
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon–chalcogen bond formation initiated by [Al(NONDipp)(E)]− anions containing Al–E{16} (E{16} = S, Se) multiple bonds†
Matthew J.
Evans
 a,
Mathew D.
Anker
*a,
Claire L.
McMullin
*b,
Samuel E.
Neale
b,
Nasir A.
Rajabi
b and
Martyn P.
Coles
*a
a,
Mathew D.
Anker
*a,
Claire L.
McMullin
*b,
Samuel E.
Neale
b,
Nasir A.
Rajabi
b and
Martyn P.
Coles
*a
aSchool of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Wellington, New Zealand. E-mail: martyn.coles@vuw.ac.nz
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
First published on 31st March 2022
Abstract
Multiply-bonded main group metal compounds are of interest as a new class of reactive species able to activate and functionalize a wide range of substrates. The aluminium sulfido compound K[Al(NONDipp)(S)] (NONDipp = [O(SiMe2NDipp)2]2−, Dipp = 2,6-iPr2C6H3), completing the series of [Al(NONDipp)(E)]− anions containing Al–E{16} multiple bonds (E{16} = O, S, Se, Te), was accessed via desulfurisation of K[Al(NONDipp)(S4)] using triphenylphosphane. The crystal structure showed a tetrameric aggregate joined by multiple K⋯S and K⋯π(arene) interactions that were disrupted by the addition of 2.2.2-cryptand to form the separated ion pair, [K(2.2.2-crypt)][Al(NONDipp)(S)]. Analysis of the anion using density functional theory (DFT) confirmed multiple-bond character in the Al–S group. The reaction of the sulfido and selenido anions K[Al(NONDipp)(E)] (E = S, Se) with CO2 afforded K[Al(NONDipp)(κ2E,O-EC{O}O)] containing the thio- and seleno-carbonate groups respectively, consistent with a [2 + 2]-cycloaddition reaction and C–E bond formation. An analogous cycloaddition reaction took place with benzophenone affording compounds containing the diphenylsulfido- and diphenylselenido-methanolate ligands, [κ2E,O-EC{O}Ph2]2−. In contrast, when K[Al(NONDipp)(E)] (E = S, Se) was reacted with benzaldehyde, two equivalents of substrate were incorporated into the product accompanied by formation of a second C–E bond and complete cleavage of the Al–E{16} bonds. The products contained the hitherto unknown κ2O,O-thio- and κ2O,O-seleno-bis(phenylmethanolate) ligands, which were exclusively isolated as the cis-stereoisomers. The mechanisms of these cycloaddition reactions were investigated using DFT methods.
Introduction
Progress in the synthesis and reactivity of main group compounds continues to legitimise comparisons between the chemistry of the p- and d-block metals.1,2 Indeed, several classes of compound that were once believed to be exclusive to transition metals have now been developed for the main group elements. Most pertinent to this study is the formation of multiple bonds involving main group metals,3,4 once considered to be ‘forbidden’ according to the double bond rule.5 Furthermore there has been an important shift in the focus of main group research away from being directed solely at the isolation of challenging synthetic targets of academic interest, and towards exploiting the reactivity of these systems in new chemical applications.6For the earth abundant metal aluminium, there are several historic reports of compounds containing Al–E{n} bonds that may be considered as including a multiple-bond component. For example, the highly anionic Zintl anions [Al2(E{15})2]6− (E{15} = P, As) contain short Al–P and Al–As terminal bonds that correspond to a Pauling bond order of 1.5.7 Power and co-workers reported the alumoxane ‘Al(Mes*)O’ (Mes* = 2,4,6-tBu3C6H2) that crystallizes as the tetramer with bridging Al–(μ-O)–Al groups.8 Related three-coordinate aluminum, gallium and indium thiolate and selenolate compounds were shown to contain short E{13}–E{16} bonds, in which a degree of π-bonding is likely.9–11
More recently, well defined examples of Al–E{n} multiple bonds have been recorded with elements from groups n = 13, 15 and 16.12,13 For Al–E{13} multiple bonds, examples of charged species containing homometallic dialuminium compounds (E{13} = Al), with an Al–Al bond order >1 have been known for almost 30 years.14–17 This pioneering work was complimented by examples of neutral compounds of ‘masked’ dialumenes that were isolated as the products of cycloaddition with aromatic solvents.18,19 This area has been expanded to include neutral dialumenes containing isolated Al–Al multiple bonds that have been structurally characterized in the solid-state.20,21 Several examples have now been synthesized and shown to activate small molecules including alkenes, alkynes, CO2 and O2.22,23
Multiply-bonded Al–E{15} species for E{15} = N have been generated transiently and identified from their products of dimerization,24 cyclization,25,26 and ligand activation.27 The first iminoalane was isolated in 2001,28 although it was not until 2012 that the first structurally characterized example of an Al–N multiple bond was reported.29 This general class of compound containing Al–E{15} multiple bonds has recently been extended to include a formal Al–N triple bond,30 and the first examples of phospha- and arsa-alumenes, with multiple Al–P and Al–As bonds, respectively.31
During the development of low-valent Al(I) chemistry, sporadic reports of compounds containing unsaturated Al–E{16} bonds (E = O,32 S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and Te34) were presented in the literature (I–III, Fig. 1).35 Despite being known for nearly two decades, inherent difficulties in the synthesis of these compounds has limited studies of these novel functional groups, and the reactivity of these bonds has remained largely unexplored.
33 and Te34) were presented in the literature (I–III, Fig. 1).35 Despite being known for nearly two decades, inherent difficulties in the synthesis of these compounds has limited studies of these novel functional groups, and the reactivity of these bonds has remained largely unexplored.
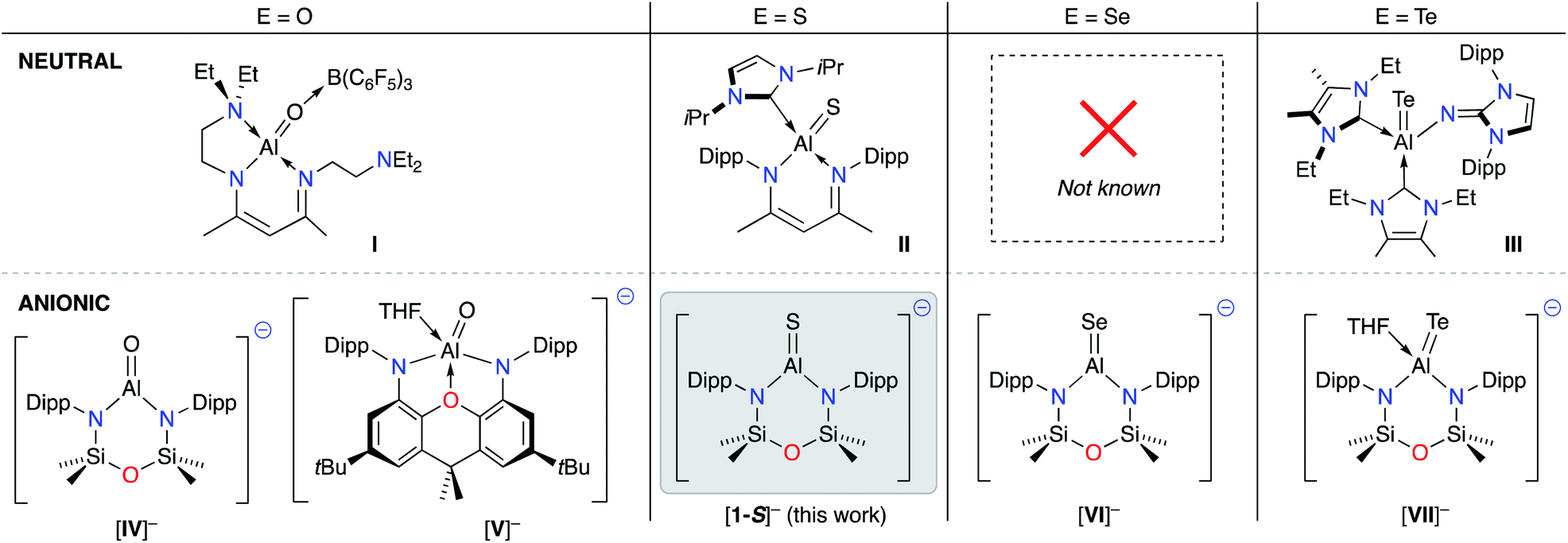 | ||
| Fig. 1 Current family of structurally characterised neutral (top) and anionic (bottom) aluminium species containing Al–E{16} multiple bonds. E{16} = chalcogen atom. | ||
In 2018, a new class of anionic Al(I) complex (the aluminyl anion) was reported,36 consisting of a three-coordinate aluminium centre supported by a dianionic, diamido(xanthene) framework. Soon afterwards we demonstrated that the NONDipp–ligand (NONDipp = [O(SiMe2NDipp)2]2−, Dipp = 2,6-iPr2C6H3), which had been successfully used in low valent bismuth37 and indium38 chemistry, was able to support the two-coordinate aluminyl anion, [K{Al(NONDipp)}]2 ([K{A}]2).39 These aluminyl species are reactive towards a number of substrates,40 and have provided a convenient entry point for the reliable syntheses of new Al–E{n} multiple bonds (n = 15, 16). To date, examples include aluminium imides (E = NR),41,42 molecular aluminium oxides (Fig. 1. E = O, K[IV]43 and K[V]44), and the terminal selenide (E = Se, K[VI])45 and telluride (E = Te, K[VII]),46 considered as heavy analogues of carbonyl groups.47
The facile access to Al–E{n} multiple bonds derived from aluminyl systems has allowed their physical and chemical properties to be assessed for the first time (Scheme 1). Studies have shown that the reactivity is dominated by cycloaddition chemistry, a pathway that was noted in the initial report of sulfido compound II that reacted with PhN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS to form the N-phenyl dithiocarbamate.33 The highly polar Al–O bonds in the anions [IV]− and [V]− show chemical reactivity consistent with ‘AlO’ character, evident from their reactions with CO2 and N2O to afford the carbonate and cis-hyponitrite ligands, respectively.43,44 An analogous reaction between [K{A}]2 and CS2 afforded the trithiocarbonate salt K[Al(NONDipp)(CS3)], postulated to proceed through [2 + 2] cycloaddition of CS2 to an intermediate (non-isolated) terminal aluminium sulfide bond.48 Cycloaddition reactivity was also observed when the aluminium telluride K[VII] was reacted with carbon dioxide, although in this case the tellurodicarbonate, [K{Al(NONDipp)}({OC(O)}2Te)] was isolated as a product of the ‘double insertion’ of CO2.46 We have also reported that the bimetallic oxide {K[IV]}2 reacts with carbon monoxide to afford a novel ethylenetetraolate ligand, [C2O4]4−, shown computationally to proceed via dimerization of an intermediate aluminium dioxocarbene.48
CS to form the N-phenyl dithiocarbamate.33 The highly polar Al–O bonds in the anions [IV]− and [V]− show chemical reactivity consistent with ‘AlO’ character, evident from their reactions with CO2 and N2O to afford the carbonate and cis-hyponitrite ligands, respectively.43,44 An analogous reaction between [K{A}]2 and CS2 afforded the trithiocarbonate salt K[Al(NONDipp)(CS3)], postulated to proceed through [2 + 2] cycloaddition of CS2 to an intermediate (non-isolated) terminal aluminium sulfide bond.48 Cycloaddition reactivity was also observed when the aluminium telluride K[VII] was reacted with carbon dioxide, although in this case the tellurodicarbonate, [K{Al(NONDipp)}({OC(O)}2Te)] was isolated as a product of the ‘double insertion’ of CO2.46 We have also reported that the bimetallic oxide {K[IV]}2 reacts with carbon monoxide to afford a novel ethylenetetraolate ligand, [C2O4]4−, shown computationally to proceed via dimerization of an intermediate aluminium dioxocarbene.48
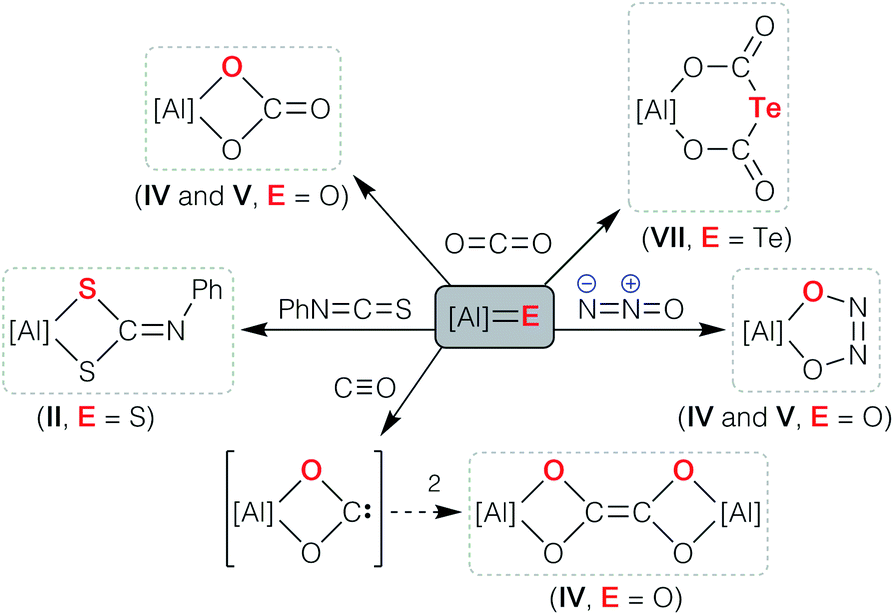 | ||
| Scheme 1 Cycloaddition reactions of Al–E{16} multiply bonded compounds (E{16} = O, S, Se, Te). The indices II, IV, V and VII refer [Al] to the parent compounds in Fig. 1. | ||
Our goal in this research is the development of new reagents of synthetic utility to the chemical community. The reproducible syntheses of K[IV], K[VI] and K[VII] allows us to conduct the first studies towards the application of Al–E{16} multiple bonds in the promotion of chemical reactions. The incorporation of element-chalcogen bonds into new molecular frameworks is of importance in several key areas, including the synthesis of pharmaceutically active molecules49,50 and materials science.51,52 We anticipate that the [Al(NONDipp)(E)]− anions will provide reactive, inorganic sources of chalcogen atoms that avoid the use of highly odoriferous and volatile species that are currently widely used for this reaction (e.g. H2E).
We report herein the isolation of the [Al(NONDipp)(S)]− anion, being the final member of the homologous series of anions containing Al–E{16} multiple bonds (E{16} = O, S, Se and Te). Alongside the previously reported selenide anion [Al(NONDipp)(Se)]−,45 we have examined the reactivity of these species with unsaturated organic substrates CO2, RC(H)O and R2CO, demonstrating the facile formation of one or more C–E bonds in the resulting products.
Results and discussion
Synthesis of K[Al(NONDipp)(S)]
The heavier selenido (K[VI]) and tellurido (K[VII]) derivatives may be obtained directly from the reaction of the potassium aluminyl [K{Al(NONDipp)}]2 ([K{A}]2) with the elemental chalcogen. However, we have previously reported that [K{A}]2 reacts with S8 to generate the spirocyclic 1,2,3,4,5-tetrathiaaluminium(III) salt, K[Al(NONDipp)(S4)] (K[B]).48 This result is obtained irrespective of the stoichiometry or the conditions of the reaction. Attempts at a more controlled delivery of sulfur using a stoichiometric amount of Ph3PS were also unsuccessful, with no reaction observed even under forcing conditions (100 °C, prolonged reaction times). Our approach to the target sulfido complex therefore involved the desulfurisation of K[B] using Ph3P (Scheme 2), a protocol that is effective for the isolation of GeS,53,54 and SnS55 bonds, and which has been recently used in the desulfurisation of (IMe4)Al(TippTer)(S5) (IMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene, TippTer = 2,6-(2,4,6-iPr3C6H2)2C6H3) to afford the tetrathiaaluminum complex, (IMe4)Al(TippTer)(S4).56
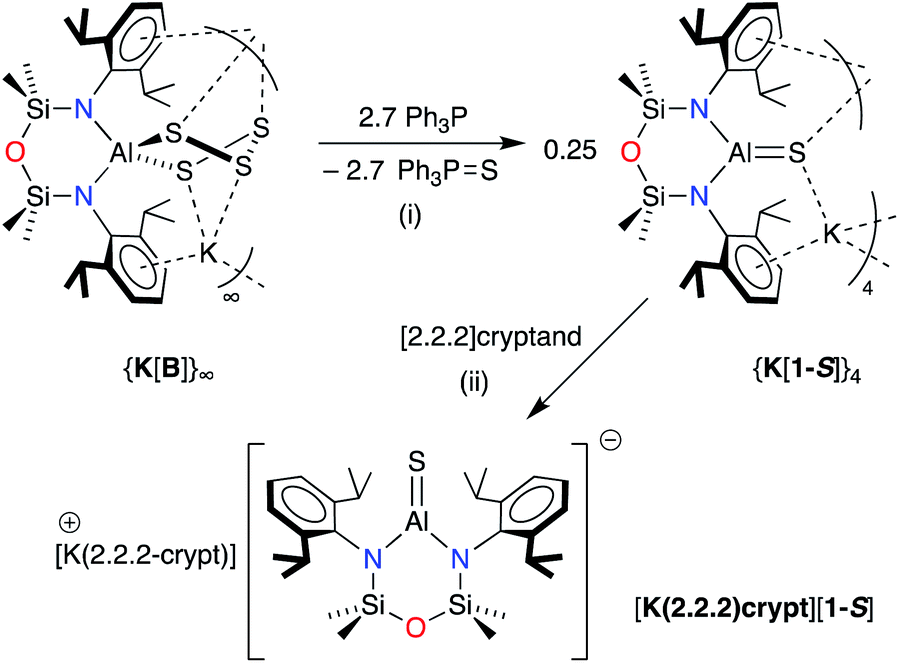 | ||
| Scheme 2 Synthesis of K[Al(NONDipp)(S)] (K[1-S]) and [K(2.2.2-crypt)][Al(NONDipp)(S)] ([K(2.2.2)crypt][1-S]). (i) THF solvent, crystallised from hexane; (ii) toluene solvent, crystallised from THF. | ||
The reaction between K[B] and PPh3 afforded the terminal sulfido compound, K[Al(NONDipp)(S)], (K[1-S], Scheme 2). Separation of the product from Ph3PS by conventional fractional crystallisation protocols was problematic due to their similar solubilities. The optimum conditions therefore involved reaction of K[B] with a sub-stoichiometric amount of PPh3 (2.7 equivalents), with separation of the product from unreacted K[B] achieved by extraction into hexane. The 1H and 13C{1H} NMR spectra of isolated crystals of K[1-S] show small changes in the chemical shifts of the NONDipp resonances (cf. [A]− and [B]−) but are otherwise uninformative.
X-ray quality crystals of K[1-S] were obtained by the slow evaporation of a hexane solution at room temperature. The compound crystallises as the non-symmetry related tetramer, {K[1-S]}4. Multiple K⋯S and K⋯π(arene) interactions are present that form a K4S4-core with a folded ladder motif (Fig. 2 and Table 1). This is in contrast to the molecular structures of [K(THF)][VI] and K[VII(THF)] that were crystallised from THF as 1-D polymeric chains [{K(THF)}{Al(NONDipp)(Se)}]∞ and [K{Al(NONDipp)(Te)(THF)}]∞, linked through K⋯Se and K⋯Te contacts.45,46 The aluminium centres in {K[1-S]}4 are three-coordinate distorted trigonal planar with Al–S bonds in the range 2.0857(7) Å–2.1039(7) Å. These distances are comparable with the Al–S multiple bond found in II (2.104(1) Å),33 and are similar to those observed in [Al(BDIDipp)(SH)(S)]− anions (Al–Sterminal = 2.116(1) Å and 2.115(1) Å).57 They are also in good agreement with the predicted value for the sum of the molecular covalent double bond radii, r2 (Σ(r2) = 2.07 Å).58
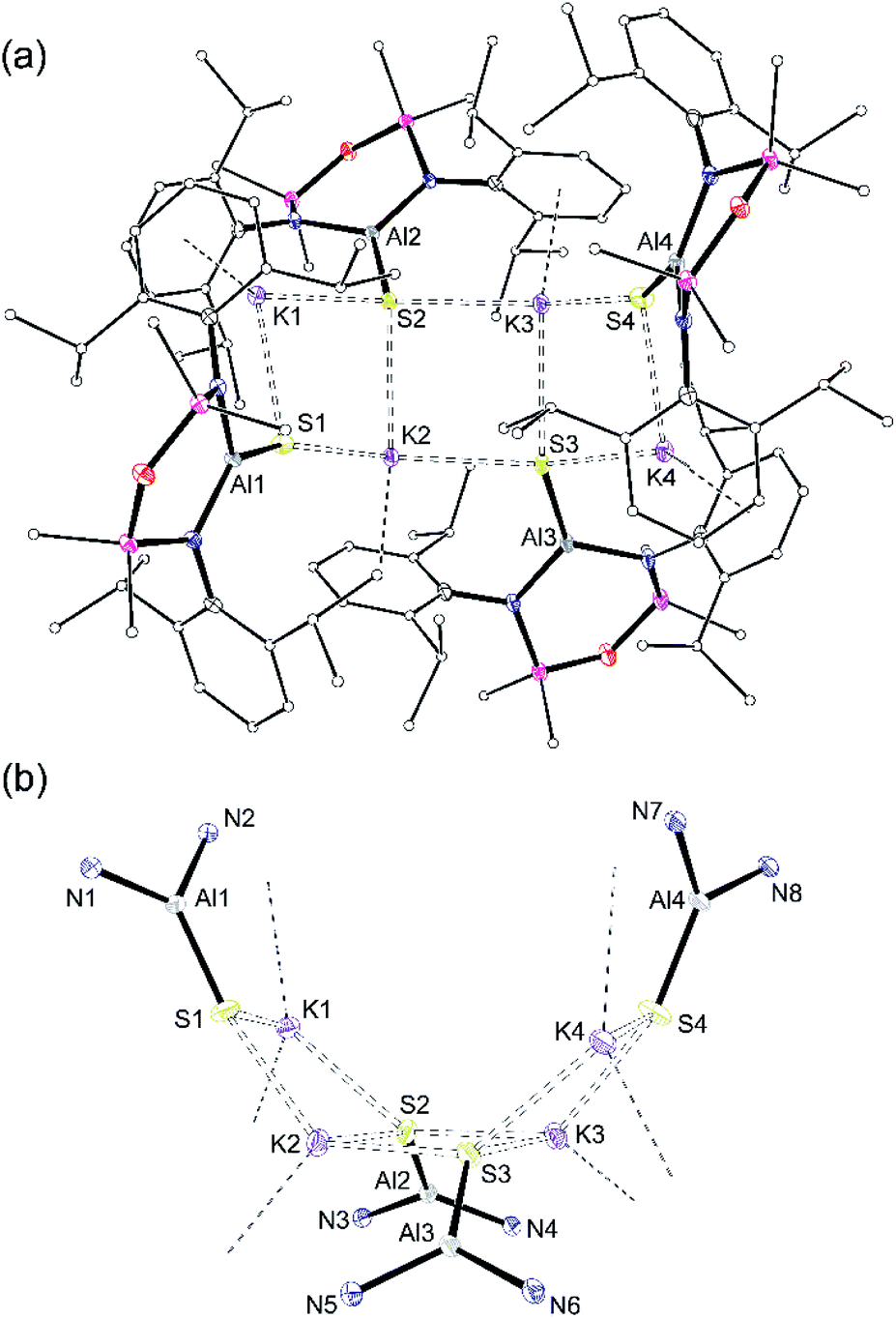 | ||
| Fig. 2 (a) Displacement ellipsoid plot (30%) of [K{Al(NONDipp)(S)}]4, {K[1-S]}4. H-atoms omitted; C-atoms reduced for clarity; (b) view illustrating the ‘S4K4’ core. | ||
| K[1-S]_(i) | K[1-S]_(ii) | K[1-S]_(iii) | K[1-S]_(iv) | |
|---|---|---|---|---|
| Al–S | 2.0873(7) | 2.0957(7) | 2.1039(7) | 2.0857(7) |
| Al–N | 1.8182(15) | 1.8234(15) | 1.8158(16) | 1.8304(16) |
| 1.8154(7) | 1.8175(15) | 1.8113(16) | 1.8170(15) | |
| S⋯K (range) | 3.0191(6)–3.0753(6) | 3.1187(6)–3.2043(6) | 3.0803(6)–3.2778(7) | 3.0247(6)–3.0521(7) |
| N–Al–N | 108.80(7) | 109.65(7) | 110.00(8) | 108.90(7) |
| N–Al–S | 128.69(6) | 127.17(5) | 126.54(6) | 127.34(6) |
| 122.45(5) | 123.17(5) | 123.44(6) | 123.68(5) |
To evaluate the percentage bond shortening in K[1-S] (ΔdAlS%),59 the Al–S bond length was compared to the corresponding distance in compounds containing the sterically non-intrusive Al–SH group. There are no structurally characterised examples of three-coordinate hydrogensulfido compounds precluding a direct comparison with K[1-S]. However, comparing with examples of four-coordinate species (range Al–SH: 2.206(2) Å–2.290(1) Å),57,60 the ΔdAlS% values for the four molecules in {K[1-S]}4 fall in the range 6.1–7.0%, showing a significant shortening of the bond.
To determine whether the intermolecular K⋯S contacts influence the Al–S bond lengths in {K[1-S]}4, a sample was reacted with [2.2.2]cryptand, affording a small number of colourless crystals on work up. An X-ray diffraction study confirmed formation of the separated ion pair [K(2.2.2-crypt)][Al(NONDipp)(S)] ([K(2.2.2)crypt][1-S], Fig. 3), composed of an isolated [Al(NONDipp)(S)]− anion with a closest S⋯K distance = 7.12 Å. The Al–S bond length (2.0760(11) Å) is significantly (within 3σ) shorter than those in {K[1-S]}4, corresponding to a ΔdAlS% of 7.4%. These data suggest that the formation of S⋯K interactions in {K[1-S]}4 causes a minor elongation of the Al–S bonds compared to the isolated [Al(NONDipp)(S)]−. A similar conclusion was made for the selenido compounds, [{K(THF)}{Al(NONDipp)(Se)}]∞[K(THF)][VI] (ΔdAlSe% 5.7%) and [K(2.2.2-crypt)][Al(NONDipp)(Se)], [K(2.2.2)crypt][VI] (ΔdAlSe% 6.6%).45
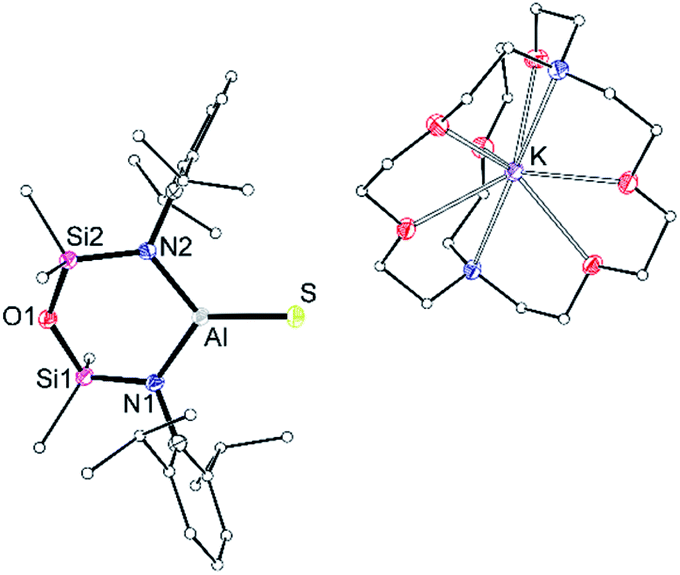 | ||
| Fig. 3 Displacement ellipsoid plot (30%) of [K(2.2.2-crypt)][Al(NONDipp)(S)], [K(2.2.2)crypt][1-S]. H-atoms and THF solvate omitted; C-atoms reduced for clarity. Selected bond lengths (Å) and angles (°): Al–S 2.0760(11), Al–N1 1.827(3), Al–N2 1.838(2). N1–Al–N2 106.22(12), N1–Al–S 126.44(9), N2–Al–S 127.34(9). | ||
Computational analysis of the Al–S bond in [Al(NONDipp)(S)]−
The terminal Al–E{16} bonds in the [Al(NONDipp)(E{16})]− anions have previously been described by two resonance structures (Scheme 3).45,46 Charge redistribution in these anionic systems does not afford a zwitterionic resonance as noted for the neutral Al–E{16} species, but localises electron density from the double bond to the chalcogenide generating a highly polar Al–E{16} bond (Scheme 3). The contribution of each resonance to the overall bonding will depend on the chalcogenide atom, and to evaluate the aluminium sulfide, density functional theory (DFT) calculations were performed on the free anion [Al(NONDipp)(S)]− [1-S]−, and the potassium included monomer, K[1-S]. Data were calculated using the BP86 functional and the SDDALL basis set with additional d-polarisation functions to describe Al (ζd = 0.190), Si (ζd = 0.284), S (ζd = 503) and 6-31G** for all other atoms. This gives a consistent data set to allow accurate comparisons between the different anions, [Al(NONDipp)(E{16})]−, [1-E]−, and their potassium salts, K[1-E] (Table 2. E{16} = O, S, Se and Te).46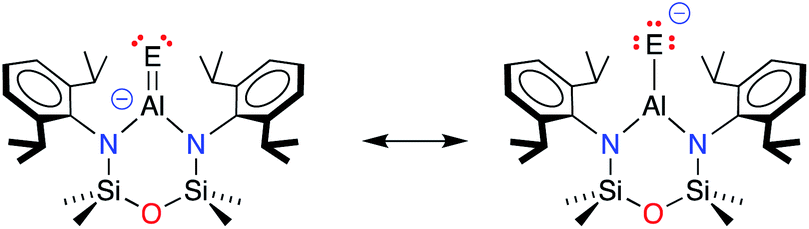 | ||
| Scheme 3 Resonance structures contributing to the terminal Al–E{16} bonds in [Al(NONDipp)(E{16})]− anions (E = E{16} = chalcogen atom). | ||
| [1-O]− | {K[1-S]}4 | [1-S]− | {K[VI]}∞ | [1-Se]− | [1-Te]− | |
|---|---|---|---|---|---|---|
| a Experimental data from [K{Al(NONDipp)(O)}]2 dimer, [K(1-O)]2. b Al–S bond length from [K(2.2.2-crypt)][Al(NONDipp)(S)], [K(2.2.2)crypt][1-S]. c Al–Se bond length from [K(2.2.2-crypt)][Al(NONDipp)(Se)], [K(2.2.2)crypt][VI]. d Al–Te bond length from THF adduct, K[Al(NONDipp)(Te)(THF)], [K(1-Te·THF)]. e Calculated using the average value of d(Al–O) = 1.74 Å from 35 entries of Al–OH bonds in the CSD. f Calculated using the average value of d(Al–S) = 2.59 Å from 12 entries of Al–SH bonds in the CSD. g Calculated using the average value of d(Al–Se) = 2.36 Å from 9 entries of Al–SeH bonds in the CSD. h Calculated using the average value of d(Al–Te) = 2.59 Å from 4 entries of Al–TePh bonds in the CSD. i ΔχP = χP(E) − χP(Al), where χP(Al) = 1.61, χP(O) = 3.44, χP(S) = 2.58, χP(Se) = 2.55, χP(Te) = 2.1, according to the Pauling electronegativity scale. j WBI values for structures without the potassium counter-ion. k WBI values for compounds containing a potassium counter-ion. | ||||||
| Al–E{16}/Å (X-ray) | 1.6362(14)a | 2.0873(7), 2.0957(7), 2.1039(7), 2.0857(7) | 2.0760(11)b | 2.2253(11) | 2.2032(6)c | 2.5039(7)d |
| Al–E{16}/Å (computed) | 1.66 | 2.13 | 2.11 | 2.27 | 2.24 | 2.46 |
| Difference: computed vs. X-ray (%) | +1.4 | +1.2 to + 2.1 | +1.6 | +2.0 | +1.6 | −1.8 |
| ΔdAlE (%) | 6.5e | Range: 6.1–6.9f | 7.4f | 5.7g | 6.6g | 3.4h |
| ΔχPi | 1.83 | — | 0.97 | — | 0.94 | 0.49 |
| NPA charges (q) | q(Al) = +1.93, q(O) = −1.23 | — | q(Al) = +1.85, q(S) = −1.26 | — | q(Al) = +1.82, q(Se) = −1.23 | q(Al) = +1.78, q(Te) = −1.19 |
| Δq = q(Al) − q(E{16}) | 3.16 | — | 3.11 | — | 3.05 | 2.97 |
| Wiberg bond index (WBI) | [1-O] − = 1.11j | — | [1-S]− = 1.30j | — | [1-Se]− = 1.38j | [1-Te]− = 1.53j |
| K[1-O] = 0.91k | — | K[1-S] = 1.15k | — | K[1-Se] = 1.24k | K[1-Te] = 1.44k | |
|
||||||
| Bond critical point (BCP) analysis | ||||||
| ρ(r)/e Å−3 | 0.115 | — | 0.076 | — | 0.068 | 0.061 |
| ∇2ρ(r)/e Å−5 | +0.990 | +0.290 | +0.183 | +0.089 | ||
| Ellipticity, ε | 0.033 | 0.080 | 0.108 | 0.139 | ||
The optimised geometry for the [1-S]− anion is in good agreement with the X-ray diffraction data, with a slight over estimation of the Al–S bond length compared with that found in {K[1-S]}4 (+1.2% to +2.1%) and [K(2.2.2)crypt][1-S] (+1.6%). The orthogonal lone pairs on the sulfur are the major contributions to the HOMO and HOMO−1 orbitals (Fig. 4a), as noted in the other members of the series. The increasing size of the orbitals for the heavier chalcogens leads to a notable extension of the principal lobes towards the aluminium, increasing according to [1-O]− < [1-S]− < [1-Se]− < [1-Te]− (Fig. S32†). The Wiberg bond indices (WBI) calculated for [1-S]− (1.30) and K[1-S] (1.15) fit within the trend O < S < Se < Te and are consistent with an increasing bond order when the heavier chalcogenides are present. As noted previously,46 this contradicts the expected mismatch in orbital energy, as the greater MO coefficients from the chalcogen atoms to the HOMO and HOMO−1 change from 2p, 3p (for E = O) to a combination of 3p, 4p and 5p for E = Te (Table S3†). This is rationalised by the significant decrease in the electronegativity of the chalcogenides as the group is descended, supported by the NPA (Natural Population Analysis) charges (q) for the aluminium and chalcogenide atoms in each anion (Table 2).
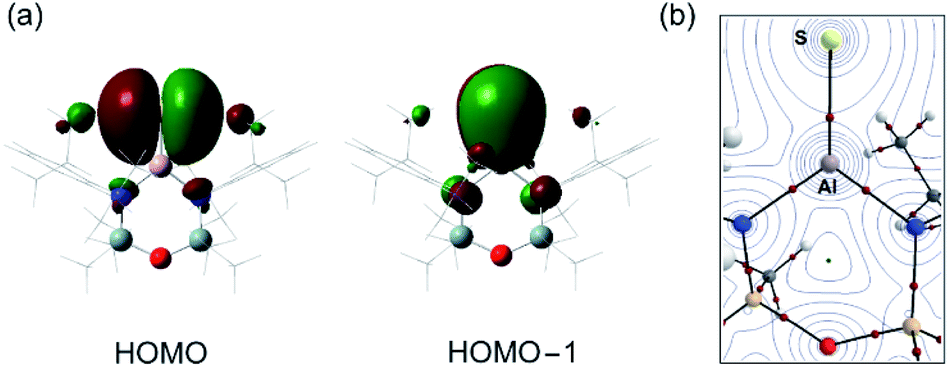 | ||
| Fig. 4 (a) Frontier orbitals (HOMO, HOMO−1) and (b) QTAIM molecular graph for the anion [Al(NONDipp)(S)]− [1-S]−. The electron density contours are computed in the {Al/E/N} planes with bond critical points (BCPs) shown as small red spheres. ρ(r) = 0.076 e Å−3, ∇2ρ(r) = 0.290 e Å−5, ε = 0.080. | ||
QTAIM (Quantum Theory of Atoms in Molecules) analysis has also been performed on [1-S]− (Fig. 4b). Compared to the remaining members of the [1-E]− series, we note that the electron density associated with the bond critical point (BCP) ρ(r) along the Al–E{16} bond path decreases as the group 16 elements increase in molecular weight, showing a weaker Al–E{16} interaction as the electronegativity of the element decreases (Table 2). This is accompanied by an increase in the bond ellipticity, ε, from 0.033 in [1-O]− to 0.139 in [1-Te]−, indicating a greater π-character of the Al–E{16} bond in the order O < S < Se < Te, consistent with the WBI analysis.61,62
Reactivity of K[Al(NONDipp)(E)] (E = S, Se)
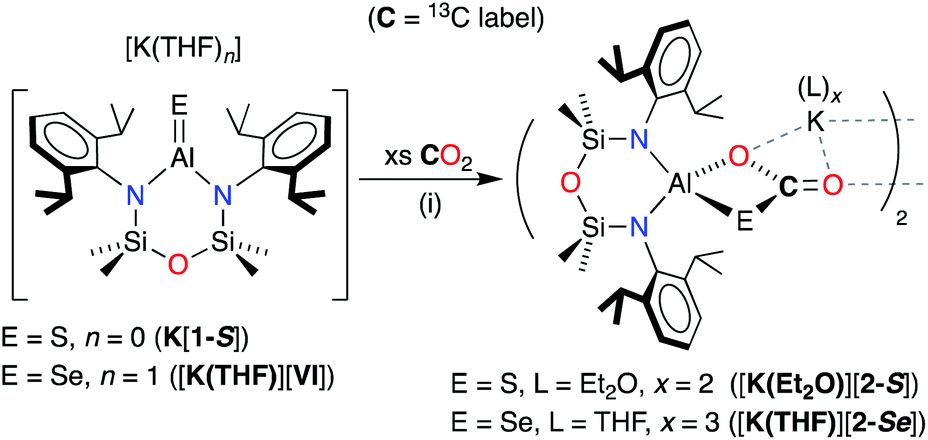 | ||
| Scheme 4 Synthesis of [K(Et2O)2][Al(NONDipp)(SC{O}O)] [K(Et2O)][2-S] and [K(THF)3][Al(NONDipp)(SeC{O}O)] ([K(THF)][2-Se]). E = S: (i) toluene solvent, crystallised from Et2O. E = Se: (i) 50:1 THF:benzene, crystallised from THF. | ||
X-ray diffraction analysis of colourless crystals isolated from an Et2O solution stored at −30 °C established the compound as [K(Et2O)2][Al(NONDipp)(SC{O}O)], [K(Et2O)][2-S] (Fig. 5a). The anion contains a four-coordinate aluminium centre supported by the chelating NONDipp-group and a planar, bidentate κ2S,O-[CO2S]2− ligand. The molecule exists as a non-symmetric dimer with each thiocarbonate ligand bridging to a potassium cation of the second unit, generating a μ3-(thio-3κS-carbonato-1κO,2κO′,3κO)-μ3-(thio-4κS-carbonato-1κO,2κO′,4κO) core.
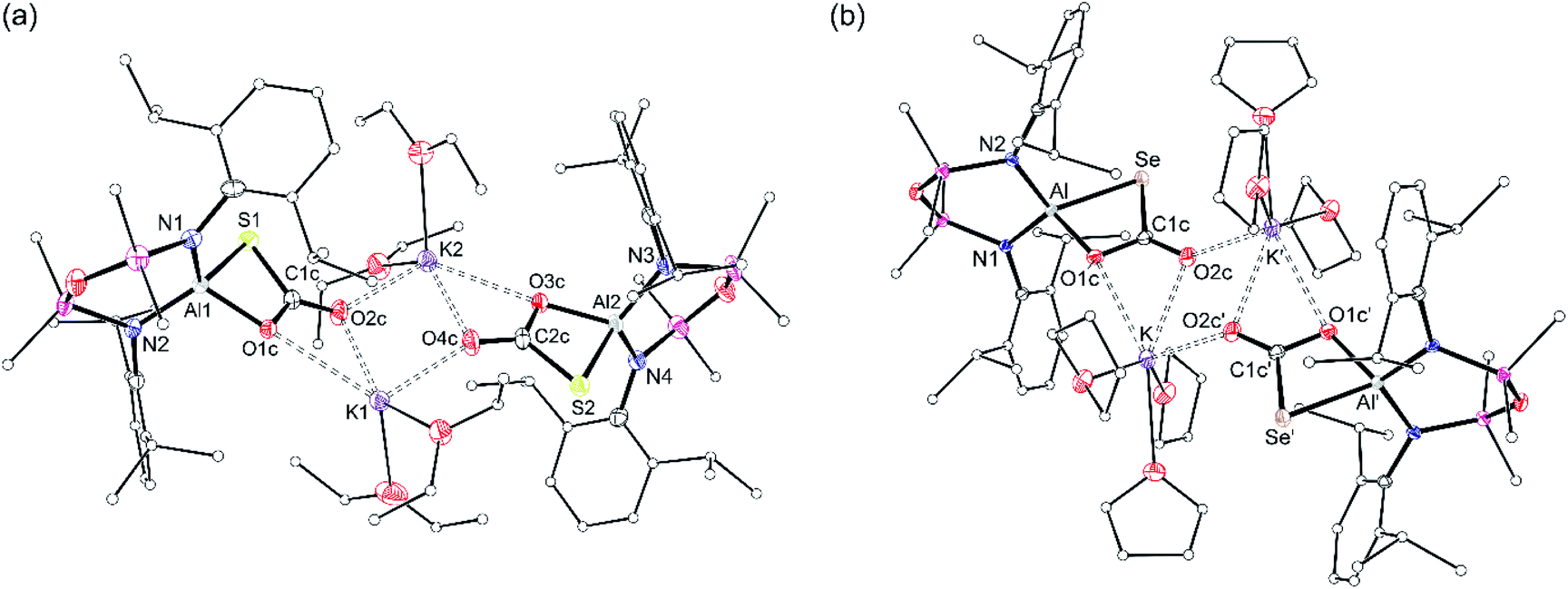 | ||
| Fig. 5 Displacement ellipsoid plot (30%) of (a) [{K(Et2O)2}{Al(NONDipp)(SC{O}O)}]2 ([K(Et2O)][2-S]) and (b) [{K(THF)3}{Al(NONDipp)(SeC{O}O)}]2 ([K(THF)][2-Se]) (′ = 1 − x, 1 − y, −z). Elipsoids 30%, H-atoms omitted; C-atoms reduced for clarity. Selected bond lengths (Å) and angles (°): [K(Et2O)][2-S] Al1–S1 2.2842(10), Al1–O1c 1.832(2), C1c–S1 1.766(3), C1c–O1c 1.317(4), C1c–O2c 1.229(4), Al2–S2 2.2812(11), Al2–O3c 1.828(3), C2c–S2 1.783(3), C2c–O3c 1.290(4), C2c–O4c 1.236(4). N1–Al1–N2 109.07(11), S1–Al1–O1c 76.50(7), S1–C1c–O1c 112.3(2), S1–C1c–O2c 124.9(2), O1c–C1c–O2c 122.9(3), N3–Al2–N4 110.17(12), S2–Al2–O3c 76.61(8), S2–C2c–O3c 112.6(2), S2–C2c–O4c 123.3(3), O3c–C2c–O4c 124.0(3). [K(THF)][2-Se] Al–Se 2.4162(6), Al–O1c 1.8295(15), C1c–Se 1.929(2), C1c–O1c 1.318(3), C1c–O2c 1.227(3). N1–Al–N2 109.39(7), Se–Al–O1c 77.80(5), Se–C1c–O1c 111.42(13), Se–C1c–O2c 125.27(18), O1c–C1c–O2c 123.3(2). | ||
The aluminium atoms are distorted tetrahedral with the thiocarbonate ligand defining the smallest angle (76.50(7)° and 76.61(8)° at Al1 and Al2, respectively). The Al–S bond lengths (2.2842(10) Å and 2.2812(11) Å) are considerably longer than in K[1-S] and [K(2.2.2)crypt][1-S], and are consistent with sum of the molecular covalent single bond radii, r1 (Σ(r1) = 2.29 Å).63
The thiocarbonate ligand is the formal product of the [2 + 2] cycloaddition of CO2 to a terminal sulfido ligand in K[1-S], and is analogous to the reaction of a terminal nickel sulfido ligand with CO2.64 It is also formed from the reaction of a bimetallic uranium μ-sulfido complex with CO2,65 and a samarium μ-oxido compound with COS.66 Although comparison of the bonding in the [CO2S]2− ligand is hampered by positional disorder of the oxygen and sulfur atoms in the Ni and Sm examples, we note that the C–O and C–S bond lengths in [K(Et2O)][2-S] differ from those in the uranium complex. For example, the C–O bonds in the bis-uranium compounds suggest electron delocalisation across the OCO unit (|ΔC–O| = 0.022 Å), whereas the corresponding distances in [K(Et2O)][2-S] indicate a more pronounced localisation as C–O single and CO double bonds , |ΔC–O| = 0.088 Å and 0.054 Å (Fig. S31†).
The corresponding reaction between 13CO2 and the selenido compound [K(THF)][VI]45 was complete after 15 minutes at room temperature (1H NMR spectroscopy). Isolation of the product and purification by crystallisation from THF afforded colourless crystals [K(THF)3][Al(NONDipp)(SeC{O}O)], [K(THF)][2-Se]. 1H NMR spectroscopy indicated a similar loss of molecular symmetry to that noted in [K(Et2O)][2-S], with the SiMe2 groups resonating at δH 0.08 and 0.04. The low field resonance in the 13C{1H} NMR spectrum (δC 166.0) showed satellites due to coupling with 77Se (I = ½), corresponding to a 1JSeC coupling of 128 Hz (Fig. S13†). This assignment was confirmed in the 77Se NMR spectrum that showed a doublet centred at δSe 150. This resonance is significantly shifted from that in the terminal selenido complex [K(2.2.2-crypt)][Al(NONDipp)(Se)] (δSe −563).45
X-ray diffraction data established formation of the selenocarbonate compound, [K(THF)3][Al(NONDipp)(SeC{O}O)] [K(THF)][2-Se] (Fig. 5b). The compound crystallises as the symmetry-generated dimer, with the planar κ2Se,O-[CO2Se]2− ligand defining the smallest angle at aluminium (77.80(5)°). As for [K(Et2O)][2-S], K⋯O interactions to an adjacent [Al(NONDipp)(SeC{O}O)]− anion generate a μ3-(seleno-3κSe-carbonato-1κO,2κO′,3κO)-μ3-(seleno-4κS-carbonato-1κO,2κO′,4κO) core.
The Al–Se bond length (2.4162(6) Å) is close to the calculated sum of the molecular covalent single bond radii, r1 (Σ(r1) = 2.42 Å),63 and is significantly longer than the terminal selenido bonds in [K(THF)][VI] 2.2253(11) Å and [K(2.2.2)crypt][VI] 2.2032(6) Å.45 The only other structurally characterised selenocarbonate ligand is in [{(AdArO)3N}U]2(CO2Se) ([(AdArO)3N]3− = trianion of tris(2-hydroxy-3-adamantyl-5-methylbenzyl)amine), isolated from the reaction of CO2 with the bimetallic μ-selenido compound.65 As for the thiocarbonate ligand in [K(Et2O)][2-S], the [CO2Se]2− bond lengths in suggest a more localised distribution of electron density within the O–C–O moiety of the ligand (Fig. S31†).
67 react rapidly with benzophenone at room temperature to afford the [2 + 2] cycloaddition products K[Al(NON)(SC{O}Ph2)] K[3-S] and K[Al(NON)(SeC{O}Ph2)] K[3-Se], respectively (Scheme 5). Crystals of K[3-S] were isolated by the slow evaporation of solvent from a Et2O solution of the compound, while K[3-Se] was crystallised from a THF solution at −30 °C. In both cases, spectroscopic analysis was hampered by the low solubility of the isolated compounds and attempts at acquiring NMR data in THF-D8 resulted in decomposition of the compound, evident from the appearance of peaks corresponding to ‘free’ benzophenone and unidentified NONDipp-containing products.
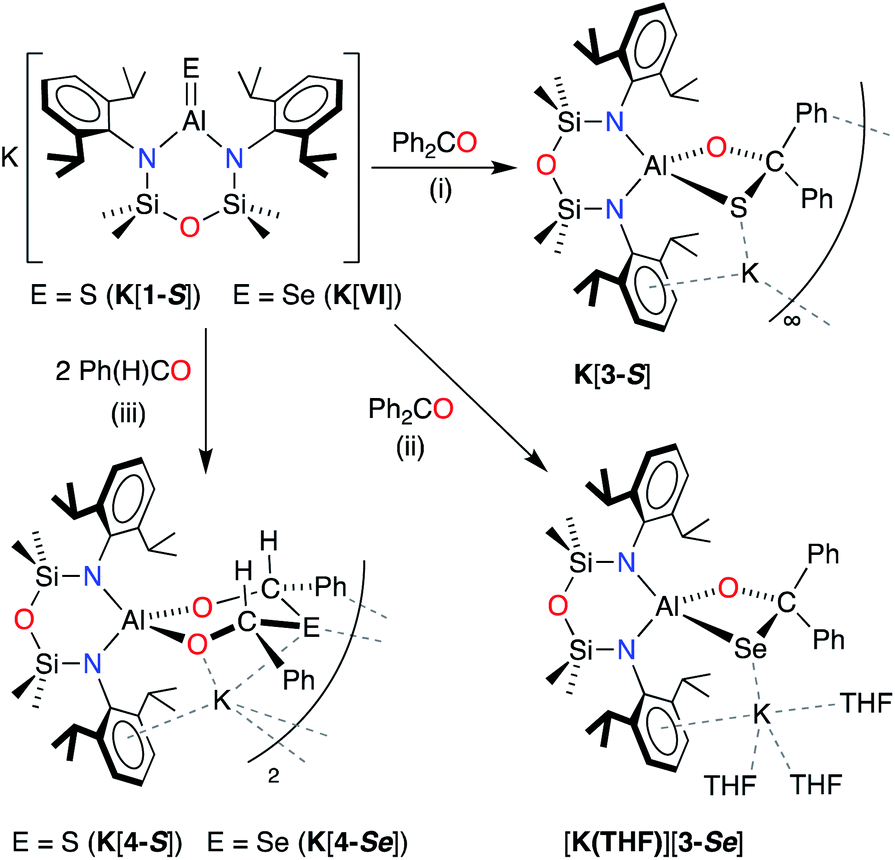 | ||
| Scheme 5 Synthesis of K[Al(NONDipp)(SC{O}Ph2)] (K[3-S]), [K(THF)3][Al(NONDipp)(SeC{O}Ph2)] ([K(THF)][3-Se]) and K[Al(NONDipp)({OCHPh}2E)] (E = S, K[4-S]; E = Se, K[4-Se]). (i) Et2O solvent, crystallised from Et2O. (ii) THF solvent, crystallised from THF. (iii) Benzene solvent, crystallised from toluene. | ||
The molecular structure of K[3-S] (Fig. 6a) consists of the [Al(NON)(SC{O}Ph2)]− anion containing the κ2S,O-diphenyl-sulfidomethanolate ligand, with a potassium cation bonded through an intramolecular K⋯S contact (3.1027(4) Å) and K⋯π(arene) interactions to a Dipp-substituent. The K[Al(NON)(SC{O}Ph2)] units are linked in a 1-D chain by additional K⋯π(arene) bonds to one of the phenyl substituents from an adjacent molecule (Fig. 6b). The four-coordinate aluminium is distorted tetrahedral with a κ2S,O-[SC{O}Ph2]2− bite angle of 78.96(3)°. The Al–S distance (2.2904(4) Å) is similar to that in [2-S]− and is consistent with a reduced bond order compared with [1-S]−. This was verified by comparing the calculated WBI of K[1-S] (1.15) with that of the Al–S single bond in the computational model of K[3-S] (WBI = 0.54), confirming a weak covalent bond between the aluminium and the sulfur atoms in the sulfidomethanolate complex. The C29–S (1.878(1) Å) and C29–O2 (1.4027(15) Å) bond lengths in the [SC{O}Ph2]2− ligand, indicate single bonds. These are notably longer than the delocalised bonding present in κ2S,O-monothiobenzoato anions, [SC{O}Ph]− (e.g. average C–S and C–O distances in Ga(SC{O}Ph)3 = 1.714(3) Å and 1.268(4) Å).68
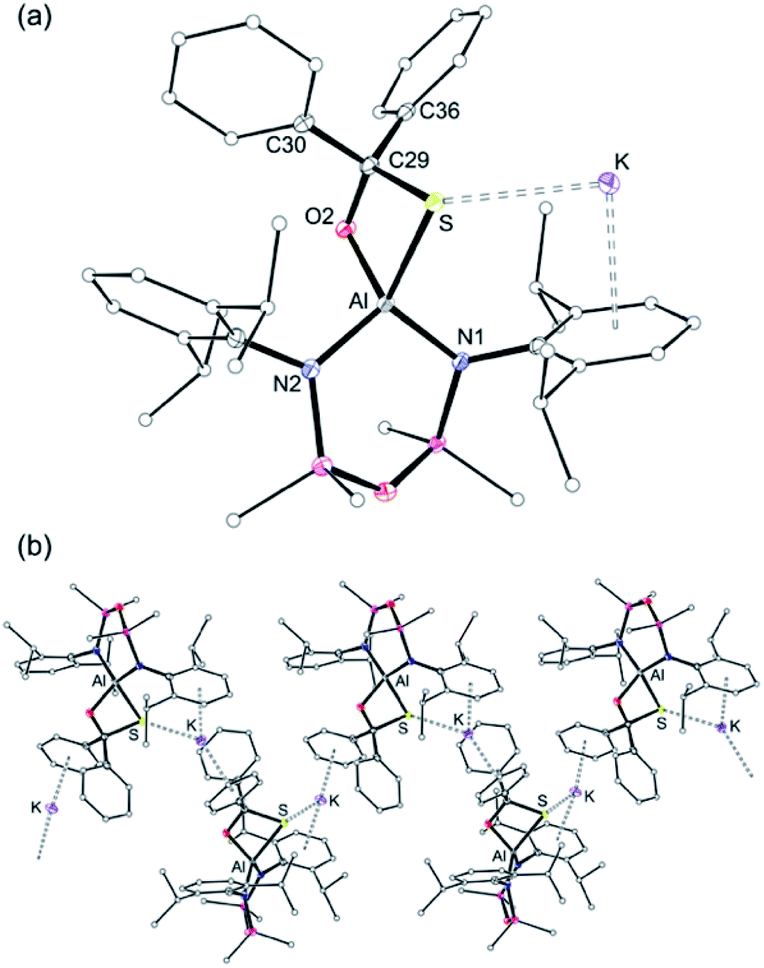 | ||
| Fig. 6 Displacement ellipsoid plot (30%) of K[Al(NONDipp)(SC{O}Ph2)] (K[3-S]). (a) Asymmetric unit. (b) Section of the polymeric chain formed by intermolecular K⋯π(arene) interactions. Et2O solvate and H-atoms omitted; C-atoms reduced for clarity. Selected bond lengths (Å) and angles (°): Al–S 2.2904(4), Al–O2 1.7788(9), C29–O2 1.4027(15), C29–S 1.878(1), Al–N1 1.8448(11), Al–N2 1.8476(11), K⋯S 3.1027(4). S–Al–O2 78.96(3), Al–S–C29 72.97(4), Al–O2–C29 103.01(7), S–C29–O2 104.88(8), N1–Al–N2 106.59(5). | ||
The corresponding diphenylselenidomethanolate complex crystallised from THF as potassium solvated compound [K(THF)3][Al(NONDipp)(SeC{O}Ph2)] [K(THF)][3-Se] (Fig. 7). The potassium atom is in an analogous position to that found in K[3-S], with a K⋯Se distance of 3.3501(4) Å. In this case, the incorporation of three THF molecules at potassium prevent formation of intermolecular contacts and limit the structure to the monomeric form. The κ2Se,O-[SeC{O}Ph2] bite angle is 79.79(4)° with an Al–Se single bond length of 2.4255(5) Å that compares well with [K(THF)][2-Se] and reflects a reduction in bond order compared with [VI]−. The C29–Se (2.0484(16) Å) and C29–O2 (1.3983(19) Å) bond lengths are consistent with localised single bonds, confirming formation of the diphenylselenidomethanolate dianion at the aluminium centre.
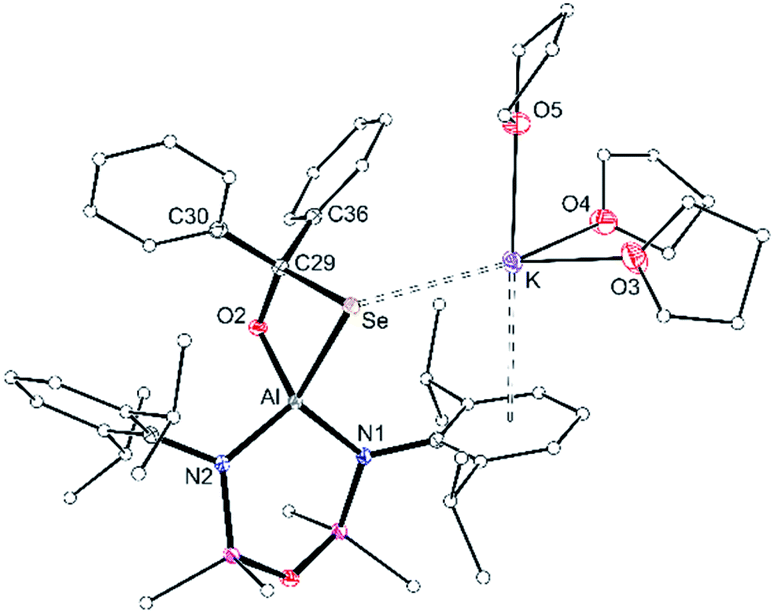 | ||
| Fig. 7 Displacement ellipsoid plot (30%) of [K(THF)3][Al(NONDipp)(SeC{O}Ph2)] ([K(THF)][3-Se]). H-atoms omitted; C-atoms reduced for clarity. Selected bond lengths (Å) and angles (°): Al–Se 2.4255(5), Al–O2 1.7687(12), C29–O2 1.3983(19), C29–Se 2.0484(16), Al–N1 1.8526(13), Al–N2 1.8477(14), K⋯Se 3.3501(4). Se–Al–O2 79.79(4), Al–Se–C29 69.19(5), Al–O2–C29 107.24(10), Se–C29–O2 103.52(10), N1–Al–N2 106.20(6). | ||
To the best of our knowledge, compounds K[3-S] and [K(THF)][2-Se] contain the first examples of the diphenylsulfidomethanolate and diphenylselenidomethanolate ligand. The related diphenylbis(oleate) ligand [O2CPh2]2− has been previously synthesised via the [2 + 2]-cycloaddition of benzophenone with metal–oxide multiple bonds, although examples are limited to in situ generated gallium-oxo69 and thorium-oxo70,71 species, and an ill-defined reaction with a lutetium methanediide-alkyl compound.72
Single crystals of each compound were grown by the slow evaporation of a toluene solution at room temperature. The structures are isomorphous and crystallise with half a dimeric unit in the asymmetric unit, located on an inversion centre (Fig. 8a, K[4-S]). The anionic components contain the thio- and seleno-bis(phenylmethanolate)dianions [{OC(H)Ph}2E]2−, from the incorporation of two equivalents of benzaldehyde.
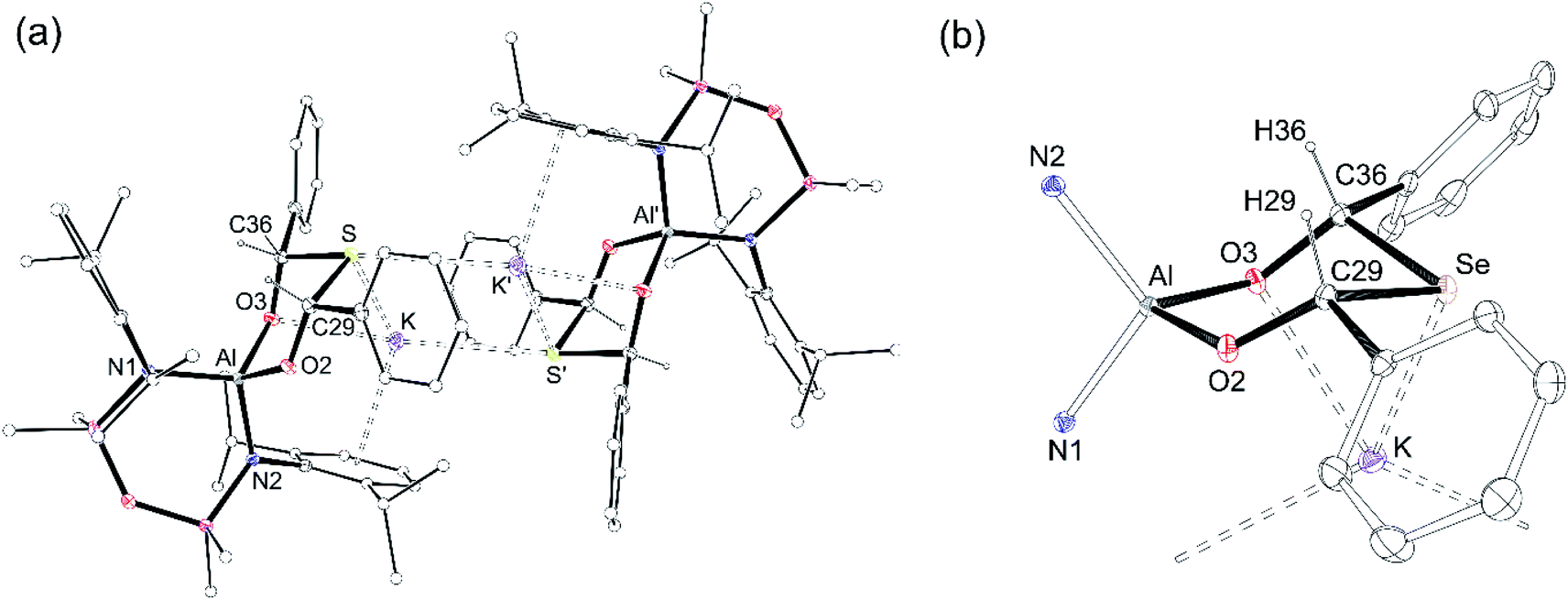 | ||
| Fig. 8 Displacement ellipsoid plot (30%) of (a) dimeric arrangement of K[{Al(NONDipp)}({OC(H)Ph}2S)] K[4-S], (′ = −x, 2 − y, −z) and core of K[{Al(NONDipp)({OC(H)Ph}2Se)] K[4-Se], (′ = 2 − x, 1 − y, −z) showing ring conformation. H-Atoms except [{OC(H)Ph}2E] and toluene solvate molecules omitted; C-atoms reduced for clarity. Selected bond lengths (Å) and angles (°): K[4-S] Al–N1 1.8537(17), Al–N2 1.8469(17), Al–O2 1.7676(15), Al–O3 1.7911(15), S–C29 1.867(2), S–C36 1.873(2), O2–C29 1.371(2), O3–C36 1.391(2), K⋯S 3.3183(7), K⋯O3 2.8193(15), K⋯S′ 3.5123(7); N1–Al–N2 107.72(8), O2–Al–O3 100.65(7), Al–O2–C29 126.63(13), Al–O3–C36 116.41(12), O2–C29–S 112.24(14), O3–C36–S 111.43(13), C29–S–C36 95.34(9). K[4-Se] Al–N1 1.8491(19), Al–N2 1.8537(19), Al–O2 1.7678(16), Al–O3 1.7935(16), Se–C29 2.023(2), Se–C36 2.036(2), O2–C29 1.364(3), O3–C36 1.378(3), K⋯Se 3.4239(6), K⋯O3 2.8304(16), K⋯Se′ 3.5264(6); N1–Al–N2 108.11(8), O2–Al–O3 101.15(7), Al–O2–C29 127.89(14), Al–O3–C36 117.83(14), O2–C29–Se 111.72(14), O3–C36–Se 111.21(14), C29–Se–C36 92.31(9). (b) view illustrating the core of K[4-Se]. | ||
The ligands adopt a κ2O,O′-bonding mode at aluminium with bite angles of 100.65(7)° and 101.15(7)°, respectively and the six-membered rings adopt chair conformations that crystallise exclusively as the cis-stereoisomer, with both Ph substituents equatorial (Fig. 8b, core of K[4-Se]). The C–S (1.867(2) Å, 1.873(2) Å) and C–Se (2.023(2) Å, 2.036(2) Å) bond lengths are similar to the C–E single bonds in K[3-S] (1.878(1) Å) and K[3-Se] (2.0484(16) Å). The acute C29–E-C36 angles (E = S, 95.34(9)°; E = Se, 92.31(9)°) are comparable to organic ring and cage structures containing the six-membered EC3O2 heterocycle (E = S, 94.71(12)°; E = Se, 90.58(6)° and 87.58(2)°).73,74 The transannular Al⋯S and Al⋯Se distances of 3.46 Å and 3.59 Å, respectively, confirm complete cleavage of the Al–E{16} multiple bond, reflected in the negligible WBI value of 0.06 calculated for the model compound K[4-S].
A survey of the literature indicates that the neutral form of the thiobis(phenylmethanolate) ligand (α-hydroxybenzylsulfide, {HOC(H)Ph}2S) is known from a single report in 1910,75 whereas the corresponding selenide is unique. This paper therefore adds the first examples of the chalcogenidobis(phenylmethanolate) ligands to the chemical library of compounds, the synthesis of which have only been made possible through the application of [Al(NONDipp)(E)]− anions.
Computational analysis of cycloaddition chemistry
During this study, we identified two experimental results that warranted mechanistic investigation by computational methods. Firstly, we wanted to examine whether a second equivalent of benzophenone could be added to K[3-S] to afford the hypothetical thio-bis(diphenylmethanolate) compound. Secondly, we wished to investigate the origins of the stereoselectivity in the formation of the cis-isomeric form of K[4-E]. The mechanisms of addition and activation of benzophenone and benzaldehyde to K[1-S] were therefore characterized by DFT at the BP86-D3BJ level of theory (see ESI† for full computational details).Initial work examined the formation of the hypothetical thio-bis(diphenylmethanolate), K[Al(NONDipp)({OCPh2}2S)] (D, Fig. 9), via the sequential addition of two equivalents of benzophenone to A (K[1-S]). The initial cycloaddition takes place viaTS(A–B) with a barrier of +8.6 kcal mol−1 to form adduct B (+5.3 kcal mol−1). Facile C–S coupling subsequently takes place viaTS(B–C) (+6.9 kcal mol−1) in an exergonic process to afford the diphenylsulfidomethanolate, C (K[3-S]) (−11.9 kcal mol−1). From C, the addition and subsequent C–S coupling of a second equivalent of Ph2CO was characterized to take place in a single step viaTS(C–D) (+8.5 kcal mol−1) with an overall barrier height of 20.4 kcal mol−1 to form the experimentally unobserved bis(diphenylmethanolate) D (−11.1 kcal mol−1). Formation of D is thus identified by DFT to be kinetically accessible yet mildly endergonic relative to the mono-activated product C by +0.8 kcal mol−1. Based on these computed energetics we propose that access to D may be feasible under the reaction conditions, although the barrier from C is of almost identical energy to the reverse reaction that regenerates A. This may explain the observation of free benzophenone by NMR spectroscopy when samples of K[3-S] are dissolved in THF. In addition, we must consider the formation of intermolecular K⋯π(arene) interactions and aggregation into a 1-D polymer for K[3-S]. This contributes to the low solubility of the mono-addition product and likely promotes crystallization from solution, which may occur before the addition of a second equivalent of benzophenone can take place.
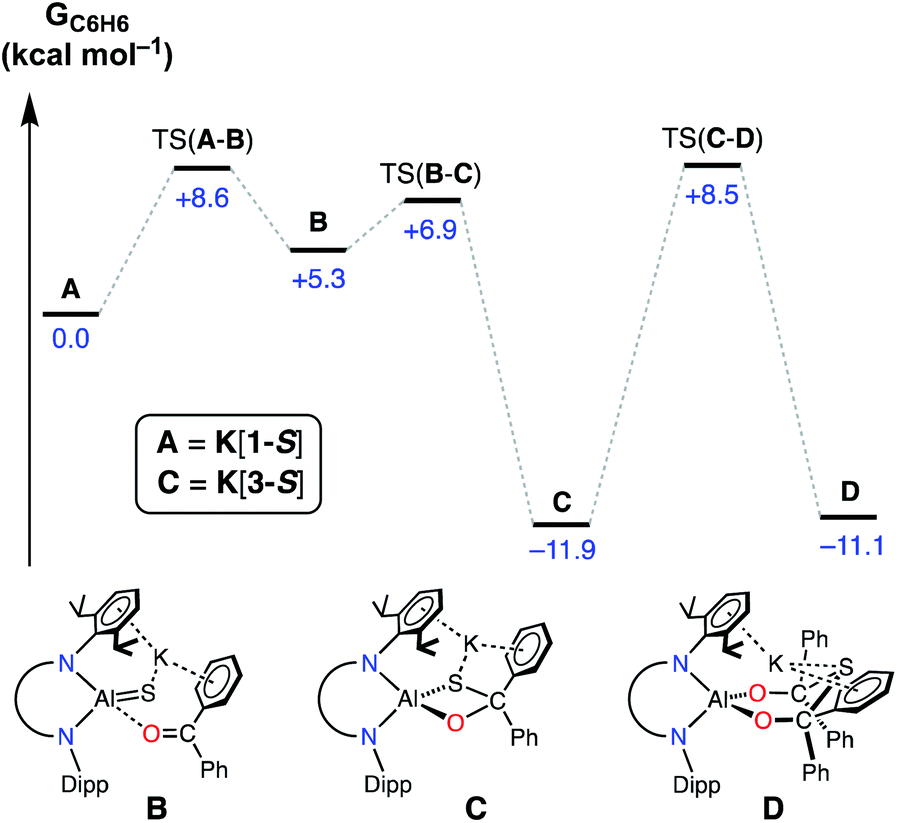 | ||
| Fig. 9 Computed free energy profile (in kcal mol−1) at the BP86-D3BJ,C6H6/6-311++G**//BP86/BS1 level for addition and activation of benzophenone to A (K[1-S]). [Al] = Al(NONDipp). | ||
To determine the origins of the cis-isomeric form of K[4-S] that is exclusively observed in the solid-state, the mechanism for the conversion of A (K[1-S]) to Hcis (K[4-S]) was also probed by DFT (Fig. 10). The initial addition of one equivalent of benzaldehyde to A (K[1-S]), and Al–O/C–S bond formation takes place via TS(A–E) (+8.3 kcal mol−1), forming the (non-observed) mono-addition product E (−14.2 kcal mol−1). Addition of a second equivalent of benzaldehyde is a low energy process that initially forms adduct F (−7.0 kcal mol−1) via TS(E–F). From F, a second Al–O/C–S bond formation occurs with concomitant breaking of the Al–S bond, to form the experimentally observed cis-isomer Hcis (−27.3 kcal mol−1) via TS(F–H)cis.76
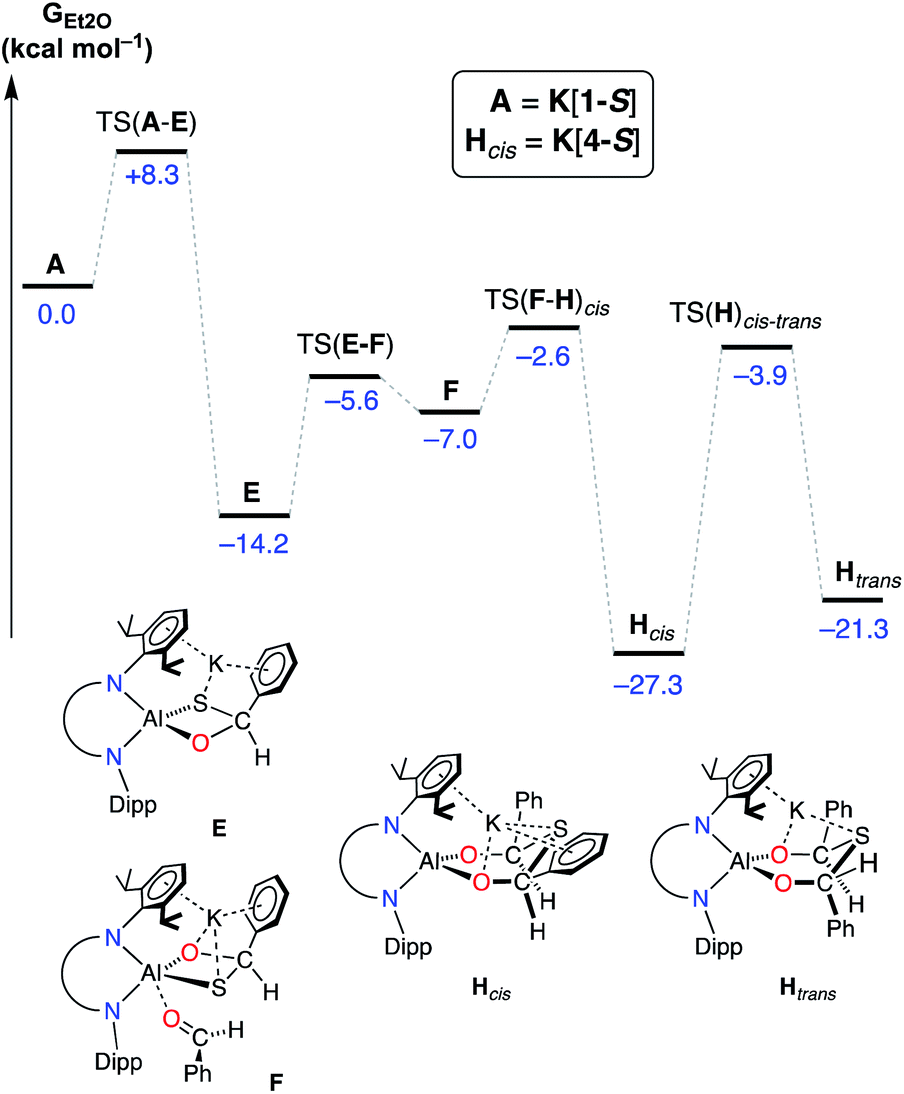 | ||
| Fig. 10 Computed free energy profile (in kcal mol−1) at the BP86-D3BJ, Et2O/6-311++G**//BP86/BS1 level for the addition and activation of benzaldehyde to A (K[1-S]). [Al] = Al(NONDipp). | ||
Formation of the cis-isomer from adduct F proceeds with an attack of the sulfur atom at the Si face of the coordinated benzaldehyde (Fig. 11a). The calculated transition-state TS(F–H)cis shows two possible features that favour Si attack (Fig. 11b). Electrostatic K⋯S interactions hold the sulfur atom ‘above’ the plane of the coordinated benzaldehyde, while the steric bulk of one of the iPr groups twists the benzaldehyde about the Al–O bond to present the Si face. To generate the trans-isomer from F, rotation of the benzaldehyde about Al–O would be required, such that the phenyl group would clash with an iPr group of the other Dipp substituent, which represents an unfavourable conformation. Such an attack at the Re face of benzaldehyde was computationally explored. However, multiple attempts to locate the equivalent isomer of F were unsuccessful, and hence a connection in the reaction pathway between E and Htrans could not be established. Furthermore, characterization of Htrans (−21.3 kcal mol−1) identifies this as an energetically disfavoured conformation with respect to Hcis (−27.3 kcal mol−1).77
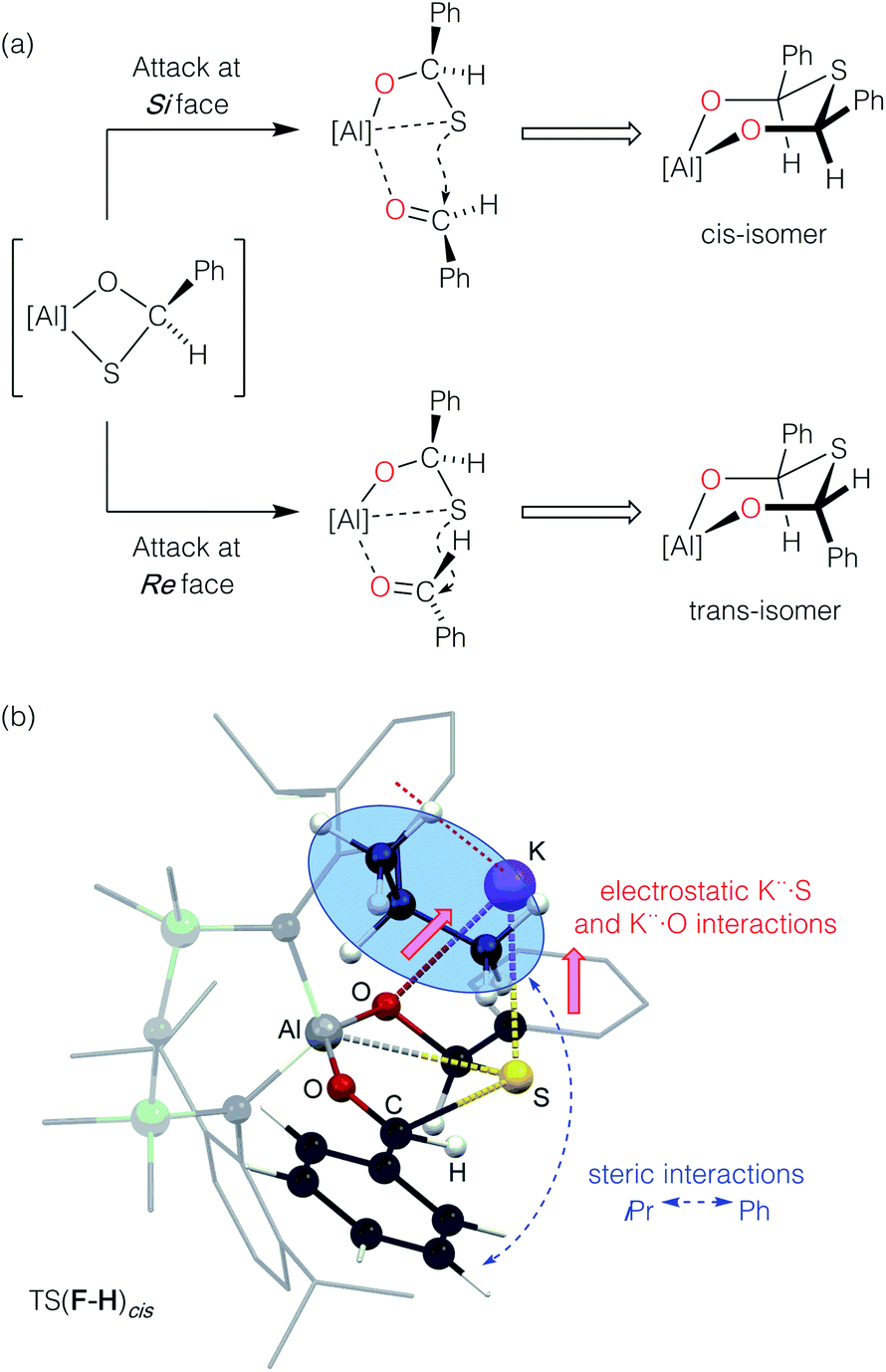 | ||
| Fig. 11 (a) Hypothetical attack of sulfur at the Si and Re faces of benzaldehyde affording the cis- and trans-isomeric forms of the thio-bis(phenylmethanolate), respectively. (b) Calculated geometry TS(F–H)cis illustrating the electrostatic K⋯S and K⋯O interactions (solid red arrows) and steric iPr ↔ Ph clash (dashed blue arrow). | ||
Interestingly, a pathway for the isomerisation of Hcis to Htrans was identified via an accessible saddle-point TS(H)cis–trans (−3.9 kcal mol−1). From a thermodynamic perspective, these results suggest that if the Htrans stereoisomer is indeed formed during the reaction, the exclusive isolation of the Hcis isomer may be a result of its lower energy (ΔΔG [Hcis–Htrans] = −6.0 kcal mol−1). However, we once again note that the observed stereoselectivity may be influenced by experimentally determined factors, including complex cation···anion and solvent interactions during the formation of K[4-S], and the crystallisation processes that afford these isolated materials.
Conclusions
We have developed a synthetic route to aluminium sulfide multiple bonds in the anion [Al(NONDipp)(S)]−. Computational analysis indicates multiple-bond character, consistent with other members of the series [Al(NONDipp)(E)]− (E = O, Se, Te). The reactivity of the sulfido and selenido anions [Al(NONDipp)(E)]− (E = S, Se) with unsaturated CO bonds in carbon dioxide, benzophenone and benzaldehyde proceeds via a [2 + 2]-cycloaddition pathway to form new C–E bonds with a reduction in the Al–E{16} bond order. With CO2 and Ph2CO, a single addition occurs to form the κ2E,O-[EC{O}O]2− and κ2E,O-[EC{O}Ph2]2− ligands, respectively. When benzaldehyde is reacted, two equivalents are consumed to afford the ligands, κ2O,O′-[{OC(H)Ph}2E]2− with complete cleavage of the Al–E{16} bond. These results demonstrate the first synthetic utility of aluminium–chalcogen multiple bonds in the formation of new C–E{16} containing organic fragments, affording new ligand architectures directly at the metal centres.
Author contributions
M. P. C. supervised the work and carried out the X-ray crystallographic analysis. M. D. A. and M. J. E. carried out the synthetic work. C. L. M., S. E. N. and N. A. R. carried out the computational studies. M. P. C. prepared the manuscript. M. P. C., M. J. E. and C. L. M. prepared the ESI.† All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. E. acknowledges the award of a Victoria University of Wellington Doctoral Scholarship. This research made use of the Balena High Performance Computing (H. P. C.) Service at the University of Bath. Financial support from the EPSRC (EP/R020752/1 – C. L. M., S. E. N. and N. A. R.) and a University Research Fund (URF) grant from Victoria University of Wellington (M. D. A.).Notes and references
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- P. P. Power, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed.
- P. Jutzi, Angew. Chem., Int. Ed. Engl., 1975, 14, 232–245 CrossRef.
- C. Weetman, Chem.–Eur. J., 2021, 27, 1941–1954 CrossRef CAS PubMed.
- M. Somer, D. Thiery, K. Peters, L. Walz, M. Hartweg, T. Popp and H. G. von Schnering, Z. Naturforsch., B: J. Chem. Sci., 1991, 46, 789–794 CrossRef CAS.
- R. J. Wehmschulte and P. P. Power, J. Am. Chem. Soc., 1997, 119, 8387–8388 CrossRef CAS.
- K. Ruhlandt-Senge and P. P. Power, Inorg. Chem., 1991, 30, 2633–2637 CrossRef CAS.
- K. Ruhlandt-Senge and P. P. Power, Inorg. Chem., 1993, 32, 3478–3481 CrossRef CAS.
- S. P. Wuller, A. L. Seligson, G. P. Mitchell and J. Arnold, Inorg. Chem., 1995, 34, 4854–4861 CrossRef CAS.
- P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed.
- F. Dankert and C. Hering-Junghans, Chem. Commun., 2022, 58, 1242–1262 RSC.
- W. Uhl, A. Vester, W. Kaim and J. Poppe, J. Organomet. Chem., 1993, 454, 9–13 CrossRef CAS.
- C. Pluta, K. R. Porschke, C. Kruger and K. Hildenbrand, Angew. Chem., Int. Ed. Engl., 1993, 32, 388–390 CrossRef.
- R. J. Wehmschulte, K. Ruhlandt-Senge, M. M. Olmstead, H. Hope, B. E. Sturgeon and P. P. Power, Inorg. Chem., 1993, 32, 2983–2984 CrossRef CAS.
- R. J. Wright, M. Brynda and P. P. Power, Angew. Chem., Int. Ed., 2006, 45, 5953–5956 CrossRef CAS PubMed.
- R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS PubMed.
- T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818–10821 CrossRef CAS PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed.
- R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 CrossRef CAS PubMed.
- C. Weetman, P. Bag, T. Szilvási, C. Jandl and S. Inoue, Angew. Chem., Int. Ed., 2019, 58, 10961–10965 CrossRef CAS PubMed.
- C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC.
- S. Schulz, A. Voigt, H. W. Roesky, L. Häming and R. Herbst-Irmer, Organometallics, 1996, 15, 5252–5253 CrossRef CAS.
- C. Cui, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2000, 39, 4531–4533 CrossRef CAS PubMed.
- H. Zhu, Z. Yang, J. Magull, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2005, 24, 6420–6425 CrossRef CAS.
- H. Zhu, J. Chai, V. Chandrasekhar, H. W. Roesky, J. Magull, D. Vidovic, H.-G. Schmidt, M. Noltemeyer, P. P. Power and W. A. Merrill, J. Am. Chem. Soc., 2004, 126, 9472–9473 CrossRef CAS PubMed.
- N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 2172–2174 CrossRef CAS PubMed.
- J. Li, X. Li, W. Huang, H. Hu, J. Zhang and C. Cui, Chem.–Eur. J., 2012, 18, 15263–15266 CrossRef CAS PubMed.
- J. D. Queen, S. Irvankoski, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2021, 143, 6351–6356 CrossRef CAS PubMed.
- M. Fischer, S. Nees, T. Kupfer, J. T. Goettel, H. Braunschweig and C. Hering-Junghans, J. Am. Chem. Soc., 2021, 143, 4106–4111 CrossRef CAS PubMed.
- D. Neculai, H. W. Roesky, A. M. Neculai, J. Magull, B. Walfort and D. Stalke, Angew. Chem., Int. Ed., 2002, 41, 4294–4296 CrossRef CAS PubMed.
- T. Chu, S. F. Vyboishchikov, B. Gabidullin and G. I. Nikonov, Angew. Chem., Int. Ed., 2016, 55, 13306–13311 CrossRef CAS PubMed.
- D. Franz, T. Szilvási, E. Irran and S. Inoue, Nat. Commun., 2015, 6, 10037 CrossRef CAS PubMed.
- D. Franz and S. Inoue, Dalton Trans., 2016, 45, 9385–9397 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2018, 57, 5885–5887 CrossRef CAS PubMed.
- R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- M. D. Anker, R. J. Schwamm and M. P. Coles, Chem. Commun., 2020, 56, 2288–2291 RSC.
- A. Heilmann, J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 4897–4901 CrossRef CAS PubMed.
- M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 18261–18265 CrossRef CAS PubMed.
- J. Hicks, A. Heilmann, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 17265–17268 CrossRef CAS PubMed.
- M. D. Anker and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 13452–13455 CrossRef CAS PubMed.
- M. J. Evans, M. D. Anker, C. L. McMullin, N. A. Rajabi and M. P. Coles, Chem. Commun., 2021, 57, 2673–2676 RSC.
- Y. K. Loh and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 8626–8648 CrossRef CAS PubMed.
- M. D. Anker, C. L. McMullin, N. A. Rajabi and M. P. Coles, Angew. Chem., Int. Ed., 2020, 59, 12806–12810 CrossRef CAS PubMed.
- M. Fourmigué and A. Dhaka, Coord. Chem. Rev., 2020, 403, 213084 CrossRef.
- N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC.
- P. C. Ho, J. Z. Wang, F. Meloni and I. Vargas-Baca, Coord. Chem. Rev., 2020, 422, 213464 CrossRef CAS.
- W. Guo, D.-Y. Wang, Q. Chen and Y. Fu, Adv. Sci., 2022, 9, 2103989 CrossRef CAS PubMed.
- N. Tokitoh, T. Matsumoto, K. Manmaru and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 8855–8856 CrossRef CAS.
- T. Matsumoto, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 1999, 121, 8811–8824 CrossRef CAS.
- M. Saito, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 2004, 126, 15572–15582 CrossRef CAS PubMed.
- H. Xu, C. Weetman, F. Hanusch and S. Inoue, Chem.–Eur. J., 2022, 28, e202104042 CAS.
- V. Jancik and H. W. Roesky, Inorg. Chem., 2005, 44, 5556–5558 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- ΔdAlS% = [1 – d(Al=S)/d(Al–S)] × 100%, with d(Al–S) equal to the average value of 2.242 Å calculated from twenty structurally determined Al–SH bonds listed in the Cambridge Structural Database (CSD).
- S. González-Gallardo, V. Jancik, R. Cea-Olivares, R. A. Toscano and M. Moya-Cabrera, Angew. Chem., Int. Ed., 2007, 46, 2895–2898 CrossRef PubMed.
- C. Silva Lopez and A. R. de Lera, Curr. Org. Chem., 2011, 15, 3576–3593 CrossRef.
- R. Hilal, S. G. Aziz, A. O. Alyoubi and S. Elroby, Procedia Computer Science, 2015, 51, 1872–1877 CrossRef.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- N. J. Hartmann, G. Wu and T. W. Hayton, Chem. Sci., 2018, 9, 6580–6588 RSC.
- O. P. Lam, S. M. Franke, F. W. Heinemann and K. Meyer, J. Am. Chem. Soc., 2012, 134, 16877–16881 CrossRef CAS PubMed.
- C. Schoo, S. V. Klementyeva, M. T. Gamer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2016, 52, 6654–6657 RSC.
- The non-solvated potassium selenide compound K[Al(NONDipp)(Se)] (K[VI]) was prepared according to the procedure for [K(THF)][VI], and isolated directly from the toluene reaction mixture without crystallization from THF.
- E. Jaśkowska, Ł. Dobrzycki, P. Rzepiński and W. Ziemkowska, J. Sulfur Chem., 2015, 36, 326–339 CrossRef.
- A. Kassymbek, S. F. Vyboishchikov, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, Angew. Chem., Int. Ed., 2019, 58, 18102–18107 CrossRef CAS PubMed.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- C. Zhang, P. Yang, E. Zhou, X. Deng, G. Zi and M. D. Walter, Organometallics, 2017, 36, 4525–4538 CrossRef CAS.
- S. Li, M. Wang, B. Liu, L. Li, J. Cheng, C. Wu, D. Liu, J. Liu and D. Cui, Chem.–Eur. J., 2014, 20, 15493–15498 CrossRef CAS PubMed.
- J. Kaschel, C. D. Schmidt, M. Mumby, D. Kratzert, D. Stalke and D. B. Werz, Chem. Commun., 2013, 49, 4403–4405 RSC.
- G. Valle, V. Busetti and M. Mammi, Cryst. Struct. Commun., 1972, 2, 167 Search PubMed.
- E. Fromm and F. Erfurt, Ber. Dtsch. Chem. Ges., 1909, 42, 3808–3812 CrossRef.
- An intermediate INT(F-H)cis at –24.4 kcal mol−1 was identified, that forms from TS(F-H)cis, although Hcis represents a lower energy conformer, and it is expected that any barriers for conformational change between Hcis and INT(F-H)cis are low.
- A corresponding transition state TS(F-H)trans leading to Htrans could not be located.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra; details of X-ray experiments and additional figures; computational details and xyz-coordinates from DFT calculations. CCDC 2038726, 2038727, 2038729, 2038730 and 2097188–2097191. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc01064j |
| This journal is © The Royal Society of Chemistry 2022 |