
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprotected amine transfer performed by non-heme iron(II) complexes†
Alizée
Boullé
,
Aminata
Doumbia
,
Jean-Pierre
Mahy
* and
Frédéric
Avenier
 *
*
Laboratoire de Chimie Bioorganique et Bioinorganique, Institut de Chimie Moléculaire et des Matériaux d’Orsay (UMR CNRS 8182), Université Paris-Saclay, 17, Avenue des Sciences, 91400 Orsay, France. E-mail: frederic.avenier@universite-paris-saclay.fr; jean-pierre.mahy@universite-paris-saclay.fr
First published on 28th November 2022
Abstract
Direct amination of C–H or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds using unprotected amino groups is very challenging, especially with earth abundant metal ions. Here we show that a bioinspired iron(II) complex catalyses the double amination of its dangling benzyl branch in the presence of hydroxylamine derivatives as the unprotected amine donor and that the replacement of the benzyl branch by a methyl group also allows the aziridination of styrene.
C bonds using unprotected amino groups is very challenging, especially with earth abundant metal ions. Here we show that a bioinspired iron(II) complex catalyses the double amination of its dangling benzyl branch in the presence of hydroxylamine derivatives as the unprotected amine donor and that the replacement of the benzyl branch by a methyl group also allows the aziridination of styrene.
Amine containing compounds are ubiquitous among natural products, pharmaceuticals or agrochemicals.1 Yet, the synthetic routes for the amination of organic molecules are not straightforward and mostly rely on the prior introduction of polar groups such as halides,2 making the access to amine containing compounds from chemical feedstocks time consuming and costly. The direct catalytic introduction of amino groups into non-activated C–H bonds would clearly appear as a more elegant and cheaper way to proceed, but such a reaction still suffers from the need for the use of rare and expensive metal ions such as rhodium and iridium as catalysts.3 Their replacement by earth abundant metal ions such as iron stands therefore as a major challenge for modern chemistry.
Inspired by iron containing monooxygenases, chemists have developed low molecular weight catalysts capable of performing selective oxidation reactions, via the formation of high valent iron-oxo intermediates which transfer their oxene group into C–H and CC bonds.4 Based on this approach, the group of Breslow was the first to demonstrate that nitrene groups could also be transferred, notably thanks to the use of high valent iodine derivatives as nitrene donating agents.5 However, all reactions developed so far have been performed using nitrene compounds stabilized by electron withdrawing substituents such as a para-toluenesulfonyl group.6 It is only about 5 years ago that hydroxylamine derivatives were introduced by the group of Morandi as a new class of unprotected amine donors and successfully used for iron catalyzed amination reactions.7–9 More recently, these hydroxylamine derivatives were also used by the group of Arnold to expand the repertoire of iron-dependent oxygenases in a very efficient way.10–12 Hence, unprotected nitrene transfer now appears as a new paradigm in the field of catalysis, just as the oxene transfer was for the last 50 years. At this stage very little is known about those reactions, especially about their mechanism. The first mechanistic studies, dealing with the aminoetherification of alkenes, catalyzed by a fully oxygenated iron(II) complex, just came out this year.13 This seminal work is paving the way to the future boom of mechanistic studies for all kinds of amination reactions (aziridination, aliphatic or aromatic amination) potentially catalyzed by a large variety of iron complexes.
In this work, we expanded the repertoire for this reaction by studying the reactivity of a previously described family of nitrogen based iron complexes14,15 (complexes 1 and 2, Scheme 1) with the unprotected amine donor PivONH2 (Scheme 1) for nitrene transfer reactions. The UV-Visible monitoring of the reaction of complex 1 with 5 equivalents of the amine donor showed the formation of a first chromophore at 850 nm (ε = 4000 M−1 cm−1) after 40 minutes of reaction, which was then evolving towards the formation of a stable chromophore at 720 nm (ε = 9000 M−1 cm−1) (Fig. 1). ESI-MS analysis of the final solution showed the presence of a peak at m/z 656.1327 corresponding to the mass of the starting complex [Fe(II)BnTPEN](OTf)+ (m/z 628.1313) + 28 amu, suggesting that 2 nitrogen atoms were transferred to the ligand during the reaction. Similar data were obtained whether the reaction was performed in acetonitrile or in trifluoroethanol (ESI,† Fig. S1). Interestingly, when the reaction was followed by ESI-MS analysis (Fig. 2), one could clearly observe the transformation of the starting material (m/z 628.1313) into a first monocation at m/z 643.1397 (+ 15 amu) after one minute of reaction, followed by its deprotonation (m/z 642.1318) after 45 minutes of reaction. Concomitantly, another monocation also appeared at m/z 656.1347 (+ 28 amu). These data clearly suggest the transfer of two nitrogen atoms into the ligand, most probably occurring on the benzyl branch, as described in Scheme 2. It is indeed important to note that when the same experiment was performed with complex 2 (Scheme 1), bearing a methyl group in place of the benzyl group, no change of the mass of the complex, nor of the absorbance of the solution could be observed, clearly evidencing that the atom transfer occurred on the benzyl branch. When the same reaction was performed with complex 1 with up to three equiv. of an amine donor, only the first chromophore was formed as a stable species (ESI,† Fig. S2). In this case, ESI-MS analysis evidenced the formation of a monocation peak at m/z 493.1773, matching with the mass of the starting complex [Fe(II)BnTPEN]2+ + 14 amu, suggesting the incorporation of one nitrogen on the ligand and the apparition of a negative charge on the nitrogen (ESI,† Fig. S3). To confirm the incorporation of nitrogen into C–H bonds of the ligand, the iron was precipitated with NaOH in aqueous solution and the ligand was extracted with dichloromethane. ESI-MS analysis of the organic layer then evidenced a peak for the remaining original ligand at m/z 424.2480 and two other peaks at m/z 439.2580 (Ligand + 15 amu) and 454.2697 (Ligand + 30 amu) corresponding to the incorporation of one and two amine groups in the ligand (Fig. 3). At this stage, one may wonder if the amination is occurring at the benzylic position rather than at the aromatic ring, but the very intense absorption band appearing at 720 nm for the complex after the reaction (Fig. 1) is in very good agreement with the formation of a para- or an ortho-quinone diimine, as was respectively observed upon oxidation of polyanilines16 or orthophenylenediamine17 (Scheme 2). Furthermore, it was previously evidenced that the coordination of benzoquinone diimine ligands to ruthenium ions induced a dramatic red-shift of absorptions with a higher molar absorption coefficient compared to the coordination of other diimine ligands.18,19 This was attributed to a ligand-to-metal charge transfer process with a very strong t2g(M)–π*(L) interaction. Thus, the rather high molar extinction coefficient of the band in the near infra-red region for the di-aminated iron complex also suggests the formation of benzoquinone diimine ligand and encouraged us to performed FT-IR spectroscopy to complete its characterization (ESI,† Fig. S4). The FT-IR spectrum of the di-aminated complex showed N–H stretching vibration modes at 3192 and 3090 cm−1, as well as a CN stretching vibration mode at 1705 cm−1 which are both in good agreement with the formation of a benzoquinone diimine ligand.20 These results obtained for the intramolecular amine transfer prompted us to test the reactivity of nitrogen based non-heme iron(II) complex for the aziridination of external substrates such as styrene. The catalytic synthesis of unprotected aziridines is indeed an important challenge for modern organic chemistry,21 but although iron catalysed aminochlorination,9 aminohydroxylation10 and aminoethrification8 of alkenes were successfully achieved using iron based catalysts, the direct formation of unprotected aziridines still relies on rhodium based complexes.22 Here, complex 2, bearing the same coordination sphere as complex 1, with a methyl group instead of the dangling benzyl group, was used in order to avoid any intramolecular reaction and was introduced as a catalyst for the reaction of PivONH2 with styrene, either in acetonitrile, or in a dichloromethane/methanol solution. In both cases, the reaction medium was left for 16 hours under stirring and filtered on silica to get rid of the catalyst, before being analysed by mass spectrometry. When the reaction was performed in acetonitrile, ESI-MS analysis showed only two ions at m/z 120.0808 and 161.1069 (ESI,† Fig. S5) matching with the aziridine 3 and another product resulting from the incorporation of acetonitrile into one of the intermediates during the aziridination process (Scheme 3, compound 4). Such a product was recently observed in the case of tosyl-protected nitrene transfer and characterized as an imidazoline compound by the group of Latour.23 These results suggest that both protected and unprotected amine transfers may follow the same mechanistic pathway and open interesting perspectives for upcoming mechanistic studies. Finally, to avoid such acetonitrile incorporation, the reaction was also performed in a dichloromethane/methanol (98/2) solution under the same conditions, and ESI-MS analysis only showed the formation of an aziridine compound (3) at m/z 120.0807 (ESI† Fig. S4).
 | ||
| Scheme 1 Chemical structures of complex 1 [Fe(II)BnTPEN](OTf)2, complex 2 [Fe(II)MeTPEN](OTf)2 and the amine donor PivONH2 used in this work.‡ | ||
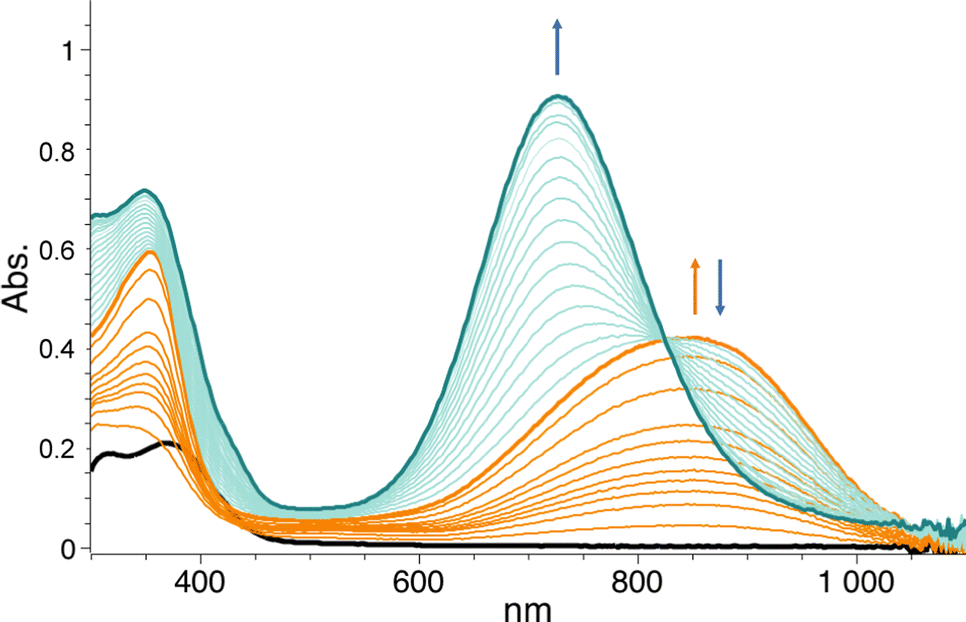 | ||
| Fig. 1 Time resolved UV-Vis. Spectrum for the reaction of complex 1 (0.1 mM) with 5 equivalents of PivONH2 in trifluoroethanol (TFE) at room temperature. The first chromophore (850 nm) is formed after 40 min and the second one (720 nm) after 3 hours. | ||
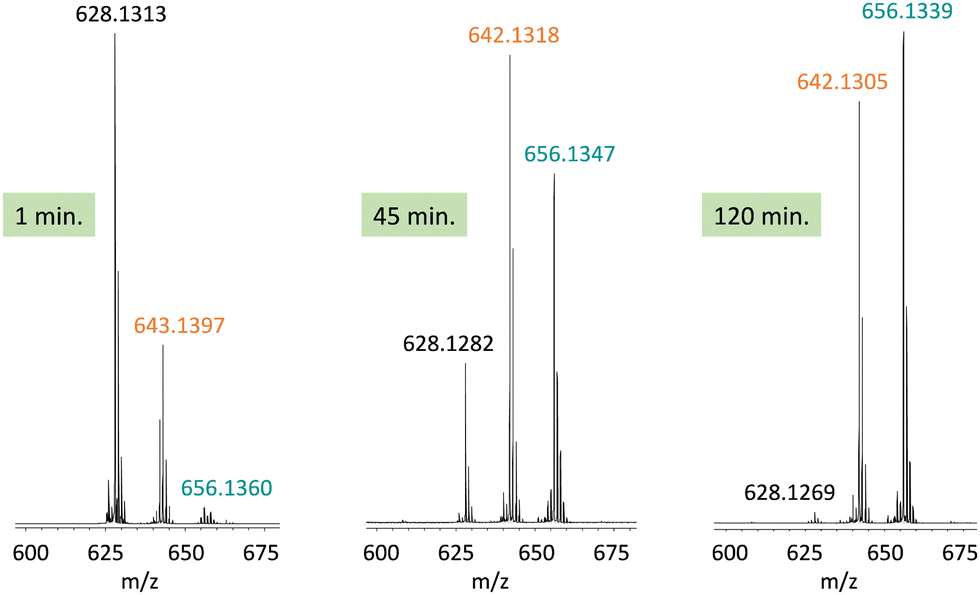 | ||
| Fig. 2 ESI-MS analysis of the solution after reaction of complex 1 with 3 equivalents of PivONH2 in acetonitrile after 1 minute, 45 minutes and 120 minutes at room temperature. | ||
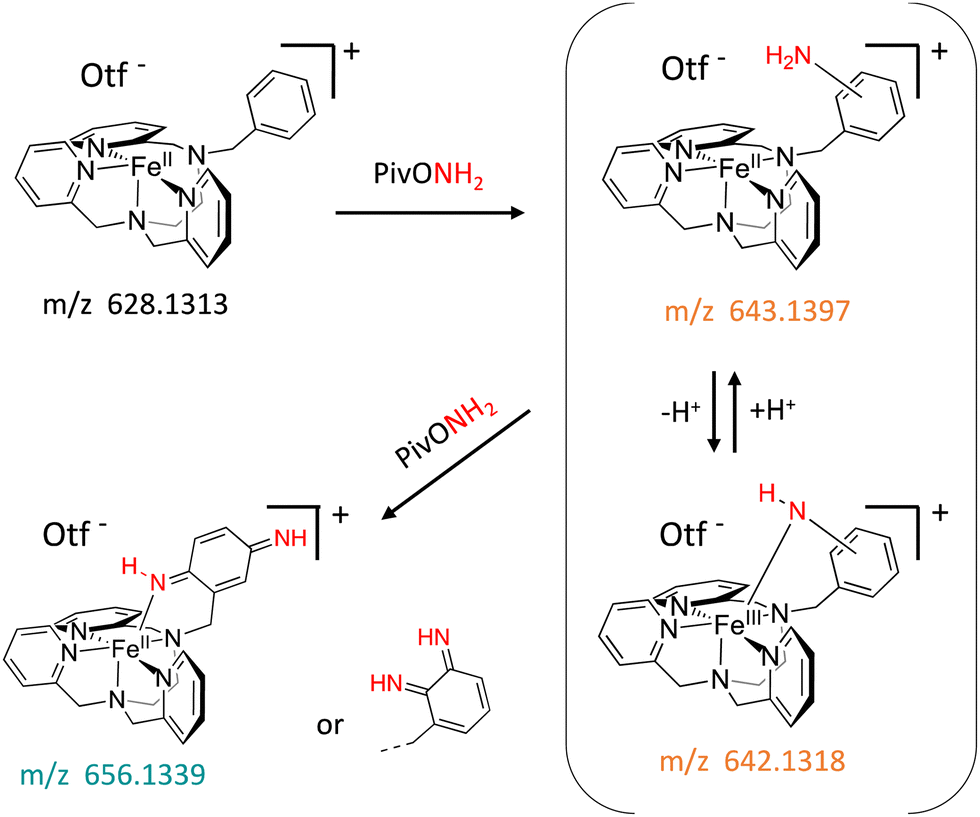 | ||
| Scheme 2 Reaction of complex 1 with 5 equivalents of PivONH2 followed by mass spectrometry. | ||
 | ||
| Scheme 3 Styrene aziridination performed by complex 2 in the presence of the unprotected amine donor either in acetonitrile or in a dichloromethane/methanol solution.§ | ||
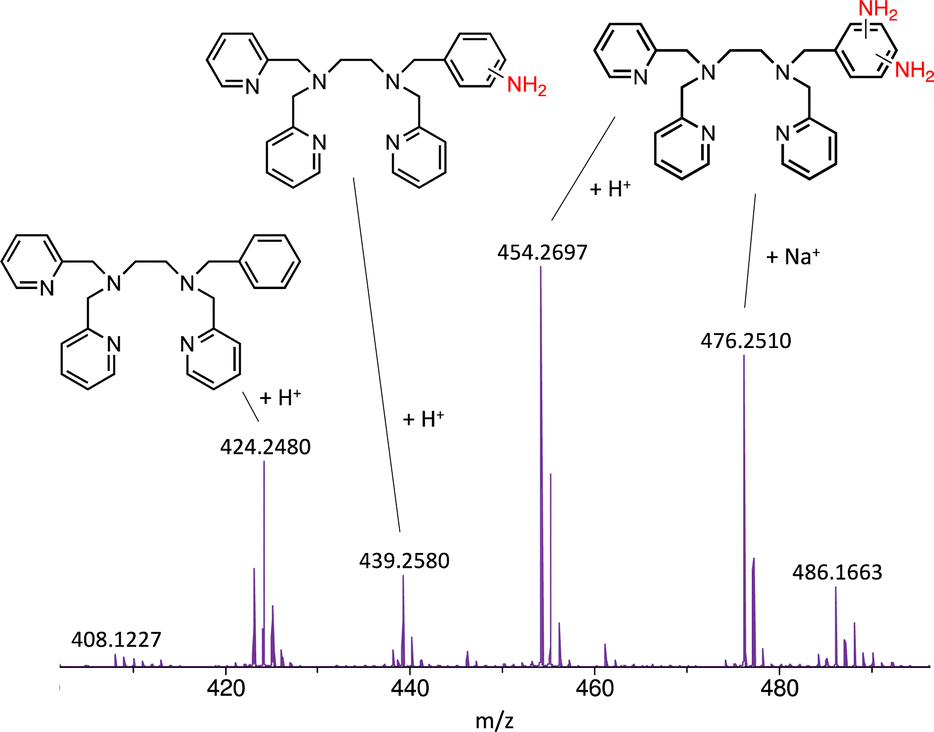 | ||
| Fig. 3 ESI-MS analysis of the organic layer obtained after reaction of complex 1 with 5 equivalents of PivONH2 in acetonitrile, followed by decomplexation of iron with NaOH and extraction with dichloromethane. | ||
In conclusion, this work demonstrates that a bioinspired nitrogen based iron(II) complex is able to perform the intramolecular amination of its benzyl branch using the unprotected amine donor PivONH2, giving rise to the formation of a benzoquinone diimine complex with very specific optical properties. We also show that a similar complex, but that bearing no internal substrate in its structure, also performs the intermolecular aziridination of styrene. We are now working on the mechanism of such an intramolecular amination reaction and on the quantification of the aziridination reaction.
We thank Hafsa Korri-Youssoufi for her help in performing FT-IR experiments.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Ricci, Amino group chemistry: from synthesis to the life sciences, Wiley-VCH, Weinheim, 2008 Search PubMed.
- M. B. Smith and J. March, March's advanced organic chemistry: reactions mechanisms and structure, John Wiley and Sons, Hoboken, 6th edn, 2007 Search PubMed.
- L. Legnani, B. N. Bhawal and B. Morandi, Synthesis, 2017, 776–789 CAS.
- L. Vicens, G. Olivo and M. Costas, ACS Catal., 2020, 10, 8611–8631 CrossRef CAS.
- R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1983, 105, 6728–6729 CrossRef CAS.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- L. Legnani, G. Prina Cerai and B. Morandi, ACS Catal., 2016, 6, 8162–8165 CrossRef CAS.
- L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248–2251 CrossRef CAS PubMed.
- L. Legnani, G. Prina-Cerai, T. Delcaillau, S. Willems and B. Morandi, Science, 2018, 362, 434–439 CrossRef CAS PubMed.
- I. Cho, C. K. Prier, Z.-J. Jia, R. K. Zhang, T. Görbe and F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 3138–3142 CrossRef CAS PubMed.
- Z.-J. Jia, S. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- Z. Liu, Z.-Y. Qin, L. Zhu, S. V. Athavale, A. Sengupta, Z.-J. Jia, M. Garcia-Borràs, K. N. Houk and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 80–85 CrossRef CAS PubMed.
- S. Chatterjee, I. Harden, G. Bistoni, R. G. Castillo, S. Chabbra, M. van Gastel, A. Schnegg, E. Bill, J. A. Birrell, B. Morandi, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2022, 144, 2637–2656 CrossRef CAS PubMed.
- A. Hazell, C. J. McKenzie, L. P. Nielsen, S. Schindler and M. Weitzer, J. Chem. Soc., Dalton Trans., 2002, 310–317 RSC.
- M. Martinho, P. Dorlet, E. Rivière, A. Thibon, C. Ribal, F. Banse and J.-J. Girerd, Chem. – Eur. J., 2008, 14, 3182–3188 CrossRef CAS PubMed.
- I. Kulszewicz-Bajer, I. Różalska and M. Kuryłek, New J. Chem., 2004, 28, 669–675 RSC.
- M. M. Bittner, S. V. Lindeman, C. V. Popescu and A. T. Fiedler, Inorg. Chem., 2014, 53, 4047–4061 CrossRef CAS.
- K. Rieder, U. Hauser, H. Siegenthaler, E. Schmidt and A. Ludi, Inorg. Chem., 1975, 14, 1902–1907 CrossRef CAS.
- S. Joss, H. Reust and A. Ludi, J. Am. Chem. Soc., 1981, 103, 981–982 CrossRef CAS.
- F. E. Prichard, Spectrochim. Acta, 1964, 20, 925–936 CrossRef CAS.
- S. Sabir, G. Kumar and J. L. Jat, Asian J. Org. Chem., 2017, 6, 782–793 CrossRef CAS.
- J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Science, 2014, 343, 61–65 CrossRef CAS.
- G. Coin, P. Dubourdeaux, P.-A. Bayle, C. Lebrun, P. Maldivi and J.-M. Latour, Dalton Trans., 2021, 50, 6512–6519 RSC.
- S. Makai, E. Falk and B. Morandi, Org. Synth., 2020, 97, 207–216 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04992a |
| ‡ Complex 1, complex 2 and the amine donor PivONH2 were synthesized as previously described in the literature.14,15,24 |
| § Conditions for catalytic experiments: [complex 2] = 1 mM, [styrene] = 10 mM and [PivONH2] = 10 mM at room temperature for 16 h in 1 mL of solvent. For the reactions performed in a CH2Cl2/MeOH mixture, PivONH2 was first solubilised in methanol before addition to the CH2Cl2 solution (CH2Cl2/MeOH = 98/2). |
| This journal is © The Royal Society of Chemistry 2023 |