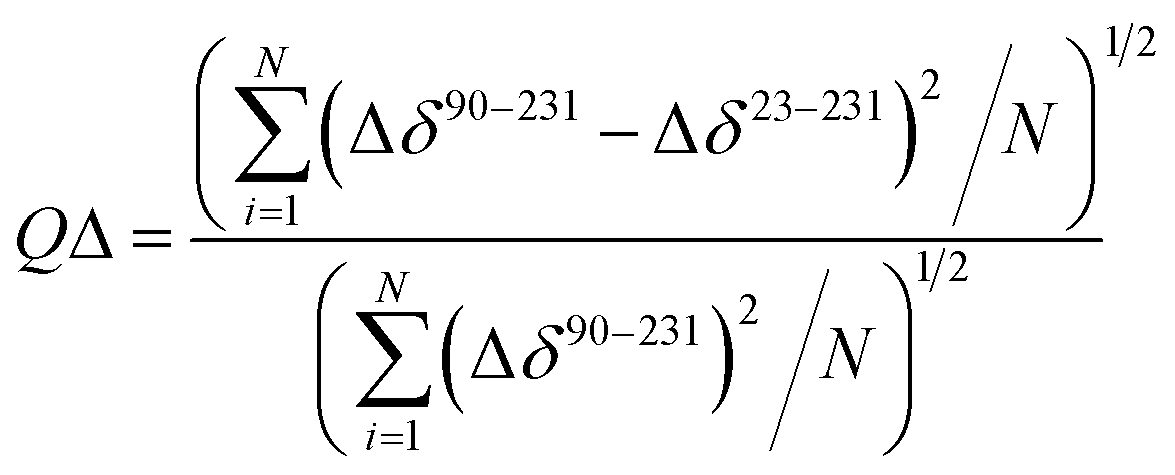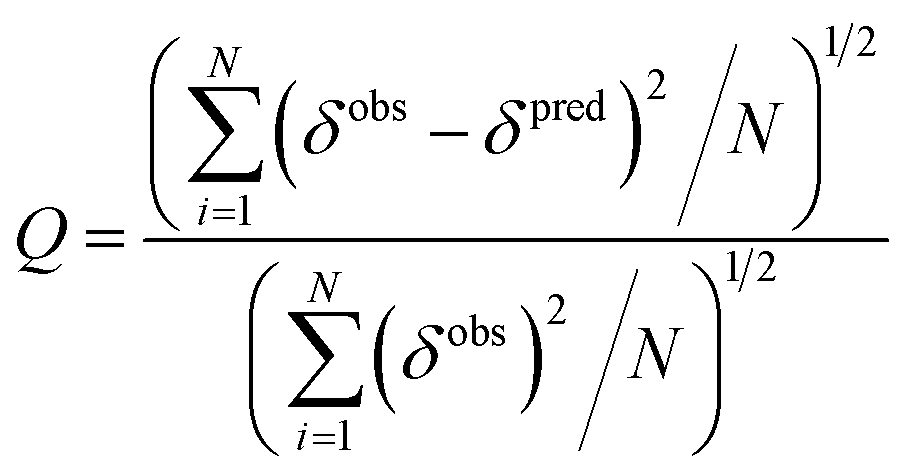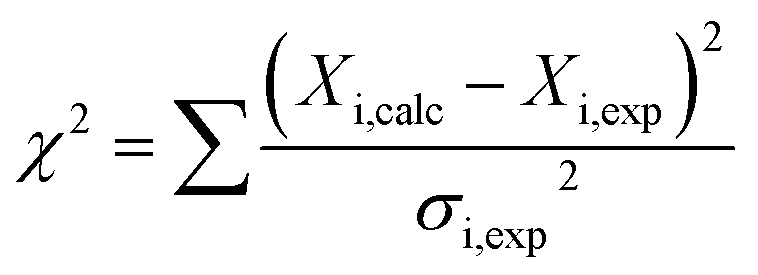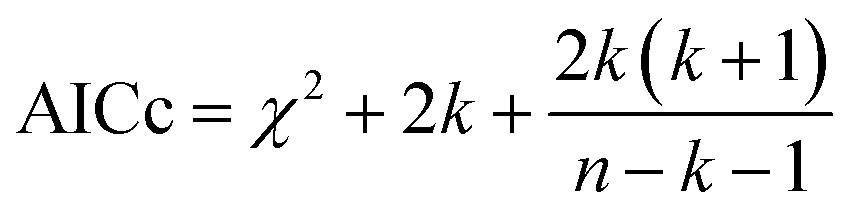DOI:
10.1039/D2SC00345G
(Edge Article)
Chem. Sci., 2022,
13, 10406-10427
Structural and dynamical determinants of a β-sheet-enriched intermediate involved in amyloid fibrillar assembly of human prion protein†
Received
18th January 2022
, Accepted 4th August 2022
First published on 5th August 2022
Abstract
The conformational conversion of the cellular prion protein (PrPC) into a misfolded, aggregated and infectious scrapie isoform is associated with prion disease pathology and neurodegeneration. Despite the significant number of experimental and theoretical studies the molecular mechanism regulating this structural transition is still poorly understood. Here, via Nuclear Magnetic Resonance (NMR) methodologies we investigate at the atomic level the mechanism of the human HuPrP(90–231) thermal unfolding and characterize the conformational equilibrium between its native structure and a β-enriched intermediate state, named β-PrPI. By comparing the folding mechanisms of metal-free and Cu2+-bound HuPrP(23–231) and HuPrP(90–231) we show that the coupling between the N- and C-terminal domains, through transient electrostatic interactions, is the key molecular process in tuning long-range correlated μs–ms dynamics that in turn modulate the folding process. Moreover, via thioflavin T (ThT)-fluorescence fibrillization assays we show that β-PrPI is involved in the initial stages of PrP fibrillation, overall providing a clear molecular description of the initial phases of prion misfolding. Finally, we show by using Real-Time Quaking-Induced Conversion (RT-QuIC) that the β-PrPI acts as a seed for the formation of amyloid aggregates with a seeding activity comparable to that of human infectious prions.
Introduction
Prions are fatal pathogenic infectious agents responsible for neurodegenerative diseases collectively known as prion diseases or transmissible spongiform encephalopathies (TSEs). Prion diseases are characterized by spongiform vacuolation throughout the cerebral grey matter, neuronal death and reactive proliferation of astrocytes and microglia. The conformational conversion of the prion protein from its physiological cellular form, PrPC, to the insoluble scrapie isoform, PrPSc, is at the basis of TSE pathogenesis. These disorders can be sporadic, inherited or acquired by infection and comprise bovine spongiform encephalopathy (BSE) in cattle, scrapie in goats and sheep, chronic wasting disease in cervids and Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker syndrome, Fatal Familial Insomnia (or FFI) and kuru in humans.1,2 The prion protein gene, PRNP in humans, is highly conserved among species and its complete open reading frame is invariably located within a single exon. PrPC is a sialoglycoprotein, predominantly found attached via a glycosylphosphatidylinositol (GPI) anchor to the outer leaflet of the plasma membrane, exposed to the extracellular space.3 PrPC is expressed mostly in the central nervous system and its proposed functions include synaptic plasticity,4 cell signaling,5 neuronal growth and differentiation, NMDA receptor modulation6–8 and brain metal homeostasis.9 These latter two functions directly involve the ability of PrPC to bind divalent cations, primarily copper.10 Nuclear Magnetic Resonance (NMR) studies11–13 revealed that the C-terminal domain of PrPC (residues 127–231) is structured, globular and largely α-helical, with three helices (α1, α2 and α3) and a short antiparallel β-sheet (β1–β2).14,15 The longest helices, α2 and α3, which are covalently linked through a disulphide bridge, form a V-shaped arrangement on which the β-sheet and α1 are anchored. The N-terminal domain (residues 23–126) is intrinsically disordered and contains two charged clusters (CC1, residues 24–30, and CC2, residues 101–110), the octarepeat region (OR, residues 59–90), the non-octarepeat region (non-OR, residues 91–110) and a hydrophobic domain (HD) (residues 111–126) containing a palindromic sequence motif (residues 113–120) (ESI Fig. 1A and B†) known to be able to initiate prion conversion.16 The N-terminal moiety of PrPC can bind Cu2+ with high affinity at four sites in the OR region and with lower affinity at two sites in the adjacent non-OR region.17–19 Recent studies,20–23 using biochemical and biophysical techniques, have shown that the flexible N-terminal domain and the C-terminal globular domain may interact with each other in a highly regulated modality playing a crucial role in the physiological and pathological functions of both prion protein and prions. In particular, as demonstrated by dynamic light scattering measurements, this cis interaction is principally dependent on charge complementarity between the positively charged residues of the N-terminal polybasic region24–26 and negatively charged amino acids of helix 3.21 Additionally, other previous studies have shown that the binding to metal ions, such as Cu2+ and Zn2+, modulates the inter-domain coupling by inducing a specific interaction between the two domains.27–29 The misfolding of PrPC into PrPSc may occur due to genetic mutations enhancing the aggregation propensity of the protein or through infection by seeding through PrPSc forms, which then act as templates for PrPC–PrPSc autocatalytic conversion. Nonetheless, most reported prionopathies are the result of spontaneous conversion of PrPC into PrPSc whose mechanism has not yet been elucidated, although several in vitro30 and computational studies31,32 indicate high PrPC conformational flexibility as a crucial factor in causing aggregation. The capability of PrPC to populate partially unfolded forms (PUFs)33,34 in equilibrium with the native state appears to be an essential step toward conversion to the β-structured toxic oligomers and successively to the fibrillar insoluble forms. In earlier studies,35,36 no PUFs were detected in hydrogen–deuterium exchange NMR analysis of human and Syrian hamster prion proteins. However, folding intermediates were successively detected in kinetic studies of HuPrP at pH 5.5, as well as of pathogenic variants of ovine PrP, but not in similar folding analysis of mouse PrP at pH 7.0.37–39 Indeed, structural characterization of PrP PUFs has been recently performed under conditions in which the PrP native state is destabilized, at lower pH and/or in the presence of denaturants.33 Moreover, a recent study has underlined that the N-terminal domain confers to the full-length mouse PrP, MoPrP(23–230), a higher chemical stability with respect to truncated MoPrP(89–230) and allows a completely reversible cooperative process of the protein thermal unfolding. Conversely, MoPrP(89–230) thermal unfolding is irreversible and characterized by an intermediate state.40 In spite of this wealth of knowledge, an exhaustive investigation at the atomic level of the molecular determinants controlling the prion protein unfolding process, including a high-resolution structural and dynamics characterization of prion protein intermediate states, is still missing. To obtain a detailed structural description of the human PrP full-length, HuPrP(23–231) and truncated HuPrP(90–231) conformational equilibria we use Circular Dichroism (CD) and NMR spectroscopy to follow their thermal unfolding. In particular, we explore at the atomic level the structural-dynamics features of the intermediate state (β-PrPI) that HuPrP(90–231) forms at 61 °C and pH 5.5 showing that, in the regions surrounding the β-sheet and α1 helix, though preserving the three native α-helices, it samples conformations with a significant tendency to increase the β-sheet folding with respect to the native state. Additionally, we provide an atomic resolution picture of the molecular mechanism by which the N-terminal domain regulates the folding process of the full-length HuPrP also highlighting how copper binding alters the prion protein folding energy landscape. Furthermore, by combining NMR relaxation dispersion and thermal unfolding data, we show that HuPrP(90–231) at pH 5.5 and 25 °C is in conformational equilibrium with a PUF that is significantly similar to the folding intermediate detected during the thermal denaturation. By means of thioflavin T (ThT) fluorescence experiments, we also show that truncated HuPrP forms amyloid fibrils more rapidly at 61 °C than at 25 °C, indicating that the documented folding intermediate plays a key role in seeding amyloid growth with a seeding activity similar to that of the pathogenic scrapie form, as revealed by RT-QuIC experiments. Finally, our study outlines a theoretical model that may be common with other multi-domain proteins, in which domain coupling regulates dynamic folding.
Results
The N-terminal domain controls the folding mechanism of human PrPC
We carried out CD thermal unfolding experiments on HuPrP(23–231) and HuPrP(90–231) at pH 6.8 and 5.5 using different protein concentrations ranging from 10 to 80 μM (only for measurements at pH 5.5) (ESI Fig. 2†); the latter pH value has been reported to facilitate PrPC to PrPSc conversion41,42 and it represents the pH environment of endosomes in which the prion protein misfolding occurs.43
At low concentration (10 μM), full-length HuPrP shows a single sigmoidal cooperative thermal unfolding with a midpoint transition temperature (Tm) of 69 ± 2 and 59 ± 1 °C at pH 5.5 and 6.8, respectively (ESI Fig. 2 and ESI Table 1†). Differently, the truncated form HuPrP(90–231) at pH 5.5 exhibits a more complex thermal unfolding curve that can be described as the convolution of two transition states, the first having a Tm of 52 ± 1 °C and the second 74 ± 2 °C (ESI Fig. 2 and ESI Table 1†). At pH 6.8, the thermal profile shows two closer transitions, the first centred at 56 ± 3 °C and the second at 70 ± 1 °C (ESI Fig. 2 and ESI Table 1†). Then, we checked the unfolding reversibility by analysing the CD spectra of the HuPrP(23–231) and HuPrP(90–231) prion protein samples that were cooled to 25 °C after heating to 90 °C. At pH 5.5, for HuPrP(23–231) and HuPrP(90–231) the CD spectrum acquired after cooling down to 25 °C is very similar to the native protein spectrum (ESI Fig. 2 and 3†) prior to thermal unfolding with a reversibility of 100 and 97%, respectively (ESI Table 2†). In contrast, at pH 6.8 for HuPrP(23–231) and HuPrP(90–231) the CD spectrum acquired after cooling the heat-denatured sample is slightly different to that of the native protein indicating that the thermal unfolding is partially irreversible with a reduction of the reversibility of ∼20% (ESI Table 2†). Therefore, to assess whether the cause of this irreversibility is aggregation of the heat-unfolded polypeptide, we performed CD thermal unfolding measurements at increasing protein concentrations (ESI Table 1†) and we evaluated the existence of oligomers and/or large aggregates in solution by comparing the aggregation index (A.I.) (ESI Table 2†) calculated before and after thermal denaturation. Interestingly, we observed that (i) the thermal unfolding process of HuPrP(23–231) and HuPrP(90–231) at both pH values is concentration independent (ESI Fig. 2 and ESI Tables 1 and 2†), (ii) the A.I. values (ESI Table 2†) obtained after cooling the heat-denatured samples are below 10 and (iii) in all cases the θ222/θ219 ratio (ESI Table 2†) calculated at 25 °C before and after melting is 0.9 which is similar to that determined using the ellipticity values predicted from the NMR structure (θ222/θ219 = 1.0) (PDB ID: 2LSB). Overall, under our experimental conditions the partial reversibility is due to loss of material and/or due to the formation of aggregates. However, as revealed by the thermal unfolding curves observed at different protein concentrations, as well as the melting temperatures and θ222/θ219 ratios, for all investigated prion protein samples the unfolding process is not influenced by the formation of a small amount of soluble aggregates which in turn may be involved in precipitation processes. Moreover, CD results support that even in HuPrP the N-terminal domain plays a key role in the thermodynamic stability of the protein and that its absence induces a more complex thermal unfolding, particularly at pH 5.5. To further understand the role of the N-terminal domain in the HuPrP stability, we recorded NMR high-resolution spectra of prion protein folding processes (Fig. 1) of HuPrP(23–231) and HuPrP(90–231) at pH 5.5 by acquiring a series of two-dimensional 1H/15N and 1H/13C HSQCs between 5 and 80 °C at intervals of 5 °C (with 2 °C intervals in the range 55–75 °C). In both cases, in the 1H–15N HSQC spectra, the position of many peaks shifts over the temperature range preceding the first thermal transition, which is indicative of fast amide proton exchange. In the 1H–15N HSQC of the full-length HuPrP most residues disappeared at around 57 °C and reappeared at 75 °C with typical unfolded chemical shift dispersion (ESI Fig. 4A and B†). This behaviour indicates that HuPrP(23–231) is characterized by a cooperative two-state thermal unfolding with folding/unfolding conformational exchanges in the micro–millisecond timescale.44,45 Conversely, for the truncated HuPrP, most of the residues exhibit a curved temperature response for the 1H and 15N resonances suggesting a multi-state process (Fig. 1A and B) with a well preserved spectral dispersion at 61 °C (Fig. 1C) with respect to the random coil spectrum simulated at the same temperature (Fig. 1D). We estimated midpoint temperatures (Tms) of each specific atom by evaluating HN and Hα chemical shift variations as a function of temperature (Fig. 1B), identifying 197 protons (84 HN and 113 Hα) that could be resolved in the range of the first transition with a sigmoidal behaviour. Interestingly, Tms analysis mapped onto the HuPrP(90–231) NMR structure (PDB ID: 2LSB) (Fig. 1E–G) reveals that all the analysed atoms experience a structural transition with suitable Tms within the 32–61 °C range. Tms occurrences as a function of temperature show Gaussian distributions centred slightly below 47 °C for HNs (Fig. 1E) and at around 52 °C for Hαs (Fig. 1F). When HN and Hα protons are grouped a more symmetrical Gaussian distribution is observed, centred at 50 °C (Fig. 1G), in accordance with the CD results. HuPrP(90–231) shows a clear hierarchical behaviour in the structural transition that leads to the formation of intermediate conformational states. Subsequently, we performed NMR thermal melt analysis at neutral pH. At pH 6.8 for HuPrP(90–231) we could not monitor the unfolding processes and detect the intermediate state due to the severe reduction of the signal-to-noise ratio of NMR signals with increasing temperature at neutral pH; differently, the instability of the NMR sample of full-length HuPrP at high protein concentration under the experimental conditions precluded the NMR thermal unfolding analysis at this pH value.
 |
| | Fig. 1 NMR thermal unfolding of the C-terminal HuPrP domain. (A) Series of 1H–15N HSQC NMR spectra of HuPrP(90–231) as a function of temperature for selected residues (Gly131, Thr199, Ile139, Phe141). Temperature increase is indicated by a gradual change in the color of the resonances from light cyan (15 °C) to red (61 °C). (B) Plot of normalized chemical shifts against temperature for 10 representative Hα protons showing mean-like behaviour in the 15–61 °C range. (C) 1H–15N HSQC spectrum of HuPrP(90–231) acquired at 600 MHz at 61 °C and pH 5.5. (D) 1H–15N HSQC spectrum of the unfolded HuPrP(90–231) at 61 °C. The NMR spectrum was reconstructed (see the ESI†) using the random coil 1H and 15N chemical shifts predicted for HuPrP(90–231) at 61 °C and pH 5.5 through a specific algorithm suitable for IDPs (Intrinsically Disordered Proteins). (E–G) Ribbon drawing representation of the HuPrP(90–231) NMR structure showing the thermal stability for HN (E), Hα (F) and both (G) protons mapped on their corresponding heavy atoms. The inset indicates the Tm scale. The Tm histogram is also reported. | |
Structural and dynamics features of the prion protein β-sheet enriched intermediate state (β-PrPI)
We assigned and analysed the backbone chemical shifts for 1H, 15N and 13C nuclei, which are sensitive probes for protein local structure.46–48 When we compared the NMR spectra (eqn (1) in Methods) of truncated HuPrP acquired at 25 °C and 61 °C (ESI Fig. 4A–E and ESI Fig. 5A and B†) significant chemical shift differences greater than the Standard Deviation (SD) were mainly observed within the β1 strand (Tyr128-Leu130) and the nearby loop (Gly131-Phe141), the N-terminus of the α1 helix (Glu146, Tyr150), the β2 strand (Val161-Tyr163) and the following loop (Arg164-Gln172), the N-terminus of the α2 helix (Gln172-Phe175) and the C-terminus of the α3 helix (Asp222, Val223) (ESI Fig. 5A and B†). Significant perturbations were observed for the residues located in the hydrophobic domain (His111-Gly126) and in the turn between α2 and α3 (Thr193-Glu200). The residues from the α2 and α3 helices flanking the disulphide bridge showed small chemical shift differences indicating that these regions are almost unaffected. These results suggest that the main structural differences between the native and the intermediate β-PrPI state are localized in the region around the two native β strands and the α1 helix and that these structural rearrangements slightly perturb only the structure of the edges of adjacent α2 and α3 helices. To identify the secondary structure changes induced by this conformational transition, the Cα secondary chemical shifts at 25 °C (ESI Fig. 4D and E†) and at 61 °C (ESI Fig. 5B†) have been compared. After that, we compared the secondary structure populations between the native and the β-PrPI state finding that in the intermediate state there is a β-sheet population increase of 30% in the region between the β1 strand/α1 helix (Gly131-Gly142) and in the loop regions flanking the β2 strand (Pro158-Ser170); the helical content for the three α-helices shows a decrease of ∼10% (Fig. 2A and B). Moreover, the Cα and Cβ chemical shift analysis of cysteines (Cys214, Cys179) (ESI Fig. 6†) and the amide temperature coefficients (ΔδNH/ΔT (ppb K−1)) (Fig. 2C) indicates that the β-PrPI is stabilized by the permanence of the disulphide bridge and by the conservation of a large part of the native hydrogen bond network that is essential for preserving the three α-helices (Fig. 2A–C). Additionally, we explored the fast ps–ns motions of the folding intermediate state at 61 °C by estimating the model-free order parameters (S2) for the backbone amide group from the backbone chemical shifts using the Random Coil Index (RCI) approach.49 To verify the ability of the assigned backbone resonances of quantifying protein flexibility we predicted S2 values for HuPrP(90–231) at 25 °C and then we compared them to the order parameters estimated from 15N NMR relaxation measurements (ESI Fig. 7†). As reported in ESI Fig. 8A–C,† the model-free order parameters obtained applying the RCI method provide for the residues located within secondary structure elements an accurate description of fast ps–ns backbone motions. In the case of the β-PrPI intermediate state, according to the structural data, the S2 values predicted from backbone chemical shifts (ESI Fig. 5B†) show that HuPrP(90–231) at 61 °C largely maintains a discrete structural rigidity in the ps–ns timescale.
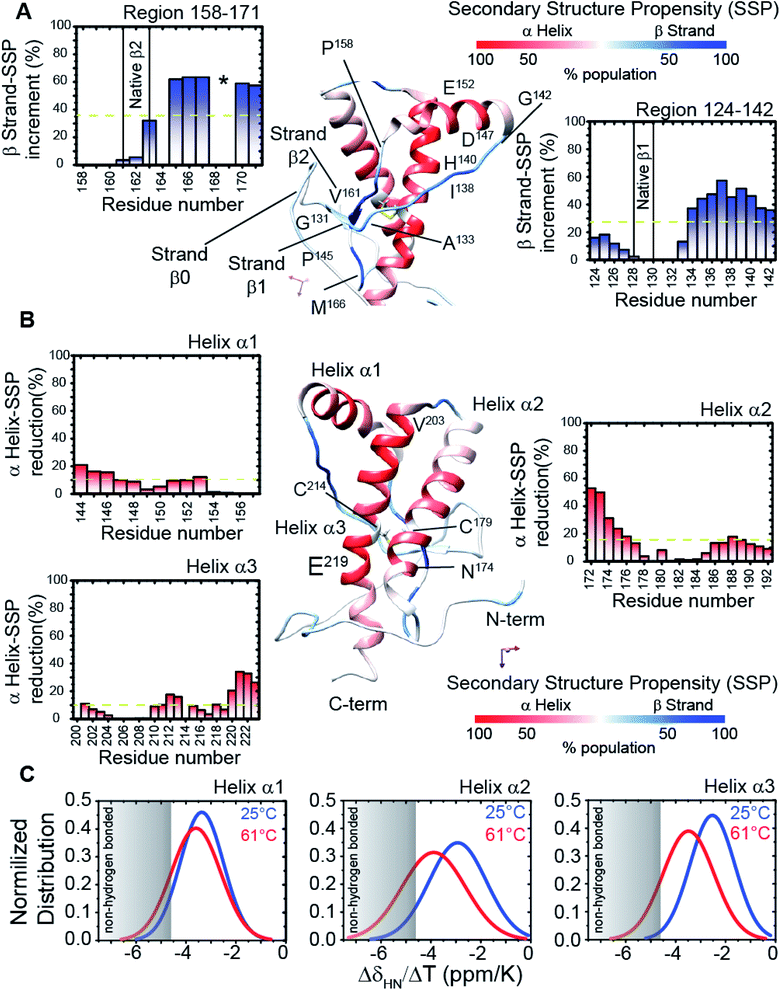 |
| | Fig. 2 Structural details of the HuPrP(90–231) conformational intermediate state. (A and B) Mapping of the SSP scores onto the representative NMR structure of HuPrP(90–231). The α-helix and β-strand populations are reported in red and blue, respectively. The insets report the increase and the reduction of β-sheet and α1 helix populations, respectively. The disulphide bridge, formed by Cys179 and Cys214, is also reported as a yellow stick. (C) Normalized distribution of 1HN temperature coefficient values (ΔδHN/ΔT) for each of the three α-helices of the β-PrPI intermediate state. | |
Identification of the key residues regulating the inter-domain coupling
The structural mechanisms underlying the different folding behaviours observed in full-length and truncated HuPrP were assessed by means of backbone (1H/15N/13C) chemical shift perturbation mapping (eqn (1) in Methods). Removal of the N-terminal domain results in substantial changes in the 1H–15N and 1H–13C HSQC correlation spectra (ESI Fig. 9A and B, ESI Fig. 4A and C, and ESI Fig. 10A–F†). Larger chemical shift perturbations (ΔH,N,C) were observed for His111, Met129, Pro137, Asn159, Asp178, Gly195 and several residues located in the α3 helix (Val209, Gln212, Cys214, Ser222-Tyr226). Important small differences were observed for a large number of residues homogeneously distributed on the globular domain surface (ESI Fig. 9A†). Notably, the perturbed residues do not create a continuous patch on the surface; they are mainly localized around negatively charged residues (Fig. 3A) and close to the disease-linked mutation sites (ESI Fig. 11A–C†). Considering N-terminal domain richness in positively charged residues (Fig. 3A), these results strongly support previous studies which suggest that the two domains cross-talk through transient electrostatic interactions.20–22 To fully address this crucial point and to identify the key amino acids of the globular domain governing the inter-domain coupling we compared the chemical shift variations observed for the full-length and truncated HuPrP upon addition of increasing amounts of sodium chloride (eqn (4) in Methods) (Fig. 3B). In the presence of 50 mM NaCl, significant chemical shift perturbations were observed for residues surrounding Glu146, Asp147, Glu152, Asp178, Glu196, Glu200 and Asp202 (Fig. 3C and D). Interestingly, upon increase of the NaCl concentration to 200 mM we observed substantial chemical shift variations for additional residues neighbouring Asp144, Glu207, Glu219 and Glu221 (Fig. 3C and D). Overall, NMR structural data clearly show that the inter-domain coupling is regulated by two patches: patch1 is formed by the acidic residues positioned in the α2 and α3 helices and it represents the primary anchor point for the N-terminal domain (Fig. 3D); patch2 is mainly composed of the negatively charged residues surrounding the α1 helix and it contributes to stabilizing the transient electrostatic inter-domain coupling (Fig. 3D). In this scenario, we hypothesized that the N-terminal domain might regulate the folding mechanism of the full-length HuPrP, avoiding the formation of intermediate states by altering the secondary and/or tertiary structure of the C-terminal domain. Thus, we first compared the Hα and Cα secondary chemical shifts of the full-length HuPrP with those of the truncated form (ESI Fig. 9B†), showing that the N-terminal domain does not affect the secondary structure organization of the globular domain. Then, we investigated the effect of the N-terminal domain on the HuPrP(90–231) tertiary structure by fitting the backbone chemical shifts measured for the full-length protein to the corresponding shift values observed for the globular domain (eqn (2) in Methods and ESI Fig. 10C and D†) and to the shifts predicted from the NMR structure of the globular domain (eqn (3) in Methods and ESI Fig. 10E and F†). The evaluation of the QΔ and Q factor values (ESI Fig. 10C–F†) clearly show that the N-terminal domain transiently interacts with the C-terminal globular domain without altering the tertiary organization of the latter.
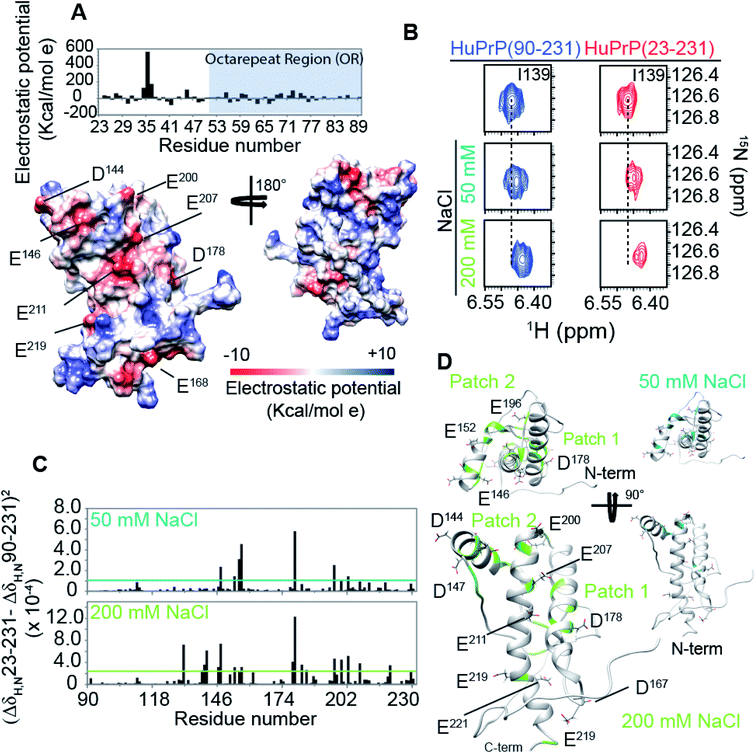 |
| | Fig. 3 The mechanism driving the inter-domain coupling. (A) (upper) Electrostatic surface potential of the N-terminal domain (23–89) of HuPrP(23–231) reported as a histogram. The OR region is also indicated as a light blue box. (lower) C-terminal domain electrostatic surface potential, HuPrP(90–231), at pH 5.5 depicted from electropositive (blue; 10 kcal mol−1) to electronegative (red; −10 kcal mol−1). The negatively charged residues are indicated. (B) Comparison of 1H–15N HSQC NMR spectra used to detect the chemical shift variations for HuPrP(90–231) (blue) and HuPrP(23–231) (red) upon addition of 50 and 200 mM NaCl. (C) (ΔδHN,N23–231 − ΔδHN,N90–231)2 values calculated for 50 and 200 mM plotted versus the primary sequence. The cyan and light green lines indicate the average value for 50 and 200 mM, respectively. (D) Mapping of the residues showing significant HN and N chemical shift changes upon addition of 50 and 200 mM NaCl, respectively. | |
Effect of copper binding on the full-length prion protein folding processes
Prompted by previous results showing that in the full-length prion protein the cis inter-domain coupling is mediated by Cu2+ binding,28,29 we carried out an atomic resolution description of the copper binding effects on the single sigmoidal cooperative unfolding mechanism observed for full-length HuPrP. First, the identification of the metal binding sites of HuPrP(23–231) was assessed by means of backbone 1H/15N chemical shift perturbation mapping and by evaluating the reduction of the height of NMR signals upon addition of 1, 4 and 6 equivalent(s) of the paramagnetic copper ion (Fig. 4A and B). At low 1 molar equivalent of Cu2+ the signal intensity ratios (Fig. 4A and B) indicate that the OR region binds the metal ion by a multiple histidine Cu2+-binding mode. At 4 molar equivalents of Cu2+ (Fig. 4A and B) the intensity ratio profile shows that in addition to the OR-region the Cu2+-binding involves both His96 and His111. Furthermore, in agreement with previous studies performed at 37 °C,29 substantial intensity ratio reduction due to the copper paramagnetic relaxation effect (PRE) is observed at three distinct regions (i.e. (i) Ile138-Gly142, (ii) Asn173-Val180, and (iii) Met206-Glu211) of the globular domain, even in the presence of 1 molar equivalent of metal ions (Fig. 4A and B). Interestingly, at both copper concentrations, chemical shift perturbation analysis shows that the recognition of the metal ion is assisted by significant long-range structural rearrangements of the globular domain that involve residues far beyond the binding sites (Fig. 4A and B). In detail, larger chemical shift perturbations (ΔHN,N) were mainly observed for the residues located in the three α-helices (Arg148-Arg151 (α1); Gln172-Ile182 (α2); Glu207-Ile215 (α3)) (Fig. 4A and B). These results indicate that in the Cu2+-loaded HuPrP(23–231) the interaction between the N-terminal and the globular domain, driven by the described electrostatic interactions, is altered independently of total copper concentration. At higher copper concentration (6 molar equivalents) we observed in the 1H–15N HSQC spectrum a homogeneous drastic reduction of all observable NMR signals indicating that the full-length HuPrP at 25 °C undergoes aggregation in the presence of a large excess of copper (Fig. 4A and B). We therefore investigated whether the modulation in the two-domain coupling by Cu2+ binding also affects the unfolding mechanism of Cu2+-loaded HuPrP(23–231). Interestingly, CD thermal unfolding data indicate that the full-length HuPrP upon copper binding (1 or 4 equivalent(s)) shifts the unfolding mechanism from a single sigmoidal cooperative model to a more complex process with two transitions (ESI Fig. 2 and ESI Table 1†). Then, we applied the thermal unfolding NMR experiments scheme used for truncated HuPrP in the range from 5 °C to 80 °C. At 61 °C (ESI Fig. 12A and B†) in the 1H–15N HSQC spectra of Cu2+-loaded HuPrP(23–231) there is considerable dispersion of signals in both proton and nitrogen dimensions indicating that the presence of copper stabilizes partially folded intermediate states. We compared the secondary HN and N shifts observed for Cu2+-loaded HuPrP(23–231) with those assigned for the truncated HuPrP intermediate state at the same temperature. As reflected by the QΔN and QΔHN factor values (eqn (2) in Methods and ESI Fig. 12C and D†), there is a discrete chemical shift correlation suggesting that the copper induced intermediate states may present structural similarities to β-PrPI. To further describe the structural details of the Cu2+-loaded HuPrP(23–231) intermediate state detected in the presence of 1 or 4 equivalent(s), we evaluated the Cα secondary chemical shifts at 61 °C and we estimated the secondary structure populations for both copper concentrations (Fig. 4C). As reported in Fig. 4C and D, in the Cu2+ induced intermediate states the three native α-helices are less conserved than β-PrPI and the regions of the globular domain comprising amino acids Gly90-Gly142 and Pro158-Ser170 are completely unfolded without showing any conformational preferences to the β-sheet structure. Additionally, both copper induced intermediate states are conformationally restricted by the conservation of the disulphide bridge as revealed by the Cα and Cβ chemical shifts of cysteines (ESI Fig. 6†). Finally, at both 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 4:1 copper ratios (Fig. 4E and F) midpoint unfolding temperatures of single HN atoms are mostly included between 37 °C and 57 °C. Overall, our results show that the copper binding in the full-length human prion protein influences the cis inter-domain coupling that in turn modifies the unfolding mechanism with the formation of a stable partially folded intermediate state.
:1 and 4:1 copper ratios (Fig. 4E and F) midpoint unfolding temperatures of single HN atoms are mostly included between 37 °C and 57 °C. Overall, our results show that the copper binding in the full-length human prion protein influences the cis inter-domain coupling that in turn modifies the unfolding mechanism with the formation of a stable partially folded intermediate state.
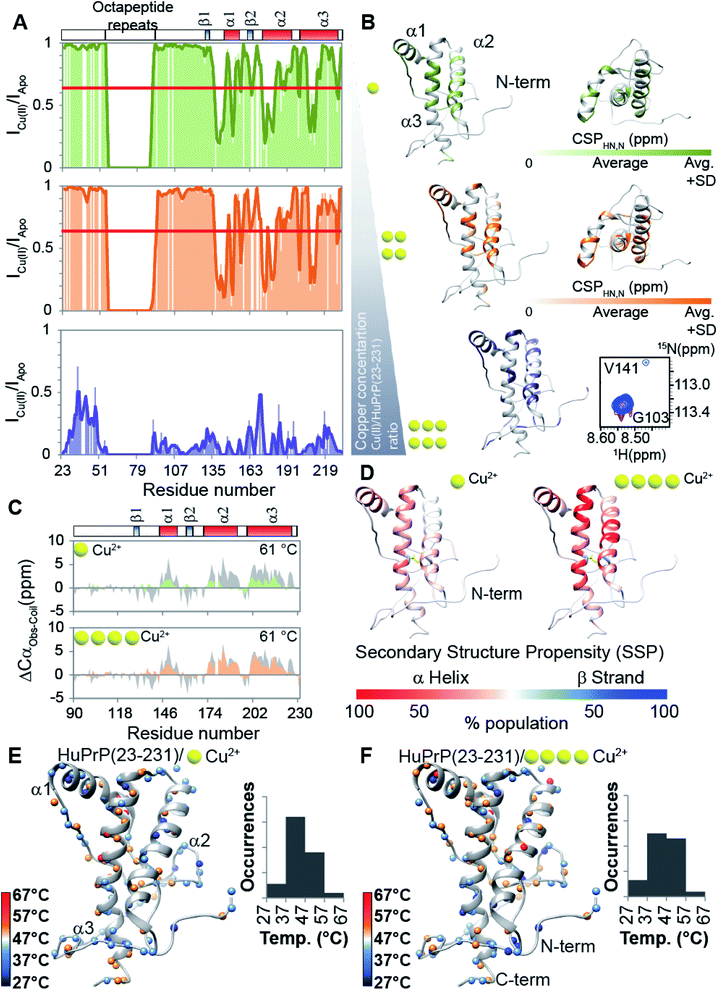 |
| | Fig. 4 Copper binding effects on the prion protein unfolding mechanisms. (A) 1H–15N signal intensity changes shown as a fraction of the starting values for the addition of 1, 4 and 6 equivalent(s) of Cu2+. I0 and I are intensities of 1H–15N cross-peaks of HuPrP(23–231) in the absence and presence of Cu2+. (B) CSPs of HuPrP(23–231) upon binding to 1 and 4 equivalent(s) of Cu2+ mapped onto the NMR structure of the C-terminal domain in two orientations. Residues for which CSPHN,N > mean + SD are shown in light green and orange for HuPrP(23–231)/Cu2+ (1:1) and (1:4), respectively. In the case of HuPrP(23–231)/Cu2+ (1:6) the residues for which the HN/N signals are not detected are reported as violet. (C) Cα secondary chemical shifts observed for the copper induced intermediate state for HuPrP(23–231) upon binding to 1 and 4 equivalent(s) Cu2+. The Cα secondary chemical shifts related to the β-PrPI intermediate state are also reported (light grey). (D) Mapping of the SSP scores obtained for HuPrP(23–231)/Cu2+ (1:1) and HuPrP(23–231)/Cu2+ (1:1) at 61 °C onto the representative NMR structure of HuPrP(90–231). The α-helix and β-strand populations are reported in red and blue, respectively. (E and F) Thermal stability of HN protons for HuPrP(23–231)/Cu2+ (1:1) (E) and (1:4) (F) mapped on their corresponding heavy atoms. | |
The N-terminal domain regulates the μs–ms dynamics of full-length prion protein
The comparison of the structural details between full-length and truncated HuPrP does not give a full explanation of the reason why the removal of the N-terminal domain induces a dramatic change in the folding process from a single sigmoidal cooperative to a more complex folding/unfolding pathway. Protein dynamics may play a role in the modulation of the unfolding mechanism. Thus, we investigated protein motions of full-length and truncated forms in the nanosecond-to-picosecond and millisecond-to-microsecond timescales. First, [1H]15N-heteronuclear NOE experiments (ESI Fig. 13A and B†), reporting on dynamics in the ns–ps timescale, show that the presence of the disordered N-terminal tail (residues 23–89) slightly affects the dynamics of the globular domain. In particular, the region from Gly90 to Asn100 shows more pronounced fluctuations, once the N-terminal domain is removed; differently, all secondary structure elements are influenced to a different extent, presenting small variations of backbone motions (ESI Fig. 13C†). Globally, these differences do not seem to result in a clear variation in rigidity between the globular domain of the two proteins in the nanosecond-to-picosecond timescale. We then analysed the dynamics of the globular domain in the millisecond-to-microsecond timescale (ESI Fig. 13D–I†), firstly evaluating 1H–15N cross-peak intensity changes over a range of temperature for both full-length and truncated HuPrP (ESI Fig. 13D and E†). In HuPrP(90–231) a discrete number of residues (Gly131, His155, Tyr149, Tyr166, Tyr218, Glu221, Ser230 and Ser231) localized in different parts of the globular domain show a remarkable signal broadening suggesting the presence of slow conformational rearrangements in these regions (ESI Fig. 13D†). In contrast, in the full-length HuPrP the signal intensities are weakly perturbed at increasing temperatures (ESI Fig. 13E†), indicating that the presence of the N-terminal domain reduces the temperature sensitivity of the residues located inside the globular domain that in turn results in a change in the conformational flexibility of these regions. Dynamic processes on the μs–ms timescale in full-length and truncated HuPrP were further identified by the excess contribution to backbone amide 15N–R2 from chemical exchange line broadening (Rex) (ESI Fig. 14A and B†). In particular, Rex values were determined as the difference between the “total R2” rates (ESI Fig. 13F–I†) and the effective R2 rates derived from R1ρ experiments50 (eqn (5) in Methods). Residues in HuPrP(90–231) showing a significant Rex (Rex ≥ 3 s−1) are mainly localized in the regions surrounding the β-sheet, the α1 and α3 helices, and the amino-acids which surround the β1-α1 and α1-β1 loops and the turn connecting the β2 strand to α2 (ESI Fig. 14A†). Interestingly, in the full-length prion protein the presence of the N-terminal domain significantly alters the chemical exchange line broadening of residues located inside the globular domain. In particular, HuPrP(23–231) shows significant Rex decreases for residues within the two β-strands, α3-helix and especially the two loops, β1-α1 and α1-β2, surrounding the β-sheet (ESI Fig. 14B†). Notably, changes in Rex for HuPrP(23–231) are not only related to the residues directly involved in the inter-domain transient interactions, but they are distributed on internal regions of the protein, suggesting that the μs–ms timescale motions are highly coupled events. Overall, these findings support the idea that the N-terminal domain controls the conformational plasticity of the globular domain on the μs–ms timescale by transient inter-domain interactions.
Conformational equilibrium of human prion protein described by relaxation dispersion experiments
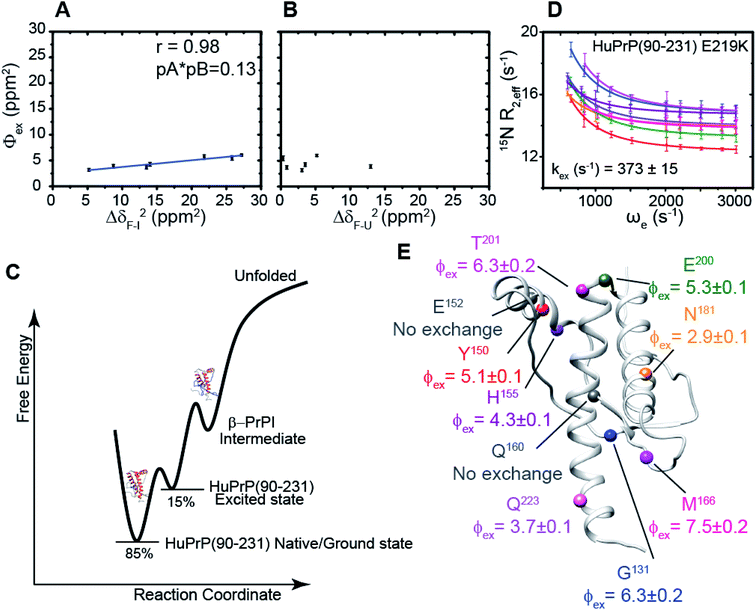
We hypothesized that the μs–ms conformational exchanging states observed for the HuPrP(90–231) might be correlated with β-PrPI. We hence performed 15N R1ρ relaxation dispersion (RD) experiments51–53 and analysed the dispersion curves of selected residues localized in the regions of HuPrP(90–231) showing significant conformational exchange. We fitted the data for 10 residues to the two-site (A and B) exchange Bloch–McConnell formalism54 (eqn (6) in Methods) for both full-length and truncated HuPrP. To understand whether the analysed residues undergo a common motion and share a common exchange rate (kex) with the same population of exchanging states (PA, PB) but with residue specific chemical shift differences ΔδAB, all residues were fitted singularly and simultaneously (ESI Tables 3 and 4†). For HuPrP(90–231) the obtained exchange rates for individual residues are consistently in the 400–3000 (s−1) range, whereas for HuPrP(23–231) the fitted kex are in the range of 200–500 (s−1). Interestingly, for truncated HuPrP the residues Gly131, Tyr150, Glu152, His155, Gln160 and Met166 are characterized by kex values higher than the residues located in the last two helices (α2 and α3), whereas for HuPrP(23–231) the residues display similar exchange rates with the exception of Glu152, His155, and Gln160, which do not show any RD profile (Fig. 5A and B and ESI Table 3†). Notably, the differences in exchange rates between truncated and full-length prion protein forms are mainly detected in the region surrounding the β-sheet and the first α-helix, where the average exchange rate drops from ∼2600 to ∼350 (s−1) (ESI Table 3†). These results indicate that the removal of the N-terminal domain in the full-length HuPrP alters the two-state conformational transition. After that, in the case of the full-length form, considering the narrow dispersion of kex (ESI Table 3†) obtained from the individual fits, we globally fitted all residues showing dispersion curves (Fig. 5A and B and ESI Fig. 15A†). As reflected by the small values of χ2global/χ2ind (<2) (eqn (7) in Methods), all analysed residues can be fitted to a common two-state exchange process with a kex of 313 ± 23 (s−1) indicating that these residues are governed by a single dynamic process (Fig. 5A and B and ESI Table 4†). In contrast, the truncated HuPrP(90–231) shows a wide dispersion of kex (ESI Table 2†) and large values of χ2global/χ2ind (>2) for residues Tyr150, Gln160, Asn181, Glu200, and Thr201 (ESI Table 4†) revealing that not all sites report on the same exchange process (kex = 2197 ± 138 (s−1)) (Fig. 5C and D). Therefore, we globally fitted all residues of cluster A (kex = 2502 ± 106 (s−1)) (Fig. 5C and ESI Table 5†) obtained by excluding the sites experiencing conformational fluctuations distinct from the global process (see the ESI†). Interestingly, the similarity between the per-site residuals estimated from the global fit (χ2global) and the values obtained from individual fits (χ2ind) (χ2global/χ2ind < 2) (Fig. 5C and ESI Table 5†) indicates that the residues included in cluster A undergo a common motion that is uncoupled from the rest of the C-terminal domain. This observation is corroborated by the lowest BIC and AICc (eqn (8) and (9) in Methods) values obtained for the global cluster fit (ESI Table 6†) revealing that the fitted parameters estimated for cluster A provide a more adequate description of the experimental data with respect to those obtained by fitting all residues simultaneously. Overall, RD data indicate that the full-length human prion protein is characterized by long-range coupled μs–ms motions between the region connecting the first β-strand with the second α-helix (Gly131-Ser170) and the rest of the protein (Asn171-Ser231) (Fig. 5D) and strongly suggest that the perturbation of these correlated dynamics plays a crucial role in the regulation of the unfolding process. We therefore analysed the residue-specific exchange parameter Φex that is correlated with the chemical shift difference (Φex = pApBΔωN2). For cluster A of HuPrP(90–231) the Φex values were in the 11000–23000 (s−2) (ESI Table 4†) range, whereas for HuPrP(23–231) Φex values obtained for the fitted residues were within 5000 (s−2) (ESI Table 4†). These results identify, in truncated HuPrP, groups of residues (Gly131, Tyr150, Glu152, His155, Gln160, Met166, and Gln223) experiencing larger structural changes in the conformational transition. Of note, residues most affected by the transition are clustered around β1, the β1-α1 loop, α1, the α1-β2 loop, and β2, which are the regions that in the first native–intermediate transition of the thermal unfolding present the greatest structural changes. Therefore, to explore whether the millisecond to microsecond local fluctuations are correlated with the unfolding transitions in HuPrP(90–231) we evaluated the correlation between the Φex values of HuPrP(90–231) (cluster A), obtained from RD measurements, versus the chemical shift difference observed between the native and β-PrPI states (Fig. 6A and B). If the line broadening in NMR spectra of truncated HuPrP is due to conformational exchange between the native and intermediate states, plots of Φexversus (ΔδF–I2) should be linear with a slope of pNpI. As reported in Fig. 6A, residues exhibiting large differences in 15N chemical shift between the native and intermediate states in NMR thermal unfolding also produce large-amplitude relaxation dispersions. Conversely, residues with 15N chemical shifts that are less perturbed during the thermal unfolding show smaller Φex values. Interestingly, the Φexversus ΔδF–I2 plot (Fig. 6A) indicates that between the parameters inferred from the RD and the thermal unfolding data exists a good correlation (Pearson correlation coefficient of 0.96) with the excited state population, calculated from the slope, of around 15 ± 2% which is in close agreement with the values obtained for human prion protein by high pressure NMR spectroscopy and kinetic measurements.55,56 We also plotted the Φex values as a function of squared differences in chemical shifts between the native state and those predicted for a random-coil chain (ΔδF–U2). As expected (Fig. 6B), there is no correlation between the two parameters confirming that the minor conformation is partially folded. These results provide evidence that truncated HuPrP, in the region connecting the first β-strand with the second α-helix, presents at 25 °C a chemical exchange in which the native (ground) state is in equilibrium with a minor conformational state (excited) that is not completely unstructured (Fig. 6C) but it is structurally similar to β-PrPI detected during the thermal unfolding. Therefore, considering the structural similarities, it is plausible that the observed low-populated state for HuPrP(90–231) might play crucial role in the formation
of the intermediate state (Fig. 6C). Nevertheless, the fact that the HuPrP(90–231) low-populated state and β-PrPI share characteristic conformational features strongly suggests that β-PrPI is an intermediate state on protein folding pathways en route to the native state.
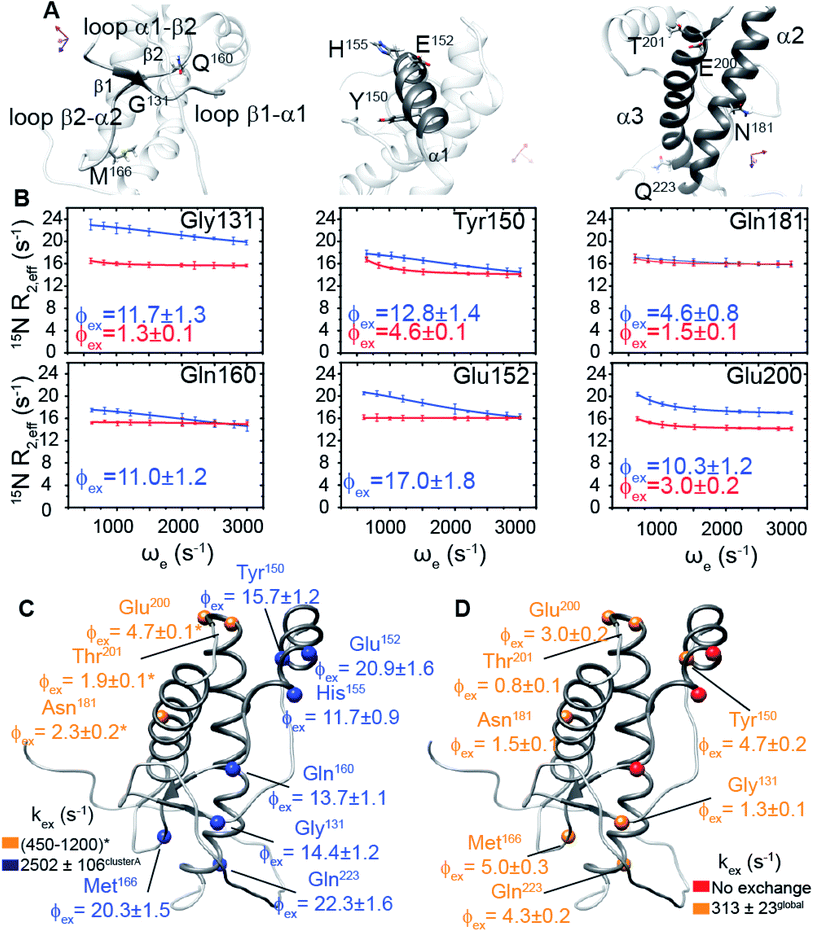 |
| | Fig. 5 Conformational exchange processes of HuPrP(90–231) and HuPrP(23–231) implicating the N-terminal domain as a dynamic switch. (A) Close-up view of HuPrP(90–231) regions showing a significant increase in Rex upon deletion of the N-terminal domain. The residues investigated by 15N R1ρ RD experiments are labelled. (B) Best fit curves to a two-site exchange model assuming a common motion for residues of HuPrP(90–231) (blue) and HuPrP(23–231) (red). (C) Residue-specific Φex (×103 (s−1)2) parameters obtained from the global fit of the HuPrP(90–231) cluster A residues (blue spheres); the sites reporting different exchange processes are reported in yellow ((*) indicates data extracted from the individual fit). (D) Residue-specific Φex parameters extracted for HuPrP(23–231) from the global fit (yellow spheres). The residues that do not show any RD profile are reported (red spheres). | |
 |
| | Fig. 6 Correlation between relaxation dispersion/thermal unfolding data. (A and B) Φex parameters (ppm2) derived from the global fit of cluster A (kex = 2502 ± 106 s−1) of HuPrP(90–231) plotted as a function of the squared differences in chemical shift between the native and the intermediate state (ΔδF–I)2 (ppm2) (A) and between the native and the denaturated state (ΔδF–U)2 (ppm2) (B). (C) Conformational landscape of HuPrP(90–231) revealed by NMR relaxation dispersion and thermal melt data. (D) Best fit curves to a two-site exchange model assuming a common motion for residues of HuPrP(90–231) E219K. (E) Mapping of the residue-specific Φex parameters extracted for HuPrP(90–231) E219K from the global fit. The residues that do not show any RD profile are reported as dark grey spheres. | |
To further show that the μs–ms conformational exchanging states play a crucial role in the conformational conversion of the prion protein we investigated the conformational equilibrium of a mutant of the HuPrP(90–231) carrying the dominant-negative E219K polymorphism57 by 15N RD experiments (Fig. 6D and ESI Tables 3 and 4†). Interestingly, for HuPrP(90–231) E219K the exchange rates for individual residues are consistently in the 300–500 (s−1) range (ESI Table 3†) indicating that the mutation causes a significant reduction of the conformational dynamics on the μs–ms timescale (please note that for HuPrP(90–231) the exchange rates are in the 400–3000 (s−1) range) (ESI Table 3†). Notably, the residues Glu152 (α1) and Gln161 (β2) do not show any RD trajectory demonstrating that for these residues the μs–ms timescale dynamics associated with conformational exchange broadening are completely quenched. In order to assess if the analysed residues undergo a common motion sharing a common exchange rate (kex), we globally fitted all residues showing dispersion curves (Fig. 6E). As reported in ESI Table 4,† the χ2global/χ2ind values are below 2 indicating that for the HuPrP(90–231) E219K mutant, different from what was observed for HuPrP(90–231), all residues can be fitted to a single two-state conformational exchange process with kex of 373 ± 15 (s−1) (Fig. 6D). Moreover, the residue-specific Φex exchange parameters predicted for HuPrP(90–231) E219K (<8000 (s−2)) (Fig. 6E) are significantly smaller than those estimated for cluster A of the wild-type HuPrP(90–231) (<23000 (s−2)).
β-PrPI activates the amyloid assembly pathway by oligomer formation having seeding activity for templated aggregation
To assess the effect of different pH values and temperatures on the amyloidogenic propensity of human PrPC we performed in vitro fibrillation experiments using ThT fluorescent dye. Both truncated and full-length HuPrP were incubated at 61 °C or 25 °C in two different buffers at pH 6.8 or pH 5.5 and aggregations were performed by subjecting the samples to several cycles of incubation and shaking. Fibril growth was monitored with the use of ThT in real time. Our results show that the aggregation of HuPrP(90–231) is significantly faster at 61 °C compared to that at 25 °C at both pH values (Fig. 7A). In particular, reactions performed at 61 °C show a lag-phase of amyloid formation of about 10 hours (Fig. 7A, light blue line for pH 6.8 and dark blue line for pH 5.5) while the truncated HuPrP at 25 °C barely aggregated after 60 hours at pH 6.8 (ESI Fig. 15B†). Regarding the full length HuPrP, all reactions performed at 61 °C and 25 °C at both pHs did not result in protein aggregation, even after 80 hours (Fig. 7A and ESI Fig. 15B†). Therefore, to gain a better understanding of the aggregation mechanism by which the β-PrPI intermediate state induces amyloid fibril formation, we investigated by NMR the molecular events occurring at the earliest stages along the aggregation pathway of β-PrPI. In particular, we compared the 1H–15N HSQC spectrum acquired at 25 °C on the β-PrPI sample, obtained by incubating HuPrP(90–231) at 61 °C for 15 hours, with that measured for the freshly prepared HuPrP(90–231) monomers (ESI Fig. 16A†). Interestingly, as reported in ESI Fig. 16A,† the 1H–15N HSQC spectrum of the β-PrPI sample exhibits, for a large number of residues, signal loss and broadening without significant chemical shift variations when compared to the spectrum of HuPrP(90–231) suggesting that the monomer is in dynamic equilibrium with β-PrPI-oligomeric (β-PrPI(o)) species. Therefore, we analysed residue-specific ratios of signal intensities after and before incubation at 61 °C (I/I0) (ESI Fig. 16B†) reporting on signal loss due to the conformational exchange and increase in rotational correlation time as a consequence of oligomerisation. The highest intensity ratio values were observed at the N-terminal part of the globular domain encompassing the portion from Gly90 to Ala113 indicating the conformational stability of this region. In contrast, intensity ratios below average ± SD and signal loss (*) were mainly observed for the residues of β1 and β2 (Met129*, Leu130*, Val161*, Tyr162*, Tyr163*), for the regions surrounding the two native β-strands (Gly131*, Ser132, Ala133, Met134, Ser135, Asn159*, Gln160*) and for the residues located within the α1 and α3 helices (Asp144, Tyr145, Glu152, His155*, Arg156*, Tyr157*, Thr201, Lys204, Met205, Ile215*, Thr216*, Gln217*, Arg220*, Glu221*) indicating that these regions are most affected by conformational exchange in the intermediate regime and therefore are most likely involved in the process of oligomerisation (ESI Fig. 16B and C†).
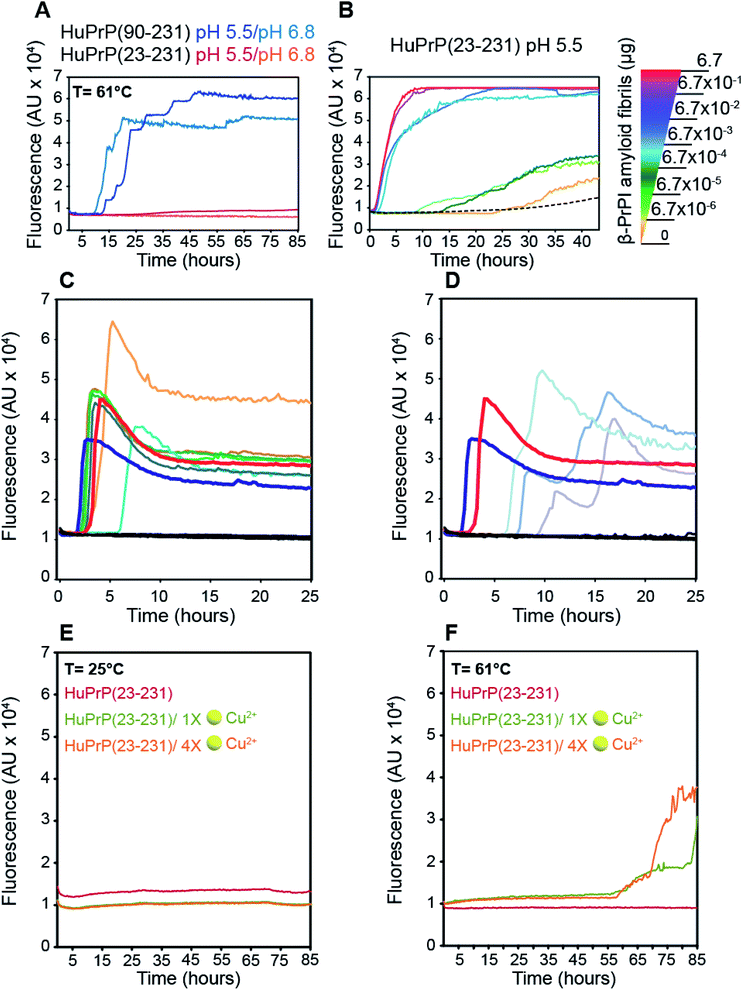 |
| | Fig. 7 Role of the β-PrPI intermediate state in amyloid fibril formation and in prion conversion. (A) HuPrP(90–231) and HuPrP(23–231) were induced to aggregate by alternating cycles of incubation and shaking at 55 °C. Average ThT fluorescence intensity was plotted against time. (B) Serial dilutions of the artificial β-PrPI(o) seeds, previously produced by incubating HuPrP(90–231) at 61 °C, were used to promote the aggregation of HuPrP(23–231). Average ThT fluorescence intensity was plotted against time. (C and D) RT-QuIC experiments monitoring the seeding activity of the β-PrPI oligomers. The experiments were conducted collecting from patients the following biological samples: brain homogenates (dilution 10−5 v/v) sCJD-129MM1 ( ), sCJD-129MM2 ( ), sCJD-129MM2 ( ), sCJD-129MV1 ( ), sCJD-129MV1 ( ), sCJD-129MV2 ( ), sCJD-129MV2 ( ), sCJD-129VV1 ( ), sCJD-129VV1 ( ), sCJD-129VV2 ( ), sCJD-129VV2 ( ), Alzheimer's disease and non-neurodegenerative disorder ( ), Alzheimer's disease and non-neurodegenerative disorder ( ); amyloid fibrillary assemblies (ng mL−1) β-PrPI amyloid fibrils pH 6.8 ( ); amyloid fibrillary assemblies (ng mL−1) β-PrPI amyloid fibrils pH 6.8 ( ) and 5.5 ( ) and 5.5 ( ), monomer HuPrP(90–231) pH 6.8 and 5.5 ( ), monomer HuPrP(90–231) pH 6.8 and 5.5 ( ); cerebrospinal fluid sCJD-129MM ( ); cerebrospinal fluid sCJD-129MM ( ), sCJD-129MV ( ), sCJD-129MV ( ), sCJD-129VV ( ), sCJD-129VV ( ), Alzheimer's disease and hydrocephalus ( ), Alzheimer's disease and hydrocephalus ( ). (E and F) Aggregation kinetics of HuPrP(23–231) at 25 °C (E) and 61 °C (F) upon addition of 1 (light green) and 4 (light orange) equivalent(s) of Cu2+ monitored by ThT fluorescence. ). (E and F) Aggregation kinetics of HuPrP(23–231) at 25 °C (E) and 61 °C (F) upon addition of 1 (light green) and 4 (light orange) equivalent(s) of Cu2+ monitored by ThT fluorescence. | |
Notably, the β-PrPI oligomerisation interface (ESI Fig. 16C†) perfectly corresponds to the regions showing a significant increase of β-sheet population. Overall, NMR and ThT data demonstrate that amyloid formation of β-PrPI occurs via a remarkably specific assembly mechanism involving the transient formation of oligomeric species (β-PrPI(o)).
Next, the β-PrPI(o) species were used as seeds to promote the conversion of full-length HuPrP. By comparison with HuPrP(23–231) (Fig. 7B, dashed line), we found that the addition of increasing concentrations of β-PrPI(o) seeds significantly enhanced the full-length HuPrP aggregation reaction, resulting in ThT positive kinetics with lag-phases shorter than the control (Fig. 7B). In addition, to test whether the oligomeric species from the β-PrPI intermediate state, obtained by incubating HuPrP(90–231) at 61 °C at pH 5.5 or 6.8, forms seeds that template aggregation we performed RT-QuIC experiments using the experimental setting employed for the analysis of biological samples collected from patients with a clinical diagnosis of sporadic Creutzfeldt–Jakob disease (sCJD) (Fig. 7C and D). Besides the aggregates, we have included in the analysis brain homogenates of patients with the six forms of sCJD as well as a few cerebrospinal fluid (CSF) samples collected from sCJD patients with MM, MV or VV polymorphisms at PRNP codon 129. The results clearly show that regardless of the pH at which they were generated, all β-PrPI induced amyloid aggregates were able to efficiently seed the RT-QuIC reaction with a kinetics very similar to that of the sCJD brain homogenates (Fig. 7C and D). All CSF samples collected from sCJD patients induced an efficient seeding activity. As expected, neither the monomeric HuPrP(90–231) nor the brain homogenates and CSF collected from patients with non-CJD patients induced seeding activity by RT-QuIC. Overall, RT-QuIC shows that the β-PrPI(o) forms active seeds which template the formation of amyloid aggregates with a seeding activity comparable to that of the infectious and pathogenic PrPSc.
Finally, we investigated the effect of Cu(II) binding on the amyloidogenic propensity of human PrPC using real-time ThT fluorescence experiments. At 25 °C, HuPrP(23–231) in the presence of either 1 or 4 equivalent(s) did not form any aggregates (Fig. 7E). In contrast, at 61 °C with 1 and 4 equivalent(s) of copper, the Cu(II) binding to HuPrP(23–231) induces the formation of ThT-positive intermediates by a double-sigmoidal aggregation kinetics (Fig. 7F). This phenomenon is more likely caused by the existence of amorphous aggregates or oligomeric intermediate species able to bind ThT. In particular, at both copper concentrations, the aggregation of HuPrP(23–231) in the presence of metal ions is significantly slower (lag-phase of about 55 hours) compared to that of HuPrP(90–231). Importantly, the double-sigmoidal curves obtained with 1 and 4 equivalent(s) of Cu(II) exhibit, at the end of the reaction, a different ThT fluorescence intensity that, in both cases, is lower than the value obtained for HuPrP(90–231). Overall, our results suggest that the presence of copper enhances the aggregation of HuPrP(23–231) through the formation of amorphous aggregates having amyloidogenic proprieties.
Discussion
The NMR structure of mouse prion protein was originally solved in 1996 and, since then, a number of PrPC structures from different mammalian species have been determined,11,58,59 mostly in solution and in one case in the solid state.60 PrPC solution dynamics have shown the co-existence of two different domains, the N-terminal which is mostly flexible and the globular C-terminal which is mostly rigid.61,62 The latter has been shown to adopt a very similar structure within all the mammalian species14 and to show only local structural perturbations when pathogenic mutations are present.63 Nonetheless, the “protein only hypothesis” predicted pathogenic conformational transitions of PrPC to PrPSc, which should be facilitated by accidental cellular conditions or by point mutations. Thus, exploring the conformational equilibria and unfolding/folding mechanism of PrPC, under different conditions of pH and temperature, represents a crucial way to investigate the molecular basis of the PrPC to PrPSc transition. Here, using an integrated approach that combines NMR and CD spectroscopies together with ThT fluorescence and RT-QuIC data, we investigated the fundamental forces driving the prion protein folding process and characterized, under physiological conditions, the prion protein conformational equilibrium by exploring the non-native states weakly populated on the folding energy landscape. Using CD and NMR techniques, we show that the N-terminal tail influences the thermal unfolding mechanism of HuPrP via monitoring temperature-induced denaturation of HuPrP(23–231) and HuPrP(90–231). CD thermal unfolding curves, independent of concentration, show that HuPrP(23–231) thermally unfolds following a single sigmoidal cooperative unfolding mechanism, both at pH 6.8 and 5.5. Conversely, the truncated HuPrP(90–231) thermal unfolding has a more complex behaviour, characterized by two successive transitions at both pH values. High-resolution NMR data, confirming the CD results, indicate for truncated HuPrP at 61 °C and pH 5.5 the presence of an intermediate conformation, named β-PrPI (Fig. 1C). Furthermore, NMR thermal melt analysis reveals an interplay between the forces driving the unfolding process, in which the breakdown of most hydrogen bonds involving the flexible N-term residues (90–125) preceded the formation of the intermediate state that in turn is followed by tertiary structure unfolding. Thus, the average backbone proton Tm (50 °C) derived by NMR well corresponds to that of the first thermally induced unfolding transition described by low-resolution CD results (ESI Table 1†). NMR characterization of β-PrPI indicates that at 61 °C the globular domain shows an increase of around 30% of its content of extended structure and reduced helical content of ∼10% (Fig. 2A–C). In detail, β1 and β2 strands extend on both sides, even though β1 enlarges predominantly towards the C-terminus and, as a consequence, α1 reduces its helical content indicating that the β-sheet expands mostly towards the loop connecting β1 and α1 and α1 itself. This β-structure extension appears to stress α2 and α3, especially within their contact surfaces including the disulphide bridge, whose absence is known to causes the global loss of the tertiary fold.64 Thus, in this non-native conformation, the gain of β-sheet character induces a reduction of the α2 and α3 helical content through the loss or remodelling of a large number of hydrophobic interactions between them and the β-sheet and α1. β-PrPI, importantly, preserves most of the native hydrogen bond network (Fig. 2D) which contributes to reducing the conformational flexibility in the ps–ns timescale as revealed by the model free order parameter analysis at 61 °C. Interestingly, ThT-based aggregation assays of HuPrP(90–231) at pH 5.5 clearly show that fibril formation becomes much faster at 61 °C than at 25 °C (Fig. 7A and B and ESI Fig. 15B†) suggesting that β-PrPI is involved in the initial stages of PrPC fibrillation. Moreover, NMR data indicate that the amyloid assembly pathway of β-PrPI involves the formation of transient oligomeric species β-PrPI(o) that, as revealed by RT-QuIC, serves as a seed that templates amyloid formation with a seeding activity similar to that observed for the infectious and pathogenic scrapie form. Very recently, a cryo-EM structure of protease-resistant fibril of (94–178)-human prion protein (r-PrP) spontaneously formed in vitro has been described.65 This human prion protein fibril contains two protofilaments linked by a tightly packed hydrophobic interface. Interestingly, each chain of r-PrP forms a β-structured arch starting at Lys106 and ending at Tyr145. This region nicely overlaps with the HuPrP(90–231) sequence (Gly131-Gly142) significantly increasing its β-character in the intermediate identified and characterized in this study, corroborating its possible involvement in the PrP fibril conversion mechanism. We therefore described the structural/dynamics peculiarities of full-length and truncated HuPrP native states and characterized the transient non-native states weakly populated on the prion protein folding energy landscape. No significant differences in dynamics behaviour have been observed for HuPrP(90–231) and HuPrP(23–231) forms in the nanosecond to picosecond timescale (ESI Fig. 14A–C†). Differently, truncated HuPrP at pH 5.5 shows that at 25 °C a significant number of residues, included in all the secondary structures (ESI Fig. 14A and 15A†), have high Rex values while in the full-length form fewer residues exhibit much smaller Rex (ESI Fig. 14B†). Interestingly, 15N R1ρ RD experiments (Fig. 5A and B and ESI Fig. 15A†) show that HuPrP(90–231) interconverts between a populated ground state and a thermally accessible conformation “excited state”. More precisely, this analysis shows that in the excited states α2 and α3 are well preserved whereas the regions surrounding β1, β2 and the first α1 helix, including the loop connecting α1 and α2, present significant structural variations with respect to the native state (Fig. 5C and D). Furthermore, these two regions experience uncoupled motions, characterized by distinct exchange processes. Importantly, RD data also indicate that the full-length human prion protein is characterized by long-range coupled μs–ms motions between the region connecting the first β-strand with the second α-helix (Gly131-Ser170) and the rest of the protein (Asn171-Ser231) (Fig. 5C and D), strongly suggesting that the perturbation of these correlated dynamics plays a crucial role in the regulation of the unfolding process. Interestingly, RD analysis of the HuPrP(90–231) E219K polymorph has shown that the change causes a significant reduction of the conformational dynamics on the μs–ms timescale (Fig. 6D), providing further evidence that the coupling of μs–ms dynamics between the β1-α1 loop, α1, α1-β2 loop, β2, β2-α2 loop and the two last α-helices represents a key mechanism to prevent prion conversion. The good correlation between Φex values and squared differences in chemical shifts between the native and the intermediate state ΔδF–I2 indicates that, at 25 °C (Fig. 6A), the truncated HuPrP is in equilibrium, in the μs–ms range, with a conformational state significantly similar to β-PrPI. Interestingly, both conformations are characterized by considerable perturbations in the β-sheet and in the α1 helix, with a substantial increase, clearly defined in the thermally induced structure, of the β-character. Our data indicate that the detected excited state may play a crucial role in facilitating the formation of the on-pathway β-PrPI intermediate state that serves as a precursor of prion protein aggregation and amyloid fibril assembly (Fig. 6).
Differently, the full-length HuPrP is characterized by a conformational equilibrium in which the low populated state exhibits a globular domain structural organization very similar to the native conformation (Fig. 5D and ESI Tables 3 and 4†). In this scenario, NMR structural data collected for both proteins identify the coupling of the two domains as a key molecular mechanism in tuning the long-range μs–ms conformational dynamics that in turn modulate the folding process. Because the removal of the N-terminal domain alters the unfolding mechanism of HuPrP(90–231), we investigated the structural mechanisms underlying such different folding behaviour. By comparing NMR structural data we show that in the human full-length prion protein the positively charged N-terminal region transiently interacts with two different patches of negatively charged C-terminus defining a more widespread interaction interface compared to that identified in previous studies18,20,21,66,67 (Fig. 3), without altering the secondary and tertiary organization of the globular domain. Of note, HuPrP patch2 interaction sites are close to the disease-linked mutations (ESI Fig. 11†) whose occurrence increases the kinetics of the misfolding process relative to HuPrP wild-type in vivo.68 Thus, our findings suggest that the electrostatic interaction network between the two domains regulates the prion folding process and a possible destabilization induced, for example, by mutations or metal-binding might increase the stability of partially folded intermediate species, which are likely to be PrPSc precursors. In this scenario, it has been reported that Cu2+ binding reduces the prion protein folding stability, making a conformation transition more thermodynamically favourable.69 Recently, Sivasankar and co-workers showed the role of Cu2+ in the misfolding, oligomerization and neurotoxicity of prion protein.70 Therefore, we explored the copper binding effects on the single sigmoidal cooperative unfolding mechanism observed for HuPrP(23–231). NMR thermal denaturation experiments show that the full-length HuPrP upon Cu2+ addition is characterized by a two-transitions unfolding mechanism with the formation of partially folded intermediate states that having secondary structural organization substantially different from β-PrPI. Accordingly, ThT measurements indicate that the copper binding induces the formation of HuPrP(23–231) amorphous aggregates by a different aggregation mechanism with respect to β-PrPI.
Very likely, this behaviour is due to a different interaction, previously described,26 between the N- and C-termini, induced by Cu2+ coordination to the octa-repeat region which, once positively charged, becomes able to transiently bind particular the negatively charged spot of α1, α2 and α3 helices (Fig. 3A–D and ESI Fig. 9A†). Therefore, the partially unfolded states, here described to be in equilibrium with the truncated protein at pH 5.5 and to prevalently constitute its structure at 61 °C, appear to be inhibited by the proper transient interaction between the N- and C-termini, since they are experimentally invisible in the full-length HuPrP, even at mildly acidic pH27 and likely substituted by diverse intermediate states formed when HuPrP binds to Cu2+. In most experimental studies, non-native conditions, induced by lowering pH or by adding chemical denaturants, have been exploited to investigate partially unfolded forms of native or pathogenically mutated PrPC, involved in fibril formation.33,56,71 Among them, recently, the Udgaonkar group has shown that the initial stages of misfolded PrPC are characterized by a loss of the α1 structure which induces a destabilization of the α2–α3 double helix, leading to an α to β structural transition.72 Here, in substantial agreement, we show that under mildly acidic conditions HuPrP(90–231) at 61 °C converts into a β-PrPI, an intermediate state characterized by a significant reduction of the α1 structure, which is accompanied by a concomitant α2–α3 structural perturbation and by a significant enlargement of β1 and β2 strands (Fig. 2A and B). This gain of β-sheet character induces a loss of α2 and α3 helical content, whose structure is stabilized by a large number of hydrophobic interactions forming a core, which has been shown to play a key role in the PrPC to PrPSc transition.73 Interestingly, β-PrPI, which appears to be induced by the structural reorganization of the HD (residues 95–110), is involved in faster fibril formation and populates a small part of the conformational preferences of HuPrP(90–231) even at 25 °C (Fig. 7A–C). Accordingly, the absence of this unstructured N-terminus makes the thermodynamic features of mouse PrP (MoPrP)(121–231) very similar to those of the full-length MoPrP(23–231) at least at pH 4.0.74 Importantly, in MoPrP(23–231) the formation of this structural intermediate appears inhibited, while another thermally induced intermediate, whose structure is not determined here, reappears in the presence of copper ions. Recently, we have shown how the folding mechanism may influence fibril formation in metal binding proteins.75 In particular, metal ion binding can stabilize an intermediate state that is energetically close to the native structure76 and being easily accessible represents a structural gate to the initial phases of fibril formation. Similarly, the presence of the disulphide bridge in the prion protein plays the key role of stabilizing the α2–α3 double helix system and therein the whole HuPrP globular domain. Once a mutation or any structural perturbation arising from, e.g., pH lowering, metal binding or truncation of the N-terminal domain induces a loss of the interaction stabilizing the core formed by α1 and α2–α3, the disulphide bridge may paradoxically represent a structural gate to the α to β conversion of the α2–α3 system. Overall, the data suggest that the N- to C-terminus interaction prevents HuPrP(23–231) from undergoing this structural conversion, at least at neutral or mildly acidic pH, unless binding to copper perturbs such interaction leading the full-length to populate a similar PUF. Interestingly, the role of inter-domain long-range transient interactions in controlling protein conformational equilibria here shown could represent a common evolutionary strategy for the stabilization of multi-domain proteins.
Conclusion
Prion diseases are a class of neurodegenerative diseases that occur due to misfolding and aggregation into amyloid fibrils of the mainly α-helical cellular prion protein (PrPC). To date, the molecular basis of prion misfolding and aggregation inducing PrP diseases is still poorly understood. Here, we identified the structural and dynamical determinants controlling the prion misfolding process by which PrPC converts into an amyloid fibril through the formation of a β-enriched intermediate state (β-PrPI) involved in the initial stages of PrPC fibrillation having a seeding activity comparable to that of human infectious prions. In this scenario, we showed that the coupling of the two N- and C-terminal domains is the key molecular mechanism in tuning long-range μs–ms conformational dynamics that in turn regulate the folding process avoiding β-PrPI formation. This study provides novel structural and dynamical insights into prion misfolding that can be used to develop molecular strategies able to inhibit aggregation of PrP amyloid fibrils.
Methods
Plasmid construction, protein expression and purification
The HuPrP(23–231) and HuPrP(90–231) sequences were inserted in pET-11a vectors (Novagen) as previously described.20,77 The constructs were expressed in Escherichia coli BL21 (DE3) cells (Stratagene). Freshly transformed overnight culture was added at 37 °C to 2 L of minimal medium (MM) plus ampicillin (100 μg mL−1). For isotope labeling 4 g L−1 [13C6] glucose and 1 g L−1 [15N] ammonium chloride were added. At 0.8 OD600 expression was induced with isopropyl β-D-galactopyranoside to a final concentration of 0.75 mM. The cells were harvested 12 h after induction. The cells were lysed using a homogenizer (PandaPLUS 2000) and the inclusion bodies were suspended in buffer containing 25 mM Tris–HCl, 5 mM EDTA, and 0.8% Triton X-100 (pH 8.0), and then in bi-distilled water several times. Inclusion bodies were dissolved in 5 volumes of 8 M guanidine hydrochloride (GndHCl), loaded onto a pre-equilibrated HiLoad 26/60 Superdex 200 pg column, and eluted in 25 mM Tris–HCl (pH 8), 5 mM ethylene-diaminetetraacetic acid (EDTA), and 6 M GndHCl at a flow rate of 2 mL min−1. Protein refolding was performed by dialysis against refolding buffers (20 mM sodium acetate and 0.005% NaN3, pH 5.5) using a SpectraPor membrane. Purified protein was analysed by SDS-polyacrylamide gel electrophoresis under reducing conditions and the western blot.
CD spectroscopy
HuPrP(23–231) and HuPrP(90–231) were prepared in 300 μL of 20 mM sodium acetate at pH 5.5 and phosphate at pH 6.8 with the same ionic strength. Copper binding experiments of HuPrP(23–231) were performed at pH 5.5 in MES buffer having the same ionic strength as that of the previously reported sodium acetate buffer.
The thermal denaturation of prion protein samples was evaluated using a JASCO J-815 CD spectropolarimeter equipped with Peltier temperature control. CD spectra were measured at 5 °C intervals in the 5–55 °C range and from 55–90 °C at regular intervals of 3 °C. After the final spectrum at 90 °C, all samples were cooled to 25 °C, and a final set of spectra were collected. For the investigation of the copper effects on the folding mechanism of HuPrP(23–231) the samples were prepared in the same way with the addition of CuCl2 to give the appropriate final Cu2+ concentrations (1 and 4 molar equivalent(s)). The experiments were conducted at different protein concentrations ranging from 10 μM to 80 μM (only for measurements at pH 5.5) of HuPrP(23–231) and HuPrP(90–231) proteins, and data were collected using a quartz cuvette with a 1 cm path length in the 200–260 nm wavelength range with a pitch of 1 nm. All data were measured with a bandwidth of 1 nm with a scanning speed of 50 min−1 and normalized against reference spectra to remove the background contribution of buffer. The data obtained were analysed using a two-state folding model. The reversibility of the thermal unfolding processes was investigated by comparing the CD spectrum acquired after cooling the heat-denatured sample; moreover, the experimental ellipticity θ222/θ219 ratios were compared to the reference value obtained from the representative NMR structure using the web-based application PDB2CD.78
Aggregation index (A.I.) determination
Aggregation of the prion protein samples was assessed using an aggregation index (A.I.) calculated from spectroscopic measurements. UV spectroscopy was performed with a SHIMADZU 1800 spectrometer. Samples of 300 μL were measured in micro-quartz cuvettes with a path length of 10 mm. UV absorbance was recorded from 240 to 360 mm using an integration time of 15 s and steps of 1 nm. The spectra were normalized for the absorbance of the particular buffer. The aggregation index (A.I.) was calculated from the UV spectra as A350/(A280 − A350) × 100, where A280 and A350 are the absorbances at 280 and 350 nm, respectively. Usually, A.I. values below 10 indicate the presence in solution of insignificant amounts of soluble aggregates.79
NMR resonance assignments
All NMR experiments were carried out at 25 °C using a Bruker AVIII HD 600 MHz spectrometer equipped with a triple resonance Prodigy N2 cryoprobe with a z-axis pulse field gradient. NMR samples for chemical shift assignment of 15N–13C labeled HuPrP(23–231), 15N–13C labeled HuPrP(90–231) and 15N–13C labeled HuPrP(90–231) E219K proteins were prepared in 20 mM sodium acetate, pH 5.5 and 90% H2O/10% D2O. The protein final concentration was 80 μM. Backbone resonances Cα, Cβ, C′, N, HN and Hα of HuPrP(23–231) and HuPrP(90–231) were assigned by analyzing the standard triple resonance experiments80 as 3D HNCA, 3D CBCANH, 3D CBCA(CO)NH, 3D HNCO and 3D HNHα using the deposited chemical shifts under the BMRB accession codes 4402, 18426 and 17780. Moreover, to further improve the previous backbone assignment11,57 two- and three-dimensional TROSY-based81 and T2-filter NMR experiments were performed after a rigorous optimization.82 At 25 °C, a nearly complete assignment of backbone and Cβ chemical shifts for HuPrP(23–231) (96%), HuPrP(90–231) (96%) and HuPrP(90–231) E219K (93%) was obtained. The chemical shift assignment for all investigated prion proteins, at different temperatures, was performed by following the peaks trajectories over a temperature range. The spectra were processed using NMRpipe83 and analyzed using SPARKY84 and CARA.85 The 1H–15N HSQC spectrum of the HuPrP(90–231) random coil conformation at 61 °C was reconstructed by using a series of in-house written NMRpipe scripts based on the random coil shifts predicted as reported below. 1H, 13C and 15N chemical shifts were calibrated indirectly to external DSS references.
NMR chemical shifts analysis
The structural rearrangements were estimated by applying a weighted combined 1H (ΔδH), 15N (ΔδN) and 13C (ΔδC) chemical shift perturbation (CSP) based on the following equation:| | | ΔH,N,C = ((ΔδHWH)2 + (ΔδNWN)2 + (ΔδCWC)2)1/2 | (1) |
where WH, WN, and WC are weighing factors for the 1H, 15N and 13C shifts defined as WH = |γH/γH| = 1; WN = |γN/γH| = 0.101; WC = |γC/γH| = 0.251. ΔδH, ΔδN and ΔδC are the chemical shift differences in ppm between the compared protein states for 1H, 15N and 13C, respectively; γH, γN and γC are the gyromagnetic ratios.
The quality factor used to evaluate the correlation between the HuPrP(23–231) and HuPrP(90–231) secondary chemical shifts (QΔ) in the region 90–231 is defined as:
| | 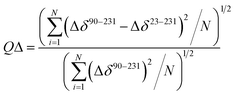 | (2) |
in which Δ
δ90–231 and Δ
δ23–231 are the secondary chemical shifts calculated for HuPrP(90–231) and HuPrP(23–231), respectively; Δ
δ is calculated as the difference between the observed chemical shift
δobs and the predicted random coil shift
δrandom coil.
The quality factor (Q) used to estimate the correlation between the measured chemical shifts (δobs) and those predicted (δpred) from the representative structure of the NMR ensemble (PDB ID code: 2LSB) is defined as:
| | 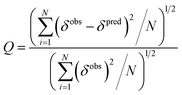 | (3) |
The random coil shifts were calculated at different temperature and pH values by using the approach defined by Poulsen and co-workers.86,87 The prediction of the chemical shifts from the representative NMR conformer was performed using Shiftx2 software.47 The secondary structure propensity (SSP) scores were calculated with the random coil chemical shifts and the average secondary shifts for the fully formed secondary structure as described previously.88 The model-free order parameters S2 for the backbone amide groups were predicted from backbone and Cβ chemical shifts using the Random Coil Index (RCI) approach.49 Data were analyzed and visualized using the software CHIMERA.89 The Adaptive Poisson–Boltzmann Solver (APBS)90 was used to calculate spatial distributions of electrostatic potentials using the linearized Poisson–Boltzmann equation and parameters from the PQR files obtained using the PDB2PQR server.91 The electrostatic map was generated using CHIMERA.89 The NaCl titration experiments were done on 80 μM HuPrP(23–231) and HuPrP(90–231) prion protein samples. A series of 1H–15N HSQC spectra were collected under the following two conditions: 50 mM and 200 mM. To evaluate the chemical shift differences between HuPrP(23–231) and HuPrP(90–231) induced by addition of sodium chloride we defined the following equation:
| | | (ΔδHN,N23–231 − ΔδHN,N90–231)2 | (4) |
where Δ
δHN,N is the weighted combined difference between
1HN and
15N chemical shifts observed upon addition of the salt concentration and the shifts obtained in the absence of NaCl for HuPrP(23–231) and HuPrP(90–231), respectively.
NMR thermal analysis
1H–15N and 1H–13C HSQC spectra were collected every 5 °C from 5 to 55 °C and every 3 °C in the 55–90 °C range for both HuPrP(90–231) and HuPrP(23–231). All NMR spectra acquired for the thermal unfolding characterization were referenced indirectly to external DSS references. 2D 1H–15N HSQC experiments were carried out with 32 scans per t1 increment, a spectral width of 1459.43 along t1 and 7211.54 along t2, 2048 × 256 complex points in t2 and t1, respectively and a 1.0 s relaxation delay for HuPrP(90–231); meanwhile, for HuPrP(23–231) the 1H–15N HSQC spectra were acquired with 32 scans per t1 increment, a spectral width of 1581.28 along t1 and 7211.54 along t2, 2048 × 256 complex points in t2 and t1, respectively and a 1.0 s relaxation delay. Spectra were apodized with a square cosine window function and a zero filling to a matrix of size 4096 × 4096 before Fourier transform and baseline correction. 2D 1H–13C CT HSQC (constant time version) spectra were acquired with 32 scans per t1 increment, a spectral width of 5131.08 Hz along t1 and 7211.54 Hz along t2, 2048 × 256 complex points in t2 and t1, respectively and a 1.0 s relaxation delay. The 1H–13C CT HSQC experiments were acquired with a heteronuclear coupling constant JHX = 145 Hz and a constant time period of 26.6 milliseconds. The 1H–13C CT HSQC were apodized with a square cosine window function and zero filled to a matrix of size 4096 × 4096 before Fourier transform and baseline correction. 1H chemical shifts at different temperatures were referenced externally to DSS,92 whereas 15N and 13C shifts were calibrated indirectly to DSS. The midpoint temperatures (Tms) were estimated by measuring HN and Hα chemical shifts as a function of temperature in the 15–61 °C range. In particular, in the case of the first transition of HuPrP(90–231), for each well-resolved proton we first normalized the observed chemical shifts with respect to the value measured at the lowest temperature (15 °C) and then we fitted for each residue the data with two models. All copper binding experiments of HuPrP(23–231) were performed at pH 5.5 in MES buffer having the same ionic strength as that of the previously reported sodium acetate buffer.
NMR relaxation measurements
15N relaxation experiments were carried out on a 600 MHz spectrometer at 25 °C. In all experiments, the concentration of HuPrP(90–231), HuPrP(90–231) E219K and HuPrP(23–231) samples was about 0.2 mM. 15N longitudinal relaxation rates (R1) were measured using relaxation delays of 11*, 161, 330, 495, 660*, 825 and 1100* ms. 15N transverse relaxation rates (“total R2”) were acquired using relaxation delays of 15.84*, 31.68, 47.56*, 63.36, 79.20*, 95.04 and 110.88 ms. Steady-state 1H–15N heteronuclear Nuclear Overhauser Enhancement (NOE) was measured by acquiring paired interleaved spectra collected with and without an initial proton saturation period (3 s) during the 5 s recycle delay. NOE values were calculated from the ratio of the peak intensities between saturated and reference spectra. For HuPrP(23–231), NOE values were mapped on a structural model of the full-length protein obtained using the software I-TASSER,93 using as template the NMR structure of the C-terminal domain (PDB ID code: 2LSB). Only for the HuPrP(90–231) were the 15N relaxation measurements also performed on a 700 MHz spectrometer at 25 °C and pH 5.5. 15N R1, 15N R2 and [1H]–15N NOE data sets acquired for HuPrP(90–231) at 500 and 600 MHz were analysed using ROTDIF and DYNAMICS software94,95 using the Lipari–Szabo approach.96,97 An axially symmetric rotational diffusion tensor was optimized against the human prion protein structure (PDB ID: 2LSB) using selected residues with [1H]–15N NOE > 0.65 within the main secondary structure elements.15N longitudinal relaxation rates in the rotating frame (R1ρ) were measured in a near-resonance mode with relaxation delays of 16*, 32, 48*, 56, 72*, and 100 ms and spin-lock field strength of 3 kHz centered on a 15N chemical shift of 116.2 ppm. The asterisks indicate duplicate measurements. R1, R2 and R1ρ relaxation rates were obtained from the best single exponential to the experimental intensity data. Effective R2 rates were determined from the following relation:| | | R2effective = (R1ρ − R1cos2θ)/sin2θ | (5) |
where θ = tan(νSL/Ω), νSL is the 15N spin-lock field strength in Hz and Ω is the resonance offset from the spin-lock carrier (Hz). Rex contributions were estimated as the difference between the “total R2” and the effective R2 rates derived from R1ρ values obtained by applying a 3 kHz spin-lock field strength.
NMR R1ρ relaxation dispersion
R
1
ρ-based relaxation dispersion experiments were performed at 25 °C and a proton Larmor frequency of 600 MHz on HuPrP(90–231), HuPrP(90–231) E219K and HuPrP(23–231) samples at a final protein concentration of 0.15 mM. R1ρ rates were obtained at 15N spin lock field strengths of 600*, 800, 1000*, 1200, 1500, 2000, 2200, 2500, 2800 and 3000 Hz with carrier centred at 116.2 ppm (the asterisks indicate duplicate measurements). The data were first analysed on a per-residue basis; a given resonance was included only when Ω ≤ |0.4 ωSL/2π| Hz. All data were fitted with a two-state (A, B) chemical exchange model.
In the fast exchange limit, the effective R2 as a function of the effective field ωe can be described by the equation reported below:
| | | R2effective (ωe) = R20 + Rex(ωe) = R20 + Φexkex/(kex2 + ωe2) | (6) |
in which
R20 is the transverse relaxation rate in the absence of conformational exchange;
Φex =
pApBΔ
ωN2;
pA and
pB are the fractional site populations with
pA +
pB = 1; Δ
ωN is the difference in
15N chemical shifts between the two states;
kex =
k1 +
k−1 is the sum of forward and reverse rate constants;
ωe2 =
ωSL2 +
Ω2. All
R2effective (
ωe) dispersion curves were fitted individually and globally to
eqn (5) using Mathematica (v9, Wolfram Research, Inc.) and CPMG_fit software (kindly provided by Dr Dmitry Korzhnev) by minimization of the following
χ2 target function:
| |  | (7) |
in which
Xi are the data sets and
σi the relative error. For group fits, all residues showing relaxation dispersion curves were grouped together based on the
χ2group/
χ2individual ratio, following the method proposed by Mulder
et al.98 The residues with
χ2group/
χ2individual > 2 were excluded from the cluster (A).
The comparison of the different models was performed using the Bayesian Information Criterion (BIC) and the corrected Akaike Information Criterion (AICc) defined as reported below:
| | | BIC = χ2 + kln(n) | (8) |
| |  | (9) |
where
k is the number of fit parameters and
n the number of data points.
Aggregation assay
The HuPrP(90–231) and HuPrP(23–231) recombinant protein solutions were allowed to thaw at room temperature and filtered through a 100kDa Nanosep centrifugal device (Pall Corporation). The final reaction volume was 100μL and the reagents (Sigma) were concentrated as follows: NaCl 170mM, PBS 10 mM, EDTA 1mM, thioflavin-T (ThT) 10μM and 0.134mg mL−1 human recombinant PrP. pH of the final reaction mixture was adjusted to 5.5 or 6.8. Five replicates for each condition were analyzed in a black 96-well optical flat bottom plate (Thermo Scientific). The plate was sealed with a sealing film (Thermo Scientific) and inserted into a FLUOstar OPTIMA microplate reader (BMG Labtech). The plate was cyclically shaken for 1minute at 600 rpm (double orbital) and incubated for 1minute at 61 °C or 25 °C. Fluorescence readings (480nm excitation) were taken every 15minutes (30 flashes per well at 450nm emission). The mean values of the five replicates of fluorescence intensity (arbitrary units, AU) were recorded and plotted in a graph against time. Real-time ThT fluorescence experiments of HuPrP(23–231) in the presence of 1 or 4 equivalents of copper were performed at 25 and 61 °C by adding CuCl2.
RT-QuIC experiments
RecHaPrP(90–231) was thawed and filtered through a 100kDa Nanosep centrifugal device (Pall Corporation). The RT-QuIC reaction mixture was composed of 10 mM PBS (pH 7.4), 150 mM NaCl, 0.13 mg mL−1 recHaPrP(90–231), 1 mM ethylenediaminetetraacetic acid tetrasodium salt (EDTA), 0.002% SDS and 10 μM ThT. Ninety-eight μL of the reaction mixture was placed in a black 96-well optical flat bottom plate (Thermo Scientific) and supplemented with 2 μL of 10% brain homogenates (weight/volume) collected from patients with six forms of sporadic Creutzfeldt–Jakob disease (sCJD: MM1, MV1, VV1, MM2, MV2 and VV2), Alzheimer's disease (AD) and non-neurodegenerative control. All samples were diluted to 10−5 (volume/volume) before testing. In parallel 98 μL of the reaction mixture was supplemented with 2 μL of HuPrP(90–231) aggregates prepared either at pH 5.5 or 6.8. Before testing all aggregates were diluted to a final concentration of about 1.35 picograms. Finally, 85 μL of the reaction mixture was supplemented with 15 μL of CSF samples collected from patients with sCJD (MM1, MV1 and VV1), Alzheimer's disease and hydrocephalus. Each sample has been analyzed in quadruplicate. The plate was sealed with a sealing film (Thermo Scientific) and underwent intermittent cycles of shaking (1 min at 600 rpm, double orbital) and incubation (1 min) at 55 °C using the OPTIMA fluorescence microplate reader (BMG Labtech). ThT fluorescence was measured every 30 minutes (wavelengths: excitation 450 ± 10 nm; emission 480 ± 10 nm). A sample was considered positive if the fluorescence value of at least 2 out of 4 replicates was higher than 10000 arbitrary units (AU) before the threshold of time set at 60hours, as described in a previous publication.99
Data availability
The datasets used and/or analysed during this study are available from the corresponding author on reasonable request.
Author contributions
GL and RF conceived the study. LR and RF designed CD and NMR experiments. LR performed CD and NMR experiments and analyzed the data. GS prepared the prion protein samples. EB and FM conducted ThT-fluorescence fibrillization assays. All authors discussed the experimental results and analyses. LR, GL and RF wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research was supported by “Programma Valere” 2018 of the University of Campania to LR and RF; by MIUR grant no. 20157WZM8A to GM and 2017WBZFHL to CI; by the Italian Ministry of Health (GR-2013-02355724 and Ricerca Corrente) to FM; by the European Community and the Italian Ministry of Health within the EuroNanoMed3 ERANET co-fund SPEEDY project to FM.
References
- C. Weissmann, M. Enari, P. C. Klöhn, D. Rossi and E. Flechsig, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(Suppl. 4), 16378–16383 CrossRef CAS PubMed.
- M. Moser, R. J. Colello, U. Pott and B. Oesch, Neuron, 1995, 14, 509–517 CrossRef CAS PubMed.
- N. Stahl, D. R. Borchelt, K. Hsiao and S. B. Prusiner, Cell, 1987, 51, 229–240 CrossRef CAS PubMed.
- M. D. Caiati, V. F. Safiulina, G. Fattorini, S. Sivakumaran, G. Legname and E. Cherubini, J. Neurosci., 2013, 33, 2973–2983 CrossRef CAS PubMed.
- A. Santuccione, V. Sytnyk, I. Leshchyns'ka and M. Schachner, J. Cell Biol., 2005, 169, 341–354 CrossRef CAS PubMed.
- L. Gasperini, E. Meneghetti, B. Pastore, F. Benetti and G. Legname, Antioxid. Redox Signaling, 2015, 22, 772–784 CrossRef CAS PubMed.
- H. Khosravani, Y. Zhang, S. Tsutsui, S. Hameed, C. Altier, J. Hamid, L. Chen, M. Villemaire, Z. Ali, F. R. Jirik and G. W. Zamponi, J. Cell Biol., 2008, 181, 551–565 CrossRef CAS PubMed.
- P. K. Stys, H. You and G. W. Zamponi, J. Physiol., 2012, 590, 1357–1368 CrossRef CAS PubMed.
- M. J. Pushie, I. J. Pickering, G. R. Martin, S. Tsutsui, F. R. Jirik and G. N. George, Metallomics, 2011, 3, 206–214 CrossRef CAS PubMed.
- E. D. Walter, D. J. Stevens, M. P. Visconte and G. L. Millhauser, J. Am. Chem. Soc., 2007, 129, 15440–15441 CrossRef CAS PubMed.
- R. Zahn, A. Liu, T. Lührs, R. Riek, C. von Schroetter, F. López García, M. Billeter, L. Calzolai, G. Wider and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 145–150 CrossRef CAS PubMed.
- R. Riek, S. Hornemann, G. Wider, M. Billeter, R. Glockshuber and K. Wüthrich, Nature, 1996, 382, 180–182 CrossRef CAS PubMed.
- T. L. James, H. Liu, N. B. Ulyanov, S. Farr-Jones, H. Zhang, D. G. Donne, K. Kaneko, D. Groth, I. Mehlhorn, S. B. Prusiner and F. E. Cohen, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10086–10091 CrossRef CAS PubMed.
- R. N. Abskharon, G. Giachin, A. Wohlkonig, S. H. Soror, E. Pardon, G. Legname and J. Steyaert, J. Am. Chem. Soc., 2014, 136, 937–944 CrossRef CAS PubMed.
- R. Abskharon, F. Wang, A. Wohlkonig, J. Ruan, S. Soror, G. Giachin, E. Pardon, W. Zou, G. Legname, J. Ma and J. Steyaert, PLoS Pathog., 2019, 15, e1008139 CrossRef PubMed.
- K. Kuwata, T. Matumoto, H. Cheng, K. Nagayama, T. L. James and H. Roder, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14790–14795 CrossRef CAS PubMed.
- G. L. Millhauser, Annu. Rev. Phys. Chem., 2007, 58, 299–320 CrossRef CAS PubMed.
- G. Salzano, G. Giachin and G. Legname, Cells, 2019, 8, 770 CrossRef CAS PubMed.
- C. Sánchez-López, G. Rossetti, L. Quintanar and P. Carloni, Int. J. Mol. Sci., 2018, 20, 18 CrossRef PubMed.
- P. D'Angelo, S. Della Longa, A. Arcovito, G. Mancini, A. Zitolo, G. Chillemi, G. Giachin, G. Legname and F. Benetti, Biochemistry, 2012, 51, 6068–6079 CrossRef PubMed.
- J. Martínez, R. Sánchez, M. Castellanos, N. Makarava, A. Aguzzi, I. V. Baskakov and M. Gasset, Sci. Rep., 2015, 5, 13623 CrossRef PubMed.
- S. P. Narayanan, D. G. Nair, D. Schaal, M. Barbosa de Aguiar, S. Wenzel, W. Kremer, S. Schwarzinger and H. R. Kalbitzer, Sci. Rep., 2016, 6, 28419 CrossRef CAS PubMed.
- A. K. Thakur, A. K. Srivastava, V. Srinivas, K. V. R. Chary and C. M. Rao, J. Biol. Chem., 2011, 286, 38533–38545 CrossRef CAS PubMed.
- J. Bremer, F. Baumann, C. Tiberi, C. Wessig, H. Fischer, P. Schwarz, A. D. Steele, K. V. Toyka, K. A. Nave, J. Weis and A. Aguzzi, Nat. Neurosci., 2010, 13, 310–318 CrossRef CAS PubMed.
- N. T. Watt, D. R. Taylor, T. L. Kerrigan, H. H. Griffiths, J. V. Rushworth, I. J. Whitehouse and N. M. Hooper, Nat. Commun., 2012, 3, 1134 CrossRef PubMed.
- H. You, S. Tsutsui, S. Hameed, T. J. Kannanayakal, L. Chen, P. Xia, J. D. Engbers, S. A. Lipton, P. K. Stys and G. W. Zamponi, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1737–1742 CrossRef CAS PubMed.
- B. Wu, A. J. McDonald, K. Markham, C. B. Rich, K. P. McHugh, J. Tatzelt, D. W. Colby, G. L. Millhauser and D. A. Harris, Elife, 2017, 6, e23473 CrossRef PubMed.
- E. G. Evans, M. J. Pushie, K. A. Markham, H. W. Lee and G. L. Millhauser, Structure, 2016, 24, 1057–1067 CrossRef CAS PubMed.
- K. M. Schilling, L. Tao, B. Wu, J. T. M. Kiblen, N. C. Ubilla-Rodriguez, M. J. Pushie, R. D. Britt, G. P. Roseman, D. A. Harris and G. L. Millhauser, J. Mol. Biol., 2020, 432, 4408–4425 CrossRef CAS PubMed.
- I. V. Baskakov, G. Legname, S. B. Prusiner and F. E. Cohen, J. Biol. Chem., 2001, 276, 19687–19690 CrossRef CAS PubMed.
- N. Okimoto, K. Yamanaka, A. Suenaga, M. Hata and T. Hoshino, Biophys. J., 2002, 82, 2746–2757 CrossRef CAS PubMed.
- E. Behmard, P. Abdolmaleki, E. B. Asadabadi and S. Jahandideh, J. Biomol. Struct. Dyn., 2011, 29, 379–389 CrossRef CAS PubMed.
- R. Moulick, R. Das and J. B. Udgaonkar, J. Biol. Chem., 2015, 290, 25227–25240 CrossRef CAS PubMed.
- I. Sengupta and J. B. Udgaonkar, Chem. Commun., 2018, 54, 6230–6242 RSC.
- G. Wildegger, S. Liemann and R. Glockshuber, Nat. Struct. Biol., 1999, 6, 550–553 CrossRef CAS PubMed.
- L. L. Hosszu, N. J. Baxter, G. S. Jackson, A. Power, A. R. Clarke, J. P. Waltho, C. J. Craven and J. Collinge, Nat. Struct. Biol., 1999, 6, 740–743 CrossRef CAS PubMed.
- A. C. Apetri, K. Surewicz and W. K. Surewicz, J. Biol. Chem., 2004, 279, 18008–18014 CrossRef CAS PubMed.
- A. C. Apetri, K. Maki, H. Roder and W. K. Surewicz, J. Am. Chem. Soc., 2006, 128, 11673–11678 CrossRef CAS PubMed.
- T. Hart, L. L. Hosszu, C. R. Trevitt, G. S. Jackson, J. P. Waltho, J. Collinge and A. R. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5651–5656 CrossRef CAS PubMed.
- F. Benetti, X. Biarnés, F. Attanasio, G. Giachin, E. Rizzarelli and G. Legname, J. Mol. Biol., 2014, 426, 3796–3810 CrossRef CAS PubMed.
- J. E. Arnold, C. Tipler, L. Laszlo, J. Hope, M. Landon and R. J. Mayer, J. Pathol., 1995, 176, 403–411 CrossRef CAS PubMed.
- M. Q. Khan, B. Sweeting, V. K. Mulligan, P. E. Arslan, N. R. Cashman, E. F. Pai and A. Chakrabartty, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19808–19813 CrossRef CAS PubMed.
- D. R. Borchelt, A. Taraboulos and S. B. Prusiner, J. Biol. Chem., 1992, 267, 16188–16199 CrossRef CAS PubMed.
- F. Huang, S. Sato, T. D. Sharpe, L. Ying and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 123–127 CrossRef CAS PubMed.
- A. N. Naganathan, J. M. Sanchez-Ruiz and V. Muñoz, J. Am. Chem. Soc., 2005, 127, 17970–17971 CrossRef CAS PubMed.
- D. S. Wishart and D. A. Case, Methods Enzymol., 2001, 338, 3–34 CAS.
- B. Han, Y. Liu, S. W. Ginzinger and D. S. Wishart, J. Biomol. NMR, 2011, 50, 43–57 CrossRef CAS PubMed.
- Y. Shen, F. Delaglio, G. Cornilescu and A. Bax, J. Biomol. NMR, 2009, 44, 213–223 CrossRef CAS PubMed.
- M. V. Berjanskii and D. S. Wishart, J. Am. Chem. Soc., 2005, 127, 14970–14971 CrossRef CAS PubMed.
- A. G. Palmer and F. Massi, Chem. Rev., 2006, 106, 1700–1719 CrossRef CAS PubMed.
- D. Ban, M. Funk, R. Gulich, D. Egger, T. M. Sabo, K. F. Walter, R. B. Fenwick, K. Giller, F. Pichierri, B. L. de Groot, O. F. Lange, H. Grubmüller, X. Salvatella, M. Wolf, A. Loidl, R. Kree, S. Becker, N. A. Lakomek, D. Lee, P. Lunkenheimer and C. Griesinger, Angew. Chem., Int. Ed. Engl., 2011, 50, 11437–11440 CrossRef CAS PubMed.
- D. Ban, T. M. Sabo, C. Griesinger and D. Lee, Molecules, 2013, 18, 11904–11937 CrossRef CAS PubMed.
- F. Munari, N. Rezaei-Ghaleh, S. Xiang, W. Fischle and M. Zweckstetter, PLoS One, 2013, 8, e60887 CrossRef CAS PubMed.
- O. Trott and A. G. Palmer, J. Magn. Reson., 2002, 154, 157–160 CrossRef CAS PubMed.
- N. Kachel, W. Kremer, R. Zahn and H. R. Kalbitzer, BMC Struct. Biol., 2006, 6, 16 CrossRef PubMed.
- R. P. Honda, M. Xu, K. I. Yamaguchi, H. Roder and K. Kuwata, Structure, 2015, 23, 1735–1742 CrossRef CAS PubMed.
- I. Biljan, G. Giachin, G. Ilc, I. Zhukov, J. Plavec and G. Legname, Biochem. J., 2012, 446, 243–251 CrossRef CAS PubMed.
- B. Christen, D. R. Pérez, S. Hornemann and K. Wüthrich, J. Mol. Biol., 2008, 383, 306–312 CrossRef CAS PubMed.
- D. A. Lysek, C. Schorn, L. G. Nivon, V. Esteve-Moya, B. Christen, L. Calzolai, C. von Schroetter, F. Fiorito, T. Herrmann, P. Güntert and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 640–645 CrossRef CAS PubMed.
- K. J. Knaus, M. Morillas, W. Swietnicki, M. Malone, W. K. Surewicz and V. C. Yee, Nat. Struct. Biol., 2001, 8, 770–774 CrossRef CAS PubMed.
- R. Riek, S. Hornemann, G. Wider, R. Glockshuber and K. Wüthrich, FEBS Lett., 1997, 413, 282–288 CrossRef CAS PubMed.
- S. H. Bae, G. Legname, A. Serban, S. B. Prusiner, P. E. Wright and H. J. Dyson, Biochemistry, 2009, 48, 8120–8128 CrossRef CAS PubMed.
- G. Giachin, I. Biljan, G. Ilc, J. Plavec and G. Legname, Molecules, 2013, 18, 9451–9476 CrossRef CAS PubMed.
- F. Benetti and G. Legname, Prion., 2015, 9, 119–124 CrossRef CAS PubMed.
- C. Glynn, M. R. Sawaya, P. Ge, M. Gallagher-Jones, C. W. Short, R. Bowman, M. Apostol, Z. H. Zhou, D. S. Eisenberg and J. A. Rodriguez, Nat. Struct. Mol. Biol., 2020, 27, 417–423 CrossRef CAS PubMed.
- T. Sonati, R. R. Reimann, J. Falsig, P. K. Baral, T. O'Connor, S. Hornemann, S. Yaganoglu, B. Li, U. S. Herrmann, B. Wieland, M. Swayampakula, M. H. Rahman, D. Das, N. Kav, R. Riek, P. P. Liberski, M. N. James and A. Aguzzi, Nature, 2013, 501, 102–106 CrossRef CAS PubMed.
- A. J. McDonald, D. R. Leon, K. A. Markham, B. Wu, C. F. Heckendorf, K. Schilling, H. D. Showalter, P. C. Andrews, M. E. McComb, M. J. Pushie, C. E. Costello, G. L. Millhauser and D. A. Harris, Structure, 2019, 27, 907–922 CrossRef CAS PubMed.
- G. Rossetti, X. Cong, R. Caliandro, G. Legname and P. Carloni, J. Mol. Biol., 2011, 411, 700–712 CrossRef CAS PubMed.
- S. S. Leal, H. M. Botelho and C. M. Gomes, Coord. Chem. Rev., 2012, 256, 2253–2270 CrossRef CAS.
- C. F. Yen, D. S. Harischandra, A. Kanthasamy and S. Sivasankar, Sci. Adv., 2016, 2, e1600014 CrossRef PubMed.
- J. Singh and J. B. Udgaonkar, J. Mol. Biol., 2016, 428, 1345–1355 CrossRef CAS PubMed.
- R. K. Singh, N. G. Chamachi, S. Chakrabarty and A. Mukherjee, J. Phys. Chem. B, 2017, 121, 550–564 CrossRef CAS PubMed.
- N. G. Chamachi and S. Chakrabarty, Biochemistry, 2017, 56, 833–844 CrossRef CAS PubMed.
- R. Moulick and J. B. Udgaonkar, Biophys. J., 2014, 106, 410–420 CrossRef CAS PubMed.
- G. Malgieri, G. D'Abrosca, L. Pirone, A. Toto, M. Palmieri, L. Russo, M. F. M. Sciacca, R. Tatè, V. Sivo, I. Baglivo, R. Majewska, M. Coletta, P. V. Pedone, C. Isernia, M. De Stefano, S. Gianni, E. M. Pedone, D. Milardi and R. Fattorusso, Chem. Sci., 2018, 9, 3290–3298 RSC.
- M. Palmieri, G. Malgieri, L. Russo, I. Baglivo, S. Esposito, F. Netti, A. Del Gatto, I. de Paola, L. Zaccaro, P. V. Pedone, C. Isernia, D. Milardi and R. Fattorusso, J. Am. Chem. Soc., 2013, 135, 5220–5228 CrossRef CAS PubMed.
- M. Kosmač, S. Koren, G. Giachin, T. Stoilova, R. Gennaro, G. Legname and V. Serbec, Mol. Immunol., 2011, 48, 746–750 CrossRef PubMed.
- L. Mavridis and R. W. Janes, Bioinformatics, 2017, 33, 56–63 CrossRef CAS PubMed.
- D. S. Katayama, R. Nayar, D. K. Chou, J. Campos, J. Cooper, D. G. Vander Velde, L. Villarete, C. P. Liu and M. Cornell Manning, J. Pharm. Sci., 2005, 94, 2703–2715 CrossRef CAS PubMed.
- S. Grzesiek, H. Döbeli, R. Gentz, G. Garotta, A. M. Labhardt and A. Bax, Biochemistry, 1992, 31, 8180–8190 CrossRef CAS PubMed.
- K. Pervushin, J. Biomol. NMR, 2001, 20, 275–285 CrossRef CAS PubMed.
- B. Farina, A. Del Gatto, D. Comegna, S. Di Gaetano, D. Capasso, C. Isernia, M. Saviano, R. Fattorusso, L. Zaccaro and L. Russo, J. Pept. Sci., 2019, 25, e3166 CrossRef PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef PubMed.
-
R. Keller, PhD thesis, Diss. ETH Nr. 15947, 2004.
- M. Kjaergaard, S. Brander and F. M. Poulsen, J. Biomol. NMR, 2011, 49, 139–149 CrossRef CAS PubMed.
- M. Kjaergaard and F. M. Poulsen, J. Biomol. NMR, 2011, 50, 157–165 CrossRef CAS PubMed.
- J. A. Marsh, V. K. Singh, Z. Jia and J. D. Forman-Kay, Protein Sci., 2006, 15, 2795–2804 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- J. L. Markley, A. Bax, Y. Arata, C. W. Hilbers, R. Kaptein, B.
D. Sykes, P. E. Wright and K. Wüthrich, J. Biomol. NMR, 1998, 12, 1–23 CrossRef CAS PubMed.
- A. Roy, A. Kucukural and Y. Zhang, Nat. Protoc., 2010, 5, 725–738 CrossRef CAS PubMed.
- J. B. Hall and D. Fushman, J. Biomol. NMR, 2003, 27, 261–275 CrossRef CAS PubMed.
- J. B. Hall and D. Fushman, J. Am. Chem. Soc., 2006, 128, 7855–7870 CrossRef CAS PubMed.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4546–4559 CrossRef CAS.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef CAS.
- F. A. Mulder, A. Mittermaier, B. Hon, F. W. Dahlquist and L. E. Kay, Nat. Struct. Biol., 2001, 8, 932–935 CrossRef CAS PubMed.
- A. Franceschini, S. Baiardi, A. G. Hughson, N. McKenzie, F. Moda, M. Rossi, S. Capellari, A. Green, G. Giaccone, B. Caughey and P. Parchi, Sci. Rep., 2017, 7, 10655 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00345g |
| ‡ Current address: Human Technopole Palazzo Italia Viale Rita Levi-Montalcini, 1 20157 Milan, Italy. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Giulia
Salzano‡
a,
Giulia
Salzano‡
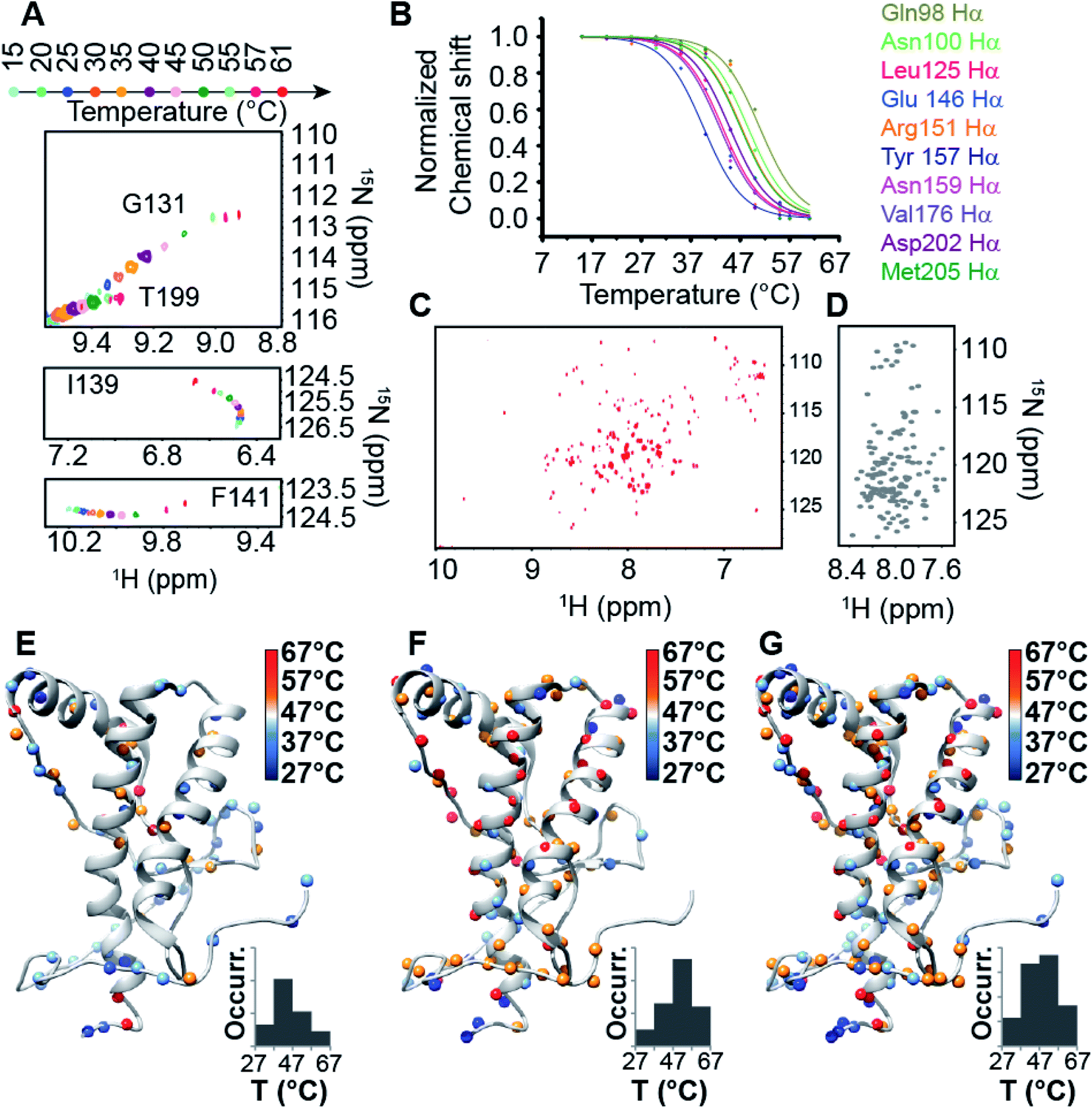
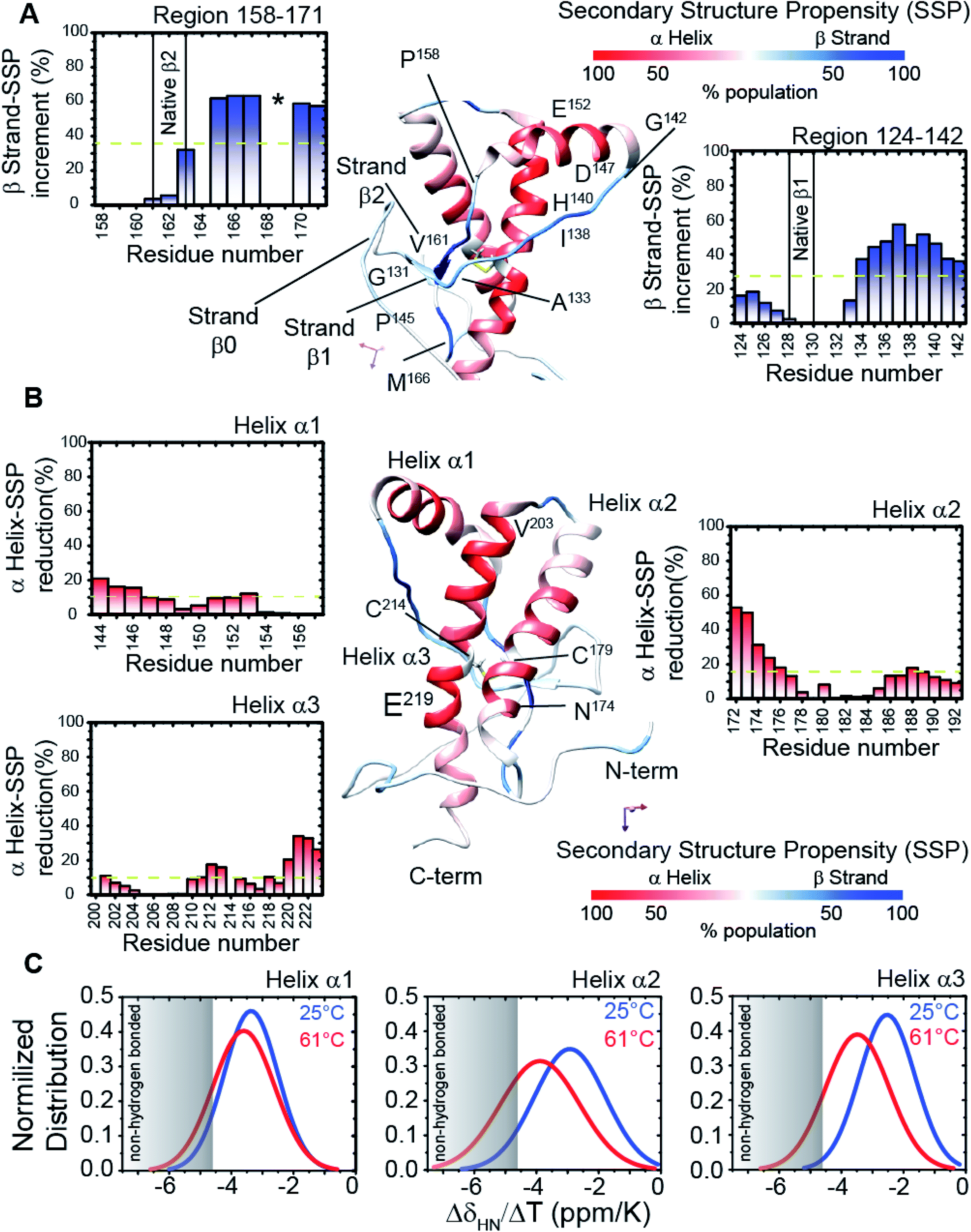
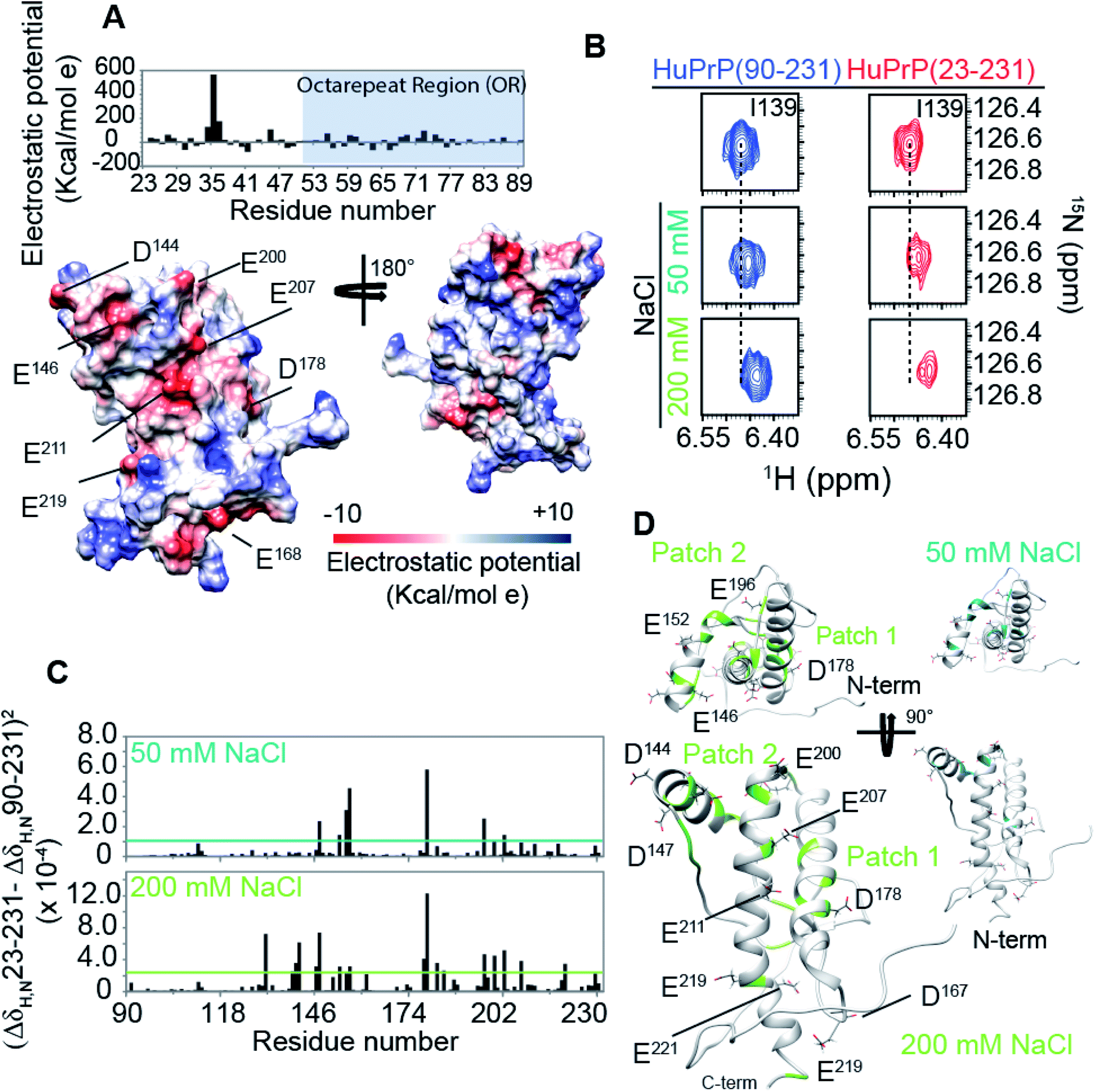

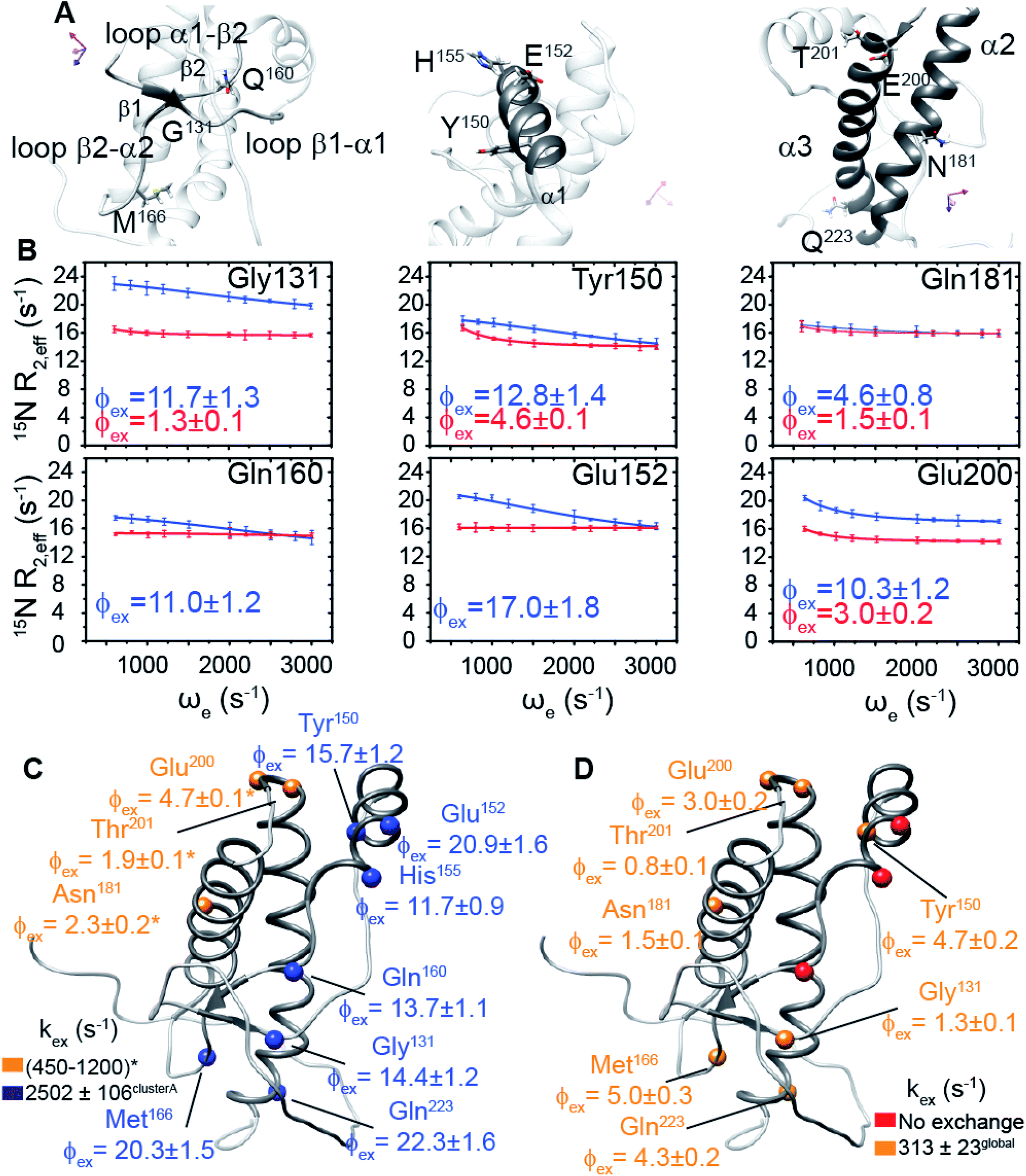
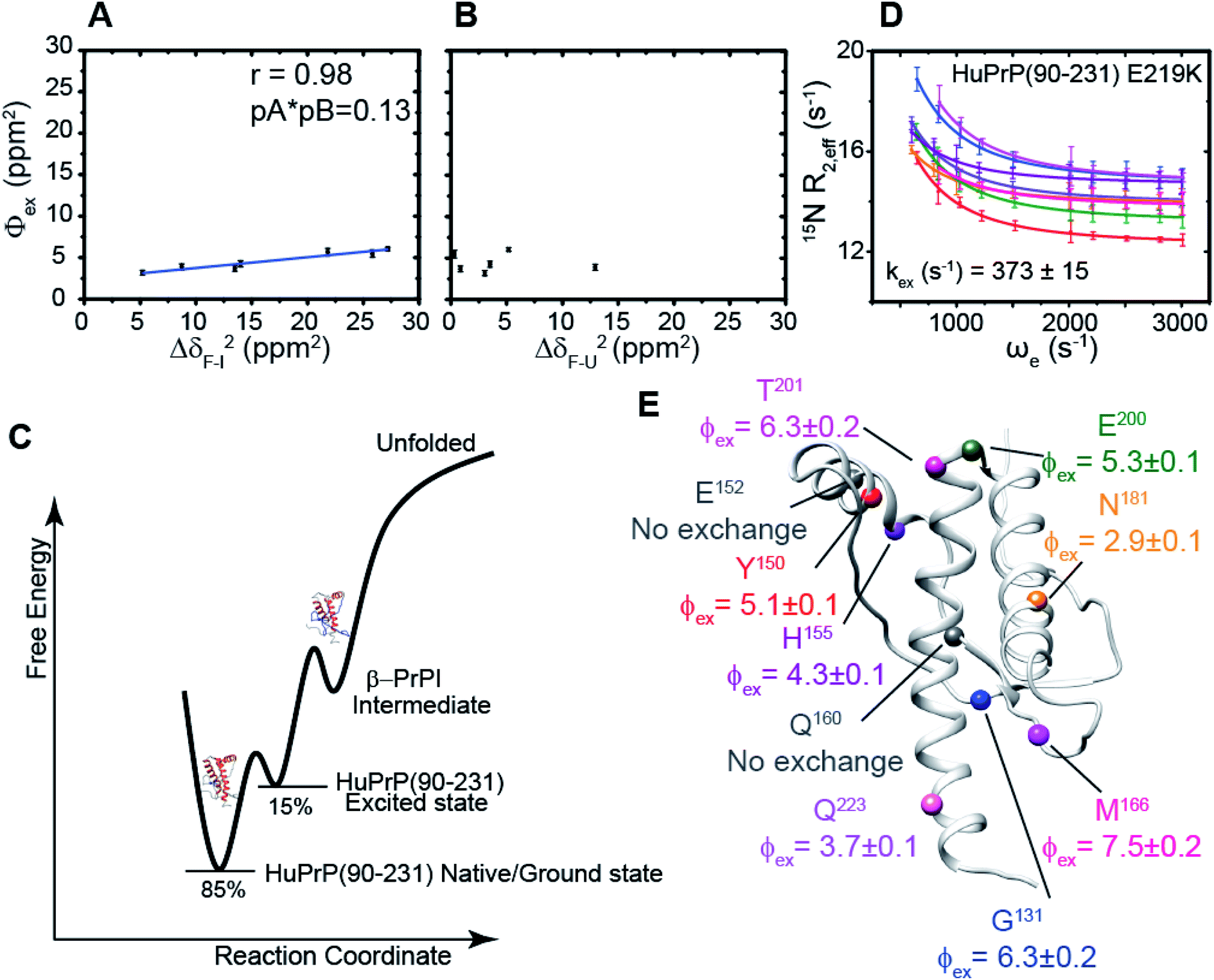
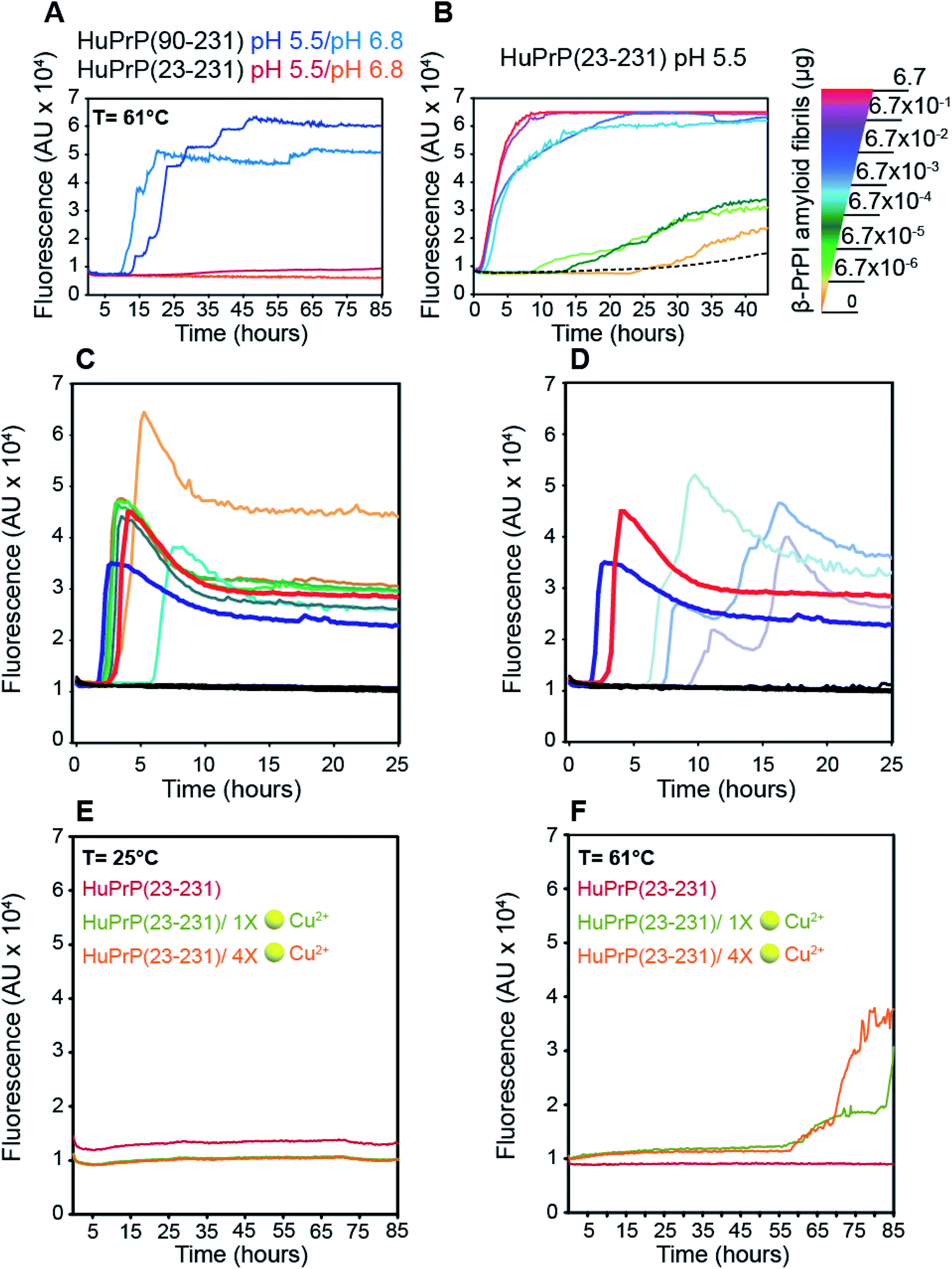
 ), sCJD-129MM2 (
), sCJD-129MM2 ( ), sCJD-129MV1 (
), sCJD-129MV1 ( ), sCJD-129MV2 (
), sCJD-129MV2 ( ), sCJD-129VV1 (
), sCJD-129VV1 ( ), sCJD-129VV2 (
), sCJD-129VV2 ( ), Alzheimer's disease and non-neurodegenerative disorder (
), Alzheimer's disease and non-neurodegenerative disorder ( ); amyloid fibrillary assemblies (ng mL−1) β-PrPI amyloid fibrils pH 6.8 (
); amyloid fibrillary assemblies (ng mL−1) β-PrPI amyloid fibrils pH 6.8 ( ) and 5.5 (
) and 5.5 ( ), monomer HuPrP(90–231) pH 6.8 and 5.5 (
), monomer HuPrP(90–231) pH 6.8 and 5.5 ( ); cerebrospinal fluid sCJD-129MM (
); cerebrospinal fluid sCJD-129MM ( ), sCJD-129MV (
), sCJD-129MV ( ), sCJD-129VV (
), sCJD-129VV ( ), Alzheimer's disease and hydrocephalus (
), Alzheimer's disease and hydrocephalus ( ). (E and F) Aggregation kinetics of HuPrP(23–231) at 25 °C (E) and 61 °C (F) upon addition of 1 (light green) and 4 (light orange) equivalent(s) of Cu2+ monitored by ThT fluorescence.
). (E and F) Aggregation kinetics of HuPrP(23–231) at 25 °C (E) and 61 °C (F) upon addition of 1 (light green) and 4 (light orange) equivalent(s) of Cu2+ monitored by ThT fluorescence.