
Abstract
A cationic terminal iminoborane [Mes*N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B ← IPr2Me2][AlBr4] (3+[AlBr4]−) (Mes* = 2,4,6-tri-tert-butylphenyl and IPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) has been synthesized and characterized. The employment of an aryl group and N-heterocyclic carbene (NHC) ligand enables 3+[AlBr4]− to exhibit both B-centered Lewis acidity and BN multiple bond reactivities, thus allowing for the construction of tri-coordinate boron cations 5+–12+. More importantly, initial reactions involving coordination, addition, and [2 + 3] cycloadditions have been observed for the cationic iminoborane, demonstrating the potential to build numerous organoboron species via several synthetic routes.
B ← IPr2Me2][AlBr4] (3+[AlBr4]−) (Mes* = 2,4,6-tri-tert-butylphenyl and IPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) has been synthesized and characterized. The employment of an aryl group and N-heterocyclic carbene (NHC) ligand enables 3+[AlBr4]− to exhibit both B-centered Lewis acidity and BN multiple bond reactivities, thus allowing for the construction of tri-coordinate boron cations 5+–12+. More importantly, initial reactions involving coordination, addition, and [2 + 3] cycloadditions have been observed for the cationic iminoborane, demonstrating the potential to build numerous organoboron species via several synthetic routes.
Introduction
Exploration of boron cations has attracted extensive attention not only due to their unique bonding environments and electronic configurations but also due to their potential applications in bond activation and catalysis.1,2 Generally, boron cations can be classified into borinium [R2B]+, borenium [R2BL]+, and boronium [R2BL2]+ cations based on the coordination number of boron (Fig. 1a). By installing π-donating ligands at the boron atom to compensate for the inherent electron deficiency, a few borinium ions have been isolated since the first report of diamido-substituted boron cations I and II by the Nöth group (Fig. 1b).3 Subsequently, Stephan and co-workers obtained the persistent borinium ion III supported by phosphoraniminato groups.4 In 2014, a seminal report on the synthesis of dimesityl boron cation IV was published by Shoji et al.5IV exhibits high Lewis acidity and electrophilic reactivity towards CO2, CS2, H2 and Et3SiH.6 In addition, twofold 1,2-carboration of diphenylacetylene with IV afforded divinylborinium ion V, in which the positive charge is delocalized over the entire π-conjugated system.7 More recently, the Inoue group demonstrated the asymmetric borinium ion VI ligated by imidazolin-2-imino and silyl substituents.8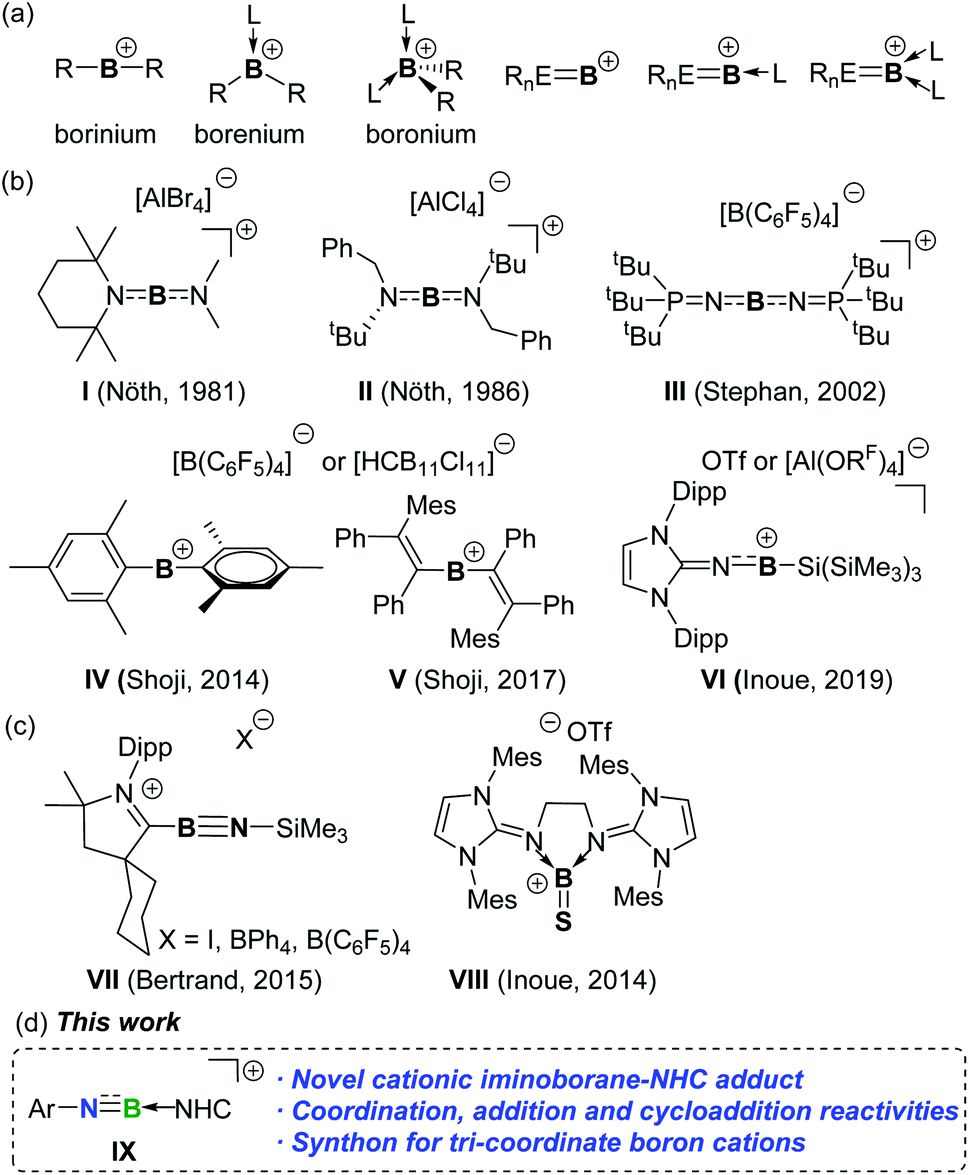 | ||
| Fig. 1 (a) Boron cations. (b) Structurally characterized borinium ions. (c) Cationic unsaturated boron compounds. (d) This work. | ||
Cationic boron species of type [RnEB]+ possess different bonding characters from classical boron cations and should be versatile reagents for organic synthesis through the functionalization of multiple bonds.9,10 Nevertheless, such species are still hitherto unknown. Even with Lewis base stabilization, only two corresponding [RnEBL]+ and [RnEBL2]+ derivatives, exemplified by iminoboryl-CAAC adducts VII and cationic thioxoborane VIII, have been described by the Bertrand, Stephan and Inoue groups, respectively (Fig. 1c).11,12VII-I with an iodide counter anion is in equilibrium with the neutral idodoiminoborane in solution, which alternatively served as a dipole to cyclize with CO2. Comparatively, VII-BPh4 and VII-B(C6F5)4 retaining the cationic structure both in the solid-state and in solutions do not exhibit dipole reactivity and are inert toward CO2. Neutral iminoboranes are extensively employed as synthons for constructing organoboron compounds through addition and cycloaddition reactions.13 Therefore, development of persistent yet highly reactive cationic counterparts and unravelling their reactivity would provide new avenues to cationic boron species. Herein, we report the access to the cationic iminoborane IX supported by an NHC, as well as its utility for constructing unusual B-containing frameworks (Fig. 1d).
Results and discussion
Synthesis and characterization of 3+[AlBr4]
Recently, we demonstrated a facile approach for the synthesis of iminoborane-NHC complexes by employing NHCs as Brønsted bases and ancillary ligands.14 In an analogous fashion, addition of two equiv. of 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (IPr2Me2) into Mes*NHBBr21 in toluene afforded compound 2 as colorless crystals in 56% yield (Scheme 1). The multi-nuclear NMR spectroscopic studies coupled with single crystal X-ray diffraction confirmed the formulation of 2 to be Mes*N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B(Br)IPr2Me2 (11B NMR: 9.4 ppm). In the solid state, the boron center of 2 adopts a planar geometry with the sum of angles at 359.9°. The Mes* and IPr2Me2 groups are located on the opposite sides of the B1N1 double bond (Fig. 2a).
B(Br)IPr2Me2 (11B NMR: 9.4 ppm). In the solid state, the boron center of 2 adopts a planar geometry with the sum of angles at 359.9°. The Mes* and IPr2Me2 groups are located on the opposite sides of the B1N1 double bond (Fig. 2a).
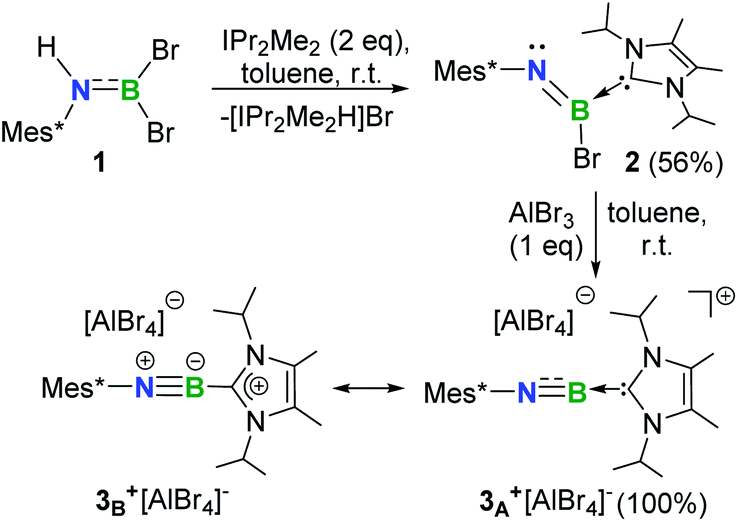 | ||
| Scheme 1 Synthesis of 2 and 3+[AlBr4]− (Mes* = 2,4,6-tri-tert-butylphenyl). | ||
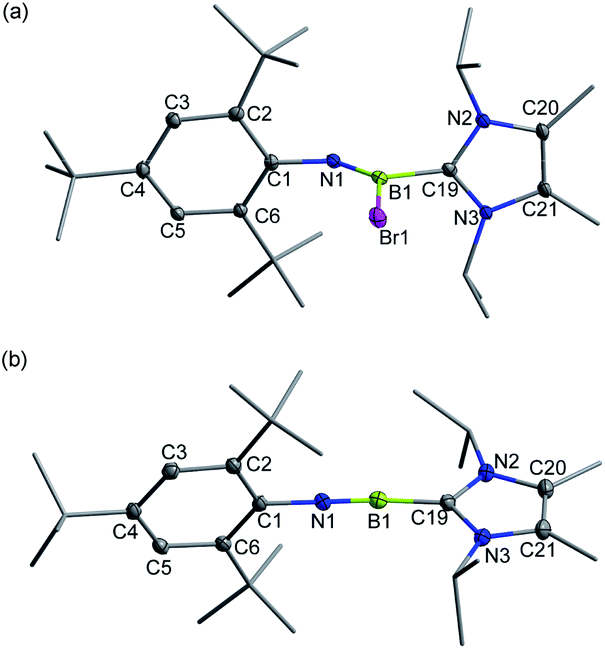 | ||
| Fig. 2 Solid-state structures of 2 (a) and 3+[AlBr4]− (b). The counterion and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
Halide abstraction of 2 was next examined. We reacted 2 with an equal molar amount of AlBr3 in toluene at ambient temperature for 5 minutes to obtain colorless solids of 3+[AlBr4]− quantitatively. The 11B NMR spectrum of 3+ shows a broad signal at 6.9 ppm, which is close to the chemical shifts seen for the iminoboranes ArBNR (2.8–4.3 ppm),13,15 borinium III (11.1 ppm)4 and cationic iminoboranes VII11 (7.7 and 7.4 ppm), but strongly upfield-shifted with respect to those of two-coordinate boron cations I, II, IV and V (36.7, 38.7, 93.3 and 74.0 ppm, respectively).3,5,7 A sharp singlet peak at 80.8 ppm assigned to [AlBr4]− was observed in the 27Al NMR spectrum, demonstrating the smooth bromide abstraction of 2 by AlBr3. Species 3+[AlBr4]− exhibits high thermal stability under an inert atmosphere and does not decompose even upon heating at 80 °C for 24 h in C6D6 (Fig. S71†).
Single crystal X-ray diffraction unambiguously confirmed the ionic nature of 3+[AlBr4]− (Fig. 2b). The B1 atom is ligated with IPr2Me2 and well-separated from the counterion [AlBr4]− with a distance of 4.2575(3) Å between the B1 and the closest bromine atom of the [AlBr4]− anion, which is much longer than the sum of the van der Waals radii (2.04 Å).16 The structure of 3+ shows an essentially linear C1–N1–B1–C19 core with slight bending at the N1 (178.6(4)°) and B1 (173.6(4)°) atoms, indicative of sp-hybridization of these two atoms. The B1–N1 bond length of 1.224(5) Å in 3+ was found to be identical to those of VII11 (1.229(3), 1.218(4) and 1.192(5) Å) within experimental uncertainty. In addition, the contracted B1–C19 (1.527(5) Å) and similar C1–N1 (1.389(4) Å) bond lengths in comparison to those in 2 (1.605(6) Å and 1.379(5) Å, respectively) were observed.
To further understand the electronic structure of 3+, DFT calculations at the B3LYP/6-311G* level of theory were performed. The calculated geometrical parameters agree well with those observed for the crystal structure of 3+. The 11B NMR chemical shift (5.8 ppm) obtained from GIAO calculations is consistent with the experimental value (6.9 ppm, Fig. S82†). The Wiberg Bond Index (WBI) for the B–N bond in 3+ is 1.98 (Table S2†), which is smaller than that of VII (2.10, Table S3†), while those for the C1–N1 and B1–C19 bonds are 1.05 and 0.94, respectively. The natural population analysis (NPA) shows that the boron atom has a positive charge of +0.75, whereas the N1 atom is negatively charged (−0.70 a.u.) (Fig. 3a and Table S4†), suggesting the pronounced polar nature of the B–N triple bond. Indeed, natural bond orbital (NBO) analysis reveals one σ- and two π-bonding features of the BN unit with significant contribution from the N atom (76%, 76% and 74%, respectively, Table S5†). The Mes* and IPr2Me2 groups bear +0.28 and +0.67 charges, respectively. Furthermore, second-order perturbation analysis confirmed the electronic conjugation between the BN triple bond and Mes* as well as imidazole rings with stabilization energies of 13.9 and 6.9 kcal mol−1, respectively (Fig. S85†).
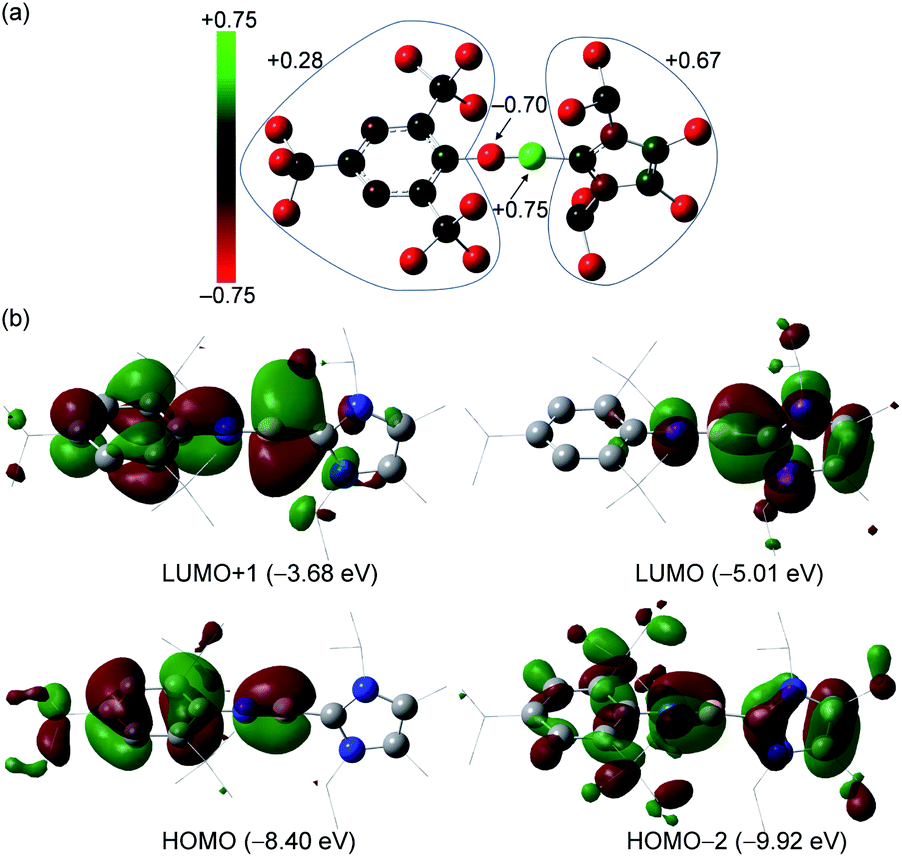 | ||
| Fig. 3 (a) Natural population analysis for 3+. (b) Selected molecular orbitals for 3+. Isovalue = 0.03. | ||
The HOMO and HOMO−2 of 3+ represent out-of-plane and in-plane B–N π-bonding orbitals, with contributions from π orbitals of Mes* or imidazole moieties, whereas the LUMO and LUMO+1 predominately involve their π*-components (Fig. 3b). The UV/Vis and IR spectroscopic analyses of 3+[AlBr4]− were also carried out. The former showed no detectable absorption between 240 and 400 nm (Fig. S15†), whereas the signal at a shorter wavelength (<240 nm) was too noisy or saturated for a reliable assignment. The latter measured in the solid state exhibited a characteristic peak at ṽ = 2004 cm−1 for the BN stretching vibration (Fig. S16†), which is comparable to that for tBuBNtBu (2018 cm−1)17 and is in good agreement with the calculated result (2008 cm−1, Fig. S84†).
Examination of the Lewis-acidity of 3+[AlBr4]−
The Lewis-acidity of 3+[AlBr4]− was first evaluated by computing its global electrophilicity index (GEI). The GEI value at the B3LYP/def2-TZVP level of theory was calculated to be 2.467 (Fig. S89†), which is slightly smaller than that for BCl3 (2.766).18 Next, the reactivity of 3+[AlBr4]− towards different Lewis bases was probed. It was demonstrated that hard halide ions such as chloride and bromide readily add to the boron center of 3+[AlBr4]−. Specifically, treatment of 3+[AlBr4]− with one equiv. of [PPN]Cl (PPN = Ph3PNPPh3) or [Ph4P]Br in toluene at ambient temperature gave the respective B-halogen substituted iminoboranes 4 and 2 smoothly (Scheme 2). The boron atom in 4 was observed at 12.8 ppm in the 11B NMR spectrum and the single-crystal X-ray analysis elucidated that 4 adopts a formal Z configuration similar to compound 2 (Fig. 4a). The B1–N1 bond length (1.305(7) Å) in 4 compares well with that for 2 (1.296(6) Å). Both values are longer than the BN triple bond length in 3+ (1.224(5) Å), illustrating the decreased B–N bond strength in iminoborane-NHC complexes 2 and 4.
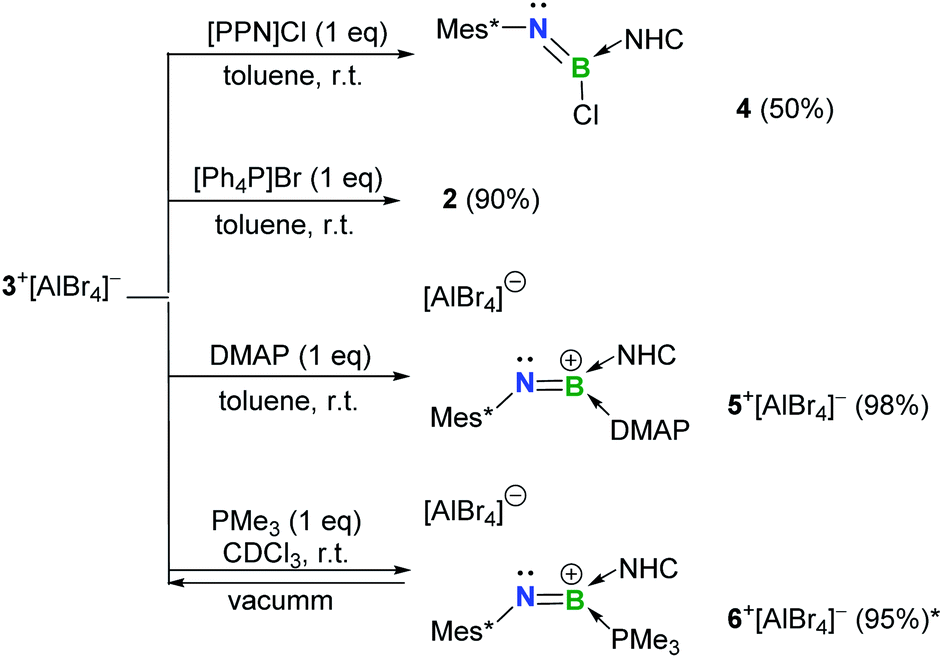 | ||
| Scheme 2 Reactivity of 3+[AlBr4]− toward bases (*NMR yield; NHC = IPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene; PPN = Ph3PNPPh3). | ||
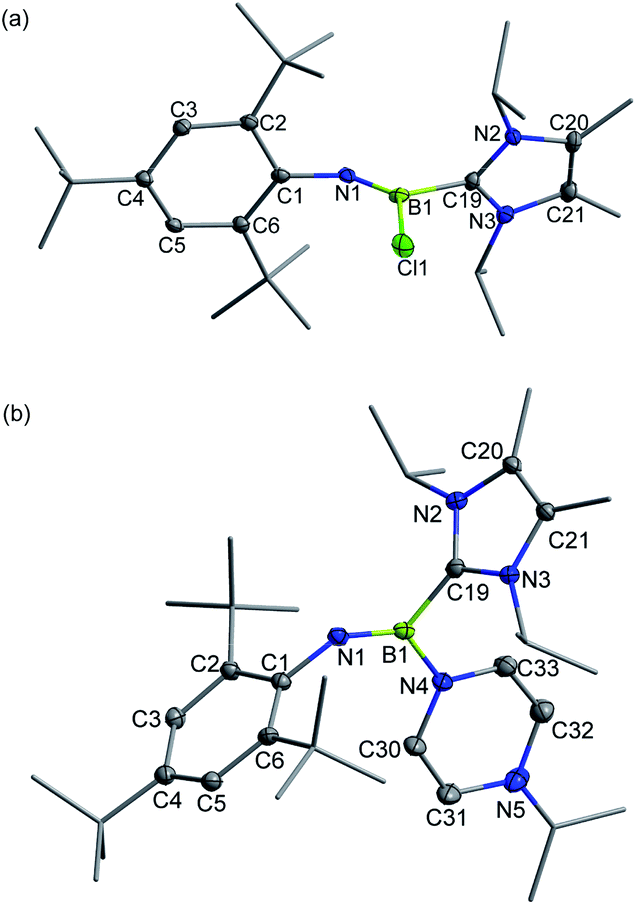 | ||
| Fig. 4 Solid-state structures of 4 (a) and 5+ (b). The counterion and hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
As for the neutral Lewis bases, 4-dimethylaminopyridine (DMAP) and trimethylphosphine (PMe3) were employed (Scheme 2). Addition of DMAP quantitatively afforded the corresponding cationic terminal iminoborane 5+[AlBr4]− (11B NMR: 15.4 ppm) in the coordination sphere of two different Lewis bases. The solid-state structure of 5+ revealed that the DMAP coordination causes the B1 and N1 atoms to be sp2-hybridized and results in a twisted C1–N1–B1–C19 skeleton (∠C1–N1–B1 = 139.9(3)°; ∠N1–B1–C19 = 123.2(3)°; Fig. 4b). Accordingly, the B1–N1 bond length of 1.323(4) Å is markedly longer than that of 3+ (1.224(5) Å) and its WBI is calculated to be 1.55 (Table S6†). In sharp contrast, the coordination of PMe3 with 3+[AlBr4]− was found to be reversible. Although multi-nuclear NMR spectroscopic investigations clearly showed the formation of an adduct 6+[AlBr4]−, evaporation of the CDCl3 solution of 6+[AlBr4]− regenerated species 3+[AlBr4]−. Indeed, PMe3 is much more fragile than NHC in 6+ judging from the corresponding bond dissociation energies (BDEs) of 3.9 and 41.8 kcal mol−1, respectively (Fig. S90†).
1,2-Addition of 3+[AlBr4]−
The addition of unactivated C–H bonds across iminoboranes to form aminoboranes is extremely challenging. It was found that in the presence of sodium cyanate (NaOCN), 3+[AlBr4]− converted into the product 7+[AlBr4]− (11B NMR: 40.8 ppm) at 50 °C (Scheme 3). Both NMR spectroscopic and X-ray single-crystal diffraction studies confirmed 7+[AlBr4]− to be a cyclic aminoborenium formed via an intramolecular C–H bond addition across the BN triple bond (Fig. 5a). NaOCN likely acts as a Lewis-base for coordination activation of the BN bond to enhance the basicity of N for capturing a proton from the methyl group. Note that a neutral three-coordinate borane enabled intramolecular C–H activation affording borocycles has been described by the Knochel research group.19 In addition, intermolecular addition proceeded smoothly between 3+[AlBr4]− and more acidic acetonitrile at ambient temperature to give the acyclic aminoborenium 8+[AlBr4]− (11B NMR: 37.2 ppm; Fig. 5b). It is proposed that this transformation may occur in successive steps involving acetonitrile complexation, proton migration and rearrangement of an aza-allene intermediate (Fig. S81†).20 Both reactions formed anti-addition products.
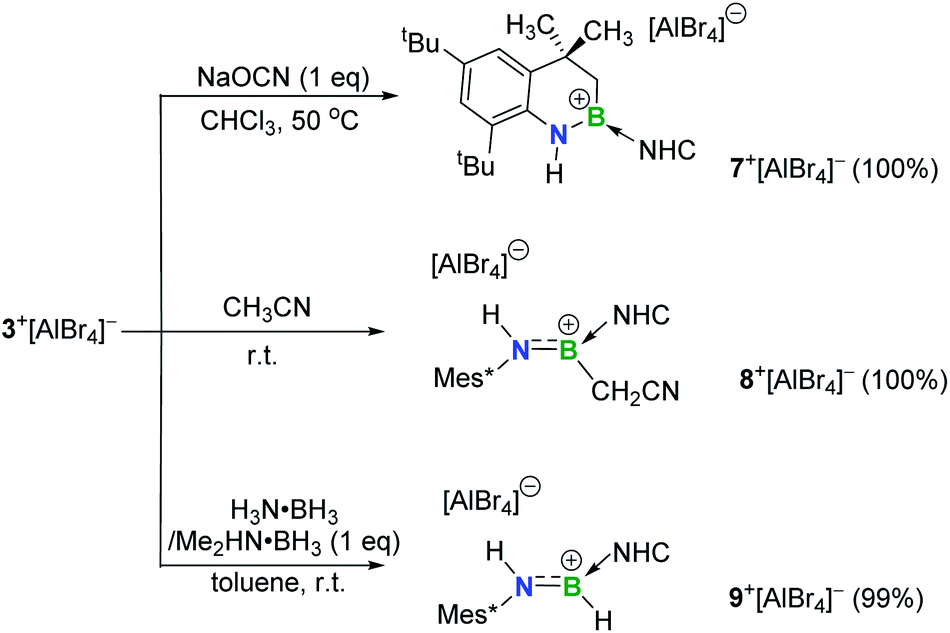 | ||
| Scheme 3 1,2-Addition of 3+[AlBr4]− (NHC = IPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). | ||
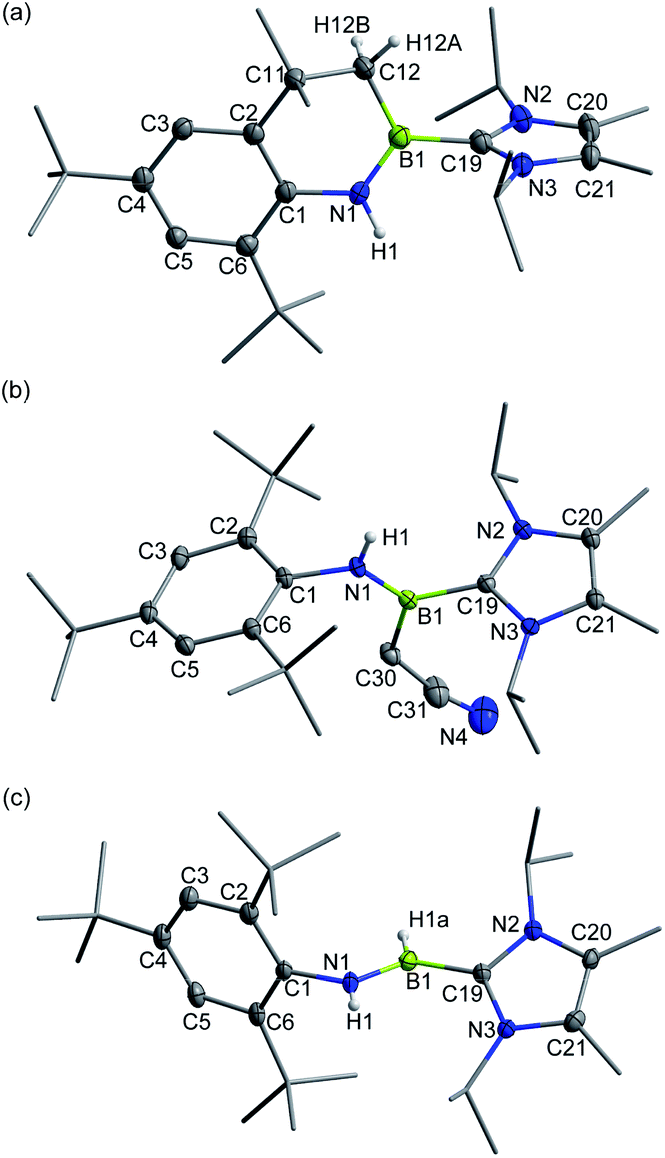 | ||
| Fig. 5 Solid-state structures of 7+ (a), 8+ (b) and 9+ (c). The counterion and hydrogen atoms except C12–H12A, C12–H12B, N1–H1 and B1–H1a are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
Inspired by a few successful examples of catalyst-free transfer hydrogenation (TH) of unsaturated compounds,21 the reactions of 3+[AlBr4]− with H3N·BH3 or Me2HN·BH3 were also conducted. The conversions were complete within half an hour at ambient temperature, generating the corresponding aminoborenium salt 9+[AlBr4]− (11B NMR: 34.1 ppm) with concomitant formation of the respective borazine and cyclodiborazane as the by-products (Fig. S72 and S73†). The solid-state structure of 9+[AlBr4]− revealed its trans stereochemistry (Fig. 5c), which is consistent with Braunschweig's results on TH of neutral iminoboranes with H3N·BH3.22 Deuterium labelling experiments using H3N·BD3, D3N·BH3 and D3N·BD3 resulted in deuterium incorporation at boron, nitrogen and both boron and nitrogen atoms, respectively, as evidenced by 2H NMR with characteristic peaks at 6.64 ppm (N–D) and 4.78 ppm (B–D) (Fig. S74†). This confirmed the polarity match mechanism of TH to 3+[AlBr4]−. This is different from the aminoborane iPr2NBH2, which readily undergoes reverse TH with Me2HN·BH3,23 and no further hydrogenation of species 9+[AlBr4]− to amineboronium could be observed in the presence of excess H3N·BH3 or Me2HN·BH3 under heating conditions (Fig. S75†).
[2 + 3] cycloaddition of 3+[AlBr4]−
The polar BN bond also enables the cycloaddition of 3+[AlBr4]−. Nitrone-, azide- and diazomethane-type 1,3-dipolar systems were thus employed.24 Treatment of 3+[AlBr4]− with one equiv. of N-tert-butyl-alpha-phenylnitrone smoothly resulted in the formation of colorless crystals of 10+[AlBr4]− (11B NMR: 25.2 ppm; Scheme 4), which were elucidated to be a cationic oxadiazaborolidine species with the newly formed C–N and B–O bonds (Fig. 6a).
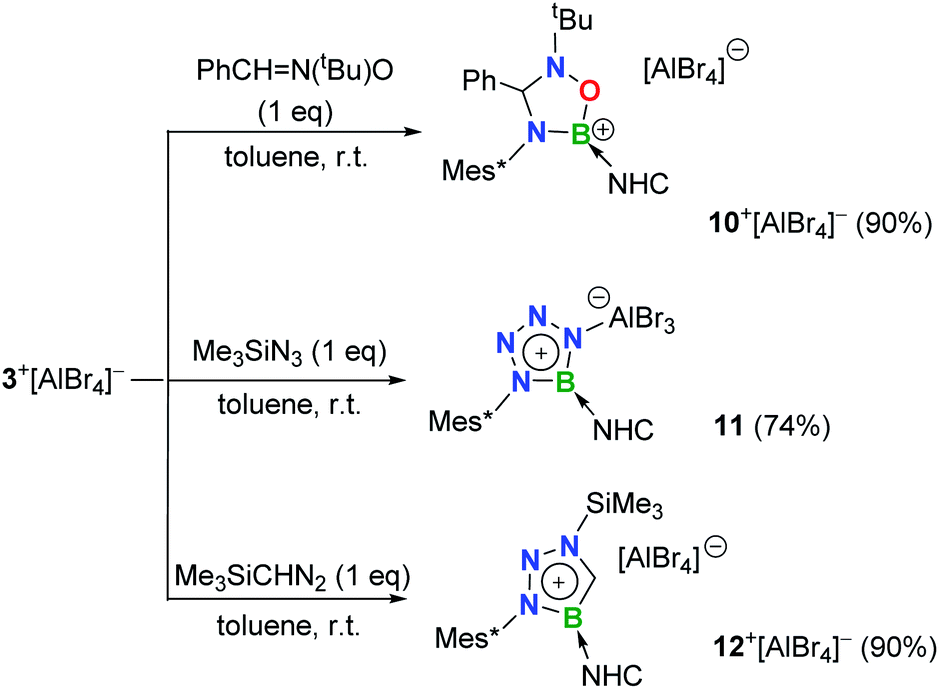 | ||
| Scheme 4 [2 + 3] cycloaddition of 3+[AlBr4]− (NHC = IPr2Me2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene). | ||
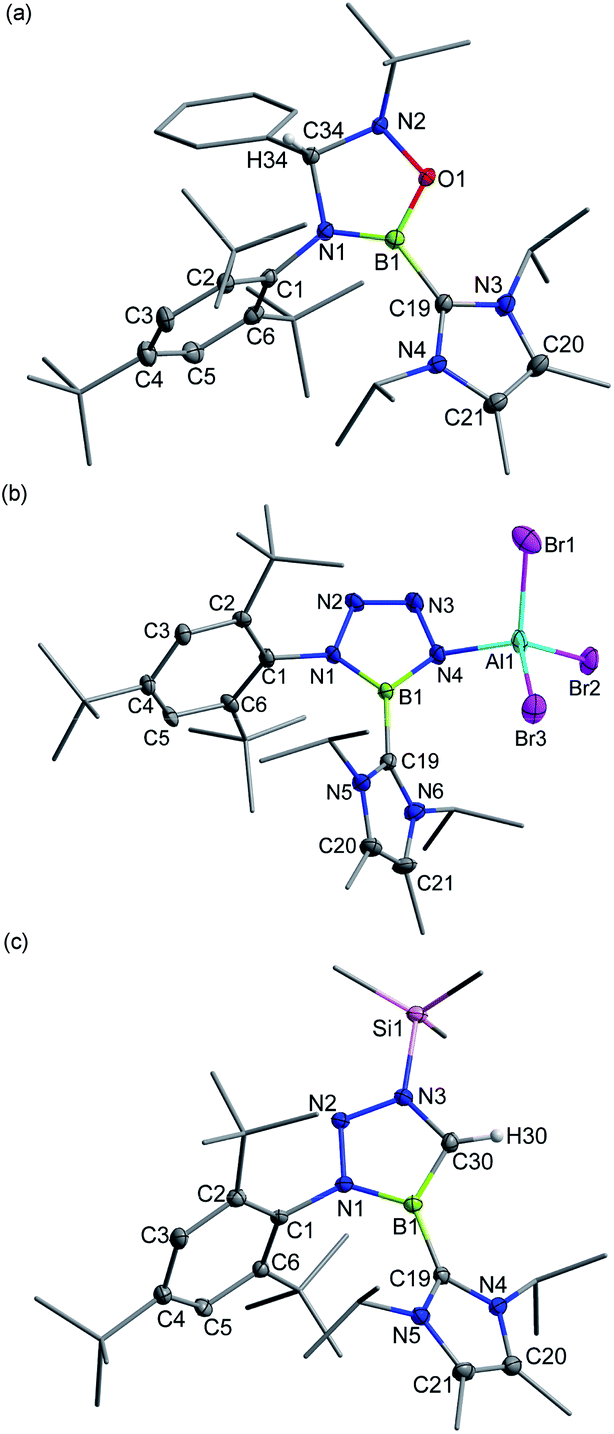 | ||
| Fig. 6 Solid-state structures of 10+ (a), 11 (b) and 12+ (c). The counterion and hydrogen atoms except C34–H34 and C30–H30 are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. | ||
When 3+[AlBr4]− was treated with trimethylsilyl azide (Me3SiN3) in toluene, a white precipitate of 11 was formed immediately. The 11B NMR of 11 showed a signal at 26.0 ppm, whereas 1H NMR revealed that no Me3Si– group was in the product. Single crystal X-ray diffraction revealed 11 to be a zwitterionic tetraazaborole species with cationic boron and anionic aluminium centers (Fig. 6b), resulting from a [2 + 3] cycloaddition and subsequent Al/Si exchange as well as Me3SiBr elimination. With respect to those of 1,4-diphenyl-5-terphenyltetraazaborole reported by the Braunschweig group,2511 features comparable B–N bond lengths (avg. 1.432 Å) and N–N double bond length (1.276(6) Å) as well as an essentially planar BN4 five-membered ring (the mean deviation from the plane is 0.0186 Å).
Moreover, 3+[AlBr4]− also cyclized with diazomethyl(trimethyl)silane to give product 12+[AlBr4]− (11B NMR: 22.6 ppm). Its solid structure confirmed the migration of the trimethylsilyl group from carbon to the nitrogen atom after cycloaddition to form an unprecedented cationic triazaborole skeleton (Fig. 6c).26 The B1–C30 bond length is 1.462(5) Å and its WBI value is 1.2978, thus confirming the typical double bond character.27 All five atoms of the BCN3 ring are perfectly coplanar with the sum of internal pentagon angles of 540°, and the calculated nucleus-independent chemical-shift values of NICS(0) and NICS(1) are −11.0 and −28.2, respectively, supporting its aromatic character.28 The computational studies reveal that the generation of aromatic 12+ from isomerization of the non-migrated intermediate is strongly exergonic (−37.1 kcal mol−1) and thus is thermodynamically favorable (Fig. S91†).
Conclusions
Collectively, this work unravels the elementary reactions of a cationic iminoborane, laying the foundation for efficient alternatives to build organoboron species. Employment of aryl and NHC ligands afforded isolable cationic iminoborane 3+. Both the boron atom and BN triple bond in 3+ could behave as reactive sites for derivatization enabling coordination, addition, and cycloaddition leading to a facile access to three-coordinate boron cations. This clearly confirms that substituent modification could boost the reactivity of cationic iminoboranes since analogous VII-BPh4 lacks the dipole reactivity toward CO2,11 CH3CN, H3N·BH3 and Me3SiN3 (Figs. S76–S78†). These systematic results indicate that unsaturated boron cations, which integrate both Lewis-acidity and multiple bond reactivities, are attractive synthons in organic synthesis. Investigations on the isolation and applications of more functional boron cations are continuously underway in our laboratory.
Data availability
Experimental and computational data has been provided as ESI.†Author contributions
L. K. designed the study and supervised the project. R. G. synthesized the complexes and conducted most of the measurements. X. Z., L. L. L. and L. K. performed the theoretical calculations. T. L. collected the crystal structures. Q. L. conducted the HRMS measurement. D. A. R., L. L. L., C.-H. T. and L. K. wrote the manuscript. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grants 21971144, 21702228 and 22101114), the Key R&D Program of Shandong Province (Grant 2019GGX102032), the Natural Science Foundation of Shandong Province (Grant ZR2019ZD46), the Qilu Youth Scholar Funding of Shandong University (Grant 11190088963021), the Multidisciplinary Research and Innovation Team of Young Scholars of Shandong University (Grant 2020QNQT007) and the Fundamental Research Funds for the Central Universities (Grant 2021JCG019). The theoretical work is supported by the Center for Computational Science and Engineering as well as CHEM High-Performance Supercomputer Cluster located in the Department of Chemistry at SUSTech. L. L. L. gratefully acknowledges the SUSTech start-up fund (Y01216248). Technical support from the SC-XRD platform of SDU SC&PP research facilities and Prof. Di Sun are acknowledged.Notes and references
- For selected reviews, see: (a) B. Rao and R. Kinjo, Chem.–Asian J., 2018, 13, 1279–1292 CrossRef CAS PubMed; (b) S. Dagorne and R. Wehmschulte, ChemCatChem, 2018, 10, 2509–2520 CrossRef CAS; (c) G. I. Nikonov, ACS Catal., 2017, 7, 7257–7266 CrossRef CAS; (d) W. Li, X. Ma, M. G. Walawalkar, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2017, 350, 14–29 CrossRef CAS; (e) P. Eisenberger and C. M. Crudden, Dalton Trans., 2017, 46, 4874–4887 RSC; (f) B. Michelet, S. Tang, G. Thiery, J. Monot, H. Li, R. Guillot, C. Bour and V. Gandon, Org. Chem. Front., 2016, 3, 1603–1613 RSC; (g) Y. Sarazin and J.-F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS PubMed; (h) C. Bour and V. Gandon, Coord. Chem. Rev., 2014, 279, 43–57 CrossRef CAS; (i) T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Widera, E. Filbeck and H.-J. Himmel, Eur. J. Inorg. Chem., 2020, 3017–3029 CrossRef CAS; (b) D. Franz and S. Inoue, Chem. –Eur. J., 2019, 25, 2898–2926 CrossRef CAS PubMed; (c) H.-J. Himmel, Angew. Chem., Int. Ed., 2019, 58, 11600–11617 CrossRef CAS PubMed; (d) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (e) Y.-F. Lin and C.-W. Chiu, Chem. Lett., 2017, 46, 913–922 CrossRef CAS; (f) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC; (g) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS; (h) S. Dagorne and D. A. Atwood, Chem. Rev., 2008, 108, 4037–4071 CrossRef CAS PubMed; (i) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed; (j) D. A. Atwood, Coord. Chem. Rev., 1998, 176, 407–430 CrossRef CAS; (k) D. Atwood and J. Jegier, Chem. Commun., 1996, 1507–1508 RSC; (l) P. Kölle and H. Nöth, Chem. Rev., 1985, 85, 399–418 CrossRef.
- (a) H. Nöth, R. Staudigl and H.-U. Wagner, Inorg. Chem., 1982, 21, 706–716 CrossRef; (b) P. Kölle and H. Nöth, Chem. Ber., 1986, 119, 313–324 CrossRef.
- S. Courtenay, J. Y. Mutus, R. W. Schurko and D. W. Stephan, Angew. Chem., Int. Ed., 2002, 41, 498–501 CrossRef CAS.
- Y. Shoji, N. Tanaka, K. Mikami, M. Uchiyama and T. Fukushima, Nat. Chem., 2014, 6, 498–503 CrossRef CAS PubMed.
- (a) K. L. Bamford, Z.-W. Qu and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 6180–6184 CrossRef CAS PubMed; (b) C. J. Major, K. L. Bamford, Z.-W. Qu and D. W. Stephan, Chem. Commun., 2019, 55, 5155–5158 RSC; (c) K. L. Bamford and D. W. Stephan, Dalton Trans., 2020, 49, 17571–17577 RSC.
- N. Tanaka, Y. Shoji, D. Hashizume, M. Sugimoto and T. Fukushima, Angew. Chem., Int. Ed., 2017, 56, 5312–5316 CrossRef CAS PubMed.
- D. Franz, T. Szilvási, A. Pöthig and S. Inoue, Chem. –Eur. J., 2019, 25, 11036–11041 CrossRef CAS PubMed.
- For ylide or carbodicarbene stabilized boron cations featuring partial BC double bonds, see:
(a) W.-C. Chen, C.-Y. Lee, B.-C. Lin, Y.-C. Hsu, J.-S. Shen, C.-P. Hsu, G. P. A. Yap and T.-G. Ong, J. Am. Chem. Soc., 2014, 136, 914–917 CrossRef CAS PubMed;
(b) T. Scherpf, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2017, 56, 3275–3279 CrossRef CAS PubMed.
- For transition metal substituted boron cations, see: (a) H. Braunschweig, K. Radacki and A. Schneider, Chem. Commun., 2010, 46, 6473–6475 RSC; (b) H. Braunschweig, K. Kraft, T. Kupfer, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2008, 47, 4931–4933 CrossRef CAS PubMed; (c) S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, D. L. Kays, N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2006, 45, 6118–6122 CrossRef CAS PubMed; (d) H. Braunschweig, K. Radacki and K. Uttinger, Angew. Chem., Int. Ed., 2007, 46, 3979–3982 CrossRef CAS PubMed; (e) H. Braunschweig, K. Radacki, D. Rais and D. Scheschkewitz, Angew. Chem., Int. Ed., 2005, 44, 5651–5654 CrossRef CAS PubMed; (f) D. L. Coombs, S. Aldridge, C. Jones and D. J. Willock, J. Am. Chem. Soc., 2003, 125, 6356–6357 CrossRef CAS PubMed.
- F. Dahcheh, D. W. Stephan and G. Bertrand, Chem. –Eur. J., 2015, 21, 199–204 CrossRef CAS PubMed.
- D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2014, 53, 14264–14268 CrossRef CAS PubMed.
- For selected reviews, see: (a) H. Nöth, Angew. Chem., Int. Ed. Engl., 1988, 27, 1603–1622 CrossRef; (b) P. Paetzold, Adv. Inorg. Chem., 1987, 31, 123–170 CrossRef CAS.
- (a) R. Guo, J. Jiang, C. Hu, L. L. Liu, P. Cui, M. Zhao, Z. Ke, C.-H. Tung and L. Kong, Chem. Sci., 2020, 11, 7053–7059 RSC; (b) P. Cui, R. Guo, L. Kong and C. Cui, Inorg. Chem., 2020, 59, 5261–5265 CrossRef CAS PubMed.
- (a) G. Elter, M. Neuhaus, A. Meller and D. Schmidt-Bäse, J. Organomet. Chem., 1990, 381, 299–313 CrossRef CAS; (b) M. Nutz, B. Borthakur, R. D. Dewhurst, A. Deißenberger, T. Dellermann, M. Schäfer, I. Krummenacher, A. K. Phukan and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- P. Paetzold, C. von Plotho, G. Schmid, R. Boese, B. Schrader, D. Bougeard, U. Pfeiffer, R. Gleiter and W. Schüfer, Chem. Ber., 1984, 117, 1089–1102 CrossRef CAS.
- (a) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Inorg. Chem., 2018, 57, 14764–14771 CrossRef CAS PubMed; (b) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 RSC.
- H. Laaziri, L. O. Bromm, F. Lhermitte, R. M. Gschwind and P. Knochel, J. Am. Chem. Soc., 1999, 121, 6940–6941 CrossRef CAS.
- A. Ansorge, D. J. Brauer, H. Bürger, T. Hagen and G. Pawelke, J. Organomet. Chem., 1993, 444, 5–14 CrossRef CAS.
- (a) S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed., 2021, 60, 14272–14294 CrossRef CAS PubMed; (b) D. H. A. Boom, A. R. Jupp and J. Chris Slootweg, Chem. –Eur. J., 2019, 25, 9133–9152 CrossRef CAS PubMed.
- L. Winner, W. C. Ewing, K. Geetharani, T. Dellermann, B. Jouppi, T. Kupfer, M. Schäfer and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 12275–12279 CrossRef CAS PubMed.
- E. M. Leitao, N. E. Stubbs, A. P. M. Robertson, H. Helten, R. J. Cox, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2012, 134, 16805–16816 CrossRef CAS PubMed.
- P. Paetzold, E. Schröder, G. Schmid and R. Boese, Chem. Ber., 1985, 118, 3205–3216 CrossRef CAS.
- (a) M. Nutz, B. Borthakur, R. D. Dewhurst, A. Deißenberger, T. Dellermann, M. Schäfer, I. Krummenacher, A. K. Phukan and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef CAS PubMed; (b) D. Prieschl, G. Belanger-Chabot, X. Guo, M. Dietz, M. Müller, I. Krummenacher, Z. Lin and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 1065–1076 CrossRef CAS PubMed.
- For cationic boroles, see: (a) T. Heitkemper and C. P. Sindlinger, Chem. –Eur. J., 2020, 26, 11684–11689 CrossRef CAS PubMed; (b) W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, A. Kaur, D. J. D. Wilson and R. J. Gilliard Jr, Chem. –Eur. J., 2019, 25, 12512–12516 CrossRef CAS PubMed; (c) C. K. Narula and H. Nöth, Inorg. Chem., 1985, 24, 2532–2539 CrossRef CAS.
- A. Berndt, Angew. Chem., Int. Ed., 1993, 32, 985–1009 CrossRef.
- B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2078017–2078022; 2109192–2109195. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00002d |
| This journal is © The Royal Society of Chemistry 2022 |