
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photolysis of nitrophenols in gas phase and aqueous environment: a potential daytime source for atmospheric nitrous acid (HONO)†
Shaoxun
Guo
 and
Hui
Li
*
and
Hui
Li
*
Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: hli@mail.buct.edu.cn
First published on 10th November 2022
Abstract
The daytime atmospheric nitrous acid (HONO) source has attracted considerable attention due to its important role in determining the concentrations of hydroxyl radicals (OH˙), O3, and secondary organic aerosols (SOAs). The widespread nitrophenols (NPs) are potential HONO sources; however, the relevant reaction mechanism has not been explored yet. We employ quantum mechanical calculations combined with the statistical RRKM rate theory to explore the photochemical reaction paths of two common NPs (2-NP and 4-NP) to form HONO in the gas and aqueous phases. It is revealed that both NPs have strong sunlight absorption, and photolysis occurs on the excited T1 state. Both NPs can easily dissociate under light in both gas and aqueous phases, where OH˙ and NO are the major products rather than HONO. Due to the higher solubility, 4-NP is found to more easily generate HONO in aqueous solution, which experiences the intermolecular excited state hydrogen transfer with vicinal water molecules. Kinetics shows that the relative humidity (RH) hardly promotes the gas-phase photolysis, and the temperature slightly affects the reaction rates. However, as a minor product, HONO generations from gas-phase 2-NP and aqueous 4-NP have high photolysis frequencies of 5.73 × 10−5 and 5.25 × 10−6 s−1, respectively, indicating that the hydrolysis of NPs can be important sources for atmospheric HONO.
Environmental significanceHONO has a great impact on atmospheric chemistry, and its origins have received intensive attention. As one of the abundant components in the heavily polluted atmosphere, nitrophenols are considered as a potential photochemical source of HONO. Using quantum mechanical calculations combined with the statistical rate theory, we unravel the possible photochemical pathway of nitrophenols, especially for HONO formation, which provides significant implications for understanding the atmospheric photochemical processes. |
1. Introduction
Nitrous acid (HONO) plays a significant role in atmospheric chemistry,1–3 which can produce 20–80% of hydroxyl radicals (OH˙) in tropospheric reactions during the heavily polluted daytime.4–8 The produced OH˙ are important oxidants in the atmosphere and have a great impact on the formation of secondary organic aerosols (SOAs).9 The sources of HONO in the atmosphere are rather diverse, and nitrogen oxides (NO3−, NO2, etc.) are commonly accepted.10–19 In recent years, researchers have observed HONO generated from atmospheric organic compounds (AOCs) in particulate matters.20 However, AOCs as the source of HONO is still not clear. As one of the typical AOCs, the aromatic compounds containing nitro groups, which widely exist in forest, rural, urban, and marine areas, have been considered as one of the important potential sources of HONO.21–24 Such compounds are also known to be harmful to living organisms.25–27As the simplest nitro-aromatic compounds, nitrophenols (NPs), which can be formed from the oxidation of phenols under high NOx conditions, as well as from the combustions of fossil fuel and biomass, have received intensive research attention due to their great contributions to the formation of SOAs.21,28–32 NPs also show highly photo-absorption properties to solar radiation, which accounts for most of the visible part (50–80%, λ > 400 nm) and ∼4% of the ultraviolet part (λ = ∼370 nm), and contribute to photochemical reactions.33–36
A high concentration of NPs has been observed in both gas phase (0.2–52 ng m−3) and particle phase (0.08–768 ng m−3).23,37–41 Furthermore, the concentration of NPs in winter is higher than it in summer.39,42,43 The nitro group of NP molecules is commonly located at the ortho- or para-position to the hydroxyl group, corresponding to 2-nitrophenol (2-NP) and 4-nitrophenol (4-NP), respectively. The concentration of 4-NP is slightly greater than that of 2-NP in the atmosphere.44,45 Pioneer studies have given strong evidence that 2-NP could be a photochemical source for HONO and OH˙. For example, Sangwan et al. found that 2-NP has strong absorption in the range of near-UV (λ = 295–400 nm),46,47 indicating that the direct photolysis of 2-NP by sunlight is possible.48 Bejan et al. detected ∼100 pptv HONO formation within an hour in the presence of ∼1.0 ppbv of 2-NP in the gas phase under irradiation (λ = 300–500 nm),49 and Nitta et al. used time-resolved photoelectron spectroscopy with 29.5 eV probe pulses to find that 2-NP can split off HONO in 0.5–1 ps.50 On the other hand, 4-NP can also be a potential photochemical source for HONO formation. Some experimental research studies have proved that 4-NP can generate HONO in the condensed phase, e.g., acidic aqueous solution and the environment with high relative humidity (RH).51,52 Since 4-NP can form an intermolecular hydrogen bond with a water molecule, it may interact with water vapor to promote the HONO formation.
Although previous observations have given evidence on HONO production from the photochemical reaction of NPs,47,49,51,52 details of the reaction mechanism, such as the reaction pathway, the rate-determining steps (RDSs), the role of water vapor in the reaction, and whether the reaction can happen in the gas phase or aqueous solution are still unclear. In recent theoretical studies, researchers have agreed that NPs easily generate triplet states after photoexcitation. As a result, the photolysis reactions and HONO formation have a high possibility of occurring in the triplet manifolds (especially the T1 state).53,54 Here, we employ quantum mechanical calculations to comprehensively reveal the possible photoreaction pathways of the two typical NPs (2-NP and 4-NP), especially for HONO generation, as well as evaluate its reaction rates in both unimolecular reaction and the reaction with the participation of water molecules (mimicking the high moist atmosphere and aqueous solution). The obtained results provide important novel mechanistic insight into the originality of atmospheric HONO.
2. Computational methods
2.1 Electronic structure calculations
The electronic energies on the unimolecular reaction paths on the S1 states are obtained by the relaxed scanning technique at the state-averaged complete active space self-consistent field (CASSCF) level of theory, in which the equal weights are used, and then followed by the single-point energy calculations of two lowest singlets and two lowest triplet states with the complete active space second-order perturbation theory (CASPT2).55,56 The active spaces for 2-NP and 4-NP are (10e, 10o) and (12e, 11o), respectively, as shown in Fig. S1, ESI,† and the rationality of such active spaces was proved by previous works.57–59 In addition, the ionization potential-electron affinity (IPEA) shift is set to zero;60 the imaginary shift technique (0.2 a.u.) is employed to avoid the intruder-state issue in the CASPT2 calculation.61 The def2-TZVPP basis sets62 are used in the CAS calculations. All of the CASPT2 and CASSCF calculations are carried out with ORCA 4.2.1 package.63,64Besides the unimolecular reactions, we also explore the more complex photochemical reactions of NPs, such as water-assisted reactions. To reduce the computational cost, density functional theory (DFT) is carried out to compute the reaction pathways in the ground state (S0) and lowest triplet excited state (T1), including optimizing the molecular geometries for the equilibrium states (ESs), transition states (TSs), and minimum energy crossing points (MECPs) between the S0 and T1 states. The TSs and MECPs are checked to connect the reactants and products using the intrinsic reaction coordinate (IRC) calculations, and the TSs are recognized to have only one imaginary frequency. Optimization of geometries for MECP between the T1 and S0 states is carried out by the sobMECP program,65 which is the modified version of the Harvey's MECP code.66 In all the DFT calculations, the B3LYP/6-311++G(d,p) level of theory67–73 is used for all the geometry optimization and energies computation, which is implemented using the Gaussian 09 program.74
In addition, linear response time-dependent density functional theory (LR-TDDFT)75 is employed to predict the spectroscopic properties of NPs, such as the vertical excitation energies (VEEs) and spin–orbit coupling (SOC) integrals. The TD-B3LYP/6-311++G(d,p) level of theory,67–73 which gives the most accurate excitation energy of NPs among several testing functionals (compared in Table S1, ESI†), is employed for these computations. The SOC integrals calculation is implemented in the PySOC program.76
2.2 Reaction rate constant calculation
Rice–Ramsperger–Kassel–Marcus (RRKM) theory is used to model the kinetics for the unimolecular photolysis channels of NPs on the T1 surface, including the photolysis of NPs and water-NP hydrated complexes. RRKM theory assumes that a transition state for passing from the reactant to the product, and rapid intramolecular vibrational energy redistribution among the degrees of freedom of a molecule. The RRKM microcanonical rate constant, k(E), which represents the statistical rate under collision-free conditions,77–80 is given by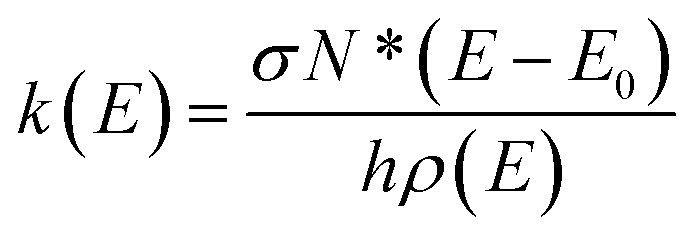 | (1) |
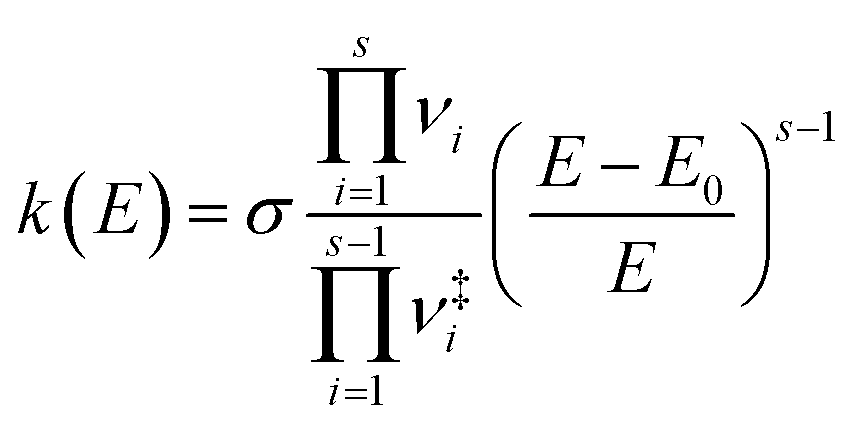 | (2) |
To further study the temperature effect of the photolysis reaction, the canonical rate constant, k(T), which assumes the energy levels are populated according to the thermal equilibrium, is followed by80
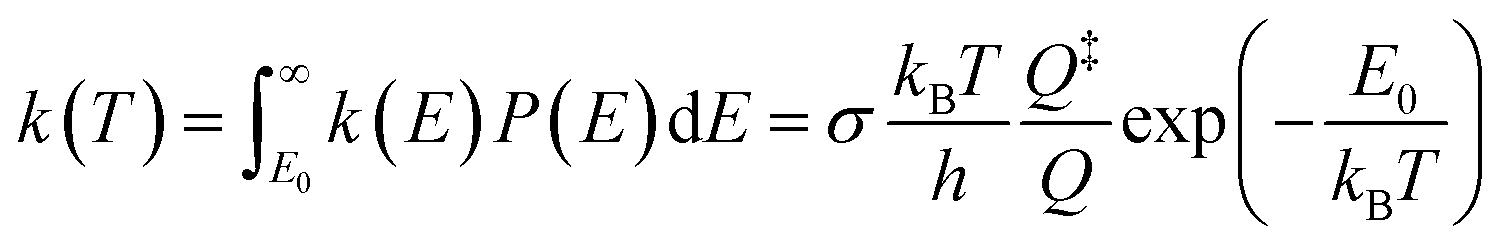 | (3) |
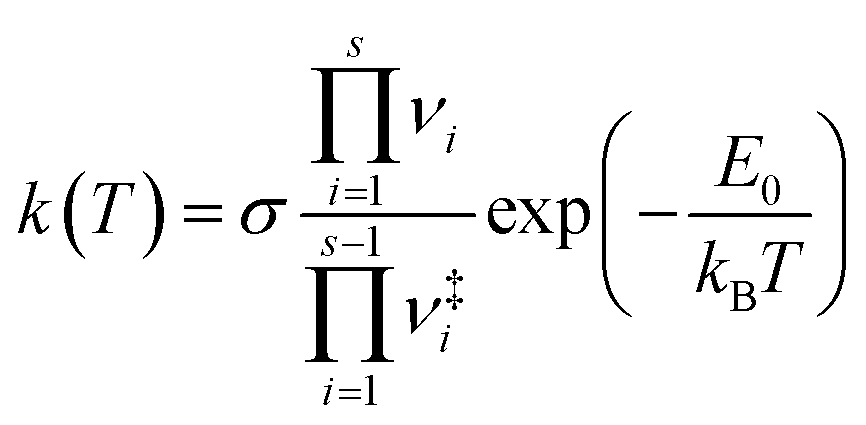 | (4) |
The rate constant of intersystem crossing (ISC) is derived from the Fermi's golden rule,82–86 which is according to the following formula
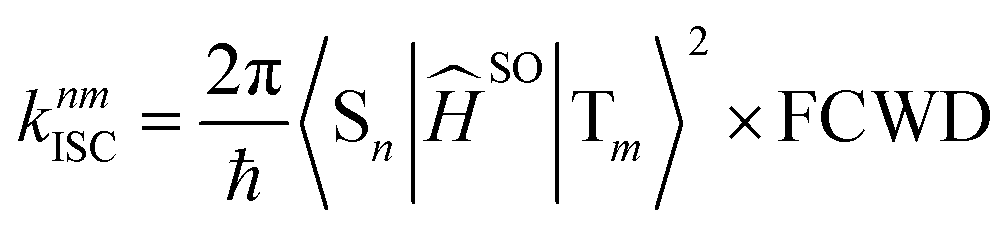 | (5) |
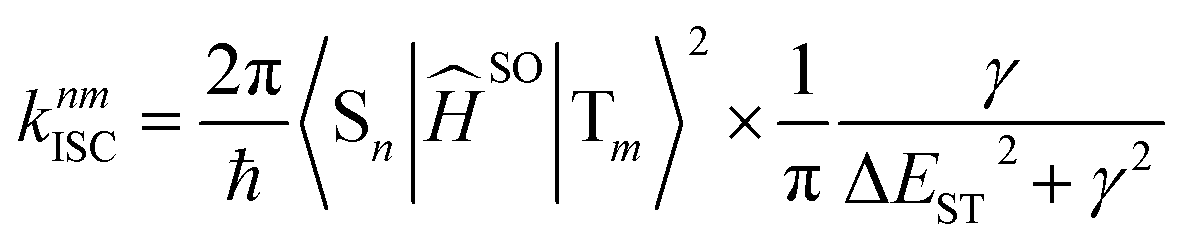 | (6) |
The total (experimental observed) ISC rate constant in state Sn is the sum of ISC to any state Tm, which is given by86
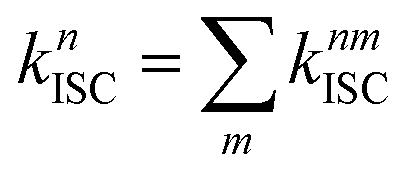 | (7) |
In addition, the bimolecular rate constants for HONO formation from 4-NP in the gas phase are evaluated using the transition state theory (TST) with the Wigner quantum tunneling correction,91–93
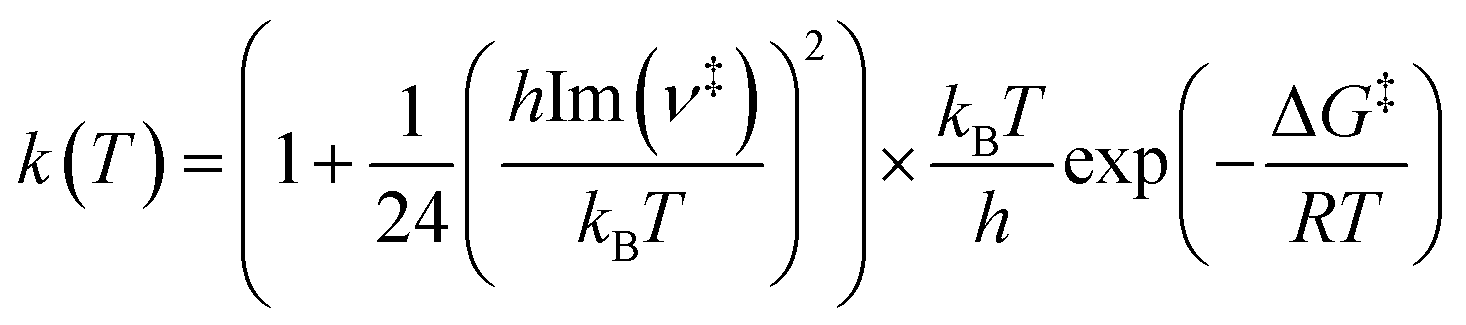 | (8) |
3. Results and discussion
3.1 Excited-state properties
To understand the possibility of a photoreaction in the atmosphere and the initial population of NPs after photo-excitation, we first study the light absorption properties of NPs. The previously reported absorption spectra of NPs showed a broad absorption band in the range of λ = 300–400 nm, indicating NPs can efficiently absorb sunlight in the lower atmosphere.46,94 At the level of theory used here (TD-B3LYP/6-311++G(d,p)), the values of VEE for S0 → S1 of 2-NP and 4-NP are predicted to be 79.8 kcal mol−1 (λ = 358.08 nm) and 87.9 kcal mol−1 (λ = 325.62 nm), respectively, which are consistent with the estimated experimental and theoretical values of ∼81.6 kcal mol−1 (λ = 350 nm).46,94 As listed in Table S2 and Fig. S2 (ESI†), the VEE of 2-NP is significantly red-shifted compared to that of 4-NP. This is because the intramolecular hydrogen bonds between the hydroxyl group and the nitro group of 2-NP lowers the relative energies of the lowest unoccupied molecular orbital (LUMO) π* and the subsequent transition.95,96 It is noteworthy to mention that a similar red shift is also observed in the spectra of nitrogen, oxygen, and halogen substituted molecules,96–98 allowing this class of species to absorb and react at the longer wavelengths of the UV range that are more available in the troposphere.Water vapors in the atmosphere may also have an impact on the excited state properties of NPs since the oxygen atoms of the NP molecules can be combined with water molecules through hydrogen bonding interaction, as shown in Fig. S3 (ESI†). However, as listed in Table S3 (ESI†), the interaction between NPs and water is relatively weak due to the positive binding free energies, as well as the small changes in VEEs (<0.2 kcal mol−1) of the hydrated NP complexes to the lowest two excited states (T1 and S1) at the FC point compared to the isolated molecules. Moreover, it is believed that a thermal fluctuation can further cause the absorption of NPs to respond to a larger wavelength range. Therefore, both the S1 and the T1 states of NPs can be efficiently populated by light with a wavelength of >300 nm (corresponding to the energy of 95.3 kcal mol−1).
In addition, it is found that the energies of the S1 and T4 excited states at the FC points are close (ΔE ≤ 1.4 kcal mol−1, TD-B3LYP/6-311++G(d,p) level, see Table S2, ESI†) for both 2-NP and 4-NP, leading to large SOC integrals of 10.46 cm−1 and 39.41 cm−1, respectively (Table S4, ESI†). Such small energy intervals and large SOC integrals indicate a high possibility for the ISC of NPs from the singlet state to triplet state.
Since the C–N fission could be a key step for the photolysis of NPs, the corresponding potential energy surfaces (PESs) on the S1 states of 2-NP and 4-NP are computed at the CASPT2//CASSCF level, as shown in Fig. 1. Due to the large dissociation barriers (>30 kcal mol−1) on the S1 state, NPs can hardly react on the S1 state, followed by a rapid ISC. In addition, a barrierless process from 2-NP to a hydrogen-shifted 2-NP in the S1 state (namely aci-2-NP) is found, as well as the large S0–T1 gaps (∼80 kcal mol−1 for 2-NP and 4-NP; ∼30 kcal mol−1 for the aci-2-NP*, CASPT2 level), indicating that the excited-state hydrogen transfer (ESHT) is coupled to ISC for 2-NP. Such result can be supported by the recent experiments and simulation observations, which showed the ISC of NPs and ESHT of 2-NP can occur within a picosecond.50,58,59,99 Besides, the aci-2-NP has two isomers on the S0 state and three isomers on the T1 state based on the geometry optimization at the B3LYP/6-311++G(d,p) level (see Fig. S4, ESI†). In addition, this barrierless ESHT only occurs intramolecularly, so that the vicinal water molecule does not affect this process (see Fig. S5, ESI†). Thus, water vapors should have little influence on the photochemical reactions (both the ESHT and C–N bond fission) of NPs due to the weak water–NP interaction.
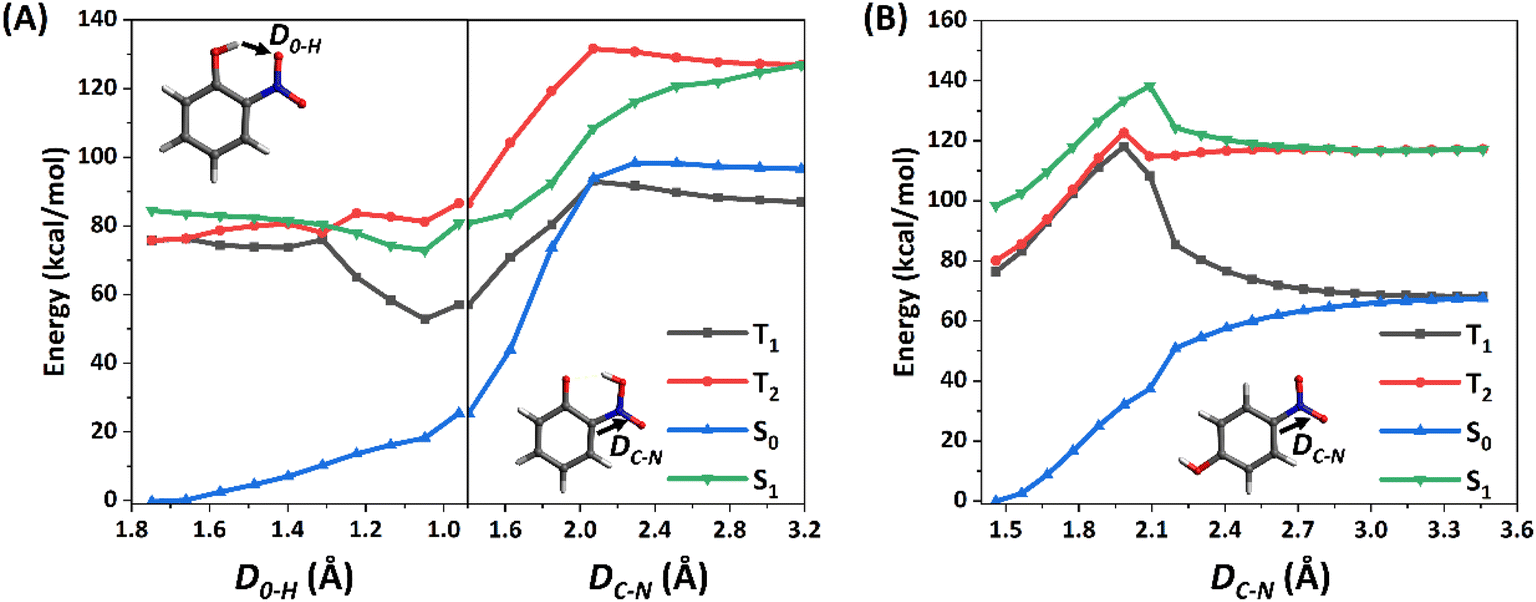 | ||
| Fig. 1 Computed energy profiles (in kcal mol−1) for the reaction pathways of (A) 2-NP, and (B) 4-NP in the lowest two singlet and triplet excited states (relaxed scan in the S1 state at CASPT2//CASSCF/def2-TZVPP level). See text for discussion. | ||
3.2 Photochemical reactions pathways of 2-NP
Both pioneer research studies and the above discussions have shown that 2-NP is favored for ISC coupled with the ESHT under the light condition. Such process can occur within a picosecond; therefore, the photoreaction of 2-NP can occur on the T1 state.50,58,59,99 As shown in Fig. 2, the energy gap between the S1 and T4 states is only ∼0.06 eV, corresponding to the ISC rate of 3.92 × 1010 s−1, which is close to the calculated total observable ISC rate kISC1(obs). This result illustrates that the S1 state of 2-NP should firstly transit to T4, and then hop back to the T1 state. Since the hopping process from T4 to T1 is fast, we consider that the photochemical reactions mainly take place on the lowest triplet state (T1). In previous studies, 2-NP has the ability to produce both HONO and OH˙.46,47,49,50,53 Thus, we separately study the photochemical reactions from the aci-2-NP isomers initiated by the C–N or O–N bond fission, and determine the possible reaction pathways in view of the energetic and kinetics aspect.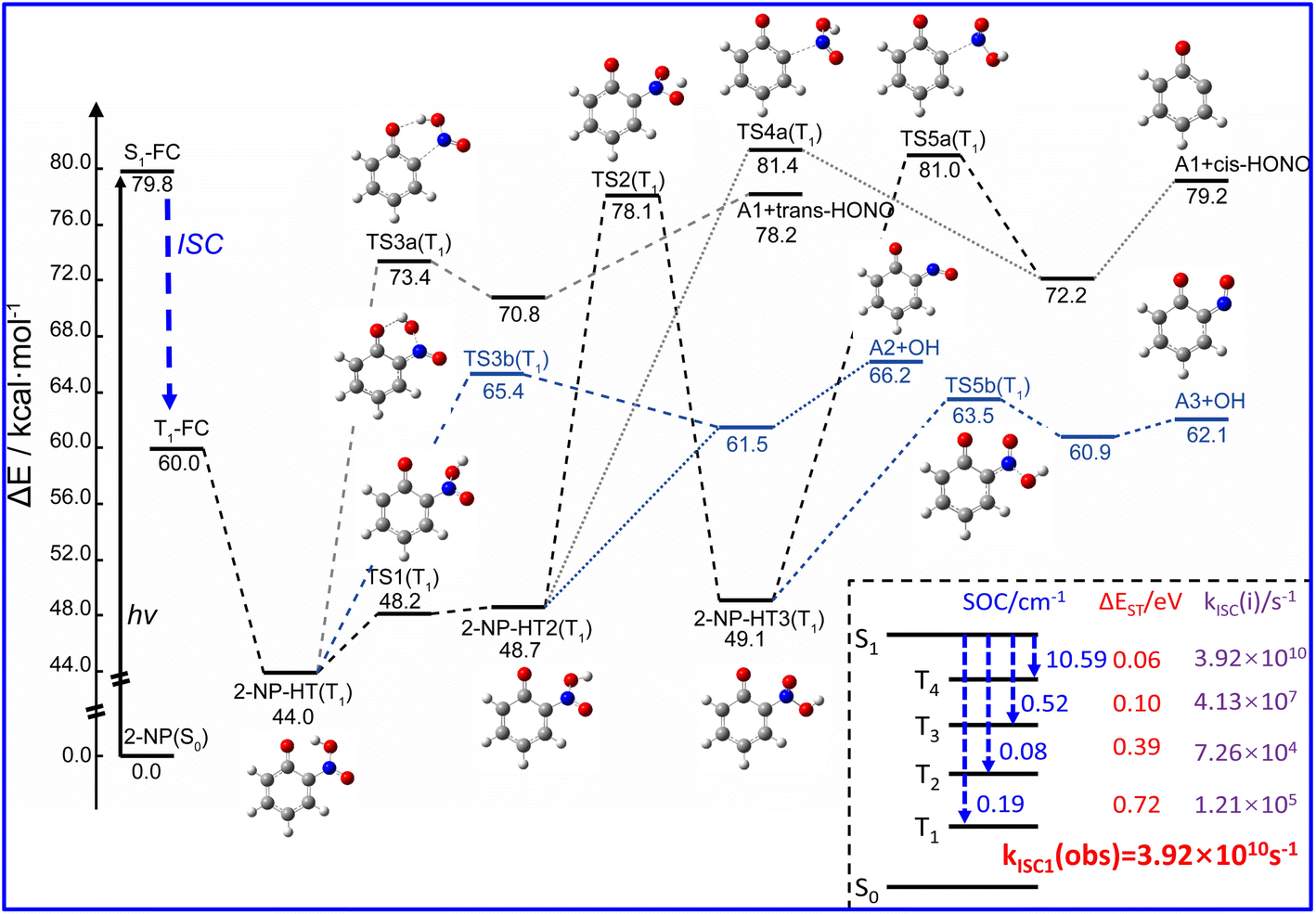 | ||
| Fig. 2 Computed photolysis pathways (at B3LYP/6-311++G(d,p) level) of the 2-NP molecule in the gas phase for the formation of HONO and OH˙. Key parameters for the ISC of 2-NP at the FC point are shown in the bottom right corner, including the SOC integrals (in cm−1), ΔEST (in eV), ISC rate constant kISC(i) (in s−1) between the lowest singlet state (S1) and lowest four triplet states (Tn, n = 1, 2, 3, 4), as well as the observed ISC rates kISC1(obs) (in s−1), which are the sums of each ISC channel. The zero-point energy (ZPE) correction is included in the energy values of ΔE. See text for discussions. | ||
In the experimental research, HONO formation from 2-NP is a pure gas-phase experiment without water.49 Therefore, the photolysis of 2-NP can be a unimolecular reaction, as shown in Fig. 2, which is examined by the intrinsic reaction coordinate (IRC) methodology. It is believed that the ESHT easily occurs. Thus, the photolysis reaction starts from the aci-2-NP isomer.
For unimolecular decomposition, both S0 and T1 states are able to generate HONO, as shown in Fig. S7 and S8.† However, although the IRC calculation proves a connection between 2-NP (or aci-2-NP) and HONO, it still cannot represent the real reaction pathway because the IRC neglects the vibrational and kinetic effects. To illustrate the rationality of the reaction pathway, we analyze the dynamical trajectories accomplished by the Born–Oppenheimer molecular dynamics (BOMD) simulation starting from the structure of the transition state. For each transition state, 25 trajectories are simulated within 400 fs. The results show that among the four isomers (2-NP, 2-NP-HT1, 2-NP-HT2, 2-NP-HT3), only 2-NP-HT2 and 2-NP-HT3 can form HONO on the T1 state, and the rest of the trajectories all roll back to the reactant, as shown in Fig. S7 and S8.† Thus, the HONO formation can only appear on the T1 state, although there is still some possibility for hopping back to the S0 state. The S0 state is found to hardly contribute to the HONO formation.
Then, we consider the unimolecular reaction starting from aci-2-NP, and find that only aci-2-NP on the T1 state can form HONO via C–N bond fission under UV light irradiation with the energy of TS being 73.4, 81.4, and 81.0 kcal mol−1 for 2-NP-HT1, 2-NP-HT2 and 2-NP-HT3, respectively (Fig. 2). This energy barrier of the TS is close to the S0 → S1 vertical excitation energy (79.8 kcal mol−1). After C–N bond decomposition, a van der Waals complex between two fragments is formed at first, and then the HONO molecule is released. Surprisingly, the energy of the isolated A1 radical and trans-HONO is still higher than that of TS3a, which illustrates that the 2-NP-HT1(T1) can hardly release HONO into the atmosphere, and only the other two isomers are available. Therefore, the 2-NP-HT1(T1) isomer is not likely to generate HONO, which is consistent with the BOMD results. After releasing HONO, as shown in Fig. S9,† biradical fragments A1 (open-shell electronic structure) may rearrange into a five-membered-ring molecule (P2), but this process is not very easy (with a barrier of ∼14.0 kcal mol−1). Since the ground state of A1 is a triplet state, a bimolecular hydrolysis reaction may occur. With the presence of a water molecule, A1 can be easily converted to catechol (P1) with a small energy barrier of ∼4.0 kcal mol−1.
Another photochemical pathway is the OH˙ formation path. Among all the TSs in the dissociation pathways shown in Fig. 2, the OH˙ formation pathways have lower energies of transition states than the HONO formation paths, which illustrate that 2-NP is more likely to produce OH˙ instead of HONO through gas phase photolysis. After producing OH˙, A2/A3 radicals (isomers) may further react with water, as shown in Fig. S10.† This reaction pathway has the chance to release NO and further react with OH˙, which was previously produced to generate HONO. However, this process seems not likely to happen due to the large energy barrier for the NO release (>38 kcal mol−1).
Water vapor plays a crucial role in atmospheric chemistry, thus we further study the photolysis channels for HONO and OH˙ formation in the presence of water as shown in Fig. 3. We find that water vapor can lower the energy of TS (ΔE < 5 kcal mol−1) in generating HONO and OH˙ compared to the unimolecular reaction. However, the kinetics results show that the participation of water vapor does not have a promoting effect on the kinetics of photolysis for 2-NP, even leading to 1–3 orders of magnitude lower reaction rate constant, as listed in Table 1. Thus, although the energy of transition state is lowered, the reaction is not accelerated by the presence of water vapor. Furthermore, the binding free energies of the 2-NP and aci-2-NP isomers with water is positive, which shows that it is difficult to combine the reactant with water. We also find that the reaction rate constants are increased with excitation wavelength, and the HONO formation rate is 2–5 order of magnitude lower than it is in OH˙ formation with and without water vapor, which agrees with the previous discussions.
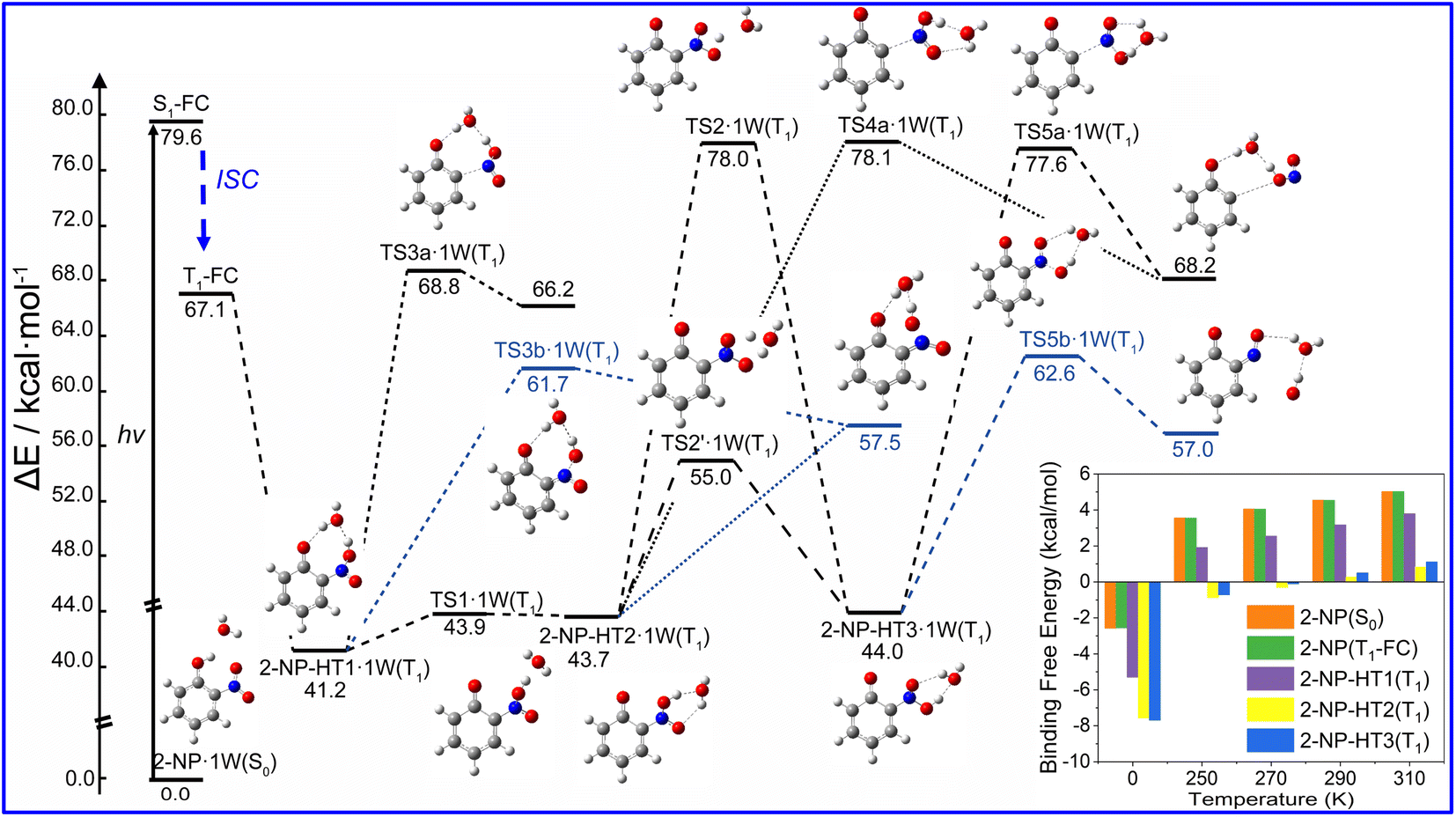 | ||
| Fig. 3 Computed photolysis pathways (at B3LYP/6-311++G(d,p) level) of the 2-NP hydrated complex for HONO and OH˙ formation. The binding free energies between the 2-NP isomers with a water molecule on the S0 and T1 states are shown in the bottom right corner. The ZPE correction is included in the values of ΔE. See text for discussions. | ||
| Reaction path | Internal energy (kcal mol−1) | |||
|---|---|---|---|---|
| 70.0 | 80.0 | 90.0 | 100.0 | |
| 2-NP-HT1 → HONO | 1.07 × 105 | 2.91 × 106 | 3.15 × 107 | 1.91 × 108 |
| 2-NP-HT2 → HONO | 1.36 × 105 | 7.11 × 106 | 1.19 × 108 | 9.85 × 108 |
| 2-NP-HT3 → HONO | 1.20 × 105 | 5.29 × 106 | 7.90 × 107 | 6.04 × 108 |
| 2-NP-HT1 → OH˙ | 5.33 × 108 | 4.12 × 109 | 1.88 × 1010 | 6.09 × 1010 |
| 2-NP-HT3 → OH˙ | 3.45 × 1010 | 1.15 × 1011 | 2.87 × 1011 | 5.88 × 1011 |
| 2-NP-HT1·1W → HONO | 3.22 × 103 | 1.25 × 105 | 1.80 × 106 | 1.37 × 107 |
| 2-NP-HT2·1W → HONO | 1.36 × 102 | 2.93 × 104 | 1.30 × 106 | 2.20 × 107 |
| 2-NP-HT3·1W → HONO | 3.44 × 102 | 5.78 × 104 | 2.18 × 106 | 3.30 × 107 |
| 2-NP-HT1·1W → OH˙ | 2.50 × 107 | 2.67 × 108 | 1.56 × 109 | 6.11 × 109 |
| 2-NP-HT3·1W → OH˙ | 4.55 × 108 | 3.65 × 109 | 1.74 × 1010 | 5.83 × 1010 |
Therefore, the direct photolysis of 2-NP for HONO and OH˙ formation experiences a unimolecular reaction. OH˙ is more likely to be generated than HONO, and all the photolysis reactions are nearly irrelevant with the participation of water vapor, which is consistent with the previous experimental results.49,50
3.3 Photochemical reactions pathways of 4-NP
Unlike 2-NP, 4-NP cannot form intramolecular hydrogen bonds, wherein the HONO-moiety cannot be produced under the light irradiation. Here, 4-NP stands for a class of molecules with non-intermolecular hydrogen bonds.The gas-phase photochemical reactions of 4-NP also follows the paths shown in Fig. 4. Similar to the reaction of 2-NP, the reaction of 4-NP starts from releasing NO2 or NO. From the energetic view, the energy of TS for NO formation (74.3 kcal mol−1) is ∼8.0 kcal mol−1 lower than that of NO2 formation (82.0 kcal mol−1), which can be further reduced by water vapor. The energies of TSs are all below the S0 → S1 transition energy (87.8 kcal mol−1). Thus, the reactions are able to occur at this energy of irradiation. ISC is also an important photophysical property for 4-NP. We do not account for the transition to the triplet states above the S1 state. Therefore, there are only three ISC channels for the gas-phase 4-NP with the total ISC rate of 8.68 × 109 s−1, which is shown in Fig. 4(c).
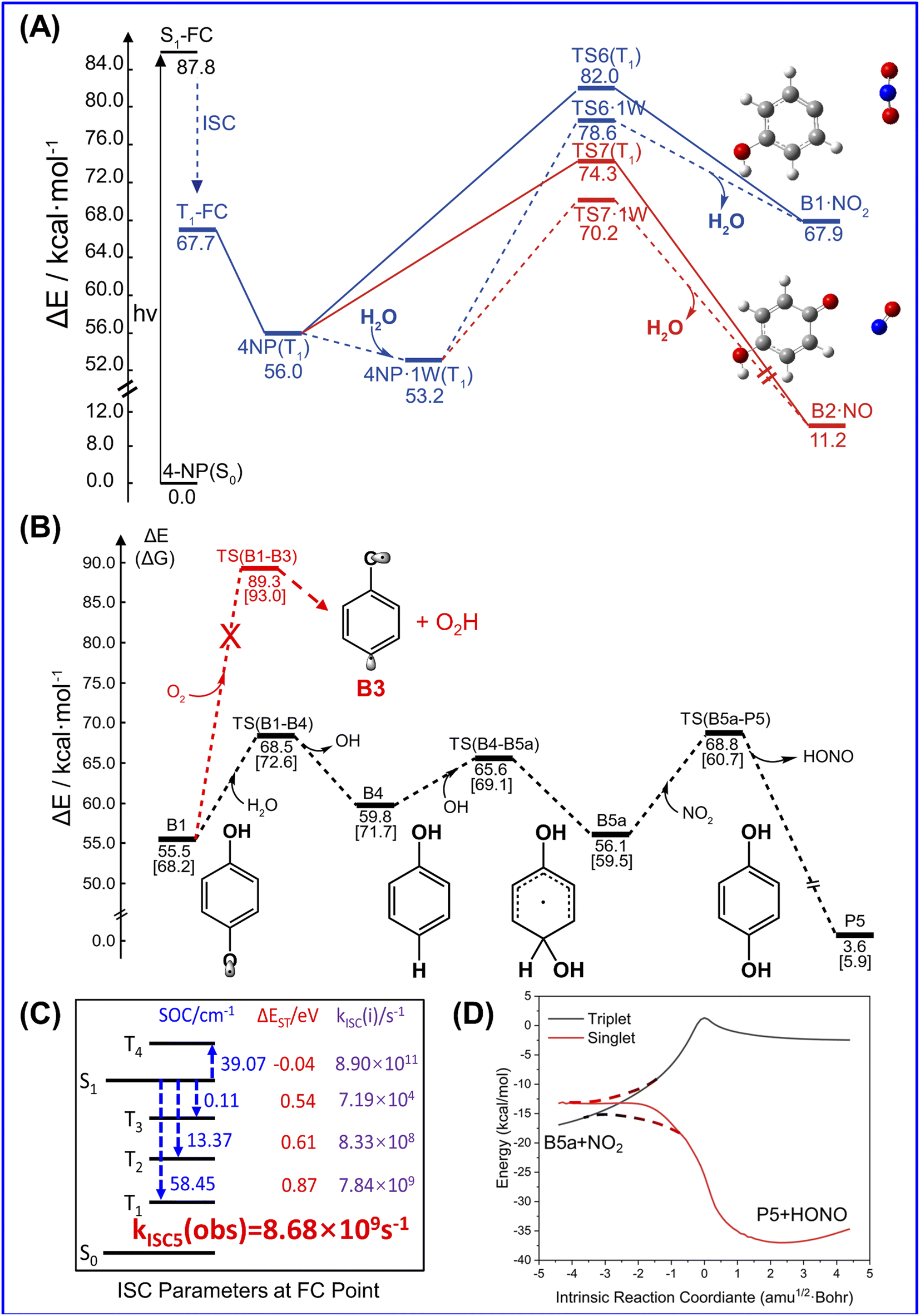 | ||
| Fig. 4 Photochemical mechanism of 4-NP in the gas phase. (A) Computed photolysis pathways (at B3LYP/6-311++G(d,p) level) of 4-NP for NO2 and NO formation in the gas phase. (B) Computed gas-phase reaction pathways (at B3LYP/6-311++G(d,p) level) for HONO formation in the presence of O2 or water vapor from the B1 radical intermediate, which is generated from the C–N bond fission of 4-NP (C) Key parameters for ISC of 4-NP (gas) at the FC point, including SOC integrals (in cm−1), ΔEST (in eV), ISC rate constant kISC(i) (in s−1) between the lowest singlet state (S1) and lowest four triplet states (Tn, n = 1, 2, 3, 4), as well as the observed ISC rates kISC5(obs) (in s−1), which are the sums of the ISC channels. (D) The IRC of the singlet and triplet states for hydrogen transfer between B5a and NO2 fragments. The ZPE correction is included in the energy values of ΔE. See text for discussions. | ||
After NO2 release, H2O vapor can react with B1 to generate phenol and OH˙, then the oxidation of phenol by OH˙ and NO2 further generate HONO (Fig. 4(b) and S13†). This reaction pathway may lead to HONO formation from 4-NP in the gas phase, but may not be highly probable to occur due to the small concentration of water in the atmosphere and the positive binding free energy between 4-NP and water molecules. Furthermore, although O2 is abundant and reactive in the atmosphere, it does not react with B1 due to the larger barrier than that in the hydrolysis of B1 (Fig. 4(b)). Moreover, as shown in Fig. S14,† in the presence of H2O and HO–C6H4 (B2) fragments, NO can be further transferred into HON, but not efficiently (with a barrier of ∼30 kcal mol−1). By contrast, the formation of HONO from B1 (with a barrier of ∼6.0 kcal mol−1) can occur. Except for hydrolysis, the oxygenation of B2 may be more reactive to form hydroquinone and O2H, but still has the energy barrier of ∼18 kcal mol−1 (Fig. S14†). Therefore, only NO2 and NO could be the major products of the gas-phase photochemistry of 4-NP.
Since 4-NP is soluble due to the intermolecular hydrogen bonding interaction with water, 4-NP can also react in the aqueous phase. In the experiments of solution photochemistry,51,52 HONO is formed with a higher rate in the acid environment, where 4-NP can maintain its neutral form, instead of anion. Unlike the gas-phase reactions, the bimolecular reaction is also possible due to the large concentration of water molecules. Fig. 5 shows the PES for photochemistry of 4-NP in solution computed by the solvation model based on density (SMD). As shown in Fig. 5(a), the properties of ISC of 4-NP in solution is similar to that of the gas phase result, and all the total ISC rates are ∼109 s−1, indicating that water molecules have little affect on the photophysical properties of 4-NPs. The results of the potential energy surfaces (Fig. 5(b)) indicate that the energy of TS for bimolecular reaction is greatly reduced (ΔE = 3–13 kcal mol−1) compared to that of the unimolecular paths, and all paths can efficiently occur under the 70–100 kcal mol−1 (λ = 280–410 nm) irradiation. Due to the large concentration of water molecules in solution, the dissolved 4-NP molecules may undergo intermolecular ESHT. It is found that at least three water molecules are needed to realize this process due to the large separation distance between the hydroxyl and nitro groups in 4-NP molecules. Here, the ISC of molecules after photoexcitation occurs in the FC region, and the subsequent reaction takes place on the T1 state. As shown in Fig. 5(c), the unimolecular ESHT of the 4-NP·3W hydrated complex has a large reaction energy barrier (>10 kcal mol−1) on all excited state energy surfaces, so the previous ISC hypothesis is reasonable. As shown in Fig. 5(c) and (d), the energy barriers for the intermolecular ESHT of 4-NP in solution are ∼10 kcal mol−1 (unimolecular reaction) and 1.1 kcal mol−1 (bimolecular reaction), which indicates that 4-NP can easily transfer a hydrogen atom to generate aci-4-NP in solution. Then, we consider the dissociation of aci-4-NP on the T1 state. It is found that the dissociation of the N–O bond of this molecule is very difficult since the energy barriers of the unimolecular and hydrated complex are ∼33 and ∼35 kcal mol−1, respectively. However, it is very easy to generate HONO through dissociating the C–N bond with the energy barrier of only ∼14 kcal mol−1, which is less than the barriers in the bimolecular generation of HONO and unimolecular formation of NO2 and NO. Thus, we can conclude that the dissociation of aci-4-NP on the T1 state is the main source of HONO generation from 4-NP.
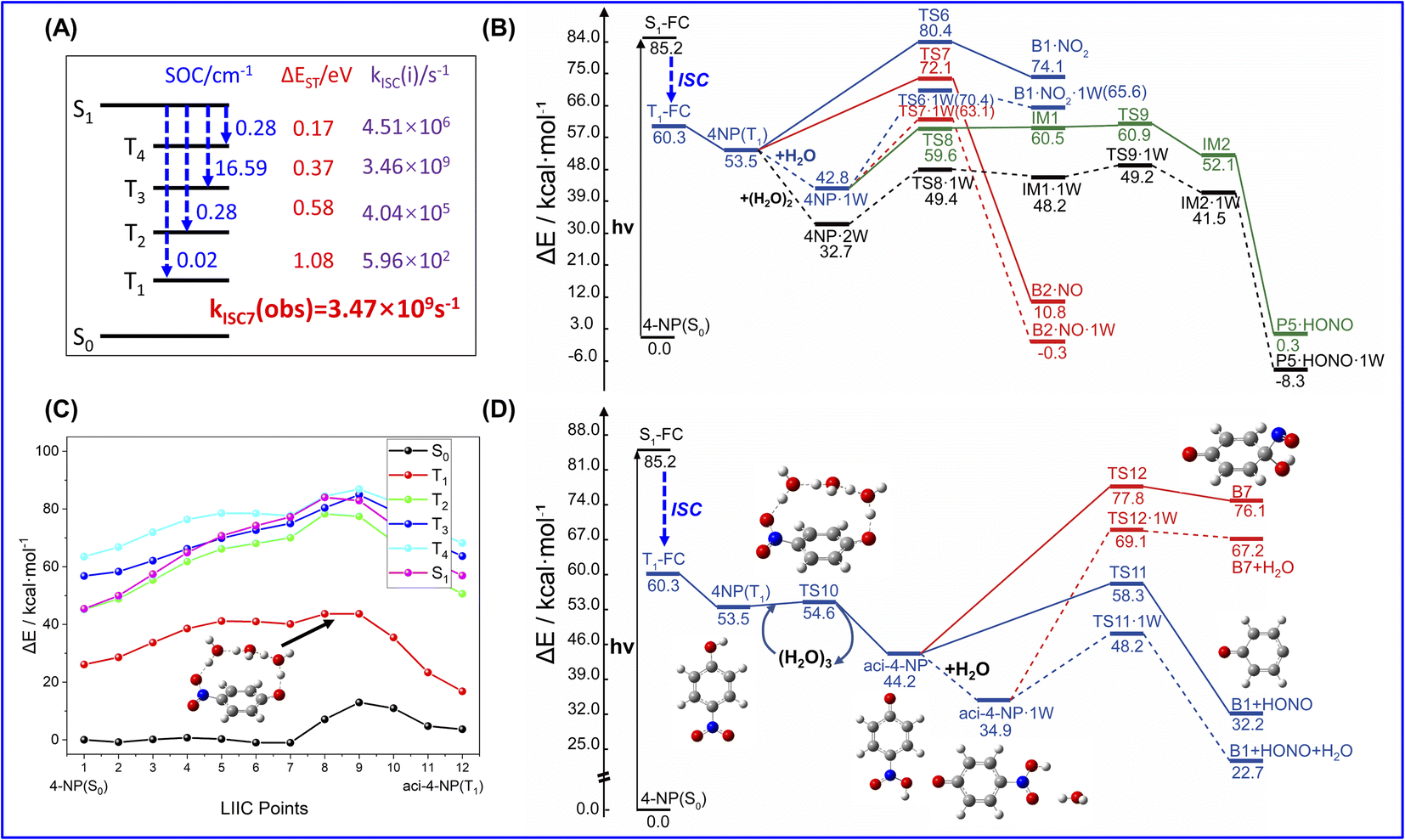 | ||
| Fig. 5 Photochemical mechanism of 4-NP in aqueous phase. (A) Key parameters for ISC of 4-NP (aq) at the FC point, including SOC integrals (in cm−1), ΔEST (in eV), ISC rate constant kISC(i) (in s−1) between the lowest singlet state (S1) and lowest four triplet states (Tn, n = 1, 2, 3, 4), as well as the observed ISC rates kISC6(obs) (in s−1), which are the sums of the ISC channels. (B) Computed photolysis pathways (at B3LYP/6-311++G(d,p) level) of 4-NP for NO2, NO and bimolecular HONO formation in aqueous phase. (C) Computed intermolecular ESHT pathway (at TD-B3LYP//B3LYP/6-311++G(d,p) level) of 4-NP in aqueous phase. Each geometry in the potential energy surfaces are obtained by the linear-interpolated internal coordinates (LIIC) technique. (D) Computed photolysis pathways (at B3LYP/6-311++G(d,p) level) of aci-4-NP in water solution initiated by intermolecular ESHT. The ZPE correction is included in the energy values of ΔE. All of the energies are calculated with the SMD solvation model. See text for discussions. | ||
We further compute the rate constants of each elementary photolysis channel in both gas and aqueous phases by RRKM theory with the excitation/internal energy of 70–100 kcal mol−1 (corresponding to light irradiation of 280–410 nm), as listed in Table 2. For gas-phase reactions, the unimolecular photolysis to form NO2 and NO have the rate constants of 106–108 s−1 and 108–109 s−1, respectively. Meanwhile, in the presence of water molecules, the rate constants of NO2 and NO formation are 104–107 s−1 and 107–108 s−1, respectively. From this issue, NO is more prone to generate from the photolysis of 4-NP, and water molecules have no apparent influence on the photolysis rates. For the photolysis in solution, as a comparison, NO2 and NO formation have the rate constants of 105–108 s−1 and 107–109 s−1, respectively. The photolysis from the hydrated complex is also unfavorable compared to the unimolecular reaction. Therefore, NO2 and NO are only favorable to generate in the gas phase rather than aqueous phase.
| Reaction path | Internal energy (kcal mol−1) | |||
|---|---|---|---|---|
| 70.0 | 80.0 | 90.0 | 100.0 | |
| Gas-phase photolysis | ||||
| 4-NP → NO2 | 1.81 × 106 | 2.72 × 107 | 1.97 × 108 | 8.97 × 108 |
| 4-NP → NO | 1.47 × 108 | 7.55 × 108 | 2.58 × 109 | 6.69 × 109 |
| 4-NP·1W → NO2 | 2.35 × 104 | 5.94 × 105 | 6.32 × 106 | 3.86 × 107 |
| 4-NP·1W → NO | 1.14 × 107 | 7.25 × 107 | 2.91 × 108 | 8.58 × 108 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Aqueous-phase photolysis | ||||
| 4-NP → NO2 | 5.87 × 105 | 1.03 × 107 | 8.37 × 107 | 4.12 × 108 |
| 4-NP → NO | 4.30 × 107 | 2.33 × 108 | 8.23 × 108 | 2.20 × 109 |
| 4-NP·1W → NO2 | 2.03 × 104 | 8.09 × 105 | 1.18 × 107 | 9.05 × 107 |
| 4-NP·1W → NO | 3.62 × 107 | 3.79 × 108 | 2.18 × 109 | 8.42 × 109 |
| 4-NP·1W → HONO | 1.55 × 103 | 4.88 × 103 | 1.12 × 104 | 2.13 × 104 |
| 4-NP·2W → HONO | 3.76 × 100 | 1.42 × 101 | 3.73 × 101 | 7.79 × 101 |
| 4-NP·3W → Aci-4-NP·3W | 3.44 × 106 | 1.27 × 107 | 3.43 × 107 | 7.52 × 107 |
| Aci-4-NP → HONO | 3.19 × 103 | 2.03 × 105 | 3.85 × 106 | 3.47 × 107 |
| Aci-4-NP → OH˙ | 8.23 × 109 | 2.69 × 1010 | 6.59 × 1010 | 1.33 × 1011 |
| Aci-4-NP·1W → HONO | 4.31 × 105 | 9.97 × 106 | 1.00 × 108 | 5.85 × 108 |
| Aci-4-NP·1W → OH˙ | 1.58 × 109 | 6.15 × 109 | 1.72 × 1010 | 3.87 × 1010 |
Then, we consider the rate constants of several possible reaction channels for the formation of HONO in solution. For the bimolecular reaction, the rate constant of HONO formation is 103–104 s−1. However, when an extra water molecule is added, its rate constant decreases to 100–101 s−1. It can be concluded that the additional water molecule does not have advantage to the reaction. In addition, the rate constant of intermolecular ESHT of 4-NP with the existence of at least three water molecules is 106–107 s−1. Meanwhile, the rate constant of the dissociation of aci-4-NP to form HONO is 103–107 s−1, and to generate OH˙ is 109–1011 s−1. With a water molecule present, the rate constant for HONO and OH˙ formation is 105–108 s−1 and 109–1010 s−1. Here, we have found that water molecules can promote the dissociation of aci-4-NP, which leads to HONO formation in solution. That is to say, the formation of HONO from 4-NP in solution does not present a bimolecular reaction, but is similar to 2-NP molecule, i.e., carries out an ESHT. This is then followed by unimolecular photolysis, which is contrary with the experimental suggestions.51,52 To summarize, these results indicate that the photochemical reaction of 4-NP preferably generates NO2 and NO in gas phase, while it generates HONO in aqueous solution.
3.4 Kinetics and atmospheric implications for HONO formation
To evaluate the contribution to HONO formation of NPs in the real atmosphere, we analyze the reaction kinetics of NPs to release HONO via the photochemical reactions. The typical concentrations of NPs in the atmosphere are ∼2.0 ng m−3 (8.60 × 106 molecules per cm3) for 2-NP and ∼5.0 ng m−3 (2.16 × 107 molecules per cm3) for 4-NP.45 It is notable that the concentration of 4-NP can be up to ∼768 ng m−3 in heavily polluted weather.100,101 The concentration of water vapor is a function of temperature and RH (see ESI† for details). Here, we regard the aqueous phase as water vapor with the concentration at 300 K and 100% RH. All of the rate constants of each elementary reaction are calculated using RRKM theory under the collision-free condition.The computational details for the time evolution of the photolysis products using the micro-kinetic analysis and related results are summarized in Section S1, ESI.† Firstly, we study the photolysis kinetics under the excitation energy of 70–100 kcal mol−1 (corresponding to the wavelength range of 280–410 nm) for 2-NP and 4-NP, as well as the reactions involving water. Almost all of the concentrations and reaction rates of the photolysis products are increased with the increase of the excitation energy (Fig. S16, S17 and S19†). For the gas-phase photolysis of 2-NP, as shown in Table S11,† the yield of OH˙ is much higher than that of HONO with and without water, demonstrating the branching ratio of ∼1:10−4. Thus, OH˙ is the main product of the gas-phase photolysis of 2-NP. The relative populations of OH˙ and HONO from the water-assisted reactions are 10−6 to 10−4 and 10−10 to 10−6, respectively. These values are much lower than the relative populations in the unimolecular reactions of 0.89–0.99 and 10−6 to 10−4, respectively, which should be attributed to the low binding free energy between 2-NP and water (Tables S3 and S10, ESI†). In addition, based on the micro-kinetics, we extract the rate constants for OH˙ and HONO formation from the unimolecular reactions (OH˙: 107–109 s−1; HONO: 104–106 s−1), which are 1–2 orders of magnitude higher than those in the water-assisted reactions. Thus, the main channel of the photolysis of 2-NP should be a unimolecular reaction, which does not rely on the humidity of the atmosphere.
The photolysis of 4-NP in the gas phase has a similar situation (results are listed in Table S12†), where the water vapors do not promote the reaction and NO is the major product with the formation rate of 107–108 s−1 (NO2: 105–107 s−1). Considering the following hydrolysis of the B1 fragment, as listed in Table 3, the final relative populations (under an excitation energy of 80 kcal mol−1) of NO2, NO, and HONO are 2.16 × 10−2, 0.6, and 1.8 × 10−17, respectively, which shows that almost no HONO can be produced from the photolysis of 4-NP in the gas phase. On the other hand, when 4-NP is dissolved in aqueous solution, the relative populations of the products are significantly changed. The final relative populations (under an excitation energy of 80 kcal mol−1) of NO2, NO, and HONO become 2.59 × 10−3, 5.85 × 10−2, and 8.50 × 10−5, respectively (Table 3), in which the content of HONO is remarkably increased, although it is still a minor product. Comparing the final relative populations of HONO formed by the aqueous bimolecular reaction of 4-NP and dissociation of aci-4-NP (Table S14, ESI†), the latter reaction path produces more HONO, because the intermolecular ESHT of 4-NP is more favorable than the bimolecular reaction. In aqueous solution, the formation rates of NO2, NO and HONO are found to be 105–108, 107–108, and 102–105 s−1, respectively (Table S14†).
| Photolysis product | ||||
|---|---|---|---|---|
| OH˙ | HONO | NO2 | NO | |
| 2-NP (gas) | 0.98 | 1.00 × 10−3 | N.A. | N.A. |
| 4-NP (gas) | N.A. | 1.80 × 10−17 | 2.16 × 10−2 | 6.00 × 10−1 |
| 4-NP (solution) | N.A. | 8.50 × 10−5 | 2.59 × 10−3 | 5.85 × 10−2 |
The photolysis frequency (J, in s−1) is often regarded as one of the key parameters in atmospheric photochemical reaction research, which is defined as
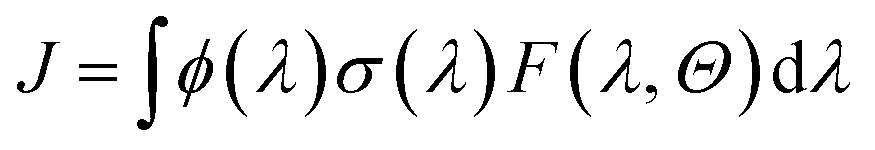 | (9) |
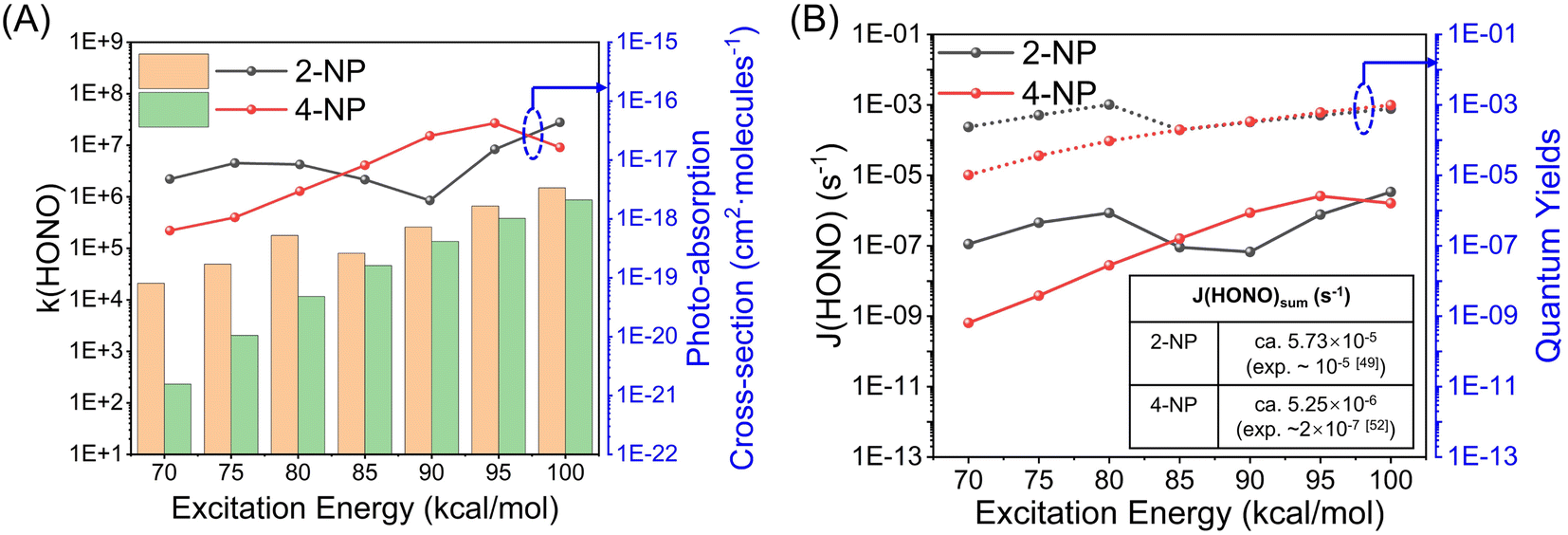 | ||
| Fig. 6 Kinetics for HONO formation. (A) Microcanonical rate constants for HONO formation, and photo-absorption cross-section of 2-NP (gas phase) and 4-NP (aqueous phase); (B) quantum yields (dash lines) and photolysis frequencies (solid lines) for the HONO formation of 2-NP (gas phase) and 4-NP (aqueous phase). These results are obtained from the micro-kinetics simulations based on the RRKM theory as a function of the excitation energy of 70–100 kcal mol−1 (corresponding to a wavelength range of 280–410 nm). Furthermore, the calculated and experimental measurement of the photolysis frequencies for HONO formation from 2-NP and 4-NP with an excitation energy range of 70–100 kcal mol−1 is listed. | ||
| Excitation energy (kcal mol−1) | |||||||
|---|---|---|---|---|---|---|---|
| 70.0 | 75.0 | 80.0 | 85.0 | 90.0 | 95.0 | 100.0 | |
| 2-NP | 2.36 × 10−4 | 5.10 × 10−4 | 1.02 × 10−3 | 1.95 × 10−4 | 3.26 × 10−4 | 5.06 × 10−4 | 7.77 × 10−4 |
| 4-NP | 1.02 × 10−5 | 3.61 × 10−5 | 9.47 × 10−5 | 1.97 × 10−4 | 3.37 × 10−4 | 6.07 × 10−4 | 9.85 × 10−4 |
We further consider the temperature effect on the photolysis reactions based on the canonical rate constants, which can be used to describe the ability to overcome the energy barriers between the minimum and transition state, including excitation energies at different temperatures. It is noteworthy that we only use this model to describe the temperature effect qualitatively, rather than provide a real population of each species at different temperatures. As listed in Table S15 (Fig. S16–S19†), a higher temperature is favorable for photolysis reaction. Therefore, more HONO originating from the photolysis of NPs should be observed at higher temperatures (i.e. summer season). In addition, the energy distribution of the reactant does not shift to higher energy levels with the increase of temperature, as shown in the calculated absorption spectra (Fig. S2†). Therefore, the temperature effect in the photolysis of nitrophenols can only affect the kinetics of the reactions.
Moreover, the air–water interface (such as the aerosol surface) is an important place for the reaction to occur. However, we just studied the photochemical reaction of NPs in gas and solution since the reaction channel at the air–water interfaces should not be different from the aqueous phase except for the concentration of water. However, based on the previous discussions, the concentration of water molecules hardly influences the photochemical reaction rates of NPs. Therefore, the reaction kinetics at the air–water interfaces should be similar to the reaction in solution.
To summarize, the kinetics analysis shows that OH˙ and NO can be efficiently produced by the gas-phase photolysis of 2-NP and 4-NP, and the temperature has minimal influence on the formation rates. Although as the minor products, the gas-phase photolysis reactions still have some contributions to generate both HONO and NO2. On the other hand, in the condensed aqueous phase, HONO can be efficiently produced from the photolysis of 4-NP with the assistance of water, and a higher temperature apparently benefits the reaction. Based on these results, we can conclude that HONO is more likely to be produced from 2-NP in the gas phase and 4-NP in the aqueous solution. In addition, according to our existing knowledge, a direct experimental comparison of these two molecules in photochemical reactions is still lacking. Therefore, we are looking forward to more precise experimental evidence to compare these two molecules directly on photochemical HONO production.
4. Conclusion
In conclusion, we employ quantum mechanical methods to predict the photoreaction pathways of HONO generation from two typical atmospheric NPs, e.g., 2-NP and 4-NP. The results show that NPs have high solar absorption efficiency in the wavelength range of 300–400 nm, as well as a high probability of ISC, indicating that the photolysis reactions of these molecules occur in the T1 state. For 2-NP, the photolysis is more likely to occur in the gas phase, where the ESHT rapidly occurs after light irradiation, and then HONO and OH˙ are released. The kinetics study shows that 2-NP and 4-NP can both easily dissociate after light irradiation in the gas phase, while OH˙ and NO are the major products rather than HONO. In addition, since 4-NP can dissolve in water, the photolysis of 4-NP in the condensed aqueous phase is much easier to produce HONO than in the gas phase, which also experiences an ESHT instead of the bimolecular reactions. However, HONO is still a minor product in the aqueous photolysis of 4-NP. These results demonstrate that HONO is a minor product from both water-insoluble NPs (WINP, such as 2-NP) and water-solvable NPs (WSNP, such as 4-NP). However, NP molecules still have considerable contributions to HONO generation in the real atmosphere. Therefore, the photolysis of WINP in the gas phase and WSNP in the aqueous phase can be a significant source for atmospheric HONO, and the gas phase photolysis may be more important.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is supported by the Beijing Natural Science Foundation (Z210016) and National Natural Science Foundation of China (21935001, 91934303). We thank the Beijing University of Chemical Technology (BUCT) for providing the High-Performance Computing (HPC) Platform.References
- J. Wang, J. Li, J. Ye, J. Zhao, Y. Wu, J. Hu, D. Liu, D. Nie, F. Shen, X. Huang, D. Huang, D. Ji, X. Sun, W. Xu, J. Guo, S. Song, Y. Qin, P. Liu, J. R. Turner, H. C. Lee, S. Hwang, H. Liao, S. T. Martin, Q. Zhang, M. Chen, Y. Sun, X. Ge and D. J. Jacob, Nat. Commun., 2020, 11, 2844 CrossRef PubMed.
- W. Zhang, S. Tong, C. Jia, L. Wang, B. Liu, G. Tang, D. Ji, B. Hu, Z. Liu, W. Li, Z. Wang, Y. Liu, Y. Wang and M. Ge, Environ. Sci. Technol., 2020, 54, 12870–12880 CrossRef.
- Y. Liu, Y. Zhang, C. Lian, C. Yan, Z. Feng, F. Zheng, X. Fan, Y. Chen, W. Wang, B. Chu, Y. Wang, J. Cai, W. Du, K. Daellenbach, J. Kangasluoma, F. Bianchi, J. Kujansuu, T. Petäjä, X. Wang, B. Hu, Y. Wang, M. Ge, H. He and M. Kulmala, Atmos. Chem. Phys., 2020, 20, 13023–13040 CrossRef.
- A. Kukui, M. Legrand, S. Preunkert, M. Frey, R. Loisil, J. Roca, B. Jourdain, M. King, J. France and G. Ancellet, Atmos. Chem. Phys., 2014, 14, 12373–12392 CrossRef.
- L. Xue, R. Gu, T. Wang, X. Wang, S. Saunders, D. Blake, P. Louie, C. Luk, I. Simpson, Z. Xu, Z. Wang, Y. Gao, S. Lee, A. Mellouki and W. Wang, Atmos. Chem. Phys., 2016, 16, 9891–9903 CrossRef CAS.
- C. Young, R. Washenfelder, J. Roberts, L. Mielke, H. Osthoff, C. Tsai, O. Pikelnaya, J. Stutz, P. Veres, A. Cochran, T. VandenBoer, J. Flynn, N. Grossberg, C. Haman, B. Lefer, H. Stark, M. Graus, J. Gouw, J. Gilman, W. Kuster and S. Brown, Environ. Sci. Technol., 2012, 46, 10965–10973 CrossRef CAS.
- J. Zheng, X. Shi, Y. Ma, X. Ren, H. Jabbour, Y. Diao, W. Wang, Y. Ge, Y. Zhang and W. Zhu, Atmos. Chem. Phys., 2020, 20, 5457–5475 CrossRef CAS.
- J. Nan, S. Wang, Y. Guo, Y. Xiang and B. Zhou, Atmos. Environ., 2017, 154, 167–178 CrossRef CAS.
- J. Brean, R. Harrison, Z. Shi, D. Beddows, W. Acton, C. Hewitt, F. Squires and J. Lee, Atmos. Chem. Phys., 2019, 19, 14933–14947 CrossRef CAS.
- C. Han, W. Yang, Q. Wu, H. Yang and X. Xue, Environ. Sci. Technol., 2016, 50, 5017–5023 CrossRef CAS.
- M. Monge, B. D'Anna, L. Mazri, A. Giroir-Fendler, M. Ammann, D. Donaldson and C. George, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6605–6609 CrossRef CAS.
- M. Brigante, D. Cazoir, B. D'Anna, C. George and D. Donaldson, J. Phys. Chem. A, 2008, 112, 9503–9508 CrossRef CAS.
- C. Ye, N. Zhang, H. Gao and X. Zhou, Environ. Sci. Technol., 2017, 51, 6849–6856 CrossRef CAS PubMed.
- K. Stemmler, M. Ammann, C. Donders, J. Kleffmann and C. George, Nature, 2006, 440, 195–198 CrossRef CAS PubMed.
- R. Ammar, M. Monge, C. George and B. D'Anna, ChemPhysChem, 2010, 11, 3956–3961 CrossRef CAS PubMed.
- L. Li, Z. Duan, H. Li, C. Zhu, G. Henkelman, J. Francisco and X. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7236–7241 CrossRef CAS.
- D. Medeiros and A. Pimentel, J. Phys. Chem. A, 2011, 115, 6357–6365 CrossRef CAS PubMed.
- M. Martins-Costa, J. Anglada, J. Francisco and M. Ruiz-López, J. Am. Chem. Soc., 2020, 142, 20937–20941 CrossRef CAS.
- M. Gen, Z. Liang, R. Zhang, B. Mabato and C. Chan, Environ. Sci.: Atmos., 2022, 2, 111–127 CAS.
- F. Bao, M. Li, Y. Zhang, C. Chen and J. Zhao, Environ. Sci. Technol., 2018, 52, 6309–6316 CrossRef CAS.
- Z. Wang, J. Zhang, L. Zhang, Y. Liang and Q. Shi, Atmos. Environ., 2021, 246, 118046 CrossRef CAS.
- M. Mulder, Y. Dumanoglu, C. Efstathiou, P. Kukučka, J. Matejovičová, C. Maurer, P. Přibylová, R. Prokeš, A. Sofuoglu, S. Sofuoglu, J. Wilson, C. Zetzsch, G. Wotawa and G. Lammel, Environ. Sci. Technol., 2019, 53, 8914–8924 CrossRef CAS.
- Y. Wang, M. Hu, Y. Wang, J. Zheng, D. Shang, Y. Yang, Y. Liu, X. Li, R. Tang, W. Zhu, Z. Du, Y. Wu, S. Guo, Z. Wu, S. Lou, M. Hallquist and J. Yu, Atmos. Chem. Phys., 2019, 19, 7649–7665 CrossRef CAS.
- J. Ahad, R. Macdonald, J. Parrott, Z. Yang, Y. Zhang, T. Siddique, A. Kuznetsova, C. Rauert, E. Galarneau, W. Studabaker, M. Evans, M. McMaster and D. Shang, Environ. Pollut., 2020, 266, 114988 CrossRef CAS PubMed.
- D. Traversi, R. Degan, R. Marco, G. Gilli, C. Pignata, S. Villani and R. Bono, Environ. Int., 2009, 35, 905–910 CrossRef CAS.
- K. Ju and R. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS.
- P. Kovacic and R. Somanathan, J. Appl. Toxicol., 2014, 34, 810–824 CrossRef CAS.
- C. Salvador, R. Tang, M. Priestley, L. Li, E. Tsiligiannis, M. Breton, W. Zhu, L. Zeng, H. Wang, Y. Yu, M. Hu, S. Guo and M. Hallquist, Atmos. Chem. Phys., 2021, 21, 1389–1406 CrossRef CAS.
- A. Kroflič, J. Anders, I. Drventić, P. Mettke, O. Böge, A. Mutzel, J. Kleffmann and H. Herrmann, ACS Earth Space Chem., 2021, 5, 1083–1093 CrossRef.
- Z. Finewax, J. Gouw and P. Ziemann, Environ. Sci. Technol., 2018, 52, 1981–1989 CrossRef CAS.
- M. Harrison, S. Barra, D. Borghesi, D. Vione, C. Arsene and R. Olariu, Atmos. Environ., 2005, 39, 231–248 CrossRef CAS.
- S. Wang and H. Li, Environ. Sci. Technol., 2021, 55, 2899–2907 CrossRef CAS PubMed.
- R. Hems and J. Abbatt, ACS Earth Space Chem., 2018, 2, 225–234 CrossRef CAS.
- J. Liu, P. Lin, A. Laskin, J. Laskin, S. Kathmann, M. Wise, R. Caylor, F. Imholt, V. Selimovic and J. Shilling, Atmos. Chem. Phys., 2016, 16, 12815–12827 CrossRef CAS.
- C. Mohr, F. Lopez-Hilfiker, P. Zotter, A. Prévot, L. Xu, N. Ng, S. Herndon, L. Williams, J. Franklin, M. Zahniser, D. Worsnop, W. Knighton, A. Aiken, K. Gorkowski, M. Dubey, J. Allan and J. Thornton, Environ. Sci. Technol., 2013, 47, 6316–6324 CrossRef CAS PubMed.
- P. Lin, N. Bluvshtein, Y. Rudich, S. Nizkorodov, J. Laskin and A. Laskin, Environ. Sci. Technol., 2017, 51, 11561–11570 CrossRef CAS PubMed.
- F. Ikemori, T. Nakayama and H. Hasegawa, Atmos. Environ., 2019, 211, 91–102 CrossRef CAS.
- M. Claeys, R. Vermeylen, F. Yasmeen, Y. Gómez-González, X. Chi, W. Maenhaut, T. Mészáros and I. Salma, Environ. Chem., 2012, 9, 273–284 CrossRef CAS.
- M. Li, X. Wang, C. Lu, R. Li, J. Zhang, S. Dong, L. Yang, L. Xue, J. Chen and W. Wang, Sci. Total Environ., 2020, 714, 136760 CrossRef CAS.
- X. Li, L. Jiang, L. Hoa, Y. Lyu, T. Xu, X. Yang, Y. Iinuma, J. Chen and H. Herrmann, Atmos. Environ., 2016, 145, 115–127 CrossRef CAS.
- J. Luttke, V. Scheer, K. Levsen, G. Wünsch, J. Cape, K. Hargreaves, R. Storeton-West, K. Acker, W. Wieprecht and B. Jones, Atmos. Environ., 1997, 31, 2637–2648 CrossRef CAS.
- Y. Zhang, L. Müller, R. Winterhalter, G. Moortgat, T. Hoffmann and U. Pöschl, Atmos. Chem. Phys., 2010, 10, 7859–7873 CrossRef CAS.
- W. Yuan, R. Huang, L. Yang, T. Wang, J. Duan, J. Guo, H. Ni, Y. Chen, Q. Chen, Y. Li, U. Dusek, C. O'Dowd and T. Hoffmann, Atmos. Chem. Phys., 2021, 21, 3685–3697 CrossRef CAS.
- M. Harrison, M. Heal and J. Cape, Atmos. Chem. Phys., 2005, 5, 1679–1695 CrossRef CAS.
- Y. Zhang, L. Muller, R. Winterhalter, G. Moortgat, T. Hoffmann and U. Poschl, Atmos. Chem. Phys. Discuss., 2010, 10, 13253–13286 Search PubMed.
- M. Sangwan and L. Zhu, J. Phys. Chem. A, 2016, 120, 9958–9967 CrossRef CAS PubMed.
- M. Sangwan and L. Zhu, J. Phys. Chem. A, 2018, 122, 1861–1872 CrossRef CAS.
- J. Chen, J. Wenger and D. Venables, J. Phys. Chem. A, 2011, 115, 12235–12242 CrossRef CAS.
- I. Bejan, Y. Aal, I. Barnes, T. Benter, B. Bohn, P. Wiesen and J. Kleffmann, Phys. Chem. Chem. Phys., 2006, 8, 2028–2035 RSC.
- Y. Nitta, O. Schalk, H. Igarashi, S. Wada, T. Tsutsumi, K. Saita, T. Taketsugu and T. Sekikawa, J. Phys. Chem. Lett., 2021, 12, 674–679 CrossRef CAS PubMed.
- F. Barsotti, T. Bartels-Rausch, E. Laurentiis, M. Ammann, M. Brigante, G. Mailhot, V. Maurino, C. Minero and D. Vione, Environ. Sci. Technol., 2017, 51, 7486–7495 CrossRef CAS PubMed.
- W. Yang, D. You, C. Li, C. Han, N. Tang, H. Yang and X. Xue, Environ. Sci. Technol. Lett., 2021, 8, 747–752 CrossRef CAS.
- S. Cheng, C. Zhou, H. Yin, J. Sun and K. Han, J. Chem. Phys., 2009, 130, 234311 CrossRef PubMed.
- L. Vereecken, H. K. Chakravarty, B. Bohn and J. Lelieveld, Int. J. Chem. Kinet., 2016, 48, 785–795 CrossRef CAS.
- K. Andersson, P. Malmqvist, B. Roos, A. Sadlej and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- K. Andersson, P. Malmqvist and B. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- C. Xu, F. Gu and C. Zhu, Phys. Chem. Chem. Phys., 2018, 20, 5606–5616 RSC.
- C. Xu, L. Yu, C. Zhu, J. Yu and Z. Cao, Sci. Rep., 2016, 6, 26768 CrossRef CAS PubMed.
- C. Xu, L. Yu, C. Zhu and J. Yu, J. Phys. Chem. A, 2015, 119, 10441–10450 CrossRef CAS PubMed.
- G. Ghigo, B. Roos and P. Malmqvist, Chem. Phys. Lett., 2004, 396, 142–149 CrossRef CAS.
- N. Forsberg and P. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- T. Lu, The sobMECP Program, 2016, website: https://sobereva.com/286, date of access: 12/05/2022 Search PubMed.
- J. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95–99 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785–789 CrossRef.
- S. H. Yosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5848 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- M. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- M. Marques, C. Ullrich, F. Nogueira, A. Rubio, K. Burke and E. Gross, Time-dependent Density Functional Theory, Springer, Berlin, 2006 Search PubMed.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515 CrossRef CAS PubMed.
- G. Cui and W.-H. Fang, ChemPhysChem, 2011, 12, 1351–1357 CrossRef CAS PubMed.
- K. Takahashi, K. L. Plath, R. T. Skodje and V. Vaida, J. Phys. Chem. A, 2008, 112, 7321–7331 CrossRef CAS PubMed.
- A. W. Harrison, M. F. Shaw and W. J. De Bruyn, J. Phys. Chem. A, 2019, 123, 8109–8121 CrossRef CAS.
- N. E. Henriksen and F. Y. Hansen, Chapter 7: Unimolecular Reactions, Theories of Molecular Reaction Dynamics: the Microscopic Foundation of Chemical Kinetics, Oxford University Press, 2008 Search PubMed.
- T. Baer and W. L. Hase, Unimolecular Reaction Dynamics: Theory and Experiments, Oxford University Press, 1996 Search PubMed.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1927, 114, 243–265 CAS.
- V. Lawetz, G. Orlandi and W. Siebrand, J. Chem. Phys., 1972, 56, 4058–4072 CrossRef CAS.
- G. W. Robinson and R. P. Frosch, J. Chem. Phys., 1963, 38, 1187–1203 CrossRef CAS.
- E. Y. T. Li, T. Y. Jiang, Y. Chi and P.-T. Chou, Phys. Chem. Chem. Phys., 2014, 16, 26184–26192 RSC.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Belijonne, J. Cornil, C. Bazzini, T. Caronna, F. Meinardi, Z. Shuai and J.-L. Bredas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS.
- X.-F. Song, Z.-W. Li, W.-K. Chen, Y.-J. Gao and G. Cui, Inorg. Chem., 2022, 6161, 7673–7681 CrossRef.
- X.-F. Song, L.-Y. Peng, W.-K. Chen, Y.-J. Gao, W.-H. Fang and G. Cui, Chem.–Eur. J., 2022, e202201782 CAS.
- X.-W. Sun, L.-Y. Peng, Y.-J. Gao, Q. Fang and G. Cui, J. Phys. Chem. A, 2022, 126, 4176–4184 CrossRef CAS PubMed.
- K. Shizu and H. Kaji, J. Phys. Chem. A, 2021, 125, 9000–9010 CrossRef CAS PubMed.
- J. L. Steinfeld, J. S. Francisco and W. L. Hase, Chemical Kinetics and Dynamics, Prentice-Hall, 2nd edn, 1999 Search PubMed.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- E. Wigner, Z. Phys. Chem., Abt. B, 1932, 19, 203–216 Search PubMed.
- P. Grammaticakis, Bull. Soc. Chim. Fr., 1951, 18, 220–226 Search PubMed.
- R. Haddon, J. Am. Chem. Soc., 1980, 102, 1807–1811 CrossRef CAS.
- P. Nagy, Int. J. Mol. Sci., 2014, 15, 19562–19633 CrossRef CAS.
- K. Mackeprang, S. Schrøder and H. Kjaergaard, Chem. Phys. Lett., 2013, 582, 31–37 CrossRef CAS.
- J. Lađarević, B. Božić and L. Matović, et al. , Dyes Pigm., 2019, 162, 562–572 CrossRef.
- H. Ernst, T. Wolf, O. Schalk, N. Gonzalez-García, A. Boguslavskiy, A. Stolow, M. Olzmann and A. Unterreiner, J. Phys. Chem. A, 2015, 119, 9225–9235 CrossRef CAS.
- S. Caumo, M. Claeys, W. Maenhaut, R. Vermeylen, S. Behrouzi, M. Shalamzari and P. Vasconcellos, Atmos. Environ., 2016, 145, 272–279 CrossRef CAS.
- X. Li, L. Jiang, L. Hoa, Y. Lyu, T. Xu, X. Yang, Y. Iinuma, J. Chen and H. Herrmann, Atmos. Environ., 2016, 145, 115–127 CrossRef CAS.
- E. Eckstein, D. Perner, Ch. Brühl and T. Trautmann, Atmos. Chem. Phys. Discuss., 2002, 2, 1939–1977 Search PubMed.
- Y. Kanaya, Y. Kajii and H. Akimoto, Atmos. Environ., 2003, 37, 2463–2475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00053a |
| This journal is © The Royal Society of Chemistry 2023 |