
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTaming the stilbene radical anion†
Grégoire
Sieg
a,
Igor
Müller
a,
Kilian
Weißer
b and
C. Gunnar
Werncke
 *a
*a
aFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35037 Marburg, Germany. E-mail: gunnar.werncke@chemie.uni-marburg.de
bInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 9th November 2022
Abstract
Radical anions appear as intermediates in a variety of organic reductions and have recently garnered interest for their role as mediators for electron-driven catalysis as well as for organic electron conductor materials. Due to their unstable nature, the isolation of such organic radical anions is usually only possible by using extended aromatic systems, whereas non-aromatic unsaturated hydrocarbons have so far only been observed in situ. We herein report the first isolation, structure and spectroscopic characterization of a simple aryl substituted alkene radical anion, namely that of stilbene (1,2-diphenyl ethylene), achieved by encapsulation between two [K{18c6}] cations. The formation of the radical anion is accompanied by Z → E isomerization of the involved double bond, also on a catalytic scale. Employing the linear iron(I) complex [Fe(NR2)2]− as a reductant and coordination site also allows for this transformation, via formation of an iron(II) bound radical anion. The use of the iron complex now also allows for Z → E isomerization of electron richer, simple alkenes bearing either mixed alkyl/aryl or even bis(alkyl) substitution.
Introduction
Organic radicals are known to play key roles in many well-established organic reactions. Radical anions, in particular, appear as intermediates in a variety of organic reductions.1–3 They are intrinsically unstable due to their ability to react subsequently in a multitude of reaction pathways, such as dimerization, as exploited in pinacol-type coupling reactions.1 In recent years, organic radical anions have garnered further importance due to the surge of photoredox catalysis.4–6 In this context, radical anions are mediators for bond transformations where the transmitted electron itself is sometimes considered as a catalyst in analogy to proton-catalysed reactions.7–9 As such there is a longstanding interest in understanding the behaviour of simple radical anions. Common methods to stabilize or even isolate such species rely on the use of extended aromatic systems or electron withdrawing functional groups, such as carbonyl units, to lower the energy of the involved π* orbitals as well as to disperse of the radical character over a larger π-system.10–14 In this instance respective radical anions play an important role in organic functional materials such as electric conductors, transistors or magnetic devices.15–19 Isolable examples of pure carbon-based radical anions are still scarce and concern only aromatic compounds with energetically accessible π*-orbitals, most prominently alkali metal anthracenes and naphthalenes. In contrast, radical anions of alkene based compounds were so far only observed in situ,20–23 but are of fundamental interest for nearly a century.24 Of those, the stilbene radical anion [S]˙− (S = stilbene/1,2-diphenylethylene) has been a particular subject of EPR25 and electronic absorption spectroscopic26–28 as well as cyclovoltammetric29 analyses. Thereby, the radical anion could only be generated in situ, either via (electro)chemical reduction,25,30,31 photolysis32 or/and radiolysis.33 It also showed that Z-stilbene is subject to Z → E isomerisation, as was extensively examined by Szwarc and others.25,30,34–36 Kinetic studies indicated that the isomerisation does not occur via the initially formed stilbene radical anion (Z-[S]˙−), but the dianion (Z-[S]2−). The latter stems from reversible disproportionation of [S]˙− (into [S]2− and [S]0), and is thought to isomerise more rapidly than the radical anion Z-[S]˙−.36–40 Despite these early reports of electron induced alkene Z → E isomerisation a use for selective formation of E-alkenes from pure Z-alkenes or n E/Z-mixtures, which are often observed in the construction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (e.g. by the Wittig reaction, McMurry coupling or alkene metathesis),3,41–45 is surprisingly lacking. In part the absence can be explained by the rather harsh reductive conditions, which would be incompatible with functional groups such as halides. With the increasing use of strongly reducing photoredox catalysts such isomerisation reactions might become nonetheless useful. Furthermore, the electrocatalytic Z → E isomerisation can pose an alternative for alkene based photoswitches, in resemblance to a recent report of electrocatalytic shifting of the photostationary equilibrium of azobenzene derivatives via their radical anions.46
C double bonds (e.g. by the Wittig reaction, McMurry coupling or alkene metathesis),3,41–45 is surprisingly lacking. In part the absence can be explained by the rather harsh reductive conditions, which would be incompatible with functional groups such as halides. With the increasing use of strongly reducing photoredox catalysts such isomerisation reactions might become nonetheless useful. Furthermore, the electrocatalytic Z → E isomerisation can pose an alternative for alkene based photoswitches, in resemblance to a recent report of electrocatalytic shifting of the photostationary equilibrium of azobenzene derivatives via their radical anions.46
Herein we report on the isolation, structure and spectroscopic characterization of the stilbene radical anion, achieved by its encapsulation between two [K{18c6}]+ cations. The radical anion readily undergoes reversible electron transfer with excess of Z-stilbene, for which accordingly catalytic Z → E isomerisation is observed. Using the linear iron(I) silylamide [Fe(NR2)2]− the catalytic Z → E isomerisation can conceptionally be extended to 1,2-alkyl/aryl and -dialkyl alkenes, not achievable by the “free” stilbene radical anion. Mechanistic insights point to involvement of an iron(II) bound alkene radical anion.
Results and discussion
Isolation of the stilbene radical anion
Z-Stilbene was reacted with 18-crown-6 and KC8 in Et2O in the presence of [K{18c6}][Fe(NR2)3]47 (R = SiMe3). This resulted in an immediate colour change from light yellow to dark red. Storage of the pentane layered reaction solution at −40 °C yielded [(K{18c6})2(E-stilbene)][Fe(NR2)3] (1) in 61% yield (Scheme 1). The use of [K{18c6}][Fe(NR2)3] is of essence as in the absence of the additional [K{18c6}] cation no reduction of stilbene was observed, while the trisamide serves as a reductively stable non-coordinating anion. Similar observations were already made during the isolation of a phenyl substituted pyridine radical anion.48 The [K{18c6}] cation is equally important moiety as the use of crypt.222 as potassium masking agent resulted in undefined decomposition. 1 is stable at room temperature under inert conditions for several hours in solution and for months in solid state at −35 °C. Attempts for the isolation of a Z-conformer of 1 were not successful, in agreement with the observed rapid isomerisation to the thermodynamically more stable E-conformer.25,30,34–36 X-Ray diffraction analysis on suitable crystals revealed the formed E-stilbene unit being encapsulated between two [K{18c6}] cations (Fig. 1 left). The potassium atoms are situated above and below the central alkene unit.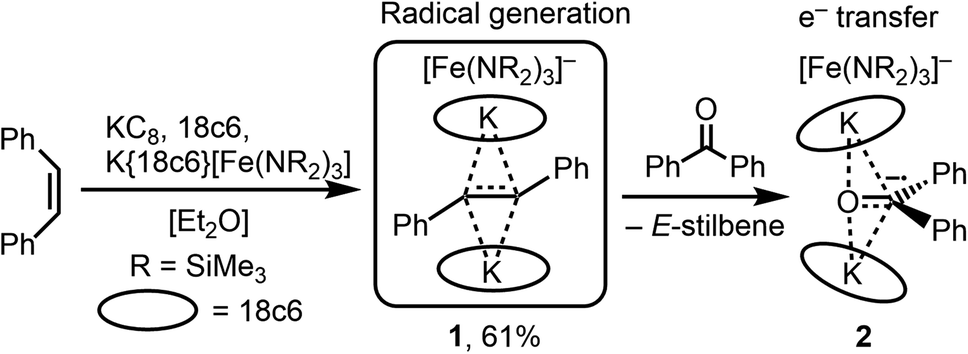 | ||
| Scheme 1 Synthesis of the bis(cation) stabilized stilbene radical anion 1 and formation of the ketyl complex 2. | ||
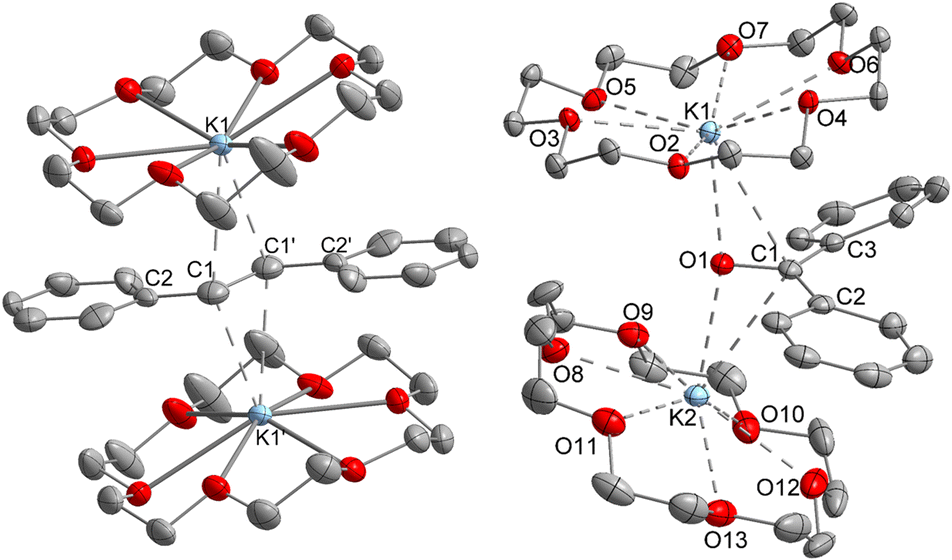 | ||
| Fig. 1 Molecular structure of 1 (left) and 2 (right) excluding the [Fe(N(SiMe3)2)3]− counter anions. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): (1) C1–C1′ 1.415(5) [1.350(7)], C1–C2 1.389(7) [1.488(7)], C2–C1–C1′ 127.6(4) [125.7(4)]. Bond metrics of the second disordered part are given in square brackets. (2) O1–C1 1.299(2), C1–C2 1.452(2), C1–C3 1.473(2), O1–K1 2.724(1), O1–K2 2.701(1), C1–K1 3.172(1), C1–K2 3.134(2), C2–C1–C3 123.3(1), O1–C1–C2 119.1(1), O1–C1–C3 117.5(1). | ||
They are pulled slightly out of the plane defined by oxygen atoms of the 18c6 ring, which is indicative of an anionic charge localized on the double bond. Due to an intrinsic, persistent crystallographic problem, unchanged upon variation of the crystallisation conditions, the whole stilbene unit is disordered lengthwise over two positions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Accordingly, this prohibits so far an in-depths evaluation of the structural metrics. For example both crystallographic parts exhibit highly different C–C bonds (1.35 Å and 1.41 Å), which are thus only in part elongated in comparison with the free stilbene (1.34 Å).49
:1). Accordingly, this prohibits so far an in-depths evaluation of the structural metrics. For example both crystallographic parts exhibit highly different C–C bonds (1.35 Å and 1.41 Å), which are thus only in part elongated in comparison with the free stilbene (1.34 Å).49
To support the notion of a stilbene radical anion, 1 was examined by X-band EPR spectroscopy in toluene. At 8 K the toluene glass of 1 exhibited a hyperfine structured singlet (g = 2.004088, Fig. 2), which is in general agreement with the presence of an organic radical anion. The hyperfine structure was however insufficiently resolved to allow for extraction of any coupling constants. The hyperfine structure further lost upon increasing the temperature to 100 K (g = 2.002396, Fig. S15†). Here, the observation of an unresolved singlet of the trans-stilbene radical anion in frozen solutions at around 100 K is in agreement with respective reports in the literature.29,50 Measuring the sample at 298 K also gave a only singlet (g = 2.002123, Fig. S14†), which is at odds with reported well resolved EPR spectra of in situ formed stilbene radical anions, that show coupling to all hydrogen atoms.25,35,51 We attribute the absence of a hyperfine structure for the radical anion in 1 at ambient conditions to persistent ion pairing with the K{18c6} units, as well as the high-spin iron(II) trisamide counter ion likely also contribute to line broadening via spin–spin interactions. Attempts for the measurement of 1 in THF to reduce the ion pairing effect led to its rapid degradation under these dilute conditions. According to its paramagnetic character, the proton NMR spectrum of 1 showed no features beyond the signals attributed to the [K{18c6}] moieties around 3.47 ppm as well as the [Fe(NR2)3]− anion at −2.54 ppm (Fig. S1†). UV-Vis spectroscopic examination of 1 in solution (Fig. 3) revealed a single absorption band at 485 nm which is in good agreement with in situ generated Na+(E-stilbene)˙− in THF (494 nm).39 The radical anionic nature of 1 was substantiated further chemically as it (incompletely) reduces [CoII(NR2)2] to the linear cobalt(I) complex [CoI(NR2)2]− (Fig. S8†) whose reduction potential (Ered = −1.45 V vs. Fc/Fc+) is less than of the stilbene (E1/2 (Z-stilbene) = −2.67 V; E1/2 (E-stilbene) = −2.70 V vs. Fc/Fc+).29,50,52
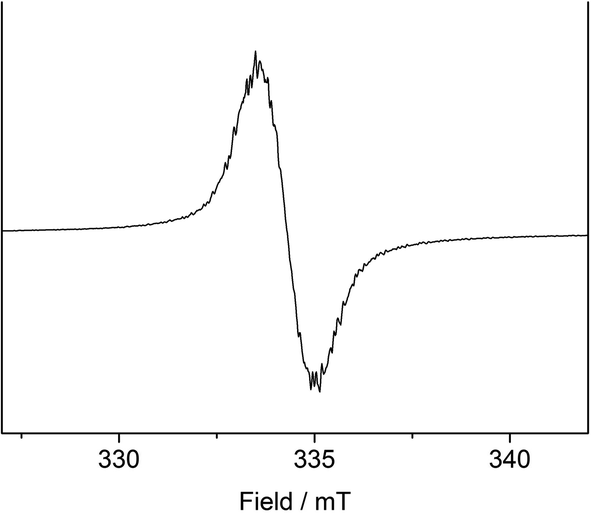 | ||
| Fig. 2 X-band EPR measurement of 1 in frozen toluene solution (9.368604 GHz) collected at 8 K. S = ½, g = 2.004088. | ||
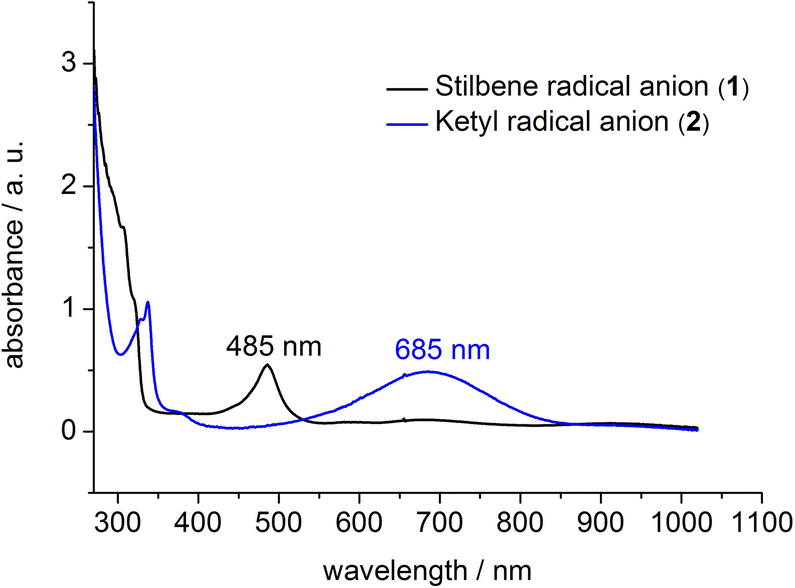 | ||
| Fig. 3 UV-Vis spectrum of 1 and 2 in THF at 300 K. | ||
Further, if 1 is subjected to benzophenone (E1/2 = −2.13 V vs. Fc/Fc+) the formation of the intensely blue coloured [(K{18c6})2(Ph2CO)]2[Fe(NR2)3] (2) is observed (Scheme 1), with a characteristic absorption band at 685 nm belonging to the π → π* transition of the ketyl radical anion.142 can also be independently obtained by reducing Ph2CO with KC8 in the presence of 18-crown-6 and [K{18c6}][Fe(NR2)2]. In analogy to 1, the ketyl radical anion in 2 is sandwiched between the [K{18c6}] cations (Fig. 1 right). These are tilted towards each other by approximately 42.5° to account for the non-planarity of the diarylketyl unit. The C–O bond length of the ketyl unit in 2 amounts to 1.299(2) Å, which is typical for ketyl radical anions and due to a reduced CO bond order by population of the antibonding π* orbital52,53 (free benzophenone: 1.23(1) Å).54 Interestingly, besides interactions with the ketyl oxygen (K1/2–O1 = 2.724(1) Å/2.701(1) Å), the potassium cations also exhibit close contacts to the ketyl carbon (K1/2–C1 = 3.172(1)/3.134(2) Å), thus overall coordinating in an asymmetric side-on fashion to the CO unit. It contrasts the typical end-on coordination of alkali metal ketyl or fluorenyl salts in solid state14,55 and is likely due to the repulsion of the opposing crown-ethers. Attempts to acquire radical anions of a more electron rich 1,2-alkyl/aryl-substituted ethylene (β-methyl styrene) or even a 1,2-dialkyl ethylene (3-hexene) were not successful.
Behaviour of the radical anion 1 towards ethylene derivatives
The unequivocal isolation of the cation stabilized stilbene radical anion 1 now offered the opportunity to study with regards to other alkenes. Quenching 1 with D2O yielded purely E-stilbene, showing that the encapsulated radical anion is not subject to substantial redox disproportionation into stilbene and its dianion as commonly observed during in situ studies of stilbene radical anions.36–40 Consequently, it implicates that the stilbene radical anion in 1 is responsible for the observed Z → E isomerisation, opposed to the isomerisation mechanism via its dianion proposed by Szwarc and others.36–40 As such we further wanted to gain insights if 1 can be used for the isomerisation of additional stilbene via electron transfer. When 1 was treated with an equimolar amount of E-stilbene in THF-d8 no signs of the added stilbene was perceivable by proton NMR spectroscopy at room temperature or at −80 °C (Fig. S6†). Increasing the amount of E-stilbene to 5 equivalents merely hinted to a broad signal centred at the median signal position of E-stilbene (7.39 ppm), which became clearly visible using a tenfold excess (Fig. S7†). This speaks to rapid electron transfer between 1 and the added stilbene, that effectuates paramagnetic line broadening. The stilbene addition is accompanied by the appearance of a minor set of signals at around 7.05 ppm as well as at 2.93 ppm, whose amount (approx. 10% with regards to employed 1) is unaffected by quenching of the reaction mixture with D2O. We tentatively attributed this to the formation of 1,2,3,4-tetraphenylbutane due to stilbene radical anion dimerization. Given that such a behaviour is absent for 1 itself, it implicates that the presence of additional stilbene leads to minor amounts of free, unstabilised stilbene radical anions, that lack any [K{18c6}] stabilisation in solution and thus allows for C–C coupling.To examine the catalytic Z → E isomerisation, Z-stilbene (Table 1) was treated with a catalytic amount of 1 (4 mol%) in [D8]THF. Conversion to E-stilbene is observed within 2.5 h (27% E-product) after which the reaction stops as perceived from a colour change from deep yellow to colourless. This is likely due to degradation of the stilbene radical anion in the presence of the substrate,37,39 as well as its general instability in [D8]THF. Given the proposed involvement of the stilbene radical anion as a reductant, we also tested KC8 as a catalyst which proved to be highly efficient under the same conditions (100% conversion in [D8]THF within 5 minutes). Interestingly, in Et2O no isomerisation is observed, showing the importance of potassium cation solvation. Expanding the catalytic isomerisation to a 1,2-alkyl/aryl-substituted (Z-β-methyl styrene) or Z-1,2-dialkyl ethylene derivative (Z-3-hexene) using 1 or KC8 as catalyst did not result in any transformation at all, which can be explained by the substrates' more electron rich nature.
| Catalyst | R1 | R2 | Cat. (mol%) | Reaction time | Conversion (%) |
|---|---|---|---|---|---|
| 1 | Ph | Ph | 4 | 2 h 30 min | 27% |
| Ph | Me | 5 | 20 h | 0% | |
| Et | Et | 10 | 7 d | 0% | |
| KC8 | Ph | Ph | 4 | 5 min | 100% |
| Ph | Me | 4 | 2 h | 0% | |
| Et | Et | 10 | 7d | 0% | |
| K{18c6}[FeI(NR2)2] | Ph | Ph | 4 | 45 min | 95% |
| Ph | Me | 10 | 6 h 30 min | 82% | |
| Et | Et | 10 | 7d | 75% | |
| K{18c6} | Ph | Ph | 10 | 3 h 45 min | 5.4% |
| [FeI(NR{Dipp})2] | Ph | Ph | 10 | 24 h | 12% |
Iron mediated Z → E isomerisation of alkenes
To overcome these shortcomings in terms of Z → E isomerisation of alkylated ethylene derivatives we contemplated on isomerisation of such species in the coordination sphere of a highly reducing metal complex (Scheme 2). For that we chose the iron(I) silylamide [FeI(NR2)2]− (Ered −2.07 V),56 which was already proven for the formation of iron(II) bound radical anions of ketones and related nitrogen derivatives (imines and aldimines), as well as of alkynes.52,57 Further, a remarkable yet slow Z → E isomerisation was observed recently for an ethylene bridged diphosphine (cis-1,2-bis(diphosphino)ethylene).47 Indeed, using 4 mol% of [K{18c6}][FeI(NR2)2] Z-stilbene is converted to E-stilbene by 95% within 45 minutes.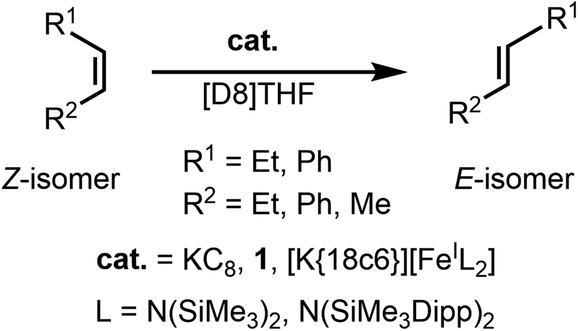 | ||
| Scheme 2 Catalytic Z to E conversion of alkenes with [FeI] as catalyst. | ||
Intriguingly, Z-β-methylstyrene as well as even Z-3-hexene are also transformed, however needing higher catalyst loadings (10 mol%) and substantially longer reaction times (Fig. 4). The slower reaction can be attributed to the increase of the π*-orbital energy in case of alkyl substituents.
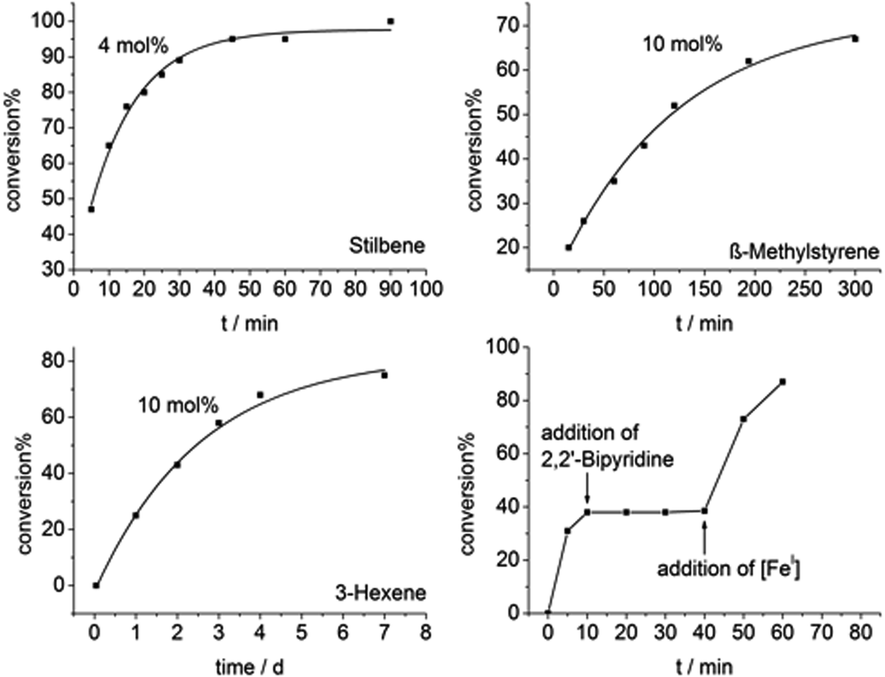 | ||
| Fig. 4 Catalytic Z to E conversion for stilbene (top left), β-methylstyrene (top right), 3-hexene (bottom left) using [FeI] as catalyst and poisoning experiment with 2,2′-bipyridine (bottom right). | ||
Mechanistic examination of these catalytic reactions gave pseudo-first order kinetics for these iron mediated transformations, with no signs of an induction phase. To further substantiate the direct involvement of [K{18c6}][FeI(NR2)2] we conducted poisoning experiments, to rule out the involvement of in situ formed iron nanoparticles. Addition of stoichiometric amounts of 2,2′-bipyridine to the reaction mixture, proven to bind tightly to [Fe(NR2)2]−,58 stopped the reaction. Upon addition of further [Fe(NR2)2]− the catalysis resumed (Fig. 4, bottom right). Conversely, the system is unaffected by the addition of an excess of PCy3 which does not interact with [Fe(NR2)2]−,56 but influences the activity of nanoparticles.59,60 To explore the steric effect of the iron(I) catalyst, the same transformations were conducted with the sterically more demanding complex [K{18c6}][FeI(NR{Dipp})2]61 (Table 1). While a Z to E isomerization is still observable for stilbene, the reaction proceeds considerably slower, and is absent in case of the alkylated ethylene derivatives.
Stoichiometric treatment of [K{18c6}][Fe(NR2)2] with the substrates in Et2O resulted in an instantaneous change of colour of the solution from green to reddish brown in case of Z-stilbene. Crystallisation from the pentane layered Et2O filtrate gave the π complex [K{18c6}][Fe(NR2)2(E-stilbene)] (3) in moderate yields (54%) (Scheme 3). Within 3 the substrate coordinates to iron in a η2 fashion (Fig. 5, left). The central C–C bond (1.384(5) Å) is elongated in comparison with free stilbene (1.338 Å).49 The elongation is in the common range for alkenes bound to a low-valent metal ion,62,63 and is usually attributed to p → π* backbonding.64–66 The alkene ligand is twisted slightly (15.6(3)°) with regards to the plane defined by the N1,Fe1 and N2 yielding a distorted square planar geometry around the metal. The Fe–N distances amount to approx. 1.99 Å. This is larger than in the iron(I) precursor (1.92 Å)56 and comparable to three coordinate iron(II) halide complexes (1.95–1.97 Å)67 as well as the previously reported related π-alkyne iron complexes [Fe(NR2)2(η2-RCCR)] (1.97–2.00 Å).68 To substantiate the oxidation state of iron in 3 zero-field 57Mössbauer spectroscopy was employed (Fig. 6).
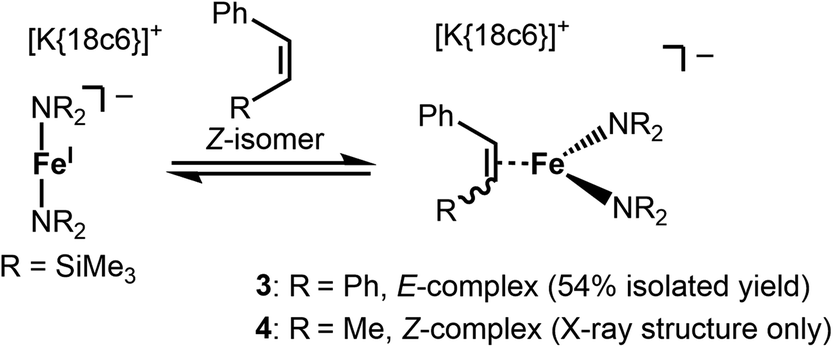 | ||
| Scheme 3 Formation of the side-on alkene complexes 3 and 4 in solution. | ||
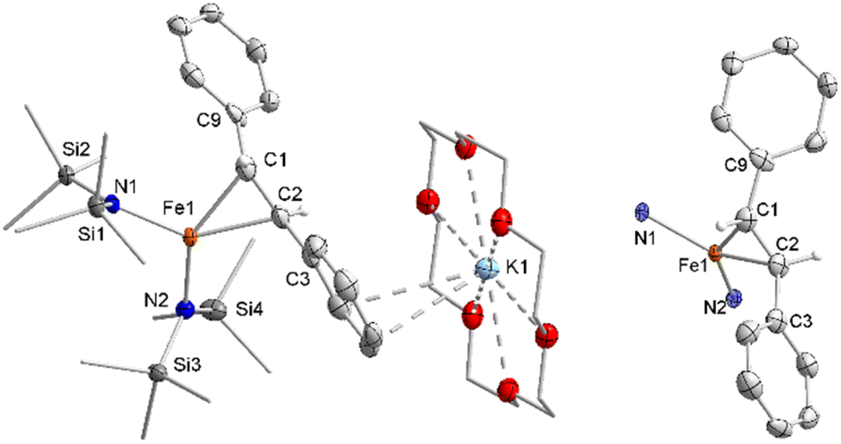 | ||
| Fig. 5 Molecular structure of 3 in the solid state (left) and depiction of the central iron-alkene unit. Unnecessary H atoms and are omitted for clarity. Selected bond lengths (Å) and angles (°): Fe1–N1 1.995(2), Fe1–N2 1.990(2), Fe1–C1 2.109(3), Fe1–C2 2.112(3), C2–C3 1.384(5), N1–Fe1–N2 115.64(10). | ||
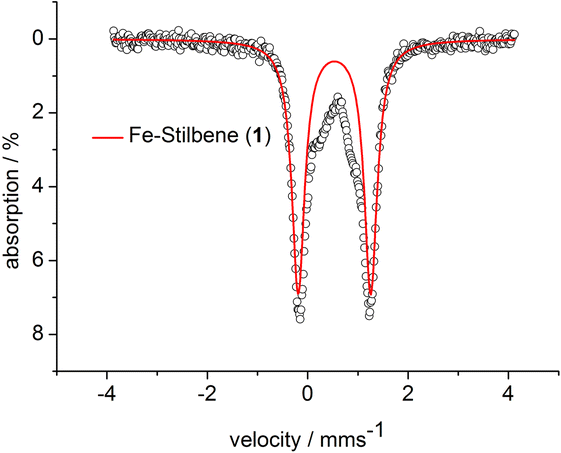 | ||
| Fig. 6 Zero-field 57Fe-Mössbauer spectrum of 3 at 13 K. δ = 0.53 mm s−1, ΔQ = 1.44 mm s−1. | ||
The spectrum of 3 at 13 K shows a doublet signal for the main species with an isomer shift of δ = 0.53 mm s−1 and a quadrupole splitting of ΔQ = 1.44 mm s−1. The isomer shift corresponds very well with low coordinate iron(II) complexes ([Fe(NR2)3]−: δ = 0.59 mm s−1, ΔQ = 0.60 mm s−1 ; [Fe(NR2)2I]−: δ = 0.63 mm s−1, ΔQ = 0.60 mm s−1; [Fe(NR2)2OCPh2(˙)](−): δ = 0.62 mm s−1, ΔQ = 1.20/1.83 mm s−1).52,56,69 The Mössbauer data implicates the formulation of 3 as an iron(II) bound alkene radical anion. Such a description is indeed plausible in view of DFT and CASSCF studies on the interaction of alkynes with [Fe(NR2)2]− (ref. 57) and a T-shaped iron(I) complex70 which were best described as metal(II) bound alkyne radical anions.
X-band EPR spectroscopic measurements at 100 K performed on 3 in THF gave no pronounced features. The absence of any signal rules out the presence of an iron(I) ion (S = 3/2) and supports the notion of a non-Kramer's iron(II) ion (S = 2) coupled to an organic radical.
1H-NMR spectroscopic examination of isolated 3 gave a resonance for the SiMe3-groups at −6.01 ppm (Fig. S3†). This signal position exhibits a slight high-field shift in comparison to either trigonal π-alkyne iron or three-coordinate iron(II) hexamethyldisilazanides (−1.88 to −4.05 ppm).52,57,67 Additional resonances at 92.7 ppm, 91.2 ppm and −25.3 ppm are attributed to aromatic substrate protons. Importantly, dissolution of pristine 3 gave rise to a signal belonging to the initially employed [Fe(NR2)2]− as well as E-stilbene. This implicated a dissociation equilibrium of 3 in solution, corroborated by independent measurement of a 1:1 mixture of Z-stilbene and [K{18c6}][Fe(NR2)2]. No evidence of the initial formation of the Z-alkene adduct [Fe(NR2)2(Z-stilbene)] by proton NMR spectroscopy was found, hinting to rapid bond isomerisation.
For the treatment of Z-β-methyl styrene with [K{18c6}][Fe(NR2)2] the colour change upon substrate addition was less pronounced. Attempts for the isolation of the adduct yielded only few crystals of the π-alkene complex 4 (see Fig. S37†) which were obtained with inseparable amounts of unreacted, crystalline [K{18c6}][Fe(NR2)2]. The structure of the anion in 4 is similar to the one found for 3 with a slightly shorter C2–C3 distance (1.416(3) Å) but exhibits otherwise comparable bond metrics. Proton NMR spectroscopic examination of a 1:1 mixture of the iron(I) precursor with equimolar amounts of Z-β-methyl styrene in [D8]THF resulted in the observation of a minor signal at −5.36 ppm belonging to the SiMe3 groups of 4 (Fig. S4†), with major amount of the iron(I) precursor and free stilbene. The latter is found only as the E-isomer. For the even more electron rich Z-3-hexene no signs of a π-complex are observed in solution, or upon crystallisation attempts. Together with the observations made for 3, it implicates an equilibrium between the free substrate as well as the iron(I) precursors and the respective adduct complex in solution, which is shifted to the former upon employing more electron rich substrates.
To probe the possible dissociation of 3 into the neutral iron(II) amide [Fe(NR2)2] and the free radical anion it was treated with KNR2 and 18c6 which however did not lead to the replacement of the alkene radical anion with the hexamethyldisilazanide and formation of 1 (Scheme 4, top). Inversely, the reaction of 1 with an excess of [FeII(NR2)2] did also not yield 3 – which could occur either by direct coordination the radical anion to [FeII(NR2)2] or via its outer-sphere reduction as seen for [CoII(NR2)2] under subsequent alkene coordination. As such the isomerisation of the ethylene derivative in 3 likely proceeds in the coordination sphere of the iron ion, and substrate dissociation occurs not as a free radical anion but as a neutral species. We thus propose for iron a catalytic cycle that starts with Z-alkene binding to the linear iron(I) silylamide (Scheme 4, bottom). The π-complex A (as in complex 4) can also be described as an iron(II) bound Z-alkene radical anion (A′). Subsequently, weakening of one Fe–C bond would allow for rotation along the C–C bond (B). Such an asymmetric substrate binding was found as a stable and energetically feasible state in case of related alkyne chromium complexes.57 This yields in an E-alkene complex (C/C′, as complex 3). Finally, the formed E-alkene is replaced by the next Z-alkene.
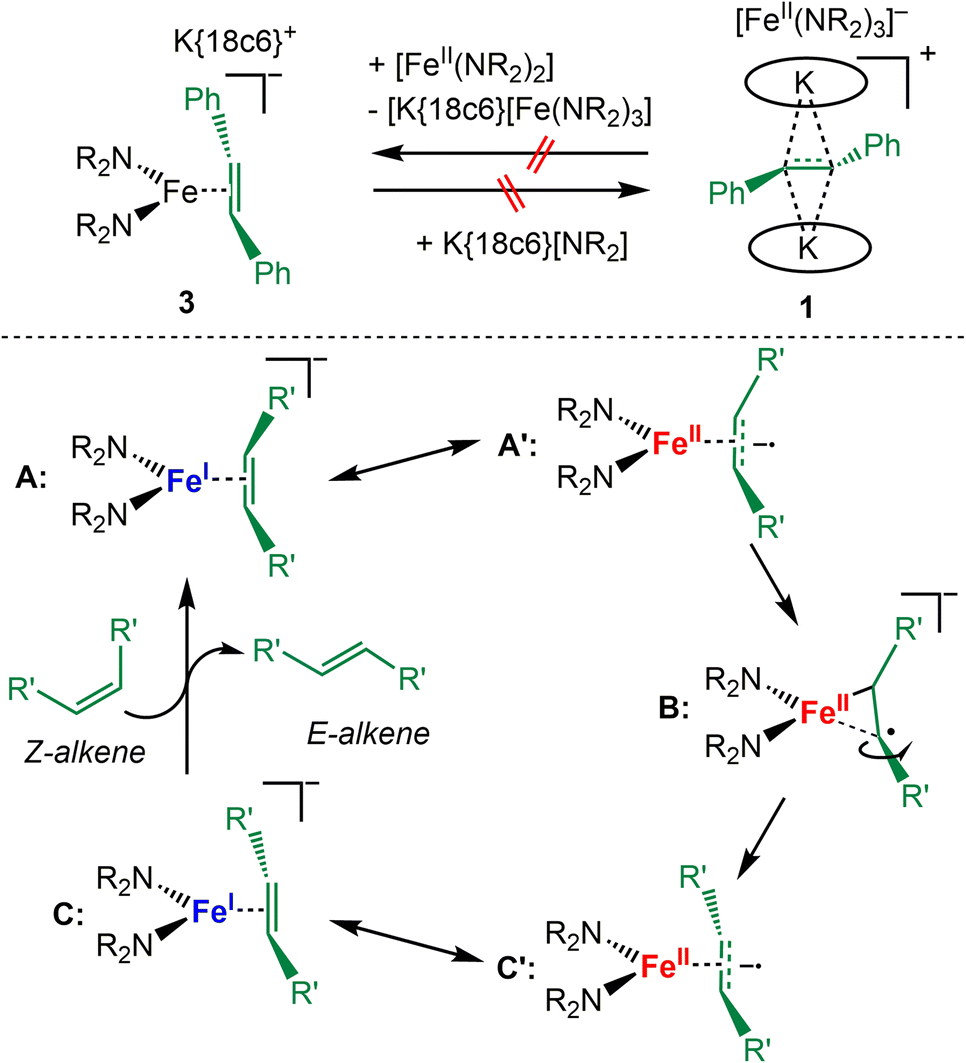 | ||
| Scheme 4 Top: Attempted conversion of 3 and 1 into each other. Bottom: Proposed mechanism for the Z to E isomerization of alkenes with [FeI] as catalyst. Coordination of the Z-alkene to the metal ion as π- (A) or radical anion (A′) complex; B: rotation along the C–C bond; C/C′E-alkene complex. | ||
Conclusions
In conclusion, we presented the isolation and characterisation of a long time elusive simple alkene radical anion, namely the E–stilbene radical anion. This otherwise fleeting species is stabilized by encapsulation between two [K{18c6}] units. Similarly, coordination of stilbene to the highly reducing iron(I) complex [Fe(NR2)2]− (R = SiMe3) leads to a situation best described as a metal(II) bound alkene radical anion. In both cases, the radical anion formation can be used for catalytic Z → E isomerisation of CC double bonds. The stilbene radical anion can only isomerize stilbene itself, whereas the transformation can be conceptionally extended to harder to reduce 1,2-alkyl/aryl and dialkyl ethylene using the iron(I) complex K{18c6}[Fe(NR2)2]. This reductively induced Z → E isomerisation of alkenes might pose a complementary approach for the photo switching of alkenes, similar to the previously mentioned report on the electrocatalytic isomerisation of azobenzenes.46
Data availability
All experimental procedures, spectral data, and computational data are included in the ESI.† NMR/EPR/IR/Mössbauer raw data is so far only available upon request to C. G. W.Author contributions
G. S. and I. M. carried out the synthetic work and analytical characterization, including the crystallographic studies. K. W. performed the 57Fe Mössbauer analysis. G. S. and C. G. W. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Christian Limberg (HU Berlin) for access to the Mössbauer spectrometer, as well as Dr Andreas Stoy, Prof. Dr Crispin Lichtenberg, Amanda O. Basilio, and Prof Dr Kallol Ray for the acquisition of EPR-spectroscopic data. The Philipps-University and the Deutsche Forschungsgemeinschaft (DFG) is acknowledged for funding (grants WE 5627/4-1 and WE 5627/4-2).Notes and references
- T. Wirth, Angew. Chem., Int. Ed., 1996, 35, 61–63 CrossRef.
- M. L. Di Vona and V. Rosnati, J. Org. Chem., 1991, 56, 4269–4273 CrossRef.
- M. Ephritikhine, Chem. Commun., 1998, 2549–2554 RSC.
- M. Zhang, W. D. Rouch and R. D. McCully, Eur. J. Org. Chem., 2012, 6187–6196 CrossRef.
- S. Okamoto, K. Kojiyama, H. Tsujioka and A. Sudo, Chem. Commun., 2016, 52, 11339–11342 RSC.
- S. Okamoto, R. Ariki, H. Tsujioka and A. Sudo, J. Org. Chem., 2017, 82, 9731–9736 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS.
- D. W. Borhani and F. D. Greene, J. Org. Chem., 1986, 51, 1563–1570 CrossRef CAS.
- D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286–6289 CrossRef CAS.
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 131, 9074–9082 CrossRef.
- M. Irwin, R. K. Jenkins, M. S. Denning, T. Krämer, F. Grandjean, G. J. Long, R. Herchel, J. E. McGrady and J. M. Goicoechea, Inorg. Chem., 2010, 49, 6160–6171 CrossRef CAS.
- M. Irwin, T. Krämer, J. E. McGrady and J. M. Goicoechea, Inorg. Chem., 2011, 50, 5006–5014 CrossRef CAS PubMed.
- M. Irwin, L. R. Doyle, T. Krämer, R. Herchel, J. E. McGrady and J. M. Goicoechea, Inorg. Chem., 2012, 51, 12301–12312 CrossRef CAS PubMed.
- T. A. Scott, B. A. Ooro, D. J. Collins, M. Shatruk, A. Yakovenko, K. R. Dunbar and H.-C. Zhou, Chem. Commun., 2009, 65–67 RSC.
- C. S. Sevov, R. E. M. Brooner, E. Chénard, R. S. Assary, J. S. Moore, J. Rodríguez-López and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 14465–14472 CrossRef CAS PubMed.
- Y. Che, A. Datar, X. Yang, T. Naddo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2007, 129, 6354–6355 CrossRef CAS.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef.
- H. Iwamura, Polyhedron, 2013, 66, 3–14 CrossRef CAS.
- S. Kumar, Y. Kumar, S. Keshri and P. Mukhopadhyay, Magnetochemistry, 2016, 2, 42 CrossRef.
- G. J. Hoijtink and P. H. Van der Meij, Z. Phys. Chem., 1959, 20, 1–14 CrossRef.
- M. Szwarc, Application of Spectroscopic and Electrochemical Techniques in Studies of Chemistry of Radical Anions and Dianions, in Characterization of Solutes in Nonaqueous Solvents, ed. G. Mamantov, Springer, Boston, MA, 1978 Search PubMed.
- J. Matsuda, J. Jagur-Grodzinski and M. Szwarc, Proc. R. Soc. London, Ser. A, 1965, 288, 212–223 Search PubMed.
- H. Muto, K. Nunome and K. Matsuura, J. Am. Chem. Soc., 1991, 113, 1840–1841 CrossRef CAS.
- W. Schlenk and E. Bergmann, Justus Liebigs Ann. Chem., 1928, 463, 1–97 CrossRef CAS.
- F. Gerson, H. Ohya-Nishiguchi, M. Szwarc and G. Levin, Chem. Phys. Lett., 1977, 52, 587–589 CrossRef CAS.
- H. Suzuki, K. Koyano, T. Shida and A. Kira, Bull. Chem. Soc. Jpn., 1982, 55, 3690–3701 CrossRef CAS.
- H. Suzuki, K. Ogawa, T. Shida and A. Kira, Bull. Chem. Soc. Jpn., 1983, 56, 66–74 CrossRef CAS.
- C. N. R. Rao, V. Kalyanaraman and M. V. George, Appl. Spectrosc. Rev., 1970, 3, 153–228 CrossRef CAS.
- O. Abdul-Rahim, A. N. Simonov, J. F. Boas, T. Rüther, D. J. Collins, P. Perlmutter and A. M. Bond, J. Phys. Chem. B, 2014, 118, 3183–3191 CrossRef CAS.
- K. Nozaki, A. Naito, T.-I. Ho, H. Hatano and S. Okazaki, Chem. Lett., 1989, 511–514 CrossRef CAS.
- G. F. Wright, J. Am. Chem. Soc., 1939, 61, 2106–2110 CrossRef CAS.
- T. Majima, M. Fukui, A. Ishida and S. Takamuku, J. Phys. Chem., 1996, 100, 8913–8919 CrossRef CAS.
- J. R. Langan and G. A. Salmon, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 3645 RSC.
- C. S. Johnson and R. Chang, J. Chem. Phys., 1965, 43, 3183–3192 CrossRef CAS.
- R. Chang and C. S. Johnson, J. Chem. Phys., 1967, 46, 2314–2316 CrossRef CAS.
- S. Sorensen, G. Levin and M. Szwarc, J. Am. Chem. Soc., 1975, 97, 2341–2345 CrossRef CAS.
- T. A. Ward, G. Levin and M. Szwarc, J. Am. Chem. Soc., 1975, 97, 258–261 CrossRef CAS.
- G. Levin, T. A. Ward and M. Szwarc, J. Am. Chem. Soc., 1974, 96, 270–272 CrossRef CAS.
- H. C. Wang, G. Levin and M. Szwarc, J. Am. Chem. Soc., 1977, 99, 2642–2647 CrossRef CAS.
- L. R. Dosser, J. B. Pallix, G. H. Atkinson, H. C. Wang, G. Levin and M. Szwarc, Chem. Phys. Lett., 1979, 62, 555–561 CrossRef CAS.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- B. E. Maryanoff, A. B. Reitz, M. S. Mutter, R. R. Whittle and R. A. Olofson, J. Am. Chem. Soc., 1986, 108, 7664–7678 CrossRef CAS.
- W.-Y. Siau, Y. Zhang and Y. Zhao, in Stereoselective Alkene Synthesis, ed. J. Wang, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, vol. 327, pp. 33–58 Search PubMed.
- J. E. McMurry, Chem. Rev., 1989, 89, 1513–1524 CrossRef CAS.
- A. Fürstner, Science, 2013, 341, 1229713 CrossRef.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS.
- C. G. Werncke and I. Müller, Chem. Commun., 2020, 56, 2268–2271 RSC.
- I. Müller and C. G. Werncke, Chem.–Eur. J., 2021, 27, 4932–4938 CrossRef.
- A. Hoekstra, P. Meertens and A. Vos, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1975, 31, 2813–2817 CrossRef.
- O. Abdul-Rahim, A. N. Simonov, T. Rüther, J. F. Boas, A. A. J. Torriero, D. J. Collins, P. Perlmutter and A. M. Bond, Anal. Chem., 2013, 85, 6113–6120 CrossRef CAS PubMed.
- R. Chang and C. S. Johnson, J. Chem. Phys., 1964, 41, 3272–3274 CrossRef CAS.
- G. Sieg, Q. Pessemesse, S. Reith, S. Yelin, C. Limberg, D. Munz and C. G. Werncke, Chem.–Eur. J., 2021, 27, 16760–16767 CrossRef CAS PubMed.
- K. C. MacLeod, I. M. DiMucci, E. P. Zovinka, S. F. McWilliams, B. Q. Mercado, K. M. Lancaster and P. L. Holland, Organometallics, 2019, 38, 4224–4232 CrossRef CAS PubMed.
- E. B. Fleischer, N. Sung and S. Hawkinson, J. Phys. Chem., 1968, 72, 4311–4312 CrossRef CAS.
- Z. Hou, A. Fujita, T. Koizumi, H. Yamazaki and Y. Wakatsuki, Organometallics, 1999, 18, 1979–1985 CrossRef CAS.
- C. G. Werncke, P. C. Bunting, C. Duhayon, J. R. Long, S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2015, 54, 245–248 CrossRef CAS PubMed.
- I. Müller, D. Munz and C. G. Werncke, Inorg. Chem., 2020, 59, 9521–9537 CrossRef.
- I. Müller, C. Schneider, C. Pietzonka, F. Kraus and C. G. Werncke, Inorganics, 2019, 7, 117 CrossRef.
- D. Gärtner, S. Sandl and A. Jacobi von Wangelin, Catal. Sci. Technol., 2020, 10, 3502–3514 RSC.
- J. F. Sonnenberg and R. H. Morris, Catal. Sci. Technol., 2014, 4, 3426–3438 RSC.
- C.-Y. Lin, J. C. Fettinger, F. Grandjean, G. J. Long and P. P. Power, Inorg. Chem., 2014, 53, 9400–9406 CrossRef CAS PubMed.
- Y. Yu, J. M. Smith, C. J. Flaschenriem and P. L. Holland, Inorg. Chem., 2006, 45, 5742–5751 CrossRef CAS PubMed.
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed.
- M. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C78 Search PubMed.
- C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, 3rd edn, 2006 Search PubMed.
- O. Eisenstein and R. Hoffmann, J. Am. Chem. Soc., 1981, 103, 4308–4320 CrossRef CAS.
- C. G. Werncke, J. Pfeiffer, I. Müller, L. Vendier, S. Sabo-Etienne and S. Bontemps, Dalton Trans., 2019, 48, 1757–1765 RSC.
- R. Weller, I. Müller, C. Duhayon, S. Sabo-Etienne, S. Bontemps and C. G. Werncke, Dalton Trans., 2021, 50, 4890–4903 RSC.
- A. Eichhöfer, Y. Lan, V. Mereacre, T. Bodenstein and F. Weigend, Inorg. Chem., 2014, 53, 1962–1974 CrossRef.
- J. C. Ott, H. Wadepohl and L. H. Gade, Inorg. Chem., 2021, 60, 3927–3938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General, synthetic, analytical and catalytic details, EPR-, 57Fe-Mössbauer-, UV/Vis-, IR-spectra, crystallographic details. CCDC 2214046, 2178651, 2178649 and 2178650. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04451j |
| This journal is © The Royal Society of Chemistry 2022 |