
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alchemical analysis of FDA approved drugs†
Markus
Orsi
 a,
Daniel
Probst
b,
Philippe
Schwaller
b and
Jean-Louis
Reymond
*a
a,
Daniel
Probst
b,
Philippe
Schwaller
b and
Jean-Louis
Reymond
*a
aDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: jean-louis.reymond@unibe.ch
bEcole Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
First published on 30th August 2023
Abstract
Chemical space maps help visualize similarities within molecular sets. However, there are many different molecular similarity measures resulting in a confusing number of possible comparisons. To overcome this limitation, we exploit the fact that tools designed for reaction informatics also work for alchemical processes that do not obey Lavoisier's principle, such as the transmutation of lead into gold. We start by using the differential reaction fingerprint (DRFP) to create tree-maps (TMAPs) representing the chemical space of pairs of drugs selected as being similar according to various molecular fingerprints. We then use the Transformer-based RXNMapper model to understand structural relationships between drugs, and its confidence score to distinguish between pairs related by chemically feasible transformations and pairs related by alchemical transmutations. This analysis reveals a diversity of structural similarity relationships that are otherwise difficult to analyze simultaneously. We exemplify this approach by visualizing FDA-approved drugs, EGFR inhibitors, and polymyxin B analogs.
Introduction
Mapping molecular databases in a chemical space where distances represent similarities between molecules helps to understand their structural similarities and identify relationships that can provide critical insights for drug development and related fields.1–15 However, molecular similarity can be computed in multiple ways,16,17 typically using various molecular fingerprints,18 resulting in a confusing multiplicity of possible chemical space representations.19,20To overcome this limitation and create a chemical space map considering various similarity measures simultaneously, we report a new approach of applying reaction informatics tools to map and analyze drug pairs, namely the differential reaction fingerprint (DRFP)21 and the Transformer-based RXNMapper model,22–24 respectively (Fig. 1). These tools were initially designed to analyze chemical reactions. However, they can also be applied to processes that do not obey Lavoisier's principle, the conservation of mass, such as the alchemical transmutation of lead into gold.25,26 Here, we apply them to transmutations between pairs of molecules selected for their similarity according to various molecular fingerprints as similarity measures, an approach related to the recent development of transformer models for drug optimization.27,28
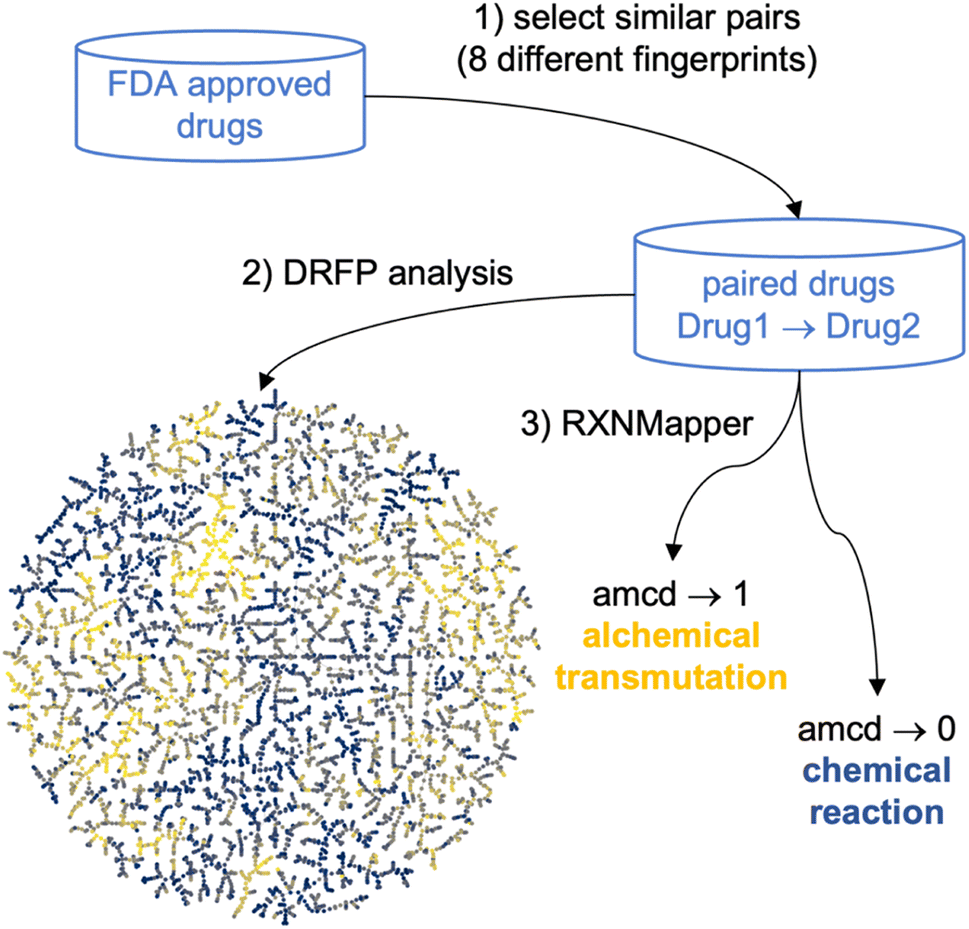 | ||
| Fig. 1 Principle of alchemical analysis of molecular sets at the example of FDA approved drugs. (1) Drugs pairs passing a similarity threshold according to eight different molecular fingerprints are selected. (2) The set of selected pairs is mapped in a TMAP computed using the differential reaction fingerprint (DRFP), color coded by the RXNmapper confidence distance (amcd). (3) The amcd distinguishes pairs of drugs related by a possible reaction (amcd → 0) from those related by an alchemical transmutation (amcd → 1). | ||
We start by using DRFP, which encodes chemical reactions by storing the symmetric difference of two sets containing the circular molecular n-grams generated from the molecules of the molecular pair as a binary fingerprint,21 to represent the chemical space of drug pairs as a TMAP (tree-map).29 A TMAP lays out the minimum spanning tree of the nearest neighbor graphs according to a selected similarity measure, here DRFP, and represents a remarkably efficient dimensionality reduction method for high-dimensional datasets. The DRFP TMAP visualization provides a global similarity perspective across drug pairs combining the selected similarity measures. We then use RXNMapper,22 a model trained on one million reactions documented in the USPTO dataset30 to pair corresponding atoms between reactants and products in a chemical reaction, to identify the structural relationship between drugs. The confidence score of this transformer appears not to correlate with any of the molecular similarity measures used. It allows us to distinguish drug pairs related by feasible chemical processes, such as matched molecular pairs corresponding to substituent exchanges,31,32 from those related by more esoteric, alchemical transmutations including scaffold-hopping changes.33,34 We demonstrate this approach with the example of FDA-approved drugs as a diversity set, as well as for a series of EGFR inhibitors and polymyxin B analogs as two high similarity sets chosen among small molecule drugs and peptide macrocyclic drugs, respectively.
Methods
Datasets
The set of FDA-approved drugs was downloaded from ZINC15,35,36 the SMILES were canonicalized and kekulized and duplicates were removed to obtain a set of 1213 unique chemical structures. For the EGFR set, all compounds binding to the tyrosine kinase erbB1 with a molecular weight <700 and an annotated IC50 value were downloaded from ChEMBL-31.37 After SMILES canonicalization and kekulization, duplicates were removed and the 1500 molecules with the highest ECFP4 Tanimoto similarity to afatinib were selected for the final set. The polymyxin B similarity set was downloaded from ChEMBL-31 by selecting compounds above the 55% ChEMBL similarity threshold with annotated MIC values. The SMILES were canonicalized and kekulized, and duplicates were removed, resulting in a final set of 274 structures.Molecular fingerprints and similarity calculations
Chemical structures were encoded as eight different fingerprints, namely extended connectivity fingerprints ECFP4 and ECFP6,38,39 the MinHashed Fingerprint MHFP6,40 the RDKit Atom-Pair Fingerprint (AP),41 the Macromolecule Extended Fingerprint (MXFP),42 the MinHashed Atom-Pair fingerprint MAP4,43 the Molecular ACCess System keys (MACCS),44 and Molecular Quantum Numbers (MQNs).45 ECFP4, ECFP6, AP, MACCS and MQN were calculated using the implementation in the RDKit package (2022.3.4., https://www.rdkit.org/). ECFPs were calculated as 2048-bit vectors. MHFP6 and MAP4 were calculated as 2048-bit vectors using the code described in https://github.com/reymond-group/mhfp and https://github.com/reymond-group/map4. MXFP was calculated using a new open-source version available at https://github.com/reymond-group/mxfp_python. The differential reaction fingerprint (DRFP)21 was calculated as 2048-bit vectors using the code available at https://github.com/reymond-group/drfp.Pairwise distances for every possible molecular pair were calculated and stored as a matrix for each fingerprint. Distances were calculated as Jaccard distances (dJ) for ECFP4, ECFP6, MHFP6, AP, MAP4 and MACCS keys, and as Taxicab distances (dT) for MXFP and MQNs, with values min–max standardized. We selected similar pairs by applying the following distance threshold: dJ < 0.6 for ECFP4, ECFP6, MHFP6, dJ < 0.5 for AP, dJ < 0.2 for MACCS, dJ < 0.8 for MAP4, dT < 0.1 for MXFP and dT < 0.05 for MQN (Taxicab distances after rescaling) for the FDA set and dJ < 0.2 for ECFP4, ECFP6, MHFP6, AP, dJ < 0.0125 for MACCS, dJ < 0.3 for MAP4, dT < 0.1 for MXFP and dT < 0.05 for MQN for the EGFR and PMB sets.
Additionally, the ranking of molecular pairs for every compound and fingerprint was calculated, resulting in 1213 ranked lists of 1213 pairs each for the FDA set, 1500 ranked lists of 1500 ranked pairs for the EGFR set and 274 ranked lists of 274 pairs for the polymyxin B similarity set for each fingerprint.
Violin plots to display the distribution of distances for every fingerprint and heatmaps to visualize correlations between fingerprints were generated using the seaborn (0.11.2) package. The pairwise distance distributions were balanced out by calculating the ranking of molecular pairs for every compound, resulting in 1213 ranked lists of 1213 pairs each for the FDA set, 1500 ranked lists of 1500 ranked pairs for the EGFR set and 274 ranked lists of 274 pairs for the polymyxin B similarity set.
Reaction informatics
A reaction SMILES in the form “SMILES1 » SMILES2” (forward reaction) as well as “SMILES2 » SMILES1” (backward reaction) was generated for every selected molecular pair. The forward reaction SMILES was generated to always have the molecule with the lower heavy atom count as a reactant and the molecule with the higher heavy atom count as a product. The reaction SMILES for each drug pair was then encoded using DRFP.21 The 20 nearest neighbors (NNs) in the DRFP feature space were extracted and the minimum spanning tree layout calculated using the TMAP package.29 The resulting layout was displayed interactively using Faerun.46 In addition, the atom-mapping and the corresponding atom-mapping confidence scores were computed for each drug pair reaction SMILES using the published model described in the RXNmapper22 GitHub repository https://github.com/rxn4chemistry/rxnmapper.Results and discussion
Datasets and selection of drug pairs
To test our reaction informatics approach to map drug space, we selected 1213 FDA-approved drugs as a representative high diversity set. As examples of a more focused series, we accessed the ChEMBL database37 and retrieved 1500 analogs of the small molecule drug afatinib, a kinase inhibitor blocking the endothelial growth factor receptor (EGFR) and used to treat non-small cell lung carcinoma (NSCLC),47 as well as 274 analogs of polymyxin B (PMB), an FDA-approved macrocyclic peptide natural product considered as a last resort antibiotic against multidrug-resistant bacteria.48To represent molecular similarities, we considered three types of molecular fingerprints. First, we selected the classical Morgan fingerprint,38 also called extended connectivity fingerprint (ECFP),39 which is a binary fingerprint encoding the presence of specific atom-centered circular substructures up to a diameter of four (ECFP4) and six (ECFP6) bonds, as well as our recently reported MinHashed fingerprint MHFP6,40 which similarly encodes circular substructures up to a diameter of six bonds using shingling and MinHashing to compress information.49 These circular substructure fingerprints are particularly efficient in virtual screening benchmarks40,50 and off-target prediction tasks.51,52 Second, we considered three pharmacophore fingerprints encoding the relative positions of atoms in a molecule and representing molecular shape, namely the RDKit atom-pair fingerprint AP,41 our recently reported macromolecule extended atom-pair fingerprint MXFP,42 and the MinHashed Atom-pair fingerprint up to a diameter of four bonds MAP4.43 Finally, we also included two composition fingerprints, namely MACCS keys44 and molecular quantum numbers (MQN),45 which encode the presence and number of features present in a molecule.
To identify relevant pairs in each of our three drug sets (FDA, EGFR and PMB), we computed all pairwise distances in each fingerprint as either Jaccard distance dJ (ECFP4, ECFP6, MHFP6, AP, MAP4, MACCS keys) or Taxicab distance dT (MXFP, MQN). For all fingerprints, distance zero indicates highest similarity. For each molecule in each set, we then selected the NN for each of the eight fingerprints, as well as any molecule appearing in at least seven of the eight lists of top-20 nearest neighbors. In addition, we selected all drug pairs having a certain similarity in each fingerprint by applying a maximum Jaccard distance (dJ) threshold (see Methods for details).
This selection corresponded to 6406 (0.87%) of the 735![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 078 possible drug pairs in the FDA set, 8932 (0.79%) of the 1124250 possible drug pairs in the EGFR set, and 8464 (22.63%) of the 37401 possible drug pairs in the PMB set. Each drug was represented in the selected pairs between 1 and 193 times in the FDA approved set, between 1 and 870 times in the EGFR set, and between 4 and 1031 times in the PMB set (Fig. S1†). Compared to the exhaustive list of drug pairs, the selected drug pairs were enriched in high similarity pairs with lower values of Jaccard distance (dJ). They spanned the entire similarity range in each fingerprint, reflecting the fact that the different fingerprints captured different similarity features (Fig. 2a/3a/4a). Distances were correlated between ECFP4, ECFP6, MHFP6, MAP4, which all encode circular substructures around atoms (r2 ∼ 0.8, Fig. 2b/3b/4b). However, correlations of MAP4 with other circular substructure fingerprints, particularly in the polymyxin B2 set, were generally lower. This can be attributed to its hybrid nature, which encodes both substructures and atom-pairs. Even so, the correlation between MAP4 and circular substructure fingerprints was notably stronger than its correlation with other fingerprint types. AP and MACCS, which both encode atomic features, were weakly correlated with each other and to a lesser extent with circular fingerprints (r2 ∼ 0.5). Finally, MQN and MXFP distances were partly correlated with each other (r2 ∼ 0.5) but not with any other fingerprints, probably because both fingerprints are size-dependent and count similar features in molecules.
078 possible drug pairs in the FDA set, 8932 (0.79%) of the 1124250 possible drug pairs in the EGFR set, and 8464 (22.63%) of the 37401 possible drug pairs in the PMB set. Each drug was represented in the selected pairs between 1 and 193 times in the FDA approved set, between 1 and 870 times in the EGFR set, and between 4 and 1031 times in the PMB set (Fig. S1†). Compared to the exhaustive list of drug pairs, the selected drug pairs were enriched in high similarity pairs with lower values of Jaccard distance (dJ). They spanned the entire similarity range in each fingerprint, reflecting the fact that the different fingerprints captured different similarity features (Fig. 2a/3a/4a). Distances were correlated between ECFP4, ECFP6, MHFP6, MAP4, which all encode circular substructures around atoms (r2 ∼ 0.8, Fig. 2b/3b/4b). However, correlations of MAP4 with other circular substructure fingerprints, particularly in the polymyxin B2 set, were generally lower. This can be attributed to its hybrid nature, which encodes both substructures and atom-pairs. Even so, the correlation between MAP4 and circular substructure fingerprints was notably stronger than its correlation with other fingerprint types. AP and MACCS, which both encode atomic features, were weakly correlated with each other and to a lesser extent with circular fingerprints (r2 ∼ 0.5). Finally, MQN and MXFP distances were partly correlated with each other (r2 ∼ 0.5) but not with any other fingerprints, probably because both fingerprints are size-dependent and count similar features in molecules.
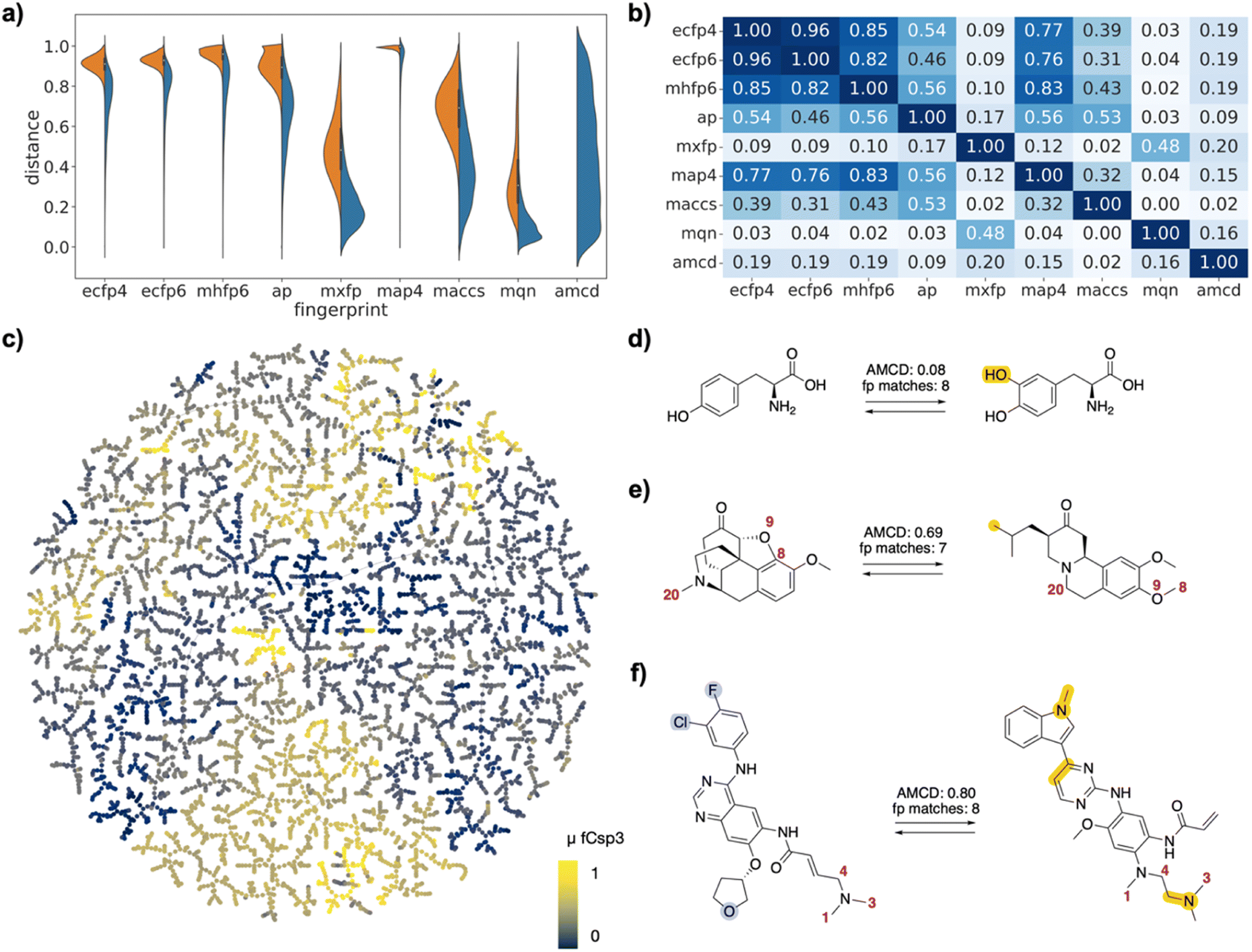 | ||
| Fig. 2 FDA-approved drugs as drug pairs. (a) Violin plot of dJ values in each of the fingerprints for all pairs (left, orange) or for selected pairs (right, blue), and for atom mapping confidence distance (amcd) of selected pairs (blue, last entry). (b) Heat map of correlation coefficients r2 between dJ values of different fingerprints, and between dJ values and amcd, calculated across all selected pairs. (c) TMAP of DRFP similarities for selected drug pairs. Each point is a different drug pair, color-coded by the fraction of sp3 atoms (Fsp3). See ESI† and https://tm.gdb.tools/map4/DRFP_FDA/ for additional color codes and for the interactive version of the map. (d) Atom-mapped drug pair L-tyrosine and L-DOPA related by a hydroxylation reaction. (e) Atom-mapped drug pair tetrabenazine and hydrocodone related by an alchemical double cyclization. (f) Atom-mapped drug pair afatinib and osimertinib related by a series of substituent and ring system changes. Atoms highlighted in blue are lost during the forward reaction, while atoms highlighted in yellow are gained. Interesting atom rearrangements as predicted by the RXNMapper are highlighted with their respective atom-mapping number. The full atom-mapping can be found in Fig. S3.† | ||
DRFP chemical space maps
To gain a closer insight into the pairwise relationships among the selected drug pairs, we represented each pair in the form of a reaction SMILES considering the conversion of one drug into the other. Form the reaction SMILES, we then computed the differential reaction fingerprint (DRFP),21 which encodes the circular substructures that occur only in either the reactant or the product. To represent the DRFP chemical space illustrating the similarities between different drug pairs, we then computed a tree-map (TMAP) providing an overview of drug pairs in each of the three datasets, using various color codes to visualize pair properties (Fig. 2c/3c/4c). The TMAP of DRFP similarities organized pairs by structural types, often series of close analogs of a reference drug. Furthermore, in the FDA-approved drug set, different compound families such as amino acids, steroids, β-lactams, catecholamines, benzodiazepines or prostaglandins appeared in different regions of the map. This was visible upon close inspection of the interactive TMAPs and is illustrated here for the FDA drug set with the color FCsp3 (Fig. 2c).Interactive browsing of the TMAPs made it very easy to inspect drug pairs with specific properties. For example, with the EGFR set, color-coding by activity differences pointed to the few similar drug pairs representing activity cliffs (Fig. 3c). Inspection of TMAPs was also key to identifying interesting pairs from the point of view of their transformations, as discussed below.
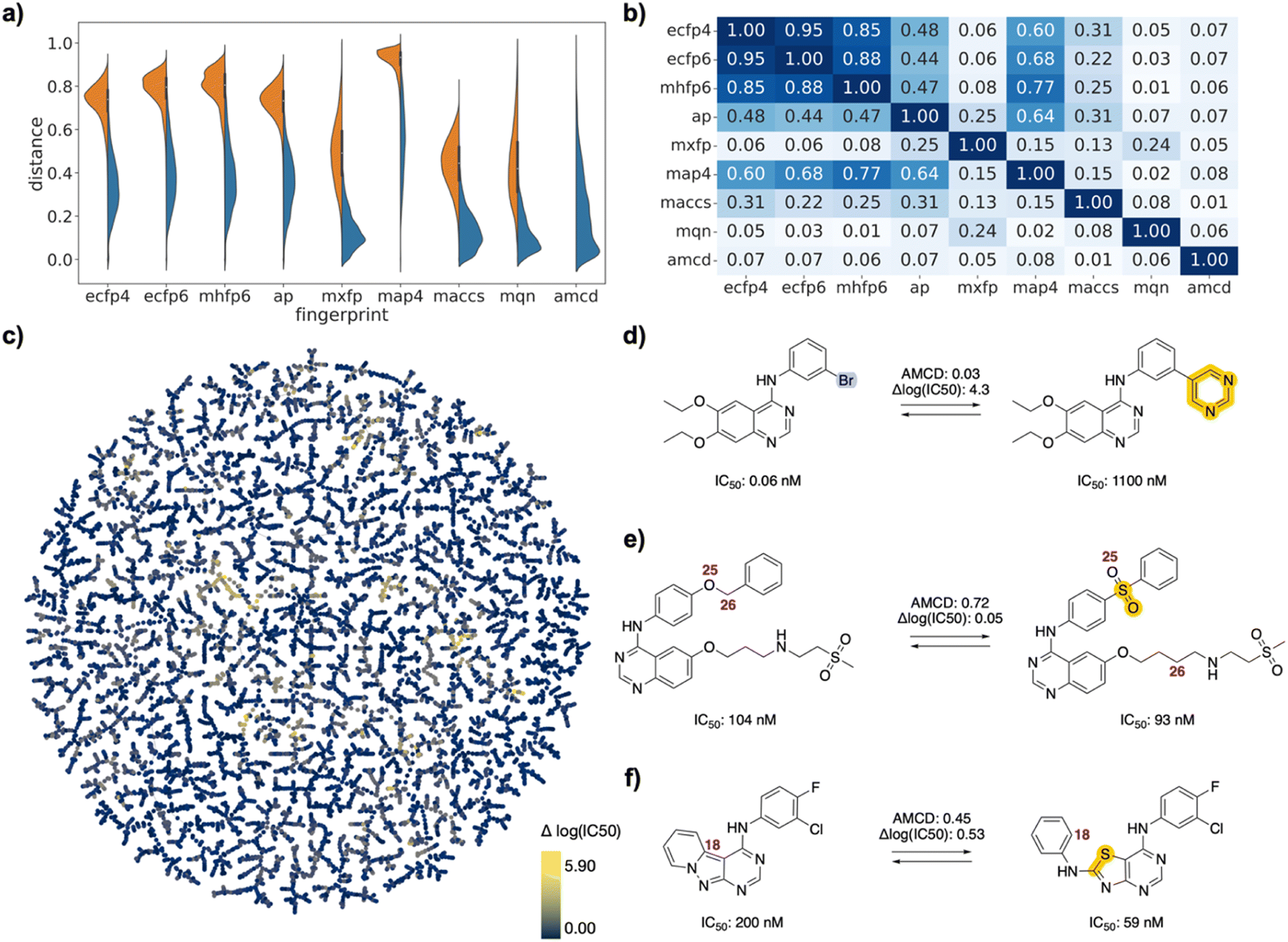 | ||
| Fig. 3 EGFR inhibitor drug pairs. (a) Violin plot of dJ values in each of the fingerprints for all pairs (left, orange) or for selected pairs (right, blue), and for atom mapping confidence distance (amcd) of selected pairs (blue, last entry). (b) Heat map of correlation coefficients r2 between dJ values of different fingerprints, and between dJ values and amcd, calculated across all selected pairs. (c) TMAP of activity differences. Each point is a different drug pair, color-coded by the activity difference. See ESI† and https://tm.gdb.tools/map4/DRFP_EGFR/for additional color codes and for the interactive version of the map. (d) Atom-mapped drug pair CHEMBL35820 and CHEMBL126974 related by a Suzuki coupling resulting in an activity cliff. (e) Atom-mapped drug pair CHEMBL460732 and CHEMBL14952 related by an alchemical double linker exchange preserving activity (f) Atom-mapped drug pair CHEMBL469997 and CHEMBL181275 related by an alchemical scaffold hopping preserving activity. Atoms highlighted in blue are lost during the forward reaction, while atoms highlighted in yellow are gained. Interesting atom rearrangements as predicted by the RXNMapper are highlighted with their respective atom-mapping number. The full atom-mapping can be found in Fig. S4.† | ||
Atom mapping
To estimate whether paired drugs were interconvertible by a feasible chemical reaction or required a more esoteric transmutation, we subjected the drug pair reaction SMILES to the Transformer-based RXNMapper model,22 which returns an atom-to-atom comparison illustrating the structural relationships within pairs, as well as an atom-mapping confidence score. Atom-mapping confidence scores were determined for the forward and backward reactions and converted to atom-mapping confidence distances (amcd), defined here as one minus the confidence score. In most cases the amcd values were similar for forward and backward reactions, however since the difference was sometimes substantial (Fig. S2†), we used the mean amcd of forward and backward reactions for our analysis. The mean amcd value spanned the entire range between low and high distance (last entry, Fig. 2a/3a/4a) except for the PMB set, which mainly contains high confidence distances as the structures are too big for the model to map with high confidence. Further, the amcd was not correlated with any of the selected molecular similarities (last entry, Fig. 2b/3b/4b).Low amcd values indicated drug pairs related by a simple and usually feasible chemical transformation, usually a functional group change or addition as those found in matched molecular pairs,31,32 illustrated in the FDA set for the hydroxylation of L-tyrosine to L-DOPA (Fig. 2d), and in the EGFR set for a Suzuki coupling resulting in a large activity change (Fig. 3d). In the case of the PMB set, low amcd values indicated pairs related by single amino acid exchange often potentially corresponding to a reaction, for example mutation of a glycine to a phenylalanine residue corresponding formally to an α-alkylation of glycine with benzyl bromide (Fig. S6†). This observation suggests that the amcd metric effectively captures chemically intuitive transformations, aligning well with the way chemists predict and perceive such changes in molecules during drug design and development.
On the other hand, high amcd values indicated alchemical transmutations that cannot be realized easily, such as scaffold-hopping changes.33,34 Note that the RXNMapper assigned corresponding atoms mostly in a correct manner even for pairs giving high amcd values. For example, tetrabenazine is paired with hydrocodone by seven of the eight molecule fingerprints used for pairing. The transformation features an exotic double-ring formation accompanied by a reshuffling of the 23 atoms (Fig. 2e). A similarly exotic alchemical change relates afatinib with osimertinib, an analog matched by all eight fingerprints used for pairing (Fig. 2f). In the EGFR set, a double linker modification preserving activity relates CHEMBL469997 to CHEMBL181275, whereby the benzyl ether linker is obtained by combining an oxygen atom of the sulfone with a methylene group of the aminobutanol second linker group (Fig. 3e). In another scaffold hopping change between CHEMBL469997 and CHEMBL181275, an aniline substituent is incorporated into the adjacent bicyclic system to form a condensed tricyclic heteroaromatic group, resulting in an interesting activity increase (Fig. 3f).
In the case of the PMB set, many pairs were generally related by high amcd values, probably because the changes corresponded to multiple amino acid exchanges, which cannot be realized on the complete molecules since each sequence analog requires a separate synthesis. Interestingly, one of the high amcd changes corresponds to a simple exchange of four aromatic aldehyde imines attached to the four diaminobutanoic acid residues, a reaction which would seem to be feasible (Fig. 4d). This imine exchange is however accompanied by a mutation of a leucine residue to a phenylalanine.
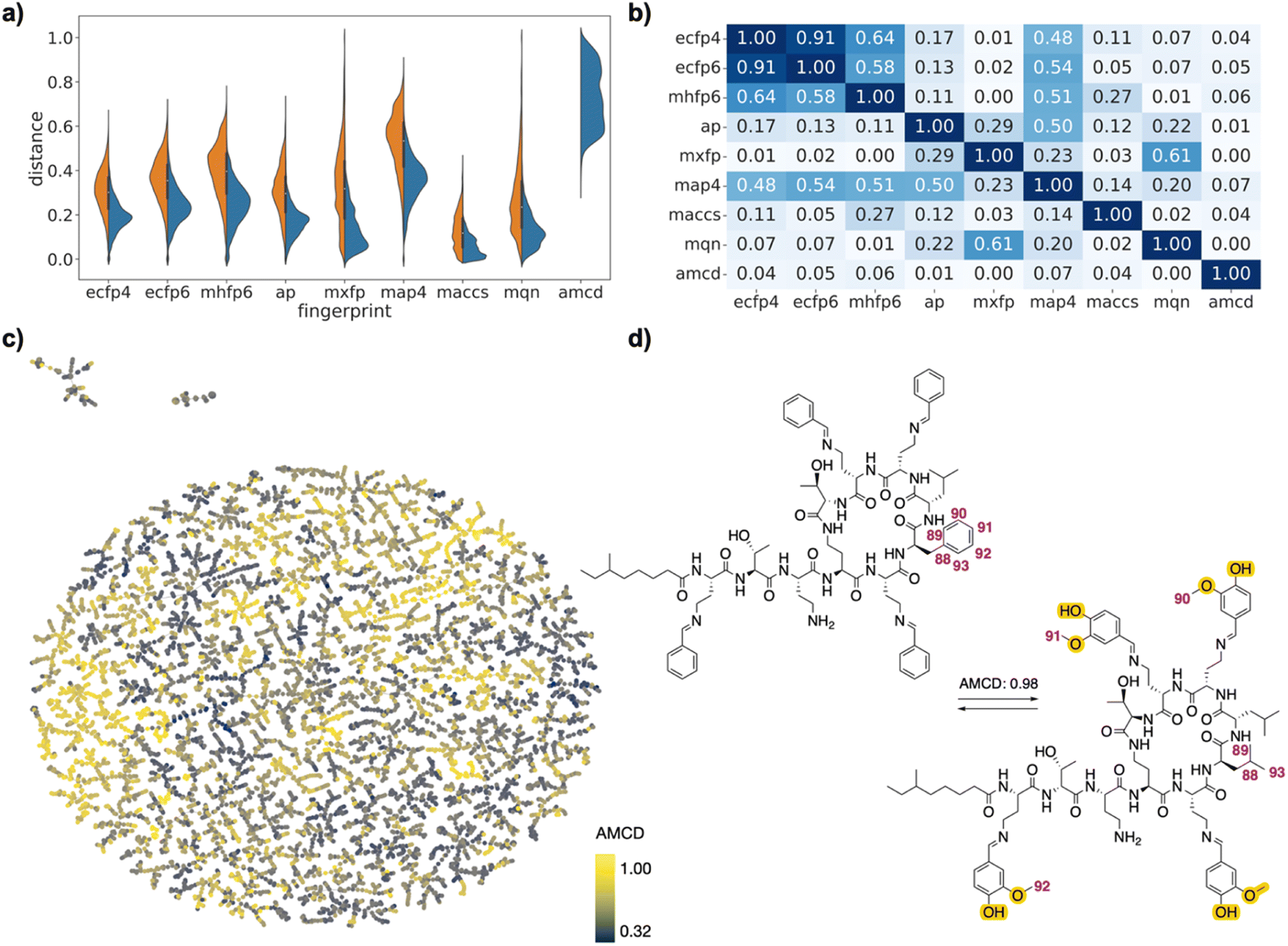 | ||
| Fig. 4 PMB analogs drug pairs. (a) Violin plot of dJ values in each of the fingerprints for all pairs (left, orange) or for selected pairs (right, blue), and for atom mapping confidence distance (amcd) of selected pairs (blue, last entry). (b) Heat map of correlation coefficients r2 between dJ values of different fingerprints, and between dJ values and amcd, calculated across all selected pairs. (c) TMAP of amcd values. Each point is a different drug pair, color-coded by the amcd value. See ESI† and https://tm.gdb.tools/map4/DRFP_PMB/for additional color codes and for the interactive version of the map. (d) Atom-mapped drug pair CHEMBL1090265 and CHEMBL2372545 related by an imine exchange and a leucine → phenylalanine mutation. Atoms highlighted in blue are lost during the forward reaction, while atoms highlighted in yellow are gained. Interesting atom rearrangements as predicted by the RXNMapper are highlighted with their respective atom-mapping number. The full atom-mapping can be found in Fig. S5.† | ||
Taken together, the analysis of the TMAP of similar drug pairs guided by DRFP similarity and amcd values allowed a rapid insight into multiple interesting comparisons between molecules in each of the three sets analyzed. Further examples of interesting pairs in the FDA approved set are provided in the ESI† (Fig. S7†).
Conclusion
In summary, we have shown that borrowing tools from reaction informatics provides an opportunity to map multiple similarity relationships between molecules simultaneously and gain insights into interesting drug pairs that are otherwise difficult to identify. Specifically, we used DRFP to map the chemical space of multiple drug pairs selected as being similar according to eight different molecular fingerprints simultaneously in the form of TMAPs. We then used RXNMapper to visualize the structural changes between drugs and identify pairs of drugs related by feasible chemical transformation from pairs related by alchemical changes corresponding to multiple and complex structural rearrangements. These tools should generally be applicable to analyze drug sets from multiple angles in the context of drug discovery. One specific case could be the analysis of analog series obtained from generative models53,54 to help identify feasible transformations or single out scaffold hopping changes.Code availability
The source codes and datasets used for this study are available at https://github.com/reymond-group/alchemical_pairs.Author contributions
MO designed and realized the project and wrote the paper. DP provided support for the DRFP implementation and wrote the paper. PS provided support for the RXNmapper implementation and wrote the paper. JLR designed and supervised the project and wrote the paper. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation (200020_178998) and the European Research Council (885076).References
- T. I. Oprea and J. Gottfries, Chemography: The Art of Navigating in Chemical Space, J. Comb. Chem., 2001, 3(2), 157–166 CrossRef CAS PubMed.
- A. Schuffenhauer, P. Ertl, S. Roggo, S. Wetzel, M. A. Koch and H. Waldmann, The Scaffold Tree – Visualization of the Scaffold Universe by Hierarchical Scaffold Classification, J. Chem. Inf. Model., 2007, 47(1), 47–58, DOI:10.1021/ci600338x.
- Y. A. Ivanenkov, N. P. Savchuk, S. Ekins and K. V. Balakin, Computational Mapping Tools for Drug Discovery, Drug Discovery Today, 2009, 14(15–16), 767–775, DOI:10.1016/j.drudis.2009.05.016.
- A. M. Wassermann, M. Wawer and J. Bajorath, Activity Landscape Representations for Structure−Activity Relationship Analysis, J. Med. Chem., 2010, 53(23), 8209–8223, DOI:10.1021/jm100933w.
- S. Sharma, A. Arya, R. Cruz and H. J. Cleaves II, Automated Exploration of Prebiotic Chemical Reaction Space: Progress and Perspectives, Life, 2021, 11(11), 1140, DOI:10.3390/life11111140.
- M. Andronov, M. V. Fedorov and S. Sosnin, Exploring Chemical Reaction Space with Reaction Difference Fingerprints and Parametric T-SNE, ACS Omega, 2021, 6(45), 30743–30751, DOI:10.1021/acsomega.1c04778.
- A. Capecchi and J.-L. Reymond, Classifying Natural Products from Plants, Fungi or Bacteria Using the COCONUT Database and Machine Learning, J. Cheminf., 2021, 13(1), 82, DOI:10.1186/s13321-021-00559-3.
- A. Vriza, I. Sovago, D. Widdowson, V. Kurlin, P. A. Wood and M. S. Dyer, Molecular Set Transformer: Attending to the Co-Crystals in the Cambridge Structural Database, Digit. Discov., 2022, 1(6), 834–850, 10.1039/D2DD00068G.
- J. L. Medina-Franco, N. Sánchez-Cruz, E. López-López and B. I. Díaz-Eufracio, Progress on Open Chemoinformatic Tools for Expanding and Exploring the Chemical Space, J. Comput.-Aided Mol. Des., 2022, 36(5), 341–354, DOI:10.1007/s10822-021-00399-1.
- C. Humer, H. Heberle, F. Montanari, T. Wolf, F. Huber, R. Henderson, J. Heinrich and M. Streit, ChemInformatics Model Explorer (CIME): Exploratory Analysis of Chemical Model Explanations, J. Cheminf., 2022, 14(1), 21, DOI:10.1186/s13321-022-00600-z.
- M. Beckers, N. Fechner and N. Stiefl, 25 Years of Small-Molecule Optimization at Novartis: A Retrospective Analysis of Chemical Series Evolution, J. Chem. Inf. Model., 2022, 62(23), 6002–6021, DOI:10.1021/acs.jcim.2c00785.
- Y. Zabolotna, F. Bonachera, D. Horvath, A. Lin, G. Marcou, O. Klimchuk and A. Varnek, Chemspace Atlas: Multiscale Chemography of Ultralarge Libraries for Drug Discovery, J. Chem. Inf. Model., 2022, 62(18), 4537–4548, DOI:10.1021/acs.jcim.2c00509.
- M. Cihan Sorkun, D. Mullaj, J. M. V. A. Koelman and S. Er, ChemPlot, a Python Library for Chemical Space Visualization, Chem.: Methods, 2022, 2(7), e202200005, DOI:10.1002/cmtd.202200005.
- M. Han, S. Liu, D. Zhang, R. Zhang, D. Liu, H. Xing, D. Sun, L. Gong, P. Cai, W. Tu, J. Chen and Q.-N. Hu, AddictedChem: A Data-Driven Integrated Platform for New Psychoactive Substance Identification, Molecules, 2022, 27(12), 3931, DOI:10.3390/molecules27123931.
- S. Moshawih, P. Hadikhani, A. Fatima, H. P. Goh, N. Kifli, V. Kotra, K. W. Goh and L. C. Ming, Comparative Analysis of an Anthraquinone and Chalcone Derivatives-Based Virtual Combinatorial Library. A Cheminformatics “Proof-of-Concept” Study, J. Mol. Graphics Modell., 2022, 117, 108307, DOI:10.1016/j.jmgm.2022.108307.
- A. Nicholls, G. B. McGaughey, R. P. Sheridan, A. C. Good, G. Warren, M. Mathieu, S. W. Muchmore, S. P. Brown, J. A. Grant, J. A. Haigh, N. Nevins, A. N. Jain and B. Kelley, Molecular Shape and Medicinal Chemistry: A Perspective, J. Med. Chem., 2010, 53(10), 3862–3886, DOI:10.1021/jm900818s.
- G. Maggiora, M. Vogt, D. Stumpfe and J. Bajorath, Molecular Similarity in Medicinal Chemistry, J. Med. Chem., 2014, 57(8), 3186–3204, DOI:10.1021/jm401411z.
- P. Willett, Similarity-Based Virtual Screening Using 2D Fingerprints, Drug Discovery Today, 2006, 11(23–24), 1046–1053, DOI:10.1016/j.drudis.2006.10.005.
- M. Awale and J. L. Reymond, Similarity Mapplet: Interactive Visualization of the Directory of Useful Decoys and ChEMBL in High Dimensional Chemical Spaces, J. Chem. Inf. Model., 2015, 55(8), 1509–1516, DOI:10.1021/acs.jcim.5b00182.
- J. Jesús Naveja and J. L. Medina-Franco, ChemMaps: Towards an Approach for Visualizing the Chemical Space Based on Adaptive Satellite Compounds, F1000Research, 2017, 6 DOI:10.5256/F1000RESEARCH.12095.D171632.
- D. Probst, P. Schwaller and J.-L. Reymond, Reaction Classification and Yield Prediction Using the Differential Reaction Fingerprint DRFP, Digit. Discov., 2022, 1(2), 91–97, 10.1039/D1DD00006C.
- P. Schwaller, B. Hoover, J.-L. Reymond, H. Strobelt and T. Laino, Extraction of Organic Chemistry Grammar from Unsupervised Learning of Chemical Reactions, Sci. Adv., 2021, 7(15), eabe4166, DOI:10.1126/sciadv.abe4166.
- J. Devlin; M.-W. Chang; K. Toutanova: Pre-Training of Deep Bidirectional Transformers for Language Understanding, arXiv, 2019, preprint, arXiv:1810.04805, DOI:10.48550/arXiv.1810.04805.
- Z. Lan; M. Chen; S. Goodman; K. Gimpel; P. Sharma; R. Soricut: A Lite BERT for Self-Supervised Learning of Language Representations, arXiv, 2020, preprint, arXiv.1909.11942, DOI:10.48550/arXiv.1909.11942.
- P. Ball, Alchemical Culture and Poetry in Early Modern England, Interdiscip. Sci. Rev., 2006, 31(1), 77–92, DOI:10.1179/030801806X84246.
- C. Wentrup, Chemistry, Medicine, and Gold-Making: Tycho Brahe, Helwig Dieterich, Otto Tachenius, and Johann Glauber, ChemPlusChem, 2023, 88(1), e202200289, DOI:10.1002/cplu.202200289.
- J. He, H. You, E. Sandström, E. Nittinger, E. J. Bjerrum, C. Tyrchan, W. Czechtizky and O. Engkvist, Molecular Optimization by Capturing Chemist's Intuition Using Deep Neural Networks, J. Cheminf., 2021, 13(1), 26, DOI:10.1186/s13321-021-00497-0.
- J. He, E. Nittinger, C. Tyrchan, W. Czechtizky, A. Patronov, E. J. Bjerrum and O. Engkvist, Transformer-Based Molecular Optimization beyond Matched Molecular Pairs, J. Cheminf., 2022, 14(1), 18, DOI:10.1186/s13321-022-00599-3.
- D. Probst and J.-L. Reymond, Visualization of Very Large High-Dimensional Data Sets as Minimum Spanning Trees, J. Cheminf., 2020, 12(1), 12, DOI:10.1186/s13321-020-0416-x.
- D. Lowe, Chemical Reactions from US Patents (1976-Sep2016). figshare. dataset, 2017, DOI: DOI:10.6084/m9.figshare.5104873.v1.
- C. Kramer, J. E. Fuchs, S. Whitebread, P. Gedeck and K. R. Liedl, Matched Molecular Pair Analysis: Significance and the Impact of Experimental Uncertainty, J. Med. Chem., 2014, 57(9), 3786–3802, DOI:10.1021/jm500317a.
- M. Awale, S. Riniker and C. Kramer, Matched Molecular Series Analysis for ADME Property Prediction, J. Chem. Inf. Model., 2020, 60(6), 2903–2914, DOI:10.1021/acs.jcim.0c00269.
- G. Schneider, W. Neidhart, T. Giller and G. Schmid, “Scaffold-Hopping” by Topological Pharmacophore Search: A Contribution to Virtual Screening, Angew Chem. Int. Ed. Engl., 1999, 38(19), 2894–2896 CrossRef CAS PubMed.
- H.-J. Böhm, A. Flohr and M. Stahl, Scaffold Hopping, Drug Discovery Today: Technol., 2004, 1(3), 217–224, DOI:10.1016/j.ddtec.2004.10.009.
- T. Sterling and J. J. Irwin, ZINC 15 – Ligand Discovery for Everyone, J. Chem. Inf. Model., 2015, 55(11), 2324–2337, DOI:10.1021/acs.jcim.5b00559.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery, J. Chem. Inf. Model., 2020, 60, 6065–6073, DOI:10.1021/acs.jcim.0c00675.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. P. Magariños, J. F. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. J. Radoux, A. Segura-Cabrera, A. Hersey and A. R. Leach, ChEMBL: Towards Direct Deposition of Bioassay Data, Nucleic Acids Res., 2019, 47(D1), D930–D940, DOI:10.1093/nar/gky1075.
- H. L. Morgan, The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service, J. Chem. Doc., 1965, 5(2), 107–113, DOI:10.1021/c160017a018.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754, DOI:10.1021/ci100050t.
- D. Probst and J.-L. Reymond, A Probabilistic Molecular Fingerprint for Big Data Settings, J. Cheminf., 2018, 10(1), 66, DOI:10.1186/s13321-018-0321-8.
- R. E. Carhart, D. H. Smith and R. Venkataraghavan, Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications, J. Chem. Inf. Comput. Sci., 1985, 25(2), 64–73, DOI:10.1021/ci00046a002.
- A. Capecchi, M. Awale, D. Probst and J. Reymond, PubChem and ChEMBL beyond Lipinski, Mol. Inf., 2019, 38(5), 1900016, DOI:10.1002/minf.201900016.
- A. Capecchi, D. Probst and J.-L. Reymond, One Molecular Fingerprint to Rule Them All: Drugs, Biomolecules, and the Metabolome, J. Cheminf., 2020, 12(1), 43, DOI:10.1186/s13321-020-00445-4.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, Reoptimization of MDL Keys for Use in Drug Discovery, J. Chem. Inf. Comput. Sci., 2002, 42(6), 1273–1280, DOI:10.1021/ci010132r.
- K. T. Nguyen, L. C. Blum, R. van Deursen and J.-L. Reymond, Classification of Organic Molecules by Molecular Quantum Numbers, ChemMedChem, 2009, 4(11), 1803–1805, DOI:10.1002/cmdc.200900317.
- D. Probst and J.-L. Reymond, FUn: A Framework for Interactive Visualizations of Large, High-Dimensional Datasets on the Web, Bioinformatics, 2018, 34(8), 1433–1435, DOI:10.1093/bioinformatics/btx760.
- Z. Yang, A. Hackshaw, Q. Feng, X. Fu, Y. Zhang, C. Mao and J. Tang, Comparison of Gefitinib, Erlotinib and Afatinib in Non-Small Cell Lung Cancer: A Meta-Analysis, Int. J. Cancer, 2017, 140(12), 2805–2819, DOI:10.1002/ijc.30691.
- P. Nordmann and L. Poirel, Plasmid-Mediated Colistin Resistance: An Additional Antibiotic Resistance Menace, Clin. Microbiol. Infect., 2016, 22(5), 398–400, DOI:10.1016/j.cmi.2016.03.009.
- M. Damashek, Gauging Similarity with N-Grams: Language-Independent Categorization of Text, Science, 1995, 267(5199), 843–848, DOI:10.1126/science.267.5199.843.
- S. Riniker and G. A. Landrum, Open-Source Platform to Benchmark Fingerprints for Ligand-Based Virtual Screening, J. Cheminf., 2013, 5(1), 26, DOI:10.1186/1758-2946-5-26.
- M. Awale and J. L. Reymond, Web-Based Tools for Polypharmacology Prediction, Methods Mol. Biol., 2019, 1888, 255–272, DOI:10.1007/978-1-4939-8891-4_15.
- M. Awale and J.-L. Reymond, Polypharmacology Browser PPB2: Target Prediction Combining Nearest Neighbors with Machine Learning, J. Chem. Inf. Model., 2019, 59(1), 10–17, DOI:10.1021/acs.jcim.8b00524.
- D. M. Anstine and O. Isayev, Generative Models as an Emerging Paradigm in the Chemical Sciences, J. Am. Chem. Soc., 2023, 145(16), 8736–8750, DOI:10.1021/jacs.2c13467.
- C. Cerchia and A. Lavecchia, New Avenues in Artificial-Intelligence-Assisted Drug Discovery, Drug Discov. Today, 2023, 28(4), 103516, DOI:10.1016/j.drudis.2023.103516.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00039g |
| This journal is © The Royal Society of Chemistry 2023 |