
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous electrochemically-triggered atom transfer radical polymerization†
Boyu
Zhao
,
Fred
Pashley-Johnson
 ,
Bryn A.
Jones
and
Paul
Wilson
*
,
Bryn A.
Jones
and
Paul
Wilson
*
University of Warwick, UK. E-mail: p.wilson.1@warwick.ac.uk
First published on 27th April 2022
Abstract
Simplified electrochemical atom transfer radical polymerization (seATRP) using CuII–N-propyl pyridineimine complexes (CuII(NPPI)2) is reported for the first time. In aqueous solution, using oligo(ethylene glycol) methyl ether methacrylate (OEGMA), standard electrolysis conditions yield POEGMA with good control over molecular weight distribution (Đm < 1.35). Interestingly, the polymerizations are not under complete electrochemical control, as monomer conversion continues when electrolysis is halted. Alternatively, it is shown that the extent and rate of polymerization depends upon an initial period of electrolysis. Thus, it is proposed that seATRP using CuII(NPPI)2 follows an electrochemically-triggered, rather than electrochemically mediated, ATRP mechanism, which distinguishes them from other CuIIL complexes that have been previously reported in the literature.
Introduction
Electrochemical intervention in synthesis and catalysis has received renewed interest over the last 5–10 years.1–6 From a synthetic point of view, the use of an applied potential/current enables accurate control over the thermodynamics and/or kinetics of electron transfer processes.7,8 This can enhance the selectivity of chemical transformations and confer spatiotemporal control over synthetic and catalytic reactions of small and macromolecular organic molecules/polymers, amongst others.In the context of reversible deactivation radical polymerization (RDRP) electrochemical intervention has been employed to regulate polymer synthesis through control of the dynamic equilibrium between dormant and active (radical) species which allows the overall radical concentration to be accurately controlled.9–11,53 In atom transfer radical polymerization (ATRP)12,13 the equilibrium (KATRP) is between a dormant alkyl (R–X) or macromolecular (Pn–X) halide and propagating radicals (R˙/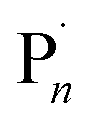 ) which undergo reversible redox reactions with transition metal complexes.
) which undergo reversible redox reactions with transition metal complexes.
In 2011, Matyjaszewski and co-workers showed that the redox nature of the Cu-mediated ATRP mechanism could lend itself to electrochemical manipulation and control.10 The active, yet oxidatively labile CuIL complex was formed in situ when a reducing potential (Eapp) was applied at the working electrode (WE) to induce a one electron reduction of an inactive CuIIL precursor. Activation of the dormant species (R–X/Pn–X) in the reaction media then generated the radical species (R˙/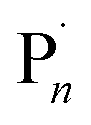 ), and the Cu-complex in a higher oxidation state (X–CuIIL). Well controlled polymerization of methyl acrylate was reported suggesting that the deactivation step of the equilibrium, between the propagating radical
), and the Cu-complex in a higher oxidation state (X–CuIIL). Well controlled polymerization of methyl acrylate was reported suggesting that the deactivation step of the equilibrium, between the propagating radical 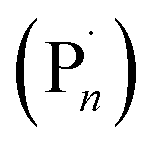 and X–CuIIL, reforming the dormant species (Pn–X) and CuIL respectively, was not perturbed by the electrochemical intervention. In fact, it was shown that by switching the Eapp at the WE to an oxidizing potential the polymerization could be completely switched off, conferring high fidelity on-off spatiotemporal control over polymer synthesis in solution.
and X–CuIIL, reforming the dormant species (Pn–X) and CuIL respectively, was not perturbed by the electrochemical intervention. In fact, it was shown that by switching the Eapp at the WE to an oxidizing potential the polymerization could be completely switched off, conferring high fidelity on-off spatiotemporal control over polymer synthesis in solution.
In the 10 years since this discovery, eATRP has been employed for the synthesis of polymers with a variety of compositions and architectures including block copolymers, bioconjugates, star and graft (co)polymers.14–22 It is compatible with aqueous23,24 and organic10 media whilst heterogeneous systems such mini-emulsion25–28 and surface-initiated (si-eATRP)29–32 polymerizations have also been reported. Furthermore, the complex reaction set-up, initially involving a 3-electrode divided electrochemical cell, has been simplified by the use of sacrificial counter electrodes (typically Al-wire), enabling undivided cells to be used in either 3-electrode (potential controlled) or 2-electrode (current controlled) configurations giving rise to simplified electrochemical atom transfer radical polymerisaiton (seATRP).33 This development is significant as it enables the chemistry to be performed using commercial, standardized hardware.24
The most widely studied systems for aqueous eATRP employ CuIIX salts with tetradentate ligands tris(2-(dimethylamino)ethyl)amine (Me6-Tren)10,34,35 or tris(2-pyridylmethyl)amine (TPMA).33,36,37 They form more active complexes, having high KATRP values.38 The ligands stabilize CuII more than CuI with cyclic voltammetry (CV) indicating that CuIMe6-Tren and CuITPMA are strongly reducing complexes, leading to fast activation (kact) of R–X/Pn–X.39,40 The kact (and KATRP) can increase by orders of magnitude when aqueous media is employed, which in the absence of appropriate conditions and/or external control of active catalyst generation, can result in high radical concentrations which has a detrimental effect on the polymerization.41,42 A great deal of discovery and optimization, of which eATRP is one example, has resulted in the development of efficient, well controlled aqueous ATRP reactions using these highly active complexes.9,43
Prior to this, less active complexes composed of bidentate ligands such as bipyridine (bpy) and N-alkyl pyridine imines (NAPI) were more suitable for aqueous ATRP.44–49 They stabilize CuI more than CuII, form less reducing CuI complexes and have lower kact and KATRP leading to lower radical concentrations. On one hand, this means that larger catalyst concentrations are required to mediate well controlled ATRP. On the other hand, it can also be beneficial for polymerizations carried out in aqueous media wherein increased kact and KATRP can lead to higher radical concentrations when more active complexes are employed. For example, 20 years ago, Haddleton and Perrier described in detail the efficient, well controlled polymerization of oligo(ethylene glycol) methyl ether methacrylate (OEGMA) using CuI(NAPI)2 complexes in water.44–46 The rates of reaction and control over the polymerization were optimized with respect to [CuI(NAPI)2]/[CuII(NAPI)2] which was controlled from the outset by using known amounts of CuIBr and CuIIBr2 to form a mixed complex system. To accurately achieve the target [CuI(NAPI)2]/[CuII(NAPI)2] ratio's careful handling of oxidatively labile CuI complexes and thoroughly deoxygenated reaction conditions were required. Looking back at this work, we considered the possibility of controlling [CuI(NAPI)2]/[CuII(NAPI)2] electrochemically, thus avoiding the need to handle the oxidatively labile CuI complexes. There are currently no reports of eATRP using Cu(NAPI)2 complexes, in either organic or aqueous media in the literature. We were inspired to investigate these complexes with a view to mediate eATRP at less reducing potentials and currents. Long term, we hope this will help to overcome some of the initial limitations associated with oxygen reduction (at more reducing potentials) in our related work in scanning electrochemical probe directed eATRP.50
To this end, herein we report for the first time the use of the N-propyl pyridineimine (NPPI) ligand to form CuII(NPPI)2 complexes for eATRP of OEGMA300. Well controlled polymerization (Đm ≈ 1.30) is possible and initial investigations into the mechanism suggest that an alternative electrochemically-triggered process is prevalent for these less-active copper complexes.
Results and discussion
Comparative CV of CuIIL complexes; CuIITPMA, CuIIMe6Tren and CuII(NPPI)2 were initially performed in solutions of the reaction mixture (10% (v/v) OEGMA300 in H2O) in the absence and presence of the initiator, hydroxyethyl-2-bromoisobutyrate (HEBiB) (Fig. S1–S3†). In the absence of HEBiB, each complex exhibited the [CuIIL]/[CuIL] redox process and as expected the standard reduction potential (Eθ ≈ E1/2 = Epc + Epa/2) shifted to less reducing potentials (vs. Ag/AgCl) going from CuIIMe6Tren (E1/2 = −0.40 V) to CuIITPMA (E1/2 = −0.21 V) to CuII(NPPI)2 (E1/2 = +0.02 V) respectively. In the presence of HEBiB the voltammograms of the CuIIMe6Tren and CuIITPMA complexes show a coupled increase in the cathodic current intensity (Epc) and decrease in the anodic current intensity (Epa). This is indicative of electrochemical reduction of CuIIL to CuIL followed by fast activation of HEBiB by the CuIL on the timescale of the CV (0.1 V s−1). In the case of CuII(NPPI)2 the coupled change in Epc and Epa was not observed. The currents decrease in both the cathodic and anodic scan suggesting that although the presence of HEBiB has an effect on the kinetics of electron transfer, the activation of HEBiB by CuI(NPPI)2 is slow on the timescale of the CV. These results are in agreement with the literature that suggests that with respect to kact, complexes with Me6Tren > TPMA ≫ NPPI.35,38Potentiostatic seATRP reactions using each complex were performed in undivided cells using an IKA ElectraSyn device. A commercial Pt-coated electrode (IKA) was employed as the cathode (WE), the anode (CE) was Al-wire and the reference electrode (RE) was Ag+/AgCl. For the bidentate NPPI ligand a ratio of [OEGMA300]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25] was used. When Eapp = E1/2 = +0.02 V the resistance in the system was too high preventing the IKA ElectraSyn from operating. However, when an overpotential of 60 mV was applied (Eapp = −0.04 V) polymerization was complete within 2 h yielding POEGMA300 with Mn,SEC = 9200 g mol−1 and Đm = 1.31 (Table 1, entry 1, Fig. S4†).
:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25] was used. When Eapp = E1/2 = +0.02 V the resistance in the system was too high preventing the IKA ElectraSyn from operating. However, when an overpotential of 60 mV was applied (Eapp = −0.04 V) polymerization was complete within 2 h yielding POEGMA300 with Mn,SEC = 9200 g mol−1 and Đm = 1.31 (Table 1, entry 1, Fig. S4†).
:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]; room temperaturea
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | E app/V | Time/h | Convk | M n,th /g mol−1 | M n,SEC /g mol−1 | Đ m |
| a [CuIIBr2] = 8.8 mM. b [OEGMA300] = 20% v/v. c [OEGMA300] = 30% v/v. d [CuIIBr2] = 4.4 mM. e [CuIIBr2] = 2.2 mM. f OEGMA500 used. g OEGMA1100 used. h [M]/[I] = 10. i [M]/[I] = 40. j [M]/[I] = 80. k Determined via1H NMR of reaction samples performed in D2O. l M n,th = [(conv./100 × DPn,th) x 300/500(e)/1100(f)] + 211. m From THF SEC. | ||||||
| 1 | −0.04 | 2 | 100% | 6211 | 9200 | 1.31 |
| 2 | −0.08 | 2 | 96% | 5971 | 11000 |
1.30 |
| 3 | −0.12 | 2 | 96% | 6211 | 12800 |
1.29 |
| 4 | −0.16 | 2 | 99% | 6211 | 10200 |
1.32 |
| 5b | −0.16 | 2 | 100% | 6211 | 10000 |
1.28 |
| 6c | −0.16 | 2 | 100% | 6211 | 9000 | 1.26 |
| 7d | −0.16 | 2 | 96% | 5971 | 18200 |
1.50 |
| 8e | −0.16 | 2 | 67% | 4231 | 21800 |
1.98 |
| 9f | −0.16 | 2 | 96% | 9811 | 14600 |
1.30 |
| 10g | −0.16 | 6.5 | 67% | 14951 |
14900 |
1.23 |
| 11h | −0.16 | 2 | 98% | 3151 | 7800 | 1.31 |
| 12i | −0.16 | 2 | 90% | 11011 |
14000 |
1.33 |
| 13j | −0.16 | 2 | 95% | 23011 |
21500 |
1.33 |
When Me6Tren and TPMA were employed as ligands ([OEGMA300]:[HEBiB]:[CuBr2]:[L] = [20]:[1]:[0.5]:[0.6]), Eapp = E1/2 was sufficient for the ElectraSyn to operate. In both cases, conversions were limited to <80% after 4 h and the control over the polymerization was poor (Đm > 4, Table S1†). This is likely due to the stoichiometry of CuIIBr2 employed which equates to [CuIIBr2] = 8.8 mM. Although, this is suitable for the less active CuII/NPPI system, it is much higher than is required for the so-called highly active complexes leading to higher than necessary [CuIL] and [R˙/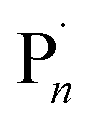 ] due to rapid over activation when Eapp = E1/2 which ultimately compromises the outcome of the polymerization.
] due to rapid over activation when Eapp = E1/2 which ultimately compromises the outcome of the polymerization.
Incrementally increasing the overpotential by 40 mV had little effect on the rate of the polymerization with conversions remaining high (>95%), and control being retained with Đm ≈ 1.30 (Table 1, entries 2–4, Fig. S5–S7†). The Eapp was then fixed at −0.16 V and [OEGMA300] was increased to 20% and 30% v/v respectively (Table 1, entries 5–6). There was no significant change in the control over the polymerization with low dispersities (Đm < 1.30) obtained (Fig. S8 and S9†). Kinetic analysis showed that quantitative conversions were obtained with 2 h. However, the semi-log plot showed that whilst the pseudo first order kinetics were observed at [OEGMA300] = 10% v/v, distinct deviations were apparent at the higher concentrations (Fig. S10†). The observed increase in rate throughout the reaction is in agreement with Haddleton and Perrier who attributed it to water and monomer completing with the ligand for coordination at the Cu centre thus affecting the CuII/CuI equilibrium.44 With this in mind, the remaining reactions were performed at [OEGMA300] = 10% v/v.
Decreasing the [Cu] from 8.8 mM to 4.4 mM and 2.2 mM had a detrimental effect on the control over the polymerization (Đm > 1.50, Table 1, entries 7–8). Increasing the length of the OEGMA monomer using OEGMA500 and OEGMA1100 had little effect on the control over the polymerization, with low dispersities retained (Đm < 1.30, Fig. S11 and S12†), though the rate of polymerization for OEGMA1100 was slower than OEGMA300/500 reaching 67% within 6.5 h (Table 1, entries 9–10).
Under the conditions established above (Eapp = −0.16 V; [M] = 10% v/v), the DPn,th was varied such that [M]:[I] = [10]/[20]/[40]/[80]:[1]. The polymerizations reached 90–98% conversion within 2 h, proceeding with good control over Mn and dispersity (Đm < 1.35; Table 1, entries 4, 11–13). An overlay of the SEC chromatograms shows the expected shift in the narrow molecular weight distributions to higher molecular weights as a function of [M]/[I] (Fig. 1A). A plot of Mn,SECvs. [M]/[I] indicates a linear increase in Mn,SEC as a function of [M]/[I], with slight deviations in Mn,SEC and Mn,th converging as [M]/[I] increased (Fig. 1B).
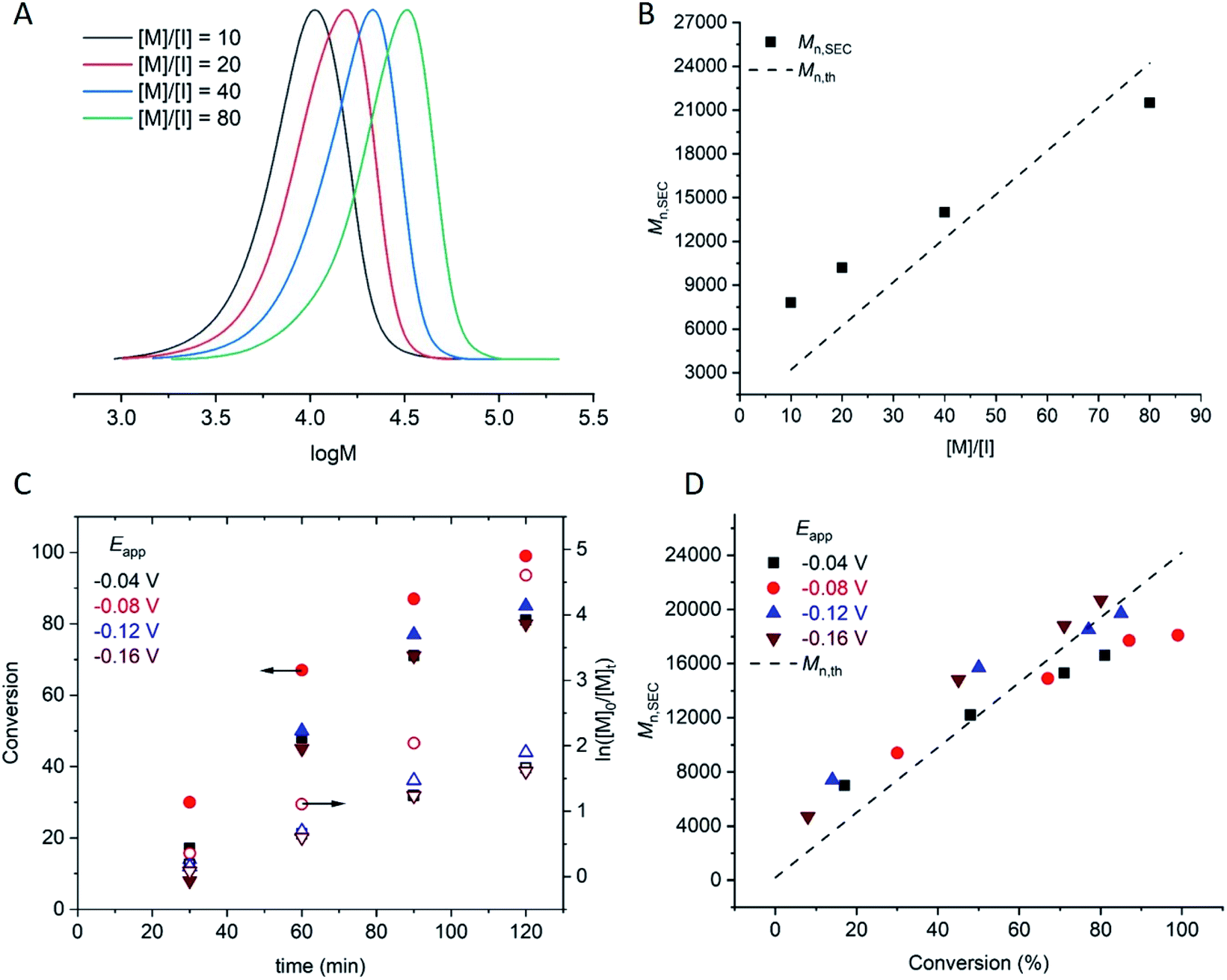 | ||
| Fig. 1 For seATRP of [OEGMA480]:[HEBiB]:[CuBr2]:[NPPI] = [M]:[1]:[0.5]:[1.25]; (A) SEC in THF showing a shift in molecular weight distributions as for [M] = 10, Mn = 7800 g mol−1, Đm = 1.31; [M] = 20, Mn = 10200 g mol−1, Đm = 1.32, [M] = 40, Mn = 14000 g mol−1, Đm = 1.33, [M] = 80, Mn = 21500 g mol−1, Đm = 1.33. (B) Plot of Mn,SEC as a function of [M]/[I] for [M] = 10, 20, 40, 80, [I] = 1, (Eapp = −0.16 V). (C) Conversion and pseudo first order kinetic plots as a function of Eapp. (D) Evolution of the Mn,SEC with conversion during polymerizations performed at different Eapp ([M]/[I] = 80). | ||
Kinetic analyses of the polymerizations performed with [M]:[I] = [80]:[1] revealed that the apparent rate constant for propagation was kappp = 0.0167 min−1 at Eapp = −0.04 V (Fig. 1C). Initially, more reducing potentials (Eapp = −0.08 V) resulted in a small increase in the rate of polymerization (kappp = 0.0456 min−1). However, at higher overpotentials (Eapp = −0.12 V, −0.16 V), the rate decreased back to kappp = 0.0201 min−1 and 0.0174 min−1 respectively. At these potentials, Eapp is close to Epc at which point the reduction of CuII/L to CuI/L is not governed by the electrode potential and is limited by the rate of diffusion of accumulated Cu/L species to and from the electrode surface to and from the bulk. Irrespective of Eapp, a linear increase in Mn,SEC as a function of conversion was observed with good agreement with the theoretical molecular weight (Mn,th) (Fig. 1D).
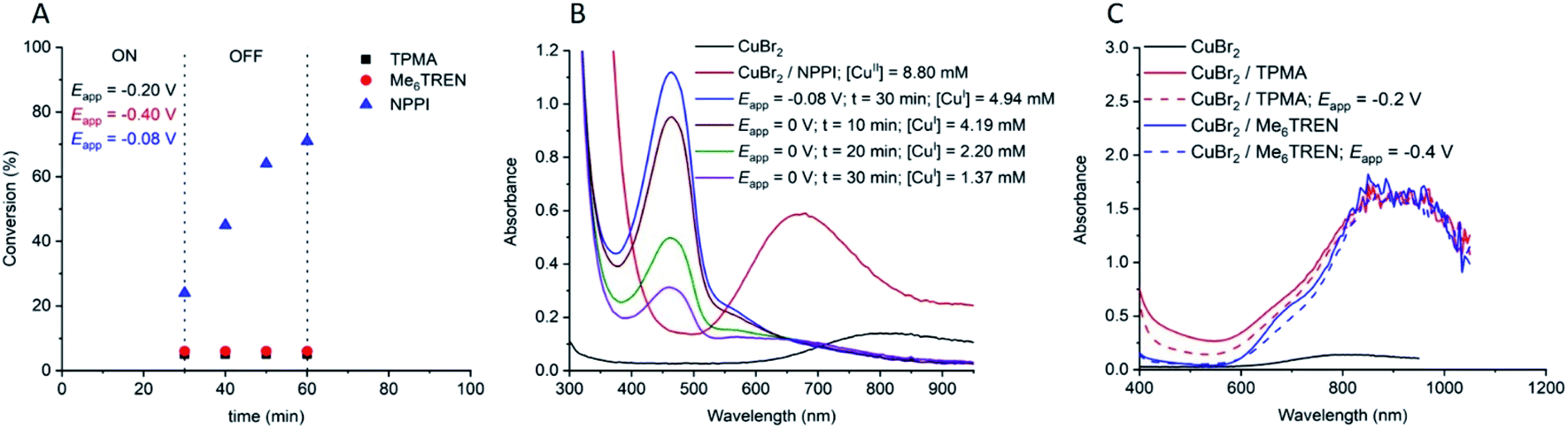
A hallmark of eATRP is the temporal control conferred by switching the potential/current on and off. At reducing potentials CuII/L is reduced to CuI/L leading to activation of dormant chains which can undergo propagation and subsequent deactivation events via the proposed ATRP mechanism. If the potential is switched off, or an oxidising potential is applied, reduction of CuII/L nolonger occurs so activation of the dormant chains stops and the polymerization is halted. Conducting this experiment using [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25] and applying Eapp = −0.16 V ([M] = 10% v/v), conversion reached >50% within 20 min (Fig. 2). The potential was then switched off (Eapp = 0 V) and stirring was continued for a further 20 min, after which conversion unexpectedly increased to >80%. The polymerization reached >90% conversion through an additional ‘on’ (20 min) and ‘off’ (20 min) cycle, indicating that CuII(NPPI)2 lacked the temporal control associated with eATRP. The lack of temporal control with this less active catalyst system is in agreement with reported differences in temporal control related to catalyst activity observed in photo-ATRP.51
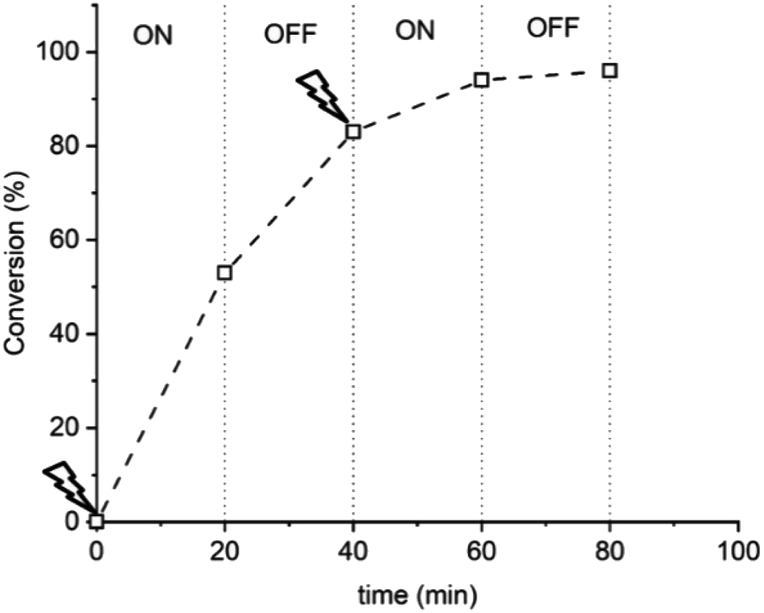 | ||
| Fig. 2 Conversion vs. time plot demonstrating a lack of temporal control afforded by seATRP with [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]:[0.15]; Eapp = −0.16 V. | ||
Unlike in eATRP reactions using CuII(Me6Tren) and CuII(TPMA) complexes, it was observed that the reaction solutions containing CuII(NPPI)2 changed colour, from green to brown, during electrolysis (Fig. S14†). The brown colour resembled that reported in early aqueous ATRP using CuI(NAPI)2 complexes.44–46 Thus, it was hypothesized that the overpotentials applied and the less activating nature of the CuI(NPPI)2 complex, resulted in accumulation of stable CuI(NPPI)2 in the reaction media which was capable of continuing to mediated the polymerization of OEGMA when Eapp was removed.
To explore this hypothesis, the temporal control experiment was repeated using [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]. After the electrolysis period (Eapp = −0.08 V; tEapp = 30 min), the reaction solution was brown, indicative of CuI(NPPI)2 accumulation, and conversion had reached 33% (Fig. S15A†). Concurrently, electrolysis was stopped and sparging with compressed air was commenced to rapidly introduce O2 into the reaction solution to stop the polymerization. The solution quickly changed colour from brown to green, indicative of oxidation of CuI(NPPI)2 to CuII(NPPI)2 and no further conversion was observed (Fig. S15B†). In an attempt to re-initiate the polymerization, the reaction solution was sparged for second time, this time with N2 to displace the O2 previously added to the solution, prior to a second period of electrolysis. Pleasingly, re-initiation was observed (Eapp = −0.08 V; tEapp = 30 min) with the polymerization reaching 64% conversion (Fig. S15C†), yielding POEGMA300 with Mn,SEC = 9800 g mol−1 and Đm = 1.25 (Fig. S16†) which is comparable to the POEGMA300 obtain during the constant electrolysis reactions.
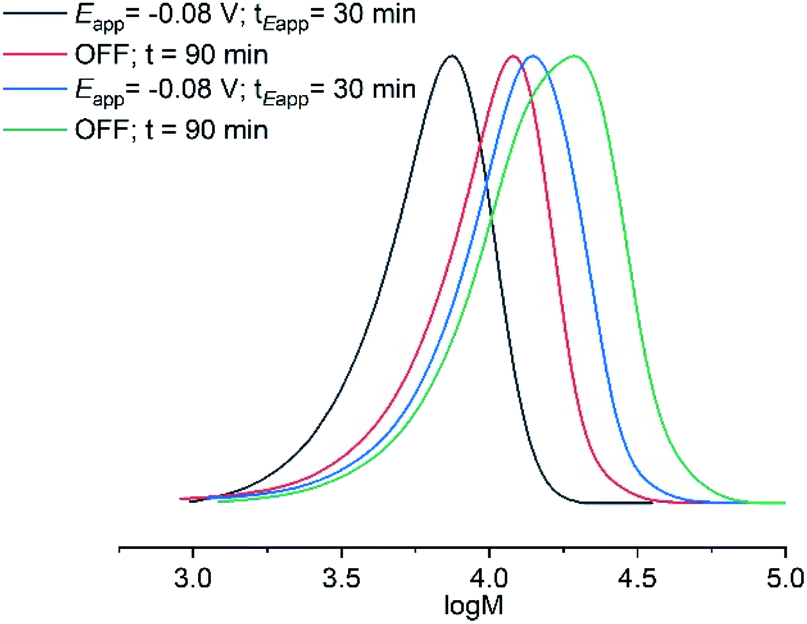
A series of experiments was then performed in which reaction solutions ([OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]) were electrolyzed at constant potential (Eapp = −0.08 V) for increasing periods of time (tEapp = 5, 10, 20, 30 min) before the potential was removed (Eapp = 0 V). Samples were taken for analysis after electrolysis and at regular intervals after the potential was removed. Increasing the initial electrolysis times led to an increase in initial conversion from 2% (Eapp = −0.08 V; tEapp = 5 min) to 22% (Eapp = −0.08 V; tEapp = 30 min). In all experiments monomer conversion continued upon removal of Eapp (Fig. 3A). At shorter electrolysis times, conversion continued up to a total reaction time of 60 min resulting in final conversions of 18% and 35% when tEapp = 5 and 10 min respectively. Increasing the initial electrolysis time to 20 min yielded initial conversions of 11% with monomer conversion continuing thereafter to reach a final conversion of 56% after a total reaction time of 70 min. When the reaction solution was electrolyzed for 30 min monomer conversion continued for a total reaction time of 90 min, reaching 93% conversion. Kinetic analysis of these reactions revealed that the rate of the reaction also increased from kappp = 0.0028 min−1 when tEapp = 5 min to kappp = 0.0425 min−1 when tEapp = 30 min (Fig. 3B). An overlay of the SEC chromatograms shows that the polymerization continues after the initial electrolysis period with the molecular weight distributions shifting to higher molecular weights as a function of time (Fig. 3C). The final polymer obtained (Eapp = −0.08 V; tEapp = 30 min) was comparable to the polymer obtained by uninterrupted electrolysis (Table 1; entry 2) with Mn,SEC = 9300 g mol−1 and Đm = 1.33.
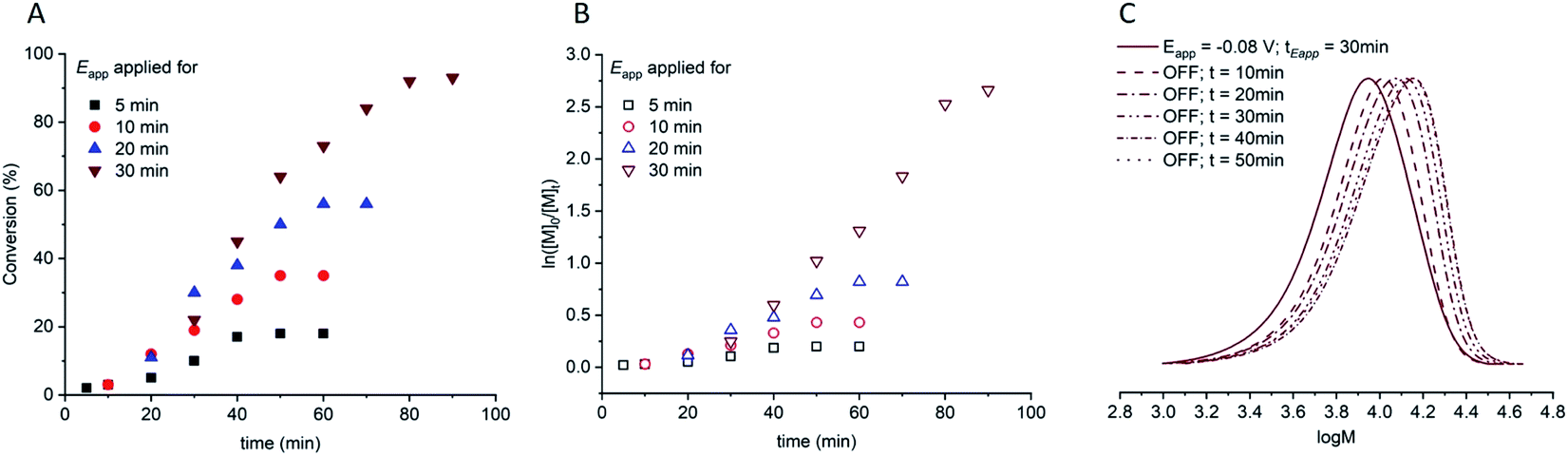 | ||
| Fig. 3 For triggered seATRP of [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]; (A) conversion vs. time plot for polymerizations with different tEapp. (B) Pseudo first order kinetic plots for polymerizations for tEapp = 5 min, kappp = 0.0028 min−1; tEapp = 10 min, kappp = 0.0046 min−1; tEapp = 20 min, kappp = 0.0218 min−1; tEapp = 30 min, kappp = 0.0425 min−1. (C) SEC in THF showing the evolution of the molecular weight distribution after electrolysis (Eapp = −0.08 V, tEapp = 30 min, solid line) and at 10 minutes intervals after the potential was removed (Eapp = 0 V, dashed lines, final Mn,SEC = 9300 g mol−1, Đm = 1.33). | ||
Quantification of the end group fidelity using conventional 1H NMR analysis was not possible as poly(methacrylates) do not contain an ω-methine proton to integrate against signals at the α-chain end. To exemplify end group fidelity, a chain extension experiment was performed. Homopolymerization of OEGMA300 was performed using [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25] (Eapp = −0.08 V). After electrolysis for 30 min (Table 2, entry 1) and stirring at room temperature in the absence of electrolysis for an additional 90 min near quantitative conversion was obtained (Table 2, entry 2, Fig. S17†). At this point a second aliquot of OEGMA300 (1 mL, [OEGMA300]:[POEGMA300-Br] = [20]:[1]) was added and electrolysis was again applied (Eapp = −0.08 V) for 30 min (Table 2, entry 3) followed by stirring in the absence of electrolysis for an additional 90 min reaching a final conversion of 81% (Table 2, entry 4). A clear shift in the molecular weight distribution was evident via SEC analysis (Fig. 4). The molecular weight of the final POEGMA300 obtained (Mn,SEC = 16600 g mol−1) was in reasonable agreement to the theoretical molecular weight (Mn,th = 12200 g mol−1) relative to the homopolymerizations performed. Although this result exemplifies good chain-end fidelity, there is scope for optimization based on a gradual increase in tailing to low molecular weight, which increased during the course of the reaction resulting in a gradual increase in dispersity (Đm = 1.27–1.45, Table 2).
:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]; followed by in situ chain extension using OEGMA300 (20 eq.); Eapp = −0.08 V; tEapp = 30 min; room temperaturea
| Entry | E app/V | Time/min (ttotal) | Convb | M n,th/g mol−1 | M n,SEC /g mol−1 | Đ m |
|---|---|---|---|---|---|---|
| a [CuIIBr2] = 8.8 mM. b Determined via1H NMR of reaction samples performed in D2O. c M n,th = [(conv./100 × DPn,th) × 300] + 211. d M n,th = [(conv./100 × DPn,th) × 300] + 6211. e From THF SEC. | ||||||
| 1 | −0.08 | 30 (30) | 33% | 2191c | 7300 | 1.26 |
| 2 | — | 90 (120) | >99% | 6211c | 11300 |
1.31 |
| 3 | −0.08 | 30 (150) | 57% | 9631d | 13800 |
1.34 |
| 4 | — | 90 (240) | 81% | 11071d |
16600 |
1.45 |
|
||||||
 | ||
| Fig. 4 SEC in THF showing the evolution of the molecular weight distribution during electrochemically triggered eATRP of OEGMA300 ([OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]; Eapp = −0.08 V; tEapp = 30 min) and chain extension ([OEGMA300]:[POEGMA300-Br] = [20]:[1]; Eapp = −0.08 V; tEapp = 30 min) (Table 2). | ||
The electrochemically triggered reaction conditions were also compatible with polymerizations of OEGMA300 targeting higher molecular weights. When DPn,th = 200, the reaction solution was electrolyzed for 30 min (Eapp = −0.08 V) leading to 13% conversion. The reaction continued in the absence of electrolysis for an additional 90 min, reaching 89% conversion (Fig. S18†) furnishing POEGMA300 with relatively low dispersity (Đm = 1.32, Fig. S19†). To expand the monomer scope, 2-N-morpholinoethyl methacrylate (DPn,th = 200) was electrolyzed for 30 min (Eapp = −0.08 V) resulting in 40% conversion. The reaction was allowed to continue in the absence of electrolysis for an additional 90 min, reaching 65% conversion (Fig. S20 and S21†).
To explore the mechanism, the polymerization using [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25] (tEapp = 30 min; Eapp = −0.08 V) was repeated and the electrochemical reduction of CuII(NPPI)2 to CuI(NPPI)2 followed by UV-vis spectroscopy (Fig. 5). Prior to electrolysis the reaction solution was green and the characteristic CuII(NPPI)2 absorbance band was present at λ = 670 nm, assigned to the d–d transitions of the CuII centre. After electrolysis the reaction solution was brown in colour, qualitatively confirming the reduction of CuII(NPPI)2 to CuI(NPPI)2. UV-vis of the reaction solution immediately after electrolysis showed disappearance of the absorbance band at λ = 670 nm and appearance of a new, strong absorbance band at λ = 465 nm, confirming reduction of CuII(NPPI)2 to CuI(NPPI)2. The absorbance at λ = 465 nm was assigned to MLCT between the CuI centre and the π* of the surrounding NPPI ligands, as reported for other bipyridyl and/or diimine based complexes of CuI.52
 | ||
| Fig. 5 (A) Conversion vs. time plot for seATRP of [OEGMA300]:[HEBiB]:[CuBr2]:[L] = [20]:[1]:[0.5]:[L]. For NPPI [L] = [1.25] and for TPMA and Me6Tren [L] = [0.6] (tEapp = 30 min). (B) UV-vis traces showing the reduction of CuII(NPPI)2 to CuI(NPPI)2 and the change in [CuI(NPPI)2] during the triggered seATRP of [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]. Using ε = 1359 M−1 cm−1 [CuI(NPPI)2] was quantified at each point (see ESI† for calibration and calculation details). (C) UV-vis traces of CuII(TPMA) and CuII(Me6Tren) before and after electrolysis. Eapp,TPMA = −0.20 V, Eapp,Me6Tren = −0.40 V, tEapp = 30 min. | ||
In order to quantify the concentration of CuI(NPPI)2 present after electrolysis, a calibration plot of CuI(NPPI)2 was used to determine the molar extinction coefficient of CuI(NPPI)2 (ε = 1359 M−1 cm−1, Fig. S22†). Prior to electrolysis, the concentration of CuII(NPPI)2 in the reaction solution was 8.8 mM. After electrolysis for 30 min, conversion reached 24% (Fig. 5A) and [CuI(NPPI)2] was measured and found to be 4.94 mM (Fig. 5B). The reaction was again allowed to continue in the absence of an applied potential (Eapp = 0 V). Though the polymerization continued, the colour of the reaction solution gradually changed from brown back to green over the course of the reaction. Further UV-vis analysis of the reaction solution allowed [CuI(NPPI)2] to be followed, revealing a steady decrease over time eventually reaching 1.37 mM after 30 min at which point the reaction had reached 72% conversion.
Identical analyses were performed during polymerization of OEGMA300 using CuIITPMA and CuIIMe6-Tren. Due to the tetradentate nature of TPMA and ME6-Tren, the reaction solutions were composed of [OEGMA300]:[HEBiB]:[CuBr2]:[TPMA/Me6-Tren] = [20]:[1]:[0.5]:[0.6]. Both CuIITPMA and CuIIMe6-Tren produced blue solutions prior to electrolysis. Qualitatively, no colour change was observed upon electrolysis. The Eapp employed was selected based on the E1/2 (−0.2 V, TPMA; −0.4 V Me6-Tren; Fig. S1 and S2†) and electrolysis was initially performed for 30 min before the potential was removed and stirring continued at room temperature. The progress of the reactions was followed by 1H NMR revealing ∼5% conversion after the initial period of electrolysis. UV-vis analysis showed very little change in the absorbance spectra of each complex (Fig. 5C) and unlike the CuII(NPPI)2 system, no further conversion of monomer to polymer was observed when the reaction was continued for 30 min at Eapp = 0 V (Fig. 5A). This is perhaps unsurprising considering the relative activity of the CuITPMA and CuIMe6-Tren complexes relative to CuI(NPPI)2. Thus we repeated the reaction using bipyridine (bipy) to form CuI(bipy)2in situ which has intermediate activity relative to the highly active complexes derived from Me6-Tren/TPMA and the less active complex derived from NPPI. Similar to CuII(NPPI)2, initial conversion in the presence of CuII(bipy)2 increased with increasing electrolysis time (Eapp = −0.08 V; tEapp = 10–30 min). However, in the absence of electrolysis polymerization was only maintained for 10–20 min reaching only moderate final conversions (< 65%, Eapp = −0.08 V; tEapp = 30 min, Fig. S23†). This suggests that the ability to conduct electrochemically triggered eATRP is directly related to the activity (kact and KATRP) of the Cu-complex employed.
Overall, these results consolidate the hypothesis that the less activating nature of the CuI(NPPI)2 complex, and its stability in water results in its accumulation in the reaction media. The accumulated CuI(NPPI)2 is then capable of mediating the polymerization of OEGMA when Eapp was removed. We therefore propose that Cu complexes containing pyridine-imine ligands (CuII(NAPI)2) follow an electrochemically-triggered, rather than electrochemically mediated, ATRP mechanism wherein Eapp is only required in order to generate the required [CuI(NAPI)2] to initiate and maintain the polymerization reaction.
Finally, to simplify the reaction set up further, current vs. time (I vs. t) graphs obtained from reactions performed under potentiostatic conditions (Fig. 6A) were used to design a step-wise current profile to enable the electrochemically triggered polymerizations to be performed using a 2-electrode current controlled configuration. Using [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25], a 3-step current profile was initially applied over 30 min (Iapp = −3.5 mA, 8 min; −1.9 mA, 7 min; −0.5 mA, 15 min) resulting in 12% conversion. At this point the reaction continued in the absence of electrolysis for a further 120 min reaching 80% conversion (Fig. S24†) with comparable control (Mn,SEC = 11600 g mol−1; Đm = 1.25, Fig. 6B) to the polymerizations performed with a potentiostatic trigger. Considering future translation to flow electrolysis, it would be beneficial to trigger these reactions using a single current, truly galvanostatic reaction configuration. With this in mind the reaction was repeated with Iapp = −2.0 mA leading to 16% conversion after the 30 min electrolysis period reaching 95% after a further 120 min stirring in the absence of electrolysis (Fig. S25†). This is very promising for intensification to flow electrolysis, though it should be noted that under these conditions, a higher overall charge is passed during the reaction which has an effect on the outcome of polymerization. Whilst the control respect to the dispersity is retained (Đm = 1.28, Fig. S26†), the Mn,SEC (14300 g mol−1) and Mn,th (6211 g mol−1) diverge relative the potentiostatic and step-wise current profile triggered reactions, leaving scope for optimisation in future. We attribute the divergence in the Mn,SEC and Mn,th to the gradual increase in the potential (required to maintain Iapp) during the initial electrolysis period. This leads to an increase in [CuI(NPPI)2] and subsequently 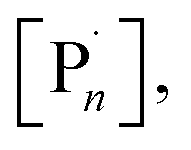 leading to increased termination and reduced initiator efficiency, relative to the potentiostatic and step-wise current profile triggered polymerizations.
leading to increased termination and reduced initiator efficiency, relative to the potentiostatic and step-wise current profile triggered polymerizations.
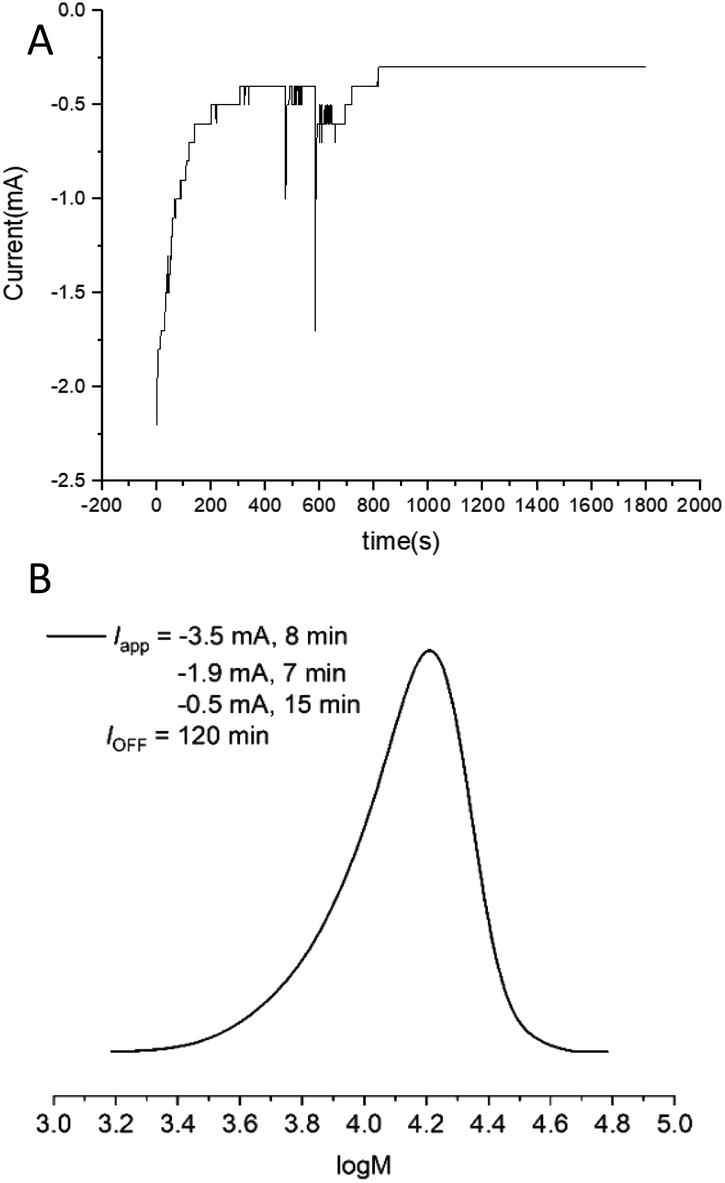 | ||
| Fig. 6 (A) I vs. t plot for the electrochemically triggered seATRP reaction of OEGMA300 performed under potentiostatic conditions [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]; Eapp = −0.08 V; tEapp = 30 min, Q = 0.78 C). (B) Molecular weight analysis of the final polymer formed from the same reaction performed using step-wise current profile min (Iapp = −3.5 mA, 8 min; −1.9 mA, 7 min; −0.5 mA, 15 min). SEC in THF Mn,SEC = 11600 g mol−1, Đm = 1.25. | ||
Conclusions
In summary, seATRP using Cu(NPPI)2 complexes in aqueous solution has been reported for the first time. Typical electrolysis conditions require less reducing potentials (Eapp = −0.08 V) than complexes derived from Me6-Tren and TPMA. Using OEGMA300 as monomer, a range of molecular weights have been targeted with the polymerizations typically complete within 2 h, yielding POEGMA300 with good control over the molecular weight distribution (Đm < 1.35). However, the defining ‘on-off’ control experiment revealed that the polymerizations were not under complete electrochemical control, as monomer conversion continued in the absence of Eapp. This is contrary to previous reports using more active CuIIL complexes. Through electrochemically triggered control experiments and UV-vis spectroscopy we have been able to propose that these less activating complexes, that stabilize CuI more than CuII, follow an alternative, previously unreported, electrochemically-triggered polymerization pathway. The polymerizations proceed with good control enabling a range of molecular weights to be targeted (DPn,th = 20–200). In situ chain extension is also possible alluding to potential application to the synthesis of block copolymers. The reaction set-up can also be further simplified to a 2-electrode, galvanostatic configuration which is promising for future intensification through translation to flow electrolysis. However, though suitable for eATRP at reduced catalyst loadings, more active ligands such as Me6-Tren, TPMA and bipy do not support the electrochemically-triggered polymerization pathway. Indeed, the ability to conduct electrochemically triggered eATRP seems to be directly related to the activity of the Cu-complex and can be related to the kact and KATRP of the complexes employed. Thus, it is possible that other ligands that stabilize CuI over CuII (e.g. other substituted NAPI and 1,4-diazabutadiene ligands) could also follow or favour this electrochemically triggered pathway.Data availability
Experimental procedures and data supporting the research, not presented in the main manuscript, is included in the ESI.† Raw data files are available from the Warwick Research Archive Portal (WRAP, https://wrap.warwick.ac.uk) and from the corresponding author on request.Author contributions
Boyu Zhao: investigation; methodology; formal Analysis; validation; visualization; writing – original draft. Fred Pashley-Johnson: investigation; methodology; formal analysis; validation; visualization; writing – original draft. Bryn Jones: Methodology; formal analysis; supervision; writing – review and editing. Paul Wilson: conceptualization; funding acquisition; investigation; methodology; project administration; resources; supervision; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Polymer Characterization Research Technology Platforms for maintenance and access to facilities. P. W. thanks the Royal Society and Tata companies for the award of a University Research Fellowship (URF\R1\180274).Notes and references
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- S. D. Minteer and P. Baran, Acc. Chem. Res., 2020, 53, 545–546 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391–11394 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- Y. Wang, M. Fantin, S. Park, E. Gottlieb, L. Fu and K. Matyjaszewski, Macromolecules, 2017, 50, 7872–7879 CrossRef CAS PubMed.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Curr. Opin. Electrochem., 2018, 8, 1–7 CrossRef CAS.
- P. Chmielarz, Polym. Adv. Technol., 2018, 29, 470–480 CrossRef CAS.
- P. Chmielarz, Polimery, 2021, 62, 642–649 CrossRef.
- P. Chmielarz, Polym. Adv. Technol., 2017, 28, 1787–1793 CrossRef CAS.
- E. Trevisanello, F. De Bon, G. Daniel, F. Lorandi, C. Durante, A. A. Isse and A. Gennaro, Electrochim. Acta, 2018, 285, 344–354 CrossRef CAS.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 603–609 CrossRef CAS.
- T. Wu, E. R. Lankshear and A. J. Downard, Chemelectrochem, 2019, 6, 5149–5154 CrossRef CAS.
- D. Li, J. Y. Wu, S. Y. Yang, W. J. Zhang, X. Q. Niu, Y. H. Chen and F. Ran, New J. Chem., 2018, 42, 2692–2701 RSC.
- I. Zaborniak, A. Macior and P. Chmielarz, Materials, 2020, 13 Search PubMed.
- I. Zaborniak, P. Chmielarz, M. R. Martinez, K. Wolski, Z. Y. Wang and K. Matyjaszewski, Eur. Polym. J., 2020, 126 Search PubMed.
- F. De Bon, S. Marenzi, A. A. Isse, C. Durante and A. Gennaro, Chemelectrochem, 2020, 7, 1378–1388 CrossRef CAS.
- B. Y. Zhao, M. Mohammed, B. A. Jones and P. Wilson, Chem. Commun., 2021, 57, 3897–3900 RSC.
- M. Fantin, S. Park, Y. Wang and K. Matyjaszewski, Macromolecules, 2016, 49, 8838–8847 CrossRef CAS PubMed.
- M. Fantin, P. Chmielarz, Y. Wang, F. Lorandi, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2017, 50, 3726–3732 CrossRef CAS PubMed.
- I. Zaborniak and P. Chmielarz, Polym. Adv. Technol., 2020, 31, 2806–2815 CrossRef CAS.
- P. Chmielarz, J. J. Yan, P. Krys, Y. Wang, Z. Y. Wang, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2017, 50, 4151–4159 CrossRef CAS.
- N. Shida, Y. Koizumi, H. Nishiyama, I. Tomita and S. Inagi, Angew. Chem., Int. Ed., 2015, 54, 3922–3926 CrossRef CAS PubMed.
- B. Li, B. Yu, W. T. S. Huck, W. Liu and F. Zhou, J. Am. Chem. Soc., 2013, 135, 1708–1710 CrossRef CAS PubMed.
- P. Chmielarz, P. Krys, Z. Wang, Y. Wang and K. Matyjaszewski, Macromol. Chem. Phys., 2017, 218, 1700106 CrossRef.
- B. Li, B. Yu, W. T. S. Huck, F. Zhou and W. Liu, Angew. Chem., Int. Ed., 2012, 51, 5092–5095 CrossRef CAS PubMed.
- S. Park, P. Chmielarz, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2015, 54, 2388–2392 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2016, 7, 5357–5365 RSC.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- A. J. D. Magenau, N. Bortolamei, E. Frick, S. Park, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 4346–4353 CrossRef CAS.
- J.-K. Guo, Y.-N. Zhou and Z.-H. Luo, AIChE J., 2015, 61, 4347–4357 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- A. A. Isse, F. Lorandi and A. Gennaro, Curr. Opin. Electrochem., 2019, 15, 50–57 CrossRef CAS.
- F. Lorandi, M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Electrochim. Acta, 2018, 260, 648–655 CrossRef CAS.
- W. A. Braunecker, N. V. Tsarevsky, A. Gennaro and K. Matyjaszewski, Macromolecules, 2009, 42, 6348–6360 CrossRef CAS.
- M. Fantin, A. A. Isse, K. Matyjaszewski and A. Gennaro, Macromolecules, 2017, 50, 2696–2705 CrossRef CAS.
- G. R. Jones, A. Anastasaki, R. Whitfield, N. Engelis, E. Liarou and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 10468–10482 CrossRef CAS PubMed.
- S. Perrier, S. P. Armes, X. S. Wang, F. Malet and D. M. Haddleton, J. Polym. Sci. Part A: Polym. Chem., 2001, 39, 1696–1707 CrossRef CAS.
- X. S. Wang, F. L. G. Malet, S. P. Armes, D. M. Haddleton and S. Perrier, Macromolecules, 2001, 34, 162–164 CrossRef CAS.
- S. Perrier and D. M. Haddleton, Macromol. Symp., 2002, 182, 261–272 CrossRef CAS.
- Y. Li, S. P. Armes, X. Jin and S. Zhu, Macromolecules, 2003, 36, 8268–8275 CrossRef CAS.
- S. B. Lee, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2003, 4, 1386–1393 CrossRef CAS PubMed.
- N. V. Tsarevsky, T. Pintauer and K. Matyjaszewski, Macromolecules, 2004, 37, 9768–9778 CrossRef CAS.
- E. E. Oseland, Z. J. Ayres, A. Basile, D. M. Haddleton, P. Wilson and P. R. Unwin, Chem. Commun., 2016, 52, 9929–9932 RSC.
- S. Dadashi-Silab, I.-H. Lee, A. Anastasaki, F. Lorandi, B. Narupai, N. D. Dolinski, M. L. Allegrezza, M. Fantin, D. Konkolewicz, C. J. Hawker and K. Matyjaszewski, Macromolecules, 2020, 53, 5280–5288 CrossRef CAS.
- M. G. Fraser, H. van der Salm, S. A. Cameron, A. G. Blackman and K. C. Gordon, Inorg. Chem., 2013, 52, 2980–2992 CrossRef CAS PubMed.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01832b |
| This journal is © The Royal Society of Chemistry 2022 |