
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed chemoselective dehydrogenation through radical translocation under visible light†
Wan-Lei
Yu
ad,
Zi-Gang
Ren
a,
Ke-Xing
Ma
a,
Hui-Qing
Yang
c,
Jun-Jie
Yang
a,
Haixue
Zheng
*b,
Wangsuo
Wu
d and
Peng-Fei
Xu
 *abd
*abd
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: xupf@lzu.edu.cn
bState Key Laboratory of Veterinary Etiological Biology, College of Veterinary Medicine, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, China. E-mail: haixuezheng@163.com
cHenan and Macquarie University Joint Centre for Biomedical Innovation, School of Life Sciences, Henan University, Kaifeng 475004, China
dFrontiers Science Center for Rare Isotopes, Lanzhou University, Lanzhou, China
First published on 15th June 2022
Abstract
The transformations that allow the direct removal of hydrogen from their corresponding saturated counterparts by the dehydrogenative strategy are a dream reaction that has remained largely underexplored. In this report, a straightforward and robust cobaloxime-catalyzed photochemical dehydrogenation strategy via intramolecular HAT is described for the first time. The reaction proceeds through an intramolecular radical translocation followed by the cobalt assisted dehydrogenation without needing any other external photosensitizers, noble-metals or oxidants. With this approach, a series of valuable unsaturated compounds such as α,β-unsaturated amides, enamides and allylic and homoallylic sulfonamides were obtained in moderate to excellent yields with good chemo- and regioselectivities, and the synthetic versatility was demonstrated by a range of transformations. And mechanistic studies of the method are discussed.
Introduction
Alkenes, a fundamental cornerstone of the organic synthetic toolbox, are present in a broad range of important biologically active compounds and extensively used as the most versatile intermediates and feedstocks in industries and fine chemicals.1 Compared with traditional synthetic methods, which relied on pre-functionalized starting materials, the development of highly selective dehydrogenation as in the desaturase enzyme system2 has been attracting considerable attention. In general, highly reactive radical species are the key intermediates for intra- or intermolecular selective HAT efficiently.3,4 In 2012, Baran et al. developed a TEMPO-mediated remote dehydrogenation with aryltriazenes as aryl radical precursors.3b Recently, Gevorgyan et al. have developed elegant strategies which employed reactive C-radical species produced from aryl or alkyl iodides to mediate the remote dehydrogenation by palladium photocatalysis.3c–e The same strategy has also been demonstrated efficiently with N-radical species by the Studer3f and Nagib3g groups (Scheme 1a). In addition, Huang,4a Sorensen4b and Morandi4cet al. reported dehydrogenation of aliphatics by intermolecular HAT strategies. However, polar effects usually control the regioselectivity of the HAT process from hydridic or neutral sites rather than acidic sites, and thus more challenging and difficult amide α,β-dehydrogenation is still underdeveloped. tamide α,β-dehydrogenation is more challenging and difficult.In addition, a series of unexpected byproducts tended to form due to low regioselectivity in the dehydrogenation process, which limited further explorations and applications along this line. As a consequence, developing a cost-effective methodology for more broadly applicable and general dehydrogenation with high regioselectivity remains an unsolved challenge in modern synthetic chemistry.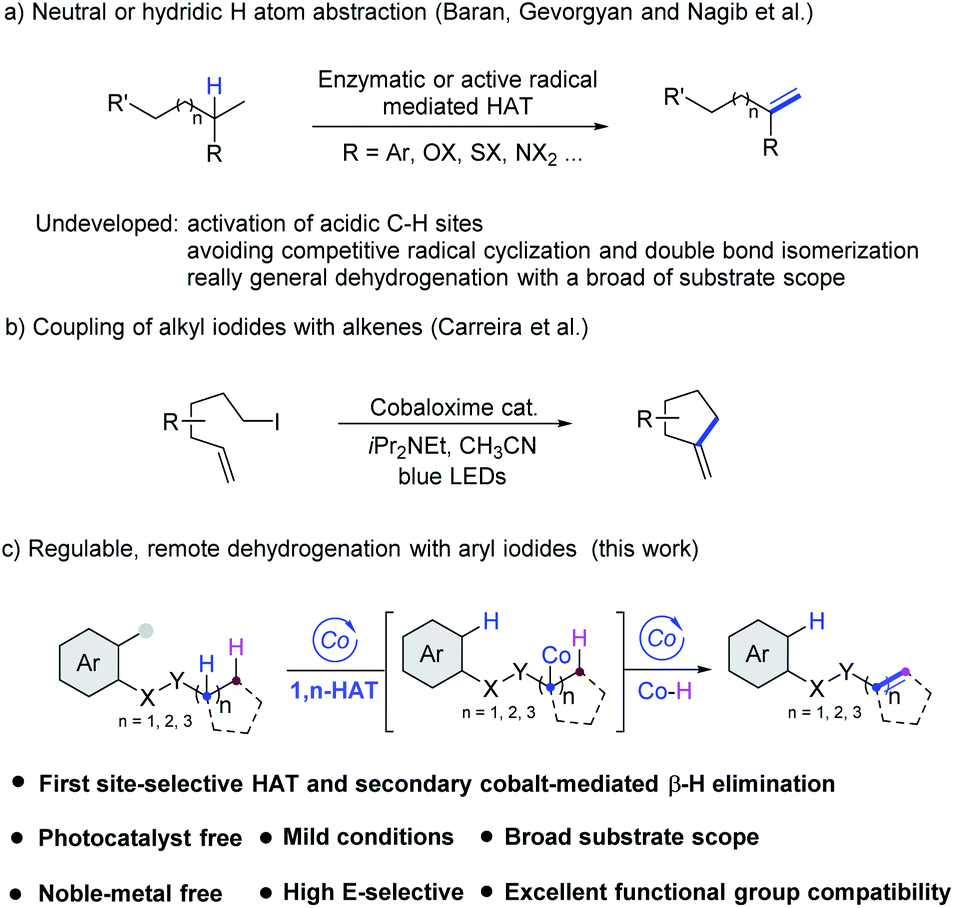 | ||
| Scheme 1 Dehydrogenation strategies for alkene synthesis. | ||
The application of earth–abundant transition metal cobalt complexes for both efficient and eco-friendly organic synthesis has been actively explored in the past several years.5 Cobaloxime as a mimic of vitamin B12, was developed as one of the powerful catalysts to carry out water splitting and chemical dehydrogenation that occur efficiently under mild conditions.6 In 2011, Carreira et al. reported cobaloxime-catalyzed intramolecular Heck reactions, and amine base conditions were demonstrated to convert the hydridocobaloxime to cobalt(I) species (Scheme 1b).7 Recently, Wu et al. reported the oxidation of H-phosphine oxide by excited cobaloxime to generate the phosphinoyl radical for subsequent addition to terminal alkenes or alkynes without using a photocatalyst.8 Subsequently, West et al. reported a vitamin B12-catalyzed alkyl halide alkenylation where the Co(I) intermediate was used to access the alkyl–Co(III) species by SN2 displacement.9 And Leonori et al. reported E2-type eliminations on alkyl halides merging halogen-atom transfer (XAT) and cobalt catalysis to obtain high olefin positional selectivity.10 In this context, taking advantage of the ability of cobaloxime catalysts to mediate transformations that allow the direct generation of unsaturated alkenes from their corresponding saturated counterparts by the dehydrogenation strategy is very attractive. Despite many reports involving alkylcobalt intermediates generated from the alkylation of cobalt catalysts with alkyl electrophiles, the application of aryl halides is still limited.11 On the basis of our previous exploration of alkene dehydrogenative silylation,12 we sought to adapt the photochemistry of cobaloxime13 into a synthetic method to achieve remote site-selective dehydrogenation (Scheme 1c). In these transformations, a variety of target compounds including α,β-unsaturated amides, enamides, allylic- or homoallylic amine derivatives, etc. could be smoothly afforded via intramolecular 1,n-HAT (n = 5–7) processes with excellent olefin positional selectivities, wide substrate scope, and without any external photosensitizers under extremely mild reaction conditions.
Results and discussion
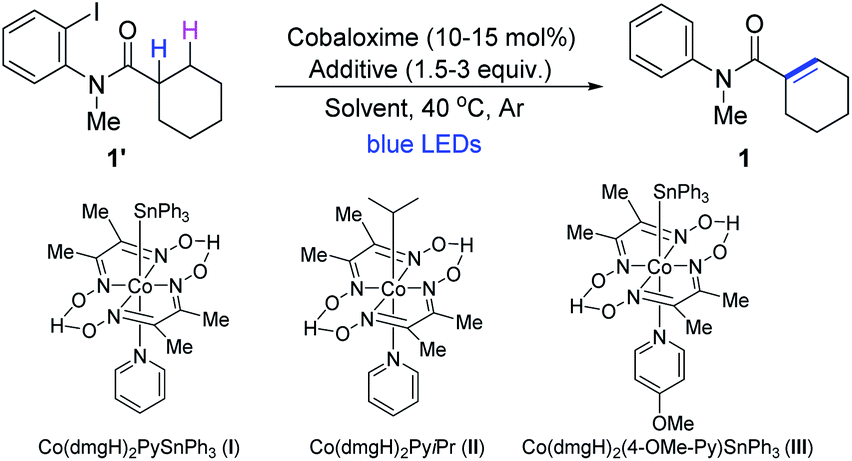
To explore the feasibility of cobaloxime catalyzed dehydrogenation reactions, we started to apply the 1,5-HAT process14 to synthesize α,β-unsaturated amides using N-(2-iodophenyl)-N-methylcyclohexanecarboxamide as a model substrate. To our delight, when the substrate was treated with a catalytic amount of the cobaloxime catalyst (I) (10 mol%), 2.0 equivalent of NEt3, at 40 °C for 24 h under irradiation with blue LEDs, the expected dehydrogenation product 1 was smoothly obtained in a yield of 68% (Table 1, entry 1). Further variations on the ligands of cobaloxime complexes showed that different cobaloximes with triphenyltins were all suitable for catalyzing the chemical transformations. However, only a trace of the target product 1 was obtained when Co(dmgH)2PyiPr(II) was used as the catalyst (Table 1, entry 2).15 Among the cobaloximes investigated, Co(dmgH)2(4-OMe-Py)SnPh3 displayed the best photocatalytic activity with the product in 79% yield (entries 3,4 and Table S1†). Next screening a series of bases showed that the amines were either able or unable to form α-aminoalkyl radicals such as iPr2NEt and DABCO could smoothly afford the product (entries 5–7 and Table S2†). Na2CO3 worked with a longer reaction time at 50 °C (entry 8 and Table S2†). And the product 1 was also afforded with the combination of Na2CO3 and the reductant Zn at 40 °C (entry 9).11,16 A survey of solvents revealed that acetone was the optimal reaction medium (entry 10). Obviously, the dehydrogenation reaction proceeded slowly at 25 °C (entry 11). Lastly, no product was detected in the absence of cobaloxime (III), or iPr2NEt, or light by the control experiments, demonstrating the need for all these components (Table S4†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cobaloxime (mol%) | Additive (equiv.) | Solvent | Time (h) | Yield (%)b |
| a Unless otherwise noted, reaction conditions are as follows: on 0.1 mmol scale, cobalt catalyst (0.01–0.015 mmol), additive (0.15–0.3 mmol), solvent (2 mL), blue LEDs (λ = 450–460 nm), 40 °C and under an Ar atmosphere. b Isolated yield. c 50 °C. d 25 °C. dmg = dimethylglyoximate. | |||||
| 1 | I (10) | NEt3 (2.0) | CH3CN | 24 | 68 |
| 2 | II (10) | NEt3 (2.0) | CH3CN | 36 | Trace |
| 3 | III (10) | NEt3 (2.0) | CH3CN | 24 | 72 |
| 4 | III (15) | NEt3 (2.0) | CH3CN | 24 | 79 |
| 5 | III (15) | iPr2NEt (2.0) | CH3CN | 24 | 83 |
| 6 | III (15) | DABCO (2.0) | CH3CN | 24 | 31 |
| 7 | III (15) | iPr2NEt (3.0) | CH3CN | 24 | 90 |
| 8c | III (15) | Na2CO3 (2.0) | CH3CN | 36 | 19 |
| 9 | III (15) | Na2CO3 (2.0) | CH3CN | 24 | 58 |
| Zn (1.5) | |||||
| 10 | III (15) | iPr2NEt (3.0) | Acetone | 20 | 96 |
| 11d | III (15) | iPr2NEt (3.0) | Acetone | 20 | 65 |
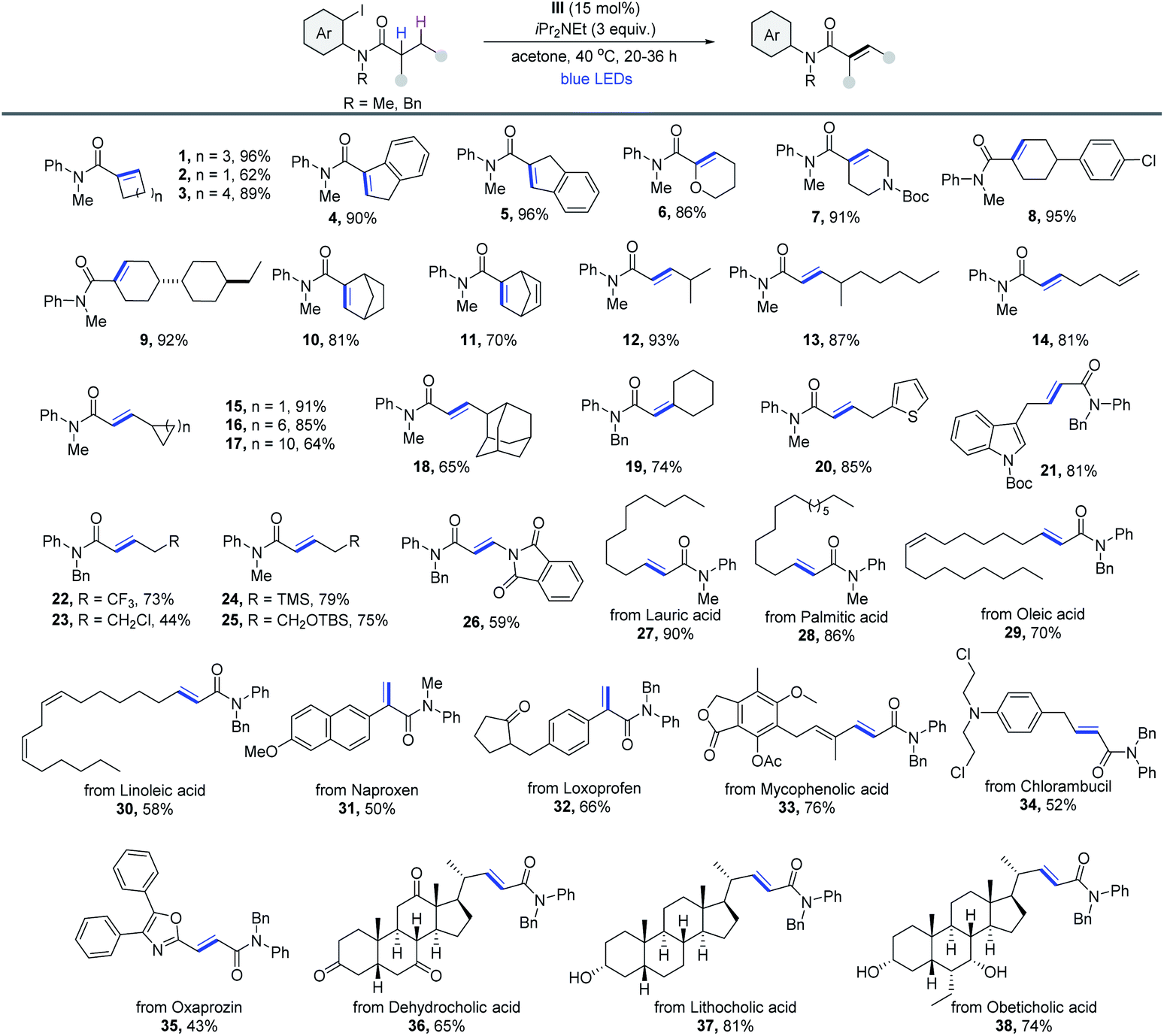
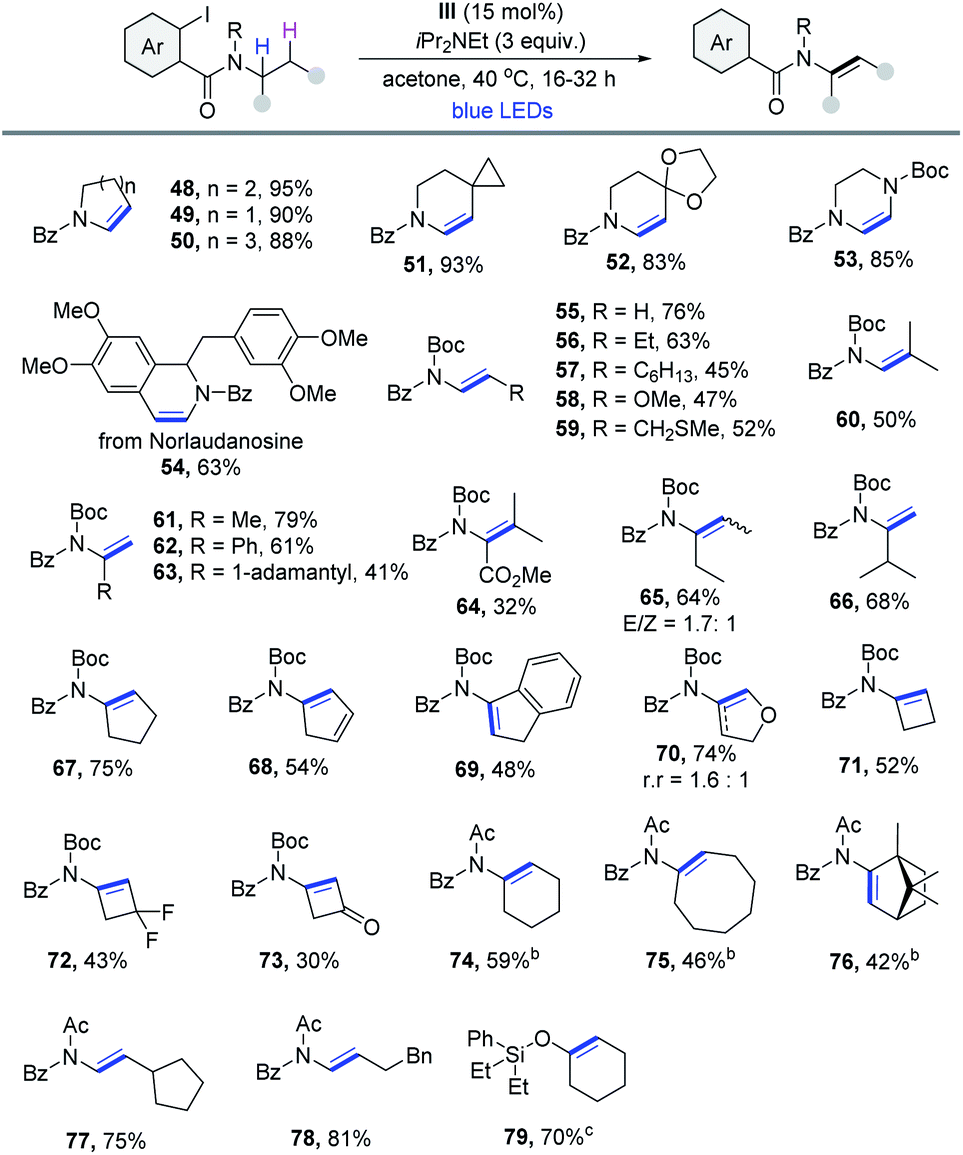
With the reaction conditions optimized, the generality of this cobaloxime catalyzed dehydrogenation method was then evaluated. As outlined in Table 2, a wide range of 2-iodoaniline derived amides were compatible with this protocol. Notably, substrates derived from cycloalkyl carboxylic acids could be dehydrogenated smoothly via the 1,5-HAT pathway in excellent yield (1–5). Heterocycles (6 and 7), cyclohexyl derivatives (8 and 9) and bridged systems such as bicyclo[2.2.1]heptane (10) and norbornene (11) formed the desired products efficiently. Moreover, the reaction was also suitable for substrates with 2° α-C–H. A variety of α,β-unsaturated amides with excellent E-selectivity were produced from readily available primary carboxylic acid precursors, such as 4-methylvaleric acid (12) and 4-methylnonanoic acid (13). Product 14 derived from the acid that contains the terminal alkenyl group was obtained in 81% yield. Substituents with varying ring sizes at the β-position (15–17), large steric hindrance (18), cyclohexyl at the α-position (19) and aromatic heterocycles (20 and 21) were all compatible with the reaction. And the medicinally important trifluoromethyl group was also suitable (22). Other heteroatom substituents bearing Cl (23), TMS (24), acid-sensitive OTBS (25) and phthalimide groups (26) were obtained in moderate yields. The scope of the reaction was further evaluated with substrates that are derived from natural products and active pharmaceutical ingredients. The dehydrogenation of fatty amides was then investigated, and products derived from lauric (27), palmitic (28), oleic (29) and linoleic (30) acids were produced in good to excellent yields. The method could also be extended to late-stage modification of naproxen (31), loxoprofen (32), mycophenolic acid (33), chlorambucil (34) and oxaprozin (35). Finally, three bile amides were subjected to the process and the desaturated products were all obtained in good yields (36-38). All cases were obtained without a regioisomer, making this reaction fully regioselective. Because of the reduced acidity of α-C–H in contrast to aldehyde and ketone, amide α,β-dehydrogenation17 is still less explored. Thus, this process is quite useful for the synthesis of α,β-unsaturated amides in terms of the atom economy and applicability.
| a Reaction conditions: on 0.1 mmol scale, [Co] (0.015 mmol), iPr2NEt (0.3 mmol), acetone (2 mL), blue LEDs (λ = 450–460 nm), 40 °C and under an Ar atmosphere. Isolated yield. |
|---|
|
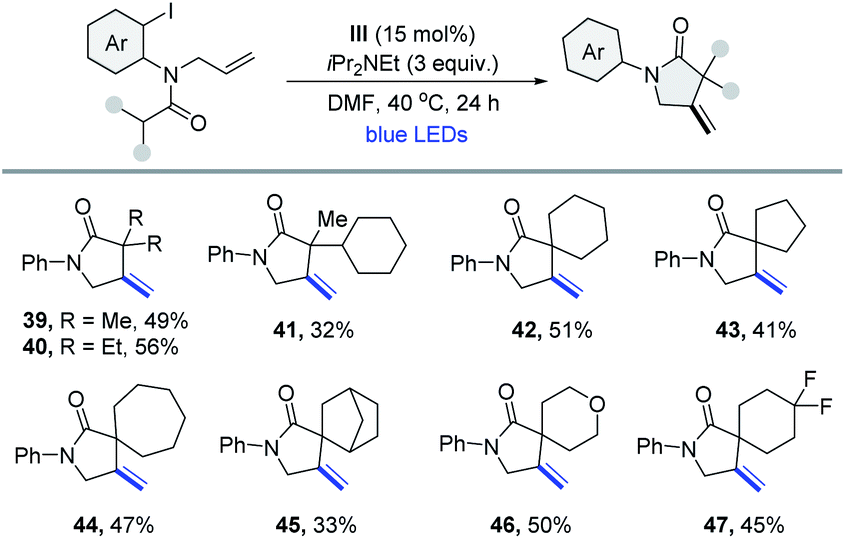
The γ-lactam ring is part of the core structure of lots of natural and non-natural compounds covering a broad spectrum of biological activities.18 The efficient synthesis of γ-lactams is still highly desirable in contemporary organic chemistry. Under similar reaction conditions, a series of β-methylene-γ-(spiro)lactams were synthesized through a tandem intramolecular 1,5-HAT and 5-exo-trig cyclization process. As shown in Table 3, the starting materials possessing various alkyl or cycloalkyl groups at the α-position of the amide could deliver the corresponding products in modest yields (39–47).
| a Reaction conditions: on 0.1 mmol scale, [Co] (0.015 mmol), iPr2NEt (0.3 mmol), DMF (2 mL), blue LEDs (λ = 450–460 nm), 40 °C and under an Ar atmosphere. Isolated yield. |
|---|
|
Enamides have proven to be extremely valuable products and chemical intermediates due to their higher stability and reaction tunability compared with enamine and are widely used in further synthetic transformations.19 However, direct dehydrogenation of carboxamide20 to construct enamide is more difficult and more inert N-benzoyl amides usually give poor yields.20a,b On the basis of the above optimized conditions, we next focused on the synthesis of enamide with diverse 2-iodobenzamides. As illustrated in Table 4, a wide range of aliphatic amine derivatives was examined, giving rise to the corresponding enamide products efficiently. First, transformation of endocyclic aliphatic amines produced the desired compounds in excellent yields (48–50). Spirocyclic (51 and 52) and piperazine-derived enamides (53) were readily synthesized in high yields. Remarkably, norlaudanosine, a dopamine metabolite, also reacted well (54). Encouraged by the study of secondary aliphatic amines, we started to investigate primary aliphatic amines, which were still not broadly explored in previous reports with low yields and limited substrate scope. To our delight, the formation of the desired enamide product 55 was observed when Boc was employed as the N-protecting group. It was found that a striking feature of this reaction was the exclusive formation of α,β-dehydrogenation products without any undesired isomer and isoindolin-1-one byproducts. Primary amines with a 2° α-C–H bond such as butylamine and octylamine, etc. were amenable, producing the desired enamides in reasonable yields (56–60). Likewise, dehydrogenation of primary amines with a 3° α-C–H bond containing different functionalities, including the methyl group (61), the phenyl group (62), the steric hindrance 1-adamantly group (63), and ester (64) proceeded well. 1-Ethylpropylamine reacted to provide the corresponding product in 64% yield, although as a mixture of stereoisomers (65). The dehydrogenative selectivity of 3-methyl-2-butanamine was conformed to Hofmann's elimination rule, leading to the expected product 66 in 68% yield with excellent regioselectivity (r.r > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Additionally, cyclopentylamine derivatives reacted smoothly to produce enamides in moderate yield (67–69). Tetrahydrofuran-3-amine could lead to the formation of a separable mixture of regioisomers, with 4,5-dihydrofuran being the major product (70). This result showed that the polar effect favored elimination of the hydridic H at the β-position rather than the neutral H. Moreover, cyclobutylamine derivatives were found to be competent substrates as well, producing the corresponding products in moderate yields (71–73). Next, we tested this method using an acetyl group as the N-protecting group. Some functional groups were found to be well tolerated under these conditions, including cycloalkyl (74–76), 2-cyclopentylethylamine (77) and 4-phenyl-1-butylamine (78). Lastly, this protocol also worked efficiently for α,β-dehydrogenation of cyclohexanol with a 2-iodophenylsilyl starting material (79).21
:1). Additionally, cyclopentylamine derivatives reacted smoothly to produce enamides in moderate yield (67–69). Tetrahydrofuran-3-amine could lead to the formation of a separable mixture of regioisomers, with 4,5-dihydrofuran being the major product (70). This result showed that the polar effect favored elimination of the hydridic H at the β-position rather than the neutral H. Moreover, cyclobutylamine derivatives were found to be competent substrates as well, producing the corresponding products in moderate yields (71–73). Next, we tested this method using an acetyl group as the N-protecting group. Some functional groups were found to be well tolerated under these conditions, including cycloalkyl (74–76), 2-cyclopentylethylamine (77) and 4-phenyl-1-butylamine (78). Lastly, this protocol also worked efficiently for α,β-dehydrogenation of cyclohexanol with a 2-iodophenylsilyl starting material (79).21
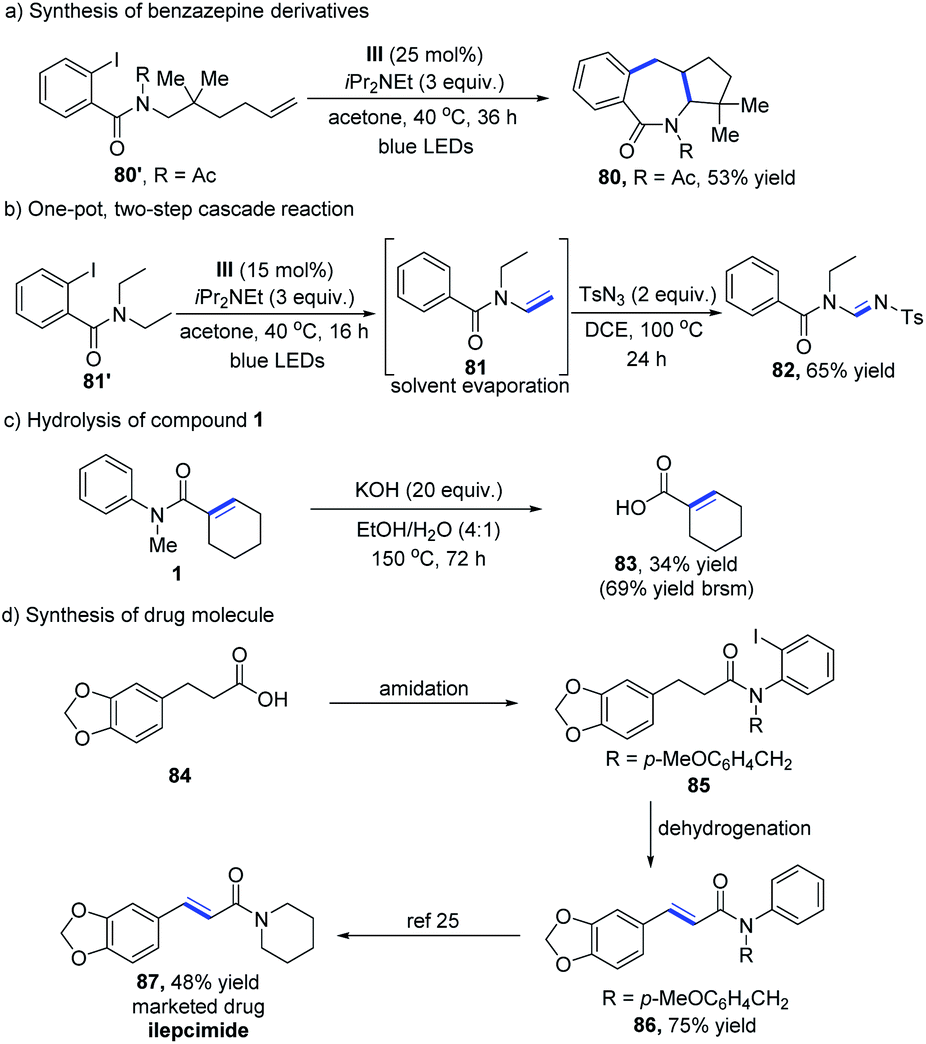
We next applied this methodology to the concise synthesis of a benzazepine derivative, which contains an important seven-membered nitrogenous heterocyclic skeleton for a wide range of biorelevant molecules. 2-Iodobenzamide substrates without a H atom at the β-position could be converted into the desired product 80 in moderate yield (Table 5a).22 The structure of 80 has been determined by X-ray crystal structure analysis. With the success of the dehydrogenative reaction for the direct construction of α,β-unsaturated systems, we continued to study the further transformations of the reaction products. Amide 81′ could be readily converted to the enamide intermediate, which was engaged in 1,3-dipolar cycloaddition with sulfonylazide for the assembly of amidine (Table 5b).23 To verify the practicability of this cobalt-catalyzed dehydrogenation process, a scale-up (4 mmol) experiment was performed, which provided α,β-unsaturated amide 1 in 83% yield (see the ESI†). On treatment of 1 under strong basic conditions, the carboxylic acid 83 was afforded with a 69% yield based on recovered starting material (Table 5c). Furthermore, this method was utilized in the synthesis of the drug ilepcimide (ICM).24 As shown in Table 5d, the dehydrogenation strategy enabled us to access the key compound 86 from commercially available carboxylic acid 84, which was then subjected to transamidation25 to provide ilepcimide 87.
|
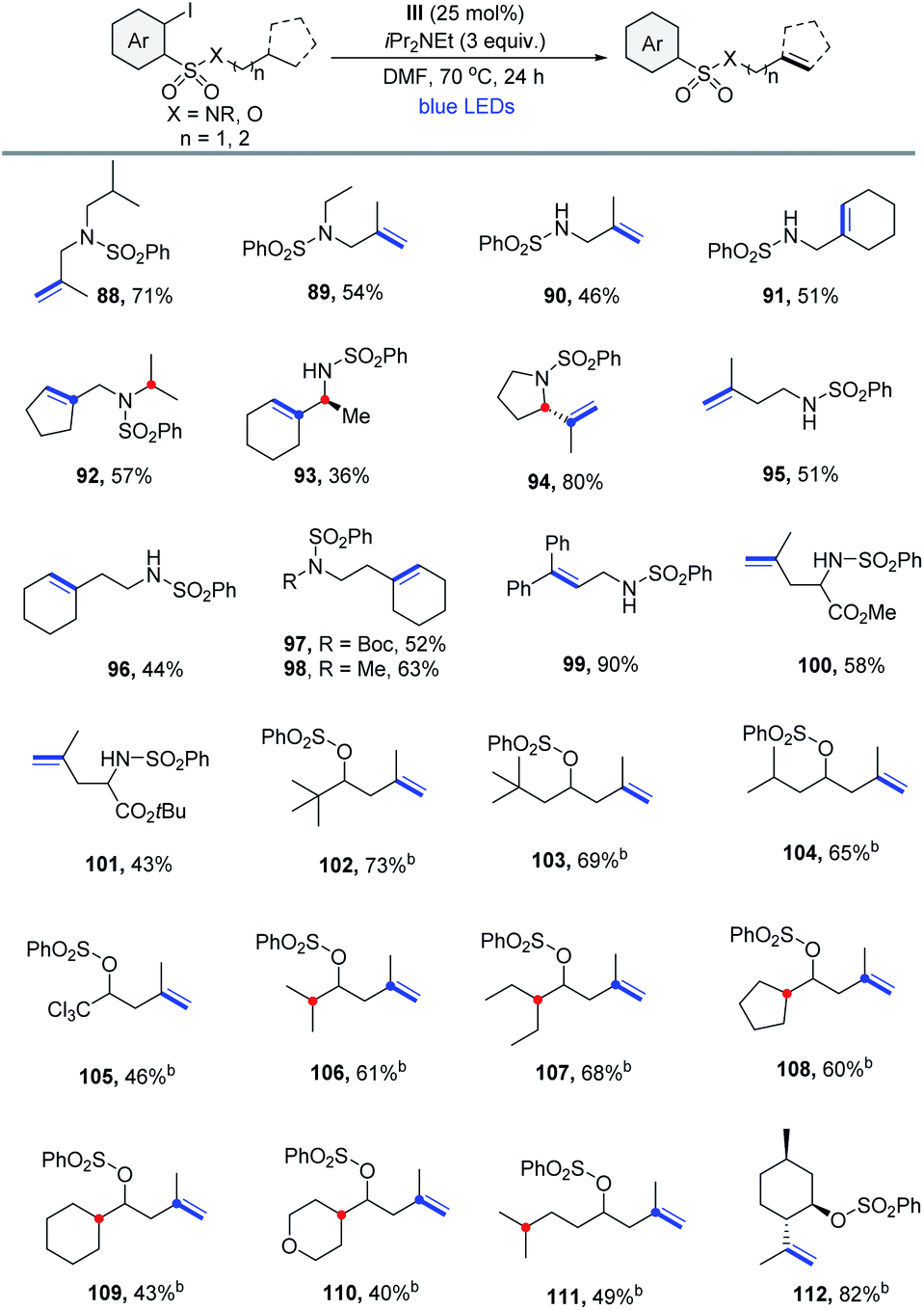
Having demonstrated the feasibility of α,β-dehydrogenation via the 1,5-HAT reaction, we next investigated the β,γ-/γ,δ-dehydrogenation of distant C–H sites via the higher order 1,n-HAT (n > 5) pathway.26 The 2-iodobenzenesulfonyl group was selected as an ideal supporting group. 1,6- or 1,7-HAT mode could be reached more easily because the longer C–S bond can readily accommodate quasi-linear geometry.27 As shown in Table 6, 2-iodobenzenesulfonyl chloride derived sulfonamides are effective substrates for remote dehydrogenation. However, the yield of the desired β,γ-dehydrogenation product 88 was poor at 40 °C. A better result was obtained when the reaction was performed at 70 °C in N,N-dimethylformamide. It was found that both the secondary and primary amines bearing an isopropyl or cycloalkyl unit, which have a 3° β-H site, underwent efficient 1,6-HAT to produce allylic sulfonamides in moderate to good yields with high regiocontrols (88-91). Substrates that possessed 3° β-H, along with competitive 3° α-H sites, reacted selectively at the β-H sites, thus producing 92 and 93 with high regioselectivities. A heterocyclic (S)-2-isopropylpyrrolidine reacted smoothly to afford 94 in 80% yield. Encouraged by the results described above, we aimed at extending this protocol to the formation of homoallylic compounds via the γ,δ-dehydrogenation process involving 1,7-HAT. For substrates with a 3° γ-H site, a series of homoallylic sulfonamide derivatives were prepared in moderate yields (95–98). The substrate bearing the benzylic C–H afforded the corresponding product in a relatively high yield (99). Reactions of leucine esters also proceeded well (100, 101). As expected, remote dehydrogenation was also effective with secondary alcohols, producing homoallylic sulfonate derivatives at 40 °C in moderate yields (102–105). Obviously, compared with 1,6- or 1,8-HAT, the regioselectivity for 1,7-HAT was very high (106–111). Finally, a natural menthol derivative gave isopulegol sulfonate in 82% yield (112), convincingly demonstrating the potential of this method.
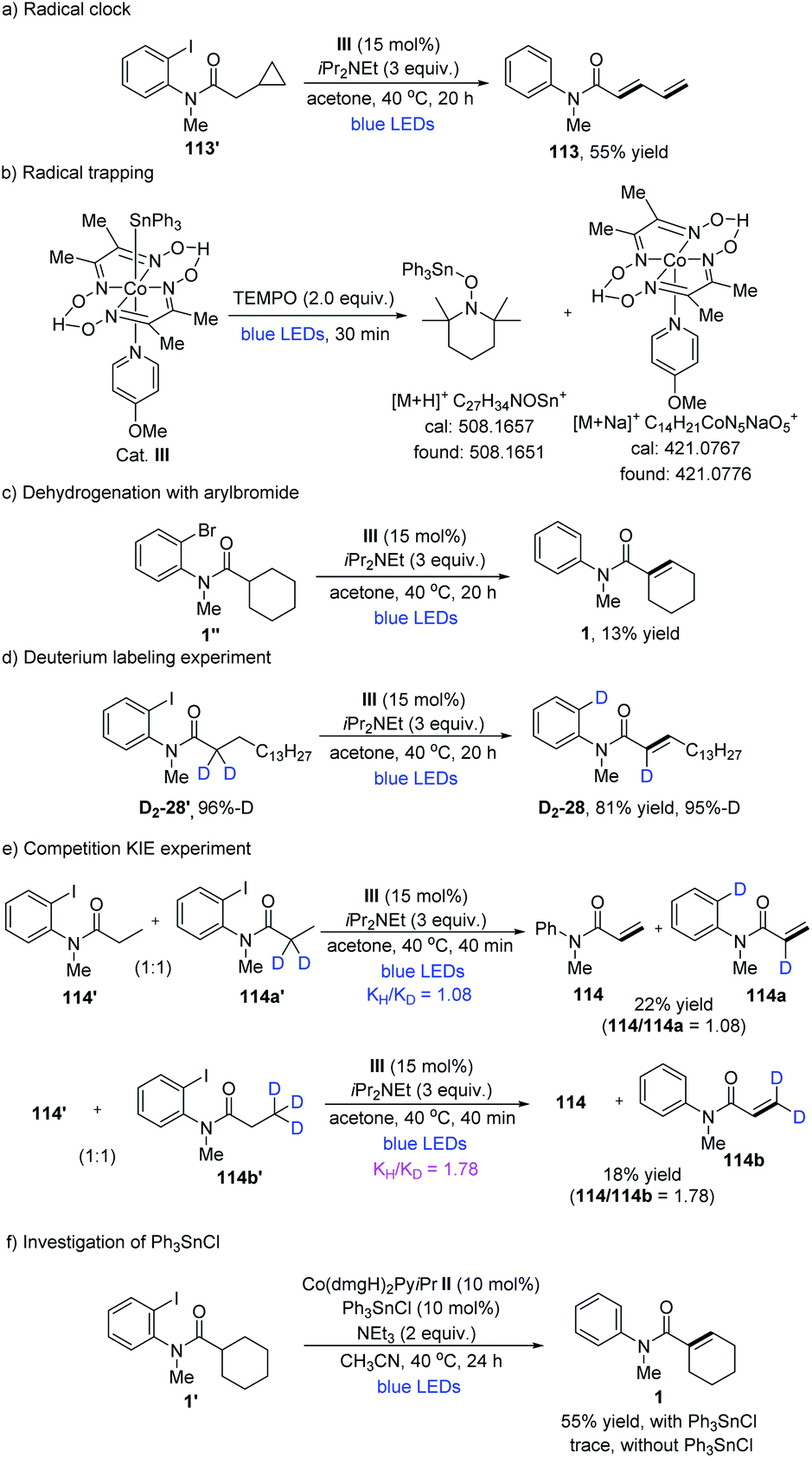
Next, some mechanistic experiments were performed. The radical nature of this transformation was preliminarily confirmed by a radical-clock experiment producing the ring-opened compound 113 in 55% yield (Table 7a). Moreover, a radical trapping experiment was performed under the irradiation of cobalt catalyst III with the addition of TEMPO. As a result, the related triphenyltin-TEMPO adduct and Co(II) species were confirmed through HRMS (Table 7b). These observations demonstrated the possibility of visible light promoting the homolysis of the Co(III)–Sn bond. The substitution of aryliodide for arylbromide derivatives led to a significant drop in yield (Table 7c). In a deuterium labeling experiment, deuterated substrate D2-28′ was prepared and subjected to the standard reaction conditions for the formation of D2-28 in 81% yield with one D atom shifted to the benzene ring (Table 7d). This experiment supported the assumption of an intramolecular 1,5-HAT process. In addition, UV-vis absorption spectra showed that Co(dmgH)2(4-OMe-Py)SnPh3 absorbs visible light with an absorption maximum at 439 nm. Further monitoring the reaction system of Co cat., 1′ and iPr2NEt revealed that an absorption band at 550–650 nm quickly appeared upon irradiation, which was consistent with the formation of CoI species (ESI Fig. S6†).28 Kinetic isotope effect (KIE) experiments were also performed. When the α-hydrogen atoms were deuterated, the intermolecular competitive reaction isotopic value (KH/KD) between 114′ and 114a′ was determined to be 1.08.29 Thereafter, when the β-hydrogen atoms were deuterated, a higher kinetic isotopic effect value (KH/KD) between 114′ and 114b′ was determined to be 1.78 (Table 7e).30 In addition, the control experiments were carried out to test alkyl cobaloximes with or without Ph3SnCl. When only Co(dmgH)2PyiPr or Co(dmgH)2Py(pentyl) was used, it appeared that only a trace of 1 was produced. Obviously, with the addition of 10 mol% Ph3SnCl, reactions proceed smoothly (Table 7f and S7†). The observation showed that the source of tin radicals was critical to the reaction for the C–I bond activation, since nucleophilic Co(I) species would react with Ph3SnCl or Ph3SnI to generate Co(dmgH)2PySnPh3, which might facilitate iodine abstraction from aryl iodide to produce an aryl radical through halogen-atom transfer.
|
Therefore, on the basis of the experimental results and previous reports,31 a plausible mechanistic pathway was proposed and shown in Scheme 2. Visible light irradiation of Co(III)SnPh3 promotes the homolysis of the Co(III)–Sn bond to facilitate iodine abstraction from aryl iodide to produce the aryl radical A and Co(II) metalloradical. This highly activated intermediate A undergoes a fast intramolecular 1,n-hydrogen atom transfer process leading to the alkyl radical B. Then, Co(II) metalloradical species can reversibly accept the radical to afford the Co(III)-alkyl adduct C (BDE < 30 kcal mol−1). With irradiation, formal β-hydride elimination or direct β-hydrogen abstraction by Co(II) species will furnish the final dehydrogenative product D and Co(III)–H (cobalt hydride equivalent) E.32 The latter is deprotonated by an amine base to generate Co(I) species F.7a Subsequently, nucleophilic Co(I) species F would react with Ph3SnI to regenerate Co(III)SnPh3 along with the completion of the catalytic cycle.33 Given the short lifetime of photoexcited Co(I)* species as well as the control experiment performed in Table 7f, it was not a major pathway that Co(I)* might undergo a single electron reduction of aryl halide G11,34,35 to generate aryl radical A in this system.
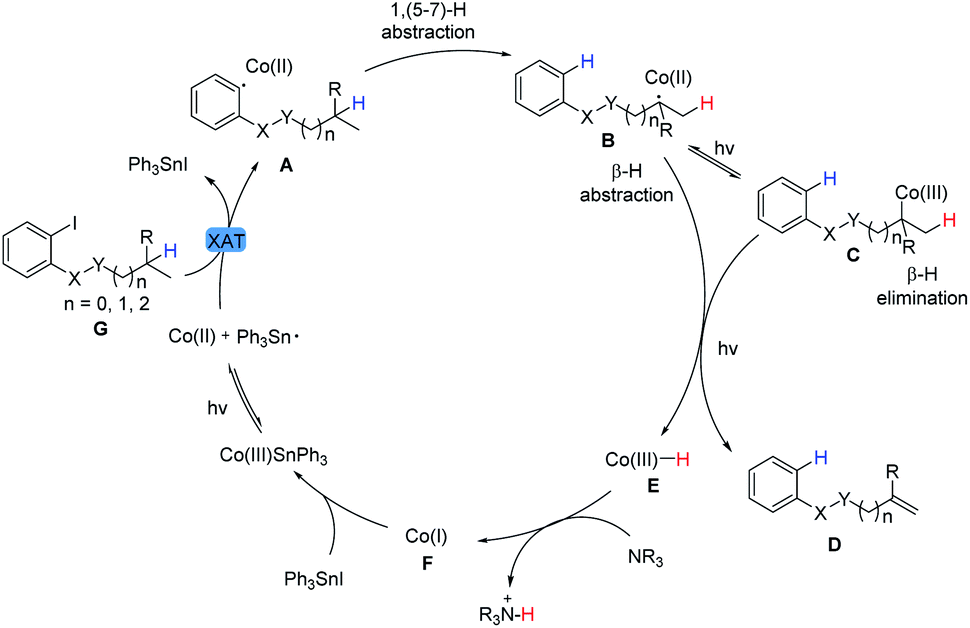 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In summary, we have described a visible-light-induced chemoselective dehydrogenation for the synthesis of a range of alkene derivatives under extremely mild reaction conditions by using inexpensive and easily accessible cobaloxime complexes. Importantly, this strategy used highly reactive aryl radical intermediates as controlling elements to mediate intramolecular 1,n-hydrogen atom transfer processes. In addition, Co-assisted efficient and selective dehydrogenation avoided competitive byproducts from radical cyclization and double bond isomerization, which showed unprecedented practicability and generality. The excellent photocatalytic activity of the cobaloxime system does not require any external photosensitizers and oxidants, which further demonstrates the potential of this method to access pharmaceutical compounds and perform late-stage functionalization.Data availability
All experimental data associated with this work are available in the ESI†.Author contributions
W.-L. Y. planned and conducted most of the experiments; Z.-G. R., K.-X. M., J.-J. Y. conducted the synthetic experiments and analyzed the experimental data. H.-Q. Y. conducted theoretical calculations; W. W. revised the manuscript; P.-F. X., H. Z. and W.-L. Y. directed the projects and wrote the manuscript. All authors contributed to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSFC (21632003, 21871116, 22071085), the Key Program of Gansu Province (17ZD2GC011), and the “111” Program from the MOE of P. R. China for funding the research.Notes and references
- K. Weissermel and H.-J. Arpe, Olefins, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2008, p. 59 Search PubMed.
- (a) C. Kim, Y. Dong and L. Que, J. Am. Chem. Soc., 1997, 119, 3635–3636 CrossRef CAS; (b) P. H. Buist, Nat. Prod. Rep., 2004, 21, 249–262 RSC; (c) M. A. Bigi, S. A. Reed and M. C. White, Nat. Chem., 2011, 3, 216–222 CrossRef CAS PubMed.
- (a) R. Breslow, B. B. Snider and R. J. Corcoran, J. Am. Chem. Soc., 1974, 96, 6792–6794 CrossRef CAS PubMed; (b) A.-F. Voica, A. Mendoza, W. R. Gutekunst, J. O. Fraga and P. S. Baran, Nat. Chem., 2012, 4, 629–635 CrossRef CAS PubMed; (c) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS PubMed; (d) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857–14860 CrossRef CAS PubMed; (e) P. Chuentragool, M. Parasram, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2018, 140, 2465–2468 CrossRef CAS PubMed; (f) Y. Xia, K. Jana and A. Studer, Chem.–Eur. J., 2021, 27, 16621–16625 CrossRef CAS PubMed; (g) L. M. Stateman, R. M. Dare, A. N. Paneque and D. A. Nagib, Chem, 2022, 8, 210–224 CrossRef CAS PubMed.
- (a) M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 16470–16485 CrossRef CAS PubMed; (b) J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093–10099 CrossRef PubMed; (c) L. Huang, A. Bismuto, S. A. Rath, N. Trapp and B. Morandi, Angew. Chem., Int. Ed., 2021, 60, 7290–7296 CrossRef CAS PubMed.
- (a) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (b) K. Pak, S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
- (a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed; (b) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed; (c) L.-Z. Wu, B. Chen, Z.-J. Li and C.-H. Tung, Acc. Chem. Res., 2014, 47, 2177–2185 CrossRef CAS PubMed; (d) B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed.
- (a) M. E. Weiss, L. M. Kreis, A. Lauber and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11125–11128 CrossRef CAS PubMed; (b) M. E. Weiss and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11501–11505 CrossRef CAS PubMed; (c) L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634–1637 CrossRef CAS PubMed; (d) M. Balkenhohl, S. Kölbl, T. Georgiev and E. M. Carreira, JACS Au, 2021, 1, 919–924 CrossRef CAS; (e) B. Morandi, B. Mariampillai and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 1101–1104 CrossRef CAS PubMed; (f) B. Gaspar and E. M. Carreira, J. Am. Chem. Soc., 2009, 131, 13214–13215 CrossRef CAS PubMed.
- (a) W.-Q. Liu, T. Lei, S. Zhou, X.-L. Yang, J. Li, B. Chen, J. Sivaguru, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 13941–13947 CrossRef CAS PubMed; (b) T. Lei, G. Liang, Y. Y. Cheng, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2020, 22, 5385–5389 CrossRef CAS PubMed; (c) Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-Y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080–10083 CrossRef CAS PubMed; (d) Q.-Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, Z.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052–19055 CrossRef CAS PubMed; (e) G. Zhang, C. Liu, H. Yi, Q.-Y. Meng, C. Bian, H. Chen, J.-X. Jian, L.-Z. Wu and A. Lei, J. Am. Chem. Soc., 2015, 137, 9273–9280 CrossRef CAS PubMed; (f) G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed; (g) G. Zhang, Y. Lin, X. Luo, X. Hu, C. Chen and A. Lei, Nat. Commun., 2018, 9, 1225–1231 CrossRef PubMed.
- (a) R. Bam, A. S. Pollatos, A. J. Moser and J. G. West, Chem. Sci., 2021, 12, 1736–1744 RSC; (b) M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391–3404 RSC; (c) S. Busato, O. Tinembart, Z.-D. Zhang and R. Scheffold, Tetrahedron, 1990, 46, 3155–3166 CrossRef CAS; (d) B. Giese, J. Hartung, J. He, O. Hüter and A. Koch, Angew. Chem., Int. Ed., 1989, 28, 325–327 CrossRef; (e) G. N. Schrauzer, L. P. Lee and J. W. Sibert, J. Am. Chem. Soc., 1970, 92, 2997–3005 CrossRef CAS PubMed; (f) D. N. Ramakrishna, R. Symons and M. Symons, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 423–434 RSC.
- (a) H. Zhao, A. J. McMillan, T. Constantin, R. C. Mykura, F. Juliá and D. Leonori, J. Am. Chem. Soc., 2021, 143, 14806–14813 CrossRef CAS PubMed; (b) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (c) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- (a) K. L. Brown and R. Legates, J. Organomet. Chem., 1982, 233, 259–265 CrossRef CAS; (b) D. Lenoir, H. Dauner, I. Ugi, A. Gieren, R. Hübner and V. Lamm, J. Organomet. Chem., 1980, 198, C39–C42 CrossRef CAS; (c) V. F. Patel and G. Pattenden, J. Chem. Soc., Chem. Commun., 1987, 871–872 RSC.
- W.-L. Yu, Y.-C. Luo, L. Yan, D. Liu, Z.-Y. Wang and P.-F. Xu, Angew. Chem., Int. Ed., 2019, 58, 10941–10945 CrossRef CAS PubMed.
- (a) G. N. Schrauzer and G. Kratel, Chem. Ber., 1969, 102, 2392–2407 CrossRef CAS; (b) M. Tada and K. Kaneko, J. Org. Chem., 1995, 60, 6635–6636 CrossRef CAS.
- J. Robertson, J. Pillaia and R. K. Lusha, Chem. Soc. Rev., 2001, 30, 94–103 RSC.
- Tin radicals might act as halogen-atom transfer (XAT) agents for activation of aryl C–I bonds (see Table 7f and S7†).
- (a) B. Giese, P. Erdmann, T. Gobel and R. Springer, Tetrahedron Lett., 1992, 33, 4545–4548 CrossRef CAS; (b) B. P. Branchaud and W. D. Detlefsen, Tetrahedron Lett., 1991, 32, 6273–6276 CrossRef CAS; (c) W. Affo, H. Ohmiya, T. Fujioka, Y. Ikeda, T. Nakamura, H. Yorimitsu, K. Oshima, Y. Imamura, T. Mizuta and K. Miyoshi, J. Am. Chem. Soc., 2006, 128, 8068–8077 CrossRef CAS PubMed.
- (a) Y. Chen, A. Turlik and T. R. Newhouse, J. Am. Chem. Soc., 2016, 138, 1166–1169 CrossRef CAS PubMed; (b) D. Huang, S. M. Szewczyk, P. Zhang and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 5669–5674 CrossRef CAS PubMed; (c) M. Chen and G. Dong, J. Am. Chem. Soc., 2017, 139, 7757–7760 CrossRef CAS PubMed; (d) Z. Wang, Z. He, L. Zhang and Y. Huang, J. Am. Chem. Soc., 2018, 140, 735–740 CrossRef CAS PubMed; (e) C. J. Teskey, P. Adler, C. R. Gonçalves and N. Maulide, Angew. Chem., Int. Ed., 2019, 58, 447–451 CrossRef CAS PubMed; (f) S. Gnaim, J. C. Vantourout, F. Serpier, P.-G. Echeverria and P. S. Baran, ACS Catal., 2021, 11, 883–892 CrossRef CAS; (g) Z. Wang, L. Hu, N. Chekshin, Z. Zhuang, S. Qian, J. X. Qiao and J.-Q. Yu, Science, 2021, 374, 1281–1285 CrossRef CAS PubMed.
- J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC.
- (a) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599–4657 CrossRef CAS PubMed; (b) F. Beltran and L. Miesch, Synthesis, 2020, 52, 2497–2511 CrossRef CAS.
- (a) G. Li, P. A. Kates, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, ACS Catal., 2019, 9, 9513–9517 CrossRef CAS; (b) N. Holmberg-Douglas, Y. Choi, B. Aquila, H. Huynh and D. A. Nicewicz, ACS Catal., 2021, 11, 3153–3158 CrossRef CAS; (c) T. Shono, Y. Matsumura, K. Tsubata, Y. Sugihara, S. Yamane, T. Kanazawa and T. Aoki, J. Am. Chem. Soc., 1982, 104, 6697–6703 CrossRef CAS; (d) P. Spieß, M. Berger, D. Kaiser and N. Maulide, J. Am. Chem. Soc., 2021, 143, 10524–10529 CrossRef PubMed.
- D. P. Curran, D. H. Kim, T. Liu and W. Shen, J. Am. Chem. Soc., 1988, 110, 5900–5902 CrossRef CAS.
- D. P. Curran, A. C. Abraham and H. Liu, J. Org. Chem., 1991, 56, 4335–4337 CrossRef CAS.
- X. Xu, X. Li, L. Ma, N. Ye and B. Wang, J. Am. Chem. Soc., 2008, 130, 14048–14049 CrossRef CAS PubMed.
- (a) S. Li, C. Wang, W. Li, K. Koike, T. Nikaido and M.-W. Wang, J. Asian Nat. Prod. Res., 2007, 9, 421–430 CrossRef CAS PubMed; (b) Y. Zeng, B. Qin, Y.-W. Shi, Y.-S. Long, W.-Y. Deng, B.-M. Li, B. Tang, Q.-H. Zhao, M.-M. Gao, N. He and W.-P. Liao, Epilepsy Res., 2021, 170, 106533–106538 CrossRef CAS PubMed.
- Z. Wang, A. Matsumoto and K. Maruoka, Chem. Sci., 2020, 11, 12323–12328 RSC.
- (a) K. A. Hollister, E. S. Conner, M. L. Mark, L. Spell, K. Deveaux, L. Maneval, M. W. Beal and J. R. Ragains, Angew. Chem., Int. Ed., 2015, 54, 7837–7841 CrossRef CAS PubMed; (b) F. W. Friese, C. Mück-Lichtenfeld and A. Studer, Nat. Commun., 2018, 9, 2808–2816 CrossRef PubMed; (c) N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561–3565 CrossRef CAS PubMed.
- M. Nechab, S. Mondal and M. Bertrand, Chem.–Eur. J., 2014, 20, 16034–16059 CrossRef CAS PubMed.
- P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576–12577 CrossRef CAS PubMed.
- The rate constant of 1,5-HAT, see: D. P. Curran and A. C. Abrahaml, Tetrahedron, 1993, 49, 4821–4840 CrossRef CAS.
- (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed; (b) K. C. Cartwright, E. Joseph, C. G. Comadoll and J. A. Tunge, Chem.–Eur. J., 2020, 26, 12454–12471 CrossRef CAS PubMed.
- (a) G. N. Schrauzer, J. W. Sibert and R. J. Windgassen, J. Am. Chem. Soc., 1968, 90, 6681–6688 CrossRef CAS PubMed; (b) C. D. Garr and R. G. Finke, J. Am. Chem. Soc., 1992, 114, 10440–10445 CrossRef CAS; (c) J. Halpern, Acc. Chem. Res., 1982, 15, 238–244 CrossRef CAS; (d) X. Sun, J. Chen and T. Ritter, Nat. Chem., 2018, 10, 1229–1233 CrossRef CAS PubMed; (e) H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 16360–16367 CrossRef CAS PubMed.
- The hypothesis for intermediate E is a Co(I) center with a protonated glyoxime ligand, serving as a cobalt hydride equivalent, see: (a) D. P. Estes, D. C. Grills and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 17362–17365 CrossRef CAS PubMed; (b) D. C. Lacy, G. M. Roberts and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 4860–4864 CrossRef CAS PubMed.
- Compared with other bases unable to form aminoalkyl radicals, iPr2NEt gave the best result. It is also possible that aminoalkyl radicals perform halogen atom transfer (XAT) from Ph3SnI to generate tin radicals (see the ESI†).
- (a) L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS; (b) A. J. Fry and R. L. Krieger, J. Org. Chem., 1976, 41, 54–57 CrossRef CAS.
- The synthesis of aryl-cobaloxime intermediates failed. And the computational experiments showed that the aryl Co-C bond had a higher BDE than the alkyl Co–C bond (see the ESI†), previous reports on aryl-cobalt, see: (a) N. A. Miller, T. E. Wiley, K. G. Spears, M. Ruetz, C. Kieninger, B. Kräutler and R. J. Sension, J. Am. Chem. Soc., 2016, 138, 14250–14256 CrossRef CAS PubMed; (b) M. Ruetz, C. Gherasim, K. Gruber, S. Fedosov, R. Banerjee and B. Kräutler, Angew. Chem., Int. Ed., 2013, 52, 2606–2610 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2117638, 2142047. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02291e |
| This journal is © The Royal Society of Chemistry 2022 |