
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An electrically conductive metallocycle: densely packed molecular hexagons with π-stacked radicals†
Mengxing
Cui
a,
Ryuichi
Murase
a,
Yongbing
Shen
 a,
Tetsu
Sato
a,
Shohei
Koyama
a,
Kaiji
Uchida
a,
Tappei
Tanabe
a,
Shinya
Takaishi
a,
Masahiro
Yamashita
ab and
Hiroaki
Iguchi‡
*a
a,
Tetsu
Sato
a,
Shohei
Koyama
a,
Kaiji
Uchida
a,
Tappei
Tanabe
a,
Shinya
Takaishi
a,
Masahiro
Yamashita
ab and
Hiroaki
Iguchi‡
*a
aDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aza-Aoba, Aramaki, Sendai 980-8578, Japan. E-mail: h-iguchi@tohoku.ac.jp
bSchool of Materials Science and Engineering, Nankai University, Tianjin 300350, P. R. China
First published on 1st April 2022
Abstract
Electrical conduction among metallocycles has been unexplored because of the difficulty in creating electronic transport pathways. In this work, we present an electrocrystallization strategy for synthesizing an intrinsically electron-conductive metallocycle, [Ni6(NDI-Hpz)6(dma)12(NO3)6]·5DMA·nH2O (PMC-hexagon) (NDI-Hpz = N,N′-di(1H-pyrazol-4-yl)-1,4,5,8-naphthalenetetracarboxdiimide). The hexagonal metallocycle units are assembled into a densely packed ABCABC… sequence (like the fcc geometry) to construct one-dimensional (1D) helical π-stacked columns and 1D pore channels, which were maintained under the liberation of H2O molecules. The NDI cores were partially reduced to form radicals as charge carriers, resulting in a room-temperature conductivity of (1.2–2.1) × 10−4 S cm−1 (pressed pellet), which is superior to that of most NDI-based conductors including metal–organic frameworks and organic crystals. These findings open up the use of metallocycles as building blocks for fabricating conductive porous molecular materials.
Introduction
Giant macrocycles have attracted a great deal of attention due to their distinctive molecular structures and diverse functions.1 Coordination-driven self-assembly is a simple but powerful approach towards synthesizing giant metal–organic macrocycles (so-called metallocycles).2 Given the high designability of organic linkers and their various combinations with metal ions, metallocycles have provided a wide variety of molecular architectures such as rings,3,4 polygons,5–8 polyhedra,7–9 and nanotubes,10–14 in tandem with their multifunctional nature. For instance, anticancer ability,15 drug delivery,16 chirality recognition,17 and other molecular responsive functions3,11,18 have been realized by the flexible cavity of metallocycles. Furthermore, organic linkers with unique electronic properties have been key to fabricating functional metallocycles such as luminescent liquid crystals,19 asymmetric catalysts,20 and artificial light-harvesting systems.21 Although some metallocycles are not isolable in solution, solid-state functions have also been a subject of active research recently. To date, single-molecule magnet behavior,10,22 solid-state luminescence,23 heterogeneous catalytic behavior,24 gas storage,25etc., have been reported in crystalline metallocycle assemblies. In this context, engendering electron transfer between assembled metallocycles is promising for providing conductivity as a new function of metallocycles, and moreover, it can be an alternative approach to design conductive porous crystals such as metal–organic frameworks (MOFs).26 However, only the hole mobility of dipyrrin-based polygons has been reported as the charge-transport property in metallocycles.27 Intrinsic conductivity, which requires both rich charge carriers and through-space conduction pathways among metallocycles, has not been realized thus far.Electrocrystallization is a reasonable method to fulfill the above requirements, because the unpaired electrons (π-radicals) generated on π-conjugated planes by a redox reaction can be charge carriers and also induce an attractive interaction between π-conjugated planes. It has been widely used for the syntheses of organic conductors28 and, more recently, porous molecular conductors (PMCs),29,30 which are porous crystals consisting of linear coordination polymers with a conductive π-stacked array of N,N′-di(4-pyridyl)-1,4,5,8-naphthalenetetra-carboxdiimide (NDI-py) linkers. The NDI core is a well-known electron acceptor used in various research fields31 such as self-assembly,32 molecule sensors33 and molecular electronics,34 whereas its application in intrinsic electron conductors is still in the early stage of development.
Herein, we report an electrically conductive metallocycle synthesized by electrocrystallization for the first time. The coordination-driven self-assembly of hexagonal metallocycle units and the columnar self-assembly of π-conjugated organic linkers occurred together in the electrocrystallization process. In this work, pyrazole was selected as the terminal group of the organic linker because of their preferential construction of various bent coordination polymers35 and metallocycles.36
Results and discussion
Synthesis and discrete metallocycle structure
N,N′-Di(1H-pyrazol-4-yl)-1,4,5,8-naphthalenetetracarboxdi-imide (NDI-Hpz) and Ni(NO3)2·6H2O were dissolved in N,N-dimethylacetamide (DMA) with a small portion of H2O. Electrocrystallization was carried out by applying a constant direct current of 20 μA to the mixed solution at room temperature (RT). Needle-like tiny black crystals of [Ni6(NDI-Hpz)6(dma)12(NO3)6]·5DMA·nH2O (hereafter abbreviated as PMC-hexagon) were obtained from the cathode in 12 hours.Single-crystal X-ray diffraction (SXRD) analysis revealed that PMC-hexagon crystallized in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (Table S1†) with merohedral twinning. Remarkably, the hexagonal metallocycle structure [Ni6(NDI-Hpz)6(dma)12(NO3)6] constructed from head-to-tail connection between Ni2+ ions and NDI-Hpz linkers was elucidated as shown in Fig. 1a and b. A Ni2+ ion is coordinated by two pyrazole groups, two DMA molecules in cis geometry, and a bidentate NO3− ion. As a result, the Ni2+ ion forms nearly octahedral coordination geometry (Fig. 1c). The positions of Ni2+ centres and the centroids of NDI cores deviate from the mean plane of the hexagon (1.967 Å and 0.402 Å, respectively), resulting in a pucker of the 6-membered metallocycle (Fig. 1b). The slightly large coordination-bond angle (∠N–Ni–N = 95.9(3)°) and the flexible dihedral angles between the NDI core and each pyrazole plane (60.39° and 62.79°) enable the puckered hexagon structure. In addition, a non-coordinating DMA molecule is located between two pyrazole groups with N–H⋯O(
(Table S1†) with merohedral twinning. Remarkably, the hexagonal metallocycle structure [Ni6(NDI-Hpz)6(dma)12(NO3)6] constructed from head-to-tail connection between Ni2+ ions and NDI-Hpz linkers was elucidated as shown in Fig. 1a and b. A Ni2+ ion is coordinated by two pyrazole groups, two DMA molecules in cis geometry, and a bidentate NO3− ion. As a result, the Ni2+ ion forms nearly octahedral coordination geometry (Fig. 1c). The positions of Ni2+ centres and the centroids of NDI cores deviate from the mean plane of the hexagon (1.967 Å and 0.402 Å, respectively), resulting in a pucker of the 6-membered metallocycle (Fig. 1b). The slightly large coordination-bond angle (∠N–Ni–N = 95.9(3)°) and the flexible dihedral angles between the NDI core and each pyrazole plane (60.39° and 62.79°) enable the puckered hexagon structure. In addition, a non-coordinating DMA molecule is located between two pyrazole groups with N–H⋯O(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)⋯H–N hydrogen bonds (Fig. 1c). Since its occupancy is 84%, the total number of hydrogen-bonded DMA molecules is calculated to be five per metallocycle, in accord with the integration ratio in the 1H NMR spectrum (Fig. S7, ESI†).
C)⋯H–N hydrogen bonds (Fig. 1c). Since its occupancy is 84%, the total number of hydrogen-bonded DMA molecules is calculated to be five per metallocycle, in accord with the integration ratio in the 1H NMR spectrum (Fig. S7, ESI†).
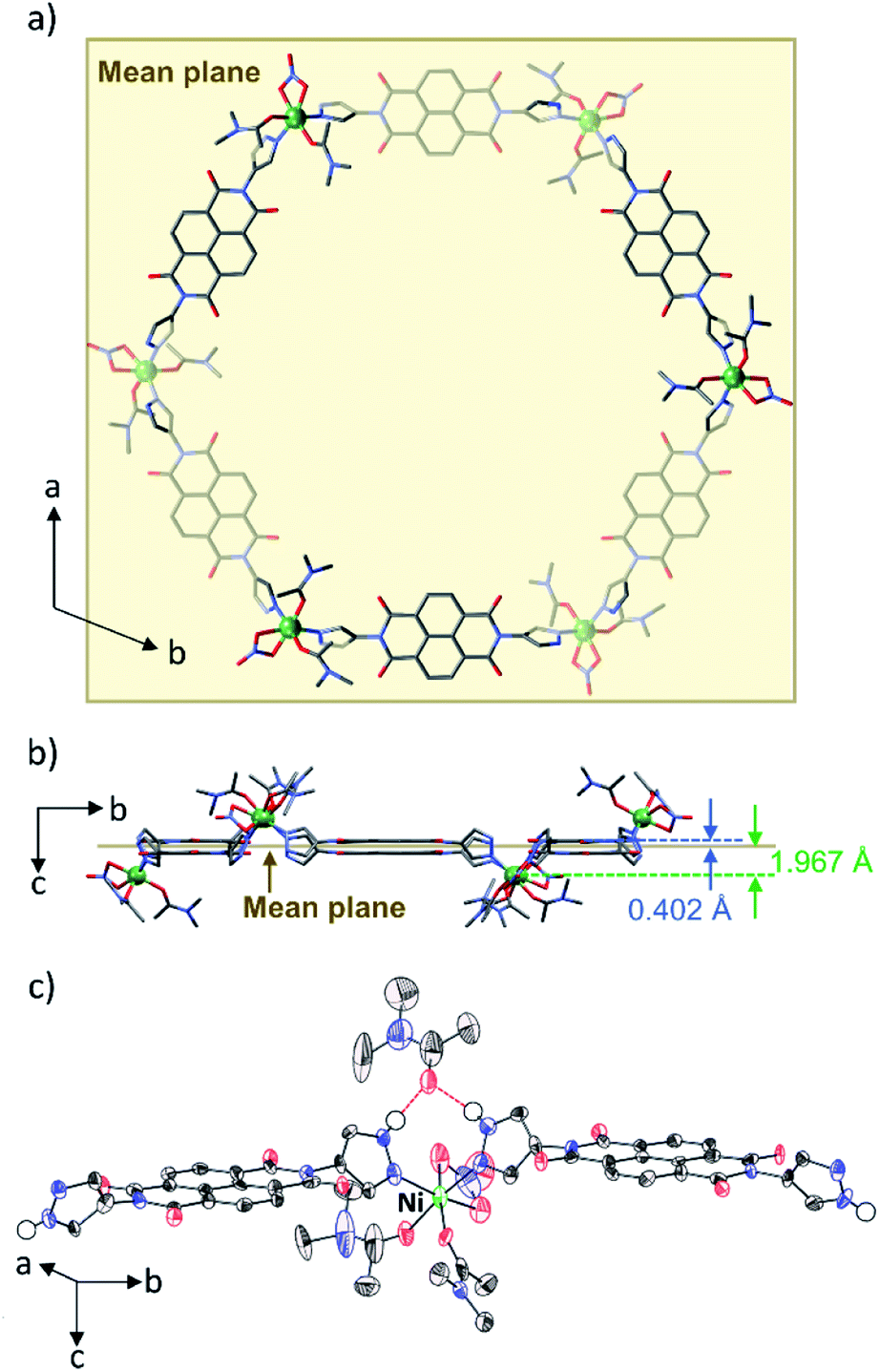 | ||
| Fig. 1 (a) Crystal structure of the hexagonal metallocycle in PMC-hexagon. The atoms under the mean plane of the hexagon are shaded. (b) Perspective view of the hexagonal metallocycle along the a axis showing the deviation of Ni2+ ions and the centroids of NDI cores from the mean plane of the hexagon. (c) Thermal ellipsoid plot of the ligands around Ni2+ ions including hydrogen-bonded DMA. Hydrogen bonds between the H atom of the pyrazole groups and the DMA are represented as red dashed lines. Other H atoms are omitted for clarity. Ni green, O red, N blue, and C black. | ||
Packing structure of the metallocycle
These discrete hexagonal metallocycles stack atop one another at the NDI cores. The crystal packing structure of PMC-hexagon (Fig. 2a) displays the hexagonal arrangement of the one-dimensional (1D) helical π-stacked columns and 1D pore channels extended along the crystallographic c axis. It is constructed from the dense packing of hexagonal metallocycles with an alternating sequence of ABCABC… (Fig. 2b). This stacking manner, which is similar to the face-centered cubic (fcc) geometry, has rarely been observed in the self-assembly of metallocycles.37 The helical stacking mode of NDI cores in 60° increments (Fig. 2c) plays a crucial role in overlapping the sides of hexagons and forming the ABCABC… sequence. Although the pyrazole group can form a variety of metallocycle-based polygons,36 hexagons were selectively crystallized probably because of their compatibility with the helical stacking mode mentioned above.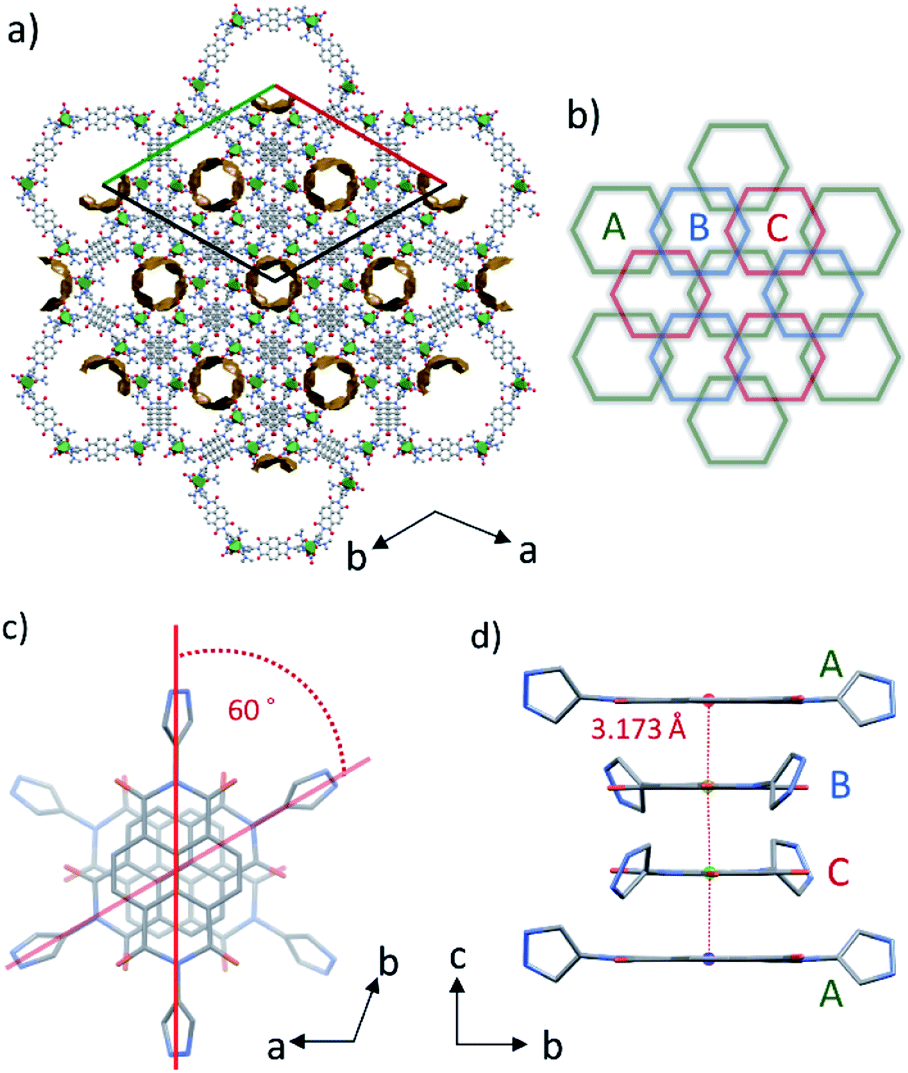 | ||
| Fig. 2 (a) Crystal packing structure along the c axis for PMC-hexagon. Coordination sphere around the Ni ion is represented as a green octahedron. The 1D voids of approximately 14.2% of the unit cell volume are mapped by the contact surface (yellow) of a probe with a radius of 1.4 Å. (b) Schematic illustration of the packing mode of hexagons densely stacked with the ABC… sequence. (c) Helical π-stacked column of NDI-Hpz projected along the c axis. (d) Side view of the 1D π-stacked column with the interplanar distance. | ||
The interplanar distance between adjacent NDI-Hpz ligands along the stacking axis is 3.173 Å (1/3 of the unit cell length c), which is shorter than those (>3.3 Å) between typical neutral NDI cores,32a,38 indicating the attractive interaction derived from NDI radicals (NDI˙−) (Fig. 2d).29,30 The transfer integral between the adjacent LUMO of NDI-Hpz linkers along the stacking axis was calculated to be 315 meV. It is significantly larger than those of neutral NDI-based organic semiconductors (typically less than 100 meV),39 reflecting the strong π-stacking interaction in PMC-hexagon.
The dense packing decreases their cavities, resulting in a small void space (14.2% of the total volume). On the basis of elemental analysis, the 1D pore channel is likely to accommodate H2O molecules (see the Experimental section), whereas their exact number was uncertain because the liberation of H2O molecules occurred even at RT (Fig. S1, ESI†). After heating PMC-hexagon at 120 °C for an hour, the powder X-ray diffraction (PXRD) pattern (Fig. 3) and the number of contained DMA molecules estimated from the integration ratio in the 1H NMR spectrum (Fig. S8, ESI†) were almost unchanged, though a weight loss was observed in thermogravimetry analysis (TGA). These results suggest that the crystal structure was maintained under the liberation of H2O molecules in the 1D pore channel. The heating of PMC-hexagon at 165 °C for an hour induced the liberation of DMA molecules and the deterioration, as suggested by the change of the 1H NMR spectrum (Fig. S9, ESI†) and the PXRD pattern (Fig. 3). Thus, it is difficult to obtain a desolvated state without deterioration in contrast to previously reported PMC with a similar π-stacked analog [Cd(NDI-py)(OH2)4](NO3)1.3±0.1·nDMA (PMC-1).29 However, it is notable that PMC-hexagon retained the crystal structure under the liberation of H2O molecules despite the lack of a coordination polymer network like MOFs. This robustness should originate from the dense packing supported by the strong π-stacking interaction.
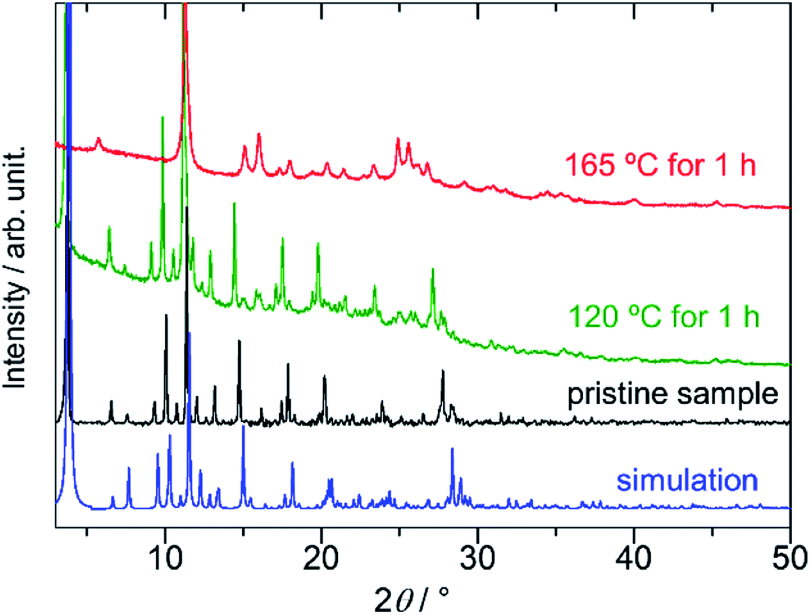 | ||
| Fig. 3 Powder X-ray diffraction (PXRD) patterns of PMC-hexagon (simulated from the crystal structure: blue, pristine sample: black, heated at 120 °C for an hour: green, and heated at 165 °C for an hour: red). | ||
Although the void volume of an individual 1D pore channel is comparable between PMC-hexagon and PMC-1 (approx. 820 Å3 par channel in the unit cell), PMC-hexagon was more air-sensitive than PMC-1, probably because the fast liberation of H2O molecules from the channel promotes the access of oxygen molecules to NDI˙− species. The nitrogen gas adsorption in the micropore of PMC-hexagon has not been observed (Fig. S10, ESI†), presumably because of the high-vacuum conditions in the activation process. The partial deterioration due to the liberation of DMA molecules probably occurred at least on the surface of the solid during the vacuuming and disturbed the insertion of nitrogen molecules into channels, as suggested by the PXRD pattern (Fig. S11, ESI†) and the 1H NMR spectrum (Fig. S12, ESI†).
Physical properties of PMC-hexagon
The existence of NDI˙− was confirmed by a sharp signal (g = 2.00354) in the electron spin resonance (ESR) spectrum (Fig. S2, ESI†). The contribution of NDI˙− species to the magnetic properties of PMC-hexagon is very small, because the Ni2+ ion has a larger spin magnetic moment (S = 1) and the antiferromagnetic interaction and/or partial dimerization along the π-stacked column decreases the total magnetic moment of NDI˙− species. We estimated the number of effective NDI˙− species in the range from 10 to 15% by fitting the temperature dependence of the χMT value (χM: molar spin susceptibility and T: temperature) with the aid of quantum calculations (see Fig. S3† and explanation in the ESI†).To investigate the electronic state in detail, the solid-state absorption spectra of PMC-hexagon and NDI-Hpz dispersed in KBr pellets were acquired. The whole process from the synthesis to the measurement was carried out under an argon atmosphere in order to avoid the air oxidation of radicals. Both spectra display strong absorption bands around 3.3 eV, which are assignable to intramolecular π–π* transition in neutral NDI (NDI0) cores (Fig. 4).29,30,40 Additional absorption bands were observed in the spectrum of PMC-hexagon as well as reported NDI˙− species. The strong band in the range of 2.5–2.8 eV and weak bands around 2.0 eV and 1.5 eV are ascribed to the intramolecular electron transition of NDI˙−.29,41,42 The weak broad band around 1 eV and the relatively strong band around 0.4 eV are ascribed to the charge transfer (CT) between two NDI˙− cores and that from NDI˙− to NDI0 cores, respectively.29,30,40,43 The existence of NDI0 species in PMC-hexagon is inconsistent with the formula, which indicates that the charge of NDI-Hpz is −1. Whilst the concentration of NDI0 in the crystal could not be determined with accessible analytical methods, we believe that partial deprotonation of the Hpz group satisfies charge neutrality in this system. The partial absence of the hydrogen-bonded DMA molecules (84% occupancy) may reflect the loss of hydrogen donors due to the partial deprotonation.
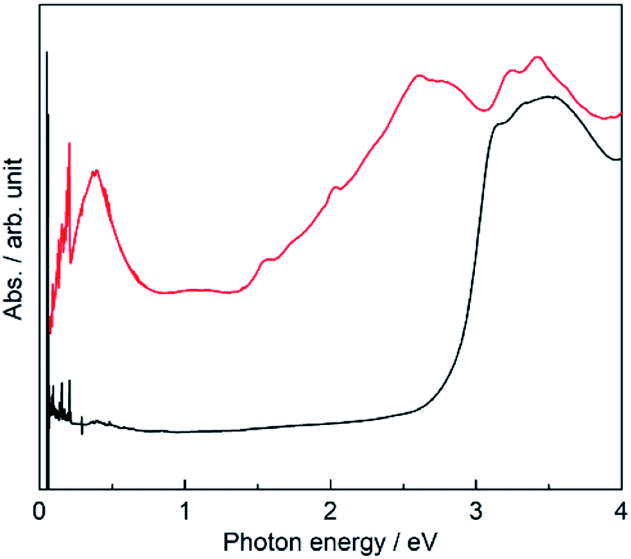 | ||
| Fig. 4 Solid-state absorption spectra of PMC-hexagon (red) and neutral NDI-Hpz (black) dispersed in KBr pellets. | ||
The low energy CT band suggests that PMC-hexagon is a narrow-gap semiconductor. Since the single crystals of PMC-hexagon were too small to measure the electrical conductivity (σ), the measurements were carried out on 3 mmϕ pressed pellets. The current–voltage (I–V) plots indicate the ohmic characteristic (V = RI, where R is the electrical resistance) as shown in Fig. 5a. The σ of PMC-hexagon at RT was calculated to be (1.2–2.1) × 10−4 S cm−1. It is 10 times higher than the σ of PMC-1, which was newly prepared and measured under the same inert conditions in this work (Fig. S4, ESI†). Note that the σ of PMC-1 in previous work ((1.5–7.6) × 10−6 S cm−1)29 was obtained under incompletely inert conditions and is lower than that obtained in the present work ((1.4–2.0) × 10−5 S cm−1 in pressed pellets), indicating that the avoidance of air oxidation is important to properly measure σ in NDI-based conductors. The π-stacked columns in PMC-hexagon are surrounded by Ni complexes and are away from 1D pore channels, freeing it from the perturbation of conduction carriers by disordered molecules in the pores. It may be one of the reasons for the 10-fold increase in conductivity compared to PMC-1. The temperature dependence of σ (Fig. 5b) shows the decrease in σ with decreasing temperature, namely semiconducting behavior. The activation energy (Ea) is determined to be 0.148 eV by fitting the data with the Arrhenius equation σ = σ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/kT), where σ0 is the prefactor, k is the Boltzmann constant and T is the temperature. The Ea of PMC-hexagon is smaller than that in PMC-1 (0.25 eV),29 supporting the highly conducting nature of PMC-hexagon. To date, the reported σ of NDI-based conductive MOFs/coordination polymers29,44 and organic crystals45 have been in the range from 10−7 to 2.0 × 10−5 S cm−1 for pressed pellets and up to 3.3 × 10−3 S cm−1 for single crystals. The σ of a pressed pellet of 1D conductors is typically smaller than that of the single crystal by two to three orders of magnitude. Therefore, PMC-hexagon is one of the most conductive NDI-based crystals. Although the σ of PMC-hexagon is lower than the record values for conductive MOFs with through-space conduction pathways (10−4 to 0.05 S cm−1 for lanthanide-hexahydroxytriphenylene-based MOFs),46 it is higher than that of other π-stacked MOFs (e.g. 10−9 to 7.6 × 10−5 S cm−1 (pressed pellet) for MOFs with a TTF moiety47). Moreover, the high σ (above 0.1 S cm−1) observed in some NDI-based polymers48 implies that we have much room to improve the conductivity of NDI-based metallocycles, which can be an alternative building block to construct conductive porous molecular crystals.
exp(−Ea/kT), where σ0 is the prefactor, k is the Boltzmann constant and T is the temperature. The Ea of PMC-hexagon is smaller than that in PMC-1 (0.25 eV),29 supporting the highly conducting nature of PMC-hexagon. To date, the reported σ of NDI-based conductive MOFs/coordination polymers29,44 and organic crystals45 have been in the range from 10−7 to 2.0 × 10−5 S cm−1 for pressed pellets and up to 3.3 × 10−3 S cm−1 for single crystals. The σ of a pressed pellet of 1D conductors is typically smaller than that of the single crystal by two to three orders of magnitude. Therefore, PMC-hexagon is one of the most conductive NDI-based crystals. Although the σ of PMC-hexagon is lower than the record values for conductive MOFs with through-space conduction pathways (10−4 to 0.05 S cm−1 for lanthanide-hexahydroxytriphenylene-based MOFs),46 it is higher than that of other π-stacked MOFs (e.g. 10−9 to 7.6 × 10−5 S cm−1 (pressed pellet) for MOFs with a TTF moiety47). Moreover, the high σ (above 0.1 S cm−1) observed in some NDI-based polymers48 implies that we have much room to improve the conductivity of NDI-based metallocycles, which can be an alternative building block to construct conductive porous molecular crystals.
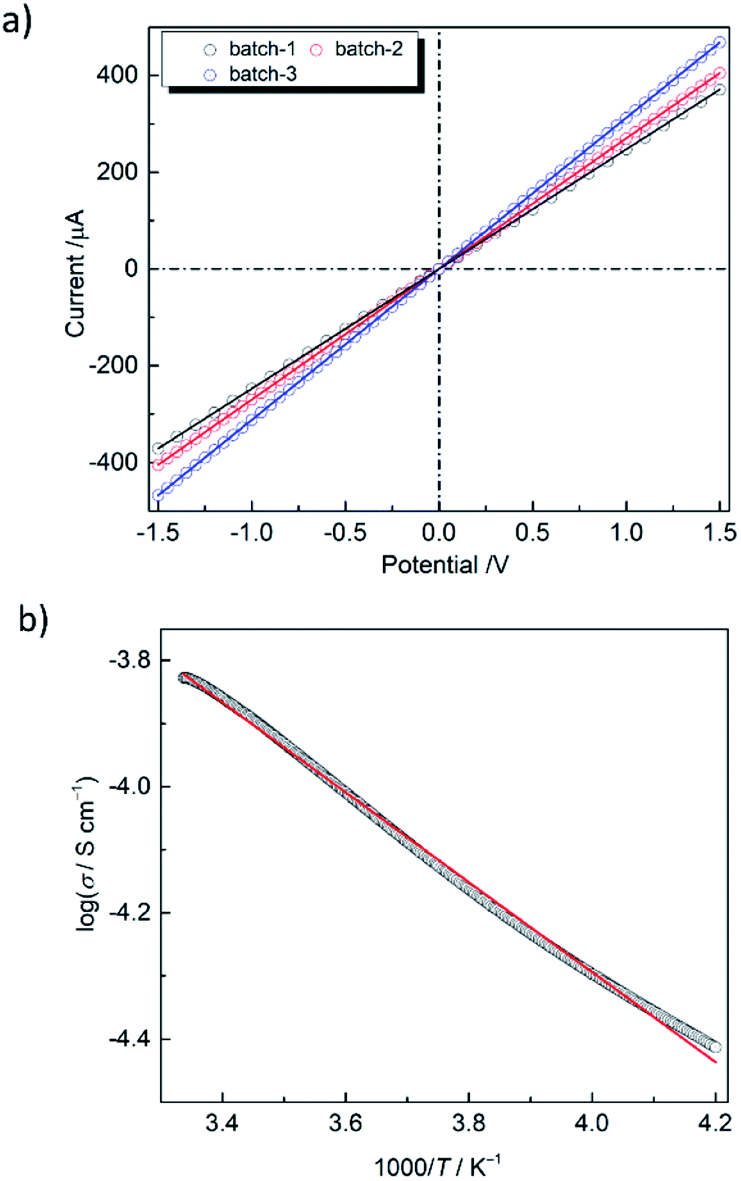 | ||
| Fig. 5 (a) Current–voltage (I–V) characteristics of PMC-hexagon in three different pellets at RT. (b) Temperature dependence of the electrical conductivity (σ) of PMC-hexagon fitted with the Arrhenius equation (red line). | ||
Conclusions
Intrinsically electron-conductive metallocycle PMC-hexagon was synthesized by using electrocrystallization. The hexagonal metallocycles were densely π-stacked in a face-centered cubic (fcc)-like geometry (ABCABC… sequence) to afford 1D helical π-stacked conductive columns and 1D pore channels. The radical at the NDI core (NDI˙−) provides quite a short interplanar distance (3.173 Å) and conducting carriers, resulting in a high electrical conductivity ((1.2–2.1) × 10−4 S cm−1 at RT) even in the polycrystalline pellet sample. Despite the lack of a coordination polymer network, the dense packing of hexagonal metallocycles provides a robust structure, which was maintained under the liberation of H2O molecules in the pore channels. The elimination of DMA molecules induced the deterioration and disturbed the nitrogen adsorption to pore channels. Although the improvement of the robustness and the conductivity remains to be addressed, the concept of using a metallocycle as a building block provides a new platform for developing electrically conductive porous molecular materials.Experimental
Materials and characterization
All purchased chemicals were used without further purification. 1H NMR spectra were recorded on a Bruker AVANCE 500 spectrometer at RT. 1H NMR chemical shifts were referenced internally to residual solvent resonances. Deuterated solvents were obtained from Wako and used as received. Electrocrystallization was performed using a direct current (DC) multisource, YAZAWA CS-12Z, and 0.3 mm electrodes of platinum-iridium alloy wires (Pt:Ir = 80:20). All elemental analyses were performed at a J-SCIENCE LAB, JM-11 at the Research and Analytical Centre, Tohoku University. Japan.
Synthetic procedures
Data availability
Detailed data including general information, characterization, crystallographic parameters, magnetism, electrical conductivity and computational methods are available in the ESI.†Author contributions
H. I. conceived and designed the project. M. C. carried out the synthesis, characterization, crystal structure analysis and measuring of the physical properties of PMC-hexagon. R. M. contributed to the design of the experiment, syntheses of ligands and PMC-hexagon, and preliminary crystal structure analysis. Y. S. contributed to the measurement and analysis of electrical conductivity. T. S. and K. U contributed to the magnetic measurements and the analyses using calculation and fitting. K. U. also contributed to the nitrogen adsorption measurement. S. K. performed the theoretical calculation of the transfer integral. T. T. contributed to measurements and characterization related to nitrogen sorption isotherms and the thermogravimetry analysis. S. T., M. Y. and H. I. supervised this work and carried out discussions. M. C. and H. I. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from Prof Akutagawa and Dr Hoshino at Tohoku University for acquiring the ESR spectra. We also thank Prof Iwamoto and Dr Ishida at Tohoku University for the preliminary SXRD analysis. This work was partly supported by JSPS KAKENHI Grant Numbers JP18H04498 (H. I.), JP19H05631 (H. I., S. T. and M. Y.), JP21H01901 (H. I.) and JP21K18971 (H. I.), by the Toyota Riken Scholar Program (H. I.), by the Kato Foundation for Promotion of Science KJ-2916 (H. I.), and by the Iketani Science and Technology Foundation 0331101-A (H. I.). M. Y. acknowledges the support from the 111 project (B18030) from China. M. C. gratefully acknowledges the financial support from the Chinese Scholarship Council (CSC).Notes and references
-
(a) K. Y. Cheung, Y. Segawa and K. Itami, Chem.–Eur. J., 2020, 26, 14791–14801 CrossRef CAS PubMed
; (b) D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed
-
(a) M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS
- S. Kawano, T. Fukushima and K. Tanaka, Angew. Chem., Int. Ed., 2018, 57, 14827–14831 CrossRef CAS PubMed
- R. Miyake, A. Ando, M. Ueno and T. Muraoka, J. Am. Chem. Soc., 2019, 141, 8675–8679 CrossRef CAS PubMed
- G.-Y. Wu, L.-J. Chen, L. Xu, X.-L. Zhao and H.-B. Yang, Coord. Chem. Rev., 2018, 369, 39–75 CrossRef CAS
- Y. Sakata, R. Yamamoto, D. Saito, Y. Tamura, K. Maruyama, T. Ogoshi and S. Akine, Inorg. Chem., 2018, 57, 15500–15506 CrossRef CAS PubMed
- H. Sepehrpour, W. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC
- M. Yamashina, Y. Tanaka, R. Lavendomme, T. K. Ronson, M. Pittelkow and J. R. Nitschke, Nature, 2019, 574, 511–515 CrossRef CAS PubMed
- J. Wu, L. Zhao, L. Zhang, X.-L. Li, M. Guo, A. K. Powell and J. Tang, Angew. Chem., Int. Ed., 2016, 55, 15574–15578 CrossRef CAS PubMed
- G. Li, W. Yu and Y. Cui, J. Am. Chem. Soc., 2008, 130, 4582–4583 CrossRef CAS PubMed
- L. Zhang, L. Lin, D. Liu, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2017, 139, 1653–1660 CrossRef CAS PubMed
- Z. Y. Chen, B. Sahli and M. J. MacLachlan, Inorg. Chem., 2017, 56, 5383–5391 CrossRef CAS PubMed
- K. Otake, K. Otsubo, T. Komatsu, S. Dekura, J. M. Taylor, R. Ikeda, K. Sugimoto, A. Fujiwara, C.-P. Chou, A. W. Sakti, Y. Nishimura, H. Nakai and H. Kitagawa, Nat. Commun., 2020, 11, 843 CrossRef CAS PubMed
- A. A. Adeyemo, A. Shettar, I. A. Bhat, P. Kondaiah and P. S. Mukherjee, Inorg. Chem., 2017, 56, 608–617 CrossRef PubMed
- W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625–631 RSC
- J. Dong, C. Tang, K. Zhang, Y. Liu, P. Low, J. Jiang and Y. Cui, J. Am. Chem. Soc., 2017, 139, 1557–1564 Search PubMed
- B. Shi, Z. Zhou, R. T. Vanderlinden, J.-H. Tang, G. Yu, K. Acharyya, H. Sepehrpour and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 11837–11841 CrossRef CAS PubMed
- L. Chen, C. Chen, Y. Sun, S. Lu, H. Huo, T. Tan, A. Li, X. Li, G. Ungar, F. Liu and M. Zhang, Angew. Chem., Int. Ed., 2020, 59, 10143–10150 CrossRef CAS PubMed
- T. Hong, Z. Zhang, Y. Sun, J.-J. Tao, J.-D. Tang, C. Xie, M. Wang, F. Chen, S.-S. Xie, S. Li and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 10244–10249 CrossRef CAS PubMed
- D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 101, 1313–1317 CrossRef PubMed
-
(a) J. Wu, X.-L. Li, M. Guo, L. Zhao, Y.-Q. Zhang and J. Tang, Chem. Commun., 2018, 54, 1065–1068 RSC
- M. El Sayed Moussa, S. Evariste, H.-L. Wong, L. Le Bras, C. Roiland, L. Le Polles, B. Le Guennic, K. Costuas, V. W.-W. Yam and C. Lescop, Chem. Commun., 2016, 52, 11370–11373 RSC
-
(a) S. Tashiro and M. Shionoya, Acc. Chem. Res., 2020, 53, 632–643 CrossRef CAS PubMed
-
(a) T. Pietrass, I. Cruz-Campa, J. Kombarakkaran, S. Sirimulla, A. M. Arif and J. C. Noveron, J. Phys. Chem. C, 2010, 114, 21371–21377 CrossRef CAS
-
(a) L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8858 CrossRef CAS PubMed
- H. Maeda, R. Akuta, Y. Bando, K. Takaishi, M. Uchiyama, A. Muranaka, N. Tohnai and S. Seki, Chem.–Eur. J., 2013, 19, 11676–11685 CrossRef CAS PubMed
-
(a) H. Anzai, J. M. Delrieu, S. Takasaki, S. Nakatsuji and J. Yamada, J. Cryst. Growth, 1995, 154, 145–150 CrossRef CAS
- L. Qu, H. Iguchi, S. Takaishi, F. Habib, C. F. Leong, D. M. D'Alessandro, T. Yoshida, H. Abe, E. Nishibori and M. Yamashita, J. Am. Chem. Soc., 2019, 141, 6802–6806 CrossRef CAS PubMed
- S. Koyama, T. Tanabe, S. Takaishi, M. Yamashita and H. Iguchi, Chem. Commun., 2020, 56, 13109–13112 RSC
-
(a) M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed
-
(a) S. K. Keshri, T. Ishizuka, T. Kojima, Y. Matsushita and M. Takeuchi, J. Am. Chem. Soc., 2021, 143, 3238–3244 CrossRef CAS PubMed
-
(a) E. Takahashi, H. Takaya and T. Naota, Chem.–Eur. J., 2010, 16, 4793–4802 CrossRef CAS PubMed
-
(a) H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed
- K. V. Domasevitch, I. Boldog, E. B. Rusanov, J. Hunger, S. Blaurock, M. Schröder and J. Z. Sieler, Z. Anorg. Allg. Chem., 2005, 631, 1095–1100 CrossRef CAS
-
(a) M. A. Halcrow, Dalton Trans., 2009, 12, 2059–2073 RSC
- E. Coronado, J. R. Galan-Mascaros, P. Gaviña, C. Martí-Gastaldo, F. M. Romero and S. Tatay, Inorg. Chem., 2008, 47, 5197–5203 CrossRef CAS PubMed
- Y. Matsunaga, K. Goto, K. Kubono, K. Sako and T. Shinmyozu, Chem.–Eur. J., 2014, 20, 7309–7316 CrossRef CAS PubMed
-
(a) T. Kakinuma, H. Kojima, M. Ashizawa, H. Matsumoto and T. Mori, J. Mater. Chem. C, 2013, 1, 5395–5401 RSC
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC
- C. F. Leong, B. Chan, T. B. Faust and D. M. D'Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS
- A. Takai, T. Yasuda, T. Ishizuka, T. Kojima and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 9167–9171 CrossRef CAS PubMed
-
(a) X. Kuang, S. Chen, L. Meng, J. Chen, X. Wu, G. Zhang, G. Zhong, T. Hu, Y. Li and C.-Z. Lu, Chem. Commun., 2019, 55, 1643–1646 RSC
-
(a) G. Heywang, L. Born, H.-G. Fitzky, T. Hassel, J. Hocker, H.-K. Müller, B. Pittel and S. Roth, Angew. Chem., Int. Ed. Engl., 1989, 28, 483–485 CrossRef
- G. Skorupskii, B. A. Trump, T. W. Kasel, C. M. Brown, C. H. Hendon and M. Dincă, Nat. Chem., 2020, 12, 131–136 CrossRef CAS PubMed
-
(a) L. Sun, S. S. Park, D. Sheberla and M. Dincă, J. Am. Chem. Soc., 2016, 138, 14772–14782 CrossRef CAS PubMed
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. A. Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: general information, characterization, crystallographic parameters, magnetism, electrical conductivity and computational method. CCDC 2091485 (PMC-hexagon). CCDC 2091485. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00447j |
| ‡ Present address: Department of Materials Chemistry, Graduate school of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: E-mail: hiroaki.iguchi@chembio.nagoya-u.ac.jp |
| This journal is © The Royal Society of Chemistry 2022 |