
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn economical approach for peptide synthesis via regioselective C–N bond cleavage of lactams†
Wataru
Muramatsu
 * and
Hisashi
Yamamoto
*
* and
Hisashi
Yamamoto
*
Peptide Research Center, Chubu University, 1200 Matsumoto-cho, Kasugai, Aichi 487-8501, Japan. E-mail: muramatsu@isc.chubu.ac.jp; hyamamoto@isc.chubu.ac.jp
First published on 26th April 2022
Abstract
An economical, solvent-free, and metal-free method for peptide synthesis via C–N bond cleavage using lactams has been developed. The method not only eliminates the need for condensation agents and their auxiliaries, which are essential for conventional peptide synthesis, but also exhibits high atom economy. The reaction is versatile because it can tolerate side chains bearing a range of functional groups, affording up to >99% yields of the corresponding peptides without racemisation or polymerisation. Moreover, the developed strategy enables peptide segment coupling, providing access to a hexapeptide that occurs as a repeat sequence in spider silk proteins.
Introduction
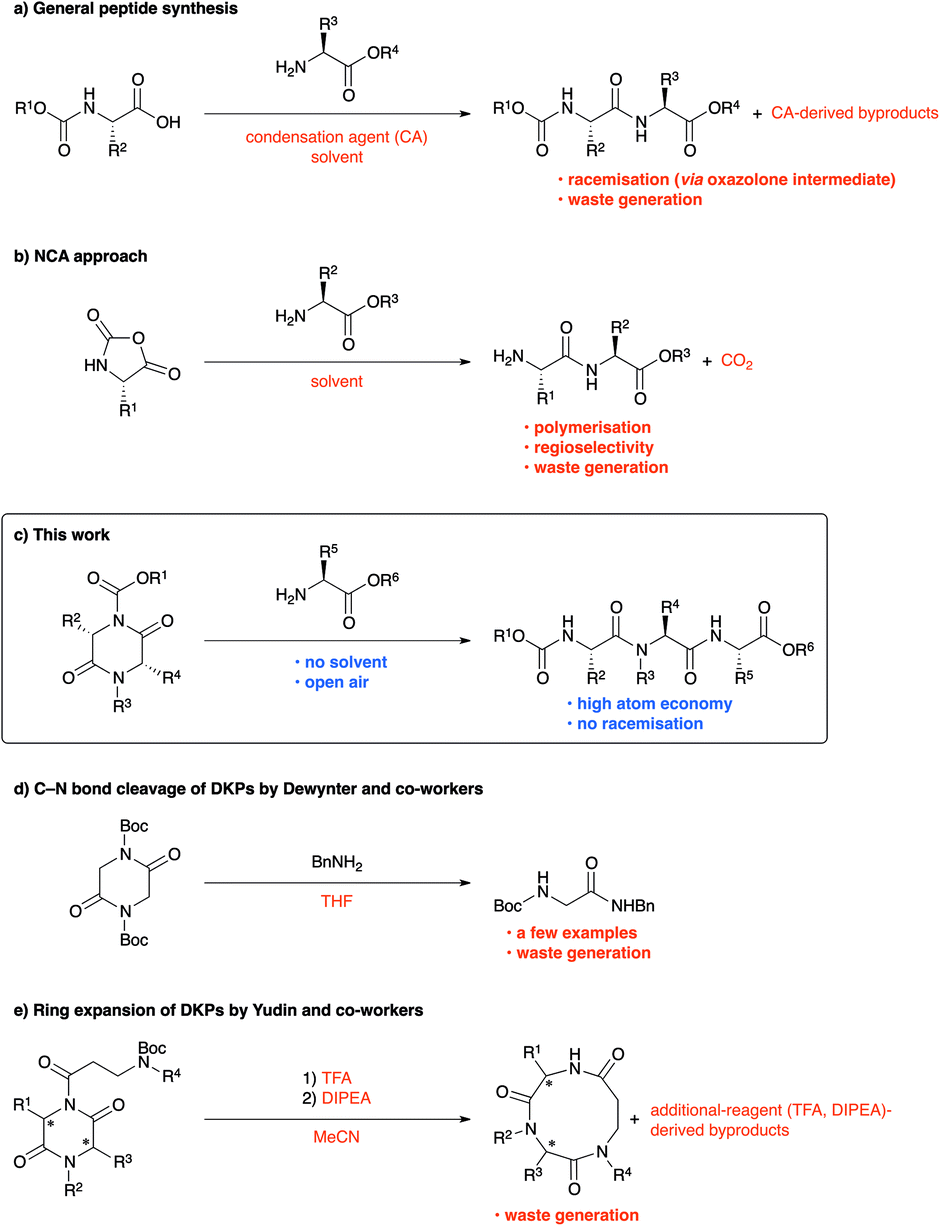
Peptide chemistry has evolved remarkably in the past century, and peptides are being extensively exploited in pharmaceuticals, materials, and cosmetics.1 Because peptide synthesis largely focuses on developing general condensation agents,2 realising high reactivity, and ensuring low risk of racemisation,3 issues such as health hazards,4 waste generation,4,5 and atom economy6 have remained neglected and must be urgently addressed. In fact, “Amide formation avoiding poor atom economy reagents,” listed as one of the 12 key green chemistry research areas in 2006, was retained as “General methods for catalytic/sustainable (direct) amide or peptide formation” and was regarded as one of the 10 key green chemistry research areas in 2018.7Peptides are usually synthesised by repeating a condensation-agent-mediated amidation reaction, followed by deprotection. Because of the importance of condensation agents, more valuable condensation-agent-mediated peptide synthesis methods are still being actively developed.2 However, condensation-agent-mediated peptide syntheses have low atom economy and occasionally face undesired racemisation (Scheme 1a).3 More importantly, the condensation agents may trigger allergic reactions in the user.4,8 Although a number of catalytic approaches have been reported in the past few decades to address these concerns,9 significant improvement is still necessary. Using amino acid N-carboxy anhydride (NCA) is among the most efficient approaches for peptide synthesis;10 however, suppression of polymerisation is quite difficult. Consequently, this method is generally used for the synthesis of polypeptides.11 Furthermore, the copious carbon dioxide generated at the industrial level is a serious concern, particularly in a modern society that is striving toward carbon neutrality. In addition, the preparation of NCAs requires considerable skill because of the use of phosgene, an extremely poisonous chemical (Scheme 1b).12
 | ||
| Scheme 1 Strategies for peptide chain elongation. | ||
In this study, we have developed a new strategy for peptide synthesis via regioselective C–N bond cleavage reaction using lactams, including diketopiperazines (DKPs), as building blocks (Scheme 1c). We believe that most concerns in peptide synthesis can be addressed by our strategy. First, our strategy requires no metals, reaction solvents, or condensation agents that present health hazards. Second, conventional peptide syntheses using condensation agents always generate wastes derived from condensation agents, whereas our developed reaction does not (high atom economy and low E factor).13 Third, although various condensation agents and catalysts have been developed to minimise racemisation, the risk of racemisation via the formation of oxazolone intermediates, which is the major cause of racemisation,3d still remains. By contrast, the risk of racemisation via the generation of oxazolone intermediates is completely eliminated in our new approach. To the best of our knowledge, similar studies have been reported so far only on amidation using BnNH2 by the Dewynter group (Scheme 1d)14 and ring expansion by the Yudin group (Scheme 1e).15 Although the two groups have conducted pioneering studies on metal-free C–N bond cleavage of lactams, reactions such as intermolecular peptide bond formation have not yet been examined, and there is significant scope for improvement in terms of substrate limitation and versatility.
Results and discussion
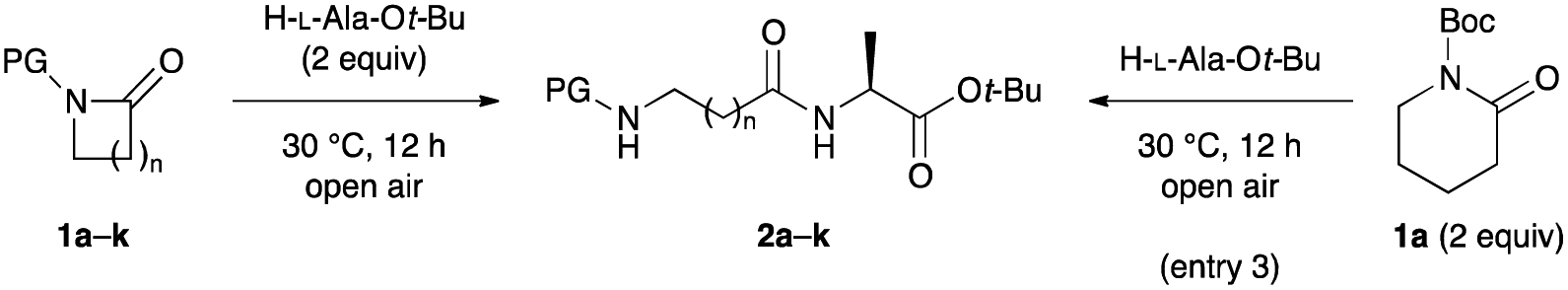
We began our investigation by reacting lactams 1 with H-L-Ala-Ot-Bu.16 Various solvents and ring sizes were tested for this reaction to determine the optimal conditions (Table 1). When 1a was reacted with H-L-Ala-Ot-Bu (2 equiv.) in the absence of solvents under air at 30 °C for 12 h, desired peptide 2a was regioselectively obtained in 99% yield, with >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er and ideal atom economy (entry 1). On a gram-scale (6 mmol), the C–N bond cleavage reaction proceeded with essentially identical yield and purity as for that conducted on a 0.50 mmol-scale (97% yield; entry 2). When 1a (2 equiv.) was reacted with H-L-Ala-Ot-Bu under the same reaction conditions, the yield of 2a decreased slightly to 92% (entry 3). When the reaction was carried out in less polar solvents, the C–N bond cleavage reaction afforded 2a in 47–74% yields, with no byproducts (entries 4–7). The use of ethers bearing sterically bulky substituents such as CPME, 2-Me-THF, or TBME, instead of toluene, led to a similar outcome (entries 8–10). The yields were lower than that in toluene when less hindered ethers such as 1,4-dioxane and DME or highly polar solvents such as DMF and DMSO were used (entries 11–13).17 The tolerance of several other protecting groups was evaluated.18 Contrary to expectation, the reaction did not proceed in some cases, while the yields were low or negligible relative to that with Boc as the protecting group (entries 14–19). This is because lactams 1b–e are unreactive, and the reaction of lactams 1b–e largely proceeds to cleave the corresponding protecting groups via C–N bond cleavage at the exocyclic amide moieties. Regardless of the ring size, lactams 1h–k gave satisfactory yields of the corresponding dipeptides involving Gly homolog residues 2h–k, without loss of stereochemical integrity and without side reactions (entries 20–23).
:1 er and ideal atom economy (entry 1). On a gram-scale (6 mmol), the C–N bond cleavage reaction proceeded with essentially identical yield and purity as for that conducted on a 0.50 mmol-scale (97% yield; entry 2). When 1a (2 equiv.) was reacted with H-L-Ala-Ot-Bu under the same reaction conditions, the yield of 2a decreased slightly to 92% (entry 3). When the reaction was carried out in less polar solvents, the C–N bond cleavage reaction afforded 2a in 47–74% yields, with no byproducts (entries 4–7). The use of ethers bearing sterically bulky substituents such as CPME, 2-Me-THF, or TBME, instead of toluene, led to a similar outcome (entries 8–10). The yields were lower than that in toluene when less hindered ethers such as 1,4-dioxane and DME or highly polar solvents such as DMF and DMSO were used (entries 11–13).17 The tolerance of several other protecting groups was evaluated.18 Contrary to expectation, the reaction did not proceed in some cases, while the yields were low or negligible relative to that with Boc as the protecting group (entries 14–19). This is because lactams 1b–e are unreactive, and the reaction of lactams 1b–e largely proceeds to cleave the corresponding protecting groups via C–N bond cleavage at the exocyclic amide moieties. Regardless of the ring size, lactams 1h–k gave satisfactory yields of the corresponding dipeptides involving Gly homolog residues 2h–k, without loss of stereochemical integrity and without side reactions (entries 20–23).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Lactam | PG | n | Solvent | Yield of 2a–k (%) | er of 2a–k |
| a Percentage represents the isolated yield. Ers were determined by HPLC. b Yield obtained via the gram-scale synthesis of 2a. c 50 °C. d H-L-Ala-Ot-Bu (3.0 equiv.) at 50 °C for 24 h. | ||||||
| 1 | 1a | Boc | 3 | — | 99 | >99:1 |
| 2b | 1a | Boc | 3 | — | 97 | >99:1 |
| 3 | 1a | Boc | 3 | — | 92 | >99:1 |
| 4 | 1a | Boc | 3 | Toluene | 74 | >99:1 |
| 5 | 1a | Boc | 3 | Benzene | 58 | >99:1 |
| 6 | 1a | Boc | 3 | CHCl3 | 47 | >99:1 |
| 7 | 1a | Boc | 3 | DCM | 52 | >99:1 |
| 8 | 1a | Boc | 3 | CPME | 88 | >99:1 |
| 9 | 1a | Boc | 3 | 2-Me-THF | 69 | >99:1 |
| 10 | 1a | Boc | 3 | TBME | 76 | >99:1 |
| 11 | 1a | Boc | 3 | DME | 58 | >99:1 |
| 12 | 1a | Boc | 3 | DMF | 52 | >99:1 |
| 13 | 1a | Boc | 3 | DMSO | 29 | >99:1 |
| 14 | 1b | H | 3 | — | 0 | — |
| 15 | 1c | Me | 3 | — | 0 | — |
| 16 | 1d | Bn | 3 | — | 0 | — |
| 17 | 1e | Ac | 3 | — | 0 | — |
| 18 | 1f | Bz | 3 | — | 5 | — |
| 19 | 1g | Cbz | 3 | — | 43 | >99:1 |
| 20c | 1h | Boc | 1 | — | 91 | >99:1 |
| 21d | 1i | Boc | 2 | — | 73 | >99:1 |
| 22 | 1j | Boc | 4 | — | 90 | >99:1 |
| 23d | 1k | Boc | 10 | — | 65 | >99:1 |
With the optimal reaction conditions in hand, lactams bearing an array of functional groups were successfully coupled with a wide variety of amino acid esters that served as the nucleophilic components for peptide formation (Scheme 2). Amino acid esters possessing a branched or relatively bulky side chain (e.g., Leu, Thr, and His) were compatible with this reaction, providing the desired dipeptides (2aa, 2ab, and 4ib, respectively) in excellent yields. When lactams comprising esters and protected amino groups 3a–d and 3f–j were employed with a variety of amino acid esters, the corresponding dipeptides 4aa, 4ab, 4ba, 4bb, 4c, 4d, 4f, 4ga, 4ha, 4ia, and 4j were successfully furnished in 60–99% yields. Fortunately, an independent parallel C–N bond cleavage reaction and our original tantalum-catalysed amidation19 could furnish the desired tripeptides 4ac, 4e, and 4gb in 96%, 80%, and 41% yields, respectively. The unsatisfactory yield of 4gb was attributed to the incomplete conversion. The activity of the metal catalyst used in peptide bond-forming reactions is often poisoned by the sulfur atoms in amino acids such as Met, Cys, and their derivatives because of their strong binding affinities.20 Such substrate limitations were not observed in our system because it did not require any additional reagents and metals. Moreover, H-L-Met-Ot-Bu was compatible under the reaction conditions, affording 4hb in 97% yield without the occurrence of either epimerisation or side reactions.
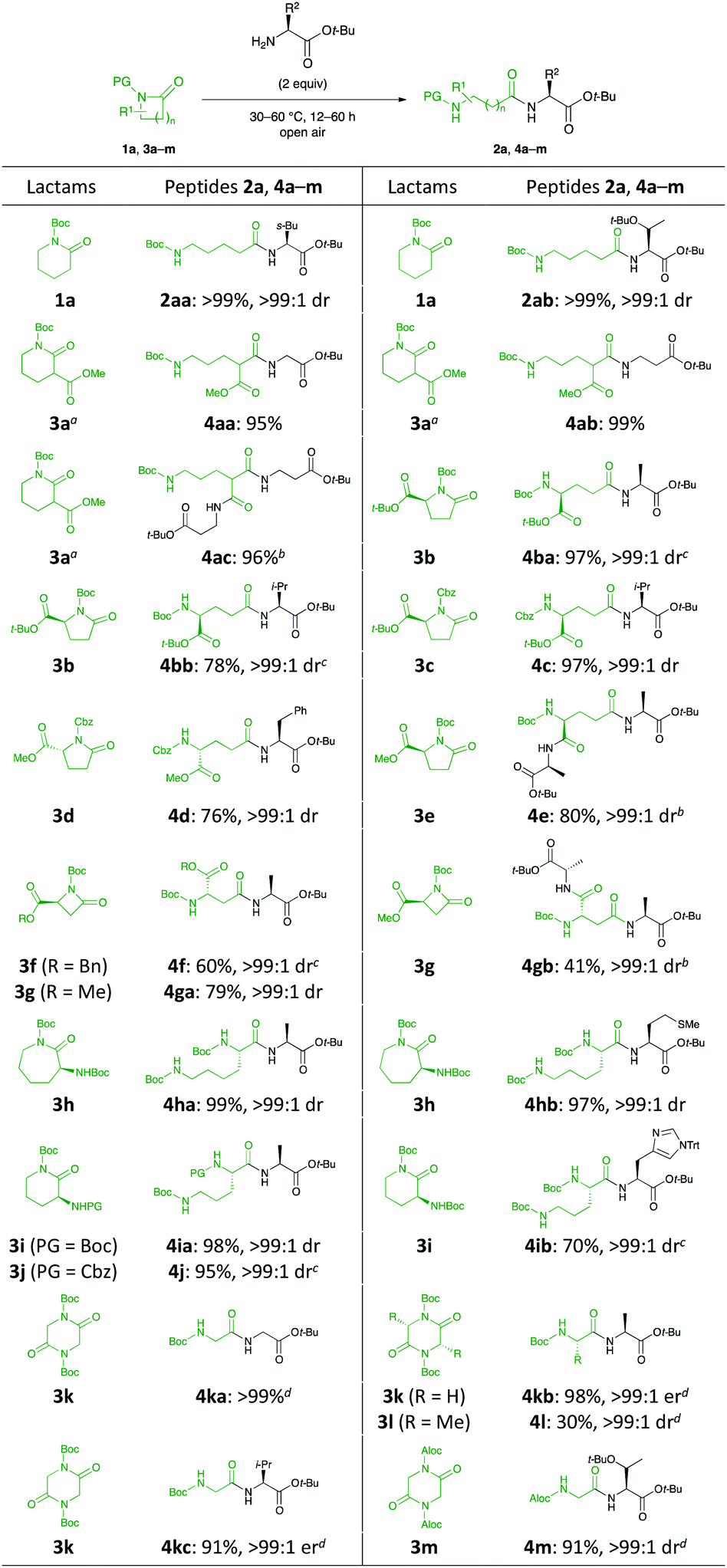 | ||
| Scheme 2 Scope and limitation of lactams. Percentage represent the isolated yield. Ers were determined by HPLC. Drs were determined by 1H NMR spectroscopy. aRacemic lactam was used as the starting material. bAmino acid tert-butyl esters (4 equiv.) and Ta(OMe)5 (10 mol%). cAmino acid tert-butyl esters (3 equiv.). dAmino acid tert-butyl esters (4 equiv.). | ||
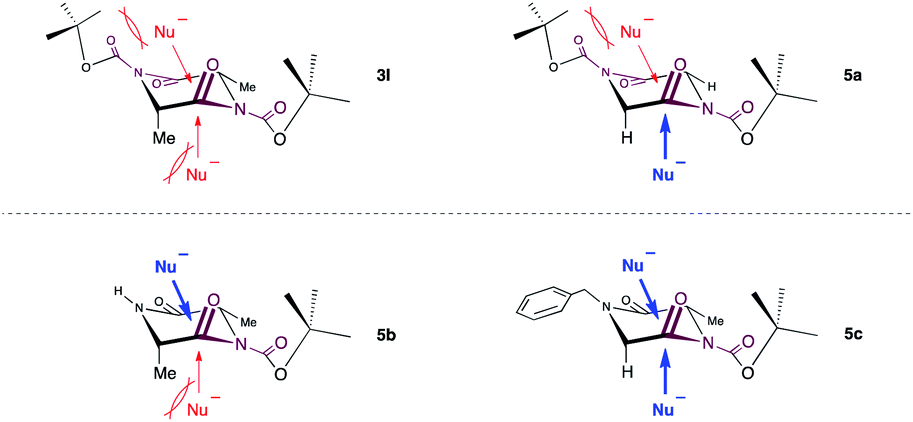
Next, the reaction was extended to the C–N bond cleavage reaction of 2,5-diketopiperazines (DKPs) for dipeptide synthesis. The C–N bond cleavage of cyclo(-PG-Gly-PG-Gly-) 3k and 3m with some amino acid esters smoothly generated the corresponding dipeptides 4ka, 4kb, 4kc, and 4m in excellent yields. Cyclo(-Boc-L-Ala-Boc-L-Ala-) 3l was less reactive than 3k and 3m.
Subsequently, we focused our attention on the applicability of DKPs bearing an array of functional groups for the formation of peptide bonds with several amino acid esters and peptides as nucleophilic components (Scheme 3).21 In contrast to 3l, whose reactivity was relatively low, 5a and 5b reacted smoothly with a variety of amino acid esters and dipeptides to afford the corresponding tri- and tetrapeptides 6aa–ae, 6ba–bc in high yields, without loss of stereochemical integrity. Similar to 5a, DKPs bearing a Bn group at the 4-position (5c and 5d) reacted to give the desired products 6ca–ce and 6da–dd in high yields. This is probably because the approach of amines to the carbonyl groups at the endocyclic amide moieties of 3l is prevented by steric hindrance of the t-Bu groups, which is caused by the twisting of the exocyclic amide moieties (Fig. 1).22
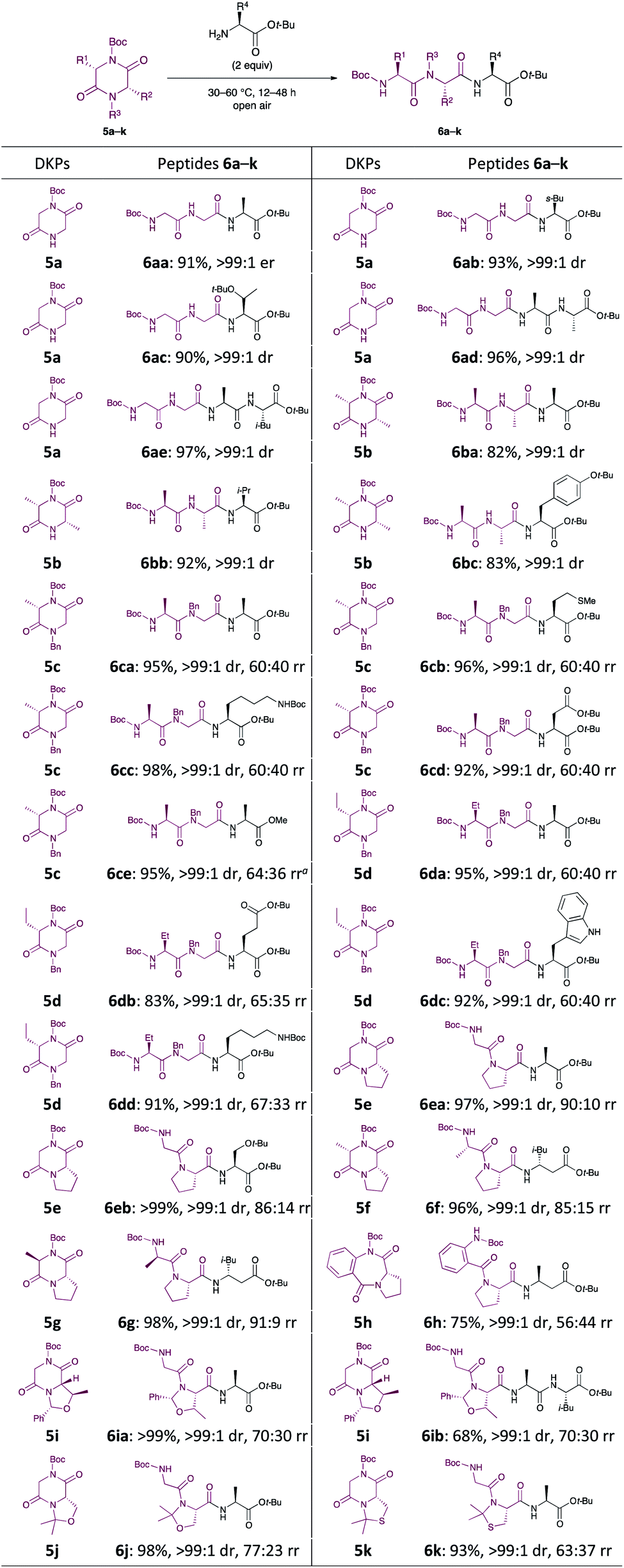 | ||
| Scheme 3 Scope and limitation of DKPs. Percentage represent the isolated yield. Er was determined by HPLC. Drs and rrs (ratio of rotation isomers)24 were determined by 1H NMR spectroscopy. aH-L-Ala-OMe·HCl (3 equiv.) and Et3N (3 equiv.) were used. | ||
 | ||
| Fig. 1 C–N bond cleavage at a twisted amide moiety. | ||
In general, hydrochloride salts of amino acid esters can be stored for a longer time period than the corresponding free amines because of the stability and ease of handling of the former. Since free amines such as H-Gly-OMe and H-L-Ala-OMe are volatile and prone to spontaneous polymerisation, it is quite difficult to handle them directly for peptide synthesis.16a Thus, when H-L-Ala-OMe, formed by the in situ neutralisation of its HCl salt using Et3N, was reacted with 5c, the desired product 6ce was obtained in 95% yield, without epimerisation. A broad variety of six-five bicyclic DKPs, serving as electrophilic counterparts, smoothly participated in this C–N bond cleavage reaction, and DKPs 5e–g reacted with α- and β-amino acid esters to form new peptides in excellent yields.23 The stereochemistry of the DKPs did not affect this C–N bond cleavage reaction, and both 5f and 5g gave 6f and 6g, respectively, in excellent yields without any apparent epimerisation. Unfortunately, a moderate yield (75%) of 6h was obtained when seven-five bicyclic compound 5h was used. This is due to the occurrence of an unexpected C–N bond cleavage reaction (cleavage of the Boc group) that produced undesired Boc-L-β-HoAla-Ot-Bu and cyclo(-Abz-L-Pro-). DKPs 5i–k, possessing ether or sulfide side chains, were also tolerated under the reaction conditions and were smoothly transformed into 6ia, 6ib, 6j, and 6k in high yields. Thus, the developed method can be used to synthesise peptides in high yields while circumventing the commonly encountered problems in peptide synthesis.
Finally, we demonstrated convergent synthesis of a hexapeptide (11: Gly-L-Ser-Gly-L-Ala-Gly-L-Ala) via this C–N bond cleavage as a key reaction because peptides that mimic parts of biologically-derived proteins or that incorporate parts of biologically-derived proteins have attracted increasing attention in recent years as new bio-based materials for potential applications due to their remarkable properties such as a balance of toughness, strength, and extensibility.25 This hexapeptide is a unique repeat sequence observed in spider, bagworm, and Bombyx mori silk proteins.25f,g,26 One-pot amidation of H-Gly-OH with H-L-Ala-Ot-Bu through transient masking with silylating agents produced 7 in 78% yield.27 In the presence of HSi[OCH(CF3)2]3,28 CsF, and imidazole,27,29 peptide segment coupling of 7 with Cbz-Gly-L-Ala-OH, which can be obtained by classical methods in two steps from commercially available Cbz-Gly-OH and H-L-Ala-OMe,30 followed by hydrogenation furnished 9 in excellent yields. The subsequent C–N bond cleavage/new amide bond formation/deprotection sequence afforded target peptide 11 with >99:1 dr (Scheme 4).
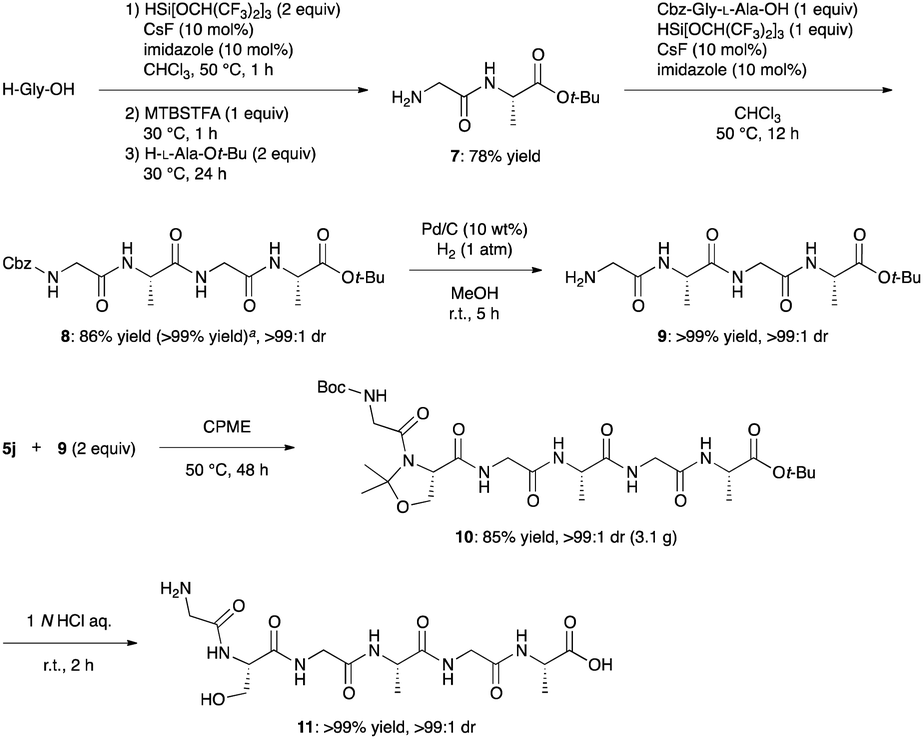 | ||
| Scheme 4 Convergent synthesis of a unique fragment of spider silk proteins. aCbz-Gly-L-Ala-OH, H-Gly-L-Ala-Ot-Bu (1.5 equiv.), HSi[OCH(CF3)2]3 (1.3 equiv.), CHCl3, 50 °C, 24 h. | ||
Conclusions
In summary, we have demonstrated that C–N bond cleavage reaction using lactams as building blocks is an economical design principle for peptide synthesis. This method tolerates amino acid side chains bearing various functional groups and can be effectively employed to avoid racemisation during peptide bond formation. Moreover, this is competent for peptide segment coupling to provide a unique fragment of spider silk proteins. Important features of our strategy are as follows: (1) no use of metals, condensation agents, auxiliaries, or reaction solvents. (2) The reactions do not produce byproducts (high atom economy) and potentially, they may be run on an industrial scale without waste generation (low E factor). (3) No racemisation. (4) Mild reaction conditions and simple operation. (5) The present synthetic approach is not only the first example of a metal-free C–N bond cleavage reaction for intermolecular peptide bond formation but also the most economical convergent peptide synthesis. (6) DKPs can be easily prepared according to known methods16b,31 and are recognised to have unique bioactivities.32 In addition, they are also attracting attention as new functional ingredients in foods and beverages.33 (7) Superfluous amino acid esters can be readily removed by aqueous extraction or using an oil rotary vacuum pump/oil diffusion pump. (8) DKPs can be stored for long periods and are easy to handle because they are relatively stable. Considering the operational simplicity and high atom economy of this approach for new peptide synthesis with high optical purity and the potential applicability of these peptides as pharmaceutically relevant scaffolds, we expect this method to be widely adopted in academia and industry.Data availability
Experimental procedures, characterisation data, and copies of 1H and 13C NMR spectra for all compounds have been included in the ESI.†Author contributions
W. M. and H. Y. conceived and designed the project. W. M. performed the experiments and collected the data. W. M. prepared the manuscript with input from all contributing authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support was provided by a grant-in-aid from the New Energy and Industrial Technology Development Organisation (NEDO, JPNP14004) and Daiko Foundation (No. 9226), Japan.Notes and references
- (a) G. Klebe, Drug Design, Springer, Berlin, Heidelberg, 2013 CrossRef; (b) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed; (c) A. Harrington and Y. Tal-Gan, Future Med. Chem., 2019, 11, 2759–2763 CrossRef CAS PubMed; (d) S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- (a) F. Albericio, R. Chinchilla, D. J. Dodsworth and C. Nájera, Org. Prep. Proced. Int., 2001, 33, 203–303 CrossRef CAS; (b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (c) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed; (d) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS; (e) F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760–772 CrossRef CAS; (f) J. B. Sperry, C. J. Minteer, J. Y. Tao, R. Johnson, R. Duzguner, M. Hawksworth, S. Oke, P. F. Richardson, R. Barnhart, D. R. Bill, R. A. Giusto and J. D. Weaver III, Org. Process Res. Dev., 2018, 2, 1262–1275 CrossRef.
- (a) M. B. A. Bodanszky, Chem. Commun., 1967, 591–593 RSC; (b) I. Antonovics and G. T. Young, J. Chem. Soc. C, 1967, 595–601 RSC; (c) N. Izumiya and M. Muraoka, J. Am. Chem. Soc., 1969, 91, 2391–2392 CrossRef CAS; (d) D. S. Kemp and J. Rebek Jr, J. Am. Chem. Soc., 1970, 92, 5792–5793 CrossRef CAS PubMed.
- (a) L. Wigman, T. Remarchuk, S. R. Gomez, A. Kumar, M. W. Dong, C. D. Medley and N. Chetwyn, Am. Pharm. Rev., 2014, 17 Search PubMed; (b) O. A. Musaimi, B. G. de la Torre and F. Albericio, Green Chem., 2020, 22, 996–1018 RSC; (c) K. Wegner, D. Barnes, K. Manzor, A. Jardine and D. Moran, Green Chem. Lett. Rev., 2021, 14, 153–164 CrossRef CAS.
- (a) C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS; (b) J. H. Rasmussen, Bioorg. Med. Chem., 2018, 26, 2914–2918 CrossRef CAS PubMed; (c) A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- (a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC; (b) B. M. Monks and A. Whiting, Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries, John Wiley & Sons, Inc., Hoboken, NJ, 2013, pp. 89–110 Search PubMed.
- K. J. McKnelly, W. Sokol and J. S. Nowick, J. Org. Chem., 2020, 85, 1764–1768 CrossRef CAS PubMed.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (b) T. Krause, S. Baader, B. Erb and L. J. Gooßen, Nat. Commun., 2016, 7, 11732 CrossRef PubMed; (c) M. T. Sabatini, L. T. Boulton, H. F. Sheddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS; (d) Handoko, S. Satishkumar, N. R. Ranigrahi and P. S. Arora, J. Am. Chem. Soc., 2019, 141, 15977–15985 CrossRef CAS PubMed; (e) M. Todorovic and D. M. Perrin, J. Pept. Sci., 2020, 112, e24210 CAS; (f) W. Muramatsu, T. Hattori and H. Yamamoto, Bull. Chem. Soc. Jpn., 2020, 93, 759–767 CrossRef CAS; (g) W. Muramatsu, T. Hattori and H. Yamamoto, Chem. Commun., 2021, 57, 6346–6359 RSC.
- V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318–9321 CrossRef CAS PubMed.
- (a) H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS PubMed; (b) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS PubMed; (c) T. J. Deming, Adv. Polym. Sci., 2013, 262, 1–37 CrossRef CAS; (d) T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed; (e) Y. Zhang, R. Liu, H. Jin, W. Song, R. Augustine and I. Kim, Commun. Chem., 2018, 1, 40 CrossRef; (f) A. R. Mazo, S. Allison-Logan, F. Karimi, N. J.-A. Chan, W. Qiu, W. Duan, N. M. O'Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737–4834 RSC; (g) Y. Wu, K. Chen, X. Wu, L. Liu, W. Zhang, Y. Ding, S. Liu, M. Zhou, N. Shao, Z. Ji, J. Chen, M. Zhu and R. Liu, Angew. Chem., Int. Ed., 2021, 60, 26063–26071 CrossRef CAS PubMed.
- (a) F. Fuchs, Ber. Dtsch. Chem. Ges. B, 1922, 55, 2943 CrossRef; (b) A. C. Farthing, J. Chem. Soc., 1950, 3213–3217 RSC; (c) R. Katakai and Y. Iizuka, J. Org. Chem., 1985, 50, 715–716 CrossRef CAS; (d) R. Wilder and S. Mobashery, J. Org. Chem., 1992, 57, 2755–2756 CrossRef CAS; (e) Y. Otake, H. Nakamura and S. Fuse, Angew. Chem., Int. Ed., 2018, 57, 11389–11393 CrossRef CAS PubMed.
- (a) R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC; (c) R. A. Sheldon, Chem. Commun., 2008, 44, 3352–3365 RSC; (d) R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- D. Farran, D. Echalier, J. Martinez and G. Dewynter, J. Pept. Sci., 2009, 15, 474–478 CrossRef CAS PubMed.
- R. Mendoza-Sanchez, V. B. Corless, Q. N. N. Nguyen, M. Bergeron-Brlek, J. Frost, S. Adachi, D. J. Tantillo and A. K. Yudin, Chem.–Eur. J., 2017, 23, 13319–13322 CrossRef CAS PubMed.
- We chose amino acid tert-butyl esters as nucleophilic components because side reactions such as the self-intermolecular condensation or intramolecular cyclisation of peptides can be avoided by the virtue of the steric hindrance from the tert-butyl moiety. See: (a) G. W. Anderson and F. M. Callahan, J. Am. Chem. Soc., 1960, 82, 3359–3363 CrossRef CAS; (b) N. Funasaki, S. Hada and S. Neya, Anal. Chem., 1993, 65, 1861–1867 CrossRef CAS.
- More data for the solvent screening is shown in ESI.†.
- We attempted to prepare N-Fmoc-2-piperidone according to the reference. Unfortunately, the desired compound could not be obtained. See, A. Barco, S. Benetti, C. D. Risi, P. Marchetti, G. P. Pollini and V. Zanirato, J. Comb. Chem., 2000, 2, 337–340 CrossRef CAS PubMed.
- W. Muramatsu, T. Hattori and H. Yamamoto, J. Am. Chem. Soc., 2019, 141, 12288–12295 CrossRef CAS PubMed.
- (a) P. Forzatti and L. Lietti, Catal. Today, 1999, 52, 165–181 CrossRef CAS; (b) C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- Asn and Gln derivatives can be applied to this method in CPME as nucleophilic components. Unfortunately, the corresponding peptides were obtained in unsatisfactory yields because of their insolubility..
- (a) C. L. L. Chai, D. C. R. Hockless and A. R. King, Aust. J. Chem., 1996, 49, 1229–1233 CrossRef CAS; (b) A. P. Mendham, J. Spencer, B. Z. Chowdhry, T. J. Dines, M. Mujahid, R. A. Palmer, G. J. Tizzard and S. J. Coles, J. Chem. Crystallogr., 2011, 41, 1323–1327 CrossRef CAS; (c) L. Lelo, V. Pace, W. Holzer, M. M. Rahman, G. Meng, R. Szostak and M. Szostak, Chem.–Eur. J., 2020, 26, 16246–16250 CrossRef PubMed; (d) G. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121, 12746–12783 CrossRef CAS PubMed.
- β-Amino acids, β-peptides, and α,β-hybrid peptides possess unique properties in biological and structural aspects. See, (a) E. A. Porter, X. Wang, H.-S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS PubMed; (b) O. M. Stephens, S. Kim, B. D. Welch, M. E. Hodsdon, M. S. Kay and A. Schepartz, J. Am. Chem. Soc., 2005, 127, 13126–13127 CrossRef CAS PubMed; (c) T. Beke, C. Somlai and A. Perczel, J. Comput. Chem., 2006, 27, 20–38 CrossRef CAS PubMed; (d) A. J. Karlsson, W. C. Pomerantz, K. J. Neilsen, S. H. Gellman and S. P. Palecek, ACS Chem. Biol., 2009, 4, 567–579 CrossRef CAS PubMed; (e) F. Kudo, A. Miyanaga and T. Eguchi, Nat. Prod. Rep., 2014, 31, 1056–1073 RSC; (f) C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed.
- Rotational isomers were observed in CDCl3. On the other hand, they were rarely observed in CD3OD. This is probably due to the effects of intramolecular hydrogen bonding..
- (a) S. Osaki, Nature, 1996, 384, 419 CrossRef CAS; (b) E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS PubMed; (c) M. Heim, D. Keerl and T. Scheibel, Angew. Chem., Int. Ed., 2009, 48, 3584–3596 CrossRef CAS PubMed; (d) A. Thathe, M. Ghodke and P. Nikalje, Int. J. Pharm. Pharm. Sci., 2010, 2, 19–23 Search PubMed; (e) F. Teulé, Y.-G. Miao, B.-H. Sohn, Y.-S. Kim, J. J. Hull, M. J. Fraser Jr, R. V. Lewis and D. L. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 923–928 CrossRef PubMed; (f) G. A. Knoll, S. M. Romanelli, A. M. Brown, R. M. Sortino and I. A. Banerjee, J. Nanosci. Nanotechnol., 2016, 16, 2464–2473 CrossRef CAS PubMed; (g) T. Yoshioka, T. Tsubota, K. Tashiro, A. Jouraku and T. Kameda, Nat. Commun., 2019, 10, 1469 CrossRef PubMed; (h) A. P. Kiseleva, P. V. Krivoshapkin and E. F. Krivoshapkina, Front. Chem., 2020, 8, 554 CrossRef CAS PubMed; (i) R. Balu, N. K. Dutta, A. K. Dutta and N. R. Choudhury, Nat. Commun., 2021, 12, 149 CrossRef CAS PubMed.
- (a) N. D. Lazo and D. T. Downing, Macromolecules, 1999, 32, 4700–4705 CrossRef CAS; (b) C. Z. Zhou, F. Confalonieri, N. Medina, Y. Zivanovic, C. Esnault, T. Yang, M. Jacquet, J. Janin, M. Duguet, R. Perasso and Z.-G. Li, Nucleic Acids Res., 2000, 28, 2413–2419 CrossRef CAS PubMed; (c) Z. Megeed, J. Cappello and H. Ghandehari, Adv. Drug Delivery Rev., 2002, 54, 1075–1091 CrossRef CAS; (d) J. G. Hardy and T. R. Scheibel, Biochem. Soc. Trans., 2009, 37, 677–681 CrossRef CAS PubMed; (e) T. Asakura, H. Nishi, A. Nagano, A. Yoshida, Y. Nakazawa, M. Kamiya and M. Demura, Biomacromolecules, 2011, 12, 3910–3916 CrossRef CAS PubMed.
- W. Muramatsu and H. Yamamoto, J. Am. Chem. Soc., 2021, 143, 6792–6797 CrossRef CAS PubMed.
- W. Muramatsu, C. Manthena, E. Nakashima and H. Yamamoto, ACS Catal., 2020, 10, 9594–9603 CrossRef CAS.
- L. Horner and J. Mathias, J. Organomet. Chem., 1985, 282, 155–174 CrossRef CAS.
- P. Singh, S. Kaur, J. Kaur, G. Singh and R. Bhatti, J. Med. Chem., 2016, 59, 3920–3934 CrossRef CAS PubMed.
- (a) H. Thajudeen, K. Park, S.-S. Moon and I. S. Hong, Tetrahedron Lett., 2010, 51, 1303–1305 CrossRef CAS; (b) Nonappa, K. Ahonen, M. Lahtinen and E. Kolehmainen, Green Chem., 2011, 13, 1203–1209 RSC; (c) J.-C. M. Monbaliu, F. K. Hansen, L. K. Beagle, M. J. Panzner, P. J. Steel, E. Todadze, C. V. Stevens and A. R. Katritzky, Chem.–Eur. J., 2012, 18, 2632–2638 CrossRef CAS PubMed; (d) H. Abe, C. Sakashita, M. Kawada, A. Nomoto, T. Watanabe and M. Shibasaki, Chem. Pharm. Bull., 2015, 63, 463–468 CrossRef CAS PubMed; (e) A. Mollica, R. Costante, S. Mirzaie, S. Carradori, G. Macedonio, A. Stefanucci and E. Novellino, J. Heterocycl. Chem., 2016, 53, 2106–2110 CrossRef CAS; (f) H. Yu, Y. Zong and T. Xu, Chem. Sci., 2020, 11, 656–660 RSC; (g) C. G. Pappas, N. K. Wijerathne, J. K. Sahoo, A. Jain, D. Kroiss, I. R. Sasselli, A. S. Pina, A. Lampel and R. V. Ulijn, ChemSystemsChem, 2020, 2, e2000013 CrossRef CAS.
- (a) A. K. Szardenings, V. Antonenko, D. A. Campbell, N. DeFrancisco, S. Ida, L. Shi, N. Sharkov, D. Tien, Y. Wang and M. Navre, J. Med. Chem., 1999, 42, 1348–1357 CrossRef CAS PubMed; (b) F. Fdhila, V. Vázquez, J. L. Sánchez and R. Riguera, J. Nat. Prod., 2003, 66, 1299–1301 CrossRef CAS PubMed; (c) A. I. Faden, S. M. Knoblach, I. Cernak, L. Fan, R. Vink, G. L. Araldi, S. T. Fricke, B. L. Roth and A. P. Kozikowski, J. Cereb. Blood Flow Metab., 2003, 23, 342–354 CrossRef CAS PubMed; (d) D. R. Houston, B. Synstad, V. G. H. Eijsink, M. J. R. Stark, I. M. Eggleston and D. M. F. van Aalten, J. Med. Chem., 2004, 47, 5713–5720 CrossRef CAS PubMed; (e) B. Nicholson, G. K. Lloyd, B. R. Miller, M. A. Palladino, Y. Kiso, Y. Hayashi and S. T. C. Neuteboom, Anti-Cancer Drugs, 2006, 17, 25–31 CrossRef CAS PubMed; (f) M. Teixidó, E. Zurita, M. Malakoutikhah, T. Tarragó and E. Giralt, J. Am. Chem. Soc., 2007, 129, 11802–11813 CrossRef PubMed; (g) S. Hirano, S. Ichikawa and A. Matsuda, Bioorg. Med. Chem., 2008, 16, 428–436 CrossRef CAS PubMed; (h) E. van der Merwe, D. Huang, D. Peterson, G. Kilian, P. J. Milne, M. V. de Venter and C. Frost, Peptides, 2008, 29, 1305–1311 CrossRef CAS PubMed; (i) R. R. King and L. A. Calhoun, Phytochemistry, 2009, 70, 833–841 CrossRef CAS PubMed; (j) Z. Song, Y. Hou, Q. Yang, X. Li and S. Wu, Mar. Drugs, 2021, 19, 403 CrossRef CAS PubMed.
- (a) T. Mizuma, S. Masubuchi and S. Awazu, J. Pharm. Pharmacol., 1997, 49, 1067–1071 CrossRef CAS PubMed; (b) N. Tsuruoka, Y. Beppu, H. Koda, N. Doe, H. Watanabe and K. Abe, PLoS One, 2012, 7, e50824 CrossRef CAS PubMed; (c) M.-K. Kwak, R. Liu, J.-O. Kwon, M.-K. Kim, A. H. Kim and S.-O. Kang, J. Microbiol., 2013, 51, 836–843 CrossRef CAS PubMed; (d) A. D. Borthwick and N. C. D. Costa, Crit. Rev. Food Sci. Nutr., 2017, 57, 718–742 CrossRef CAS PubMed; (e) T. Shimamura, K. Yamamoto, Y. Otsuka, M. Hayashi, T. Kashiwagi and H. Ukeda, Food Preserv. Sci., 2017, 43, 227–233 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, and copies of 1H and 13C NMR spectra for all compounds. See https://doi.org/10.1039/d2sc01466a |
| This journal is © The Royal Society of Chemistry 2022 |