Hydrogen evolution reaction of Ben + H2O (n = 5–9) based on density functional theory†
Received
5th October 2022
, Accepted 28th November 2022
First published on 29th November 2022
Abstract
The structural evolution of Ben clusters with n = 5–9, the adsorption energy created by the Ben@H2O (n = 5–9) complex, and the mechanism of the hydrogen evolution reaction of Ben + H2O (n = 5–9) were all studied using DFT calculations based on the PBE0-D3/Def2TZVP level. Excluding the Be7 cluster, the global minimum structures of beryllium clusters with n = 5–9 showed a higher point group pair formation. Be7 clusters’ high point group symmetry is unstable. Be9@H2O released the greatest energy during the complex's creation (−1.45 eV). Exothermic hydrogen evolution occurs in Ben + H2O (n = 5–9), and all transition states, intermediate stages, and products have energies lower than the equilibrium constant (EC). More energy is released when an O–H bond in the Ben@H2O (n = 5–9) complex is broken, and the energy release results in a change in the cluster structure, which is more pronounced in the Be7 + H2O reaction. Interestingly, there are eight transition states in the Be6 + H2O hydrogen evolution reaction, with the second O–H bond break requiring more energy than the first. There are only three transition states in the Be8 + H2O hydrogen evolution reaction, and the reaction energy is the greatest (−4.13 eV).
1. Introduction
Burning fossil fuels produces carbon dioxide emissions, and they are the primary drivers of global warming (coal, oil, and natural gas). Intensification of the greenhouse effect will cause serious consequences to the earth. Carbon dioxide emissions and the search for clean energy to replace fossil fuels have become extremely urgent.1 One of the most perfect renewable energy sources to replace conventional fossil fuels is hydrogen. Due to its high energy density (i.e., a low heating value of ∼120 MJ kg−1), the combustion product of hydrogen is water, and power plants that can use the existing industrial infrastructure or fossil fuel power generation have attracted unprecedented attention around the world.2–4 Separate water molecules are dissociated into H+ and OH−, and then hydrogen is generated,5 which requires 286 KJ mol−1 of energy to generate hydrogen from liquid water.6
Beryllium metal exhibits many excellent properties, such as low density, high stiffness, and a low coefficient of thermal expansion, leading to alloys of beryllium metal that also have many excellent properties and exhibit unique quantum effects as the size of beryllium metal is reduced to the scale of clusters.7–11 Special quantum size effects and high specific surface areas have considerable application potential in the realm of catalysis because of the distinct electronic structure of beryllium clusters. The molecular geometry and characteristics of pure beryllium clusters and doped beryllium clusters have drawn a lot of interest because beryllium clusters have a distinctive electronic structure.12–17 Srinivas et al.12 investigated the geometric structure and electronic properties of small beryllium clusters in the gradient-corrected density functional theory. The advantage of higher multiplicity was found in the low-energy isomers of larger clusters, and the influence of cluster size on the structure and electronic properties was analyzed. Abyaz et al.13,14 used the USPEX method combined with density functional theory (DFT) calculations to obtain the global minimum structure of beryllium (Ben, n = 3–25) clusters. Thermodynamic stability, photoelectric and photocatalytic capabilities, and bonding qualities are taken into account for the most stable clusters. Based on ELF (electronic localization function) analysis, it can be concluded that the Be–Be bond in small clusters is mainly van der Waals type, while the Be–Be bond in large clusters has metal properties. Zhang et al.15 obtained the global minimum structure of carbon-doped Ben(n = 1–12) clusters at the B3PW91/6-311+G (d) level and compared it with that of pure beryllium clusters. The results show that the introduction of carbon doping strengthens the intra-cluster interaction of beryllium clusters. In addition, Be3C and Be8C clusters have special stability and are considered to be magic clusters in the BenC series. Li et al.16 obtained the lowest energy structure of oxygen-doped Ben(n = 1–12) clusters at the B3PW91 level based on density functional theory. The energy and electronic properties of phenoxy clusters were systematically studied using the QCISD(T) method and compared with those of pure Ben+1 clusters. Based on density functional theory, Ge et al.17 determined the minimum energy structure and electronic properties of MgBen (n = 2–12) clusters. It is found that MgBe3 and MgBe9 clusters have a higher binding energy and a larger HOMO–LUMO gap, which make them more stable than adjacent clusters. The stability of Be clusters is decreased by Mg doping, according to an analysis of the electrical characteristics of MgBen (n = 2–12) clusters.
In addition to investigating the lowest energy structure and electronic properties of beryllium clusters and doped beryllium clusters, the adsorption structure of beryllium clusters and doped beryllium clusters for small molecules and their related properties have been explored.18–21 Orlando et al.18 studied the adsorption of hydrogen molecules on (BeO)n (n = 2–7) clusters based on density functional theory. The effect of hydrogen doping on the beryllium cage structure was studied by Naumkin et al.19 Javad et al.20 reported the adsorption energy, structure, energy gap (Eg), charge transfer and electronic properties of several adsorption states of carbon monoxide (CO) on primary, cationic Li−, Li− and two kinds of Li− coated fullerene beryllium oxides (Be16O16, Li+@Be16O16, Li@Be16O16, and 2Li@Be16O16, respectively), and the results have been explained by DFT calculations. As doped, Be atoms effectively improve the NLO reaction, thermodynamics, and hydrogen adsorption properties of Al12N12 nanoclusters, which were found in the study of Mehboob et al.21
Recent studies on two-dimensional structures, clusters and visible-light-driven photocatalytic hydrogen evolution reactions are also effective methods for developing hydrogen energy.22–27 Vos et al.28,29 reported a high level of single-reference and multi-reference ab initio calculations showing that Be4 clusters exhibit very efficient Lewis acids when interacting with conventional Lewis bases such as ammonia, water, or hydrogen fluoride. The corresponding acid–base interactions trigger the sequential dissociation of all Lewis bases. Chakraborty et al.30 investigated the possibility of CO oxidation on the Al12Be cluster, dissociation, and addition of CH3F and C2H5F, decomposition of the NAH bond in NH3, dissociation of NO, and hydrogenation of C2H2.
Based on the above research, we studied the interaction between Ben(n = 5–9) clusters and H2O, including the lowest energy structure of Ben(n = 5–9) clusters, the lowest energy structure of H2O adsorbed by Ben(n = 5–9) clusters, and the hydrogen evolution reaction (HER) of water molecules on the surface of beryllium clusters.
2. Computational methodology
The structure of many isomers of Ben(n = 5–9) clusters was optimized, and the stability of the structure was analyzed by frequency analysis. The adsorption process of H2O is carried out based on the lowest energy structure obtained by the above operation. In the adsorption process, the adsorption sites of H2O were considered based on the symmetry of the lowest energy structure of the cluster. The structures after water molecule adsorption were optimized and frequency analyzed to determine the low-energy cluster adsorption structure. The HER of water molecules on the surface of beryllium clusters is conducted based on the lowest energy structure obtained after adsorption, which serves as the reactant of HER. The TS approach is used to determine the transition state in the hydrogen evolution reaction, and the intrinsic reaction coordinate (IRC) is used to determine the transition state. Considering the unique electronic structure of beryllium clusters, Perdew, Burke, and Ernzerhof (PBE) proposed in 1996 a pure functional,31 and Adamo created a hybrid functional PBE032 density functional theory for simulation calculations. The PBE0 functional uses 25% exchange and 75% correlation weight. Focus is on the weak interaction between water molecules and beryllium clusters, increasing the functional dispersion correction. To make the calculation structure more accurate, the base sets were selected as def2-TZVP.33–35 Finally, all the above operations were performed at the PBE0-D3/def2-TZVP level of Gaussian16.36
To determine the mechanism underlying the hydrogen event, we thoroughly investigated the changes in electron distribution of the two interacting systems. For this purpose, we use the interaction region indicator function (IRI)37 in the Multiwfn38 program to provide the information we are looking for. Visualization of the information is completed by the VMD39 and Gnuplot40 programs.
During the formation of the Ben@H2O (n = 5–9) complex, the adsorption energy is a physical quantity that we are very concerned about. The formula for calculating the adsorption energy is as follows:
| | | Ea = EBen@H2O − (EBen + EH2O) | (1) |
where
Ea is the adsorption energy,
EBe is the total energy of the Be
n clusters,
EH2O is the energy of H
2O, and
EBen@H2O is the total energy of the Be
n cluster after adsorbing water.
The calculation formula for the energy barrier involved in the reaction process is as follows:
where
Eb is the energy barrier,
ETS is the total energy of a transition state (
e.g.: TS-A
1-B
1, TS-B
1-C
1…), and
Er represents the reactant's energy in this transition state. The formula for calculating the reverse energy barrier is as follows:
where
Erb is the reverse energy barrier and
ETS is the total energy of a transition state (
e.g.: TS-A
1-B
1, TS-B
1-C
1…), where
Ep represents the energy generated in this transition state. The calculation formula for the reaction energy of Be
n@H
2O (
n = 5–9) is as follows:
| | | ER = Eproduct − Ereaction | (4) |
where
ER is the reaction energy,
Eproduct is the energy of the reaction product (
e.g.: E
1, I
2, F
3…), and
Ep represents the energy of the reactant in this transition state.
Ereaction represents the energy of the whole reaction reactant (
e.g.: A
1, A
2, A
3…). The formula for calculating the Gibbs free energy is as follows:
| | | G = ε0 + Hcorr − TStot | (5) |
where
ε0 is the total electronic energy,
Hcorr is the thermal correction to enthalpy,
T represents temperature, and
Stot is the entropy.
3. Results and discussion
3.1. Lowest-energy structures of Ben(n = 5–9) clusters
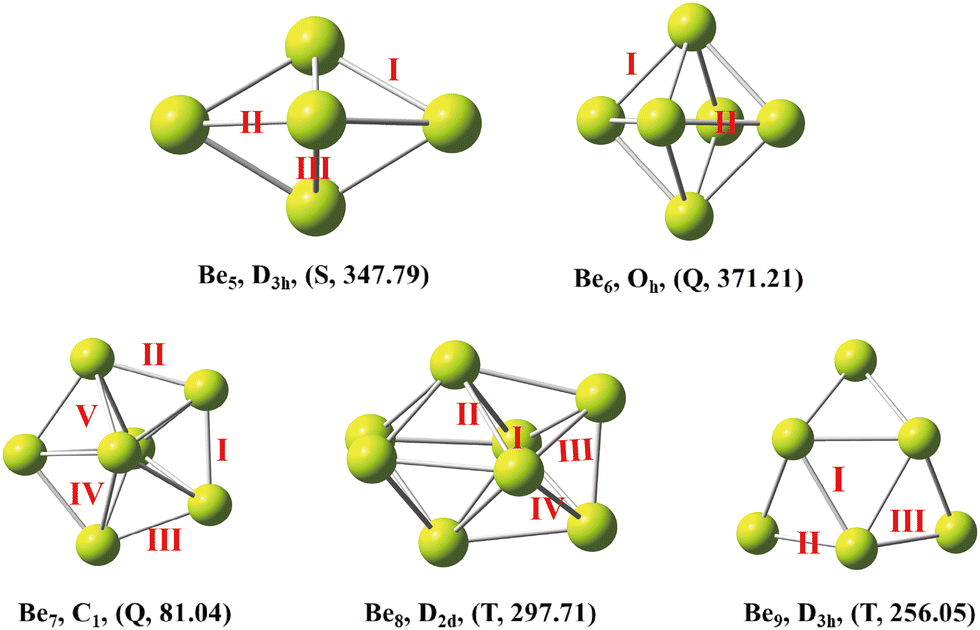
The low-energy isomers of beryllium clusters (n = 5–9) and their symmetry and lowest frequency are shown in Fig. 1. The most stable structure of beryllium clusters and the magnetism of low-energy isomers were further studied. The spin-polarized DFT calculations show that the stable structures of beryllium clusters with n = 6,7 and n = 8,9 are the quintet ground state and triplet ground state, respectively, except that the clusters with n = 5 are favorable for the singlet ground state. These results are summarized in Fig. 1. As shown in Fig. 1, the Be5 cluster has D3h symmetry, and the Be–Be bond lengths are 2.01 Å and 2.06 Å, respectively (for bond length information see Table 1). The Be6 cluster has Oh symmetry, and the Be–Be bond lengths are 2.91 Å and 2.06 Å, respectively. The Be5 cluster has a tripartite structure and Be6 has a diamond structure, which is consistent with the calculation results of Abyaz et al13 and Sulka et al.41 Our calculation results show that Be7 has a pentagonal bipyramid shape with low symmetry, where the Be–Be bond lengths are 2.01 Å to 2.09 Å. It is worth noting that the highest symmetry of the pentagonal bipyramid structure of the Be7 cluster should be D5h, but our calculation results show that the symmetry of D5h is unstable, which is consistent with the results of Abyaz et al13 and Srinivas et al.12 According to our prediction, Be8 has D2d symmetry, and lateral bond lengths in the range of 1.89 Å to 2.72 Å. Be9 is a tricapped trigonal prism with D3h symmetry, in which the average Be–Be bond length in the trigonal prisms is about 2.15 Å.
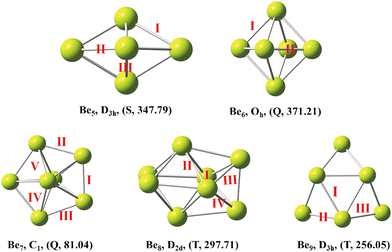 |
| | Fig. 1 The lowest-energy structures of Ben(n = 5–9) clusters with exact symmetries, states (S = singlet, T = triplet and Q = quartet) and minimum vibrational frequency (cm−1) in parentheses. | |
Table 1 The bond length (Å) of the most stable beryllium cluster. The structural performance was calculated using the PBE0-D3 method (see Fig. 1 for different bonds)
| Cluster size n |
I |
II |
III |
IV |
V |
| 5 |
2.01 |
2.06 |
|
|
|
| 6 |
2.91 |
2.06 |
|
|
|
| 7 |
2.05 |
2.03 |
2.02 |
2.01 |
2.09 |
| 8 |
2.72 |
2.18 |
2.05 |
1.98 |
|
| 9 |
2.22 |
2.15 |
2.07 |
|
|
3.2. Ben@H2O (n = 5–9) complexes
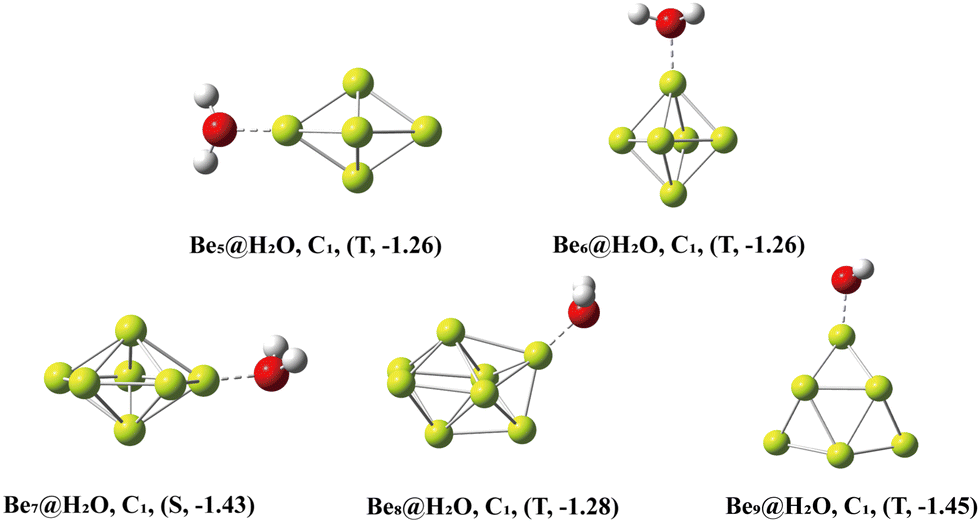
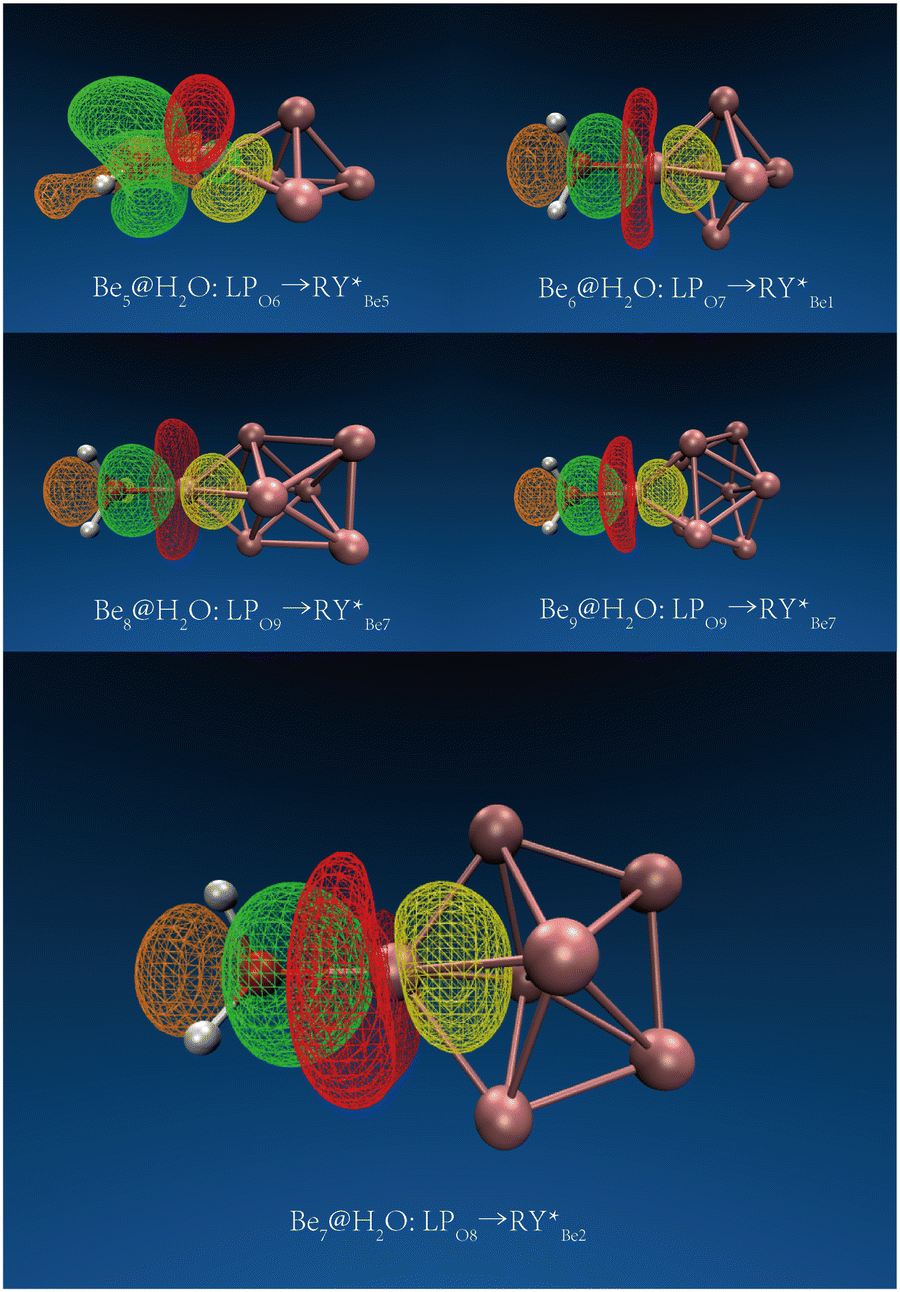
The Ben@H2O (n = 5–9) complex structures formed by beryllium clusters and water molecules are shown in Fig. 2. On the one hand, the adsorption sites of water molecules, the spin multiplicity of the complex and the electron transfer between beryllium clusters and water molecules are discussed using NBO analysis (see Fig. 3 for specific information). Furthermore, the adsorption energy of the adsorption process is also sorted out (see Table 2 for specific information).
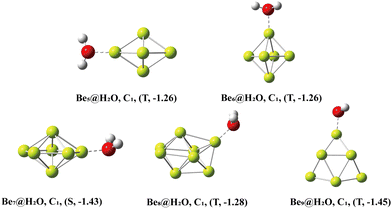 |
| | Fig. 2 The lowest-energy structures of Ben@H2O (n = 5–9) clusters with exact symmetries, states (S = singlet, T = triplet and Q = quartet) and adsorption energy (eV) in parentheses. | |
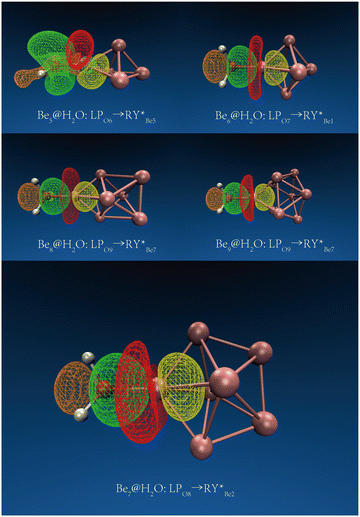 |
| | Fig. 3 The main NBO donor–acceptor overlaps between H2O and Ben (n = 5–9). The orbit and direction of electron transfer. | |
Table 2 Adsorption and reaction energy of Ben (n = 5–9) and H2O molecule reactions
| Cluster size n |
5 |
6 |
7 |
8 |
9 |
| Adsorption Energy (eV) |
−1.26 |
−1.26 |
−1.43 |
−1.28 |
−1.45 |
| Reaction Energy (eV) |
−2.82 |
−2.86 |
−3.55 |
−4.13 |
−3.17 |
In NBO analysis, it was found that electron transfer occurred between two different OH bonds and an O atom to the clusters. The electron transfer between the O atom and Be atom at the adsorption site accounts for the main part of the electron transfer between water molecules and beryllium clusters. The second-order stabilization energy (E(2)) was positively correlated with the adsorption energy in the adsorption process.42 The adsorption energies of Be5@H2O and Be6@H2O complexes are about −1.26 eV. In NBO analysis, the E(2)s of Be5@H2O and Be6@H2O complexes are 2.00 kcal mol−1 and 3.33 kcal mol−1, respectively. The adsorption energies of Be7@H2O and Be9@H2O complexes are about −1.44 ev, and the E(2)s in the NBO analysis are 4.34 kcal mol−1 and 3.99 kcal mol−1, respectively. In NBO analysis, Be8@H2O has adsorption energy (−1.43 eV) and E(2) (3.84 kcal mol−1). Our calculation results are consistent with the rule that the second-order stabilization energy and adsorption energy are positively correlated.
3.3. The reaction path between Ben (n = 5–9) and H2O molecule
We concentrate on the modifications of chemical bonds in Ben@H2O (n = 5–9) complexes, the cleavage of OH bonds, the production of hydrogen molecules, the energy barriers in the dissociation process, and the reaction energy (see Table 2 for specific information) when predicting the reaction channels of Ben (n = 5–9) clusters and H2O molecules.
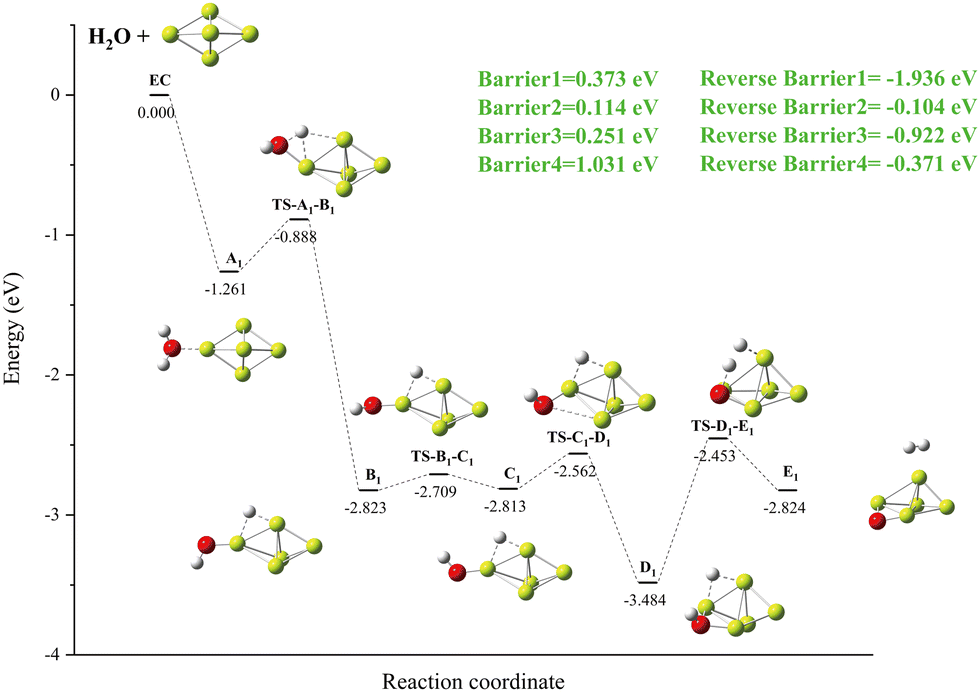
3.3.1. Reaction: Be5 + H2O.
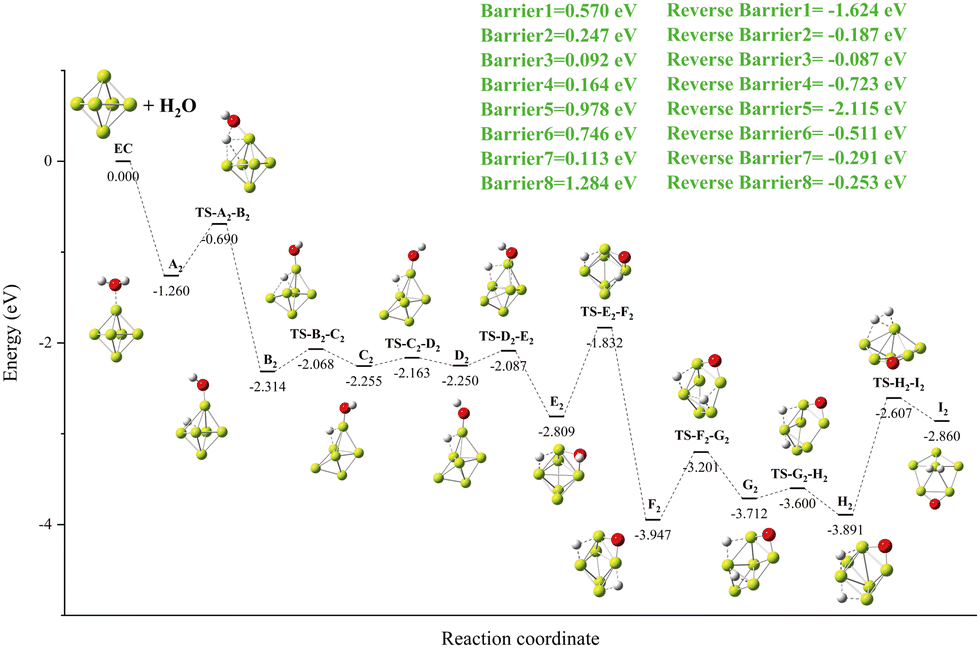
A new local minimum value A2 with a Be–H–Be three-center bond results when an O–H bond breaks within the local minimum value of A1. In this case, the H atom bridged between the two Be atoms of the Be5 group (as shown in Fig. 4). The important finding is that, as we have just mentioned, the energy of the transition state (TS-A1-B1) and the new local minimum (B1) related to this process are lower than those of the import channel EC. The OH group rotates 180° on the Be–O bond to obtain the local minimum C1. A global minimum D1 is produced when the OH group is deflected and drawn to another Be atom as a result of the H–O bond's spin. At D1, the H–O bond breaks, drawing two hydrogen atoms to the surface of Be5, where they combine to form hydrogen molecules.
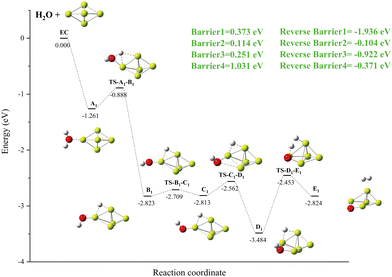 |
| | Fig. 4 The energy diagram and the structures of the reactant A1, intermediate states B1, C1, and D1, and transition states TS-A1-B1, TS-B1-C1, TS-C1-D1, and TS-D1-E1, and product B1, involved in the Be5 + H2O reaction. The energies are calculated at the PBE0-D3/Def2TZVP level. | |
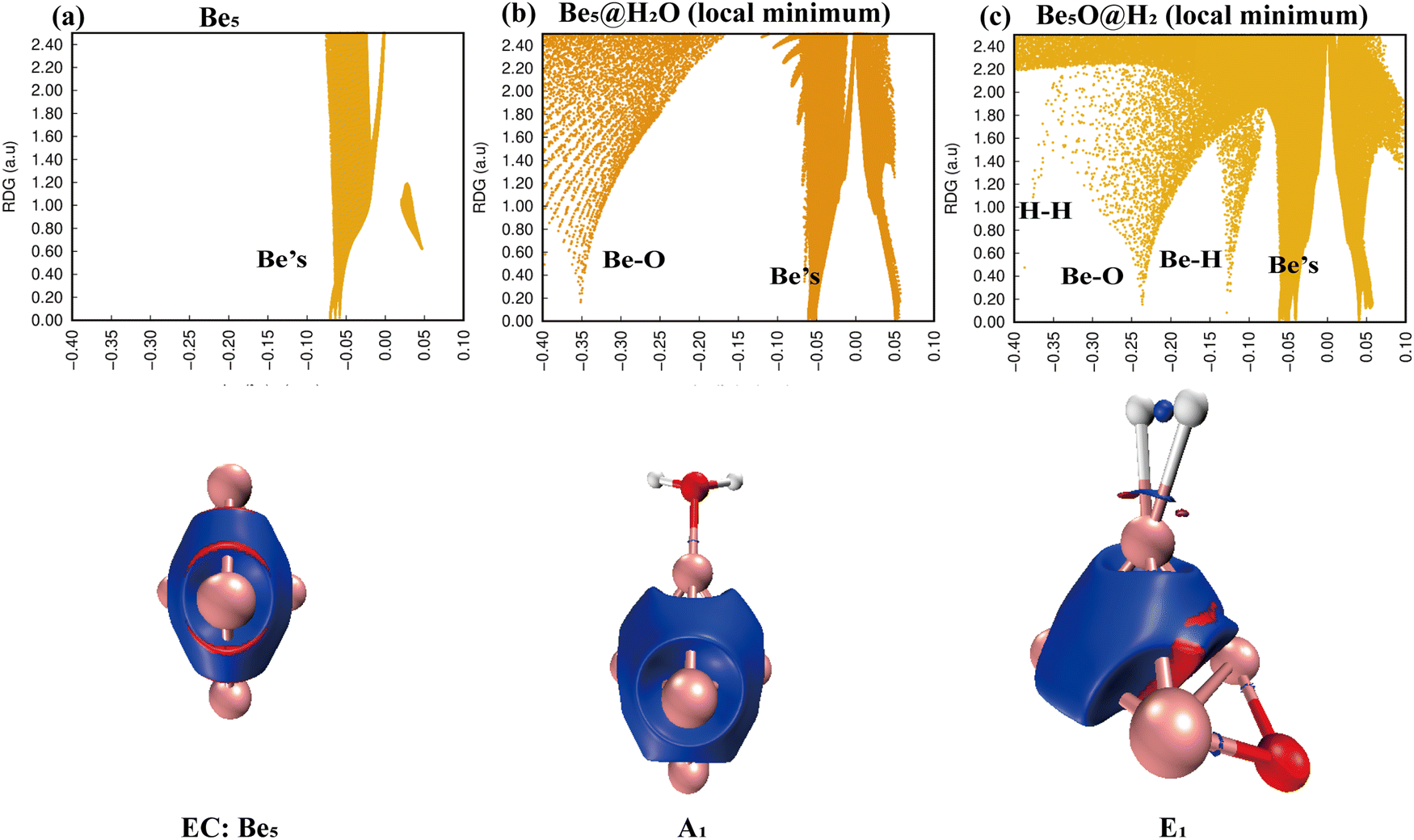
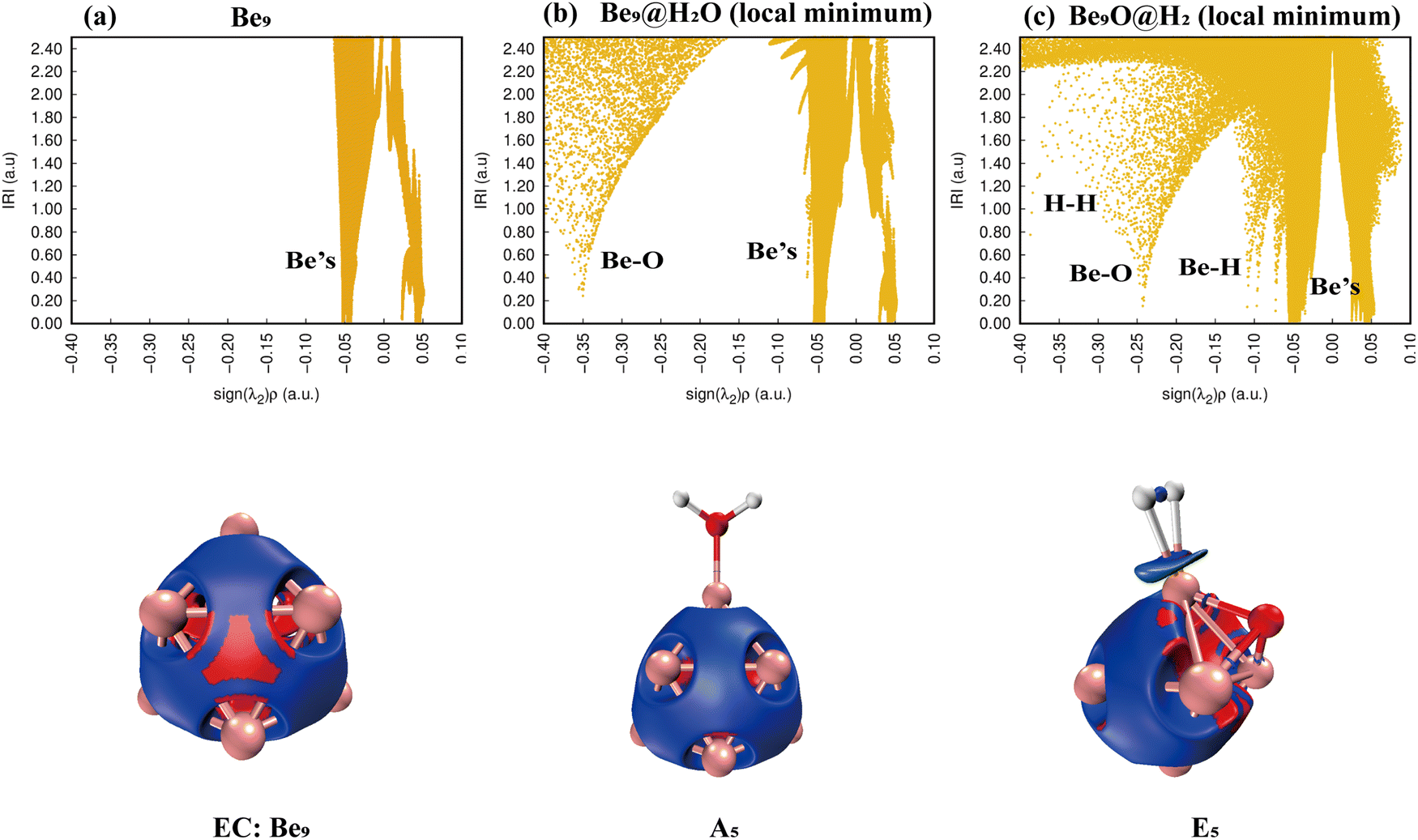
Simply looking at the results shown in Fig. 5, we can clearly see that the electronic density of H2O has undergone profound changes, because its initial geometric structure with two O–H bonds has been completely destroyed, and eventually formed a stable Be–O–Be three-center bond and H–H bond. The IRI 3D plot in Fig. 5(a) shows that the Be5 double triangular pyramid cluster exhibits a structure, and five Be atoms share valence electron pairs, forming a typical metal bonding arrangement.
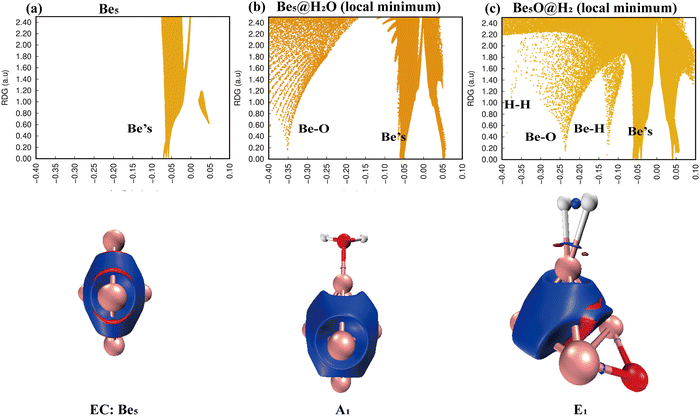 |
| | Fig. 5 Bonding characteristics of Be5, the Be5@H2O complex and product Be5O@H2. The first line contains IRI 2D plots. The second line shows IRI 3D plots. In the IRI 3D plots, the blue color of the isosurface represents the chemical bond action, and the bright red color represents the strong steric hindrance here. | |
The symmetry of distribution is also reflected in the single spike in the corresponding IRI 2D plot. The IRI 3D plot around the Be5 group is not significantly disturbed by the attachment of H2O (Fig. 5(b)). This is well reflected in the IRI 2D plot. Two spikes have been clearly observed, one corresponding to the new Be–O bond and the other to the Be–Be interaction. The IRI 2D plot (Fig. 5(c)) clearly shows that the single spike observed by the Be5 cluster (Fig. 5(a)) has now been split into several spikes: one Be–Be, two Be–H and Be–O spikes, and one H–H spike. It is very important that the intensity of these new Be–H, Be–O, and H–H spikes is much higher than that of the corresponding Be–Be bonds, indicating that the driving force for the systematic and total dissociation of the initial O–H bond of H2O comes from the special intensity of the newly formed Be–O and Be–H bonds relative to the initial Be–Be bonds.
3.3.2. Reaction: Be6 + H2O.
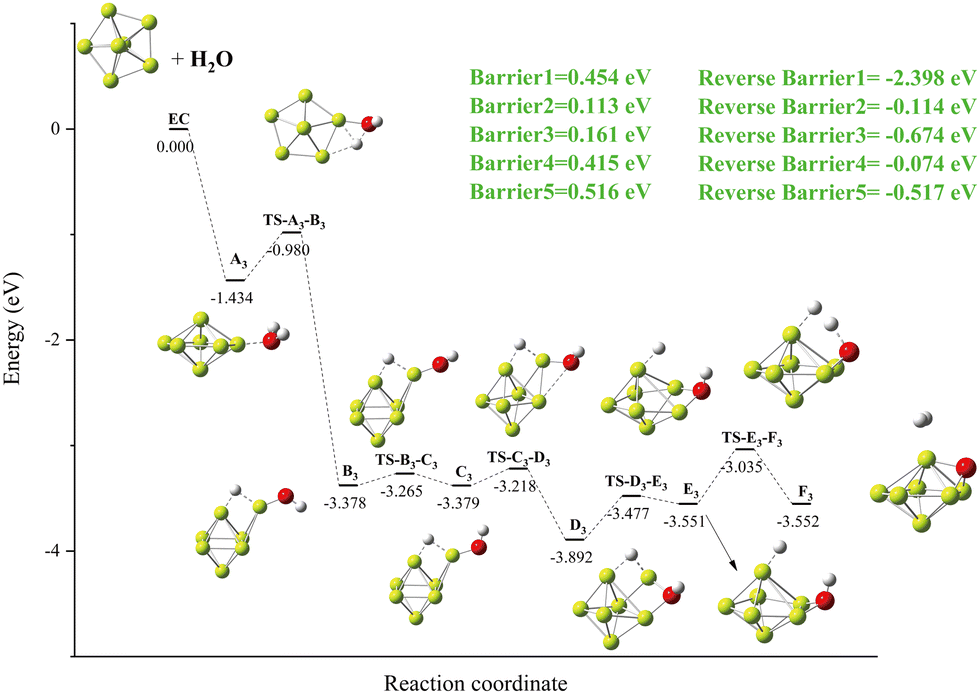
Details of the Be6 + H2O reaction channel are shown in Fig. 6. O–H bond breaking in A2 also creates a new local minimum (B2) with a tricentric bond. Due to the movement of the H atom on the surface of the Be6 cluster and the rotation of OH groups, the geometric shape of the Be6 cluster is changed, and the local minimum E2 with a stable Be–O–Be three-center bond is formed. This process (B2 to E2) undergoes three transition states (TS-B2-C2, TS-C2-D2, TS-D2-E2) with lower energy barriers. The second O–H bond breaks, forming a global minimum F2. In the global minimum F2 to product I2 process, after three transition states (TS-F2-G2, TS-G2-H2, TS-H2-I2), the geometric structure of the Be6 cluster changes from biquarangular pyramid to pentagonal pyramid due to the formation of a hydrogen molecule by hydrogen atoms.
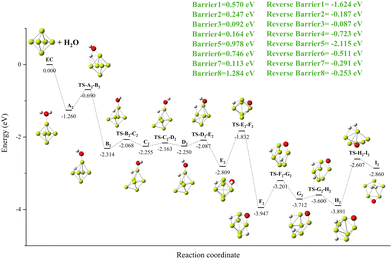 |
| | Fig. 6 The energy diagram and the structures of the reactant A2, intermediate states B2, C2, D2, E2, F2, G2, and H2, transition states TS-A2-B2, TS-B2-C2, TS-C2-D2, TS-D2-E2, TS-E2-F2, TS-F2-G2, TS-G2-H2, and TS-H2-I2, and product I2, involved in the Be6 + H2O reaction. The energies are calculated at the PBE0-D3/Def2TZVP level. | |
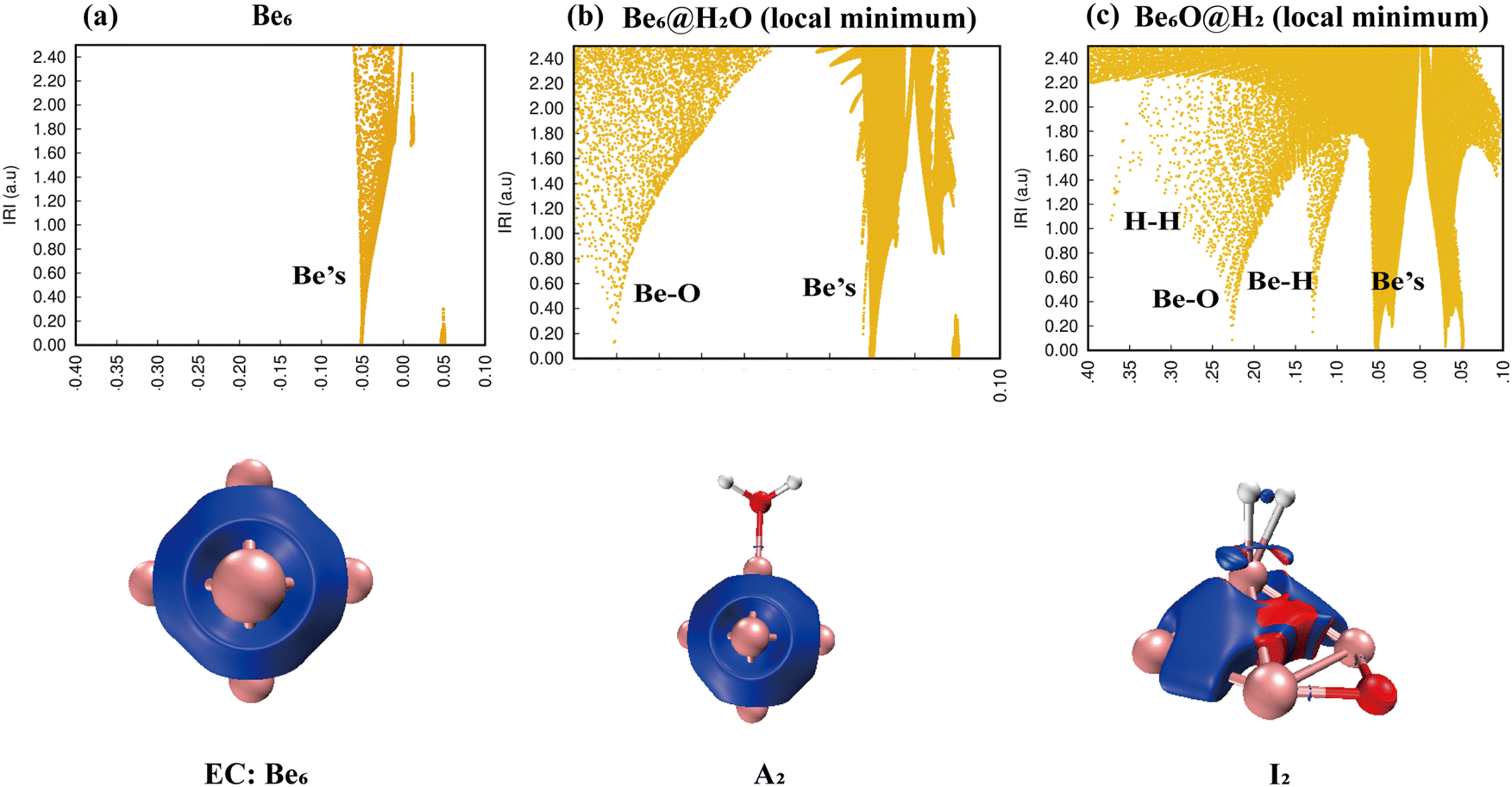
IRI analysis of the Be6 + H2O reaction is shown in Fig. 7. The electronic density of H2O also changed dramatically on the surface of the Be6 cluster. The cleavage of O–H bonds resulted in the formation of a stable Be–O–Be three-center bond and H–H bond. The IRI 3D plot in Fig. 7(a) shows that the Be6 cluster presents a biquadrangular pyramid structure and forms a typical metal bond. It can be clearly seen in the IRI 3D plot (see Fig. 7(b)) that H2O attached to the Be6 cluster has little interference with the surroundings. This shows two spikes (Be–O, Be) in the IRI 2D plot.
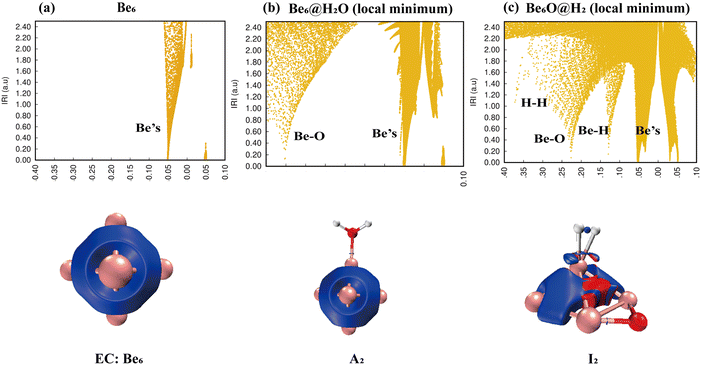 |
| | Fig. 7 Bonding characteristics of Be6, the Be6@H2O complex and product Be6O@H2. The first line contains IRI 2D plots. The second line shows IRI 3D plots. In the IRI 3D plots, the blue color of the isosurface represents the chemical bond action, and the bright red color represents the strong steric hindrance here. | |
The dissociation of H2O on the surface of the Be6 cluster leads to changes in the structure of the Be6 cluster. This is evident in the IRI analysis. The single spike of Be splits into almost double spikes in the IRI 2D plot (Fig. 7(c)), and the geometry changes caused by the dissociation of H2O can be clearly seen in the IRI 3D plot.
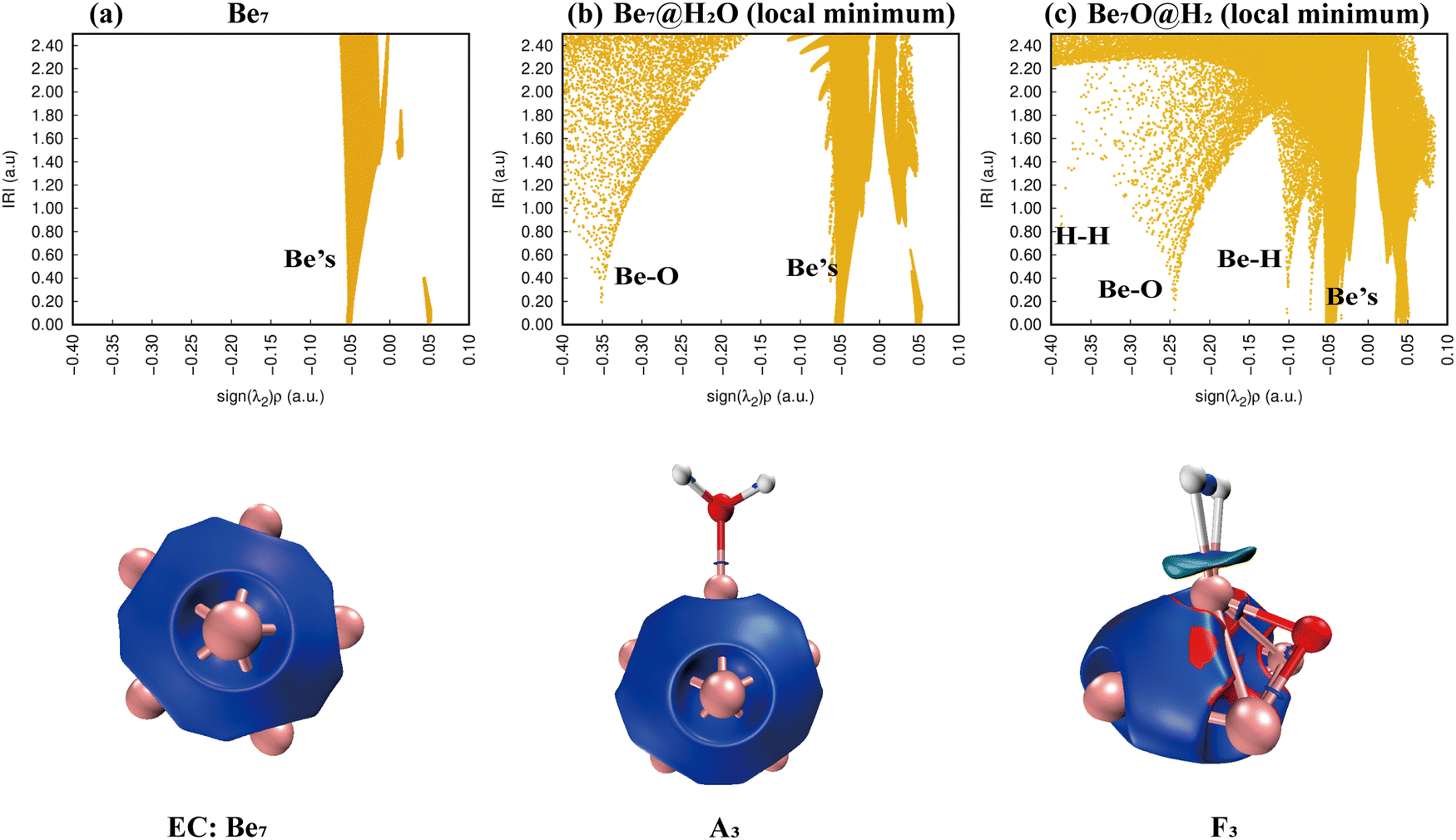
3.3.3. Reaction: Be7 + H2O.
Details of the reaction mechanism of Be7 + H2O are shown in Fig. 8. In this reaction, there are five transition states, and there is no high energy barrier. The local minimum B2 is created though an O–H bond in the local minimum A3 breaks, which releases energy of around 2.398 eV (Fig. 8, Reverse Barrier 1). The reason for the change in the structure of B3 may be the release of energy. In the process of B3 to D3, the OH group moves to form a relatively stable Be–O–Be three-center bond, and D3 is the global minimum. In the process of D3 to F3, the second O–H bond was broken, and the hydrogen molecule was formed on the surface of the cluster. It is worth noting that the structure of the Be7 cluster was restored to the pentagonal bipyramid structure, and O atom and one surface of the cluster formed a stable tetrahedral structure (F3).
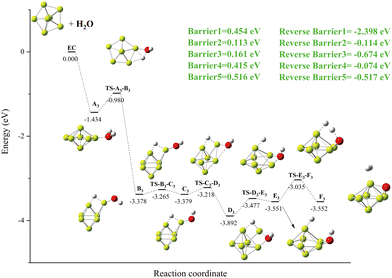 |
| | Fig. 8 The energy diagram and the structures of the reactant A3, intermediate states B3, C3, D3, E3, and F3, and transition states TS-A3-B3, TS-B3-C3, TS-C3-D3, TS-D3-E3, TS-E3-F3, and product F3, involved in the Be7 + H2O reaction. The energies are calculated at the PBE0-D3/Def2TZVP level. | |
According to Fig. 9(a), only Be–Be bonds exist. Fig. 9(b) shows that the adsorption of a water molecule has little effect on the overall disturbance of Be7 clusters. Fig. 9(c) shows that H–H bonds appear in the IRI plots of product F3, whereas Be–H has two peaks, and the IRI 3D plot shows that the interaction between hydrogen molecules and the Be7O cluster is weaker than chemical bonds. This shows that F3 is easier to dehydrogenate than E1 and I2.
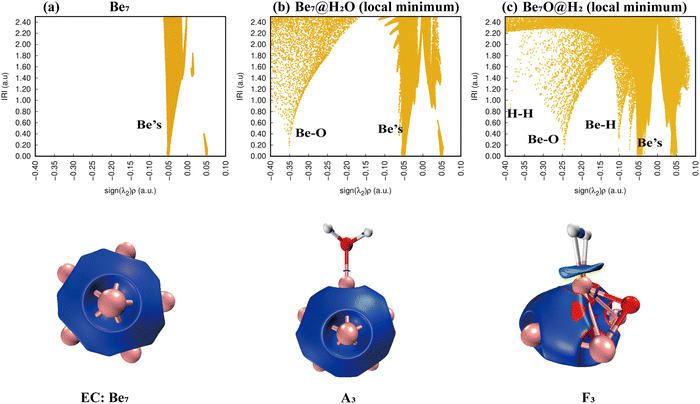 |
| | Fig. 9 Bonding characteristics of Be7, the Be7@H2O complex and product Be7O@H2. The first line contains IRI 2D plots. The second line shows IRI 3D plots. In the IRI 3D plots, the blue color of the isosurface represents the chemical bond action, and the bright red color represents the strong steric hindrance here. Patina green indicates weaker chemical bonds. | |
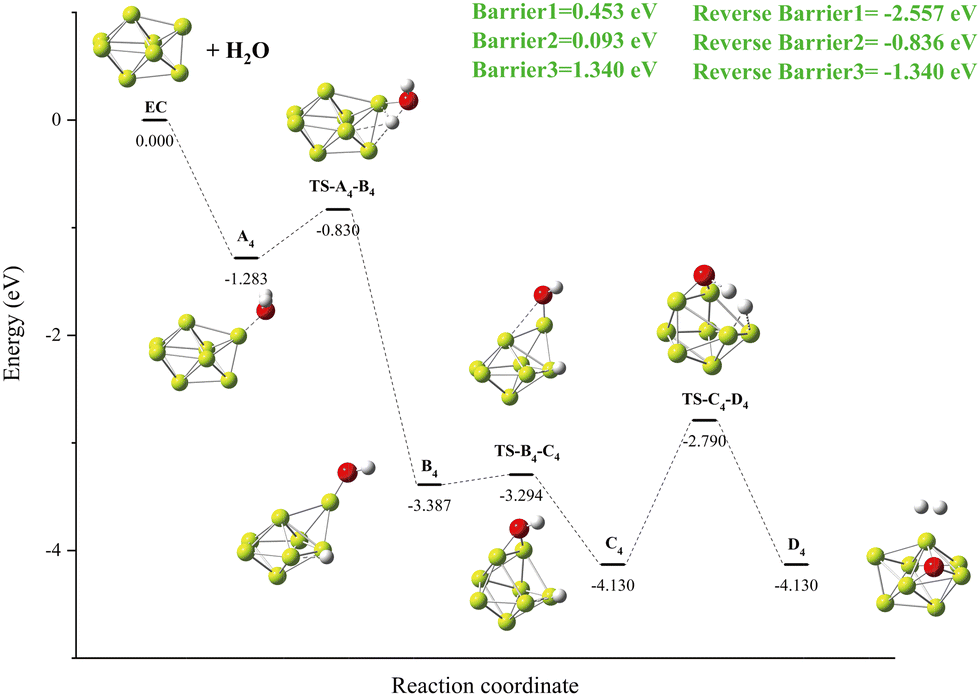
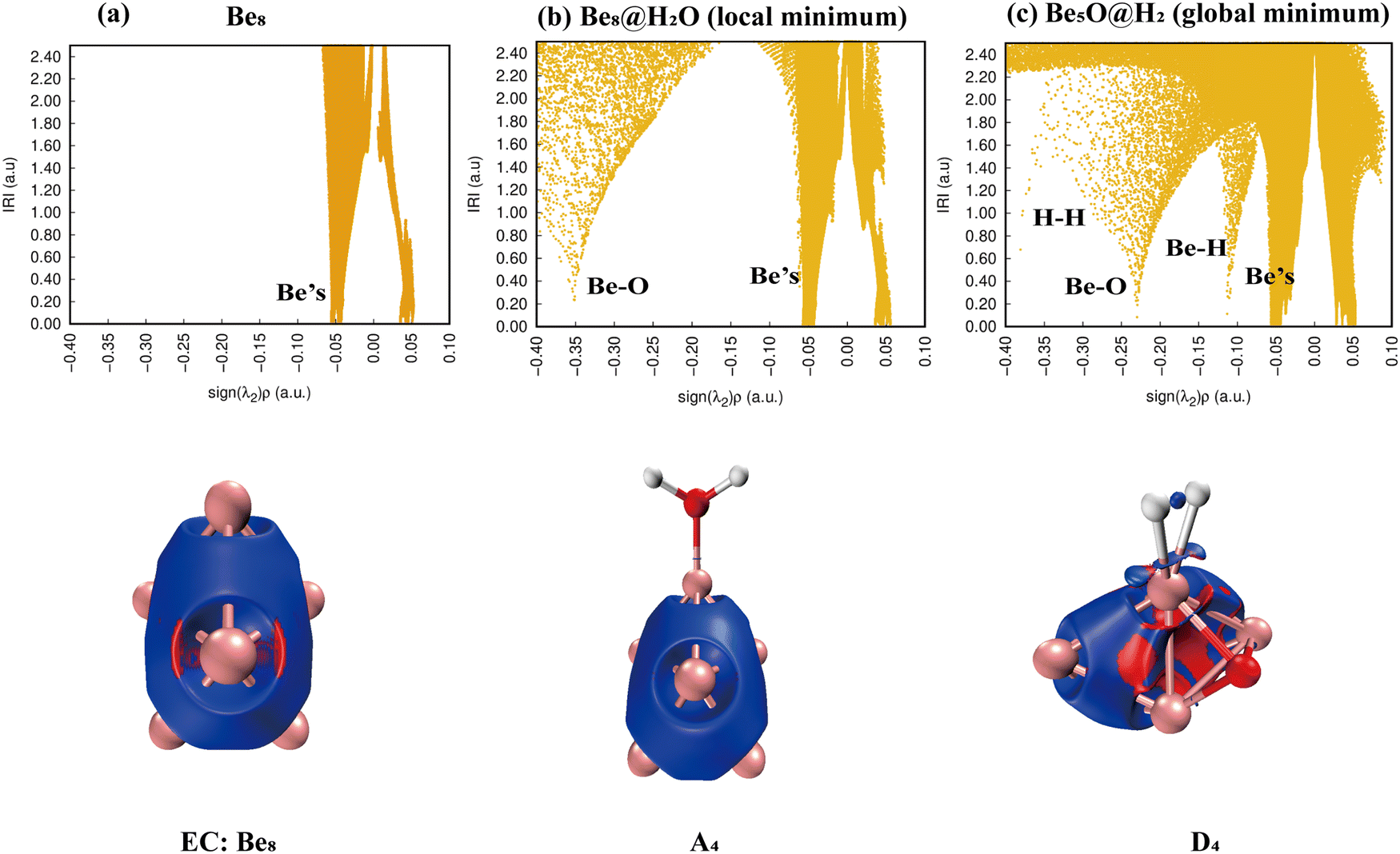
3.3.4. Reaction: Be8 + H2O.
The reaction mechanism of Be8 + H2O is shown in Fig. 10. Breaking of the O–H bond in A4 releases an energy of −2.557 eV (Fig. 10, Reverse Barriers1) and forms a local minimum B4 with the Be–H–Be triple center bond. The deflection of the remaining OH groups also forms the global minimum C4 with the Be–O–Be triple center bond. The cleavage of the second O–H bond forms hydrogen molecules and the product D4 at the same energy level as the C4 structure. The energy barrier for the cleavage of O–H is the highest at 1.340 eV in the Be8 + H2O reaction (Fig. 10, Barrier3).
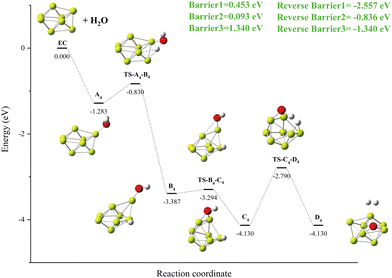 |
| | Fig. 10 The energy diagram and the structures of the reactant A4, intermediate states B4, C4, and transition states TS-A4-B4, TS-B4-C4, TS-C4-D4, and product D4, involved in the Be8 + H2O reaction. The energies are calculated at the PBE0-D3/Def2TZVP level. | |
As shown in Fig. 11, the IRI 2D plot shows that Be8 has a continuous peak belonging to the Be bond (Fig. 11(a)), and Be8@H2O has one more spike belonging to the Be–O bond (Fig. 11(b)). There are several obvious spikes for the product D4 (Be8O@H2O), belonging to Be, Be–H, Be–O, and H–H, respectively, and the specific situation is shown in the IRI 3D plot (Fig. 11(c)). It is worth noting that the chemical bonding of the Be–H bond should be of a strong type, and the desorption of hydrogen molecules should be relatively difficult.
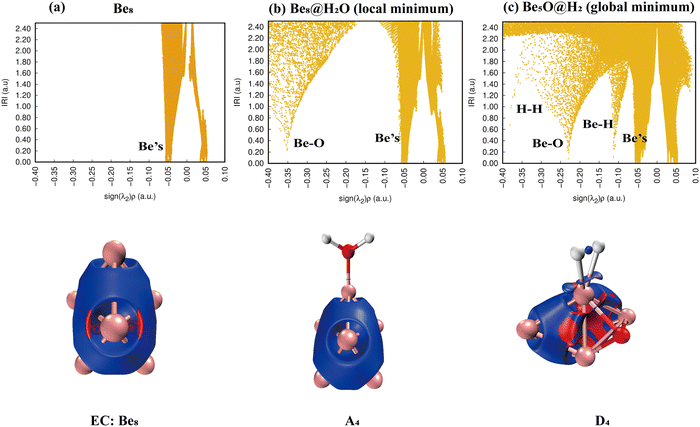 |
| | Fig. 11 Bonding characteristics of Be8, the Be8@H2O complex and product Be8O@H2. The first line contains IRI 2D plots. The second line shows IRI 3D plots. In the IRI 3D plots, the blue color of the isosurface represents the chemical bond action, and the bright red color represents the strong steric hindrance here. | |
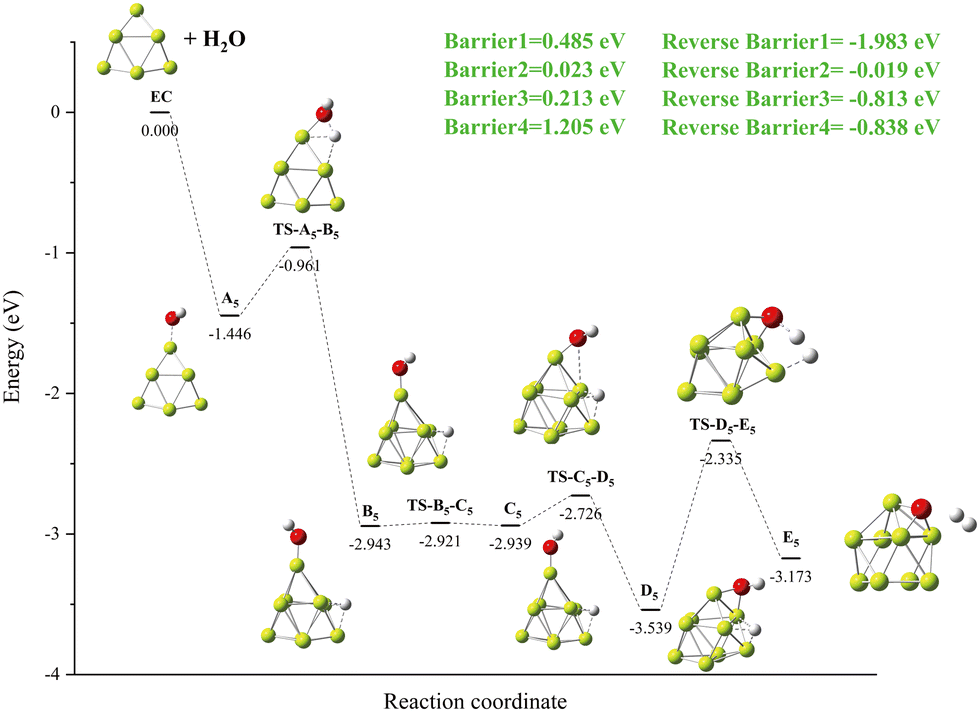
3.3.5. Reaction: Be9 + H2O.
The specific details of the Be9 + H2O reaction mechanism are shown in Fig. 12. A local minimum B5 is produced with one O–H bond in A5 breaks, releasing −1.983 eV energy (Fig. 12 Reverse Barrier1). The process from B5 to D5 undergoes deflection of the OH group, forming the global minima D5 structure of the Be–O–Be triple center bond. The second O–H bond is broken, and hydrogen molecules are formed on the surface of the cluster to give the product E5. It is worth noting that the energy barrier of 1.205 eV to be overcome for the breaking of the second O–H bond is higher than that of the first O–H bond at 0.485 eV.
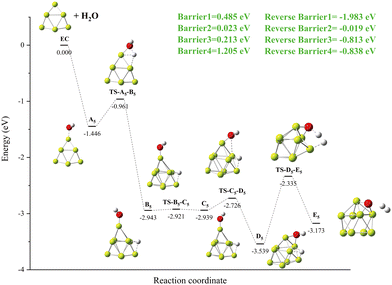 |
| | Fig. 12 The energy diagram and the structures of the reactant A5, intermediate states B5, C5, and D5, and transition states TS-A5-B5, TS-B5-C5, TS-C5-D5, and TS-D5-E5, and product E5, involved in the Be8 + H2O reaction. The energies are calculated at the PBE0-D3/Def2TZVP level. | |
A strong steric hindrance effect appears in the center of the Be9 cluster, as shown in Fig. 13(a). When an H2O molecule is adsorbed on the Be9 cluster, the steric hindrance effect in the face center disappears (Fig. 13(b)). The IRI 2D plot in Fig. 13(c) shows that there are multiple peaks, including Bes bonds, Be–O bonds, H–H bonds, and Be–H bonds. Notable among them is the Be–H bond, which has three peaks in the IRI 2D plot and a bronze isosurface in the IRI 3D plot, indicating that the interaction is weaker than the chemical bond.
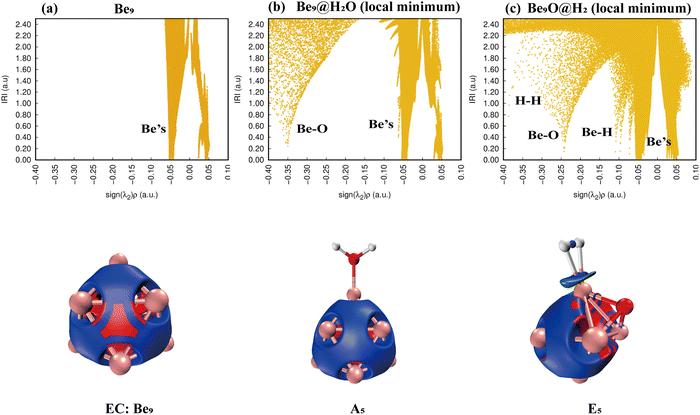 |
| | Fig. 13 Bonding characteristics of Be9, the Be9@H2O complex and product Be9O@H2. The first line contains IRI 2D plots. The second line shows IRI 3D plots. In the IRI 3D plots, the blue color of the isosurface represents the chemical bond action, and the bright red color represents the strong steric hindrance here. Patina green indicates weaker chemical bonds. | |
3.3.6. Gibbs free energy.
We provide the Gibbs free energy of the reaction process so that it is feasible to directly compare with potential experiments. The specific outcomes are displayed in Table 3. The results show that the Gibbs free energy of reactants, transition states, intermediate states, and products in the Ben + H2O (n = 5–9) reaction are consistent with the changing trend of electron energy (energy above).
Table 3 Gibbs free energy of hydrogen evolution reaction of beryllium clusters (temperature 298.150 K, pressure 1.00000 Atm, n = 1–5, and the unit is electron volt)
| Path |
Be5 |
Be6 |
Be7 |
Be8 |
Be9 |
| EC |
0.000 |
0.000 |
0.000 |
0.000 |
0.000 |
| An |
−0.868 |
−0.877 |
−0.747 |
−0.825 |
−1.048 |
| TS-An-Bn |
−0.637 |
−0.433 |
−0.400 |
−0.497 |
−0.667 |
| Bn |
−2.549 |
−2.080 |
−2.735 |
−3.065 |
−2.649 |
| TS-Bn-Cn |
−2.482 |
−1.828 |
−2.655 |
−2.908 |
−2.621 |
| Cn |
−2.540 |
−2.000 |
−2.738 |
−3.701 |
−2.648 |
| TS-Cn-Dn |
−2.235 |
−1.937 |
−2.547 |
−2.487 |
−2.370 |
| Dn |
−3.083 |
−1.997 |
−3.182 |
−3.200 |
−3.123 |
| TS-Dn-En |
−2.191 |
−1.794 |
−2.829 |
|
−2.053 |
| En |
−2.605 |
−2.462 |
−2.914 |
|
−2.845 |
| TS-En-Fn |
|
−1.641 |
−2.439 |
|
|
| Fn |
|
−3.713 |
−2.970 |
|
|
| TS-Fn-Gn |
|
−2.993 |
|
|
|
| Gn |
|
−3.463 |
|
|
|
| TS-Gn-Hn |
|
−3.355 |
|
|
|
| Hn |
|
−3.642 |
|
|
|
| TS-Hn-In |
|
−2.502 |
|
|
|
| In |
|
−2.643 |
|
|
|
4. Conclusions
To investigate the structural evolution of Ben clusters with n = 5–9, the adsorption energy produced by the complex generated by Ben@H2O (n = 5–9), and the mechanism of the hydrogen evolution process of Ben + H2O (n = 5–9), DFT calculations were conducted based on the PBE0-D3/Def2TZVP level. Our calculation results show that Be5 and Be9 clusters have D3h point group symmetry, the Be6 cluster has Oh point group symmetry, the Be8 cluster has D2d point group symmetry, and the Be7 cluster has low C1 point group symmetry. During the formation of the Ben@H2O (n = 5–9) complex, the formation and adsorption energies of Be5@H2O and Be6@H2O were −1.26 eV, −1.28 eV, −1.43 eV and −1.45 eV, respectively. The adsorption reaction between Be clusters and water molecules are a barrierless reaction, which indicates that water molecules are more easily adsorbed on the surface of Be clusters, and the adsorption energy is larger, which can also reflect that the adsorption structure is more stable.
The hydrogen evolution reaction in Ben + H2O (n = 5–9) is an exothermic reaction, and the energy of all transition states, intermediate states, and products is below EC.
When an O–H bond in the Ben@H2O (n = 5–9) complex is broken, more energy is released, and the release of energy brings about a change in the cluster structure, which is more obvious in the Be7 + H2O reaction. When the second O–H bond breaks, a high energy barrier needs to be overcome. It is worth noting that the hydrogen evolution reaction of Be6 + H2O has 8 transition states, and the second O–H bond breaks more energy than the first O–H bond breaks. The hydrogen evolution reaction of Be8 + H2O has only 3 transition states, and the reaction heat is the highest (−4.13 eV). It can be seen from the IRI analysis that the hydrogen molecules in the products of Ben + H2O (n = 5–9) are all adsorbed on the surface of the cluster in the form of chemical bonds. The weaker Be–H bond interaction in Be7O@H2 and Be9O@H2 is beneficial for the desorption of hydrogen molecules. The Gibbs free energy of the Ben + H2O (n = 5–9) reaction is consistent with the trend of electron energy change, revealing the spontaneity of the reaction to some extent, and can be compared with potential experiments.
Author contributions
Kai Diao: conceptualization, formal analysis, investigation, writing – original draft. Shun Ping Shi: conceptualization, writing – review & editing, supervision. Yong Song: supervision. Leilei Tang: data curation. Jiabao Hu: data curation. Zhanjiang Duan: writing – review & editing. Jing Jiang: writing – review & editing. De Liang Chen: software, resources.
Conflicts of interest
The authors declare that they have no known competing financial interests or relationships that could have appeared to influence the work reported in the paper.
Acknowledgements
This project is supported by the Open Research Fund of Computational Physics Key Laboratory of Sichuan Province, Yibin University (Grant No. YBXYJSWL-ZD-2020-005). This work is also supported by the Student's Platform for Innovation and Entrepreneurship Training Program (No. S202110616084 and G202210616030).
References
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- Y.-Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- X. Liu, P. Wang, X. Liang, Q. Zhang, Z. Wang, Y. Liu, Z. Zheng, Y. Dai and B. Huang, Mater. Today Energy, 2020, 18, 100524 CrossRef CAS.
- P. Liao and E. A. Carter, Chem. Soc. Rev., 2013, 42, 2401–2422 RSC.
- W.-L. Xie, Z.-H. Zhang, C.-L. Yang, M.-S. Wang and X.-G. Ma, Int. J. Hydrogen Energy, 2017, 42, 4032–4039 CrossRef CAS.
- S. Dong, Z. Wang, Y. Wang and J. zhang, J. Alloys Compd., 2017, 727, 1157–1164 CrossRef CAS.
- N. Gong, C. Deng, L. Wu, B. Wan, Z. Wang, Z. Li, H. Gou and F. Gao, J. Alloys Compd., 2019, 781, 371–377 CrossRef CAS.
- C. Li, Z. Fan, H.-L. Jia, C. Wang, P.-K. Ma, M.-W. Ren and H.-Y. Wang, J. Alloys Compd., 2021, 888, 161477 CrossRef CAS.
- M. D. Milosavljević, U. Burkhardt, A. Leithe-Jasper, Y. Grin and H. Borrmann, J. Alloys Compd., 2022, 890, 161420 CrossRef.
- D. Zhu, Q. Zeng, Y. Liu, C. Li, L. Lu, T. Han, G. Xie and X. Shu, J. Alloys Compd., 2022, 919, 165780 CrossRef CAS.
- S. Srinivas and J. Jellinek, J. Chem. Phys., 2004, 121, 7243–7252 CrossRef CAS PubMed.
- B. Abyaz, Z. Mahdavifar, G. Schreckenbach and Y. Gao, Phys. Chem. Chem. Phys., 2021, 23, 19716–19728 RSC.
- R. Kawai and J. H. Weare, Phys. Rev. Lett., 1990, 65, 80–83 CrossRef CAS PubMed.
- D.-D. Zhang, D. Wu, H. Yang, D. Yu, J.-Y. Liu, Z.-R. Li and Y. Li, Eur. J. Inorg. Chem., 2017, 2428–2434 CrossRef CAS.
- J.-Y. Li, D. Wu, Y. Li and Z.-R. Li, Chem. Phys. Lett., 2017, 674, 1–5 CrossRef CAS.
- G.-x Ge, Y.-l Yan, F.-z Ren, X.-l Lei, Z. Yang, W.-j Zhao, Q.-l Wang and Y.-h Luo, Chin. J. Chem. Phys., 2007, 20, 518–524 CrossRef CAS.
- O. Roberto-Neto and E. F. V. de Carvalho, Theor. Chem. Acc., 2020, 139, 93 Search PubMed.
- F. Y. Naumkin and D. J. Wales, Int. J. Quantum Chem., 2012, 112, 3068–3075 CrossRef CAS.
- J. Beheshtian and I. Ravaei, Struct. Chem., 2018, 29, 231–241 CrossRef CAS.
- M. Y. Mehboob, R. Hussain, Z. Irshad, U. Farwa, M. Adnan and S. Muhammad, J. Comput. Biophys. Chem., 2021, 20, 687–705 CrossRef CAS.
- R. Sun, C. L. Yang, M. S. Wang and X. G. Ma, J. Power Sources, 2022, 547 DOI:10.1016/j.jpowsour.2022.232008.
- Q. Fu, F. Xin, X. Yin, Y. Song and Y. Xu, Int. J. Hydrogen Energy, 2021, 46, 22446–22453 CrossRef CAS.
- Q. K. Yin, C. L. Yang, M. S. Wang and X. G. Ma, J. Mater. Chem. C, 2021, 9, 12231–12238 RSC.
- S. Jayachitra, D. Mahendiran, P. Ravi, P. Murugan and M. Sathish, Appl. Catal., B, 2022, 307 DOI:10.1016/j.apcatb.2022.121159.
- L. L. Liu, B. W. Jiang, D. Sun, H. Y. Liu and Y. Xie, J. Mater. Chem. A, 2022, 10, 14060–14069 RSC.
- M. Ge, C.-L. Yang, M.-S. Wang and X.-G. Ma, Int. J. Hydrogen Energy, 2022, 47(100) DOI:10.1016/j.ijhydene.2022.09.249.
- E. Vos, I. Corral, M. M. Montero-Campillo, O. Mó, J. Elguero, I. Alkorta and M. Yáñez, Phys. Chem. Chem. Phys., 2021, 23, 6448–6454 RSC.
- E. Vos, I. Corral, M. M. Montero-Campillo and O. Mó, J. Chem. Phys., 2021, 154, 044302 CrossRef CAS PubMed.
- D. Chakraborty and P. K. Chattaraj, J. Comput. Chem., 2018, 39, 535–545 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14(47), 16187–16191 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and Q. Chen, Chem.: Methods, 2021, 1, 231–239 CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS PubMed.
-
C. K. Thomas Williams Gnuplot. An Interactive Plotting Program.
- M. Šulka, D. Labanc, M. Kováč, M. Pitoňák, I. Černušák and P. Neogrády, J. Phys. B: At., Mol. Opt. Phys., 2012, 45, 085102 CrossRef.
- B. Samanta, T. Sengupta and S. Pal, J. Phys. Chem. C, 2018, 122, 28310–28323 CrossRef CAS.
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Shunping
Shi
*a,
Yong
Song
*a,
Leilei
Tang
a,
Jiabao
Hu
a,
Jing
Jiang
a,
Zhanjiang
Duan
a and
Deliang
Chen
b
a,
Shunping
Shi
*a,
Yong
Song
*a,
Leilei
Tang
a,
Jiabao
Hu
a,
Jing
Jiang
a,
Zhanjiang
Duan
a and
Deliang
Chen
b