
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic acid formation in the gas-phase ozonolysis of α,β-unsaturated ketones†
Niklas
Illmann
 *,
Iulia
Patroescu-Klotz
and
Peter
Wiesen
*,
Iulia
Patroescu-Klotz
and
Peter
Wiesen
Institute for Atmospheric and Environmental Research, Bergische Universität Wuppertal, Gaußstraße 20, 42119 Wuppertal, Germany. E-mail: illmann@uni-wuppertal.de
First published on 7th December 2022
Abstract
Organic acids are key species in determining the radiative properties of the atmosphere due to their contribution to particle formation. Reported discrepancies between field measurements and modelling suggest significant missing sources. Herein, we present a mechanistic investigation on the gas-phase ozonolysis of ethyl vinyl ketone (EVK, 1-penten-3-one), which we chose as a model compound for α,β-unsaturated ketones. Experiments were performed in a 1080 L quartz-glass reaction chamber (QUAREC) at 990 ± 15 mbar and 298 ± 2 K (r. h. ≪ 0.1%) and analysed via long-path FTIR spectrometry and PTR-ToF-MS. The experiments were performed in the presence of an excess of CO to suppress the chemistry of OH radicals. For a comprehensive picture, in selected experiments, SO2 was also added to the reaction system to scavenge the stabilized Criegee intermediates (sCIs) and to investigate their formation yield. Combining the results of both set-ups allowed us to quantify 2-oxobutanal, for which we report vapour-phase FTIR spectra. In addition, we introduce the first-ever infrared spectra of perpropionic acid, which was also positively identified in the EVK + O3 system. A detailed analysis of the experimental findings allowed us to link the identified reaction products (acetaldehyde, ethyl hydroperoxide, and perpropionic acid) to known bimolecular reactions of RO2 radicals. Thereby, it is shown that the EVK + O3 reaction yields formic acid, HC(O)OH, and propionic acid, C2H5C(O)OH, and their formation is not covered by mechanisms reported in the literature. Three different pathways accounting for their formation from chemically activated CIs are proposed and possible implications for the ozonolysis of α,β-unsaturated ketones in the atmosphere are discussed.
Introduction
Organic acids are a class of atmospheric compounds that, with the exception of formic acid, HC(O)OH, and acetic acid, CH3C(O)OH, determined also in the gas-phase, are mainly found in aerosol particles, which explains their importance for rainwater pH, cloud growth and chemistry and hence the radiative properties of the atmosphere.1–3However, a series of studies have shown significant discrepancies between model predictions concerning their chemical formation and ground-based and airborne observations.4–6 For example, models under-predict formic acid concentrations by up to a factor of 10,5,6 indicating a large gap in our understanding of carboxylic acid forming mechanisms. Several investigations revealed candidates for the missing sources. These contained the OH chemistry of isoprene oxidation products including isoprene nitrates7 or α-hydroxy carbonyls like glycolaldehyde8 and hydroxyacetone,9 the OH oxidation of enols10 or reactions of Criegee intermediates (mainly CH2OO) with water vapour.11,12 Yet, even when these “candidate sources” were included in models, HC(O)OH remains underestimated by up to a factor of 3.5 Recently, the formation of ketene-enols in the OH oxidation of aromatic species and their further reactions with OH and O3 were suggested to account for the rapid photochemical formation of organic acids in urban areas.13 However, modelling studies revealed also that the interplay of different sources is needed to account for the missing levels of small organic acids, many of them associated though with isoprene chemistry or the oxidation of biogenic volatile organic compounds (BVOCs) in general.5
Ozonolysis reactions were considered mainly as a source of larger carboxylic acids (derived from larger alkenes and terpenes), easily partitioning into the aerosol phase. In many cases, the overall mechanism and the reaction products of ozonolysis reactions remain poorly characterized, which is often reflected by a low overall carbon balance. This is partly due to the nature of ozonolysis reactions, which proceed almost exclusively (with some exceptions, e.g. epoxide formation) via a 1,3-dipolar cycloaddition yielding initially a five-membered primary ozonide (POZ). This, in turn, is formed loaded with a substantial amount of energy due to the exothermicity of ozonolysis reactions. As a consequence, the POZ will decompose immediately following two possible pathways (see Fig. 1), where only 50% results initially in the formation of stable carbonyl products (primary carbonyls), if the POZ channel represents the only pathway of the ozonolysis reaction. The remaining fraction yields Criegee intermediates (CIs), which are described formally as zwitterionic carbonyl oxides (see Fig. 1). Since the excess energy is necessarily distributed among the initially formed reaction products, this results in nascent CIs, which are subject to further decomposition, whereas a fraction might be thermalized (stabilized CIs = sCIs) and is prone to bimolecular reactions.14 Extensive work during the last few decades has shown that sCIs react very fast with SO2 and organic acids (e.g. ref. 14 and references therein), although under atmospheric conditions, sCIs will react preferentially with H2O or the water dimer. This suggests that the different levels of H2O present in the experimental systems might be one reason for the difference in the product distributions reported by various authors. Furthermore, ozonolysis reactions are also experimentally challenging, as they were shown to produce OH radicals (e.g. ref. 14 and references therein), which infer a competition between the O3 and OH reactions of the target species. In order to overcome this issue, OH scavengers like cyclohexane or CO are often used to investigate the ozonolysis system without OH interference in atmospheric simulation chamber experiments. However, in the case of CO, almost any OH radical is converted into HO2, which might imply a significant increase of the HO2 level in the reaction system.
| CO + OH + O2 → CO2 + HO2 | (R1) |
| c-C6H12 + OH + M (+O2) → c-C6H11O2 + H2O + M* | (R2) |
| NO + O3 → NO2 + O2 | (R3) |
| RO2 + HO2 → products | (R4) |
| RO2 + RO2 → products | (R5) |
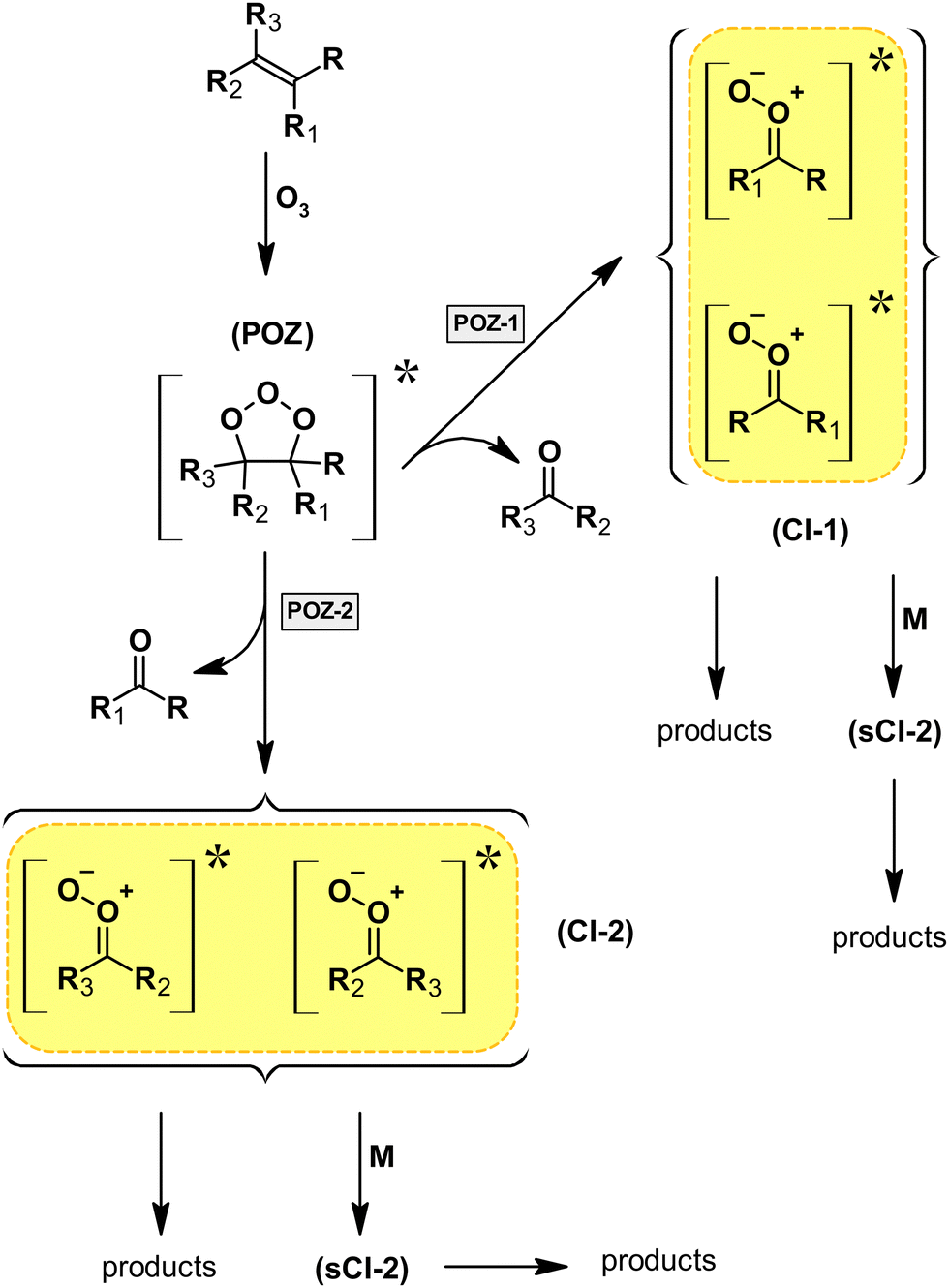 | ||
| Fig. 1 Generalized mechanism for the gas-phase ozonolysis of unsaturated organic species proceeding through the initially formed primary ozonide (POZ) and subsequent possible decomposition pathways (POZ-1 and POZ-2), yielding a stable carbonyl species and a Criegee intermediate (CI). Two stereoisomers are possibly formed for each CI, which results in four possible CIs in total. | ||
It was shown that α,β-unsaturated ketones are formed in the oxidation of BVOCs (e.g. methyl vinyl ketone from isoprene) or are directly released by vegetation under stress conditions (e.g. ethyl vinyl ketone was reportedly emitted upon leave wounding15). Despite the importance of α,β-unsaturated ketones in terms of their atmospheric burden, their ozonolysis reactions were scarcely studied. Furthermore, the existing studies report rather contradictory results, for example, in the case of the primary carbonyls obtained from methyl vinyl ketone + O3.16,17 For the longer-chain analogue ethyl vinyl ketone (EVK, 1-penten-3-one), only two studies were reported previously by Grosjean et al.16 and O’Dwyer et al.,18 both targeting the primary carbonyl formation. Grosjean et al.16 identified also acetaldehyde, although its formation remained unexplained. Current recommendations by the IUPAC (International Union of Pure and Applied Chemistry) Task Group on Atmospheric Chemical Kinetic Data Evaluation are limited to small alkenes and terpene-derived CIs.14 Combining this, the fate of carbonyl-substituted CIs, where the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is close to the CO+–O− moiety, seems to be generally poorly understood. Considering the available kinetic data for the OH and O3 reaction of MVK19 (see ref. 20 for a list of available literature data on the MVK + O3 rate coefficient) and EVK,21,22 the ozonolysis reactions account for about 17% of the MVK and about 15% of the EVK daytime loss, respectively (for [OH] ≈ 1 × 106 cm−3 and [O3] ≈ 35 ppbV). In an attempt to close the existing gaps in the α,β-unsaturated ketone ozonolysis mechanism, we studied in the present work the reaction of ozone with EVK, chosen as a model species for the title compounds.
O bond is close to the CO+–O− moiety, seems to be generally poorly understood. Considering the available kinetic data for the OH and O3 reaction of MVK19 (see ref. 20 for a list of available literature data on the MVK + O3 rate coefficient) and EVK,21,22 the ozonolysis reactions account for about 17% of the MVK and about 15% of the EVK daytime loss, respectively (for [OH] ≈ 1 × 106 cm−3 and [O3] ≈ 35 ppbV). In an attempt to close the existing gaps in the α,β-unsaturated ketone ozonolysis mechanism, we studied in the present work the reaction of ozone with EVK, chosen as a model species for the title compounds.
To keep the reaction system as simple as possible, two experimental approaches were followed. First, all experiments were performed with an excess of CO to suppress OH reactions and exclude the formation of RO2 radicals other than formed from EVK + O3. In a second series of experiments, sufficient SO2 was also added to the reaction mixture to favour the sCI + SO2 reaction and hence suppress potential reactions of sCIs with other reaction products. Combining the results of both experimental set-ups provided a more comprehensive perspective on the ozonolysis mechanism. Clear evidence was found for the formation of organic acids and pathways accounting for their formation are proposed. In pursuing this issue, we were able to obtain the infrared absorption features of gaseous perpropionic acid.
Experimental
In order to investigate the EVK + O3 system, experiments were carried out in a 1080 L quartz reaction chamber (QUAREC) under dry conditions (r.h. ≪ 0.1%) at 990 ± 15 mbar of synthetic air and a temperature of 298 ± 2 K. The chamber is coupled to a Fourier-transform infrared (FTIR) spectrometer (Nicolet iS 50) by means of a White-type mirror system. FTIR spectra were recorded in the spectral range of 4000–700 cm−1 at a resolution of 1 cm−1. The system is operated at an optical path length of 484.7 ± 0.8 m. The current set-up of the QUAREC chamber is described in greater details elsewhere.23The initial mixing ratios were 1.5–2.3 ppmV for EVK (Alfa Aesar, 97%), 3.1–4.0 ppmV for SO2 (Air Liquide, 99.9%), and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 ppmV for CO (Air Liquide, 99.97%), where 1 ppmV = 2.46 × 1013 cm−3 (at 298 K and 1 atm).
000–20000 ppmV for CO (Air Liquide, 99.97%), where 1 ppmV = 2.46 × 1013 cm−3 (at 298 K and 1 atm).
EVK and the reaction products were quantified via FTIR spectroscopy using calibrated reference spectra from the internal laboratory database. A list of the used integrated absorption cross sections is given in the ESI† (Table S1). In the experiments, typically 50–70 interferograms were co-added per spectrum, which results in averaging recording periods of about 80–115 s. In addition to the quantification by long-path FTIR spectroscopy, a qualitative identification of reaction products was performed using a PTR-ToF-MS 8000 instrument (Ionicon Analytik GmbH, Innsbruck, Austria) in experiments without SO2 injection. The drift tube was operated at a temperature of 70 °C, 2.2 mbar pressure and 500 V drift voltage. Accordingly, the E/N value was about 120 Td (1 Td = 1 × 10−17 V cm2), where E is the electric field in the drift tube, and N is the gas number density. The sampling line of the instrument was operated at a temperature of 70 °C and coupled to a heated stainless steel line, which was mounted on the middle flange of the 1080 L chamber. The sample flow was 200 mL min−1. In order to obtain stable signals with the PTR-MS instrument, the fan mounted on the middle flange inside the chamber was switched on during the whole experiment. The PTR-ToF-MS data were recorded and processed using the TOFDAQ data acquisition software and the PTR-MS VIEWER software (Ionicon Analytik GmbH, Innsbruck, Austria), respectively.
The reaction mixtures were observed over a period of 15–50 min without an oxidant in order to determine the wall loss in each individual experiment. After that, the reaction was started by the injection of O3, which was generated by passing a stream of pure oxygen (Messer, 99.995%) through an electrical discharge in a homemade device. Typically, the formation of reaction products was observed over a period of about 20 min.
Simulations of the temporal evolution of reaction products, possibly formed from peroxy radical reactions, were performed following the approach outlined previously.23 A detailed description of the sensitivity analysis and model validation can be found elsewhere.24 In contrast to previous applications, an explicit chemical mechanism was used for the relevant RO2 reactions, in order to prove if the temporal behaviour of selected reaction products can be described by known reaction sequences. For this purpose, the source of the parent RO2 was included in a simplified reaction (EVK + O3 → x1RO2 + x2R′O2) as well as an average HO2 concentration. Both the branching ratio towards the RO2 formation and [HO2] were varied to check for a match between the experimental data and the modelled time profiles for all relevant species.
Results and discussion
The wall loss of EVK was found to be in the range (1–5) × 10−5 s−1. The loss due to the O3 reaction was about (6–8) × 10−4 s−1 and hence at least one order of magnitude faster than the wall loss in all performed experiments. The EVK + O3 reaction was observed until about 50–80% of the EVK was consumed. Table 1 summarizes the yields of the quantified reaction products based on the FTIR measurements together with the assigned ion signals from the PTR-MS data. A detailed discussion on the assignment of ion signals is presented in Section B of the ESI.†| Species | Yielda | Yieldb | Yielda | Yieldc | Assigned m/za |
|---|---|---|---|---|---|
|
a Experiments were performed in the presence of CO as an OH radical scavenger.
b Experiments were performed in the presence of cyclohexane as an OH radical scavenger.
c Experiments were performed in the presence of CO as an OH radical scavenger and SO2.
d The yields were obtained following a semi-quantitative approach by assuming the infrared cross section of the CO band to equal the cross section of the structural analogue methyl glyoxal.
e The formation is not unequivocally proven due to interfering fragment ions.
f Sum of CO2 formed from the reaction of EVK + O3 and CO + OH.
|
|||||
| HCHO | 0.28 ± 0.10 | 0.548 ± 0.069 | 0.37 ± 0.02 | 0.65 ± 0.12 | 31.018 (CH3O+) |
| 2-Oxobutanal | 0.29 ± 0.10d | 0.444 ± 0.025 | 0.49 ± 0.03 | 0.53 ± 0.12d | 87.045 (C4H7O2+), 59.050 (C3H7O+) |
| Formic acid | 0.012 ± 0.008 | 0.12 ± 0.06 | 47.013 (CH3O2+) | ||
| Formic anhydride | 0.14 ± 0.08 | 47.013 (CH3O2+) | |||
| Formic propionic anhydride | 0.11 ± 0.04 | 75.045 (C3H7O2+), 47.013 (CH3O2+) | |||
| Acetaldehyde | 0.10 ± 0.04 | 0.097 ± 0.001 | 0.15 ± 0.05 | 45.034 (C2H5O+) | |
| Ethyl hydroperoxide | 0.14 ± 0.05 | 45.034 (C2H5O+) | |||
| Perpropionic acid | 0.065 ± 0.043 | n.q. | 91.040 (C3H7O3+), 75.045 (C3H7O2+) | ||
| Propionic acid | 0.17 ± 0.03 | ||||
| Propionaldehydee | 59.050 (C3H7O+) | ||||
| Methyl ketenee | 57.034 (C3H5O+) | ||||
| CO2f | 0.90 ± 0.21 | 0.54 ± 0.08 | |||
| sCI | 0.40 ± 0.09 | ||||
| Detection method | FTIR | LC-UV | FTIR | FTIR | PTR-MS |
| Ref. | This work | Grosjean et al.16 | O’Dwyer et al.18 | This work | This work |
a. EVK + O3 in the presence of SO2
The FTIR spectra show unambiguously the formation of formaldehyde (HCHO), HC(O)OH, propionic acid, acetaldehyde and carbon dioxide (CO2). In all cases, the yield plots are highly linear in the initial stages of the ozonolysis reaction, where the product formation is large, suggesting their formation as first-generation closed-shell products. For HC(O)OH, a non-linear behaviour is observed at long reaction times (Fig. 2), which is likely the result of a significant wall loss at higher HC(O)OH concentrations. The reported errors (Table 1) are a combination of the 2σ statistical error and the overall accuracy of the measurements. Due to the usage of carbon monoxide, CO, as an OH radical scavenger, the reported CO2 yield consists of CO2 formed from both the ozonolysis reaction and from the reaction of CO with OH radicals.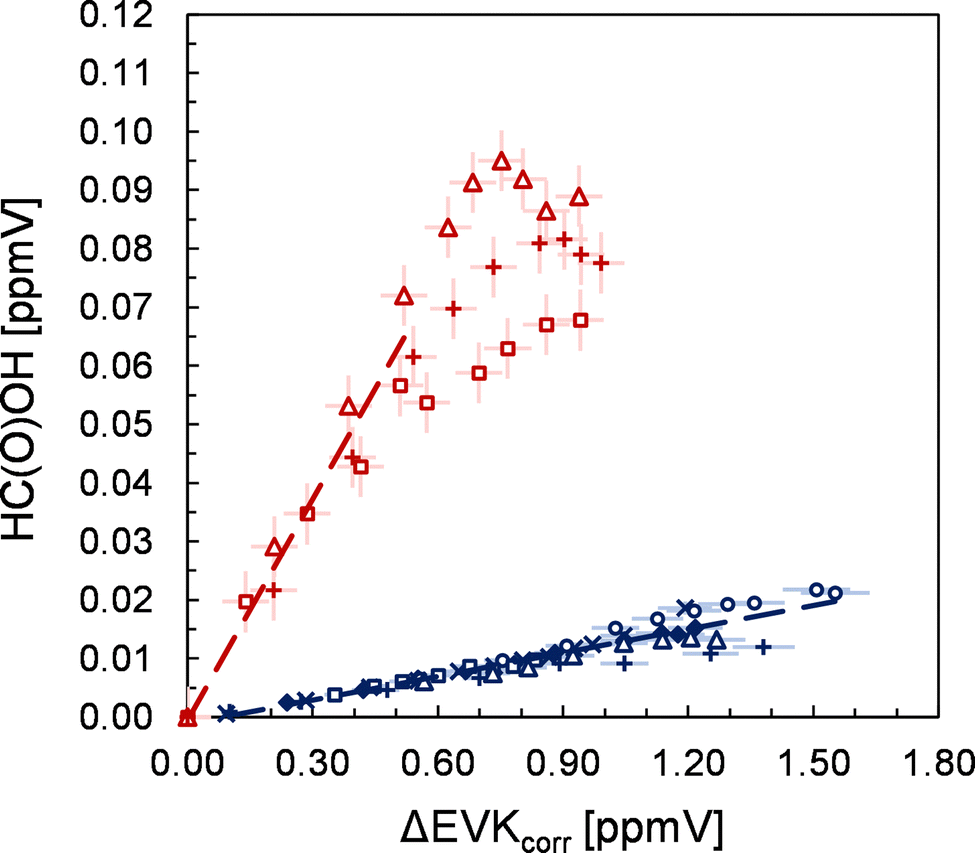 | ||
| Fig. 2 Yield plots obtained for formic acid in the presence (red) and absence (blue) of SO2. Different experimental runs are denoted using different symbols. The error bars represent a precision error as a combination of a relative error plus the corresponding detection limit under the experimental conditions. | ||
Up to 20% of SO2 initially added is consumed during the EVK + O3 reaction. A plot of the consumed SO2vs. the consumed EVK (Fig. S1, ESI†) yields a slope of 0.40 ± 0.09, where the error represents the overall accuracy as mentioned above. The results of different experimental runs were reproducible within 4%. Since only the overtone of SO2 in the range 2530–2440 cm−1 could be used for the evaluation of the FTIR spectra, we prefer to assign an expanded uncertainty of 20%.
The decomposition of the primary ozonide (POZ), which is formed through the 1,3-dipolar cycloaddition according to the well-established ozonolysis mechanism,25 is expected to proceed via two pathways, forming either HCHO and a carbonyl-substituted Criegee intermediate (CI) or 2-oxobutanal and formaldehyde oxide, CH2OO. The consumption of SO2 during the ozonolysis reaction indicates the formation of stabilized CIs in the reaction system. Since the CH2OO + SO2 reaction results mainly in the production of formaldehyde,26 the HCHO yield observed in the presence of SO2 represents the sum of the primary HCHO and the bimolecularly formed HCHO.
Theoretical calculations by Vereecken et al.27 suggest that for a large fraction of stabilized CIs, unimolecular loss might be still faster than bimolecular reactions. Based on the SAR approach presented in the same publication,27 the rate coefficients for the unimolecular loss (1,3-cyclisation) are calculated to be 0.3 s−1 (CH2OO), 0.04 s−1 (E-C2H5C(O)CHOO), and 20 s−1 (Z-C2H5C(O)CHOO) for the sCIs possible in the present reaction system. Since rate coefficients for the sCI + SO2 reaction are expected to be in the range 10−11–10−10 cm3 s−1,27 it is likely that unimolecular loss is not competitive with the SO2 reaction under the present experimental conditions. In addition, the linearity of the plots (Fig. S1, ESI†) over the whole experimental duration suggests that the level of SO2 was sufficiently high to suppress the reaction of sCIs with the acids formed in the reaction system. Nevertheless, we prefer the sCI yield obtained here to be regarded as a lower limit.
Trace (a) in Fig. 3 shows a residual FTIR spectrum of an experiment after the subtraction of all undoubtedly identified species mentioned above. In all experiments with added SO2, a non-linear baseline shift is observed, which strongly indicates the formation of particles in the experimental system. This can be rationalized through the size-dependent scattering of IR radiation when particle diameters are similar to the wavenumber of the radiation and the particle concentration is sufficiently high. The particle-based scattering lowers the IR radiation intensity entering the detector of the spectrometer. Hence, a broad absorption becomes visible in the FTIR spectra. Given that this effect is not observed in the absence of SO2, this is interpreted as the formation of H2SO4 due to the reaction of SO3, formed from the reaction of SO2 with sCIs, with a trace amount of H2O present in the reaction system.
Trace (b) shows the same residual spectrum after applying a baseline correction. Since all other identified reaction products were subtracted at this point and secondary reactions were largely minimized due to the addition of SO2, the residual absorption bands visible in the range of 3500–1600 cm−1 should correspond mainly to 2-oxobutanal. For comparison, trace (c) shows a reference spectrum of gas-phase methyl glyoxal, which possesses characteristic absorption features for the CO stretching vibrations centred at 1730 cm−1 and for the C–H stretching vibration of the aldehyde moiety in the range 2860–2800 cm−1. Both position and relative intensity of these absorption bands are similarly found in the residual spectrum in panel (b). Additionally, the absorption pattern around 3000 cm−1 (C–H stretching vibration of alkyl groups) is quite similar to the gas-phase FTIR spectrum of propionaldehyde presented in panel (d). This all supports the assignment of the residual spectrum to 2-oxobutanal.
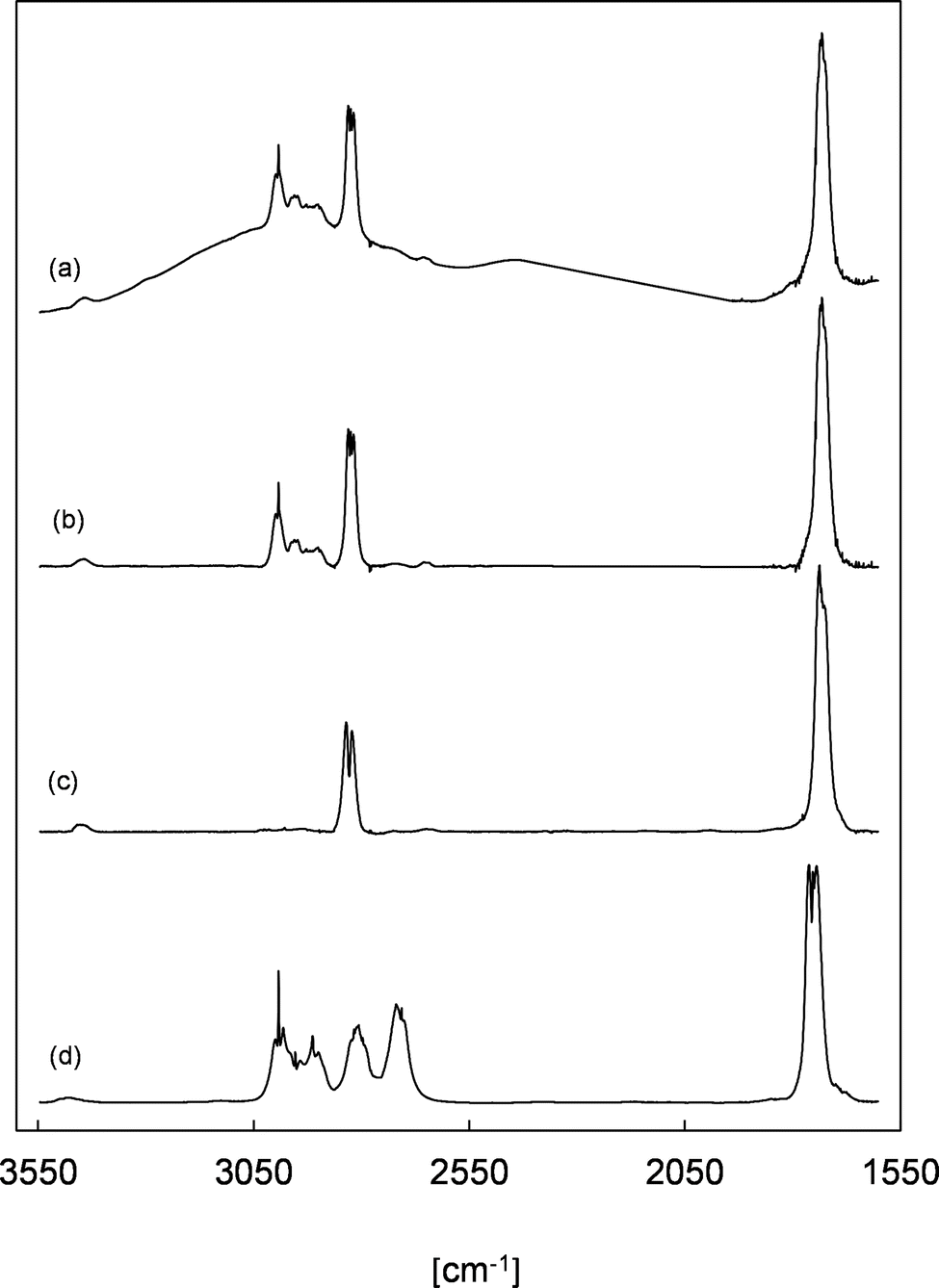 | ||
| Fig. 3 (a) Residual spectrum assigned to 2-oxobutanal (ethyl glyoxal) from an EVK + O3 experiment in the presence of CO used as an OH radical scavenger and SO2 used as a sCI scavenger (the spectrum was cut in the range of 2400–1900 cm−1), (b) spectrum (a) after baseline correction, (c) reference spectrum of methyl glyoxal, and (d) reference spectrum of propionaldehyde. | ||
Taking into account that absorption cross sections of characteristic vibrations usually do not show a large variability for structurally similar compounds, a quantitative FTIR spectrum of 2-oxobutanal was obtained by applying the infrared cross section of the methyl glyoxal CO absorption band28 to the baseline-corrected residual spectrum, which allowed to calculate the dicarbonyl yield as listed in Table 1.
b. EVK + O3 in the absence of SO2
In the absence of SO2, the FTIR spectra show the formation of HCHO, 2-oxobutanal, HC(O)OH, formic anhydride (FA), formic propionic anhydride (FPA), acetaldehyde, ethyl hydroperoxide and CO2. Through separate experiments, in which we irradiated propionaldehyde/methanol/Cl2 mixtures, we obtained reference spectra of gas-phase perpropionic acid (Fig. 4, see the ESI† for details). The characteristic absorption band of the O–H stretching vibration centred on about 3303 cm−1 allowed us to identify the peracid as a reaction product of the EVK + O3 reaction. Applying available cross sections for peracetic acid29 provides an estimate of the perpropionic acid yield as listed in Table 1. Due to this semi-quantitative approach, an expanded uncertainty of 50% was added to the statistical error.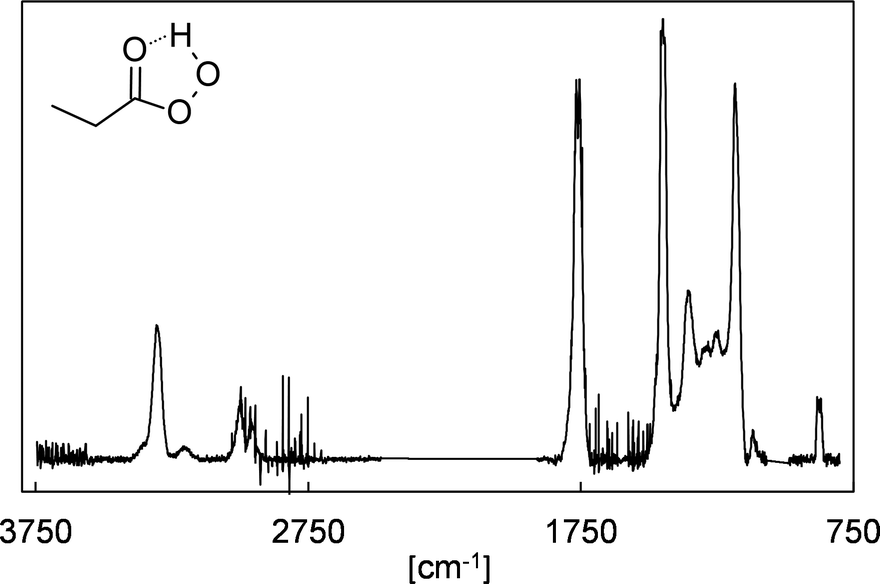 | ||
| Fig. 4 Gas-phase infrared spectrum assigned to perpropionic acid (peroxypropionic acid), CH3CH2C(O)OOH, obtained from irradiating a propionaldehyde/methanol/Cl2-mixture. The spectrum was baseline-corrected and cut in the range of 2400–1900 cm−1 and 1070–990 cm−1. | ||
The formation of all organic species is further supported by the presence of characteristic signals in the PTR mass spectra (see the ESI,† for details). The yield plot of FA, combining the FTIR data of all experimental runs, is shown in panel (a) of Fig. 5. In each experiment, the correlation is linear, but different slopes are observed, which is reflected in the large error assigned to the FA yield. In fact, when considering all experimental data, the plot suggests a larger FA yield at higher EVK consumption levels, hence at long reaction times.
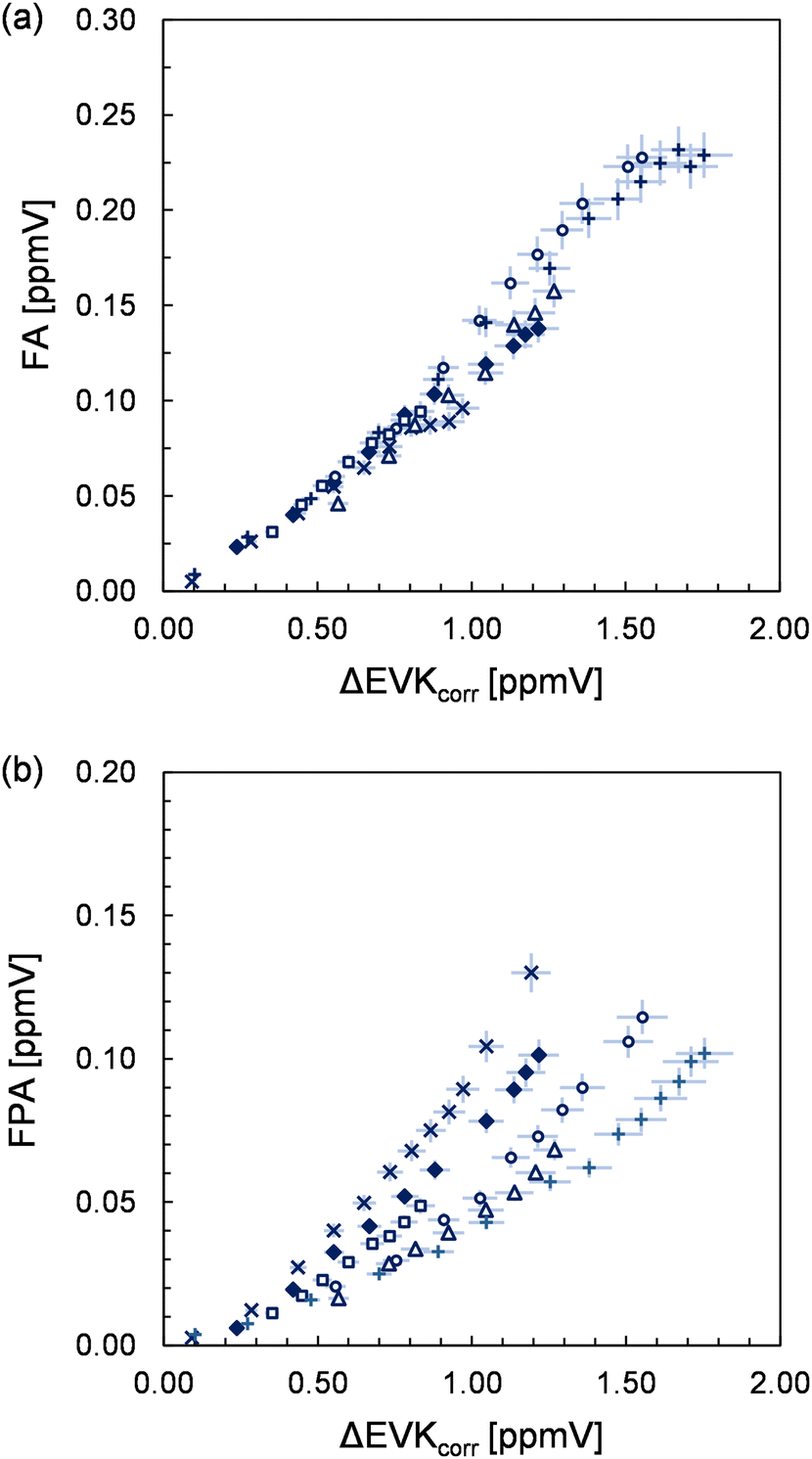 | ||
| Fig. 5 Yield plots obtained for (a) formic anhydride (FA) and (b) formic propionic anhydride (FPA). Different experimental runs are denoted using different symbols. The error bars represent a precision error as a combination of a relative error plus the corresponding detection limit under the experimental conditions. | ||
As presented in panel (b) of Fig. 5, the FPA data of each single experiment do not exhibit significant scatter, indicating a small precision error in the FPA quantification via the FTIR spectra. The profiles of all experimental runs evidence a non-linear behaviour suggesting also an increase in the FPA yield with longer reaction times. It appears that although the profiles are precisely non-linear, significant differences are found for correlations of different experimental runs.
However, when the FPA yields are determined from the data at higher EVK consumption levels, they are found to be reproducible within a factor of 1.2 with the exception of one experiment. This argues against systematic errors in the FPA quantification in different experimental runs. Thus, it appears that on the one hand, both anhydrides are formed delayed (although this is less obvious in the case of FA) and hence do not result directly from the CI further reactions. On the other hand, the delay seems to differ from one to the other experiment in the case of formic propionic anhydride.
FA cannot result directly from the ozonolysis reaction itself, which is indicated by the yield plot (Fig. 5) as discussed above. Its formation is expected to proceed through the bimolecular reaction of formaldehyde oxide with formic acid resulting in the generation of hydroperoxymethyl formate (HPMF), as observed in previous studies.30,31 The spectral features observed here do not support the existence of HPMF. However, it was reported that HPMF decomposition leads to H2O and formic anhydride,30,31 which is, therefore, indirect evidence for HC(O)OH formation in the system. Nearly all formic acid is consumed via the fast reaction of HC(O)OH + sCI. This is confirmed, on the one hand, by the large increase of the HCHO yield in the presence of SO2, indicating a large fraction of thermally equilibrated formaldehyde oxide prone to bimolecular reactions. On the other hand, the consumption of HC(O)OH through sCIs is being suppressed in the presence of SO2, which leads to the observed substantial increase in the HC(O)OH yield. Since both formic acid and HPMF influence the formation of FA, the FA yield is highly sensitive to the further fate of both precursor species. This supposedly explains the variation observed for the FA yield (Fig. 5). For instance, if HC(O)OH exhibits a significantly larger wall loss in one experiment, this will result necessarily in a lowering of HPMF and consequently in a lower FA yield. This appears to be even more important in the case of FPA. Since the yield plots do unambiguously suggest its formation from secondary reactions, we can only imagine a reaction sequence in analogy to HC(O)OH + CH2OO to explain the formation of the mixed anhydride. Accordingly, FPA is expected to result from the decomposition of hydroperoxymethyl propionate (HPMP) formed following a reaction of propionic acid with CH2OO. Overall, the formation of both anhydrides and the shape of their yield plots evidence the formation of HC(O)OH and propionic acid in the EVK + O3 reaction.
c. Comparison
The primary carbonyl yields observed in the absence of SO2 suggest a 1:1 ratio for the POZ decomposition channels. However, this would imply that in the present investigation, the POZ decomposition accounts solely for about 57 ± 14% of the EVK + O3 reaction. The experimental results do not provide any hint for other reactions, e.g. the formation of an epoxide. At the same time, the increase of the HCHO + 2-oxobutanal yield employing large SO2 concentrations is significantly larger than can be explained by sCI + SO2 reactions. We do not find any conclusive explanation for this behaviour (the error analysis is provided in Section E of the ESI†) and can only speculate that an unidentified secondary consumption of the primary carbonyls exists in the experimental system in the absence of any sCI scavenger.
The gas-phase ozonolysis of EVK has been formerly investigated by Grosjean et al.16 and O’Dwyer et al.18 Grosjean and co-workers16 reported product yields of 0.548 ± 0.069 and 0.444 ± 0.025 for HCHO and 2-oxobutanal, respectively. The quantification of the carbonyls was done by liquid chromatography, after the collection of samples onto DNPH-coated cartridges.16 Both the HCHO and 2-oxobutanal yields we determined are significantly lower than those reported by Grosjean et al.16 In contrast to the present work, the former study has been carried out at a relative humidity of 55 ± 10%. Although the bimolecular reaction of formaldehyde oxide with H2O is quite slow14 (≈10−16 cm3 s−1), at least a fraction of the stabilized CH2OO will react with H2O under these experimental conditions. The CH2OO + H2O as well as the CH2OO + (H2O)2 reactions have been shown to form initially the chemically activated hydroxymethyl hydroperoxide, OHCH2OOH,32 which is either thermalized or decomposes into HC(O)OH and H2O or HCHO and H2O2. Therefore, an additional source of HCHO might have been present under the humid conditions of the former study. However, since at least for the reaction with the water dimer, other studies provided contradictory conclusions on the relative importance of each reaction channel,12,14,33 no clear statement can be made. On the other hand, no data are available for the carbonyl-substituted CI. Due to the lack of a standard, Grosjean et al.16 used a response factor estimated from the results of other α-dicarbonyls, where standards were available, for the quantification of 2-oxobutanal. Accordingly, they assigned a 20% relative error to the 2-oxobutanal response factor. Therefore, the relative error of <6% reported for the 2-oxobutanal yield under humid conditions16 is at least surprisingly low.
By contrast, O’Dwyer et al.18 worked at a relative humidity <1%. They reported product yields of 0.37 ± 0.02 and 0.49 ± 0.03 for HCHO and 2-oxobutanal, respectively. The present value for HCHO is, thus, within the assigned uncertainty, identical to that of O’Dwyer et al.18 Similar to the present approach, O’Dwyer and co-workers18 quantified 2-oxobutanal by assuming the cross section of CO absorption to be the same as for a structurally similar compound, namely n-butanal. Following this rationale, they obtained a 2-oxobutanal yield which is about 60% larger than that determined here. Comparing the cross section of the integrated carbonyl absorption band of methyl glyoxal (e.g. Talukdar et al.28) with a value for the CO absorption band of n-butanal from the Wuppertal laboratory database, it is found that the cross section is about ≈65% larger in the case of the α-dicarbonyl. Consequently, the experimentally observed correlation between the integrated carbonyl absorption of 2-oxobutanal and the consumption of EVK is nearly the same in the present study and that of O’Dwyer et al.18 The authors argue solely that the cross section of n-butanal was chosen due to the similarities between the residual spectrum and that of n-butanal. By contrast, they do not state why they expect the intensity of the CO absorption of an α-dicarbonyl to be the same as for an aldehyde containing only one carbonyl group, which is intriguing.
d. Mechanistic interpretation
The formation of perpropionic acid and ethyl hydroperoxide can solely be rationalized through the reaction of their parent RO2 radical with HO2. Accordingly, the level of HO2 in the system must be sufficiently high to allow a competition between RO2 + HO2 and RO2 + RO2 reactions. This is likely the result of OH radicals formed from the EVK + O3 reaction, which in the presence of an excess of CO are converted nearly quantitatively into HO2 radicals, or the result of H atoms following CI fragmentation.At the same time, the hydroperoxides evidence the formation of both propionyl peroxy, C2H5C(O)O2, and ethyl peroxy radicals, C2H5O2. Following available literature data,34–37 the presence of C2H5C(O)O2, C2H5O2 and HO2 suggests the RO2 reaction sequence to evolve in the reaction system as presented in Fig. 6. This in turn accounts for the acetaldehyde formation through RO2 permutation reactions of the ethyl peroxy radical.35 The acetaldehyde yield observed in the absence of SO2 is in good agreement with the value reported by Grosjean et al.16 Since RO2 reactions were not considered in the former study, the authors did not provide a conclusive explanation for acetaldehyde formation.
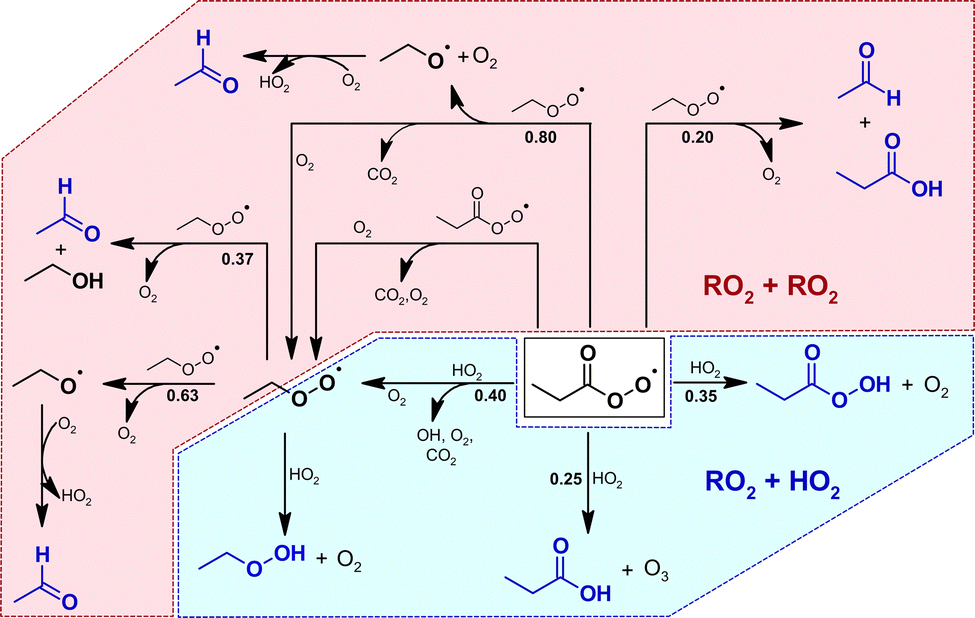 | ||
| Fig. 6 Chemistry of the propionyl peroxy and ethyl peroxy radical based on the identified reaction products (blue structures). For clarity, the reaction sequences are only shown considering a prompt propionyl peroxy radical source in the experimental system. The blue area represents the RO2 + HO2 reactions, the rose area marks the RO2 + RO2 domain. The corresponding branching ratios, given in bold numbers, are the current IUPAC recommendations (2021). For the C2H5C(O)O2 + HO2 reaction, the values are taken from Hasson et al.37 | ||
In the presence of SO2, a small curvature around 3300 cm−1 might indicate the formation of perpropionic acid. However, a reliable quantification is not possible due to the baseline shift during the reaction, as discussed above. At the same time, no ethyl hydroperoxide was observed, whereas the acetaldehyde yield increased. This suggests both the RO2 and HO2 levels to be lower when the sCI scavenger is present, which would imply that the RO2 species result partly from sCI.
Simulations on the experiments without SO2 addition were performed considering the reaction sequence shown in Fig. 6. Since ethyl peroxy radicals might result solely from C2H5C(O)O2 further reactions or as well from CI decomposition, we simulated both scenarios for all experiments. The details of this analysis are presented in Section D of the ESI† and only the main conclusions are briefly summarized here.
The temporal evolution of acetaldehyde, ethyl hydroperoxide and perpropionic acid can be reproduced using experimentally plausible HO2 concentrations (2.3 × 1010 cm−3 on average). Accordingly, there is no reason to argue against their time profiles being properly described by the peroxy radical reaction sequence presented in Fig. 6, e.g. an additional source of acetaldehyde. At the same time, it is not possible to conclude whether the ethyl peroxy radical evolves solely from the propionyl peroxy radical further chemistry or also promptly from the CI chemistry. For the latter case, all model runs suggest the C2H5O2 source to be a factor of ≈5 smaller than the C2H5C(O)O2 source with an overall RO2 radical yield below 40%.
The simulations predict an upper limit of 7% for the propionic acid yield in the absence of SO2. The acid appears to be entirely consumed via the reaction with stabilized CH2OO, as discussed above, which finally converts propionic acid into FPA. However, considering the FPA yield of 0.11 ± 0.04 at the end of the experiments, this indicates an additional source of propionic acid. In the presence of SO2, we find a reproducible propionic acid/acetaldehyde ratio of 1.1 ± 0.2, which is more than a factor of 2 larger than possible from the RO2 reaction sequence (see Section D of the ESI† for further analysis). Apparently, there is an additional source for propionic acid in the EVK ozonolysis mechanism besides the RO2-based formation route. In addition, since propionic acid is observed under conditions, where sCI reactions are drastically minimized, this suggests strongly its formation from chemically activated carbonyl oxides.
The formation of stabilized acids was reported for larger CIs following the 1,3-ring closure and subsequent isomerization according to the dioxirane route.38–40 This may be interpreted in terms of a larger probability of collisional stabilization due to a larger number of vibrational modes comparative to smaller CIs, which potentially allows the excess energy to be distributed below a dissociation threshold. However, since the POZ decomposition yields initially a C1- and a C4-fragment, the above mentioned mechanism cannot account for the formation of C3-acid (propionic acid). This, in turn, would result into 2-oxobutanoic acid, for which no evidence is observed in the PTR mass spectra, nor in the FTIR spectra, assuming a similar spectral structure as the infrared spectra of gas-phase pyruvic acid (2-oxopropanoic acid).
Nevertheless, the prompt formation of the C2H5C(O)O2 radical (and possibly of the C2H5O2 radical) and the amount of propionic acid, not accounted for by RO2 further reactions, can only originate from the C4-fragment, namely the carbonyl-substituted CI. The carbonyl group in α-position prohibits the possibility of an 1,4-H shift isomerization and the intermediate formation of a vinyl hydroperoxide. Therefore, this CI is expected to undergo mainly an isomerization following a 1,3-ring closure (dioxirane route), which yields probably the vibrationally excited oxobutanoic acid, [C2H5C(O)C(O)OH]*, irrespective of the E- or Z-configuration of the CI (reaction (R6a)). This implies that both the propionyl peroxy radical and the subsequent reaction products as well as propionic acid, not accounted for by RO2 reactions, arise from the decomposition of [C2H5C(O)C(O)OH]*. Alternatively, the carbonyl-substituted CI might isomerize into vibrationally excited formic propionic anhydride, [C2H5C(O)OC(O)H]*:
| [C2H5C(O)CHOO]* → [C2H5C(O)C(O)OH]* | (R6a) |
| [C2H5C(O)CHOO]* → [C2H5C(O)OC(O)H]* | (R6b) |
One should note that at least some of the reaction products could also result from the decomposition of the excited anhydride. However, it is unclear to which extent the migration of a C2H5C(O) group competes with the migration of an H atom in the intermediately formed bis(oxy) biradical. Therefore, since a direct detection of the excited intermediates (both acid and anhydride) is not possible, we postulate the reaction products to arise from the decomposition of the oxoacid.
Fragmentation routes of vibrationally excited acids were reported only for [HC(O)OH]* and [CH3C(O)OH]*, although the data level remains relatively scarce14 and no such data are available for substituted acids. Based on the observed reaction products, we propose here 7 possible distinct fragmentation routes for the vibrationally excited oxobutanoic acid. Accordingly, 4 fragmentation pathways are following a bond cleavage, which, in turn, result in the formation of the radical species confirmed by the observed products. The bond scission may occur either between the H and O atom of the OH group or between the OH and the carbonyl group. The remaining radical will readily eliminate either CO2 or CO. However, in both cases, a propionyl radical is formed, which, in the presence of O2, is immediately converted into a propionyl peroxy radical.
| [C2H5C(O)C(O)OH]* → C2H5C(O) + CO2 + H | (R7a) |
| [C2H5C(O)C(O)OH]* → C2H5C(O) + CO + OH | (R7b) |
Theoretically, fragmentation might also occur by scission of the bond between both carbonyl groups yielding, in the presence of O2, the same products as reaction (R7a), namely propionyl peroxy + CO2 + HO2.
| [C2H5C(O)C(O)OH]* → C2H5C(O) + C(O)OH | (R7c) |
| [C2H5C(O)C(O)OH]* → C2H5 + CO + C(O)OH | (R7d) |
As discussed above, it is presently not clear, if ethyl peroxy radicals are formed promptly from the EVK + O3 reaction or result only from the propionyl peroxy radical further chemistry. Regardless of the relative branching ratios for the above-mentioned decomposition channels, the formation of propionyl peroxy radicals would dominate, as concluded from the experimental results.
Since an additional source of propionic acid is necessarily related also to the chemically activated carbonyl-substituted CI, propionic acid appears to arise from a molecular fragmentation pathway.
| [C2H5C(O)C(O)OH]* → C2H5C(O)OH + CO | (R7e) |
Another possible molecular fragmentation route were the elimination of CO2 and the consecutive formation of propionaldehyde.
| [C2H5C(O)C(O)OH]* → C2H5C(O)H + CO2 | (R7f) |
A molecular fragmentation route similar to the ketene formation as observed in the case of the vibrationally excited acetic acid might also occur. Accordingly, one would expect the formation of methyl ketene and HC(O)OH.
| [C2H5C(O)C(O)OH]* → CH3CHCO + HC(O)OH | (R7g) |
In principle, the formation of formic acid is also possible from the CH2OO + SO2 reaction. Hatakeyama et al.41 suggested this reaction to proceed initially via the reversible formation of an adduct, which decomposes into HC(O)OH and SO2. It was further shown that higher SO2 levels lower the HC(O)OH yield. Theoretical calculations indicate that the further fate of the initially formed adduct evolves mainly into HCHO + SO3, whereas only 17% result into the formation of a singlet bis(oxy) biradical.26 This, in turn, might isomerize and yield at least partly stabilized formic acid. Accordingly, a contribution of CH2OO + SO2 to the overall HC(O)OH yield is actually possible. However, since there is strong evidence for formic acid formation in the absence of SO2 (formic acid and FA), this cannot be the major source of formic acid.
Aside from that, the chemically activated fraction of CH2OO is expected to isomerize into vibrationally excited formic acid, [HC(O)OH]*, for which decomposition was reported exclusively.14 The only closed-shell reaction products are CO and CO2.14 The latter one therefore contributes to the CO2 yield observed. However, due to the unexpectedly high HC(O)OH yield found here, it appears possible that [HC(O)OH]* is partly stabilized. This may be interpreted in terms of a low vibrational excitation that allows a competition between decomposition and collisional stabilization.
Overall, the HC(O)OH observed in both experimental set-ups (FA and HC(O)OH in the absence of SO2, HC(O)OH in the presence of SO2) proves its formation from chemically activated CIs. However, it does not prove unequivocally any of the above discussed formation routes. The overall ozonolysis mechanism suggested for EVK based on our findings is presented in Fig. 7.
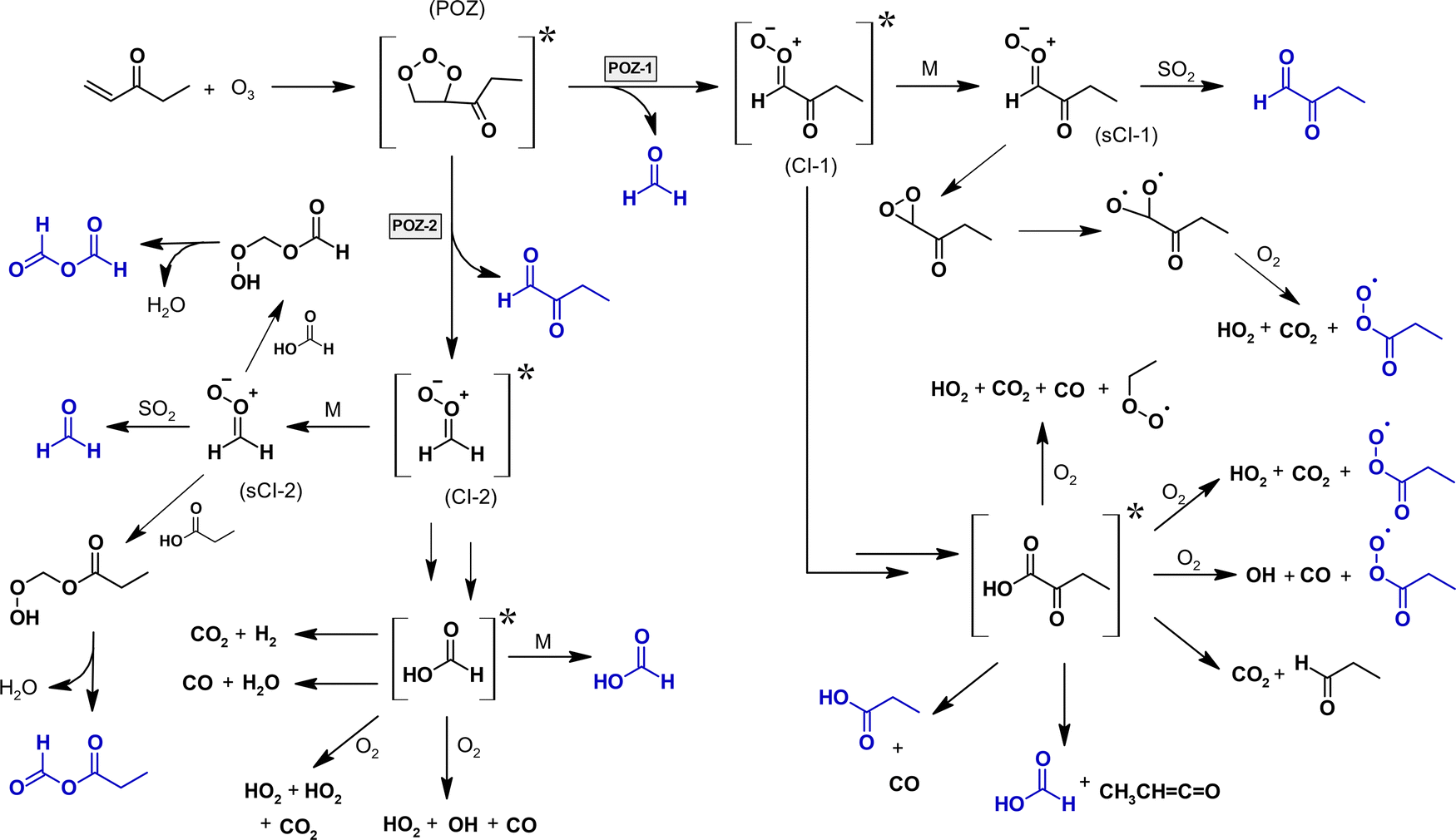 | ||
| Fig. 7 Mechanism of the EVK + O3 reaction built on the results of the present investigation. The structures are marked in blue for species quantified within this work and for radical species, whose presence is indirectly proven by the quantification of subsequently formed species. For clarity, only the E-isomer is drawn for the carbonyl-substituted CI. The pathways of (CI-1) are developed in detail in the text. | ||
Conclusions
The present work revealed that the ozonolysis reaction of EVK in the gas-phase is a source of formic and propionic acid with an overall carboxylic acid yield of about 29 ± 7% under these experimental conditions. Bimolecular reactions of the propionyl peroxy radical did contribute to the formation of propionic acid. However, our analysis revealed that >50% of the propionic acid observed is not accounted for by RO2 reactions. We proposed three different pathways, as summarized in Fig. 8 explaining the formation of both formic and propionic acid.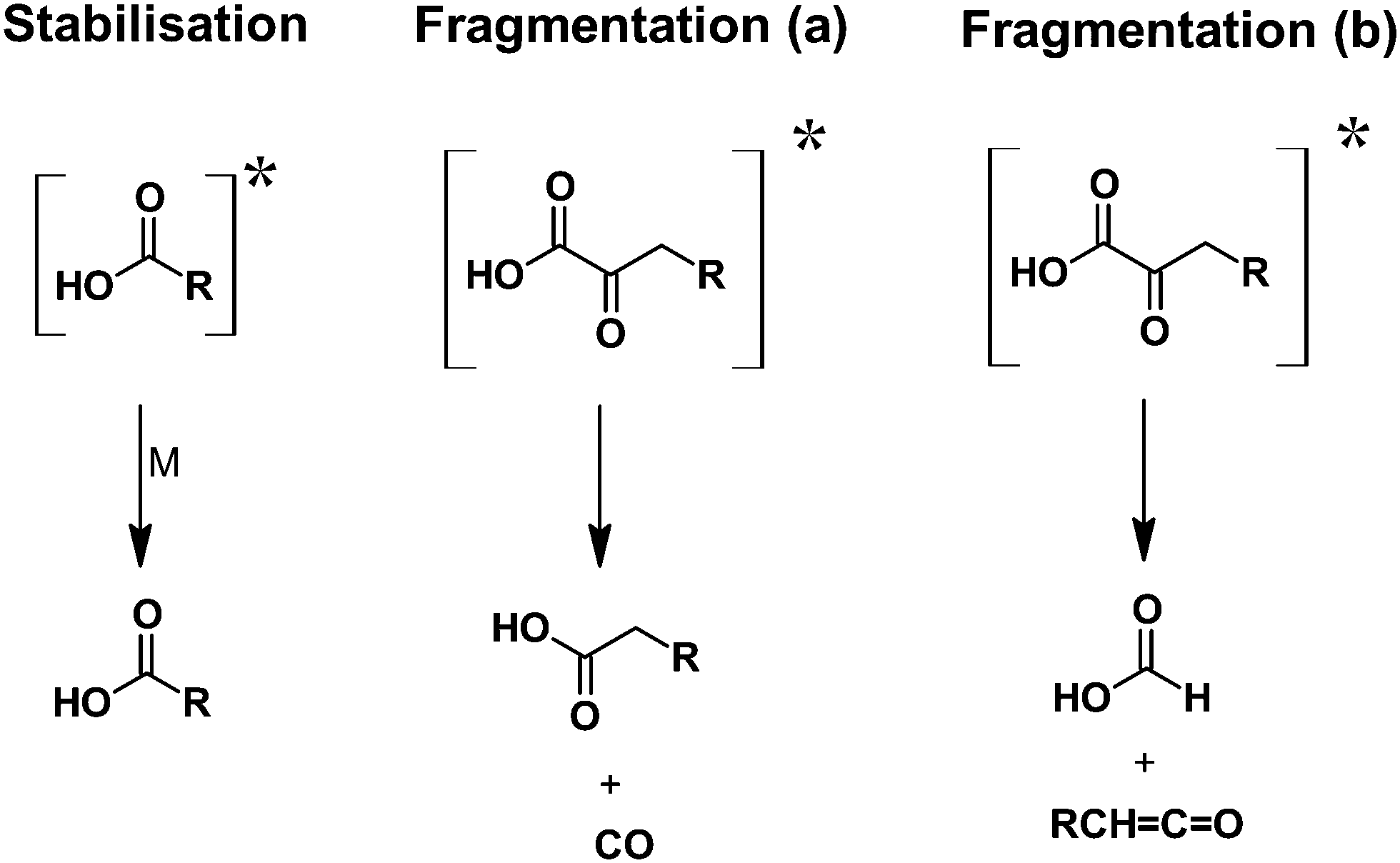 | ||
| Fig. 8 Acid-forming pathways proposed within this work from chemically activated acids. | ||
The SO2 consumption indicated that about 40 ± 9% of the CIs formed in the EVK + O3 reaction are stabilized and hence prone to bimolecular reactions, particularly with H2O and (H2O)2 under tropospheric conditions. Irrespective of the pathway yielding HC(O)OH, the present study provides strong evidence that both acids are formed from chemically activated CIs. Accordingly, both formic and propionic acid should also be formed under the humid conditions present in the troposphere.
The impact on the acid budget might be limited in the case of EVK. However, this will change dramatically if the same mechanism applies to MVK ozonolysis (accordingly yielding formic and acetic acid). Under low-NO conditions, the OH-initiated oxidation of isoprene results in about 50% MVK.42 As stated in the introduction, under typical daytime conditions about 17% of MVK is consumed via the ozonolysis reaction. Considering the yearly emission strength of isoprene43 and assuming a similar overall carboxylic acid yield from MVK + O3, the acid production might be as high as about 4 Tg a−1 for HC(O)OH and 7 Tg a−1 for acetic acid. Clearly, there is an urgent need for better characterization of the MVK ozonolysis reaction, which will be covered in follow-up work.44
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG) through the grant agreement WI 958/18-1.References
- A. Chebbi and P. Carlier, Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- P. Khare, N. Kumar, K. M. Kumari and S. S. Srivastava, Atmospheric formic and acetic acids: an overiew, Rev. Geophys., 1999, 37, 227–248 CrossRef CAS.
- S. Yu, Role of organic acids (formic, acetic, pyruvic and oxalic) in the formation of cloud condensation nuclei (CCN): a review, Atmos. Res., 2000, 53, 185–217 CrossRef CAS.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. González Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. de Gouw, E. J. Dunlea, M. De Mazière, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11, 1989–2013 CrossRef CAS PubMed.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, A large and ubiquitous source of atmospheric formic acid, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef CAS.
- S. Chaliyakunnel, D. B. Millet, K. C. Wells, K. E. Cady-Pereira and M. W. Shephard, A Large Underestimate of Formic Acid from Tropical Fires: Constraints from Space-Borne Measurements, Environ. Sci. Technol., 2016, 50, 5631–5640 CrossRef CAS PubMed.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, J. H. Kroll, J. H. Seinfeld and P. O. Wennberg, Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 2009, 9, 1479–1501 CrossRef CAS.
- N. I. Butkovskaya, N. Pouvesle, A. Kukui and G. Le Bras, Mechanism of the OH-Initiated Oxidation of Glycolaldehyde over the Temperature Range 233–296 K, J. Phys. Chem. A, 2006, 110, 13492–13499 CrossRef CAS PubMed.
- N. I. Butkovskaya, N. Pouvesle, A. Kukui, Y. Mu and G. Le Bras, Mechanism of the OH-Initiated Oxidation of Hydroxyacetone over the Temperature Range 236–298 K, J. Phys. Chem. A, 2006, 110, 6833–6843 CrossRef CAS PubMed.
- S. So, U. Wille and G. da Silva, Atmospheric Chemistry of Enols: A Theoretical Study of the Vinyl Alcohol + OH + O2 Reaction Mechanism, Environ. Sci. Technol., 2014, 48, 6694–6701 CrossRef CAS PubMed.
- J. S. Francisco and W. Eisfeld, Atmospheric oxidation mechanism of hydroxymethyl hydroperoxide, J. Phys. Chem. A, 2009, 113, 7593–7600 CrossRef CAS PubMed.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Miller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 2016, 18, 10241–10254 RSC.
- S. Wang, M. J. Newland, W. Deng, A. R. Rickard, J. F. Hamilton, A. Muñoz, M. Ródenas, M. M. Vázquez, L. Wang and X. Wang, Aromatic Photo-oxidation, A New Source of Atmospheric Acidity, Environ. Sci. Technol., 2020, 54, 7798–7806 CrossRef CAS PubMed.
- R. A. Cox, M. Ammann, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, A. Mellouki, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 2020, 20, 13497–13519 CrossRef CAS.
- R. Fall, T. Karl, A. Jordan and W. Lindinger, Biogenic C5 VOCs: release from leaves after freeze-thaw wounding and occurrence in air at high mountain observatory, Atmos. Environ., 2001, 35, 3905–3916 CrossRef CAS.
- E. Grosjean, D. Grosjean and J. H. Seinfeld, Gas-Phase Reaction of Ozone with Trans-2-Hexenal, Trans-2-Hexenyl Acetate, Ethylvinyl Ketone, and 6-Methyl-5-Hepten-2-One, Int. J. Chem. Kinet., 1996, 28, 373–382 CrossRef CAS.
- Y. Ren, B. Grosselin, V. Daële and A. Mellouki, Investigation of the reaction of ozone with isoprene, methacrolein and methyl vinyl ketone using the HELIOS chamber, Faraday Discuss., 2017, 200, 289–311 RSC.
- M. A. O’Dwyer, T. J. Carey, R. M. Healy, J. C. Wenger, B. Picquet-Varrault and J. F. Doussin, The Gas-phase Ozonolysis of 1-Penten-3-ol, (Z)-2-Penten-1-ol and 1-Penten-3-one: Kinetics, Products and Secondary Organic Aerosol Formation, Z. Phys. Chem., 2010, 224, 1059–1080 CrossRef.
- A. Mellouki, M. Ammann, R. A. Cox, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms ( ≥ C4), Atmos. Chem. Phys., 2021, 21, 4797–4808 CrossRef CAS.
- J. N. Illmann, I. Patroescu-Klotz and P. Wiesen, Gas-phase reactivity of acyclic α,β-unsaturated carbonyls towards ozone, Phys. Chem. Chem. Phys., 2021, 23, 3455–3466 RSC.
- M. B. Blanco and M. A. Teruel, Atmospheric photodegradation of ethyl vinyl ketone and vinyl propionate initiated by OH radicals, Chem. Phys. Lett., 2011, 502, 159–162 CrossRef CAS.
- D. Grosjean, E. Grosjean and E. L. Williams, II, Rate constants for the gas-phase reactions of ozone with unsaturated alcohols, esters, and carbonyls, Int. J. Chem. Kinet., 1993, 25, 783–794 CrossRef CAS.
- N. Illmann, R. G. Gibilisco, I. G. Bejan, I. Patroescu-Klotz and P. Wiesen, Atmospheric oxidation of α,β-unsaturated ketones: kinetics and mechanism of the OH radical reaction, Atmos. Chem. Phys., 2021, 21, 13667–13686 CrossRef CAS.
- N. Illmann, I. Patroescu-Klotz and P. Wiesen, Biomass burning plume chemistry: OH-radical-initiated oxidation of 3-penten-2-one and its main oxidation product 2-hydroxypropanal, Atmos. Chem. Phys., 2021, 21, 18557–18572 CrossRef CAS.
- R. Criegee, Mechanism of Ozonolysis, Angew. Chem., 1975, 14, 745–752 CrossRef.
- L. Vereecken, H. Harder and A. Novelli, The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- L. Vereecken, A. Novelli and D. Taraborrelli, Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- R. K. Talukdar, L. Zhu, K. J. Feierabend and J. B. Burkholder, Rate coefficients for the reaction of methylglyoxal (CH3COCHO) with OH and NO3 and glyoxal (HCO)2 with NO3, Atmos. Chem. Phys., 2011, 11, 10837–10851 CrossRef CAS.
- J. J. Orlando, G. S. Tyndall, L. Vereecken and J. Peeters, The Atmospheric Chemistry of the Acetonoxy Radical, J. Phys. Chem. A, 2000, 104, 11578–11588 CrossRef CAS.
- P. Neeb, O. Horie and G. K. Moortgat, The nature of the transitory product in the gas-phase ozonolysis of ethene, Chem. Phys. Lett., 1995, 246, 150–156 CrossRef CAS.
- P. Neeb, O. Horie and G. K. Moortgat, Gas-Phase Ozonolysis of Ethene in the Presence of Hydroxylic Compounds, Int. J. Chem. Kinet., 1996, 28, 721–730 CrossRef CAS.
- P. Neeb, F. Sauer, O. Horie and G. K. Moortgat, Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour, Atmos. Environ., 1997, 31, 1417–1423 CrossRef.
- L. Sheps, B. Rotavera, A. J. Eskola, D. L. Osborn, C. A. Taatjes, K. Au, D. E. Shallcross, M. A. H. Khan and C. J. Percival, The reaction of Criegee intermediate CH2OO with water dimer: primary products and atmospheric impact, Phys. Chem. Chem. Phys., 2017, 19, 21970–21979 RSC.
- A. S. Hasson, G. S. Tyndall and J. J. Orlando, A Product Yield Study of the Reaction of HO2 Radicals with Ethyl Peroxy (C2H5O2), Acetyl Peroxy (CH3C(O)O2), and Acetonyl Peroxy (CH3C(O)CH2O2) Radicals, J. Phys. Chem. A, 2004, 108, 5979–5989 CrossRef CAS.
- J.-P. Le Crâne, E. Villenave, M. D. Hurley, T. J. Wallington and J. C. Ball, Atmospheric Chemistry of Propionaldehyde: Kinetics and Mechanisms of Reactions with OH Radicals and Cl Atoms, UV Spectrum, and Self-Reaction Kinetics of CH3CH2C(O)O2 Radicals at 298 K, J. Phys. Chem. A, 2005, 109, 11837–11850 CrossRef PubMed.
- A. C. Noell, L. S. Alconcel, D. J. Robichaud, M. Okumura and S. P. Sander, Near-Infrared Kinetic Spectroscopy of the HO2 and C2H5O2 Self-reactions and Cross-Reactions, J. Phys. Chem. A, 2010, 114, 6983–6995 CrossRef CAS PubMed.
- A. S. Hasson, G. S. Tyndall, J. J. Orlando, S. Singh, S. Q. Hernandez, S. Campbell and Y. Ibarra, Branching Ratios for the Reaction of Selected Carbonyl-Containing Peroxy Radicals with Hydroperoxy Radicals, J. Phys. Chem. A, 2012, 116, 6264–6281 CrossRef CAS PubMed.
- T. L. Nguyen, R. Winterhalter, G. Moortgat, B. Kanawati, J. Peeters and L. Vereecken, The gas-phase ozonolysis of β-caryophyllene (C15H24). Part II: A theoretical study, Phys. Chem. Chem. Phys., 2009, 11, 4173–4183 RSC.
- T. L. Nguyen, J. Peeters and L. Vereecken, Theoretical study of the gas-phase ozonolysis of β-pinene (C10H16), Phys. Chem. Chem. Phys., 2009, 11, 5643–5656 RSC.
- R. Winterhalter, F. Herrmann, B. Kanawati, T. L. Nguyen, J. Peeters, L. Vereecken and G. K. Moortgat, The gas-phase ozonolysis of of β-caryophyllene (C15H24). Part I: an experimental study, Phys. Chem. Chem. Phys., 2009, 11, 4152–4172 RSC.
- S. Hatakeyama, H. Kobayashi, Z.-Y. Lin, H. Takagi and H. Akimoto, Mechanism for the Reaction of CH2OO with SO2, J. Phys. Chem., 1986, 90, 4131–4135 CrossRef CAS.
- M. E. Jenkin, J. C. Young and A. R. Rickard, The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 2015, 15, 11433–11459 CrossRef CAS.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- N. Illmann, I. Patroescu-Klotz and P. Wiesen, Methyl vinyl ketone + O3: A Source of Carboxylic Acids in the Troposphere, manuscript in preparation.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03210d |
| This journal is © the Owner Societies 2023 |