
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA CRISPR/Cas12a-responsive dual-aptamer DNA network for specific capture and controllable release of circulating tumor cells†
Dong-Xia
Wang
 a,
Jing
Wang
*ab,
Ya-Xin
Wang
ac,
Jia-Yi
Ma
a,
Bo
Liu
a,
An-Na
Tang
a and
De-Ming
Kong
*a
a,
Jing
Wang
*ab,
Ya-Xin
Wang
ac,
Jia-Yi
Ma
a,
Bo
Liu
a,
An-Na
Tang
a and
De-Ming
Kong
*a
aState Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Biosensing and Molecular Recognition, Research Centre for Analytical Sciences, College of Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: wangjing822@tmu.edu.cn; kongdem@nankai.edu.cn
bSchool of Medical Laboratory, College of Medical Technology, Tianjin Medical University, Guangdong Road, Tianjin, 300203, P. R. China
cSchool of Pharmacy, Binzhou Medical University, Yantai, Shandong 264003, PR China
First published on 12th August 2022
Abstract
The separation and detection of circulating tumor cells (CTCs) have a significant impact on clinical diagnosis and treatment by providing a predictive diagnosis of primary tumors and tumor metastasis. But the responsive release and downstream analysis of live CTCs will provide more valuable information about molecular markers and functional properties. To this end, specific capture and controllable release methods, which can achieve the highly efficient enrichment of CTCs with strong viability, are urgently needed. DNA networks create a flexible, semi-wet three-dimensional (3D) microenvironment for cell culture, and have the potential to minimize the loss of cell viability and molecular integrity. More importantly, responsive DNA networks can be reasonably designed as smart sensors and devices to change shape, color, disassemble, and giving back to external stimuli. Here, a strategy for specifically collecting cells using a dual-aptamer DNA network is designed. The proposed strategy enables effective capture, 3D encapsulation, and responsive release of CTCs with strong viability, which can be used for downstream analysis of live cells. The programmability of CRISPR/Cas12a, a powerful toolbox for genome editing, is used to activate the responsive release of captured CTCs from the DNA network. After activation by a specified double-strand DNA (dsDNA) input, CRISPR/Cas12a cleaves the single-stranded DNA regions in the network, resulting in molecular to macroscopic changes in the network. Accompanied by the deconstruction of the DNA network into fragments, controllable cell release is achieved. The viability of released CTCs is well maintained and downstream cell analysis can be performed. This strategy uses the enzymatic properties of CRISPR/Cas12a to design a platform to improve the programmability and versatility of the DNA network, providing a powerful and effective method for capturing and releasing CTCs from complex physiological samples.
Introduction
Circulating tumor cells (CTCs) are important biomarkers for tumor recurrence and prognosis prediction.1,2 Although CTCs are extremely scarce in physiological samples, the rapid development of micro/nanomaterials and microfluidics has overcome the technical challenges of capturing CTCs, achieving efficient enrichment and sensitive detection.3–5 However, damage-free release and downstream analysis of live CTCs is still a challenging task in molecular characterization and functional analysis and nowadays has attracted widespread attention. Existing techniques such as electrokinetic release, enzymatic cleavage of DNA and biological signal stimulation (for example, ATP) enable efficient release of CTCs.6–9 However, the released cells are often affected by fluidic shear force, electrochemical repulsion, and temperature oscillation associated with these devices. Therefore, it is necessary to develop a controllable and defined bio-responsive stimulus to release live CTCs, while maximally maintaining cell viability and molecular integrity without damaging subsequent culture and live cell analysis.Hydrogel-like bulky DNA/RNA networks have emerged as promising substrates for three-dimensional (3D) cell capture and show the potential to minimize loss of cell viability and molecular integrity.6,10 Besides, the programmability, broad biocompatibility, and precise molecular recognition capabilities of DNA/RNA can confer DNA/RNA networks with fascinating bio-responsive behaviors.11,12 In recent years, clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein (CRISPR/Cas) nuclease systems have quickly become a powerful toolbox for genome engineering, in vivo and in vitro editing and nucleic acid detection.13–16 These systems utilize the special functions of various Cas proteins, including RNA-guided endonuclease activity, sequence-specific recognition, and multiple turnover trans-Cas12 and Cas13 cleavage activity.17–20 In the CRISPR/Cas12a system, correct base pairing activates Cas12a's RuvC nuclease and thus cleaves target double-stranded DNA (dsDNA), along with initiating deoxyribonuclease (DNase) activity to indiscriminately cleave single-stranded DNA (ssDNA).21–23 Taking advantage of this, the CRISPR-associated nuclease Cas12a has been extended into smart responsive materials. The programmability of CRISPR-associated nucleases has been reported to drive DNA networks in response to Cas/crRNA activation, altering the network properties through user-defined input materials including Cas/crRNA complexes and dsDNA triggers.24 However, CRISPR/Cas-based smart-responsive DNA networks still require the participation of polymers (e.g., polyethylene glycol and polyacrylamide), which inevitably affect biocompatibility and cause irreversible toxicity.25 Therefore, there is an urgent need to develop a CRISPR/Cas-responsive pure DNA network with high biocompatibility, low toxicity, and intelligent response to external stimuli.
Here, we report dual-aptamer DNA networks that enable efficient capture, 3D encapsulation and CRISPR/Cas-responsive release of CTCs for live-cell downstream analysis (Scheme 1). The DNA network is composed of winding and self-assembly of two ultra-long DNA strands generated by rolling circle amplification (RCA). Each of the two DNA strands contains alternately arranged aptamer units and “glue sequences” that are complementary to those in the other strand. The dual-aptamer strategy has a higher degree of cellular anchoring compared to the single-aptamer strategy. The cross-linking of two cell-anchored DNA strands forms a 3D DNA network, resulting in encapsulation and separation of target cells. Subsequent cell release is triggered by CRISPR/Cas12a-induced network disassembly. After the addition of the specified Cas12a/crRNA complex and dsDNA initiator, Cas12a cleaves the ssDNA regions in the DNA network, leading to a macroscopic change in the DNA network. As a result, the DNA network is deconstructed into fragments, enabling artificially controlled cellular release. Using the proposed strategy, selective isolation of different target cells from cell mixtures was achieved using different combinations of aptamers, providing highly alive CTCs for downstream analysis. This strategy uses the ssDNA cleavage properties of CRISPR/Cas12a to specifically release cells, improving the high degree of programmability of DNA networks and providing a powerful and effective method for capturing and releasing CTCs from complex physiological samples.
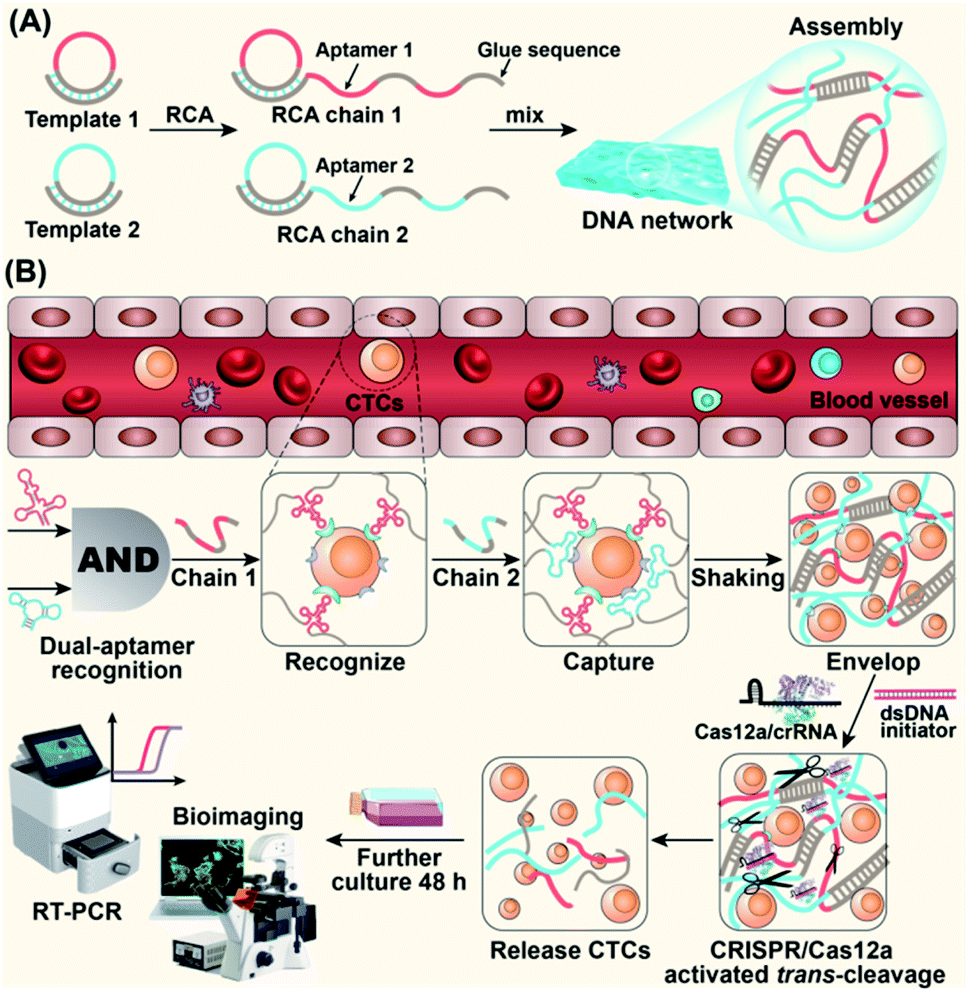 | ||
| Scheme 1 Schematic of a CRISPR/Cas12a-responsive DNA network for CTC capture and release. (A) Formation of a 3D DNA network via the cross-linking of ultra-long DNA strands prepared by RCA. (B) The process of cell capture and release is as follows: single-aptamer recognition, dual-aptamer synergetic recognition, DNA network capture, and CRISPR/Cas12a-responsive release of CTCs. | ||
Results and discussion
Synthesis and characterization of the DNA network
We tried to combine dual-aptamer synergetic recognition with DNA network encapsulation to accurately isolate cancer cells from cell mixtures and physiological samples with enhanced specificity and improved sensitivity.26,27 The DNA network was generated by RCA. The DNA sequences used to synthesize the network were carefully designed and are listed in Table S1.† After forming circular templates by T4 DNA ligase, long single-stranded RCA products (RCA chain 1 and RCA chain 2) were synthesized under the catalysis of phi29 DNA polymerase. By the modular design of the RCA template, every RCA chain contains one kind of aptamer that recognizes CTCs and glue sequences that can hybridize with the other RCA chains. The aptamer sequences targeting CTCs and the glue sequences forming cross-linked networks are spaced in the RCA products (Fig. 1A). When two RCA chains are mixed and gently shaken, glue sequences can hybridize together to form a soft 3D DNA network visible to the naked eye. The 3D DNA network contains two kinds of aptamers that work together to identify CTCs. This integrated dual-aptamer approach will provide greater advantages than the current single-aptamer approach due to its accuracy and sensitivity.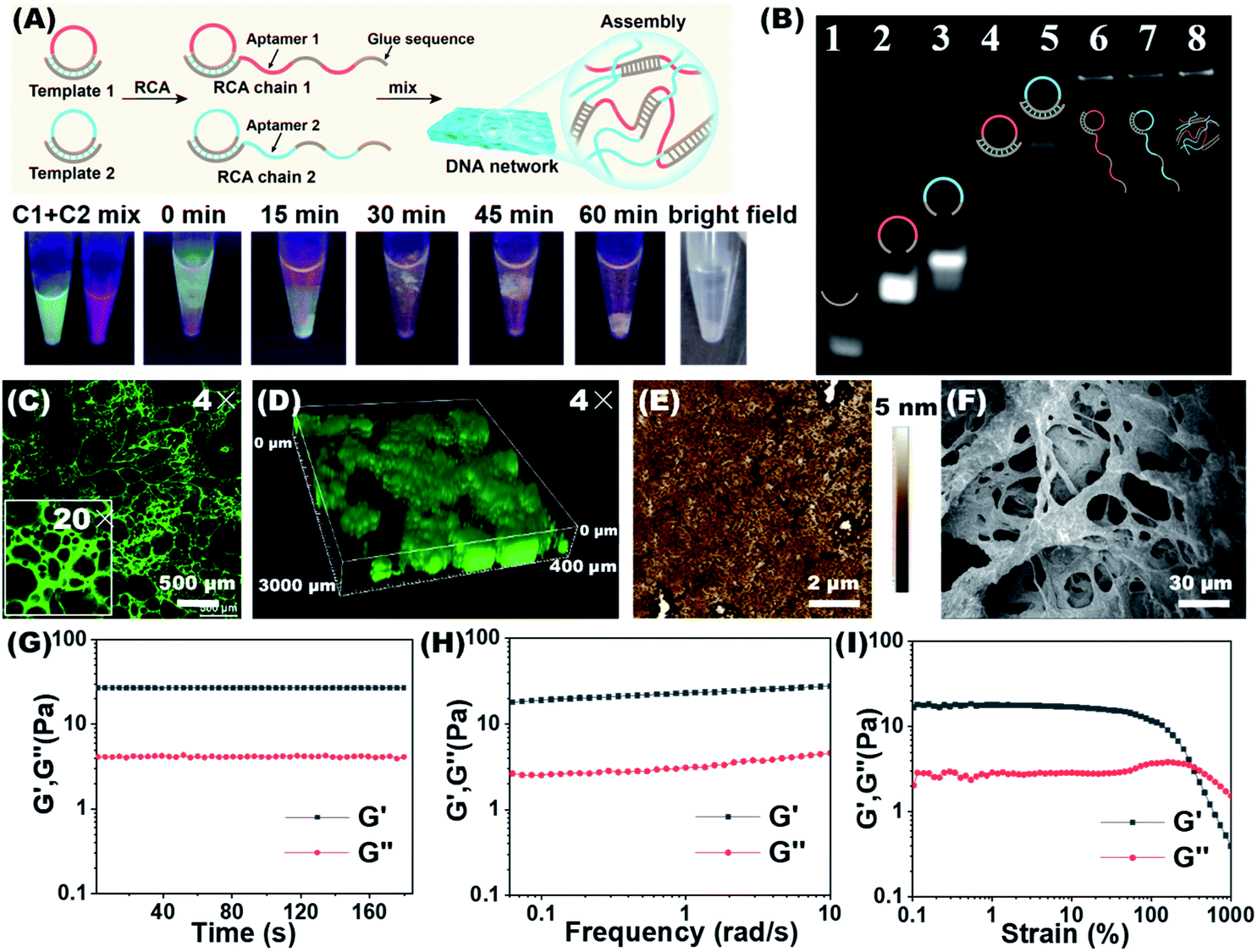 | ||
| Fig. 1 Synthesis and characterization of the DNA network. (A) The upper figure shows the synthesis route of a cross-linked DNA network by the RCA reaction. The figure below shows time-dependent changes of the mixture of two RCA chains, from which the formation of the DNA network can be visualized. RCA chain 1 and RCA chain 2 were stained with SYBR Green I and Gel Red, respectively. (B) Non-denaturing PAGE characterization of primer1, padlock1, padlock2, the primer1/circle1 hybrid, the primer2/circle2 hybrid, RCA chain 1, RCA chain 2, and the DNA network. (C) Confocal laser scanning microscopy (CLSM) images of the freeze-dried DNA network (stained with SYBR Green I). (D) CLSM 3D image of the DNA network in an aqueous solution. (E) AFM image of the freeze-dried DNA network. (F) SEM image of the freeze-dried DNA network. (G) Dynamic time sweep rheological test of the DNA network with a fixed strain of 1%. Storage modulus (G′) and loss modulus (G′′) of the DNA network from frequency sweep (H) and strain sweep (I) tests. | ||
The properties of the DNA network were first studied in the absence of cells. The synthesis of RCA chains can be verified by non-denaturing polyacrylamide gel electrophoresis (PAGE). As shown in Fig. 1B, after the hybrid formation by different circular templates and primers, new bands with lower mobility were observed (Lanes 4, 5). The RCA products could not move from the loading sites, demonstrating the formation of extra-long products (Lanes 6, 7). To demonstrate that the DNA network was formed by the base complementary pairing of two RCA chains, we used DNA dyes to visualize this process. The two RCA chains were stained with the nucleic acid dyes SYBR Green I (green) and GelRed (red), respectively. To promote molecular diffusion and increase collision probability, we used a shaking dry bath to perform the reaction at 37 °C and 600 rpm. As shown in Fig. 1A, when RCA chain 1 and RCA chain 2 were first mixed, the interface between the green and red layers could be observed. With time, the interface between the two layers gradually melted into one color, and yellow filaments appeared when the DNA network aggregates were formed. The bright field image of the reaction system demonstrates the same result. In contrast, when the glue sequences in the two RCA chains were replaced by non-complementary random sequences, the DNA network could not be formed (Fig. S1†).
The DNA network was stained with SYBR Green I and lyophilized for confocal laser scanning microscopy (CLSM) observation. As shown in Fig. 1C and S2,† the porous network structure was shown using both 4× and 20× lenses. When the DNA network was placed in an aqueous solution, its 3D images also showed flocculent sparse porous structures (Fig. 1D, S2 and Video S1†). The porous fibrous structures were further verified by atomic force microscopy (AFM) and scanning electron microscopy (SEM) (Fig. 1E and F). In the SEM images, as the magnification of the freeze-dried DNA network gradually increased, the porous and lattice packing structure of the DNA network could be clearly seen (Fig. S3†). We further investigated the rheological properties of the DNA network. As shown in Fig. 1G and 1H, the storage modulus (G′) was significantly higher than the loss modulus (G′′) in the time sweep and frequency sweep tests, demonstrating the formation of a soft and bulky DNA network. In addition, strain scan analysis revealed a relatively low critical strain point for the formation of a DNA network (Fig. 1I), implying that the prepared DNA network would have low mechanical damage to cells. The above results prove that we have successfully synthesized the RCA chains and constructed the DNA network. This porous and loose structure may facilitate cell capture and viability preservation, provide good nutrient transport for the enveloped cells, and remove waste products from the cells.
Aptamer-specific anchoring of CTCs
Multiple aptamer units are embedded at intervals in our synthesized RCA chains, and the synergy of multivalent aptamers would enhance the accuracy and efficiency of cell capture. To validate aptamer-specific anchoring of cells, CCRF-CEM (abbreviated as CEM), a T lymphoblast-like cell line, was used as a model cell type. CEM cells can be recognized by the aptamers sgc8c and sgc4f. The sgc8c aptamer targets the surface receptor tyrosine protein kinase (PTK7)28 and the sgc4f aptamer targets another unknown membrane receptor.29 Ramos cells, human Burkitt lymphoma cells with low PTK7 expression, were used as the control.30 To establish a fluorescent signal for indicating membrane anchoring, the 6-carboxy-fluorescein (FAM) fluorophore was modified to the 5′-end of the aptamer sgc8c (FAM-sgc8c) (Fig. 2A).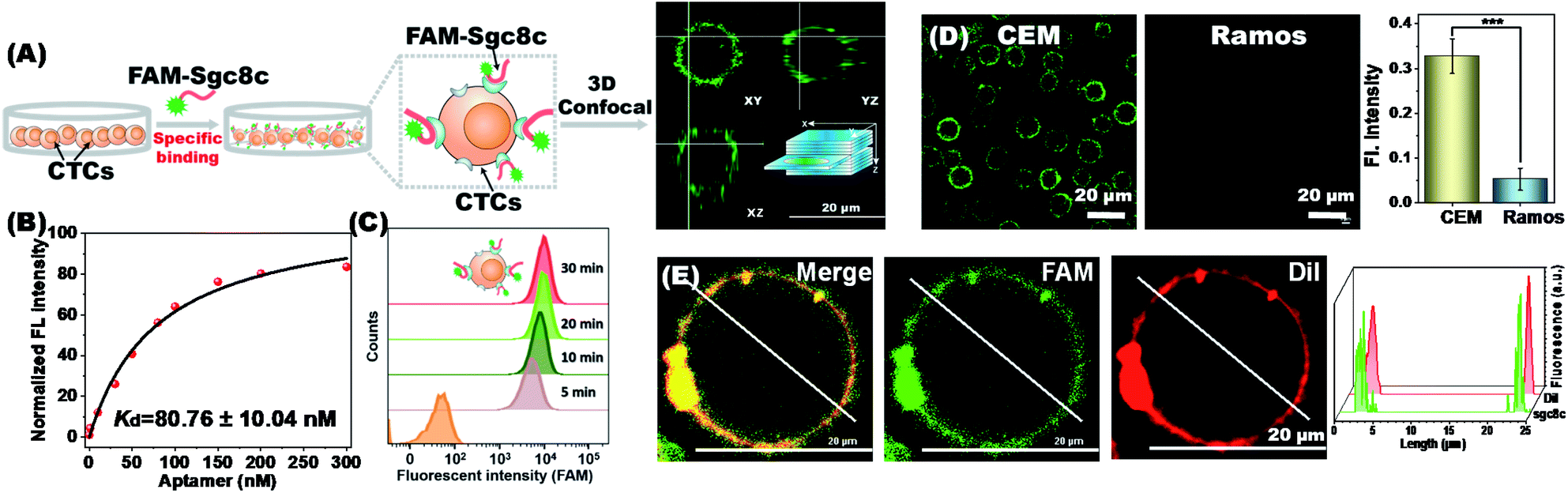 | ||
| Fig. 2 Aptamer-specific anchoring of CTCs. (A) Targeted recognition and binding of FAM-sgc8c (green) toward CEM cells. (B) Binding affinity of the sgc8c aptamer to CEM cells in a working environment for cell capture. (C) Flow cytometry analysis showed that FAM-sgc8c can be stably immobilized on the CEM cell surface. (D) CLSM and fluorescence analysis images of FAM-sgc8c binding on CEM cells. Ramos cells were used as the control. (Right) Statistical analysis was performed using an unpaired two-tailed t test; ***, P <0.001. The bars represent mean ± SD (n = 3). (E) Colocalization of FAM-sgc8c (green) and cell membrane dye CM-Dil (red). (Right) A representative line was drawn through the center of the cell to show the fluorescence distribution. | ||
Under the working conditions for cell capture, the sgc8c aptamer maintained good target cell-binding ability with a dissociation constant (Kd) value of 80.76 ± 10.04 nM (Fig. 2B). Flow cytometry analysis showed that the recognition of CEM cells by the sgc8c aptamer would be completed within 10 minutes (Fig. 2C). Correspondingly, the membrane surface of CEM cells showed green fluorescence, demonstrating the strong binding ability of sgc8c to CEM cells. Confocal 3D stacks of FAM-sgc8c-anchored CEM cells were scanned every 3 μm for a total of 18 μm. The results showed that the aptamer was accurately anchored on the cell membrane surface instead of entering the cell (Fig. S4 and Video S2†). As a control, non-target Ramos cells almost showed no fluorescence after incubation with FAM-sgc8c. The mean optical density values of CEM and Ramos cells were significantly different (Fig. 2D), demonstrating that sgc8c can be specifically anchored to CEM cells. This process can be further demonstrated using the membrane localization dye CM-DiI (red) (Fig. 2E and S5†). Cells were first conjugated with the FAM-sgc8c aptamer to give green fluorescence, and then the cell membrane was stained with CM-DiI to show red fluorescence. The overlapping images showed the co-localization of red and green fluorescent signals, confirming that sgc8c was anchored to the membrane of CEM cells. Similar anchoring results were also seen for aptamer sgc4f, and the mean optical density values of CEM and Ramos cells were also significantly different (Fig. S6†). The above results demonstrate that the two aptamers, sgc8c and sgc4f, can accurately anchor target cells and be used in the dual-aptamer cell capture strategy.
Capture of CTCs by the DNA network
After verifying that the aptamers can be specifically anchored to the cell membrane of CEM cells, we then used the DNA network to capture CTCs. CLSM and SEM analysis were used to evaluate the 3D spatial distribution of the captured cells. CEM cells were used as the model cells and pre-stained with the red fluorescent dye CM-DiI. We named the two RCA chains (containing sgc8c and sgc4f aptamers) used to capture CEM cells as C1 and C2, respectively. Three samples were prepared: pure CEM cells (Fig. 3A, D, S7A and S7D†), CEM + C1 (Fig. 3B, E, S7B and S7E†), and CEM + C1 + C2 (Fig. 3C, F, S7C and S7F†). As shown in Fig. 3A, when only CEM cells were present in the sample, the cells could settle to a flat surface on the bottom of the confocal dish after a few minutes (red box), proving that the cells were not affected by any external material. When C1 was added to the system, sgc8c recognized and anchored the suspended cells, but the DNA network could not be formed. Therefore, the cells were still in a free state, and CEM + C1 would also settle on a plane (Fig. 3B). When C2 was also added to the system, CEM cells could be specifically recognized by both sgc8c and sgc4f. Moreover, the two RCA chains undergo base complementary pairing via molecular diffusion, and the DNA network was gradually formed over approximately 1 h, during which CEM cells were anchored and encapsulated in it. As shown in Fig. 3C, cells were evenly distributed in each layer of the stack, showing a multi-layered spatial topology. Unlike before, the cells in the network could remain suspended at the bottom of the confocal dish without settling due to the 3D support and spatial distribution provided by the DNA network (Video S3†). | ||
| Fig. 3 CLSM and SEM characterization of CTC capture by the DNA network. (A–C) 3D stacking CLSM images of CEM cells only (A), CEM + C1 (B) and CEM + C1 + C2 (C). A total of 15 layers were scanned every 10 μm to obtain a 3D stack. (D–F) SEM images of CEM cells only (D), CEM + C1 (E) and CEM + C1+C2 (F). (G and H) 3D stacking CLSM image (region of interest: 600 × 600 × 160 μm3) of CTCs (stained with CM-DiI, red) enveloped in the DNA network (stained with SYBR Green I, green). (H) The 3D image of CTCs in the DNA network. (I) 3D stacking image showing the uniform spatial distribution of CTCs in the DNA network. | ||
SEM images also verified this process. The presence of only CEM cells showed good monodisperse properties (Fig. 3D). When C1 was added, the DNA chain began to recognize and anchor the cells. Because consecutively spaced aptamer units in C1 could recognize and anchor to different CEM cells, the cells showed a state of being close to each other and interconnected (Fig. 3E). When C2 was also added to the system, the CEM cells were synergistically recognized by the two aptamers and wrapped by the DNA network, showing a densely packed and tightly wrapped state (Fig. 3F). We further observed the spatial distribution of the DNA network and captured CEM cells under a confocal microscope. The DNA strands were stained with the green fluorescent dye SYBR Green I. The DNA network showed a fibrillar structure that entangled the cells within the network (Fig. 3G–I; S7; S8 and Video S4†). Almost all CEM cells were tightly cross-linked to the DNA network, rather than adhering to the surface.
Dual aptamers, regarded as the two inputs of an AND logic gate, can anchor CTCs more efficiently (Fig. 4A). The capture rates and captured numbers of CTCs using the dual-aptamer DNA network were further evaluated. As shown in Fig. 4B, the CEM cell capture efficiencies of a single aptamer (sgc8c or sgc4f), in which the aptamer sequence in C1 or C2 was replaced by a non-aptamer random sequence, were about 53.7% and 48.3%, while the dual-aptamer strategy showed an overwhelming cell capture efficiency of around 75.5%. To exclude the possibility that the different cell capture efficiencies are caused by the number difference of aptamer units, we incorporated the same aptamer (e.g., sgc8c) into both C1 and C2 to prepare a single-aptamer DNA network with a comparable total number of aptamer units to the dual-aptamer DNA network. As shown in Fig. 4C, the capture efficiency of this single-aptamer DNA network was only marginally higher than that of the single-aptamer DNA network formed by C1 with the sgc8c aptamer and C2 with a random sequence (named R2, Fig. 4C). Statistical analysis revealed no statistical difference. This suggests that C1 can provide enough aptamers to fully occupy the target proteins on CEM cells. Increasing the aptamer unit number will not further strengthen the recognition reaction, since no more sites can be recognized. However, when two aptamers, which recognize different sites on the target cell, are used to perform dual-aptamer recognition, the recognition efficiency can be further promoted. When no aptamer sequence was present in the two RCA chains, the DNA network was unable to accurately anchor the target cells, resulting in a very low cell capture efficiency of around 20.4%. It was observed through a microscope that the cells captured by the non-aptamer chains were still in a monodisperse state, while the cells captured by aptamers showed a clustered state (Fig. S9†). These results prove that the strategy based on the dual aptamers to capture cells has a higher cell capture rate.
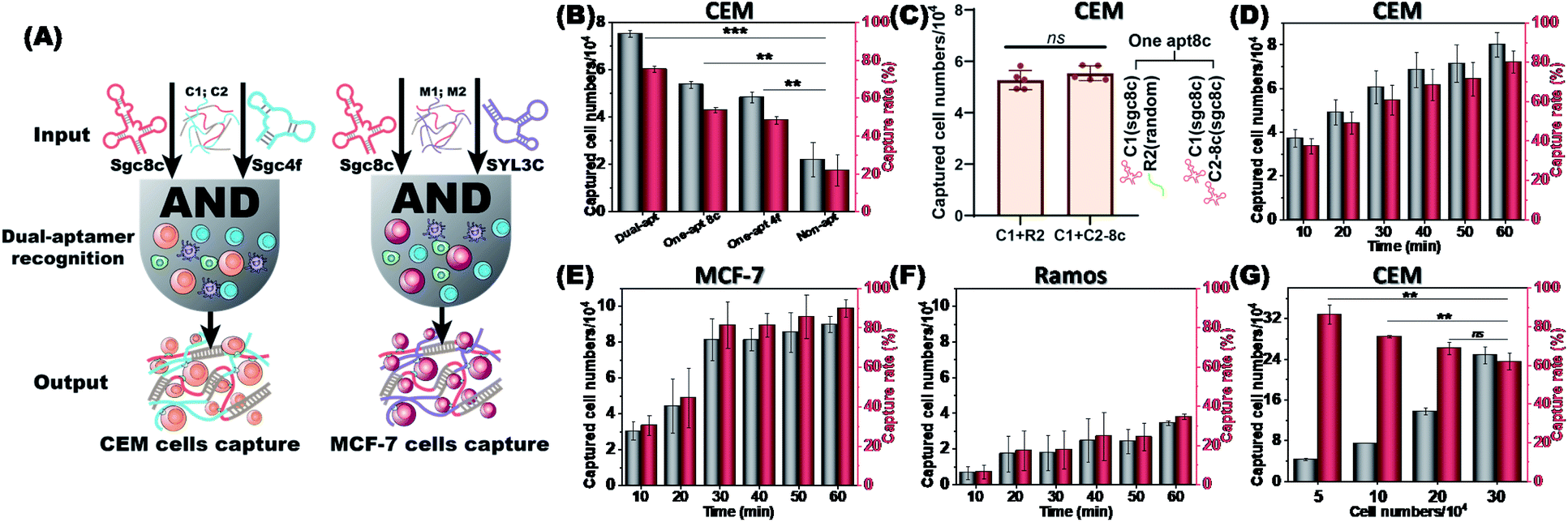 | ||
| Fig. 4 Target cell capture efficiency of the dual-aptamer DNA network. (A) The AND logic gate mechanism of the dual-aptamer DNA network for capturing CTCs. (B) Capture rates of CEM cells under different conditions: dual aptamers (sgc8c and sgc4f), single aptamer (sgc8c or sgc4f) and non-aptamer sequence. (C) Capture of CEM cells by two single-aptamer DNA networks: C1 (sgc8c) + R2 (random); C1 (sgc8c) + C2-8c (sgc8c). (D–F) Time dependent-capture rate and captured cell numbers of CEM, MCF-7, and Ramos cells by the DNA network. In (F), the representative results of Ramos cell capture by C1/C2 are given. (G) Cell number-dependent capture rate and captured cell numbers of CEM cells. Incubation time was 60 min. The bars represent mean ± SD (n = 3); statistical analysis was performed using an unpaired two-tailed t-test; ***, P <0.001. | ||
To demonstrate the universality of the dual-aptamer strategy, another type of tumor cells, human breast adenocarcinoma MCF-7 cells, were tested. MCF-7 cells overexpress both PTK7 and EpCAM, an epithelial cell adhesion molecule that has been widely used as a CTC biomarker.31 To specifically capture MCF-7 cells, the sgc8c aptamer (targeting PTK7) and SYL3C aptamer (targeting EpCAM) were introduced into the two RCA chains to give M1 and M2, respectively. Similar to the sgc8c aptamer, the SYL3C aptamer also maintained good target-binding ability in the working environment for cell capture (Fig. S10†). M1 and M2 could efficiently capture MCF-7 cells with high specificity via the formation of a dual aptamer-containing DNA network. By recording the capture efficiency of 1 × 105 CEM, MCF-7 and Ramos cells as a function of incubation time, it could be found that CEM and MCF-7 cells could be rapidly captured by C1/C2 and M1/M2, respectively. The capture efficiency and the number of captured cells increased and nearly reached a plateau after 40 and 30 minutes, respectively. Capture rates of approximately 75% of CEM cells (Fig. 4D) and approximately 85% of MCF-7 cells (Fig. 4E) were achieved, reflecting the rapid capture kinetics and high capture capability of the dual aptamer DNA network strategy. This high-efficiency capture results from the multivalent binding between the target cell and the multiple repeat aptamer sequences. Neither C1/C2 nor M1/M2 was able to efficiently capture the negative control Ramos cells, only showing a low capture efficiency of <30% after 40 min (Fig. 4F). We further investigated the relationship between the cell number and capture rate. As the number of cells increased, the number of captured cells gradually increased, but the capture rate slowly decreased (Fig. 4G). This is because there is a maximum capacity limit for fishing CTCs by a fixed-quality DNA network.
CRISPR/Cas12a-responsive disassembly of the DNA network and release of captured cells
After verifying the successful capture of CTCs using the dual-aptamer DNA network, we then investigated the release of captured CTCs from the DNA network. To make the released CTCs keep high viability for downstream cell analysis, a CRISPR/Cas12a-responsive release strategy was designed. In such a strategy, cell release was triggered and controlled by a dsDNA in a sequence-specific manner (Fig. 5A). Via sequence-specific base pairing between one strand of the dsDNA and the crRNA of effector nuclease Cas12a, dsDNA could bind with the complex formed by Cas12a and crRNA, thus activating the single-stranded deoxyribonuclease (ssDNase) activity of Cas12a, which would indiscriminately cleave ssDNA regions in the DNA network,32–34 resulting in the collapse of the DNA network and thus the release of captured cells. Since the ssDNA cleavage by activated Cas12a is sequence-independent, the designed Cas12a system can work for any DNA network, which contains different aptamer combinations for the capture of different cancer cells, without the need to redesign crRNA and dsDNA. In addition, Cas12-catalyzed cleavage of the DNA network can perform multiple turnovers (∼17 turnover events per second),17 suggesting that low concentrations of the Cas12a system are required to achieve the release of captured cells. | ||
| Fig. 5 CRISPR/Cas12a-responsive release of CTCs for downstream analysis of cells. (A) Working mechanism of CRISPR/Cas12a-controlled disassembly of the DNA network: after activation by the dsDNA initiator, Cas12a cleaves the ssDNA regions in the DNA network. (B) PAGE images of the DNA network after treatment with activated CRISPR/Cas12a for different times. (C) Visual observation of DNA network disassembly by activated CRISPR/Cas12a or DNase I. The DNA network was stained with SYBR Green I. Sample 1: 1 μM Cas12a + 1 μM crRNA + 1 μM dsDNA; sample 2: 0.5 μM Cas12a + 0.5 μM crRNA + 0.5 μM dsDNA; sample 3: 40 U mL−1 DNase I. (D) Visual observation of DNA network-driven cell capture and CRISPR/Cas12a-responsive cell release. (E) Released CEM cell numbers and release rate under different conditions. The bars represent mean ± SD (n = 3). (F) Incubation time-dependent cell release rate of CEM cells from the DNA network. (G) Schematic diagram of using released cells for downstream analysis: fluorescence analysis and genetic analysis. (H) Fluorescence microscopy image of released CEM cells after culturing for 48 h. Alive and dead cells were stained with Calcein-AM (green) and PI (red), respectively. On the right is the average optical density of the fluorescence images. The bars represent mean ± SD (n = 3); statistical analysis was performed using an unpaired two-tailed t-test; ***, P <0.001. (I) CLSM images of FAM-sgc8c binding on released CEM cells. Cell membranes were stained with CM-Dil (red). (J) The comparison of CK19 mRNA and EGFR mRNA expression levels between original MCF-7 and released MCF-7 cells. R: relative fluorescence. The released MCF-7 cells were incubated for 48 hours before RT-PCR analysis. | ||
PAGE was used to verify CRISPR/Cas12a-responsive disassembly of the DNA network. As shown in Fig. 5B, the DNA network was unable to move down due to its large size and gave a bright band at the sample-loading site. When the Cas12a/crRNA complex was added and its ssDNA cleavage activity was activated by the dsDNA initiator, the DNA network was cleaved into small-sized fragments, which could migrate from the sample-loading site, resulting in the weakness of the DNA network band. In contrast, the DNA network band barely changed when crRNA, Cas12a, or the dsDNA initiator was missed (Fig. S11†). Herein, Cas12a/crRNA and target dsDNA need to match lock–key pairs, and non-initiator dsDNA, whose sequence is not complementary to crRNA, could not trigger the disassembly of the DNA network. CRISPR/Cas12a-responsive disassembly of the DNA network could be clearly observed by the naked eye using DNA-embedding dyes. As shown in Fig. 5C, when the DNA network was exposed to a CRISPR/Cas12a system activated by a 1 μM dsDNA initiator, the network's fluorescence dissipated in about 30 minutes. When the concentration of the CRISPR/Cas12a system was decreased to 0.5 μM, DNA network breakdown was observed within two hours, indicating that the disassembly rate could be easily controlled by adjusting the CRISPR/Cas12a system concentration. Using deoxyribonuclease I (DNase I), DNA network disassembly and cell release could also be achieved.5 Compared to DNase I-based cell release, our CRISPR/Cas12a-based strategy can be easily controlled by the dsDNA initiator and thus might provide a good choice for the design of systems, in which controllable release of cells is needed.
Via CRISPR/Cas12a-responsive disassembly of the DNA network, controllable cell release could be achieved. In fact, the DNA network formation-driven cell capture and CRISPR/Cas12a-responsive cell release could also be directly observed by the naked eye. As shown in Fig. 5D and S12,† with the addition of two RCA chains in the cell suspension, the capture of target cellswas triggered immediately. Correspondingly, the cell culture medium became light in color. With the extension of incubation time, a white flocculent precipitate, visible to the naked eye, was deposited at the bottom of the tube due to the DNA network formation. In this way, specific capture of target cells was achieved by discarding the cell culture medium. To achieve the cell release, the collected DNA network was resuspended in the fresh cell culture medium, and the Cas12a/crRNA complex was added together with the dsDNA initiator. Due to the disassembly of the DNA network by activated CRISPR/Cas12a, captured cells were gradually released and re-dispersed in the cell culture medium. Corresponding microscopy photographs also confirmed this process (Fig. S13†). Subsequently, the cell release rate was evaluated. Activated CRISPR/Cas12a could result in the highly efficient release of captured cells (Fig. 5E). By recording the cell number released in the cell culture medium, it could be found that the cell release rates of CEM and MCF-7 cells monotonously increased with time, eventually reaching approximately 80% after 60 min (Fig. 5F and S14†). In contrast, it was difficult for cells to be released from the network when either crRNA, Cas12a or dsDNA was missing, or non-initiator dsDNA was added instead of the dsDNA initiator.
High viability is of great importance for the downstream analysis of captured cells. To test the survival rate of the released cells, the cells were further cultured and their proliferation capability was evaluated. Excitingly, the released cells exhibited comparable viability to pristine cells without any treatment (Fig. S15†), indicating that there is no damage to the cells during cell capture and release. In addition, the released cells could proliferate well. After 48 h of incubation, the released MCF-7 cells spread out and attached to the surface of the culture dish and appeared spindle-shaped (Fig. S16†). In addition, only the released cells could attach to the culture dish surface after proliferation and maintain their integrity and homogeneity, while the unreleased cells failed to grow even after 48 h incubation and could not attach to the culture dish surface at all. After 48 h of incubation, we stained CEM cells using Calcein-AM (green) and propidium iodide (PI, red), respectively. As shown in Fig. 5H, the images show that the majority of CEM cells were living cells (green). The percentages of dead (red) and alive (green) cells were 8.2% and 91.8%, respectively, indicating that CEM cells released from the DNA network still retained excellent viability. Similar results were given by MCF-7 cells (Fig. S17†).
DNA network formation-driven cell capture and subsequent CRISPR/Cas12a-controlled cell release were gentle, with little damage to cells. Therefore, we further evaluated the feasibility of the released cells for downstream analysis, including fluorescence analysis and genetic analysis. After incubating the released CEM cells for another 48 hours, we used the FAM-sgc8c aptamer to identify CEM cells again (Fig. 5I). The sgc8c aptamer could stably anchor to the cell membrane again, whose fluorescence co-localized well with that of the membrane dye DiI. This proves that characteristic proteins can also be expressed by released CEM cells and the cell surface properties are well maintained, which is helpful for downstream cell analysis.
Due to the characteristics of high frequent metastasis and strong potential invasion, CTCs generally highly express the two mRNAs of cytokeratin 19 (CK19) and the epidermal growth factor receptor (EGFR).6 To evaluate whether the released cancer cells could maintain these typical characteristics of CTCs, the real-time polymerase chain reaction (RT-PCR) was used to detect the CK19 and EGFR mRNA expression of MCF-7 cells. As shown in Fig. 5J, the differences in EGFR and CK19 mRNA expression before and after DNA network-based capture and release appeared to be negligible, suggesting that these treatments did not affect the physiological properties of the cells. The above results indicate that our proposed cell capture and release strategy is highly biocompatible. The obtained CTCs can perfectly keep their original surface and interior features and can be used for subsequent applications and downstream analysis.
The above experiments demonstrate that our CRISPR/Cas12a-responsive strategy can achieve rapid and highly efficient release of captured cells, giving high fidelity CTCs. It has been reported that a DNA-supported network could also be destructed by DNAzyme,8 deoxyribonuclease (e.g., DNase I),35 or an aptamer substrate (e.g., ATP).6,7 Our CRISPR/Cas12a-responsive release strategy can serve as a perfect supplement to these methods. Compared to that used in the DNAzyme-based method, the DNA network used in our method is more cost-effective and easier to prepare. Ribonucleotides (e.g., rA) need to be included in the DNA network to work as the substrates of DNAzymes, which will certainly increase the cost and difficulty of DNA-supported network preparation in a DNAzyme-based strategy. Compared to the ATP aptamer-based method, our method is more flexible in the design of the DNA network. The DNA network used in the ATP aptamer-based method contains lots of ATP aptamer units. To achieve the specific capture of different kinds of CTCs, different aptamers, which specifically recognize different CTCs, might be used. If the CTC aptamers interact with the ATP aptamer to fold into undesirable secondary structures, the CTC-capturing efficiency will certainly decrease. Our method uses CRISPR/Cas12a-catalyzed cleavage of ssDNA to achieve the release of captured CTCs. Since the ssDNA cleavage is sequence-unspecific, its sequence can be arbitrarily adjusted according to the sequences of CTC aptamers. Thus, the formation of undesirable secondary structures can be efficiently avoided, which makes our method easy to use for different CTCs. Compared to the deoxyribonuclease or ATP aptamer-based method, our method may obtain reduced interference from biological endogenous substances. Since deoxyribonuclease and ATP are biological endogenous molecules and ubiquitous in biological systems, it is very possible that the difference in deoxyribonuclease or the ATP concentration in different biological systems and different patients might give fluctuant CTC-capturing efficiencies. In contrast, CRISPR/Cas12a is exogenous, and the effects caused by biological endogenous substances can be efficiently overcome.
Evaluation of target cell enrichment from a cell mixture and complex samples
Then, the ability of our method to identify and isolate CTCs from a cell mixture was investigated. To this end, we stained CEM cells and MCF-7 cells with CellTracker Green CMFDA (green) and Ramos cells with CytoTraceTM Red fluorescent probe (red). Equal amounts of CEM and Ramos cells were mixed (Fig. S18†) and subjected to C1 capture, C2 envelope and CRISPR/Cas12a-responsive release. The amount of CEM and Ramos in the cell mixture was calculated separately using fluorescence microscopy. After cell capture and release, CEM cells resulted in an approximate 6-fold enrichment (85%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15%), indicating the high capture selectivity of the dual-aptamer strategy towards the target cells. Because the content of CTCs in all cells is extremely low in real situations, we then mixed CEM and Ramos cells at a ratio of 1:10. As shown in Fig. 6A, even after the target cell percentage was greatly reduced, CEM cells could be enriched by about 27 times (73%:27%). Our method could also work when the CEM to Ramos ratio was decreased to 1:1000. As shown in Fig. 6B, after capture and release treatments, the proportion of CEM cells was increased from 0.1% to 32%. To further validate the applicability of our method, specific capture of MCF-7 cells from the cell mixture was conducted by M1/M2. MCF-7 cells (green) were mixed with Ramos cells (red) in equal amounts (Fig. S19†). After the capture and release, an approximate 6-fold enrichment (86%:14%) of MCF-7 cells was obtained. When MCF-7 and Ramos cells were mixed in a ratio of 1:10, a 30-fold enrichment effect (75%:25%) could be obtained (Fig. 6C). These results suggest that our proposed dual-aptamer cell capture and CRISPR/Cas12a-responsive cell release method can be used for highly efficient and selective enrichment of target CTCs from cell mixtures.
:15%), indicating the high capture selectivity of the dual-aptamer strategy towards the target cells. Because the content of CTCs in all cells is extremely low in real situations, we then mixed CEM and Ramos cells at a ratio of 1:10. As shown in Fig. 6A, even after the target cell percentage was greatly reduced, CEM cells could be enriched by about 27 times (73%:27%). Our method could also work when the CEM to Ramos ratio was decreased to 1:1000. As shown in Fig. 6B, after capture and release treatments, the proportion of CEM cells was increased from 0.1% to 32%. To further validate the applicability of our method, specific capture of MCF-7 cells from the cell mixture was conducted by M1/M2. MCF-7 cells (green) were mixed with Ramos cells (red) in equal amounts (Fig. S19†). After the capture and release, an approximate 6-fold enrichment (86%:14%) of MCF-7 cells was obtained. When MCF-7 and Ramos cells were mixed in a ratio of 1:10, a 30-fold enrichment effect (75%:25%) could be obtained (Fig. 6C). These results suggest that our proposed dual-aptamer cell capture and CRISPR/Cas12a-responsive cell release method can be used for highly efficient and selective enrichment of target CTCs from cell mixtures.
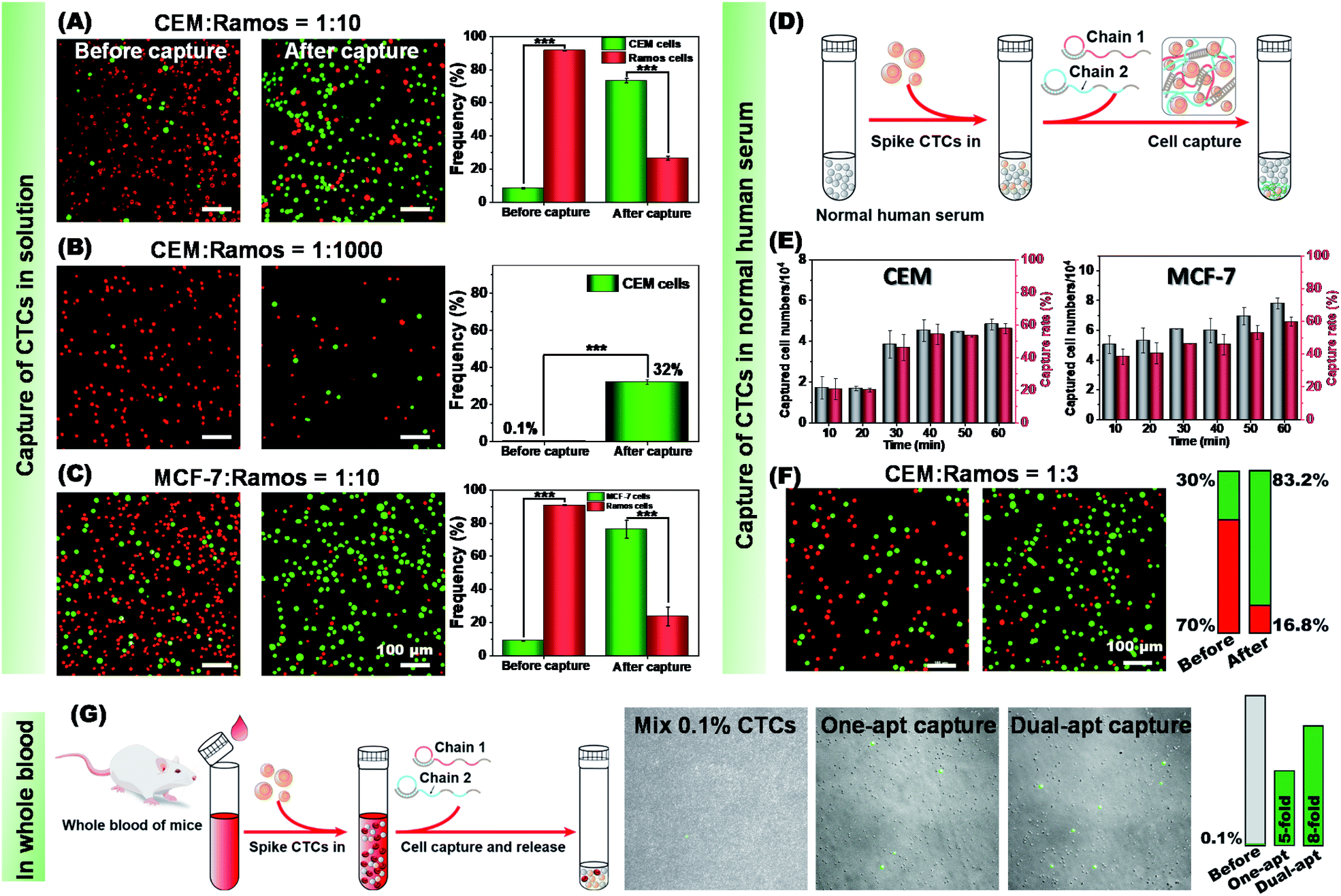 | ||
| Fig. 6 Specific enrichment of target cancer cells from cell mixtures, human serum and mice whole blood samples. (A) Target CEM cells (green) and non-target Ramos cells (red) were mixed at 1:10. After the specific capture and controllable release, target CEM cells were significantly enriched. Statistical analysis on the right showed approximate 27-fold enrichment of target CEM cells. (B) Target CEM cells (green) and non-target Ramos cells (red) were mixed at 1:1000. The proportion of CTCs was increased from 0.1% to 32% after enrichment. (C) Target MCF-7 cells (green) and non-target Ramos cells (red) were mixed at 1:10. After the specific capture and controllable release, targeted MCF-7 cells were significantly enriched. Statistical analysis on the right showed approximate 30-fold enrichment of target MCF-7 cells. Statistical analysis was performed using an unpaired two-tailed t test; ***, P <0.001. (D) Schematic of targeted capture of CTCs from human serum samples. (E) Time-dependent capture rate and captured cell numbers of CEM cells and MCF-7 cells from human serum samples. The bars represent mean ± SD (n = 3). (F) Targeted capture and enrichment of CEM cells from human serum samples containing CEM cells (green) and control Ramos cells (red) with a ratio of 1:3. After the specific capture and controllable release, target CEM cells were significantly enriched. Statistical analysis on the right showed approximate 9-fold enrichment of target CEM cells. (G) Schematic of targeted capture of CTCs from mice whole blood samples. Target CEM cells (green) were mixed into whole blood with a ratio to blood cells of 1:1000. Single- and dual-aptamer DNA networks gave 5 and 8 time enrichment of CEM cells, respectively. | ||
To verify whether our method could work in a more complex environment, a standard addition experiment was further carried out on a human serum sample (Fig. 6D). Human serum contains abundant plasma proteins, peptides, lipids and other substances. To test the feasibility of our method in this complex sample, 1 × 105 CEM, MCF-7 and Ramos cells were individually added to normal human serum, and then captured using the dual-aptamer DNA network. By monitoring the cell capture efficiency as a function of incubation time, it could be found that about 59% of CEM cells and about 60% of MCF-7 cells were captured by C1/C2 and M1/M2 at 60 min, respectively (Fig. 6E). In contrast, only about 30% of Ramos cells were captured by C1/C2 or M1/M2 (Fig. S20†). Compared with that from buffer solution, the CTC capture from human serum needed longer time to reach the plateau and gave a little lower capture efficiency due to the influence of the suspended proteins in this complex system.
To simulate the blood samples of cancer patients that contain CTCs, target cancer cells (CEM or MCF-7) and non-target cells (Ramos cells) were added to normal human serum samples in a 1:3 ratio. As shown in Fig. 6F, the microscope images showed that CEM cells were specifically captured from the samples by the dual-aptamer DNA network, thus achieving highly efficient enrichment after CRISPR/Cas12a-controlled cell release. By comparing the number of cells per unit area (mm2) before and after enrichment, it could be calculated that the enrichment efficiency of CEM cells reached about 9-fold, indicating the high efficiency and good reliability of this method for CTC isolation and enrichment from blood samples. In the same way, the enrichment efficiency of MCF-7 cells could also reach 7.5 times (Fig. S21†). To further verify the feasibility of our method in practical applications, mouse whole blood was used to simulate real clinical samples. CTCs (pre-stained in green) were mixed into mouse whole blood with a ratio to blood cells of 1:1000. As shown in Fig. 6G, the network formed with single-aptamer can achieve a 5-fold enrichment of CTCs, and the network formed with a dual-aptamer can achieve an 8-fold enrichment. At the same time, non-target erythrocytes are greatly reduced by three orders of magnitude, which proves that our capture strategy is accurate and efficient. Thus, we foresee that this strategy could be a good supplement to current methods for the efficient capture and lossless release of CTCs.
Conclusions
In summary, we have developed a dual-aptamer DNA network-based cell capture and release strategy for cell enrichment and collection. The proposed strategy was demonstrated to work well for effective capture, 3D encapsulation, and CRISPR/Cas12a-controlled release of CTCs for downstream analysis of live cells. Compared with traditional cell enrichment strategies, this method has advantages in the following aspects. Firstly, due to the synergetic recognition and anchoring of dual aptamers to target cells, the proposed method can capture target CTCs with high specificity, accuracy and efficiency. Compared with that of a single aptamer-based method, the cell capture efficiency of a dual-aptamer strategy is approximately doubled. In addition, repeated aptamer units on each chain can simultaneously bind to multiple target proteins of the cell, thus giving further improved capture efficiency. By using different aptamer combinations, the proposed method can be used for the highly specific separation and enrichment of different types of CTCs. Secondly, CRISPR/Cas12a-responsive disassembly of the DNA network provides a rapid and cell-friendly way for the cell release because it works only on DNA networks rather than on cells. dsDNA can act as a cost-effective, easily obtained and highly biocompatible initiator to control the disassembly of DNA networks and the release of captured cells, thus providing a good choice for the design of systems, in which controllable release of cells is highly desirable. In addition, activated CRISPR/Cas12a may work for any DNA network targeting the capture of different cancer cells, without the requirement for the redesign of crRNA and dsDNA. Thirdly, the DNA network can provide a gentle and semi-wet 3D microenvironment for captured CTCs, and a softly wrapped DNA network around the cell surface has almost no damage to the cells. Moreover, the DNA network is visible to the naked eye, thus making a physical separation of the enclosed cells from the cell mixture possible, which not only simplifies the separation operation but can also further reduce the damage to cells. These, together with cell-friendly cell release, endow the released cells with high viability for subsequent applications and downstream analysis. We expect this approach to have great potential for early tumor diagnosis and the collection of reliable information about primary tumors.Data availability
Experimental details and additional data can be found in the attached ESI and video.†Author contributions
Dong-Xia Wang: conceptualization, methodology, investigation, writing-original draft. Jing Wang: visualization, investigation, supervision. Ya-Xin Wang: investigation. Jia-Yi Ma: investigation. Bo Liu: validation. An-Na Tang: supervision and reviewing. De-Ming Kong: supervision, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22074068 and 21874075) and the Fundamental Research Funds for Central University, Nankai University (No. 63201043).Notes and references
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- M. Poudineh, E. H. Sargent, K. Pantel and S. O. Kelley, Nat. Biomed. Eng., 2018, 2, 72–84 CrossRef CAS PubMed.
- W. Qin, L. Chen, Z. Wang, Q. Li, C. Fan, M. Wu and Y. Zhang, Adv. Sci., 2020, 2000647 CrossRef CAS PubMed.
- L. Wu, H. Ding, X. Qu, X. Shi, J. Yang, M. Huang, J. Zhang, H. Zhang, J. Song, L. Zhu, Y. Song, Y. Ma and C. Yang, J. Am. Chem. Soc., 2020, 142, 4800–4806 CrossRef CAS PubMed.
- J. Zhang, B. Lin, L. Wu, M. Huang, X. Li, H. Zhang, J. Song, W. Wang, G. Zhao, Y. Song and C. J. Yang, Angew. Chem., Int. Ed., 2020, 59, 14115–14119 CrossRef CAS PubMed.
- P. Song, D. Ye, X. Zuo, J. Li, J. Wang, H. Liu, M. T. Hwang, J. Chao, S. Su, L. Wang, J. Shi, L. Wang, W. Huang, R. Lal and C. Fan, Nano Lett., 2017, 17, 5193–5198 CrossRef CAS PubMed.
- D. Ye, M. Li, T. Zhai, P. Song, L. Song, H. Wang, X. Mao, F. Wang, X. Zhang, Z. Ge, J. Shi, L. Wang, C. Fan, Q. Li and X. Zuo, Nat. Protoc., 2020, 15, 2163–2185 CrossRef CAS PubMed.
- M. Hou, X. Yin, J. Jiang and J. He, ACS Appl. Mater. Interfaces, 2021, 13, 15031–15039 CrossRef CAS PubMed.
- C. Yao, C. Zhu, J. Tang, J. Ou, R. Zhang and D. Yang, J. Am. Chem. Soc., 2021, 143, 19330–19340 CrossRef CAS PubMed.
- T. Gao, T. Chen, C. Feng, X. He, C. Mu, J. I. Anzai and G. Li, Nat. Commun., 2019, 10, 2946 CrossRef PubMed.
- F. Mo, K. Jiang, D. Zhao, Y. Wang, J. Song and W. Tan, Adv. Drug Delivery Rev., 2020, 168, 79–98 CrossRef PubMed.
- F. Li, D. Lyu, S. Liu and W. Guo, Adv. Mater., 2020, 32, e1806538 CrossRef PubMed.
- S. C. Knight, L. Xie, W. Deng, B. Guglielmi, L. B. Witkowsky, L. Bosanac, E. T. Zhang, M. El Beheiry, J. B. Masson, M. Dahan, Z. Liu, J. A. Doudna and R. Tjian, Science, 2015, 350, 823–826 CrossRef CAS PubMed.
- Y. Zhang, Y. Wu, Y. Wu, Y. Chang and M. Liu, Trends Anal. Chem., 2021, 137, 116210 CrossRef CAS.
- H. Yue, M. Huang, T. Tian, E. Xiong and X. Zhou, ACS Nano, 2021, 15, 7848–7859 CrossRef CAS PubMed.
- W. Feng, A. M. Newbigging, J. Tao, Y. Cao, H. Peng, C. Le, J. Wu, B. Pang, J. Li, D. L. Tyrrell, H. Zhang and X. C. Le, Chem. Sci., 2021, 12, 4683–4698 RSC.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- D. X. Wang, J. Wang, Y. C. Du, J. Y. Ma, S. Y. Wang, A. N. Tang and D. M. Kong, Biosens. Bioelectron., 2020, 168, 112556 CrossRef CAS PubMed.
- S. Y. Wang, Y. C. Du, D. X. Wang, J. Y. Ma, A. N. Tang and D. M. Kong, Anal. Chim. Acta, 2021, 338882 CrossRef CAS PubMed.
- S. Y. Li, Q. X. Cheng, J. K. Liu, X. Q. Nie, G. P. Zhao and J. Wang, Cell Res., 2018, 28, 491–493 CrossRef CAS PubMed.
- S. Y. Li, Q. X. Cheng, J. M. Wang, X. Y. Li, Z. L. Zhang, S. Gao, R. B. Cao, G. P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef PubMed.
- Y. C. Du, S. Y. Wang, Y. X. Wang, J. Y. Ma, D. X. Wang, A. N. Tang and D. M. Kong, Biosens. Bioelectron., 2021, 171, 112734 CrossRef CAS PubMed.
- R. V. Gayet, H. de Puig, M. A. English, L. R. Soenksen, P. Q. Nguyen, A. S. Mao, N. M. Angenent-Mari and J. J. Collins, Nat. Protoc., 2020, 15, 3030–3063 CrossRef CAS PubMed.
- M. A. English, L. R. Soenksen, R. V. Gayet, H. de Puig, N. M. A. Mari, A. S. Mao, P. Q. Nguyen and J. J. Collins, Science, 2019, 365, 780–785 CrossRef CAS PubMed.
- Z. Liu, L. Sun, F. Shen, J. Xu, X. Han and C. Fan, Angew. Chem., Int. Ed., 2020, 59, 14842–14853 CrossRef PubMed.
- J. Li, K. Xun, L. Zheng, X. Peng, L. Qiu and W. Tan, J. Am. Chem. Soc., 2021, 143, 4585–4592 CrossRef CAS PubMed.
- L. Wu, Y. Wang, L. Zhu, Y. Liu, T. Wang, D. Liu, Y. Song and C. Yang, ACS Appl. Bio Mater., 2020, 3, 2743–2764 CrossRef CAS PubMed.
- M. You, G. Zhu, T. Chen, M. J. Donovan and W. Tan, J. Am. Chem. Soc., 2015, 137, 667–674 CrossRef CAS PubMed.
- R. Peng, X. Zheng, Y. Lyu, L. Xu, X. Zhang, G. Ke, Q. Liu, C. You, S. Huan and W. Tan, J. Am. Chem. Soc., 2018, 140, 9793–9796 CrossRef CAS PubMed.
- Y. Song, Z. Zhu, Y. An, W. Zhang, H. Zhang, D. Liu, C. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS PubMed.
- K. H. Ooi, M. M. Liu, J. W. D. Tay, S. Y. Teo, P. Kaewsapsak, S. Jin, C. K. Lee, J. Hou, S. Maurer-Stroh, W. Lin, B. Yan, G. Yan, Y. G. Gao and M. H. Tan, Nat. Commun., 2021, 12, 1739 CrossRef CAS PubMed.
- S. X. Kai Shi, R. Tian, S. Wang, Q. Lu, D. Gao, C. Lei, H. Zhu and Z. Nie, Sci. Adv., 2021, 7, eabc7802 CrossRef PubMed.
- Y. Xiong, J. Zhang, Z. Yang, Q. Mou, Y. Ma, Y. Xiong and Y. Lu, J. Am. Chem. Soc., 2020, 142, 207–213 CrossRef CAS PubMed.
- C. Yao, H. Tang, W. Wu, J. Tang, W. Guo, D. Luo and D. Yang, J. Am. Chem. Soc., 2020, 142, 3422–3429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03374g |
| This journal is © The Royal Society of Chemistry 2022 |