
Guanidinium–amino acid hydrogen-bonding interactions in protein crystal structures: implications for guanidinium-induced protein denaturation†
Indu
Negi‡
a,
Raman
Jangra‡
 a,
Amit
Gharu
a,
John F.
Trant§
*bc and
Purshotam
Sharma
*ab
a,
Amit
Gharu
a,
John F.
Trant§
*bc and
Purshotam
Sharma
*ab
aComputational Biochemistry Laboratory, Department of Chemistry and Centre for Advanced Studies in Chemistry, Panjab University, Chandigarh, 160014, India. E-mail: psharma@pu.ac.in
bDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Ave. Windsor ON, N9B 3P4, Canada. E-mail: j.trant@uwindsor.ca
cBinary Star Research Services, LaSalle, ON N9J 3 X 8, Canada
First published on 8th December 2022
Abstract
In the present work, 86 available high resolution X-ray structures of proteins that contain one or more guanidinium ions (Gdm+) are analyzed for the distribution and nature of noncovalent interactions between Gdm+ and amino-acid residues. A total of 1044 hydrogen-bonding interactions were identified, of which 1039 are N–H⋯O, and five are N–H⋯N. Acidic amino acids are more likely to interact with Gdm+ (46% of interactions, 26% Asp and 20% Glu), followed by Pro (19% of interactions). DFT calculations on the identified Gdm+–amino acid hydrogen-bonded pairs reveal that although Gdm+ interacts primarily with the backbone amides of nonpolar amino acids, Gdm+ does interact with the sidechains of polar and acidic amino acids. We classified the optimized Gdm+–amino acid pairs into parallel [p], bifurcated [b], single hydrogen bonded [s] and triple hydrogen bonded [t] types. The [p] and [t] type pairs possess higher average interaction strength that is stronger than that of [b] and [s] type pairs. Negatively charged aspartate and glutamate residues interact with Gdm+ ion exceptionally tightly (−76 kcal mol−1) in [p] type complexes. This work provides statistical and energetics insights to better describe the observed destabilization or denaturation process of proteins by guanidinium salts.
1 Introduction
Properly folded protein tertiary conformations are an emergent phenomenon arising from a combination of the primary amino acid sequence, the formation of secondary structures, and the influence of chaperones.1–3 But it can be easy to forget that this all takes place in the context of a surrounding solvation shell; transient and conserved interactions with water molecules mediate both the folding process and the final structure.4–6 Protein–protein interactions, and the consequent changes in conformation are driven as much by the reorganization and disruption of the solvation shell as by interactions between the two proteins.7,8 This means that the presence of other molecules in solution that both bind tightly to the protein and disrupt the semiorganized first hydration sphere can have outsized effects on the conformation of the protein as a whole.3,9,10 In this context, Hofmeister and coworkers suggested that increased charge density on an interacting cation escalates protein denaturation, whereas low charge density promotes protein folding.11,12 In this respect, guanidinium (Gdm+) is exceptional as it acts as a chaotrope despite its low charge density. As a result, Gdm+ is widely used as a protein denaturant in protein folding studies.13–15 In fact, Gdm+ is one of the most effective chemical denaturants, meaning it significantly reduces protein stability in aqueous media.15 However, the exact mechanism of Gdm+ induced protein destabilization remains unclear.There are two non-exclusive mechanistic proposals to explain why the low charge density Gdm+ so effectively disrupts protein structure: it either works indirectly by disrupting the hydration sphere—weakening the interchain hydrophobic interactions that hold the architecture together16–18—or works directly as a competitive H-bond donor by forming protein–ion noncovalent interactions that weaken internal protein hydrogen bonds.19–22 Gdm+ only forms weak interactions with water, so a “pure” indirect mechanism is unlikely.16,18 On the other hand, several experimental21–29 and computational19,20,30–35 studies have observed that Gdm+ collapses onto the protein surface, supporting a direct mechanism (this still disrupts protein hydration and would clearly induce the indirect effects). Although it is well established that Gdm+ complexation increases aqueous solubility,36–41 the identities, and mode of interaction, of the amino acids that preferentially adsorb Gdm+ are not clear. Nevertheless, studies hint at a preference for specific residues.28,42 In particular, 2D-IR spectroscopy hypothesizes that Gdm+ selectively disrupts the secondary structure of proteins due to its direct interactions with the hydrogen bond acceptors on residue side chains.24 The peptide backbone is also likely an important interaction site for Gdm+,28 although affinity appears indifferent to the identity of the residues.24 Another recent 2D-IR spectroscopic study suggests that Gdm+ does not bind strongly to aromatic side chains through π–cation interactions.25 It does appear clear, if anodyne, that Gdm+ mainly destabilizes protein through interaction with backbone amides or charged side chains.25,26,29,43
In addition to these significant experimental efforts, many computational studies have studied Gdm+-induced protein denaturation, to identify the mechanism of action.19,20,31–33,44–47 MD simulations suggest that the low charge density of Gdm+ primarily induces protein destabilization following direct hydrogen bonding (HB) to the carbonyl oxygen of the backbone or the carboxylate of aspartate or glutamate.20 MD simulations on a small model peptide suggest that Gdm+ prefers to interact with planar amino acids such as Arg, Trp and Gln side chains through stacking interactions, thereby disrupting other packing effects, and leveraging open gaps between secondary structures to drive protein unfolding.19 Further, an unexpected attractive affinity was observed between positively charged Arg side chains and Gdm+ (mediated by chloride counterions) in aqueous solution using MD simulations coupled with experimental capillary electrophoresis analysis, which was suggested by the authors as the cause of the observed enhanced effect of guanidinium on destabilizing Arg-rich proteins.32 However, no direct interactions can be logically expected between the positively charged side chains (i.e., Lys, Arg and protonated His side chain headgroups) and Gdm+. In contrast, like the interaction between main-chain NH3+ and side-chain COO− groups observed in the Asp–Asp dimer,48 Gdm+ can favorably interact with the COO− groups of acidic amino acids. Similarly, DFT calculations suggested that Gdm+ can form both cation–π and HB interactions with the aromatic Phe, His, Trp and Tyr, but that His forms only weak complexes due to its lower aromaticity.33 Analysis of available protein crystal structures revealed that Gdm+ forms a wide variety of contacts,49 but the work did not describe the energetics of these interactions to allow this information to be used to predict how Gdm+ would induce protein destabilization.
Although significant progress has been achieved in our understanding of the mechanism of Gdm+ induced protein destabilization, a series of unanswered questions remains: (1) is there any preference for polar or nonpolar residues when Gdm+ interacts with the backbone amides; (2) are backbone interactions a significant site for Gdm+ contacts; and (3) what are the relative strengths of the interactions between Gdm+ and specific residues. These questions might be answerable from an analysis of the high-resolution crystal structures deposited in the Protein Data Bank (PDB). Quantum chemical calculations could describe the causes of intrinsic structures and stabilities of Gdm+:protein residue contacts, and this could then help predict where these interactions would first occur in any given protein. However, only a few quantum mechanical (QM) studies have analyzed the role of Gdm+ induced noncovalent interactions in the proteins.33,35,46 As these were all case studies, none provide a comprehensive model of the interaction between Gdm+ and proteins.
In this present work, we analysed available PDB structures to understand the frequency and nature of HB interactions between Gdm+ and crystallized proteins. Classifying all interactions into different patterns, we calculated the intrinsic stability of all observed noncovalently bonded Gdm+–amino acid pairs using quantum chemical methods on a representative example from each class. These energies should assist in understanding the mechanism of Gdm+-mediated protein denaturation.
2 Computational details
Preparation of the dataset
To analyze the Gdm+–protein HB interactions, all X-ray crystal structures of proteins that contain at least one Gdm+ deposited before August 24, 2021 were extracted from the PDB. With one exception (PDB code: 7LOX, resolution: 3.2 Å), the X-ray resolution of all the analyzed structures was between 1.2 Å and 2.9 Å. This search was accomplished by setting the ‘Chemical ID’ and ‘Polymer Entity Type’ options to ‘GAI’ (i.e., Gdm+) and ‘Protein’ respectively in the ‘Advanced Search Query Builder’ utility available on the PDB website (https://www.rcsb.org). The ‘Release Date’ parameter was set to ≤24th August 2021. This provided a total of 86 crystal structures (Table S1, ESI†), which can be grouped into 4 non-redundant clusters of Gdm+–protein complexes (Table S2, ESI†). However, we did not attempt to further remove redundancies within the dataset, since the same protein can interact differently with Gdm+ in different crystal structures.Identification of HB interactions
HB interactions between Gdm+ and protein residues were identified using HBPLUS (version 3.2)50 after cleaning the PDB files for alternate positions of some atoms using the ‘Clean’ script of HBPLUS. Since the default version of HBPLUS does not recognize Gdm+ in PDB files, HB interactions were identified by defining Gdm+ as a residue, using the ‘−f’ option. The hydrogen bonds were detected using the default criteria of HBPLUS, except the donor–acceptor distance (D–A) cut-off, which was changed from the default (3.90 Å) to 3.35 Å, in accordance with previous studies.51–55 The output file obtained for all HB interactions (including those within the proteins) present in each PDB file were filtered to retain only the HB interactions involving Gdm+. For further verification of the identified interactions, protein residues located within 10 Å of Gdm+ were visually inspected using PyMOL.56 Although we also searched for the occurrence of cation−π interactions between Gdm+ and aromatic protein residues (Phe, Tyr, His and Trp) using visual inspection, no significant instances of these interactions were observed in the dataset, despite predictions for their existence. It is important to note that only conserved and strong interactions would be observable in a PDB dataset, transient interactions would not be expected to be represented.QM calculations
All amino acid–Gdm+ pairs identified in the crystal structures were used as initial models for QM analysis. For QM geometry optimizations, the missing hydrogen atoms were added to the heavy-atom coordinates of the models, to complete the necessary covalency requirements. Subsequently, the C- and N-terminals of the amino acids were capped with methyl groups, to eliminate their non-native interactions with Gdm+. Further, to understand the stability of the HB network at neutral pH, the structures were optimized after ionizing the side-chain carboxylate group of Asp and Glu to COO−. Similarly, the guanidinium group of the Arg side chain, the Nδ atoms of the His imidazole ring, and lysine's side chain amino groups were protonated before carrying out QM geometry optimizations.Geometry optimizations were carried out at the DFT level, using ωB97X-D/6-31G(d,p), an effective tool for dealing with weak noncovalent interactions.57,58 We chose ωB97X-D, since it is an improved, range-separated version of the Becke's original 97 functional59 that includes both long range correction to the non-Coulomb part of exchange functional and second order dispersion correction, and has been used in a previous similar study on biologically-relevant hydrogen-bonded complexes.60 Furthermore, a benchmarking of the performance of DFT functionals for studying the optimized geometry, stability and cooperativity of DNA triplexes has shown that the results of the parent B97-D functional are consistent with higher theoretical levels.61 The dispersion correction within the ωB97X-D was implemented using the Grimme's ‘DFT-D2’ model,62 albeit with a different damping function and unscaled dispersion correction to model the correct asymptotic pairwise vdW potentials.63 Further, the choice of medium-sized 6-31G(d,p) basis set stems from our requirement of geometry optimization of a large number of crystallographic motifs studied in this work, as well as its proven utility in previous studies on hydrogen-bonded complexes.64,65 Furthermore, in accordance with a previous study,66 the HB interactions in the optimized complexes were considered using the donor acceptor (D–A) distance cut-off of 3.0 Å, and donor–hydrogen–acceptor (D–H–A) angle cut-off of 140°. Basis set superposition error (BSSE) corrected interaction energy was calculated for each structure at the ωb97D/6-311 + g(2df,2p) level using the standard counterpoise method.67 This theoretical level was chosen since the combination of ωb97X-D and a triple-zeta basis set provides reasonable binding energies estimation of weakly-bonded systems.68 The choice of a reasonably-sized valence triple zeta basis set containing diffuse functions on heavy atoms for interaction energies ensures that the major components of interaction energies are better captured, compared to the smaller 6-31G(d,p) basis set that was used for optimizations. Although ωb97X-D generally provides slightly long hydrogen bond lengths and slightly over-estimated relative energies, we do not expect significant differences in the interaction energy trends for Gdm+ with different amino-acid classes, which is the focus of the current work. Further, since we focused on deriving the intrinsic interaction energy trends of different amino acid groups, a dielectric continuum was not implemented in our calculation. All QM calculations were carried out using Gaussian 16, rev. B.01.69
3 Results
Constitution of the dataset
The dataset contains a variety of protein types. It is important to note that it does not constitute a representative sample of the proteome: X-ray structures are normally obtained to provide information about the native conformation of proteins. Further, the data has been collected for reasons other than denaturation, since the number of proteins that are amenable to crystallization following denaturation is likely far smaller than the number that crystallize in their native state. Thus, it appears, from our analysis, that this subset is highly biased towards certain families. Most of the proteins in this dataset are either hydrolases (45.3%) or oxidoreductases (20.9%, Fig. 1A and Table S3, ESI†), which also constitute the highest and third-highest classes of protein crystal structures available in the PDB (https://www.rcsb.org/stats/explore/enzyme_classification_name). However, despite constituting only one-fifth of the dataset, oxidoreductases contribute the most (62%) Gdm+–protein interactions, which can be ascribed to their large size offering more opportunities for Gdm+ interactions (Table S3, ESI†). Hydrolases contribute only about 26% of the total Gdm+–protein interactions (Table S3, ESI†).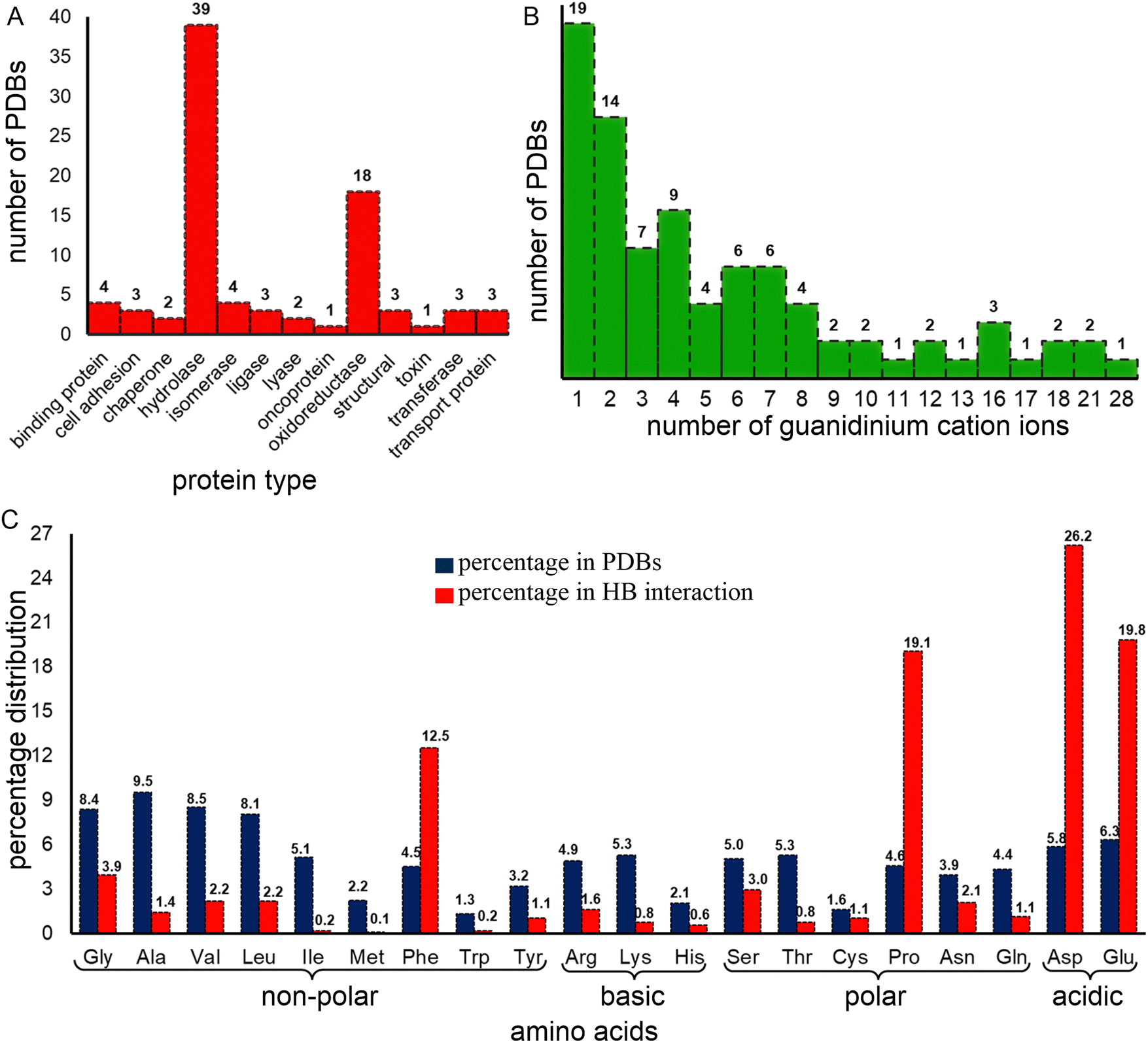 | ||
| Fig. 1 Distribution of the X-ray crystal structures based on (A) protein type and (B) number of guanidinium ions present. (C) Percent contribution of each amino acid to the proteins present in the structure dataset (red) and to the total number of hydrogen bonds involving Gdm+ (blue). | ||
From these 86 structures we identified 693 individual residues, including at least one example of all 20 proteogenic amino acids, interacting with 382 distinct Gdm+ ions, for a total of 1044 Gdm+–amino acid HBs. A single crystal structure (PDB code: 2ONP), human mitochondrial aldehyde dehydrogenase, accounts for 101 (∼10% of the total) HB interactions; this involves 67 of its 4000 residues and all 28 Gdm+ present in the crystal. 57 (66%) of the crystal structures contain at least 10 Gdm+–amino acid HB interactions, constituting 2/3 of interactions. However, 7 (8%) of the crystal structures with Gdm+ do not contain any Gdm+–protein HB interactions at all (Table S4, ESI†). 19 crystal structures contain only one Gdm+, 42% of which are hydrolases (Fig. 1B and Table S3, ESI†). †However, no preference for a particular protein type occurs for the 14 crystal structures that contain two Gdm+ (Fig. 1B and Table S3, ESI†). Finally, although hydrolases have three to six Gdm+ ions, crystal structures containing a higher number of Gdm+ ions are almost exclusively oxidoreductases.
Overall, the chosen crystal-structure dataset is varied, representing oligopeptides to large proteins (50 to 6000 residues, Table S3, ESI†), and anywhere between 1 and 28 Gdm+ ions. This represents significant diversity and so the dataset, although biased, is expected to be generalizable to Gdm+–protein HB interactions.
Each Gdm+ forms up to four HB interactions
77.8% of the Gdm+ ions present form HB interactions with proteins (Tables S5–S23, ESI†). One quarter form three HBs, accounting for 35.9% of total Gdm+–protein HB interactions. The next most common numbers of HBs per Gdm+ are four, two and one (18.9%, 18.3% and 13% respectively, Tables S5–S23, ESI†). 31.7% of the interacting Gdm+ ions HB with a single amino acid, the remainder interact with multiple amino acids (Table S24, ESI†). On average, the interacting residues have lower B-factors than the corresponding non-interacting entities due to hydrogen bonding, although the B-factors of interacting and non-interacting Gdm+ are similar (Table S25, ESI†). However, due to limited number of available crystal structures containing Gdm+–protein interactions, it is difficult to establish a relationship between B-factors and HB interactions. Regardless, examples of the classes of Gdm+ HB interactions with one or more amino acids, are provided in Fig. 2.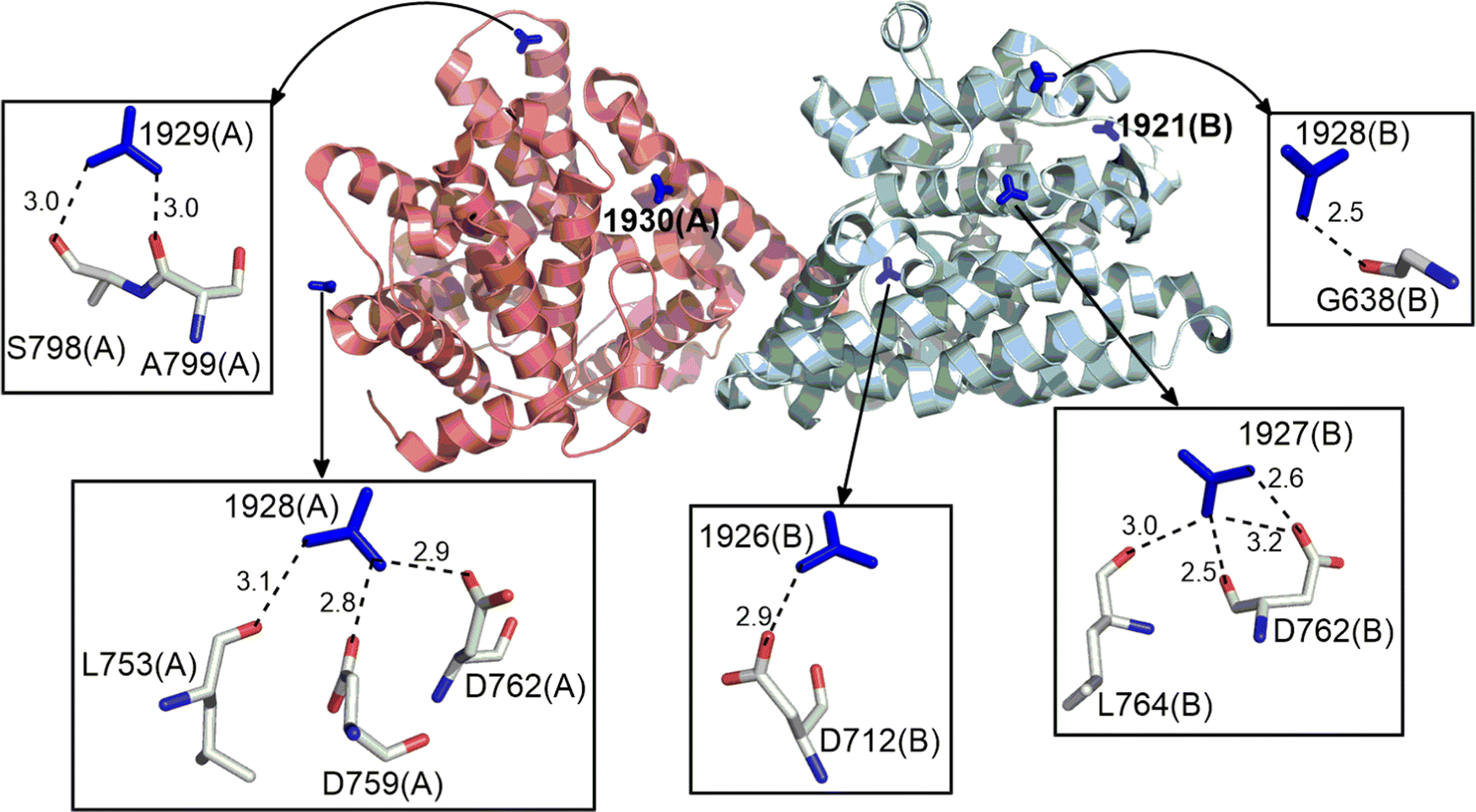 | ||
| Fig. 2 Examples of distinct HB interactions formed by Gdm+ ion with amino acid residues in the crystal structure of the catalytic domain of the PDE-B1 protein of Trypanosoma Brucei (PDB Code:5G57). Gdm+ are shown in stick form around the protein represented in cartoon form. Individual HB interactions between Gdm+ and amino acids are represented with stick models. Donor–acceptor distance (Å) is provided for each hydrogen bond. | ||
Despite their low frequency, acidic amino acids are more likely to interact with Gdm+, especially in crystal structures with few Gdm+ ions
Around half (52.2%) of the amino-acid residues in the crystal structures are non-polar (Ala, Gly, Leu, Ile, Met, Phe, Pro, Trp, and Val). They account for 41.9% of the Gdm+–protein HBs (Fig. 1C). Another 23.4% residues are polar uncharged (Asn, Cys, Gln, Ser, Thr, and Tyr) but are involved in only 9.1% of the Gdm+–protein interactions (Fig. 1C). In contrast, although both acidic (Asp and Glu) and basic (Arg, His and Lys) amino acid residues each constitute only 12% of residues, the two acidic amino acids are involved in 46% of Gdm+–protein HB interactions, while charged basic amino acids contribute only 3% (Fig. 1C). The insignificant contribution of charged basic amino acids is understandable, since Gdm+ is not expected to interact with their positively charged side chains.Approximately 26% of Gdm+–protein HB interactions involve Asp, followed by Glu (20%), Pro (19%) and Phe (12.5%). These four account for over three quarters of the total contacts (Fig. 3 and Table S26, ESI†). The remaining sixteen residues each form less than 50 contacts (<5%, Fig. 3 and Table S26, ESI†). As expected, the acidic residues clearly dominate due to the formation of strong ion-pair interactions. This is because many of the Gdm+ interactions are likely transient during solution phase denaturation, and only the strongest bonds, such as those formed by Gdm+ and acidic amino acids, would be conserved in the different unit cells, and will be detected by X-ray analysis.
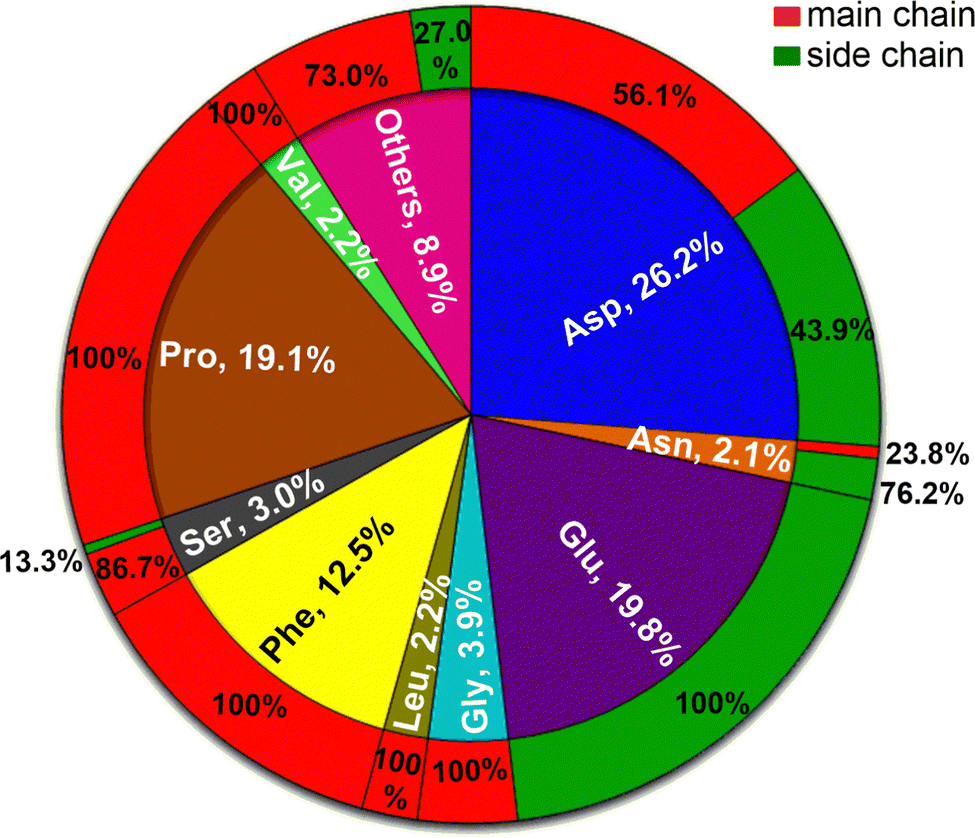 | ||
| Fig. 3 Statistical distribution of amino acids that hydrogen bond with Gdm+ in the analyzed crystal structures. Outer ring represents percentage distribution of the HB contacts as a function of amino acid chain where red represents main chain and green represents side chain of amino acid. | ||
It would be reasonable to believe that the percentage of interactions with any given residue would be proportional to the frequency of the residue of interest in the protein. For long proteins, this would likely regress to the mean values. Surprisingly, this was not the case. For example, constituting only 6.8% of the dataset, Asp is involved in 89% of the Gdm+ interactions (Table 1). Despite a frequency of ∼10%, Glu does not form even a single interaction with Gdm+ in structural proteins (Table 1). Further, at least one of Asp, Glu, Pro, Ser, and Asn possesses the highest interaction propensity in each protein class (Table 1 and Table S27, ESI†). The interactions involving these five amino acids can be divided into two mutually exclusive groups. Although interactions in chaperones, hydrolases, isomerase, lyases, oxidoreductases, structural proteins and transport proteins are dominated by Asp, Glu, and Pro; Ser and Asn interactions are more prominent for binding proteins, ligases, toxins, and transferases (Table 1). Thus, the predominance of interactions involving Ser and Asn in certain protein classes, despite the relatively smaller contribution of these residues towards protein constitution, as well as to the total number of interactions with Gdm+ in the total dataset (5.1%, Table 1) is noteworthy, and points towards the dependence of denaturation mechanism on the structural characteristics of each protein class. In terms of amino acid–Gdm+ hydrogen-bonded pairing motifs, Asp–Gdm+ and Glu–Gdm+ pairs are the most abundant (24.2% and 23.5% respectively), followed by Pro–Gdm+ (15.3%) and Phe–Gdm+ (10%) pairs. Altogether, these are responsible for around three-quarters of all amino acid–Gdm+ pairs (Table S28, ESI†).
| Protein type | # of PDBs | % hydrogen bonding interactions (occurrence frequency, percent occurrence in crystal structures) | ||||
|---|---|---|---|---|---|---|
| Asp | Glu | Pro | Ser | Asn | ||
| a Comma separated values in parentheses depict occurrence frequency and percent occurrence of each of the five residues within each protein class, respectively. | ||||||
| Binding protein | 4 | 7.7 (69, 4.7) | 0(97, 6.6) | 0(56, 3.8) | 0(55, 3.8) | 46.2(49, 3.3) |
| Cell adhesion | 3 | 37.5 (39, 9.5) | 0(12, 2.9) | 0(18, 4.4) | 0(24, 5.9) | 0(21, 5.1) |
| Chaperone | 2 | 25(35, 5.1) | 50(89, 12.9) | 25(28, 4.1) | 0(21, 3) | 0(10, 1.4) |
| Hydrolases | 39 | 37.2(1937, 6.5) | 28.5(2158, 7.3) | 0(1246, 4.2) | 5.5(1711, 5.8) | 3.6(1103, 3.7) |
| Isomerase | 4 | 19.2(167, 7) | 69.2(204, 8.6) | 0(115, 4.8) | 0(120, 5) | 0(44, 1.8) |
| Ligase | 3 | 22.2(56, 5.8) | 0(70, 7.3) | 0(29, 3) | 22.2(52, 5.4) | 44.4 (43, 4.5) |
| Lyase | 2 | 63.6(58, 6.1) | 18.2(65, 6.9) | 0(45, 4.8) | 0(48, 5.1) | 0(49, 5.2) |
| Onco-protein | 1 | 0(6, 3.1) | 100(8, 4.2) | 0(10, 5.2) | 0(10, 5.2) | 0(6, 3.1) |
| Oxido-reductase | 18 | 21.2(3772, 5.7) | 15.2(3689, 5.6) | 30.5(3285, 5) | 0(2911, 4.4) | 0.3(2762, 4.2) |
| Structural | 3 | 88.9(109, 6.8) | 0(154, 9.6) | 0(46, 2.9) | 0(97, 6.1) | 0(40, 2.5) |
| Toxin | 1 | 0(5, 2.4) | 0(5, 2.4) | 0(15, 7.1) | 50(20, 9.5) | 0(10, 4.7) |
| Transferase | 3 | 9.1(250, 5.9) | 13.6(333, 7.8) | 0(171, 4) | 36.4 (200, 4.7) | 0(142, 3.3) |
| Transport | 3 | 35.7(41, 5) | 21.4(51, 6.2) | 0(27, 3.3) | 35.7 (52, 6.3) | 0(19, 2.3) |
| Average values | 26.2(6544, 6) | 19.8(6935, 6.3) | 19.1(5091, 4.6) | 3(5321, 4.8) | 2.1(4298, 3.9) | |
Our analysis further reveals that the interactions involving Asp and Glu dominate in the crystal structures containing up to 6 Gdm+. On the other hand, in crystal structures containing 7, 8, 9 or 13 Gdm+ ions, interactions with Pro outnumber the interactions involving Asp and Glu (Table S29, ESI†). However, with a further increase in the number of Gdm+, interactions involving Phe exceed those of Glu and numerically compete with interactions involving Asp, and Pro (Table S29, ESI†). In the lone case of the crystal structure containing 28 Gdm+, interactions involving Gly dominate over all others, though as this is a single example, caution must be taken in overinterpretation (Table S29, ESI†). Together this data suggests a hierarchy of strengths of interaction. This order likely emerges from the context of how the dataset was collected.
Main-chain carbonyl groups interact more commonly with Gdm+ than sidechains
Gdm+–protein HB interactions are primarily mediated through the peptide bond carbonyl oxygens (64.3%, Fig. 3 and Tables S26 and S30, ESI†). The only residues where the side chains are important are the highly polar Glu (100%), His (83.3%), Tyr (82%), Asn (77.3%), Gln (58.3%), Thr (50%) and Asp (43.8%, Fig. 3 and Table S26, ESI†). In these side-chain interactions, the HBs are made with the oxygens (98.7%, Table S31, ESI†), with the obvious exception of His that interacts through its side-chain Nδ atom, albeit with negligible occurrence (0.5%, Tables S30–S32, ESI†). The sidechains of non-polar amino acids do not meaningfully interact with Gdm+ (Fig. 3 and Table S26, ESI†). Four polar residues do not interact at all with Gdm+. It is not surprising that positively charged Arg, and Lys do not interact with Gdm+, but that Cys and Met, whose sulfur is often a HB acceptor and donor (for Cys), do not is intriguing.Optimal geometries and interaction energies of Gdm+:amino acid pairs
We conducted QM geometry optimizations of each Gdm+–amino acid pair to determine the optimal, intrinsically-stable, HB patterns that drive Gdm+–protein interactions (Fig. 4 and Tables S33–S101, ESI†). Gdm+–amino acid pairs can be divided into four broad categories. The first category includes pairs that form bifurcated ([b] type) HB, which can be further divided in two classes – acceptor-bifurcated [ab] HB, which involves the interaction of two of the Gdm+ H atoms present on different nitrogens with a single amino acid acceptor atom, and donor bifurcated HB ([db] type), which involves two of the Gdm+ H atoms present on the same nitrogen with two different acceptor atoms of the amino acid (Fig. 4A and F). The [ab] complexes can be further categorized as either the [abm] subtype, involving a main chain carbonyl oxygen, or the [abs] subtype, involving an acceptor atom present on a side chain (O in case of Asn, Gln, Tyr, Glu and Asp, N in His and S in Met, Fig. 4A). Similarly, the [db] type complexes can be split into the [dbms] subtype, corresponding to a donor-bifurcated interaction involving the acceptor atoms of both main chain and side chain, and the [dbs] subtype which involves only the amino-acid side chain atoms as acceptors (Fig. 4B and F).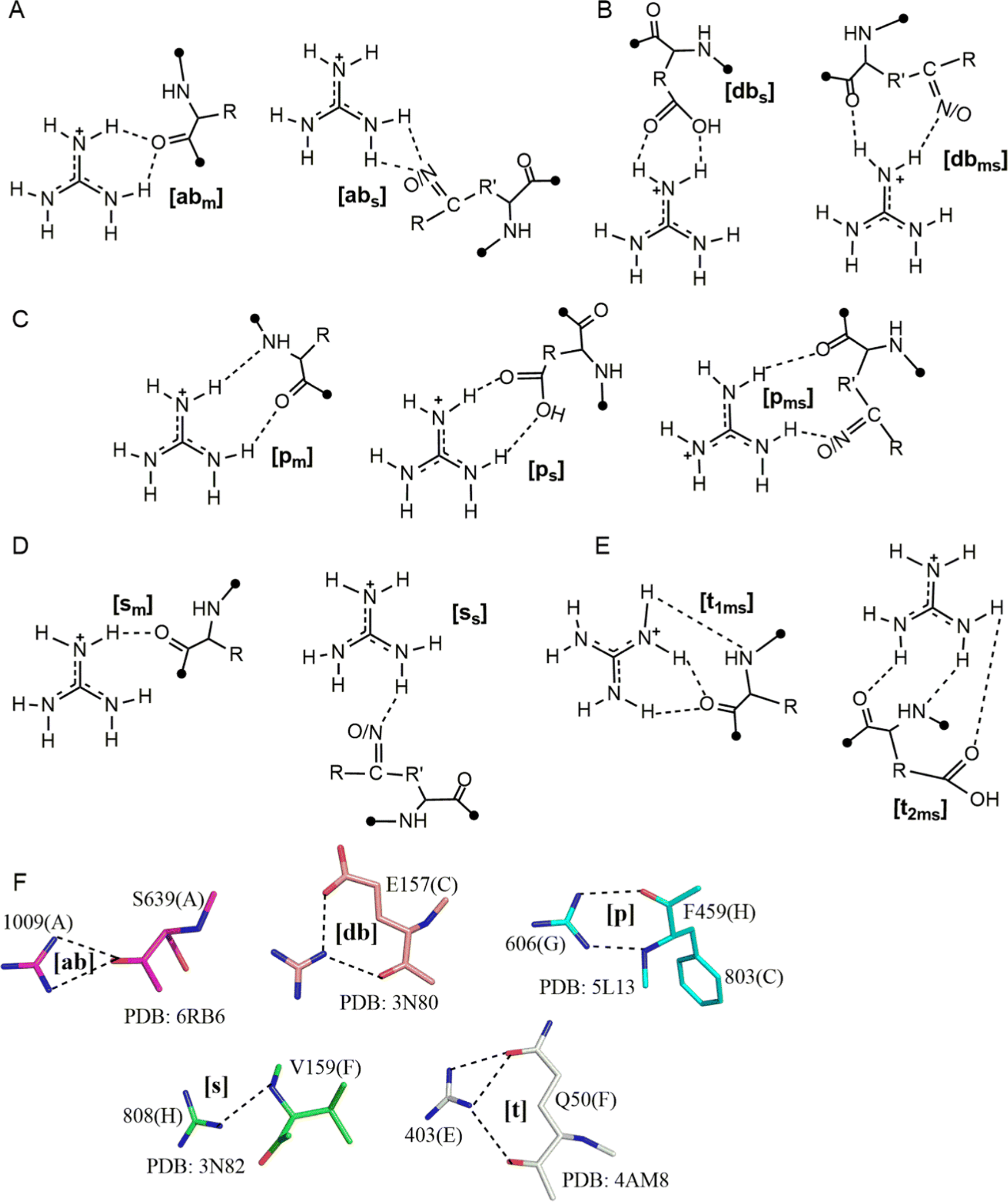 | ||
| Fig. 4 Different types of HB interaction patterns observed in optimized structures of Gdm+:amino acid, (A) represents [b] type acceptor-bifurcated HB pattern [abs] and [abm], (B) donor-bifurcated type [dbms] and [dbs], (C) represents [p] type HB interaction subtypes [pm][ps] and [pms] type, (D) represents [s] HB pattern [sm] and [ss] types and (E) represents [t] type, [t1ms] and [t2ms] subtypes. (F) Examples of different patterns obtained in crystal structure. This figure does not imply that multiple Gdm+ are interacting with a given residue at the same time; compression of the different modes onto a single structure is an abstraction for visualization purposes only, and values are based on only one Gdm+ per molecule. | ||
The second category involves a parallel HB interaction pattern ([p], Fig. 4C and F) where hydrogens on two different nitrogens of the Gdm+ ion act as donors. There are three types within this category: the [pm] type, involving two main chain HB acceptors; the [pms] type, involving one O/N acceptor from the backbone amide, with the second O/N acceptor on the side chain; and the [ps] type that involves two side-chain HB-acceptors (Fig. 4C). The third category ([s] type) involves pairs stabilized by a single Gdm+–amino acid hydrogen bond (Fig. 4D). Using the same nomenclature, this can be subdivided into [sm] and [ss] types, depending on whether the HB-acceptor atom (O or N) belongs to the main chain or the side chain of the amino acid (Fig. 4D and F). Finally, the fourth category ([t] type) involves three Gdm+ H atoms (two H atoms belonging to the same nitrogen and one H atom belonging to a different N, Fig. 4E and F). Based on the number of HB-acceptor atoms participating in HB, the [t] category can be further divided into the [t1ms] type, where one of the acceptors forms a bifurcated HB interaction while the other forms a conventional single HB, and the [t2ms] type where three separate acceptor atoms participate through conventional hydrogen bonds (Fig. 4E). The [t] category interactions inevitably involve both side chain and backbone atoms, and so are restricted to only a few amino acids.
Due to their preference for main chain interactions, nonpolar amino acids predominantly form [abm] and [pm] type complexes with Gdm+
QM optimizations reveal that [abm] and [pm] are the most prominent patterns in complexes of nonpolar amino acids with Gdm+. Further, interaction energies reveal that [pm] type complexes (up to −45.0 kcal mol−1) are more stable than their [abm] counterparts (up to −27.1 kcal mol−1, Fig. 5). However, although the participation of the Met side chain leads to [pms] type interaction with a significant strength (−24.2 kcal mol−1), both Trp and Met do not form [abm] type complexes. On the other hand, Ile lacks a [pm] type interaction. Further, an additional [sm] type complex was observed in case of Val with a significant (−27.1 kcal mol−1) stabilization energy (Fig. 5).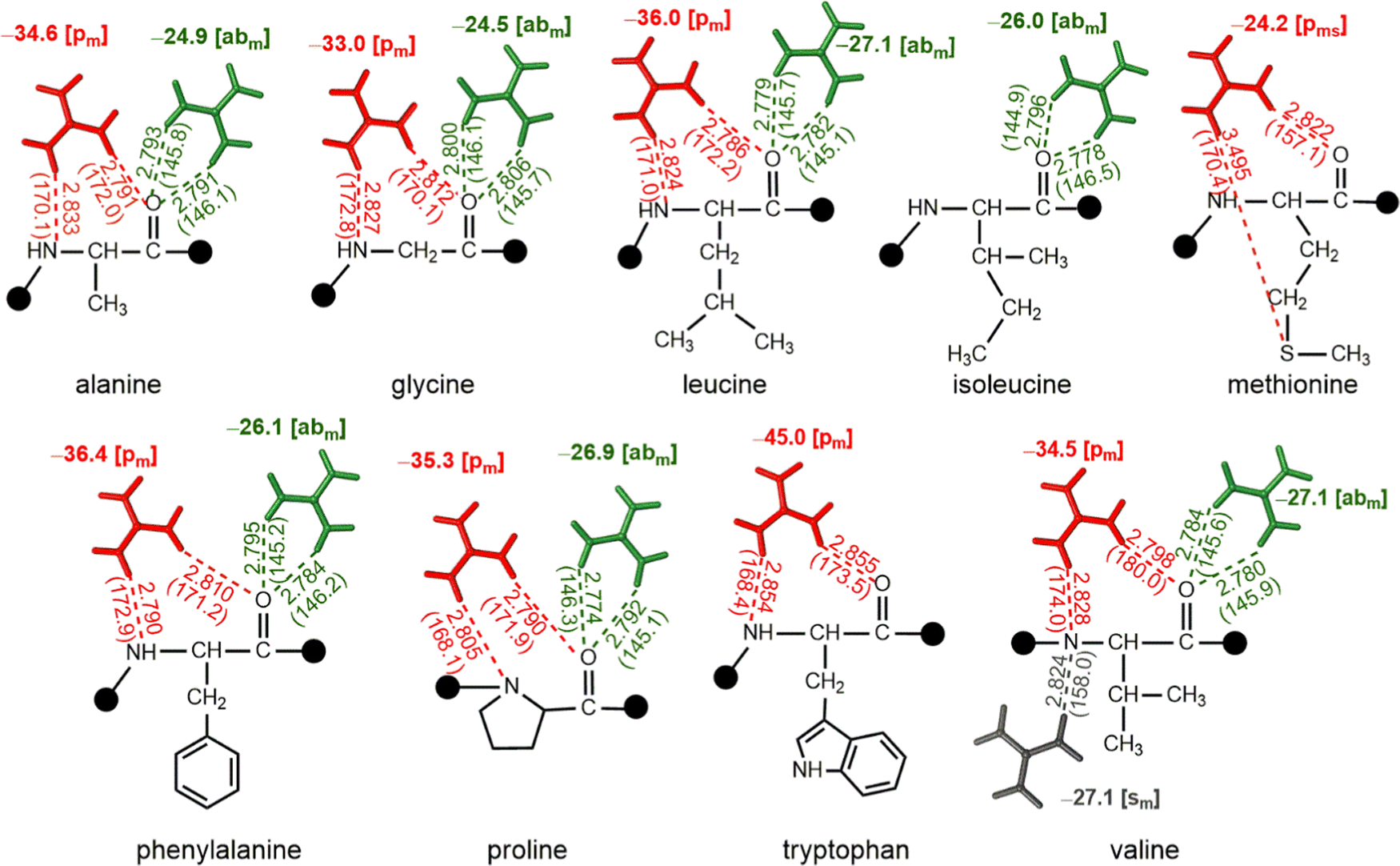 | ||
| Fig. 5 Interaction energies (bold, kcal mol−1) obtained from QM optimized hydrogen-bonded complexes at the ωB97XD/6-311 + G(2df,2p) level, involving the interaction of nonpolar amino acids with Gdm+. D–A distances (Å) and D–H–A angles (deg) are provided in parentheses, and the type of HB pattern is provided in brackets. This figure does not imply that multiple Gdm+ are interacting with a given residue at the same time; compression of the different modes onto a single structure is an abstraction for visualization purposes only, and values are based on only one Gdm+ per molecule. | ||
Interactions involving polar uncharged amino acids involve both main chain and side chain atoms
Except Cys, all polar uncharged amino acids interact with Gdm+ through both their main chain and side chain atoms. As a result, polar uncharged amino acids form a variety of interaction patterns. The [abm] and [abs] type interactions are preferred in Asn, Gln and Tyr, with the interaction energy ranging between −29.1 kcal mol−1 and −35.5 kcal mol−1 respectively (Fig. 6). The [abs] type interaction is notably weaker in the case of Tyr, with interaction energy of only −19.6 kcal mol−1, although the [dbms] type complex is only observed for Asn with interaction strength of −35.7 kcal mol−1 (Fig. 6). However, of all the possibilities, the [pm] type pattern is the most common; it occurs in five of the six polar uncharged amino acids, with interaction energies ranging from −32.7 kcal mol−1 to −45.7 kcal mol−1. The greatest stabilization occurs for Gln (Fig. 6). [pms] type complexes involving either O and N acceptor or two O atoms, are prevalent among Asn, Thr, Ser and Tyr, with up to −40.2 kcal mol−1 stabilization energy (Fig. 6). The [sm] type of pattern was observed in Ser, whereas the [ss] type was observed in case of Thr and Tyr, with interaction energies ranging from −21 kcal mol−1 to −35 kcal mol−1 (Fig. 6). The [t1ms] type complex was observed only in the case of Gln (which includes two carbonyl group of main chain and amide moiety of side chain) with a stabilization energy of −39.6 kcal mol−1 (Fig. 6).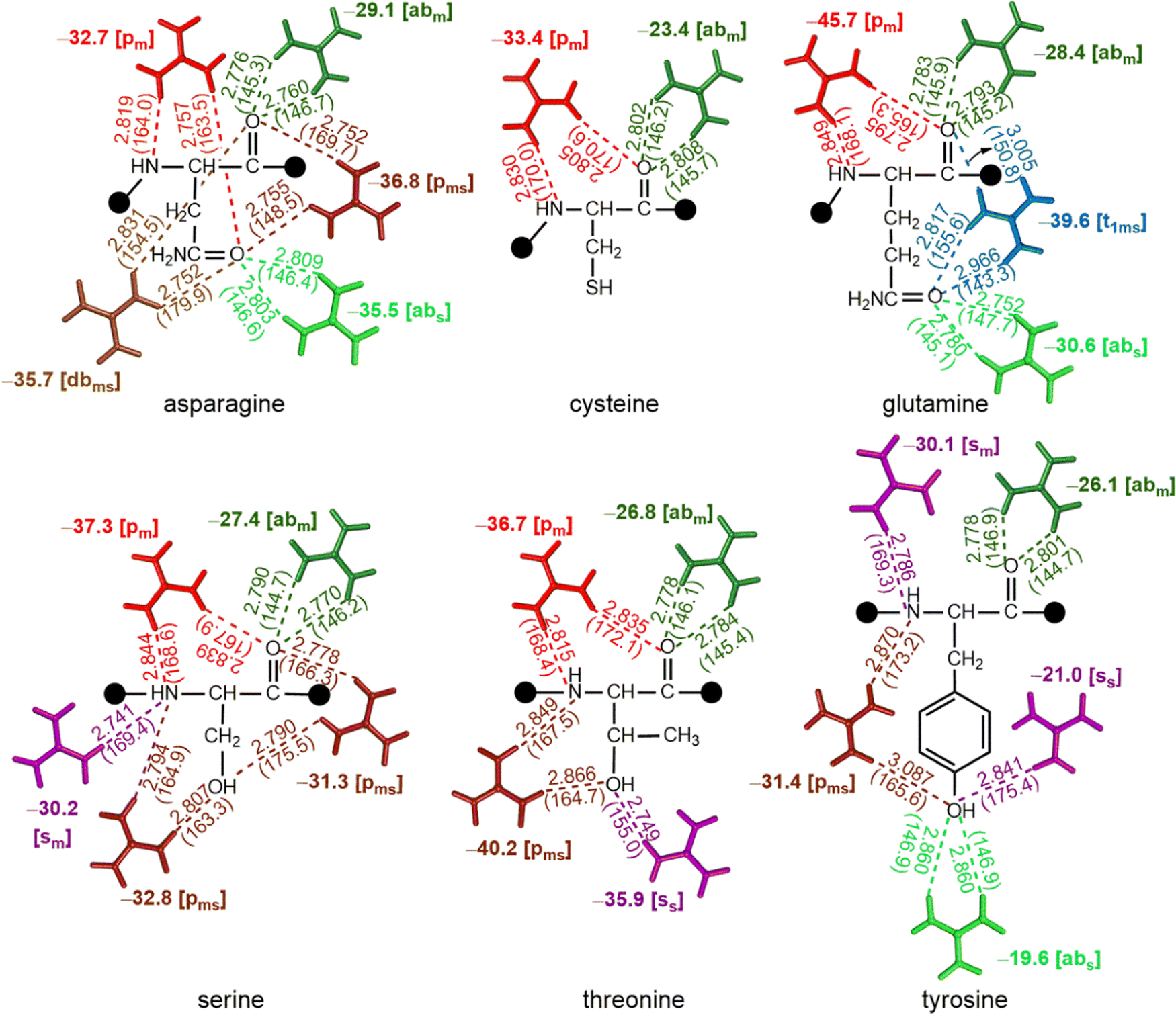 | ||
| Fig. 6 Interaction energies (bold, kcal mol−1) obtained from QM optimized hydrogen-bonded complexes at the ωB97XD/6-311 + G(2df,2p) level, involving the interaction of polar uncharged amino acids with Gdm+. D–A distances (Å) and D–H–A angles (deg) are provided in parentheses, and the type of HB pattern is provided in brackets. This figure does not imply that multiple Gdm+ are interacting with a given residue at the same time; compression of the different modes onto a single structure is an abstraction for visualization purposes only, and values are based on only one Gdm+ per molecule. | ||
Oppositely charged amino acid-Gdm+ pairs are highly stabilized, while like charged pairs are stabilized through contact pair interactions
At physiological pH, the acids on the side chains of Asp and Glu are negatively charged, whereas the side chain nitrogens of Lys, Arg and His are positively charged. Optimizations reveal that the [pms] type interaction dominates Gdm+–Glu and Gdm+–Asp complexes, although there are a few [b] type interactions (Fig. 7). These interactions either remain ion pairs or undergo a complete transfer of the hydrogen atom from Gdm+ ion to the HB acceptor atom of the amino acid, leading to a very strong acid-guanidine HB (Fig. 7). These are by far the strongest interactions we see, with stabilization energies of up to −134.3 kcal mol−1 (Fig. 7). These strongly attractive interactions could be the dominant mechanism by which Gdm+ destabilizes compact protein structures. However, the structures involving neutral guanidinium and neutral Asp that result from localization of H atom on carboxylic O of Asp instead of Gdm+ possess decreased interaction strength (up to −26 kcal mol−1, Fig. 7), mainly due to loss of strong electrostatic interactions that occur between positively charged Gdm+ and negatively charged amino acids. In addition, some weak (−17 kcal mol−1) [ss] type interactions were observed for both Asp and Glu (Fig. 7). Although the [pm] and [abm] type patterns were not observed, the [pms] type pattern strongly stabilizes the carboxylate–Gdm+ pairs (Fig. 7).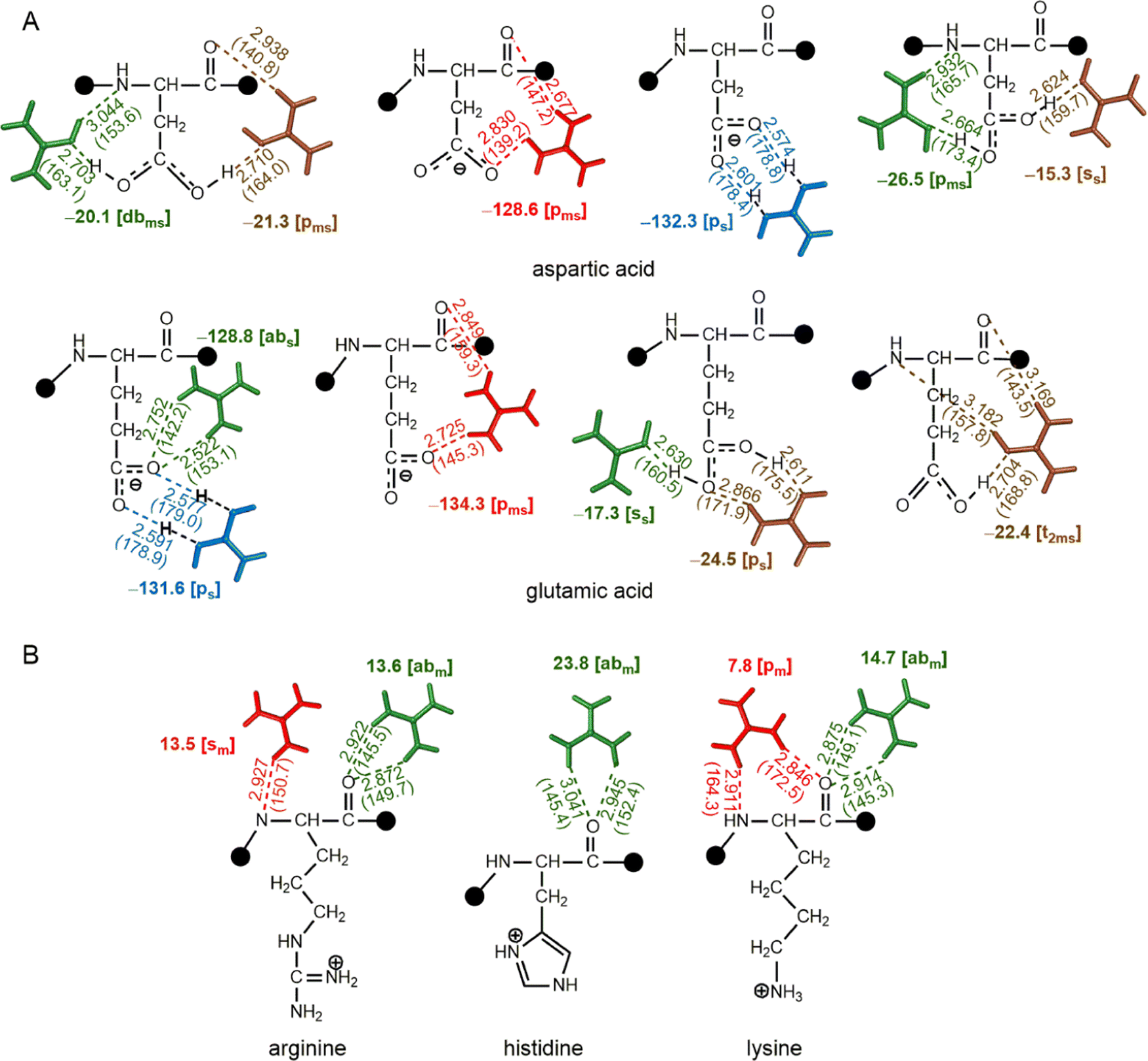 | ||
| Fig. 7 Interaction energies (bold, kcal mol−1) obtained from QM optimized hydrogen-bonded complexes at the ωB97XD/6-311 + G(2df,2p) level, involving the interaction of (A) deprotonated and (B) protonated amino acids with Gdm+. D–A distances (Å) and D–H–A angles (deg) are provided in parentheses, and the type of HB pattern is provided in brackets. This figure does not imply that multiple Gdm+ are interacting with a given residue at the same time; compression of the different modes onto a single structure is an abstraction for visualization purposes only, and values are based on only one Gdm+ per molecule. | ||
In contrast, protonated His, Lys, and Arg form optimized pairs only through their backbone atoms. However, the protonated amino acid–Gdm+ complexes must contend with repulsive interaction energies, ranging from +17 kcal mol−1 to +23 kcal mol−1 (Fig. 7). This means that the Gdm+ must localize as far from the side chain as possible to make a reasonable complex; this is clearly feasible, as the His, Lys, and Arg complexes were still found to optimize in intrinsically stable [abm] and [pm] type complexes (Fig. 7). These complexes align with the pairs involving guanidinium groups of protonated Arg and side chains of protonated His in protein structures.70,71
4 Discussion and conclusion
In the present work, we surveyed all available high-resolution X-ray PDB structures, and identified 1044 Gdm+–amino acid HB contacts involving 693 amino acid residues. Of these, 1039 contacts belong to N–H⋯O type, the remaining 5 are N–H⋯N type contacts involving the His side chain. Of all PDB entries, Gdm+–protein complexes were most abundant in oxidoreductases, which may be because of their long protein chains. Approximately 22% of Gdm+ present in crystal structures do not form HB contacts, as observed in a previous study,49 and are mainly located at the proteins interface or between two different chains of a protein. Further, Gdm+ commonly interacts with the main chain carbonyl group of amino acids, and the side-chain oxygen atoms of amino acids, although the side chains of nonpolar amino acids are not involved in Gdm+–amino acid contacts.A plethora of Asp–Gdm+ and Glu–Gdm+ motifs were found in the crystal structure dataset. This suggests that strong ionic interactions between Gdm+ and negatively charged side chains of acidic amino acids are preferred. In contrast, since the positive charge of charged basic amino acids repels the positively charged Gdm+, motifs involving basic amino acids are highly infrequent. Pro–Gdm+ pairs are frequently observed. Since Pro does not occur in α-helical regions, this suggests that a significant number of the Gdm+ in crystal structures interact at locations away from the α-helices of protein chains.
When working with protein X-ray crystal structures, it is important to always consider that they are only a model of the protein. Many highly flexible proteins cannot be crystallized without significant truncation or the formation of chimeras. Consequently, the available dataset is inherently enriched in highly rigid, symmetrical proteins as these pack better and are more likely to generate crystals. Similarly, many proteins function through dynamic flexibility, but one conformer is obtained in the crystal. Consequently, what we are seeing in all these structures is not the absolute “true” nature of the denatured protein, but rather a snapshot of the pathway from properly folded (likely 0 Gdm+) to fully denatured (effectively saturated with Gdm+). The crystals would be inherently biased towards the “properly folded” side of this continuum as these would be more ordered. Thus, we would expect to see a sequence of structures, starting with those with only the strongest Gdm+–protein interactions, fading through those with moderately-strong interactions, to those that are so stable and symmetric that they can crystallize with even moderate Gdm+ interactions. We would, however, not anticipate detecting weakly held, or fast exchanging, Gdm+–residue interactions in this dataset. Thus, when the crystal only contains the strongest bonds, Glu and Asp dominate as would be expected, whereas when the crystal also contains less tightly bound Gdm+, the interactions with Pro, then Phe, and finally Gly are present.
Our QM calculations reveal that all Gdm+–amino acid pairs undergo rearrangement and adjust their HB pattern on optimization. Regardless, four distinct types of stable interaction patterns were observed: bifurcated [b], parallel [p], single [s], and triple [t] HB type. The average interaction energies of the [b] and [s] type complexes are weaker (by 26.8 kcal mol−1) than [p] type complexes. The [t] type interaction further possesses 16.3 kcal mol−1 weaker average interaction than [p] type indicating the strong preference for parallel type HB interaction contacts (Fig. 8A). Further, the complexes of non-polar amino acids interact with Gdm+ through their main-chain acceptor atoms, and form [b] and [p] type complex, except in case of Met where the side chain S acceptor atom also participates in HB and forms a [p] type interaction. Since the side chains of non-polar amino acids are repelled from water and thereby form hydrophobic interactions that help maintain the protein tertiary structure, direct interaction of Gdm+ with backbone atoms of nonpolar amino acids may decrease the hydrophobic effect of these side chains and thus may promote solvation of hydrophobic side chains and may thereby aid in protein denaturation. Regardless, the nonpolar amino acids exhibit a moderate average interaction of up to −31 kcal mol−1, (Fig. 8B), except in case of Trp that possesses an unusually high average interaction strength of −45 kcal mol−1. It is important to recognize that these, and all other discussed, energy values are not benchmarked in this study, and are best considered relative strengths of interactions and care must be taken in interpreting the absolute impact of these interactions.
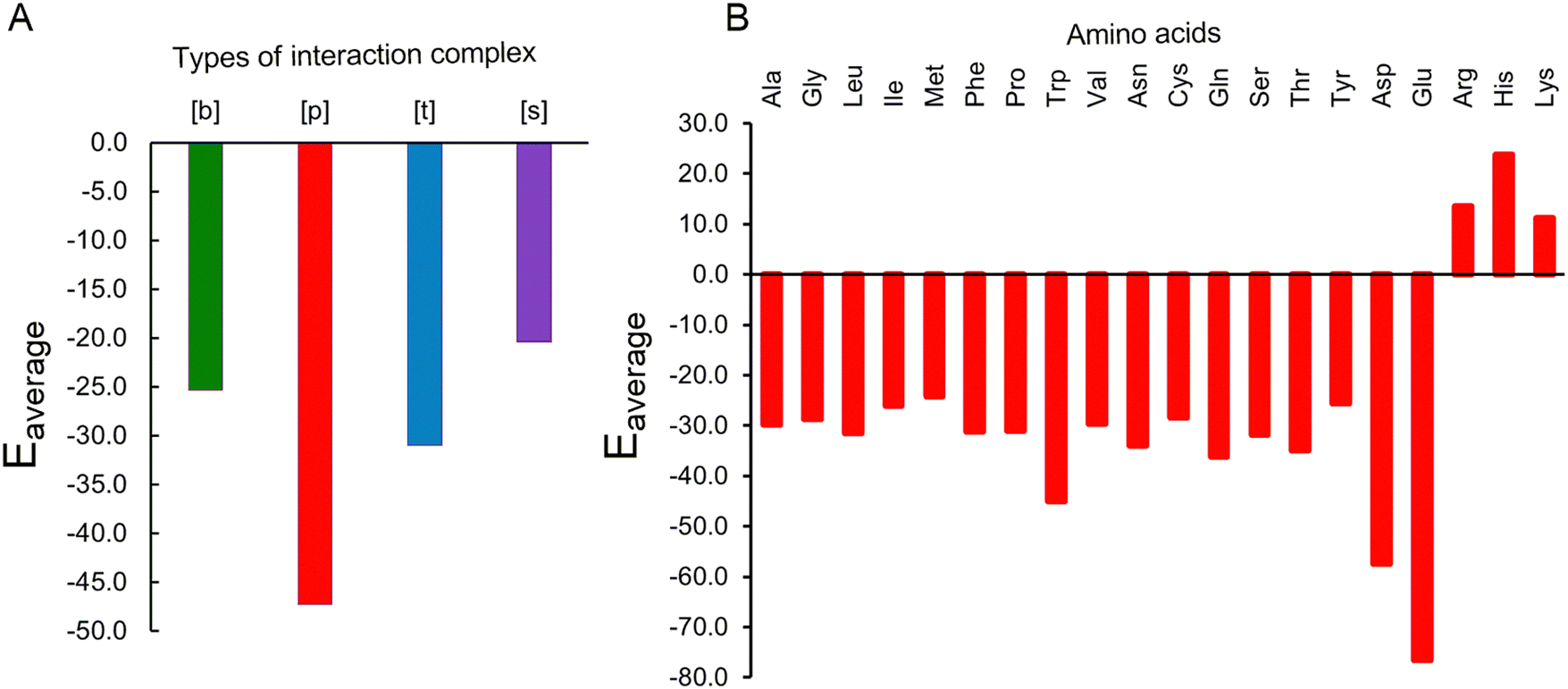 | ||
| Fig. 8 Interaction energies (kcal mol−1) obtained from averaging over (A) different types of interaction complexes and (B) different amino acids. | ||
In case of polar uncharged amino acids, apart from backbone atoms, side chains also participate in forming complexes. Since the polar uncharged residues are mostly present at the surface of proteins, they mostly interact within proteins through their side chain functional groups and the inter-residue HB involving these residues is vital for maintaining the protein structure. However, interaction with Gdm+ may disrupt the internal hydrogen bonds involving polar uncharged amino acids, which may cause the proteins to destabilize. In this context, it is worth mentioning that the complexes of nonpolar amino acids with Gdm+ exhibit significant average interaction energy ranging from −25.6 kcal mol−1 to −36.1 kcal mol−1 (Fig. 8B). These interactions thus appear to be strong enough to disrupt the native interactions that stabilize the protein tertiary structure.
Though the pairs involving Gdm+ and acidic amino acids exhibit diverse interactions, the complexes involving the charged forms of acidic amino acids form [p] type interactions and have higher interaction energy (−76.5 kcal mol−1) than complexes involving the neutral form of the acidic amino acids. This significant stabilization is due to enhanced electrostatic interactions between Gdm+ and negatively charged side chains of Asp and Glu. Such strong interactions have the propensity to loosen up the protein residue side chains participating in the formation of parent protein strands. On the other hand, despite being positively charged, protonated amino acids (His, Arg and Lys) form optimized pairs through their backbone atoms with similarly charged Gdm+. However, these complexes exhibit an average repulsive interaction energy of up to (+22 kcal mol−1), which correlates with the insignificant occurrence of such interactions in the crystal structures.
In conclusion, our calculations yield important insights into the abundance and strength of protein residue–Gdm+ interactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PS gratefully acknowledges the Department of Science and Technology (DST) and University Grants Commission (UGC), New Delhi, for financial support through the DST INSPIRE (IFA14-CH162) and the UGC FRP (F.4-5(176F-RP/2015(BSR)) programs, respectively. JFT thanks the Natural Sciences and Engineering Research Council of Canada (JFT: grant # 2018-06338), and the Canadian Tricouncil (JFT: NFRFE-2018-00075). PS and JFT wish to recognize that this work was made possible by the facilities of Compute Ontario (https://www.computeontario.ca) and the Digital Research Alliance of Canada (http://www.alliancecan.ca). IN and RJ thank UGC for research fellowship.References
- A. R. Fersht, Nat. Rev. Mol. Cell Biol., 2008, 9, 650–654 CAS.
- J. Frydman, Annu. Rev. Biochem., 2001, 70, 603 CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CAS.
- D. Reichmann, Y. Phillip, A. Carmi and G. Schreiber, Biochemistry, 2008, 47, 1051–1060 CAS.
- H. G. Wallnoefer, S. Handschuh, K. R. Liedl and T. Fox, J. Phys. Chem. B, 2010, 114, 7405–7412 CAS.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CAS.
- M. Heyden and D. J. Tobias, Phys. Rev. Lett., 2013, 111, 218101 Search PubMed.
- B. Halle, Philos. Trans. R. Soc., B, 2004, 359, 1207–1224 CAS.
- K. A. Dill and D. Shortle, Annu. Rev. Biochem., 1991, 60, 795–825 CAS.
- J. L. England and G. Haran, Annu. Rev. Phys. Chem., 2011, 62, 257–277 CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- K. D. Collins, Biophys. J., 1997, 72, 65–76 CAS.
- K. H. Dooley and F. J. Castellino, Biochemistry, 1972, 11, 1870–1874 CAS.
- C. Tanford, in Adv. Protein Chem., ed. C. B. Anfinsen, M. L. Anson, J. T. Edsall and F. M. Richards, Academic Press, 1968, vol. 23, pp. 121–282 Search PubMed.
- J. A. Schellman, Biophys. Chem., 2002, 96, 91–101 CAS.
- P. Mason, G. Neilson, C. Dempsey, A. Barnes and J. Cruickshank, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4557–4561 CAS.
- I. M. Pazos and F. Gai, J. Phys. Chem. B, 2012, 116, 12473–12478 CAS.
- S. T. van der Post, K.-J. Tielrooij, J. Hunger, E. H. Backus and H. J. Bakker, Faraday Discuss., 2013, 160, 171–189 CAS.
- P. E. Mason, J. W. Brady, G. W. Neilson and C. E. Dempsey, Biophys. J., 2007, 93, L04–L06 CAS.
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2007, 129, 7346–7353 Search PubMed.
- W. K. Lim, J. Rösgen and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2595–2600 CAS.
- V. Balos, M. Bonn and J. Hunger, Phys. Chem. Chem. Phys., 2015, 17, 28539–28543 CAS.
- J. K. Myers, C. Nick Pace and J. Martin Scholtz, Protein Sci., 1995, 4, 2138–2148 CAS.
- A. Huerta-Viga and S. Woutersen, J. Phys. Chem. Lett., 2013, 4, 3397–3401 CAS.
- B. Ding, D. Mukherjee, J. Chen and F. Gai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1003–1008 CAS.
- G. I. Makhatadze and P. L. Privalov, J. Mol. Biol., 1992, 226, 491–505 CAS.
- H. Meuzelaar, M. R. Panman and S. Woutersen, Angew. Chem., 2015, 127, 15470–15474 Search PubMed.
- A. Möglich, F. Krieger and T. Kiefhaber, J. Mol. Biol., 2005, 345, 153–162 Search PubMed.
- A. Q. Mohammad and M. Taha, Protein Pept. Lett., 2013, 20, 87–191 Search PubMed.
- C. Camilloni, A. G. Rocco, I. Eberini, E. Gianazza, R. Broglia and G. Tiana, Biophys. J., 2008, 94, 4654–4661 CAS.
- V. Balos, B. Marekha, C. Malm, M. Wagner, Y. Nagata, M. Bonn and J. Hunger, Angew. Chem., Int. Ed., 2019, 58, 332–337 CAS.
- A. Kubíčková, T. S. Křížek, P. Coufal, E. Wernersson, J. Heyda and P. Jungwirth, J. Phys. Chem. Lett., 2011, 2, 1387–1389 Search PubMed.
- C. Trujillo, A. A. Rodriguez-Sanz and I. Rozas, Molecules, 2015, 20, 9214–9228 CAS.
- D. Shukla, C. Shinde and B. L. Trout, J. Phys. Chem. B, 2009, 113, 12546–12554 CAS.
- K. Balamurugan, M. Prakash and V. Subramanian, J. Phys. Chem. B, 2019, 123, 2191–2202 CAS.
- J. N. Scott, N. V. Nucci and J. M. Vanderkooi, J. Phys. Chem. A, 2008, 112, 10939–10948 CAS.
- N. Samanta, D. D. Mahanta and R. K. Mitra, Phys. Chem. Chem. Phys., 2014, 16, 23308–23315 CAS.
- Y. Nozaki and C. Tanford, J. Biol. Chem., 1970, 245, 1648–1652 CAS.
- Y. Nozaki and C. Tanford, J. Biol. Chem., 1971, 246, 2211–2217 CAS.
- D. B. Watlaufer, S. K. Malik, L. Stoller and R. L. Coffin, J. Am. Chem. Soc., 1964, 86, 508–514 Search PubMed.
- D. R. Robinson and W. P. Jencks, J. Am. Chem. Soc., 1965, 87, 2462–2470 CAS.
- E. S. Courtenay, M. W. Capp and M. T. Record Jr, Protein Sci., 2001, 10, 2485–2497 CAS.
- O. D. Monera, C. M. Kay and R. S. Hodges, Protein Sci., 1994, 3, 1984–1991 CAS.
- F. Vanzi, B. Madan and K. Sharp, J. Am. Chem. Soc., 1998, 120, 10748–10753 CAS.
- W. Zheng, A. Borgia, K. Buholzer, A. Grishaev, B. Schuler and R. B. Best, J. Am. Chem. Soc., 2016, 138, 11702–11713 CAS.
- C. Trujillo, V. Previtali and I. Rozas, Theor. Chem. Acc., 2016, 135, 1–12 Search PubMed.
- F. Mehrnejad, M. Khadem-Maaref, M. M. Ghahremanpour and F. Doustdar, J. Comput.-Aided Mol. Des., 2010, 24, 829–841 CAS.
- B. Hernández, F. Pflüger and M. Ghomi, J. Comput. Chem., 2020, 41, 1402–1410 Search PubMed.
- S. Cozzolino, N. Balasco, M. Vigorita, A. Ruggiero, G. Smaldone, P. Del Vecchio, L. Vitagliano and G. Graziano, Int. J. Biol. Macromol., 2020, 163, 375–385 CAS.
- I. K. McDonald and J. M. Thornton, J. Mol. Biol., 1994, 238, 777–793 CAS.
- M. Treger and E. Westhof, J. Mol. Recognit., 2001, 14, 199–214 CAS.
- J. J. Ellis, M. Broom and S. Jones, Proteins: Struct., Funct., Genet., 2007, 66, 903–911 CAS.
- D. Kagra, R. Jangra and P. Sharma, Chem. Phys. Chem., 2022, 23, e202100731 CAS.
- S. Jones, D. T. Daley, N. M. Luscombe, H. M. Berman and J. M. Thornton, Nucleic Acids Res., 2001, 29, 943–954 CAS.
- A. Barik and R. P. Bahadur, Proteins: Struct., Funct., Genet., 2012, 80, 1866–1871 CAS.
- L. Schrodinger, Version, 2010, 1, 0 Search PubMed.
- K. S. Thanthiriwatte, E. G. Hohenstein, L. A. Burns and C. D. Sherrill, J. Chem. Theory Comput., 2011, 7, 88–96 CAS.
- L. M. da Costa, S. R. Stoyanov, S. Gusarov, X. Tan, M. R. Gray, J. M. Stryker, R. Tykwinski, J. W. D. M. Carneiro, P. R. Seidl and A. Kovalenko, Energy Fuels, 2012, 26, 2727–2735 CAS.
- A. D. Becke, Chem. Phys., 1997, 107, 8554–8560 CAS.
- A. Halder, A. Jhunjhunwala, D. Bhattacharyya and A. Mitra, bioRxiv. 2021, 426061.
- J. Antony, B. Brüske and S. Grimme, Phys. Chem. Chem. Phys., 2009, 11, 8440–8447 CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 CAS.
- P. Bhutani, D. J. Nikkel, K. A. Wilson and S. D. Wetmore, Chem. Res. Toxicol., 2019, 32, 2107–2117 Search PubMed.
- S. Kaur, P. Sharma and S. D. Wetmore, Phys. Chem. Chem. Phys., 2017, 19, 30762–30771 CAS.
- A. Ohri, P. P. Seelam and P. Sharma, J. Biomol. Struct. Dyn., 2021, 39, 5411–5426 CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CAS.
- L. A. Burns, Á. V. Mayagoitia, B. G. Sumpter and C. D. Sherrill, J. Chem. Phys., 2011, 134, 084107 Search PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, H. N. G. A. Petersson, X. Li, M. Caricato, A. V. Marenich, B. G. J. J. Bloino, R. Gomperts, B. Mennucci, H. P. Hratchian, A. F. I. J. V. Ortiz, J. L. Sonnenberg, D. Williams-Young, F. L. F. Ding, F. Egidi, J. Goings, B. Peng, A. Petrone, D. R. T. Henderson, V. G. Zakrzewski, J. Gao, N. Rega, W. L. G. Zheng, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, K. T. T. Vreven, J. A. Montgomery Jr., J. E. Peralta, M. J. B. F. Ogliaro, J. J. Heyd, E. N. Brothers, K. N. Kudin, T. A. K. V. N. Staroverov, R. Kobayashi, J. Normand, A. P. R. K. Raghavachari, J. C. Burant, S. S. Iyengar, M. C. J. Tomasi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, R. L. M. J. W. Ochterski, K. Morokuma, O. Farkas and A. D. J. F. J. B. Foresman, Gaussian 16, rev. B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Vazdar, J. Heyda, P. E. Mason, G. Tesei, C. Allolio, M. Lund and P. Jungwirth, Acc. Chem. Res., 2018, 51, 1455–1464 CAS.
- J. Heyda, P. E. Mason and P. Jungwirth, J. Phys. Chem. B, 2010, 114, 8744–8749 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04943k |
| ‡ Both authors contributed equally to this work. |
| § Dr John F. Trant, a co-corresponding author on this work, is also affiliated to Binary Star Research Services as a Chief Executive Officer. However, Binary Star had no input into the paper. |
| This journal is © the Owner Societies 2023 |