
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe simplest structure of a stable radical showing high fluorescence efficiency in solution: benzene donors with triarylmethyl radicals†
Yohei
Hattori
 *a,
Ryota
Kitajima
a,
Wataru
Ota
b,
Ryota
Matsuoka
cd,
Tetsuro
Kusamoto
cde,
Tohru
Sato
fg and
Kingo
Uchida
a
*a,
Ryota
Kitajima
a,
Wataru
Ota
b,
Ryota
Matsuoka
cd,
Tetsuro
Kusamoto
cde,
Tohru
Sato
fg and
Kingo
Uchida
a
aMaterials Chemistry Course, Faculty of Advanced Science and Technology, Ryukoku University, Seta, Otsu, Shiga 520-2194, Japan. E-mail: hattori@rins.ryukoku.ac.jp
bMOLFEX, Inc., Takano-Nishibiraki-cho 34-4, Kyoto 606-8103, Japan
cDepartment of Life and Coordination-Complex Molecular Science, Institute for Molecular Science, 5-1, Higashiyama, Myodaiji, Okazaki, Aichi 444-8787, Japan
dSOKENDAI (The Graduate University for Advanced Studies), Shonan Village, Hayama, Kanagawa 240-0193, Japan
eJST-PRESTO, 4-1-8, Honcho, Kawaguchi, Saitama 332-0012, Japan
fFukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishibiraki-cho 34-4, Kyoto 606-8103, Japan
gDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 24th October 2022
Abstract
Donor–radical acceptor systems have recently attracted much attention as efficient doublet emitters that offer significant advantages for applications such as OLEDs. We employed an alkylbenzene (mesityl group) as the simplest donor to date and added it to a diphenylpyridylmethyl radical acceptor. The (3,5-difluoro-4-pyridyl)bis[2,6-dichloro-4-(2,4,6-trimethylphenyl)phenyl]methyl radical (Mes2F2PyBTM) was prepared in only three steps from commercially available reagents. A stable radical composed of only one pyridine ring, four benzene rings, methyl groups, halogens, and hydrogens showed fluorescence of over 60% photoluminescence quantum yield (PLQY) in chloroform, dichloromethane, and PMMA. The key to high fluorescence efficiency was benzene rings perpendicular to the diphenylpyridylmethyl radical in the doublet ground (D0) state. The relatively low energy of the β-HOMO and the electron-accepting character of the radical enabled the use of benzenes as electron donors. Furthermore, the structural relaxation of the doublet lowest excited (D1) state was minimized by steric hindrance of the methyl groups. The reasons for this high efficiency include the relatively fast fluorescence transition and the slow internal conversion, both of which were explained by the overlap density between the D1 and D0 states.
Introduction
Recently, stable radicals have attracted much interest as luminescent materials.1,2 The key to their luminescence is the fluorescence from the D1 state to the D0 state. In contrast to the singlet lowest excited (S1) state of closed-shell molecules, which is higher in energy than the triplet lowest excited state (T1), it is normal for the energy of the D1 state to be lower than that of the quartet lowest excited (Q1) state. Therefore, the fluorescence of radicals can avoid unfavorable quenching from higher multiplicity excited states produced by the recombination of electrons and holes or intersystem crossing from the S1 state. Stable luminescent radicals are one of the most promising substance groups of emitters suitable for highly efficient electroluminescent (EL) devices.3,4 A relatively basic application would be use in a heavy atom environment.5,6Problems due to thermal stability of the radicals had been resolved with the invention of polychlorotriphenylmethyl radicals such as perchlorotriphenylmethyl radical (PTM)7 and tris(2,4,6-trichlorophenyl)methyl radical (TTM).8 For a luminescent material, higher stability, that is, stability under photoexcitation conditions (photostability), is necessary; however, the photodecomposition of PTM and TTM had been reported.9,10 We have reported that the introduction of a pyridyl group instead of a phenyl group has greatly improved the stability of the radical under photoirradiation.11 Compared with TTM, the (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl radical (PyBTM) showed ca. 70 times higher photostability in dichloromethane.
Dilute PyBTM doped in (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methane (αH-PyBTM) crystal had an excellent feature of high fluorescence efficiency (Φf = 89%).12 In molecular solids of stable radicals, spin-derived properties such as magnetism are important properties,13 and PyBTM has been used to investigate interesting photophysical phenomena such as the coherent coupling between spin ensembles,14 a magnetic field effect on luminescence (magnetoluminescence),12,15,16 and photoluminescence anisotropy amplified by exciton funneling.17 However, the fluorescence efficiency of PyBTM was low in liquid solutions, similar to other simple triarylmethyl radicals. The PLQYs of PyBTM and TTM were both 2% in dichloromethane and 3% in chloroform.11 Introduction of fluorine atoms on the pyridine ring slightly improved the PLQY in solution, and the (2,5-difluoro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl radical (F2PyBTM) showed PLQY of 4% in dichloromethane and 6% in chloroform.18 Utilizing the coordination ability of a nitrogen atom on the pyridine ring, we have developed AuI complexes of PyBTM derivatives and improved the fluorescence efficiency to 36%.19–21
On the other hand, the PLQYs of PTM or TTM derivatives have been improved by constructing a donor–acceptor system using nitrogen-containing electron donors such as carbazoles22–26 or triphenylamines.25,27 Highly fluorescent radicals showing PLQY above 50% in solution were first reported as a carbazole donor–TTM radical acceptor system showing 53% PLQY in cyclohexane.22 However, its fluorescence was quenched by the polarity of solvent molecules to 2% in chloroform. By adding electron-withdrawing groups on carbazole, strong fluorescence was maintained in chloroform.24 It is noteworthy that efficient fluorescence in more polar solvents has recently been reported in completely different systems: pyrene-dithiadiazolyl radical (Φf = 50% in acetonitrile)28 and the π-radical stabilized with boron (Φf = 67% in DMF).29
Here, we report that simple aromatic hydrocarbons, mesityl groups, work as donors, and significantly enhance the fluorescence efficiencies of PyBTM and F2PyBTM in dichloromethane and chloroform solutions. We explain the reasons for the high efficiencies of these nonplanar π-electron systems by photophysical theories and calculations using DFT and TD-DFT.
Results and discussion
From commercially available reagents, αH-PyBTM11 and (3,5-difluoro-4-pyridyl)bis(2,4,6-trichlorophenyl)methane (αH-F2PyBTM)18 were prepared in one step (Scheme 1). In the second step, the Suzuki–Miyaura coupling reaction with a micellar catalysis30 and 2,4,6-trimethylphenylboronic acid selectively yielded mesityl substituents at the para position. The stable radicals MesPyBTM, Mes2PyBTM, and Mes2F2PyBTM were synthesized by deprotonation and oxidation processes. Thus, these radicals were all prepared in three steps from commercially available reagents. ESR spectra revealed that these radicals have S = 1/2 spin on one molecule (Fig. S1†).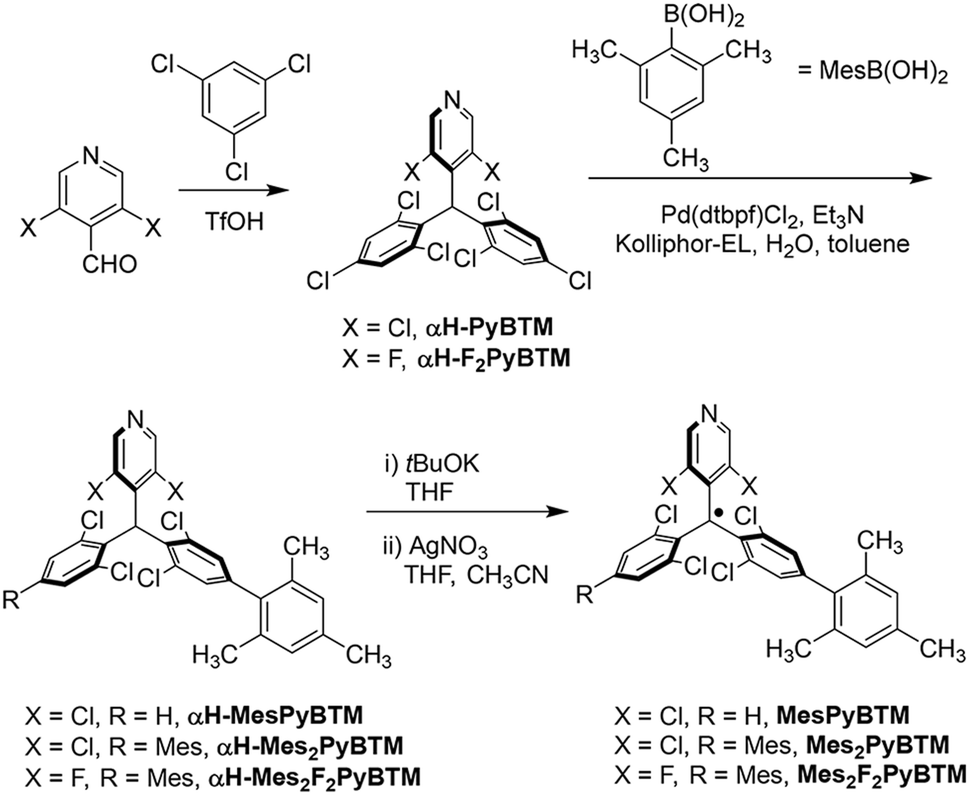 | ||
| Scheme 1 Synthesis of MesPyBTM, Mes2PyBTM, and Mes2F2PyBTM. | ||
These radicals were stable under ambient conditions similar to other triarylmethyl radicals protected by halogen atoms.7,8 Actually, these radicals were purified by chromatography on silica gel under ambient conditions, and their melting points could be determined. All the spectroscopic measurements were conducted under ambient conditions as no effect due to oxygen was observed similar to the other PyBTM derivatives,5,11,18–21 probably due to short fluorescence lifetimes. As with PyBTM,11 no change was observed in the solutions stored in the dark.
Absorption and emission spectra of MesPyBTM and Mes2PyBTM in dichloromethane are compared with those of PyBTM and (3,5-dichloro-4-pyridyl)bis(2,6-dichloro-4-phenylphenyl)methyl radical (PyPBTM)30 in Fig. 1a. The absorption spectrum of PyPBTM was clearly different from that of PyBTM, and both the α-HOMO–α-LUMO band (λmax = 370 → 401 nm) and the β-HOMO–β-LUMO band (541 → 564 nm) were significantly redshifted. On the other hand, the shapes of the absorption spectra of MesPyBTM and Mes2PyBTM were rather similar to that of PyBTM with the same absorption maxima at λmax = 370 and 541 (±1) nm. This result is attributed to the only slightly changed energy levels of the frontier orbitals.
 | ||
| Fig. 1 (a) Absorption (solid line) and emission (broken line, λex = 450 nm) spectra of MesPyBTM (orange), Mes2PyBTM (red), PyPBTM (light green), and PyBTM (pink) in dichloromethane. Enlarged portions of the absorption spectra (10 fold) are shown from λ = 450 to 650 nm (dotted lines). The concentration of MesPyBTM, Mes2PyBTM, PyPBTM, and PyBTM was 7.2 × 10−5, 7.7 × 10−5, 2.0 × 10−5, and 2.5 × 10−5 M, respectively. (b) Absorption (solid line) and emission (broken line, λex = 450 nm) spectra of Mes2F2PyBTM (purple) and F2PyBTM (light blue) in dichloromethane. Enlarged portions of the absorption spectra (10 fold) are shown from λ = 450 to 650 nm (dotted lines). The concentration of Mes2F2PyBTM and F2PyBTM was 6.2 × 10−5 and 2.5 × 10−5 M, respectively. | ||
This occurs because the mesityl group has bulky methyl groups at ortho positions and can barely conjugate with π-orbitals on the neighboring phenyl group. In order to estimate structures using DFT, we adopted the UB3LYP level of theory with 6-31G(d, p) basis sets, since they closely reproduced the experimental absorption and emission spectra from previous studies.11,19–21,31 The solvent effect of dichloromethane was taken into account by using a polarizable continuum model (PCM).32,33 The dihedral angle between the mesityl and dichlorophenyl groups was 83° for MesPyBTM and 84° for Mes2PyBTM in the DFT optimized D0 state model (Table S1†). These are nearly perpendicular in contrast to the rather flat angles (34°) between the phenyl and dichlorophenyl groups in PyPBTM.
Redshifts of emission from PyBTM (λem = 585 nm) were seen in MesPyBTM (λem = 645 nm) and Mes2PyBTM (λem = 628 nm), although these shifts were smaller than that of PyPBTM (λem = 654 nm). In the TD-DFT (UB3LYP/6-31G(d, p)) optimized D1 structure, the mesityl groups under the β-HOMO are electron-deficient, and the dihedral angle decreased to 50° to conjugate to the relatively electron-rich dichlorophenyl group (Table S1†). This structural relaxation was the cause of this redshift. The dihedral angle became even smaller in PyPBTM, decreasing to 25°.
Absorption and emission spectra of Mes2F2PyBTM and F2PyBTM are shown in Fig. 1b. The shape of the absorption spectrum of Mes2F2PyBTM (λmax = 352 nm) resembles that of F2PyBTM (λmax = 351 nm). The dihedral angle between the mesityl and dichlorophenyl groups was 87° in the DFT optimized D0 state (Table S1†). The redshift of emission from F2PyBTM (λem = 566 nm) was seen in Mes2F2PyBTM (λem = 623 nm), similar to the case of Mes2PyBTM. The optimized dihedral angle between the mesityl group under the β-HOMO and the neighboring dichlorophenyl group was 49° in the DFT optimized D1 state.
Bright reddish-orange fluorescence was observed when the solutions of new radicals were irradiated with a UV lamp. The PLQYs in dichloromethane are shown in Table 1, and the PLQYs in chloroform are shown in Table S2.† As observed, the addition of mesityl groups dramatically increased the fluorescence efficiency of the radical, and the effect was much larger than that by the phenyl groups. The PLQY of 2% for PyBTM was elevated to 30% by the addition of a mesityl group and to 47% by double substitution of mesityl groups. In particular, Mes2F2PyBTM displayed as much as 66% PLQY in dichloromethane, and 69% in chloroform (Fig. S2†). As far as we know, a higher PLQY of the fluorescent radical in a liquid solution has only been reported for pyridoindole donor–TTM acceptor systems in 202034 and 202235 and the π-radical stabilized with boron in 2022.29
| λ em (nm) | Φ f (%) | τ/ns | k f/107 s−1 | k nr/107 s−1 | |
|---|---|---|---|---|---|
| a Cited from ref. 11. b Cited from ref. 18. All Φfs were obtained by absolute PLQY measurement. | |||||
| PyBTMa | 585 | 2 | 6.4 | 0.3 | 14 |
| F2PyBTMb | 566 | 4 | 12.5 | 0.3 | 7.7 |
| PyPBTM | 654 | 9.5 | 12 | 0.8 | 7.5 |
| MesPyBTM | 645 | 30 | 26 | 1.2 | 2.7 |
| Mes2PyBTM | 628 | 47 | 38 | 1.2 | 1.4 |
| Mes2F2PyBTM | 623 | 66 | 44 | 1.5 | 0.8 |
The major structural difference between pyridoindole donor–TTM acceptor systems34,35 and Mes2F2PyBTM is that the former has two nitrogen atoms in the donor and the latter has one nitrogen atom in the radical. In contrast, the TTM radical is made of a carbon skeleton and the mesityl group is a hydrocarbon. In terms of organic chemistry of nitrogen-containing aromatics, an indole ring is electron-rich and a pyridine ring is electron-deficient. A pyridoindole is thought to cancel out the two effects internally. The use of mesityl groups simplifies the situation and provides important scientific or economic insights.
The nitrogen atom in the radical was introduced to improve the photostability of the radical as described in the Introduction. The photostabilities of the new radicals were measured in dichloromethane under UV light (370 nm) irradiation. The decay of fluorescence is plotted in Fig. S3,† and the stabilities of the new radicals were of about the same order as that in PyBTM (Table S3†). The photostability of Mes2F2PyBTM was slightly higher than that of PyBTM. The photostability of PyBTM was ca. 70 times that of TTM and similar to that of TIPS pentacene.36 Therefore, it was found that a reasonable degree of photostability of PyBTM could be maintained.
The availability of the radicals as fluorophores in polar solvents means that they are also useful in polymers having polar substituents. Poly(methyl methacrylate) (PMMA) is one of the most useful polymers in optical applications, and it has polar carboxyl groups that can quench the fluorescence of some donor–acceptor type fluorophores. Mes2F2PyBTM in a PMMA film displayed PLQY of 62%, proving its usefulness in this polymer.
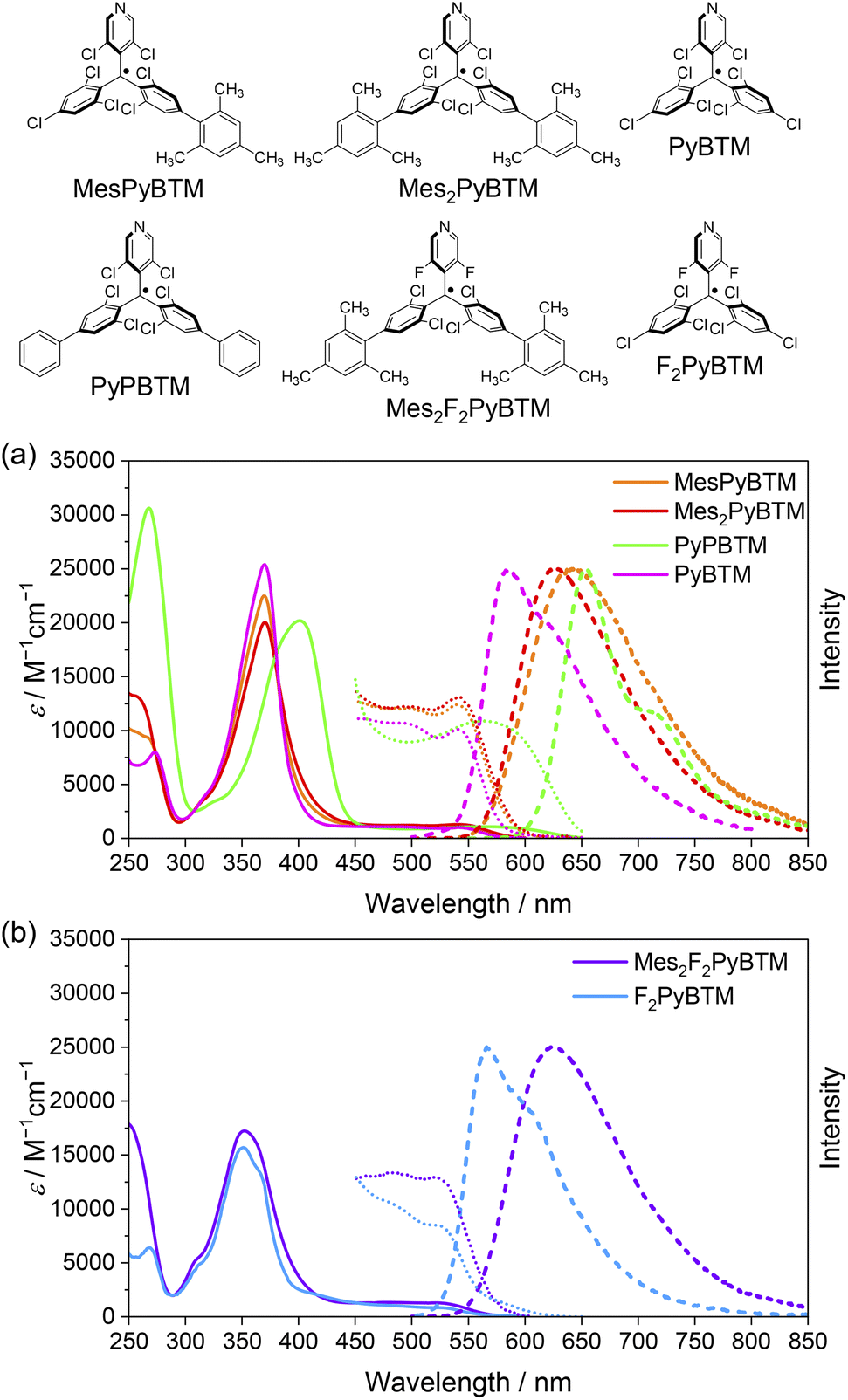
As mentioned in the Introduction, the Q1 state is higher in energy, and all of the excited states are thought to converge to the D1 state in a short time. The PLQYs of the radicals are determined by competition between the rate of fluorescence (kf) and the rate of nonradiative decay (knr) from the D1 state (Fig. 2). Here, kf and knr were calculated from the PLQYs and the fluorescence lifetimes (τ, Fig. S4†) of the radicals, and they are shown in Table 1.
 | ||
| Fig. 2 Energy diagram for the D1–D0 fluorescence of the radicals and the β-HOMO and β-LUMO of MesPyBTM, Mes2PyBTM, and Mes2F2PyBTM at the D1 optimized structure calculated using UB3LYP/6-31G(d, p). | ||
One cause of the low PLQY of triarylmethyl radicals was the small kf. The small transition dipole moment between D1 and D0 is interpreted as being caused by the C3 symmetry of the molecule37 or cancellation of the HOMO–SOMO and SOMO–LUMO transition dipole moments.34 This barrier for emission is partially eliminated by constructing a donor–acceptor system. The β-HOMOs and β-LUMOs calculated using the TD-DFT (UB3LYP/6-31G(d, p)) at the optimized D1 structure are shown in Fig. 2 (see also Fig. S5†). Since the D1 states are mainly generated via the β-HOMO–β-LUMO transition, the radicals are regarded as a mesityl donor–PyBTM acceptor system. Experimentally, kf in MesPyBTM, Mes2PyBTM, and Mes2F2PyBTM was enhanced to a level 4 or 5 times that of PyBTM.
In 2020, Abdurahman et al. suggested that the large oscillator strength of a donor–acceptor system is due to intensity borrowing from the intense high-lying transition of the radical,34 while Cho et al. showed that the kf values are dominated by the coupling between the CT and ground state, and nearly independent of the donor strength.38 We propose another view of the increase in kf from visualization of the overlap density distribution by TD-DFT. Consideration of interactions with local excitations is no longer necessary for intuitive understanding.
Within the crude adiabatic approximation,39,40 we consider transitions from the initial vibronic state |Φmν〉 = |Ψm〉|χmν〉 to the final one |Φnν′〉 = |Ψn〉|χnν′〉, where |Ψm〉 (|Ψn〉) and |χmν〉 (|χnν′〉) are the initial (final) electronic and vibrational states, respectively. The initial and final vibronic energies are denoted as Emν and Enν′, respectively. According to Fermi's golden rule, the rate constant of fluorescence from |Ψm〉 to |Ψn〉 is given by eqn (1).41
 | (1) |
A density form of the transition dipole moment (τ10(x)) is written as a product of a three-dimensional Cartesian coordinate (x = (x, y, z)) and an overlap density between the D1 and D0 states (ρ10(x)) as in eqn (2) and (3).41,42
| μ10 = ∫dxτ10(x) | (2) |
| τ10(x) = −exρ10(x) | (3) |
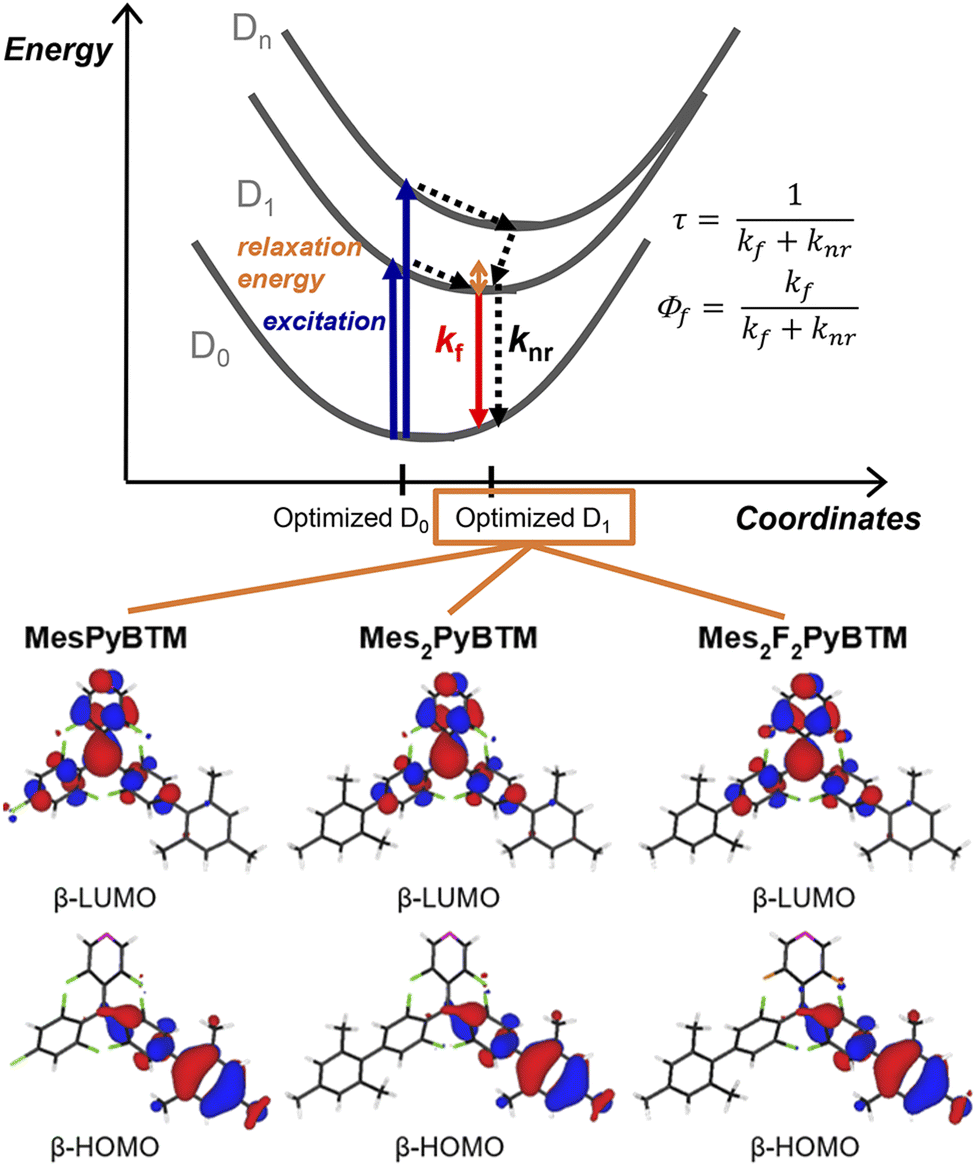
Thus, an overlap density having widespread distribution generally gives a large transition dipole moment. The calculated transition dipole moments of Mes2PyBTM and Mes2F2PyBTM were larger than that of PyBTM (Table 2). This result was attributed to the overlap densities of Mes2PyBTM and Mes2F2PyBTM delocalizing on both the PyBTM moiety and the mesityl group (Fig. 3).
| |μ10|/a.u. | |||
|---|---|---|---|
| x | y | z | |
| PyBTM | 0.7012 | 0.3654 | 0.0725 |
| Mes2PyBTM | 1.3525 | 0.7567 | 0.0451 |
| Mes2F2PyBTM | 1.2884 | 0.7722 | 0.0132 |
 | ||
| Fig. 3 Overlap densities between the D1 and D0 states of PyBTM, Mes2PyBTM, and Mes2F2PyBTM. These were approximately given by the product of the β-HOMO and β-LUMO at the D1 optimized structure. | ||
The suppressed knr contributed to the high PLQY no less than the enhanced kf. Since the only state lower in energy than the D1 state is the ground state (D0), the rate of intersystem crossing (kISC) to the other spin multiplet states is substantially zero, and 100% of knr is the rate constant of internal conversion (kIC) to the D0 state. Compared to the phenyl groups (PyPBTM), the relaxation of the D1 state with the mesityl groups is sterically hindered by the methyl groups at the ortho positions. As a result, the energy of the D1 state was raised and the internal conversion from D1 to D0 was slowed by the energy-gap law.43 The fluorine atoms on the pyridine ring also have the effect of widening the D1–D0 gap,18,20 and the record-small knr among diphenylpyridyl radicals was achieved in Mes2F2PyBTM.
In addition, the kIC can also be discussed from the overlap density distribution. The rate constant of internal conversion from |Ψm〉 to |Ψn〉 is given by eqn (4).41
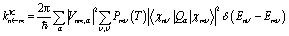 | (4) |
A density form of the off-diagonal VCC (η10,α) is written as a product of the overlap density and potential derivative (vα(x)) as in eqn (5) and (6).41,42
| V10,α = ∫dxη10,α(x) | (5) |
| η10,α(x) = ρ10(x) × vα(x) | (6) |
Note that the density forms of both the transition dipole moment and the off-diagonal VCC are expressed using the overlap density. Mes2PyBTM and Mes2F2PyBTM have smaller off-diagonal VCCs than PyBTM (Fig. S6†) because their overlap densities are delocalized on the PyBTM moiety and on the mesityl group, which couple weakly to potential derivatives.
Conclusions
Enhancement of fluorescence by the addition of mesityl groups to PyBTM is explained from the viewpoint of the overlap density between the D1 and D0 states. In particular, Mes2F2PyBTM shows high fluorescence efficiency above 60% in dichloromethane, chloroform, and PMMA at room temperature under ambient conditions. Although the PLQY of 69% in chloroform is not the highest among those of luminescent radicals today,29,34,35 this value is comparable to those of famous fluorescent dyes such as rhodamine B (Φf = 0.70 in methanol,44 Fig. S7†). This paper has raised many important points: Simple benzene rings can behave as an electron-donor for electron-accepting PyBTM radicals, their fluorescence is not quenched in polar solvents due to their non-excessive electron-donating nature, and steric hindrance of the ortho methyl groups plays an important role in reducing internal conversion. Introducing a nitrogen atom into a radical without introducing a nitrogen atom into the donor is a reversal of the previous highly efficient stable luminescent radicals. This study will provide important clues about donor selection in donor–acceptor systems or non-planar fluorescent π-systems. We also expect that easily synthesized and highly fluorescent Mes2PyBTM and Mes2F2PyBTM will be used in a variety of applications.Experimental
Materials and methods
All reactions were carried out under an argon atmosphere. The starting materials, αH-PyBTM11 and αH-F2PyBTM,18 were prepared according to the reference. Commercially available compounds were used as received without further purification. Preparative recycling gel permeation chromatography was performed with a recycling preparative HPLC, LaboACE LC-5060, Japan Analytical Industry Co., Ltd. 1H (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a JEOL JNM-ECS 400 spectrometer using CDCl3. The residual solvent signals (1H NMR: δ 7.26, 13C NMR: δ 77.16) were used as the internal standards. Elemental analysis was conducted at the Center for Organic Elemental Microanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University. ESR spectra were recorded with a JEOL JES-FR30EX spectrometer with X-band microwave. Sample solutions were charged in a 2.5 mmϕ sample tube. Magnetic field was calibrated with the Mn2+/MgO standard. Mass spectrometry was performed with a JEOL-JMS-S3000 (MALDI-Spiral-TOF MS) mass spectrometer with DCTB (20 mg mL−1 in CHCl3) as a matrix and TFANa (1 mg mL−1 in THF) as a cationization agent. Melting points were measured on a Yanaco MP-500D. Absorption and emission spectra were monitored on a Hitachi U-4150 spectrophotometer and a Hitachi F-7100 fluorescence spectrophotometer, respectively. Photostability under 370 nm light was recorded with a JASCO FP-8600KS spectrofluorometer. Absolute luminescence quantum yields were measured using a Hamamatsu Photonics Quantaurus QY. Photoluminescence decay curves were measured using a measurement system with a picosecond diode laser with the emission wavelength of 375 nm (Advanced Laser Diode Systems PIL037X) as the light source, a single grating spectrometer (Andor Kymera193i-B1), and a photon counting detector (MPD SPD-050-CTE) operated using a time-correlated single photon counting (TCSPC) technique.Preparation of emulsion
An emulsion of toluene in aqueous 2 wt% Kolliphor EL (K-EL) is prepared by mixing a 2 wt% aqueous dispersion of K-EL (1.8 g of K-EL in 88.2 mL of deionized water) with 10 mL of toluene until a stable, milky dispersion is obtained. The emulsion and triethylamine were deoxygenated by bubbling argon before use.Synthesis of αH-MesPyBTM and αH-Mes2PyBTM
In a Schlenk tube, αH-PyBTM (783 mg, 1.50 mmol), 2,4,6-trimethylphenylboronic acid (746 mg, 4.55 mmol), and Pd(dtbpf)Cl2 (79.3 mg, 0.122 mmol), were put under an argon atmosphere. The degassed K-EL 2 wt%: toluene (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) emulsion (3.5 mL) was added, and the mixture was heated at 70 °C. Degassed triethylamine (1.3 mL, 9.3 mmol) was finally added, and the reaction mixture was stirred at 70 °C overnight. The reaction mixture was cooled down to room temperature, dichloromethane was added, and filtered on a celite pad. The solvent was evaporated and purified by silica gel column chromatography (CHCl3:hexane = 1:1). The crude product (742 mg) was separated by GPC (CHCl3) to obtain pure αH-MesPyBTM (351 mg, 0.581 mmol, 39%, mp 192–195 °C) and αH-Mes2PyBTM (252 mg, 0.367 mmol, 24%, mp 122–124 °C).
:1 v/v) emulsion (3.5 mL) was added, and the mixture was heated at 70 °C. Degassed triethylamine (1.3 mL, 9.3 mmol) was finally added, and the reaction mixture was stirred at 70 °C overnight. The reaction mixture was cooled down to room temperature, dichloromethane was added, and filtered on a celite pad. The solvent was evaporated and purified by silica gel column chromatography (CHCl3:hexane = 1:1). The crude product (742 mg) was separated by GPC (CHCl3) to obtain pure αH-MesPyBTM (351 mg, 0.581 mmol, 39%, mp 192–195 °C) and αH-Mes2PyBTM (252 mg, 0.367 mmol, 24%, mp 122–124 °C).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 mixture of two conformers).
1H NMR (400 MHz, CDCl3, ppm): δ 8.50 (s, 0.5H), 8.49 (s, 0.5H), 8.38 (s, 0.5H), 8.37 (s, 0.5H), 7.41 (d, J = 2.2 Hz, 0.5H), 7.39 (d, J = 2.2 Hz, 0.5H), 7.29 (d, J = 2.2 Hz, 0.5H), 7.25 (d, 0.5H), 7.16 (d, J = 1.7 Hz, 0.5H), 7.15 (d, J = 1.7 Hz, 0.5H), 7.04 (d, J = 1.7 Hz, 0.5H), 7.01 (d, J = 1.7 Hz, 0.5H), 6.93 (s, 2H), 6.80 (s, 1H), 2.32 (s, 3H), 2.02–2.00 (m, 6H).
:1 mixture of two conformers).
1H NMR (400 MHz, CDCl3, ppm): δ 8.50 (s, 0.5H), 8.49 (s, 0.5H), 8.38 (s, 0.5H), 8.37 (s, 0.5H), 7.41 (d, J = 2.2 Hz, 0.5H), 7.39 (d, J = 2.2 Hz, 0.5H), 7.29 (d, J = 2.2 Hz, 0.5H), 7.25 (d, 0.5H), 7.16 (d, J = 1.7 Hz, 0.5H), 7.15 (d, J = 1.7 Hz, 0.5H), 7.04 (d, J = 1.7 Hz, 0.5H), 7.01 (d, J = 1.7 Hz, 0.5H), 6.93 (s, 2H), 6.80 (s, 1H), 2.32 (s, 3H), 2.02–2.00 (m, 6H).
13C NMR (100 MHz, CDCl3, ppm): δ 149.7, 149.5, 147.9, 147.8, 144.2, 144.2, 143.1, 143.1, 138.1, 138.0, 137.8, 137.5, 137.4, 137.4, 137.4, 136.8, 135.8, 135.7, 135.6, 134.6, 134.6, 134.2, 134.2, 133.7, 133.6, 133.3, 132.2, 132.0, 131.4, 131.1, 130.5, 130.0, 129.7, 129.5, 128.8, 128.5, 128.4, 50.0, 21.2, 20.7.
Elemental analysis calcd for C27H18Cl7N: C 53.64, H 3.00, N 2.32; found: C 53.92, H 3.04, N 2.25.
HRMS (MALDI-TOF MS positive mode) m/z: [MH]+ calcd for C27H19Cl7N+ 601.93317; found 601.93316.
13C NMR (100 MHz, CDCl3, ppm): δ 149.6, 147.8, 144.8, 142.9, 142.8, 137.7, 137.7, 137.6, 137.4, 137.0, 136.9, 135.9, 135.7, 134.7, 133.8, 132.9, 132.5, 131.5, 131.0, 129.6, 129.5, 128.4, 50.3, 21.2, 20.7.
Elemental analysis calcd for C36H29Cl6N: C 62.82, H 4.25, N 2.03; found: C 63.06, H 4.32, N 1.98.
HRMS (MALDI-TOF MS positive mode) m/z: [MH]+ calcd for C36H30Cl6N+ 686.05039; found 686.05075.
Synthesis of MesPyBTM
Under an argon atmosphere, αH-MesPyBTM (76.2 mg, 0.126 mmol) was dissolved in dry THF (∼4 mL), and tBuOK in THF (1M solution, 0.2 mL, 1.6 eq.) was added. The reaction mixture was stirred overnight in the dark. Silver nitrate (61.3 mg, 0.361 mmol) in acetonitrile (1.5 mL) was added and stirred for 2.5 h. The reaction mixture was filtered, evaporated, and purified by flash chromatography on silica gel (CHCl3:hexane = 1:1) and dried in vacuo to afford MesPyBTM (74.1 mg, 0.123 mmol, 97%) as a red solid (mp 99–100 °C).
HRMS (MALDI-TOF MS negative mode) m/z: [M]− calcd for C27H17Cl7N− 599.91862; found 599.91850.
Synthesis of Mes2PyBTM
Under an argon atmosphere, αH-Mes2PyBTM (60.7 mg, 0.0882 mmol) was dissolved in dry THF (∼3 mL), and tBuOK in THF (1M solution, 0.15 mL, 1.7 eq.) was added. The reaction mixture was stirred overnight in the dark. Silver nitrate (59.6 mg, 0.351 mmol) in acetonitrile (1.5 mL) was added and stirred for 2 h. Chloroform was added to the mixture and filtered on a celite pad. The solvent was evaporated and the reaction mixture was purified by silica gel column chromatography (CHCl3:hexane = 2:1) and dried in vacuo to afford Mes2PyBTM (52.4 mg, 0.0762 mmol, 86%) as a red solid (mp 122–125 °C).
HRMS (MALDI-TOF MS negative mode) m/z: [M]− calcd for C36H28Cl6N− 684.03584; found 684.03660.
Synthesis of αH-Mes2F2PyBTM
In a Schlenk tube, αH-F2PyBTM (488 mg, 1.00 mmol), 2,4,6-trimethylphenylboronic acid (494 mg, 3.01 mmol), and Pd(dtbpf)Cl2 (53.1 mg, 0.0815 mmol), were put under an argon atmosphere. The degassed K-EL 2 wt%:toluene (9:1 v/v) emulsion (3.6 mL) was added, and the mixture was heated at 70 °C. Degassed triethylamine (0.85 mL, 6.1 mmol) was finally added, and the reaction mixture was stirred at 70 °C overnight. The reaction mixture was cooled down to room temperature, dichloromethane was added, and filtered on a celite pad. The solvent was evaporated and purified by silica gel column chromatography (CHCl3:hexane = 1:1). The mixture (402 mg) was separated by GPC (CHCl3), and recrystallization from dichloromethane–methanol gave pure αH-Mes2F2PyBTM (114 mg, 0.174 mmol, 17%, mp 219–221 °C).
1H NMR (400 MHz, CDCl3, ppm): δ 8.36 (s, 1H), 8.24 (s, 1H), 7.12 (s, 4H), 6.93 (s, 4H), 6.85 (s, 1H), 2.32 (s, 6H), 2.04 (s, 12H).
13C NMR (100 MHz, CDCl3, ppm): δ 142.8, 137.7, 136.6, 135.9, 135.8, 135.4, 135.1, 133.7, 133.5, 132.2, 130.5, 128.4, 125.4, 125.3, 125.1, 42.3, 21.2, 20.7.
Elemental analysis calcd for C36H29Cl4F2N: C 65.97, H 4.46, N 2.14; found: C 66.00, H 4.44, N 2.10.
HRMS (MALDI-TOF MS positive mode) m/z:[MH]+ calcd for C36H30Cl4F2N+ 654.10949; found 654.10945.
Synthesis of Mes2F2PyBTM
Under an argon atmosphere, αH-Mes2F2PyBTM (28 mg, 0.043 mmol) was dissolved in dry THF (3 mL), and tBuOK in THF (1M solution, 0.1 mL, 2.3 eq.) was added. The reaction mixture was stirred overnight in the dark. Silver nitrate (35.5 mg, 0.209 mmol) in acetonitrile (0.7 mL) was added and stirred for 3 h. Chloroform was added to the mixture and filtered on a celite pad. The solvent was evaporated and the reaction mixture was purified by silica gel column chromatography (CHCl3:hexane = 1:1) and dried in vacuo to afford Mes2F2PyBTM (25.6 mg, 0.0391 mmol, 90%) as a red solid (mp 107–109 °C).
HRMS (MALDI-TOF MS positive mode) m/z: [M]+ calcd for C36H28Cl6NF2+ 652.09384; found 652.09347.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Y. H. conceived the project. Y. H. and R. K. prepared the compounds. W. O. and Y. H. conducted the DFT calculations. Y. H., R. M. and R. K. carried out the photophysical measurements. T. S. and W. O. contributed to the theoretical interpretation. Y. H. and W. O. wrote the original draft, and R. M., T. K., T. S. and K. U. reviewed and edited.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Takehiro Kawauchi, Ryukoku University for absolute luminescence quantum yield measurements. We acknowledge support by Ms. Yoshiko Nishikawa and Ms. Mieko Yamagaki for HRMS (MALDI-TOF MS) conducted in NAIST. This work was partly supported by the ARIM Program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. This research was supported by Cooperative Research by Institute for Molecular Science (IMS program 22IMS1222), 2022 Ryukoku University Science and Technology Fund, JSPS KAKENHI Grant Number JP20H02759, 22H02157 in Scientific Research (B), 22K05253 in Scientific Research (C), and CREST program grant JPMJCR17N2 of the Japan Science and Technology Agency. Numerical calculations were partly performed at the Supercomputer System, Institute for Chemical Research, Kyoto University, Academic Center for Computing and Media Studies (ACCMS), Kyoto University, the information initiative center, Hokkaido University, and Research Center for Computational Science, Okazaki (Project: 22-IMS-C065).Notes and references
- P. Murto and H. Bronstein, J. Mater. Chem. C, 2022, 10, 7368–7403 RSC.
- R. Matsuoka, A. Mizuno, T. Mibu and T. Kusamoto, Coord. Chem. Rev., 2022, 467, 214646 CrossRef.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- Y. Hattori, S. Kimura, T. Kusamoto, H. Maeda and H. Nishihara, Chem. Commun., 2018, 54, 615–618 RSC.
- C.-H. Liu, E. Hamzehpoor, Y. Sakai-Otsuka, T. Jadhav and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 23030–23034 CrossRef CAS PubMed.
- M. Ballester and G. de la Fuente, Tetrahedron Lett., 1970, 11, 4509–4510 CrossRef.
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Casteñer, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608–5616 CrossRef CAS.
- M. A. Fox, E. Gaillard and C.-C. Chen, J. Am. Chem. Soc., 1987, 109, 7088–7094 CrossRef CAS.
- S. R. Ruberu and M. A. Fox, J. Phys. Chem., 1993, 97, 143–149 CrossRef CAS.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed.
- S. Kimura, T. Kusamoto, S. Kimura, K. Kato, Y. Teki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 12711–12715 CrossRef CAS PubMed.
- S. Kumar, Y. Kumar, S. K. Keshri and P. Mukhopadhyay, Magnetochemistry, 2016, 2, 42 CrossRef.
- A. Ghirri, C. Bonizzoni, F. Troiani, N. Buccheri, L. Beverina, A. Cassinese and M. Affronte, Phys. Rev. A, 2016, 93, 063855 CrossRef.
- K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Angew. Chem., Int. Ed., 2019, 58, 2606–2611 CrossRef CAS PubMed.
- S. Kimura, S. Kimura, K. Kato, Y. Teki, H. Nishihara and T. Kusamoto, Chem. Sci., 2021, 12, 2025–2029 RSC.
- Z. Zhou, C. Qiao, J. Yao, Y. Yan and Y. S. Zhao, J. Mater. Chem. C, 2022, 10, 2551–2555 RSC.
- Y. Hattori, T. Kusamoto and H. Nishihara, RSC Adv., 2015, 5, 64802–64805 RSC.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto, T. Sato and H. Nishihara, Chem. Commun., 2016, 52, 13393–13396 RSC.
- Y. Hattori, R. Kitajima, R. Matsuoka, T. Kusamoto, H. Nishihara and K. Uchida, Chem. Commun., 2022, 58, 2560–2563 RSC.
- V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305–2309 CrossRef CAS.
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed.
- S. Castellanos, D. Velasco, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2008, 73, 3759–3767 CrossRef CAS PubMed.
- S. Dong, W. Xu, H. Guo, W. Yan, M. Zhang and F. Li, Phys. Chem. Chem. Phys., 2018, 20, 18657–18662 RSC.
- H. Guo, Q. Peng, X.-K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J.-L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- A. Heckmann, S. Dümmler, J. Pauli, M. Margraf, J. Köhler, D. Stich, C. Lambert, I. Fischer and U. Resch-Genger, J. Phys. Chem. C, 2009, 113, 20958–20966 CrossRef CAS.
- Y. Beldjoudi, M. A. Nascimento, Y. J. Cho, H. Yu, H. Aziz, D. Tonouchi, K. Eguchi, M. M. Matsushita, K. Awaga, I. Osorio-Roman, C. P. Constantinides and J. M. Rawson, J. Am. Chem. Soc., 2018, 140, 6260–6270 CrossRef CAS PubMed.
- M. Ito, S. Shirai, Y. Xie, T. Kushida, N. Ando, H. Soutome, K. J. Fujimoto, T. Yanai, K. Tabata, Y. Miyata, H. Kita and S. Yamaguchi, Angew. Chem., Int. Ed., 2022, e202201965 CAS.
- S. Mattiello, F. Corsini, S. Mecca, M. Sassi, R. Ruffo, G. Mattioli, Y. Hattori, T. Kusamoto, G. Griffini and L. Beverina, Mater. Adv., 2021, 2, 7369–7378 RSC.
- S. Kimura, M. Uejima, W. Ota, T. Sato, S. Kusaka, R. Matsuda, H. Nishihara and T. Kusamoto, J. Am. Chem. Soc., 2021, 143, 4329–4348 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- R. Improta, V. Barone, G. Scalmani and M. J. Frisch, J. Chem. Phys., 2006, 125, 054103 CrossRef PubMed.
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224–1229 CrossRef CAS PubMed.
- Y. Zhao, A. Abdurahman, Y. Zhang, P. Zhang, M. Zhang and F. Li, CCS Chem, 2022, 4, 722–731 CrossRef CAS.
- S. Kimura, A. Tanushi, T. Kusamoto, S. Kochi, T. Sato and H. Nishihara, Chem. Sci., 2018, 9, 1996–2007 RSC.
- T. L. Chu and S. I. Weissman, J. Chem. Phys., 1954, 22, 21–25 CrossRef CAS.
- E. Cho, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2020, 142, 17782–17786 CrossRef CAS PubMed.
- G. Fisher, Vibronic Coupling: The Interaction between the Electronic and Nuclear Motions, Academic Press, London, 1984 Search PubMed.
- T. Azumi and K. Matsuzaki, Photochem. Photobiol., 1977, 25, 315–326 CrossRef CAS.
- M. Uejima, T. Sato, D. Yokoyama, K. Tanaka and J.-W. Park, Phys. Chem. Chem. Phys., 2014, 16, 14244–14256 RSC.
- T. Kato, N. Haruta and T. Sato, Vibronic coupling density: Understanding molecular deformation, Springer, Singapore, 2014 Search PubMed.
- R. Englman and J. Jortner, Molecular. Phys., 1970, 18, 145–164 CrossRef CAS.
- R. A. Velapoldi and H. H. Tønnesen, J. Fluoresc., 2014, 14, 465–472 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05079j |
| This journal is © The Royal Society of Chemistry 2022 |