
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh-catalyzed regio-switchable cross-coupling of gem-difluorinated cyclopropanes with allylboronates to structurally diverse fluorinated dienes†
Yaxin
Zeng
a,
Hui
Yang
a,
Jiayi
Du
a,
Qin
Huang
b,
Guoliang
Huang
 b and
Ying
Xia
*a
b and
Ying
Xia
*a
aWest China School of Public Health and West China Fourth Hospital, West China-PUMC C.C. Chen Institute of Health, and State Key Laboratory of Biotherapy, Sichuan University, Chengdu 610041, China. E-mail: xiayingscu@scu.edu.cn
bDepartment of Biomedical Engineering, School of Medicine, Tsinghua University, Beijing 100084, China
First published on 5th October 2022
Abstract
The control of linear/branched selectivity is one of the major focuses in transition-metal catalyzed allyl–allyl cross-coupling reactions, in which bond connection occurs at the terminal site of both the allyl fragments forming different types of 1,5-dienes. Herein, terminal/internal regioselectivity is investigated and found to be switchable in allyl–allyl cross-coupling reactions between gem-difluorinated cyclopropanes and allylboronates. The controlled terminal/internal regioselectivity arises from the fine-tuning of the rhodium catalytic system. Fluorinated 1,3-dienes, 1,4-dienes and 1,5-dienes are therefore produced in good yields with respectively isomerized terminal, internal, and terminal regioselectivity.
Introduction
Allyl–allyl cross-coupling reactions catalyzed by a transition-metal complex play an important role in organic synthesis, not only because they constitute a robust and efficient method for the construction of C(sp3)–C(sp3) bonds, but also because the two olefin moieties in the products enable diversified downstream transformations.1–4 Typically, allyl–allyl cross-coupling reactions proceed between allylic electrophiles and allylmetal reagents, and the two allyl moieties are connected together at the terminal site of the allyl fragments through reductive elimination, resulting in the formation of different types of 1,5-dienes via linear2 or branched3,4 selectivity (Scheme 1a). Furthermore, the introduction of chiral ligands in allyl–allyl cross-coupling reactions,4 mainly contributed by Morken and coworkers,4a–4f allows the generation of enantioenriched 1,5-diene structures, which further enhances the importance of this methodology in synthetic chemistry.5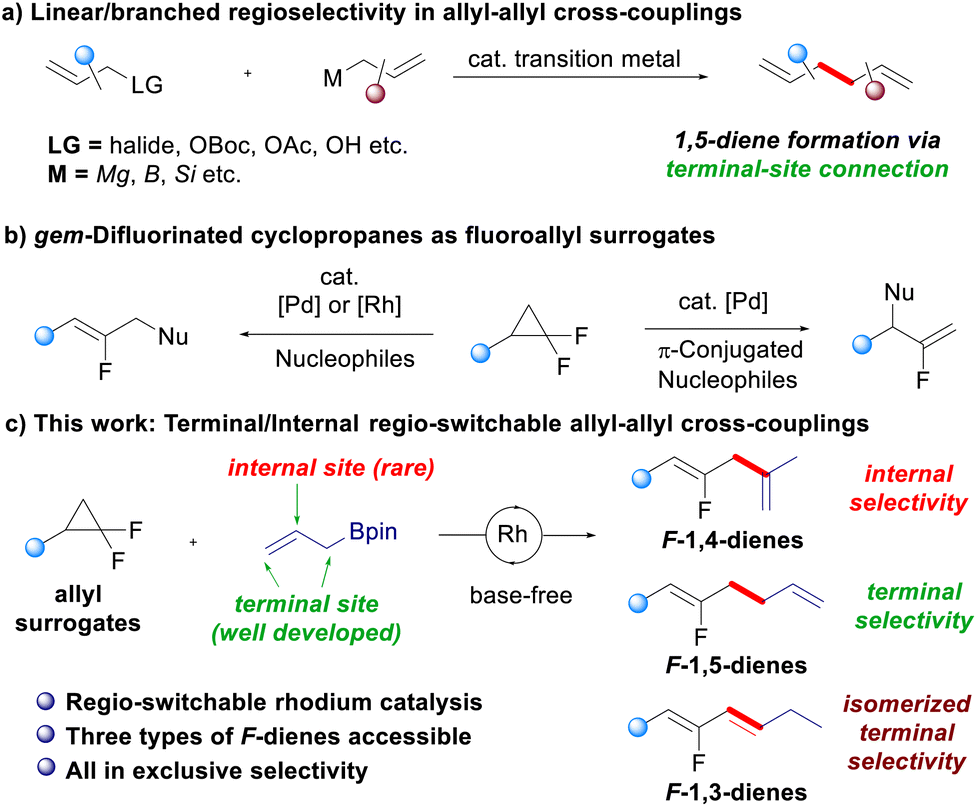 | ||
| Scheme 1 Regioselectivity in transition-metal catalyzed allyl–allyl cross-coupling reactions and its background. | ||
On the other hand, gem-difluorinated cyclopropanes6 have been explored as novel fluoroallyl surrogates to access fluoroalkenes through transition-metal catalyzed C–C bond activation.7–12 The pioneering work from Fu's group showed the Pd-catalyzed ring-opening functionalization of gem-difluorinated cyclopropanes with various nucleophiles, providing fluoroallyllic skeletons with linear selectivity.7 Subsequently, the reaction scope was extensively expanded by Gong, Fu8 and other research groups,9 including our work using rhodium catalysis that realized the fluoroallylation of simple arenes via aryl C–H activation and the site-divergent alkenyl C–H fluoroallylation of olefins10 (Scheme 1b, left). In addition, Lv and Li developed a branched selective alkylation of gem-difluorinated cyclopropanes with π-conjugated ambident nucleophiles (including hydrazones and ketones), which may involve 3,3′-reductive elimination to deliver α-fluoroalkene skeletons (Scheme 1b, right).11 During the preparation of this manuscript, Lv, Chen, Li and coworkers reported that the linear/branched selectivity in the reaction of gem-difluorinated cyclopropanes can be controlled by the steric effects of the Pd–N-heterocyclic carbene complex, which was also achieved by Wang and Shi using a Pd-monophosphine complex, producing both linear and branched fluorinated 1,5-dienes with excellent regioselectivity (Scheme 1b).12
Switchable reactivity allows the production of two or more structurally diverse products starting from the same substrates, which has been a constant quest in synthetic chemistry.13 In the aspect of allyl–allyl cross-coupling reactions, the control of linear and branched selectivity has been well-developed, in which the C–C bond was formed at the terminal site of the allyl moiety.1–4,12 While there are great advancements in the linear/branched control of allyl–allyl cross-coupling reactions, a strategy that can form a bond connection at the internal site of an allyl moiety to achieve terminal/internal-switchable regioselectivity remains elusive.14 As continuous research interest on the development of new reactivity of gem-difluorinated small rings,10,15 we envisioned that this challenge can be potentially addressed by the use of gem-difluorinated cyclopropanes as fluoroallyl surrogates under rhodium catalysis. Herein, we developed regio-switchable rhodium-catalyzed cross-coupling reactions between gem-difluorinated cyclopropanes and allylboronates,16 which provides a practical and modular approach to structurally diverse fluorinated 1,n-dienes (n = 3, 4, and 5). The internal selectivity gives fluorinated 1,4-dienes, while the terminal selectivity furnishes fluorinated 1,3-dienes and 1,5-dienes respectively, depending on whether the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond migrates or not (Scheme 1c). Remarkably, fine-tuning the rhodium precursor and the ligand enables the diversity-oriented synthesis of fluorinated dienes in excellent regioselectivity.
C bond migrates or not (Scheme 1c). Remarkably, fine-tuning the rhodium precursor and the ligand enables the diversity-oriented synthesis of fluorinated dienes in excellent regioselectivity.
Results and discussion
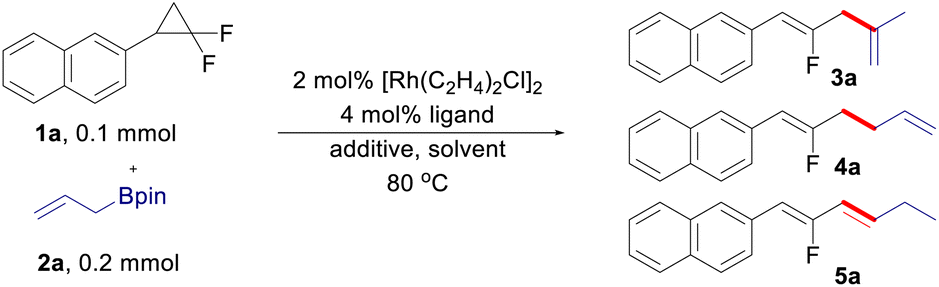
At the outset of the study, (2,2-difluorocyclopropyl)benzene 1a and allyl-Bpin 2a were selected as the model substrates to optimize the reaction conditions (Table 1). With a combination of 2 mol% [Rh(C2H4)2Cl]2 and 4 mol% monodentate phosphine ligand as the catalytic system, internal-selective fluorinated 1,4-diene 3a was observed in 8–15% yield with low conversion of the substrates (entries 1–3), in which (4-ClC6H4)3P turned out to be the best ligand in this transformation (entry 3). The reaction conversion was sharply increased with the addition of H2O, in which the product 3a can be isolated in 88% yield with exclusively internal selectivity (entries 4–5). It is noteworthy that the regioselectivity can be totally reversed when AgBF4 was used as the additive, leading to the formation of fluorinated 1,5-diene 4a in 20% yield with exclusively terminal selectivity (entry 6). While increasing the reaction temperature can push the substrate 1a to full conversion, the yield of 4a was only slightly improved (entry 7). Replacing the pre-catalyst of [Rh(C2H4)2Cl]2 with [Rh(CO)2Cl]2 can improve the yield of 1,5-diene to some extent (entry 8). The yield of 4a could be further improved to 42% when DME (1,2-dimethoxyethane) was used as the solvent (entry 9). Taking a more electron-deficient ligand and reducing the reaction concentration to 0.2 M are both beneficial to the reaction (entries 10 and 11). The best result can be gained by modulating the loading of the ligand to 6 mol%, in which the terminal-selective product 4a can be isolated in 72% yield (entry 12). Interestingly, it was found that the use of BINAP (1,1′-binaphthyl-2,2′-diphenyl phosphine) as the ligand can lead to the formation of a conjugated diene 5a (entry 13), which is likely generated through CC bond isomerization from the terminal coupling product 4a. The development of an efficient approach for CC bond migration in a site- and stereoselective manner is highly demanded in organic synthesis and has drawn considerable attention recently.17 [Rh(C2H4)2Cl]2 was proved to be a more efficient pre-catalyst for the generation of 1,3-diene (entry 14). The yield of 5a can be further improved by using THF (tetrahydrofuran) as the solvent (entry 15), and finally lowering the reaction temperature to 80 °C led to the optimized conditions for isomerized terminal selectivity (entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent (mL) | Additive (mol%) | Yieldb (%) of dienes 3a/4a/5a |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), [Rh(C2H4)2Cl]2 (2 mol%), ligand (4 mol%), and additive (x mol%) in solvent (x mL) at 80 °C for 24 h. b Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard. c Isolated yield. d Reaction was carried out at 100 °C for 12 h. e [Rh(CO)2Cl]2 was used instead of [Rh(C2H4)2Cl]2. f 6 mol% ligand was used. g Reaction was carried out at 80 °C for 12 h. BINAP, 1,1′-binaphthyl-2,2′-diphenyl phosphine; DME, 1,2-dimethoxyethane. | ||||
| 1 | PPh3 | Dioxane (0.3) | — | 8/0/0 |
| 2 | (4-MeC6H4)3P | Dioxane (0.3) | — | 11/0/0 |
| 3 | (4-ClC6H4)3P | Dioxane (0.3) | — | 15/0/0 |
| 4 | (4-ClC6H4)3P | Dioxane (0.3) | H2O (10 μL) | 56/0/0 |
| 5 | (4-ClC6H4)3P | Dioxane (0.3) | H2O (30 μL) | 88c/0/0 |
| 6 | (4-ClC6H4)3P | Dioxane (0.3) | AgBF4 (5) | 0/20/0 |
| 7d | (4-ClC6H4)3P | Dioxane (0.3) | AgBF4 (5) | 0/26/0 |
| 8d,e | (4-ClC6H4)3P | Dioxane (0.3) | AgBF4 (5) | 0/35/0 |
| 9d,e | (4-ClC6H4)3P | DME (0.3) | AgBF4 (5) | 0/42/0 |
| 10d,e | (4-CF3C6H4)3P | DME (0.3) | AgBF4 (5) | 0/51/0 |
| 11d,e | (4-CF3C6H4)3P | DME (0.5) | AgBF4 (5) | 0/63/0 |
| 12d,e,f | (4-CF3C6H4)3P | DME (0.5) | AgBF4 (5) | 0/72c/0 |
| 13d,e | BINAP | DME (0.5) | AgBF4 (5) | 0/0/10 |
| 14d | BINAP | DME (0.5) | AgBF4 (5) | 0/0/27 |
| 15d | BINAP | THF (0.5) | AgBF4 (5) | 0/0/50 |
| 16g | BINAP | THF (0.5) | AgBF4 (5) | 0/0/68c |
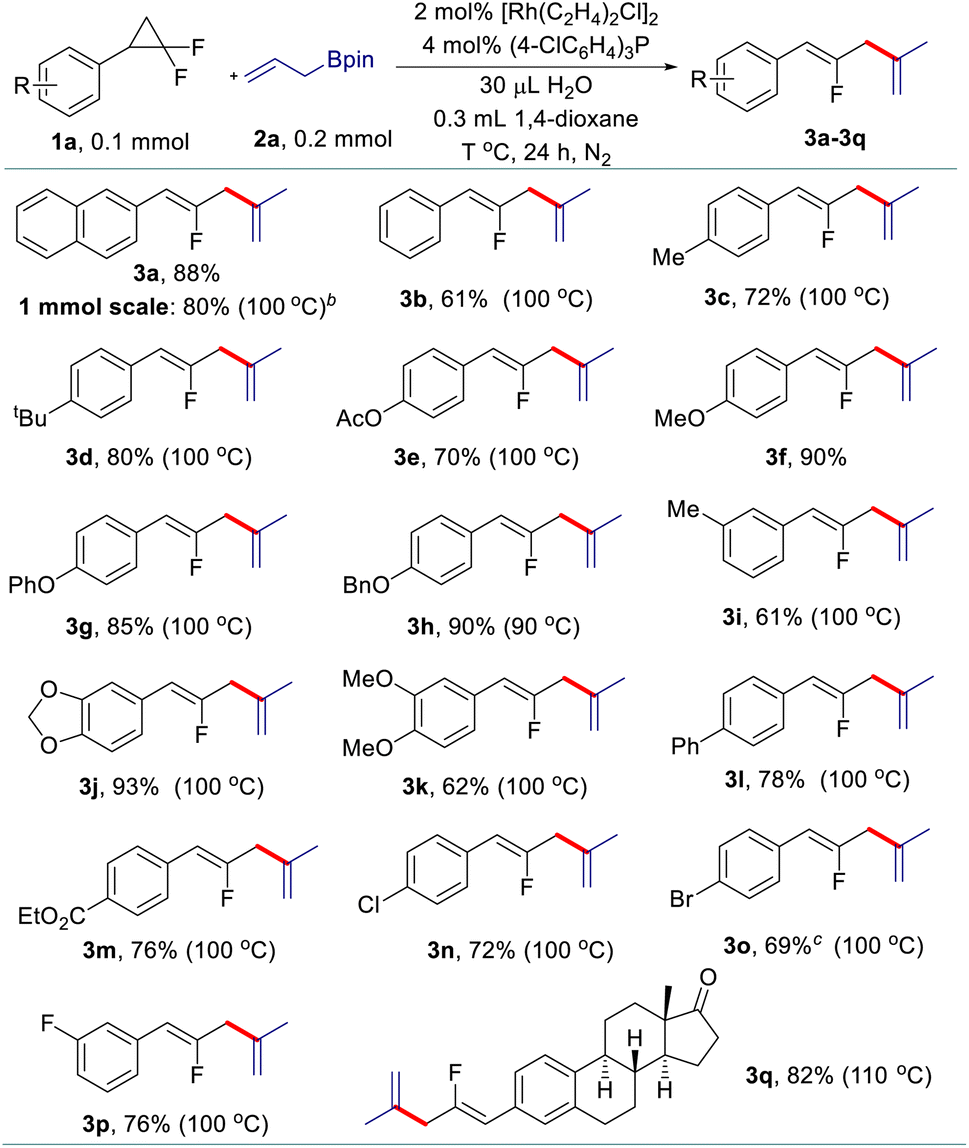
Having identified the three sets of optimized reaction conditions, we then evaluated the generality of the allyl–allyl cross-coupling reactions with different regioselectivity. Firstly, we tested the substrate scope for the synthesis of fluorinated 1,4-dienes with internal selectivity (Table 2). The model product 3a can be obtained in 80% yield in a scale-up reaction. A variety of gem-difluorinated cyclopropanes bearing electron-donating (3a–3i) or electron-withdrawing (3l–3p) aryl moieties all react smoothly, delivering the corresponding fluorinated 1,4-dienes in good to excellent yields with internal selectivity. gem-Difluorinated cyclopropanes containing di-substituted aryls also work well to provide the desired products in good yield (3j and 3k). Besides, a gem-difluorinated cyclopropane derived from estrone can be successfully transformed into the complex 1,4-diene (3q) in 82% yield. Overall, substrates with electron-donating groups showed relatively better reactivity than those bearing electron-withdrawing groups.
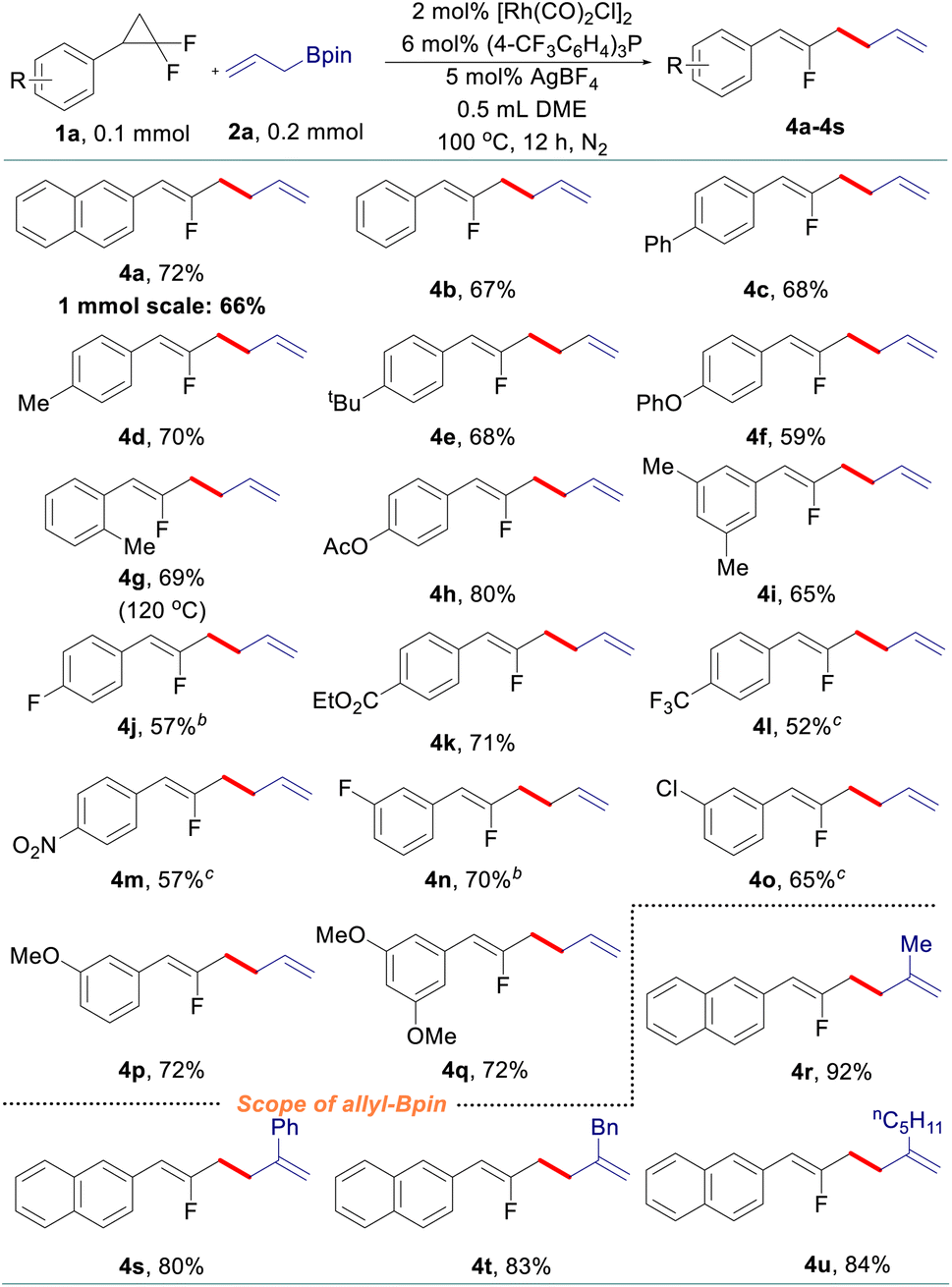
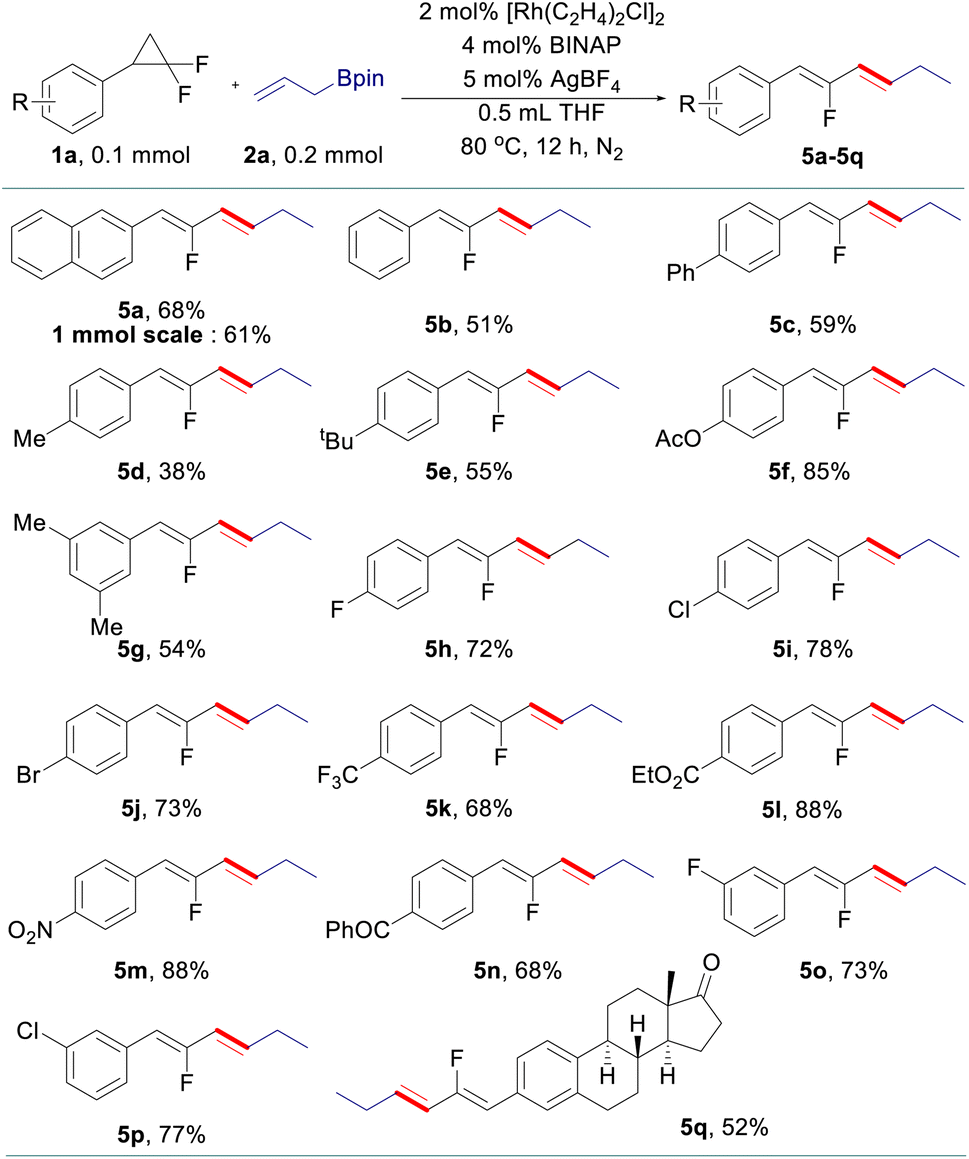
After that, various gem-difluorinated cyclopropanes were employed for the construction of fluorinated 1,5-dienes with terminal selectivity. As shown in Table 3, the model fluorinated 1,5-diene 4a can be produced in good yield in a 1 mmol scale reaction. It was found that fluorinated 1,5-dienes bearing aryls substituted with phenyl (4c), alkyl (4d, 4e, 4g, and 4i), phenoxy (4f), acetoxy (4h), methoxy (4p and 4q), halogen (4j, 4n, and 4o), ester (4k), trifluoromethyl (4l) or nitro (4m) groups were produced in 52–80% yields. Among them, increasing the loading of the rhodium catalyst was generally adopted to gain a satisfactory result for gem-difluorinated cyclopropanes bearing an electron-withdrawing group. A sterically hindered substrate with an ortho-substituted aryl moiety also works well by elevating the reaction temperature to 120 °C (4g). Besides, we also tested the reactivity of different allylboronate derivatives under the optimized reaction conditions. Allylboronates with methyl (4r), phenyl (4s), benzyl (4t) or amyl (4u) groups at the 2-position undergo this transformation to afford the corresponding fluorinated 1,5-dienes in excellent yields. Meanwhile, unsymmetric 1-methyl-substituted allylboronate could provide two different 1,5-dienes in 58% combined yields with 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 site-selectivity; 1,1-disubstituted and 3-substituted allylboronates did not react under the reaction conditions (see details in the ESI†).
:1 site-selectivity; 1,1-disubstituted and 3-substituted allylboronates did not react under the reaction conditions (see details in the ESI†).
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), [Rh(CO)2Cl]2 (0.002 mmol), (4-CF3C6H4)3P (0.006 mmol), and AgBF4 (0.005 mmol) in DME (0.5 mL) at 100 °C for 12 h. Isolated yields are presented. b [Rh(CO)2Cl]2 (0.003 mmol), (4-CF3C6H4)3P (0.009 mmol), and AgBF4 (0.0075 mmol) were used. c [Rh(CO)2Cl]2 (0.004 mmol), (4-CF3C6H4)3P (0.012 mmol), and AgBF4 (0.01 mmol) were used. |
|---|
|
The scope of allyl–allyl cross-coupling in isomerized terminal selectivity was then explored to produce a wide range of fluorinated 1,3-dienes (Table 4). Our protocol tolerates the presence of a variety of functional groups, including alkyl (5d and 5e), phenyl (5c), halogen (5h–5j), ester (5l), nitro (5m), and benzoyl (5n). In general, substrates bearing electron-withdrawing groups exhibit better reactivity than those with electron-donating groups. gem-Difluorinated cyclopropanes with di-substituted (5g) and meta-substituted groups (5o and 5p) on the benzene ring are also suitable substrates in this reaction, giving the desired products in moderate to good yields with isomerized terminal selectivity. Again, the synthesis of estrone-derived 1,3-diene 5q can be smoothly conducted via this coupling reaction.
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), [Rh(CO)2Cl]2 (0.002 mmol), BINAP (0.004 mmol), and AgBF4 (0.005 mmol) in THF (0.5 mL) at 80 °C for 12 h. |
|---|
|
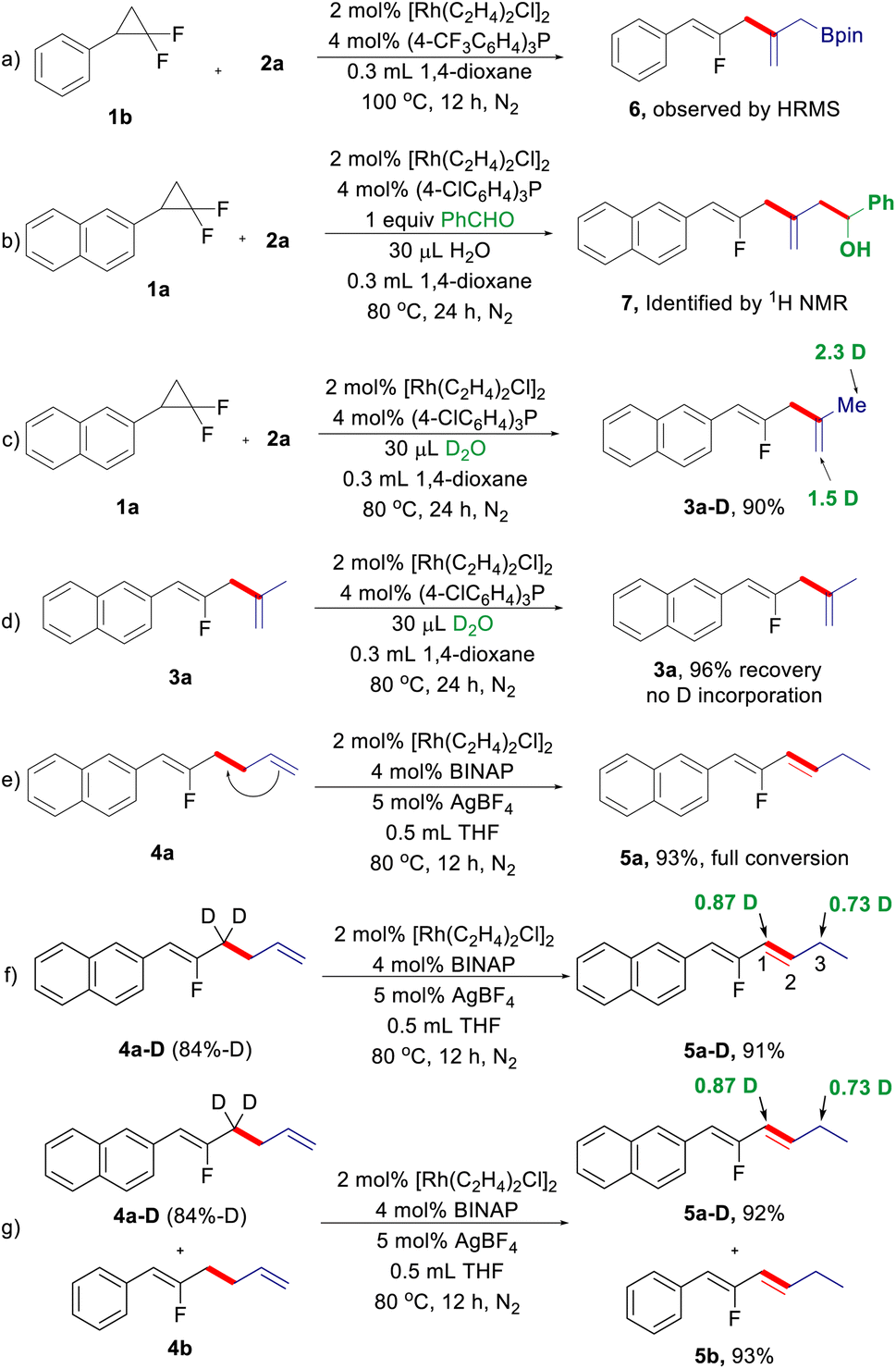
Next, we turned our attention to the reaction mechanism, particularly regarding the process of 1,4-diene formation via internal selectivity (Scheme 2). During the optimization of the reaction conditions for 1,4-dienes, the putative dienyl-Bpin 6 can be detected by GC-MS analysis, which was further supported by HRMS (Scheme 2a). Benzaldehyde was used to capture the dienyl-Bpin intermediate under the optimized conditions, and the allylation product 7 was indeed produced (Scheme 2b). A deuterium experiment using D2O as the additive under the standard conditions can deliver deuterated 1,4-diene 3a-D in excellent yield with a high degree of deuterium incorporation at the allyl moiety derived from allyl-Bpin, while 1,4-diene 3a cannot be deuterated under the same reaction conditions (Scheme 2c and d). The above results suggest that a Heck-type process followed by protodeboronation of the resulting dienyl-Bpin accounts for the internal selectivity. Then, we also investigated the origin of the 1,3-diene formation (Scheme 2d). As expected, 1,5-diene 4a can be smoothly isomerized to give 1,3-diene 5a under the reaction conditions for isomerized-terminal selectivity, indicating that 1,3-diene is generated by CC double bond migration from 4a (Scheme 2e). To elucidate the process of this alkene isomerization,18 deuterium labelling experiments were conducted. When isotopically labelled fluorinated 1,5-diene 4a-D was treated under cationic rhodium/BINAP conditions, a 1,3-deuterium shift was observed (Scheme 2f). Furthermore, a H/D crossover experiment shows that no intermolecular deuterium is exchanged between 4a-D and 4b, which indicates that the 1,3-hydrogen migration is exclusively intramolecular (Scheme 2g). These results suggest that a π-allyl mechanism may be involved in this alkene isomerization.
 | ||
| Scheme 2 Mechanistic investigations. | ||
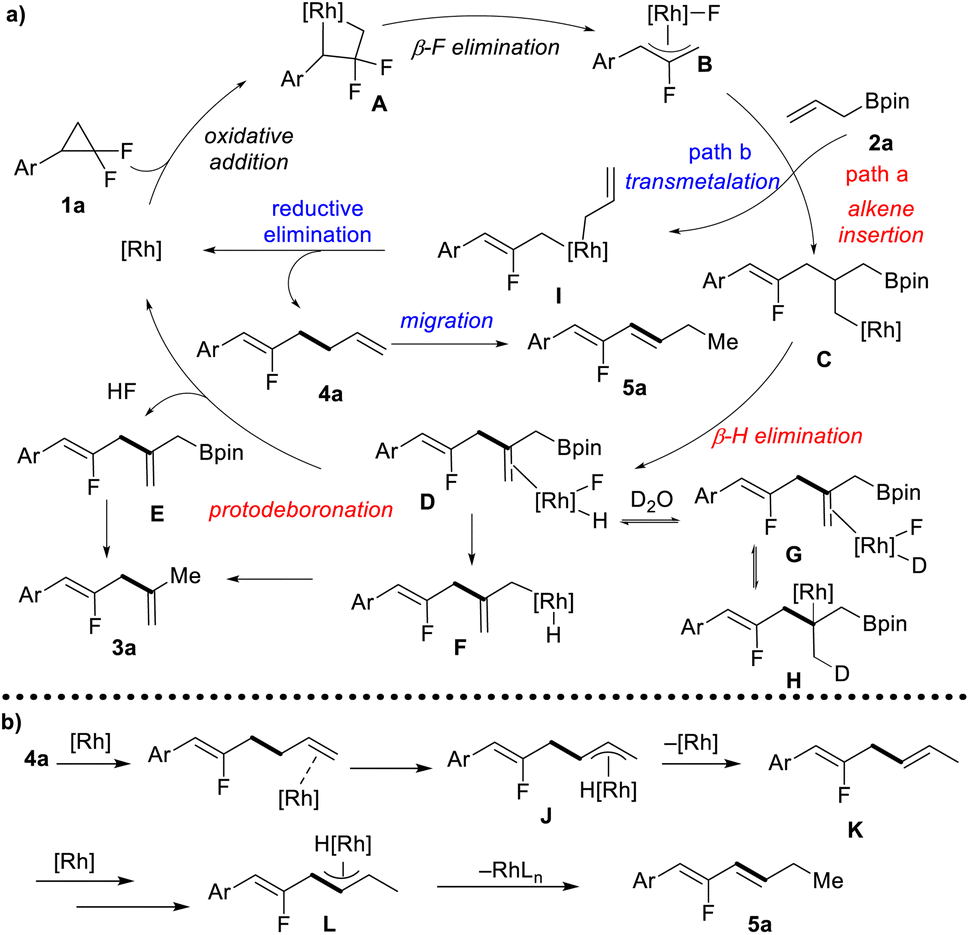
Based on our mechanistic investigations and previous reports,10 a plausible mechanism is proposed (Scheme 3a). Firstly, the oxidative addition of gem-difluorinated cyclopropane with a Rh(I) complex gives four-membered rhodacycle A, followed by β-F elimination to form the key allyl-Rh(III) complex B. At this stage, there are two reaction pathways for the Rh-complex B. In path a, allyl-Bpin serves as an alkene functionality that inserts into the Rh-allyl bond to afford intermediate C, followed by β-H elimination to give a dienyl-Bpin-bound rhodium complex D. The dissociation of the rhodium complex D would give dienyl-Bpin E and F-[Rh]-H species. The reductive elimination of F-[Rh]-H would regenerate the rhodium catalyst and release one molecule of HF. Finally, dienyl-Bpin E would undergo protodeboronation with in situ generated HF to form 1,4-diene 3a as the coupling product. Meanwhile, the protodeboronation can also occur through a sequence of intramolecular transmetallation and reductive elimination via intermediate F. The rationalization of the high degree of deuterium incorporation may include deuterium exchange from D to G, multiple times of olefin migratory insertion and β-H elimination (G to H), and protodeboronation with in situ generated DF. In path b, the allyl-Rh(III) complex B would undergo transmetallation with allyl-Bpin to give a di-allyl rhodium complex I, and allyl–allyl reductive elimination furnishes fluorinated 1,5-diene 4a. When using BINAP as the ligand, further CC double migration can occur in a site- and stereoselective manner forming conjugated diene 5a as the final product. Mechanistic studies support that this alkene migration follows the π-allyl pathway, as shown in Scheme 3b. The terminal olefin coordinates to the rhodium catalyst, followed by C–H bond addition at the allylic site to form a η3-allyl rhodium intermediate J. Then, C–H bond reductive elimination at the terminal site gives 1,4-diene K, and a second migratory process would provide the thermodynamically more stable 1,3-diene 5a.
 | ||
| Scheme 3 Proposed mechanism for the formation of fluorinated 1,n-dienes. | ||
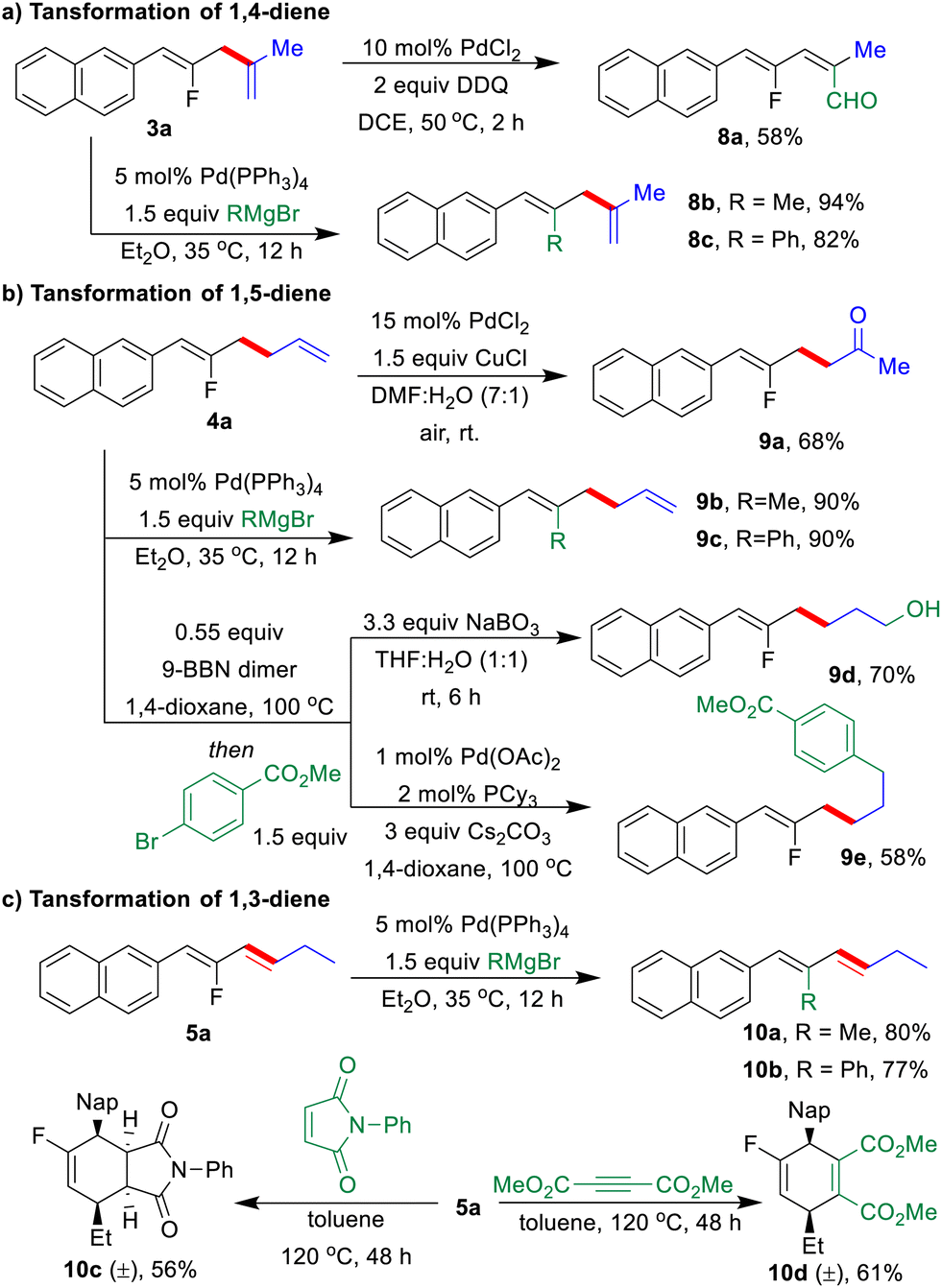
The synthetic practicability of the regio-switchable allyl–allyl coupling strategy was further demonstrated by a series of post-functionalization of three types of fluorinated dienes (Scheme 4). Under palladium catalysis, F-based Kumada-coupling between 1,n-dienes (3a, 4a, and 5a) with Grignard reagents as the nucleophiles gave highly functionalized 1,n-dienes in excellent yields, in which the configuration of the double bonds remain unchanged (8b, 8c, 9b, 9c, 10a, and 10b).19 A Pd-catalyzed oxygenation of the allylic C–H bond was performed with 3a to generate fluorinated alkenyl aldehyde 8a in 58% yield (Scheme 4a).20 Furthermore, Wacker oxidation of 4a in the presence of Pd/Cu under air gave fluorinated γ,δ-unsaturated ketone 9a in 68% yield.21 The fluorinated 1,5-diene 4a underwent a highly regioselective hydroboration with 9-BBN, followed by oxidation with NaBO3 or Suzuki coupling with methyl 4-bromobenzoate to deliver 9d or 9e in moderate to good yields, respectively (Scheme 4b).22 Finally, Diels–Alder reactions of conjugated diene 5a with 1-phenyl-1H-pyrrole-2,5-dione or dimethyl but-2-ynedioate produced fluorinated cyclic compounds 10c and 10d in moderate yields (Scheme 4c).
 | ||
| Scheme 4 Synthetic applications. | ||
Conclusions
In conclusion, we have developed a facile protocol to access structurally diverse fluorinated dienes through rhodium-catalyzed region-switchable cross-coupling of gem-difluorinated cyclopropanes with allylboronates. The regioselectivity pattern could be dominated by an appropriate choice of the rhodium catalyst and phosphine ligand. The internal selectivity that gives fluorinated 1,4-dienes can be achieved in the presence of a neutral rhodium catalyst and a monodentate phosphine ligand, while the use of a cationic rhodium catalyst and monodentate phosphine ligand ensures terminal selectivity to produce fluorinated 1,5-dienes. When BINAP serves as the ligand, the terminal-selective transformation undergoes an additional CC bond migration to give conjugated dienes. Mechanistic investigations indicate that the internal selectivity comes from a Heck-type process/protodeboronation sequence, while 1,3-dienes are derived from the isomerization from 1,5-dienes via the π-allyl mechanism. The practicability of regio-switchable allyl–allyl coupling reactions is also demonstrated by a series of downstream transformations of three types of dienes for the synthesis of polysubstituted and fluorine-containing molecules. Future work will focus on understanding how the combination of Rh/ligand controls the regioselectivity.
Data availability
All experimental data in this manuscript are available in the ESI.†Author contributions
Y. Z. and Y. X. conceived and designed the experiments. Y. Z., H. Y., J. D., and Q. H. performed the experiments, compound characterization, and data analysis. Y. Z., G. H. and Y. X. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the “Thousand Young Talents Program” (Grant 15-YINGXIA), the National Natural Science Foundation (Grant 22001180), the Key Research and Development Program of Sichuan Province (Grant 2021YFQ0060), the start-up funding from Sichuan University (Grant YJ201965), and the Tsinghua Laboratory Innovation Fund (100020019).References
- For early reports and mechanistic studies, see: (a) A. Goliaszewski and J. Schwartz, J. Am. Chem. Soc., 1984, 106, 5028–5030 CrossRef CAS; (b) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef; (c) M. Méndez, J. M. Cuerva, E. Gómez-Bengoa, D. J. Cárdenas and A. M. Echavarren, Chem.–Eur. J., 2002, 8, 3620–3628 CrossRef; (d) D. J. Cárdenas and A. M. Echavarren, New J. Chem., 2004, 28, 338–347 RSC.
- (a) B. M. Trost and E. Keinan, Tetrahedron Lett., 1980, 21, 2595–2598 CrossRef CAS; (b) H. Nakamura, M. Bao and Y. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 3208–3210 CrossRef CAS; (c) P. Srihari, A. P. Singh, A. K. Basak and J. S. Yadav, Tetrahedron Lett., 2007, 48, 5999–6001 CrossRef CAS; (d) Y. Sumida, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 1629–1632 CrossRef CAS PubMed; (e) E. Ferrer Flegeau, U. Schneider and S. Kobayashi, Chem.–Eur. J., 2009, 15, 12247–12254 CrossRef PubMed; (f) A. Jiménez-Aquino, E. Ferrer Flegeau, U. Schneider and S. Kobayashi, Chem. Commun., 2011, 47, 9456–9458 RSC; (g) Q. Yuan, K. Yao, D. Liu and W. Zhang, Chem. Commun., 2015, 51, 11834–11836 RSC; (h) J. S. Marcum, T. N. Cervarich, R. S. Manan, C. C. Roberts and S. J. Meek, ACS Catal., 2019, 9, 5881–5889 CrossRef CAS; (i) D.-W. Ji, Y.-C. Hu, H. Zheng, C.-Y. Zhao, Q.-A. Chen and V. M. Dong, Chem. Sci., 2019, 10, 6311–6315 RSC; (j) D.-W. Ji, G.-C. He, W.-S. Zhang, C.-Y. Zhao, Y.-C. Hu and Q.-A. Chen, Chem. Commun., 2020, 56, 7431–7434 RSC; (k) M. Estaitie and D. G. Hall, Chem. Commun., 2022, 58, 1370–1373 RSC.
- S. Porcel, V. López-Carrillo, C. García-Yebra and A. M. Echavarren, Angew. Chem., Int. Ed., 2008, 47, 1883–1886 CrossRef CAS PubMed.
- (a) P. Zhang, L. A. Brozek and J. P. Morken, J. Am. Chem. Soc., 2010, 132, 10686–10688 CrossRef CAS PubMed; (b) P. Zhang, H. Le, R. E. Kyne and J. P. Morken, J. Am. Chem. Soc., 2011, 133, 9716–9719 CrossRef CAS PubMed; (c) L. A. Brozek, M. J. Ardolino and J. P. Morken, J. Am. Chem. Soc., 2011, 133, 16778–16781 CrossRef CAS PubMed; (d) M. J. Ardolino and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 7092–7100 CrossRef CAS PubMed; (e) H. Le, A. Batten and J. P. Morken, Org. Lett., 2014, 16, 2096–2099 CrossRef CAS PubMed; (f) V. Hornillos, M. Pérez, M. Fañanás-Mastral and B. L. Feringa, J. Am. Chem. Soc., 2013, 135, 2140–2143 CrossRef CAS PubMed; (g) J. Y. Hamilton, N. Hauser, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 10759–10762 CrossRef CAS PubMed; (h) Y. Yasuda, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2016, 55, 10816–10820 CrossRef CAS PubMed; (i) X. Wang, Z. Han, Z. Wang and K. Ding, Angew. Chem. Int. Ed., 2017, 56, 1116–1119 CrossRef CAS PubMed; (j) Y. Zheng, B. Yue, K. Wei and Y. Yang, Org. Lett., 2018, 20, 8035–8038 CrossRef CAS PubMed.
- For the application of 1,5-dienes see: (a) E. Breitmaier, Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, Wiley-VCH, Weinheim, 2006 CrossRef; (b) Y. Zhao, S. Chng and T. Loh, J. Am. Chem. Soc., 2007, 129, 492–493 CrossRef CAS PubMed; (c) H. Lee, Acc. Chem. Res., 2015, 48, 2308–2319 CrossRef CAS PubMed; (d) P. K. Dornan, D. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2016, 138, 6372–6375 CrossRef CAS PubMed; (e) B. Schmidt, M. H. Petersen and D. Braun, J. Org. Chem., 2018, 83, 1627–1633 CrossRef CAS PubMed; (f) L. Chuang, C. Wen, Y. Lee, Y. Lin, L. Hsu, S. Wang and F. Chu, J. Nat. Prod., 2018, 81, 1162–1172 CrossRef CAS PubMed; (g) A. C. Huang, Y. J. Hong, A. D. Bond, D. J. Tantillo and A. Osbourn, Angew. Chem., Int. Ed., 2018, 57, 1291–1295 CrossRef CAS PubMed.
- For reviews on gem-difluorinated cyclopropanes, see: (a) W. R. Dolbier and M. A. Battiste, Chem. Rev., 2003, 103, 1071–1098 CrossRef CAS PubMed; (b) M. Fedoryński, Chem. Rev., 2003, 103, 1099–1132 CrossRef PubMed; (c) K. S. Adekenova, P. B. Wyatt and S. M. Adekenov, Beilstein J. Org. Chem., 2021, 17, 245–272 CrossRef CAS PubMed; (d) L. Lv, H. Qian and Z. Li, ChemCatChem, 2022 DOI:10.1002/cctc.202200890; (e) Y. Zhu, Y. Zeng, Z.-T. Jiang and Y. Xia, Synlett, 2022 DOI:10.1055/a-1912-3059.
- J. Xu, E.-A. M. A. Ahmed, B. Xiao, Q.-Q. Lu, Y.-L. Wang, C.-G. Yu and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 8231–8235 CrossRef CAS PubMed.
- (a) E.-A. M. A. Ahmed, A. M. Y. Suliman, T.-J. Gong and Y. Fu, Org. Lett., 2019, 21, 5645–5649 CrossRef CAS PubMed; (b) E.-A. M. A. Ahmed, A. M. Y. Suliman, T.-J. Gong and Y. Fu, Org. Lett., 2020, 22, 1414–1419 CrossRef CAS PubMed; (c) A. M. Y. Suliman, E.-A. M. A. Ahmed, T.-J. Gong and Y. Fu, Chem. Commun., 2021, 57, 6400–6403 RSC; (d) A. M. Y. Suliman, E.-A. M. A. Ahmed, T.-J. Gong and Y. Fu, Org. Lett., 2021, 23, 3259–3263 CrossRef CAS PubMed.
- (a) J. Ni, B. Nishonov, A. Pardaev and A. Zhang, J. Org. Chem., 2019, 84, 13646–13654 CrossRef CAS PubMed; (b) Z. Fu, J. Zhu, S. Guo and A. Lin, Chem. Commun., 2021, 57, 1262–1265 RSC; (c) P. Zhou, X. Yang, J. Wang, C. Ge, W. Feng, Y.-M. Liang and Y. Zhang, Org. Lett., 2021, 23, 4920–4924 CrossRef CAS PubMed; (d) B. Xiong, X. Chen, J. Liu, X. Zhang, Y. Xia and Z. Lian, ACS Catal., 2021, 11, 11960–11965 CrossRef CAS; (e) W. Yuan, X. Li, Z. Qi and X. Li, Org. Lett., 2022, 24, 2093–2098 CrossRef CAS PubMed ; For recent work using co-catalysis, see:; (f) Y. Ai, H. Yang, C. Duan, X. Li and S. Yu, Org. Lett., 2022, 24, 5051–5055 CrossRef CAS PubMed.
- (a) Z.-T. Jiang, J. Huang, Y. Zeng, F. Hu and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 10626–10631 CrossRef CAS PubMed; (b) Z.-T. Jiang, Y. Zeng and Y. Xia, Synlett, 2021, 32, 1675–1682 CrossRef CAS; (c) Y. Zeng, H. Gao, Y. Zhu, Z.-T. Jiang, G. Lu and Y. Xia, ACS Catal., 2022, 12, 8857–8867 CrossRef CAS.
- (a) L. Lv and C.-J. Li, Angew. Chem., Int. Ed., 2021, 60, 13098–13104 CrossRef CAS PubMed; (b) L. Lv, H. Qian, Y. Ma, S. Huang, X. Yan and Z. Li, Chem. Sci., 2021, 12, 15511–15518 RSC.
- (a) L. Lv, H. Qian, A. B. Crowell, S. Chen and Z. Li, ACS Catal., 2022, 12, 6495–6505 CrossRef CAS; (b) L. Wu, M. Wang, Y. Liang and Z. Shi, Chin. J. Chem., 2022, 40, 2345–2355 CrossRef CAS.
- (a) Z. Huang and G. Dong, Acc. Chem. Res., 2017, 50, 465–471 CrossRef CAS PubMed; (b) C. Nájera, I. P. Beletskaya and M. Yus, Chem. Soc. Rev., 2019, 48, 4515–4618 RSC; (c) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Soc. Rev., 2020, 49, 7101–7166 RSC.
- (a) G. Hilt and S. Lüers, Synthesis, 2002, 5, 0609–0618 CrossRef; (b) M. Arndt, A. Reinhold and G. Hilt, J. Org. Chem., 2010, 75, 5203–5210 CrossRef CAS PubMed.
- J. Jia, F. Yuan, Z. Zhang, X. Song, F. Hu and Y. Xia, Org. Lett., 2022, 24, 1985–1990 CrossRef CAS PubMed.
- C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2–14 CrossRef CAS PubMed.
- (a) C. R. Larsen, G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2014, 136, 1226–1229 CrossRef CAS PubMed; (b) Q. Meng, T. E. Schirmer, K. Katou and B. König, Angew. Chem., Int. Ed., 2019, 58, 5723–5728 CrossRef CAS PubMed; (c) A. Kapat, T. Sperger, S. Guven and F. Schoenebeck, Science, 2019, 363, 391–396 CrossRef CAS PubMed; (d) X. Yu, H. Zhao, P. Li and M. J. Koh, J. Am. Chem. Soc., 2020, 142, 18223–18230 CrossRef CAS PubMed; (e) S. Scaringi and C. Mazet, ACS Catal., 2021, 11, 7970–7977 CrossRef CAS.
- For mechanistic studies on olefin isomerization, see: (a) S. Biswas, Z. Huang, Y. Choliy, D. Y. Wang, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2012, 134, 13276–13295 CrossRef CAS PubMed; (b) S. Biswas, Comments Inorg. Chem., 2015, 35, 300–330 CrossRef CAS.
- W. Dai, J. Xiao, G. Jin, J. Wu and S. Cao, J. Org. Chem., 2014, 79, 10537–10546 CrossRef CAS PubMed.
- H. Chen, H. Jiang, C. Cai, J. Dong and W. Fu, Org. Lett., 2011, 13, 992–994 CrossRef CAS PubMed.
- G. Reginato, A. Mordini, M. Verrucci, A. Degl'Innocenti and A. Capperucci, Tetrahedron: Asymmetry, 2000, 11, 3759–3768 CrossRef CAS.
- (a) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS; (b) G. W. Kabalka, T. M. Shoup and N. M. Goudgaon, J. Org. Chem., 1989, 54, 5930–5933 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04118a |
| This journal is © The Royal Society of Chemistry 2022 |